

Abstract
On December 19, 2018, the U.S. Food and Drug Administration (FDA) granted approval to olaparib monotherapy for first‐line maintenance treatment of BRCA‐mutated (BRCAm) advanced ovarian cancer and, on May 8, 2020, expanded the indication of olaparib to include its use in combination with bevacizumab for first‐line maintenance treatment of homologous recombination deficient (HRD)–positive advanced ovarian cancer. Both these approvals were based on randomized, double‐blind, placebo‐controlled trials. Approval for olaparib monotherapy was based on the SOLO‐1 trial, comparing the efficacy of olaparib versus placebo in patients with BRCAm advanced ovarian, fallopian tube, or primary peritoneal cancer after surgical cytoreduction and first‐line platinum‐based chemotherapy. Two companion diagnostic (CDx) tests were approved with this indication: BRACAnalysis CDx, for germline BRCA1/2 alterations, and FoundationOne CDx, for BRCA1/2 alterations in tissue specimens. Approval for olaparib in combination with bevacizumab was based on the results of the PAOLA‐1 trial that compared olaparib with bevacizumab versus placebo plus bevacizumab in patients with advanced high‐grade epithelial ovarian cancer, fallopian tube, or primary peritoneal cancer after first‐line platinum‐based chemotherapy and bevacizumab. Myriad myChoice CDx was designated as a companion diagnostic device for use of olaparib plus bevacizumab combination for ovarian cancer associated with HRD‐positive status. Both trials demonstrated clinically meaningful improvements in progression‐free survival and favorable benefit‐risk profiles for the indicated populations. This article summarizes the FDA thought process and data supporting the approval of olaparib as monotherapy and in combination with bevacizumab for maintenance therapy in this setting.
Implications for Practice
These approvals represent the first poly (ADP‐ribose) polymerase inhibitor, alone or in combination with bevacizumab, approved in first‐line maintenance treatment of women with advanced ovarian cancer after cytoreductive surgery and chemotherapy. In patients with BRCA‐mutated tumors, olaparib monotherapy demonstrated a 70% reduction in the risk of disease progression or death compared with placebo, and olaparib in combination with bevacizumab demonstrated a 67% reduction in the risk of disease progression or death compared with bevacizumab alone in homologous recombination deficient–positive tumors. These approvals represent a major advance for the treatment of women with advanced ovarian cancer who are in complete or partial response after their initial platinum‐based chemotherapy.
Keywords: Olaparib, Ovarian cancer, PARP inhibitors, Olaparib in combination with bevacizumab
Short abstract
Despite the best standard of care for newly diagnosed advanced ovarian cancer, approximately 70% of women relapse within the first 3 years. This article presents the FDA's rationale for approval of olaparib for the maintenance therapy of patients with deleterious or suspected deleterious germline or somatic BRCA‐mutated advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first‐line platinum‐based chemotherapy.
Introduction
Ovarian cancer is the leading cause of death from gynecological cancers in the U.S., ranking as the fifth most common cause of cancer death in women [1]. In 2020, an estimated 21,750 new cases of ovarian cancer will be diagnosed in the U.S., and approximately 13,940 people will die from ovarian cancer [1]. Nearly 75% of women with ovarian cancer present with advanced disease (stage III or IV), and most of these will die from their disease, with 5‐year survival rates around 30% for advanced stages [2, 3].
Deleterious mutations of the breast cancer susceptibility gene (BRCA) 1 and BRCA2 are important risk factors for ovarian cancer [4]. BRCA1 and BRCA2 are tumor suppressor genes that encode proteins involved in repair of double‐stranded DNA breaks by homologous recombination [5]. Women with BRCA1/2 mutation have an estimated 15% to 40% lifetime risk of developing ovarian cancer and are more likely to develop ovarian cancer earlier in their life than those without the mutation [6]. Mutations in BRCA1/2 may be heritable (germline) or acquired during tumorigenesis (somatic). Data from The Cancer Genome Atlas Research Network and the medical literature suggest that approximately 25% of the overall population of patients with ovarian cancer have BRCA1/2 mutations at diagnosis, with ∼18% being germline and ∼ 7% somatic in origin [7, 8].
BRCA mutation disrupts the DNA repair pathway known as homologous recombination repair (HRR), which is used by tumor cells when the repair of single‐strand breaks is blocked by poly (ADP‐ribose) polymerase (PARP) inhibition [5]. PARP inhibitors take advantage of this susceptibility in homologous recombination deficient (HRD) tumor cells, leading to cell death through synthetic lethal interaction [5]. HRD is not limited to tumors with BRCA mutations and also includes other mechanisms of genomic instability, such as germline mutations, somatic mutations, and epigenetic modifications of other genes involved in the HRR pathway, thus supporting the biologic plausibility for efficacy of PARPs [5].
The majority of women with newly diagnosed advanced ovarian cancer undergo primary surgical cytoreduction of their disease, whereas women with extensive disease may receive neoadjuvant chemotherapy when up‐front complete resection of gross disease is not considered feasible. The extent of cytoreduction is prognostic: increased residual disease has been correlated with poorer survival outcomes [9]. Surgery is followed by adjuvant chemotherapy with a platinum agent in combination with a taxane, usually carboplatin, and paclitaxel [10]. The vascular endothelial growth factor inhibitor, bevacizumab, in combination with carboplatin and paclitaxel followed by bevacizumab as a single agent, was approved in first‐line and maintenance setting in the U.S. for treatment of patients with stage III or IV epithelial ovarian, fallopian tube, or primary peritoneal cancer after initial surgical resection. The approval was based on a 6.2‐month improvement in median progression‐free survival (PFS) (hazard ratio, 0.62; 95% confidence interval [CI], 0.52–0.75) for carboplatin and paclitaxel plus bevacizumab followed by bevacizumab maintenance versus chemotherapy alone in the GOG‐218 trial [11, 12]. Despite high initial response rates to standard‐of‐care treatment for newly diagnosed advanced ovarian cancer, approximately 70% of women relapse within the first 3 years [13].
Herein, we summarize key review findings that supported the two approvals (Table 1) of olaparib monotherapy and olaparib in combination with bevacizumab for two related but overlapping maintenance settings in patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first‐line platinum‐based chemotherapy: first, as monotherapy in patients with deleterious or suspected deleterious germline or somatic BRCA mutations, and second, in combination with bevacizumab, in patients whose cancer is associated with HRD‐positive status.
Table 1.
Olaparib: Background Information
| Variables | Information |
|---|---|
| Chemical name | 4‐[(3‐{[4‐(cyclopropylcarbonyl) piperazin‐1‐yl]carbonyl}‐4‐fluorophenyl)methyl]phthalazin‐1 (2H)‐one |
| Mechanism of action | Olaparib is an inhibitor of PARP enzymes, including PARP1, PARP2, and PARP3. |
| Other approvals |
December 19, 2014: As monotherapy in patients with deleterious or suspected deleterious germline BRCA‐mutated advanced ovarian cancer that have been treated with three or more prior lines of chemotherapy. August 17, 2017: As monotherapy for the maintenance treatment of adult patients recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete or partial response to platinum‐based chemotherapy. January 12, 2018: As monotherapy for the treatment of adult patients with gBRCAm HER2‐negative metastatic breast cancer who have previously been treated with chemotherapy. These patients could have received chemotherapy in the neoadjuvant, adjuvant, or metastatic setting. December 27, 2019: As monotherapy for the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first‐line platinum‐based chemotherapy regimen. May 19, 2020: As monotherapy for the treatment of adult patients with deleterious or suspected deleterious germline or somatic HRR gene‐mutated mCRPC who have progressed after prior treatment with enzalutamide or abiraterone. |
Abbreviations: FDA, U.S. Food and Drug Administration; gBRCAm, BRCA mutated in the germline; HRR, homologous recombination repair; mCRPC, metastatic castration‐resistant prostate cancer; PARP, poly (ADP‐ribose) polymerase.
Materials and Methods
Clinical Trial Designs
The supplemental New Drug Applications for olaparib monotherapy and olaparib in combination with bevacizumab for patients with ovarian cancer were supported by SOLO‐1 and PAOLA‐1, respectively. Each was a randomized, double‐blind, placebo‐controlled trial designed to demonstrate an improvement in PFS. Although these trials shared some common features, such as similar patient population, efficacy endpoints, and evaluation in the maintenance setting after response to first‐line platinum‐based therapy, they also had several differences (Table 2) [14].
Table 2.
Key features of the SOLO‐1 and PAOLA‐1 trial designs
| Trial name | Patient population studied | Key demographics | Biomarker subgroups | Key clinical characteristics | Randomization | Stratification factors | Treatment duration | Endpoints |
|---|---|---|---|---|---|---|---|---|
| SOLO‐1 (n = 391) | Serous or endometrioid carcinomas with BRCA mutation |
ECOG 0: 78% ECOG 1: 22% Stage III: 83% Stage IV: 17% |
gBRCA: 388/391 sBRCA: 2/391 BRCA1: 72% BRCA2: 27% Both BRCA1/2: 1.2% |
PSC: 63% IDS: 35% CR: 82% PR: 18% |
2:1 olaparib 300 mg BID maintenance vs. placebo | Response to first‐line platinum‐based chemotherapy | Until disease progression or up to 2 years |
Primary: PFS Secondary: OS |
| PAOLA‐1 (n = 806) | Serous or endometrioid carcinomas |
ECOG 0: 70% ECOG 1: 28% Stage III: 70% Stage IV: 30% |
HRD positive: 48% tBRCAm: 29% HRD negative: 34% HRD unknown: 18% |
NED at PSC: 32% NED/CR at IDS: 31% NED/CR with incomplete resection/no debulking surgery: 15% PR: 22% |
2:1 olaparib 300 mg BID + bevacizumab maintenance vs. placebo + bevacizumab maintenance | tBRCAm status determined by prospective local testing and first‐line treatment outcome (timing and outcome of cytoreductive surgery and response to platinum‐based chemotherapy) | Until disease progression or up to 2 years |
Primary: PFS Secondary: OS |
Abbreviations: BID, twice daily; CR, complete response; ECOG, Eastern Cooperative Oncology Group; gBRCA, germline BRCA; HRD, homologous recombination deficient; IDS, interval debulking surgery; NED, no evidence of disease; OS, overall survival; PFS, progression‐free survival; PR, partial response; PSC, primary surgical cytoreduction; sBRCA, somatic BRCA; tBRCAm, BRCA mutation detected in the tumor.
SOLO‐1 compared the efficacy of olaparib maintenance monotherapy with placebo in patients with BRCA‐mutated (BRCAm) advanced ovarian, fallopian tube, or primary peritoneal cancer who had responded after first‐line platinum‐based chemotherapy. After completion of platinum‐based chemotherapy (minimum of four and maximum of nine9 treatment cycles; patients did not receive bevacizumab, either in combination or as maintenance therapy), patients were randomized 2:1 to receive olaparib tablets 300 mg orally twice daily (n = 260) or placebo (n = 131) for up to 2 years or until disease progression or unacceptable toxicity; however, patients with evidence of disease at 2 years, who could derive further benefit from continuous treatment at the discretion of the treating clinician, were treated beyond 2 years. Randomization was stratified by response to first‐line platinum‐based chemotherapy (complete response [CR] vs. partial response [PR]). Patients enrolled in SOLO‐1 were tested with a Myriad BRACAnalysis test (Myriad, Salt Lake City, UT) to identify patients carrying a loss of function (deleterious or suspected deleterious) mutation in either BRCA1 or BRCA2. Tumor tissue was also requested for all randomized patients where possible, and retrospectively tested with FoundationOne CDx (F1CDx; Foundation Medicine, Inc. [FMI], Cambridge, MA) in a bridging study to support the companion diagnostic approval. The primary efficacy endpoint was PFS, defined as the time from randomization to the earliest date of objective progression (per RECIST criteria) as assessed by the investigator or death by any cause (in the absence of disease progression). This primary analysis of PFS was supported by the analysis of PFS assessed by blinded independent central review of patient scans. Overall survival (OS) was a secondary endpoint.
PAOLA‐1 compared the efficacy of olaparib plus bevacizumab (O+B) versus placebo plus bevacizumab (P+B) for the maintenance treatment of advanced high‐grade epithelial ovarian cancer, fallopian tube, or primary peritoneal cancer after first‐line platinum‐based chemotherapy and bevacizumab. Randomization stratification factors included first‐line treatment outcome (timing and outcome of cytoreductive surgery and response to platinum‐based chemotherapy) and tBRCAm (BRCA mutations detected in the tumor; may suggest presence of germline or somatic BRCA mutation) status, determined by prospective local testing. Myriad myChoice CDx was used to test all available clinical samples, retrospectively. Patients were required to have CR or PR after completion of first‐line platinum‐containing chemotherapy and bevacizumab or to have no evidence of disease (NED) because of complete surgical resection. Patients were randomized (2:1) to receive olaparib tablets 300 mg orally twice daily in combination with bevacizumab (n = 537) 15 mg/kg every 3 weeks or P+B (n = 269). Bevacizumab was continued in the maintenance setting and olaparib was started after a period of 3 to 9 weeks after completion of the last dose of chemotherapy. Olaparib treatment was continued for up to 2 years or until progression of the underlying disease or unacceptable toxicity. Treatment could be extended beyond 2 years if patients could derive further benefit from continuous treatment, based on the treating physician's opinion. Duration of treatment with bevacizumab totaled up to 15 months, including the period given with chemotherapy and as maintenance. The primary endpoint was investigator‐assessed PFS evaluated according to RECIST version 1.1, and OS was a secondary efficacy endpoint. Exploratory subgroup analyses based on stratification factors, predefined clinical characteristics, and biomarkers per Myriad myChoice HRD Plus test were conducted comparing PFS between treatments to further understand the consistency of olaparib treatment effect across potential predictive and prognostic factors.
After randomization, patients enrolled in PAOLA‐1 were tested using the Myriad myChoice HRD Plus clinical trial assay prior to database lock to identify whose cancer was associated with HRD‐positive status. All available clinical trial samples were retrospectively tested by the Food and Drug Administration (FDA)–approved Myriad myChoice CDx test after the completion of the clinical trial. Myriad myChoice CDx defines HRD‐positive status as deleterious or suspected deleterious mutations in BRCA1 and BRCA2 genes and/or positive genomic instability, which is calculated by the Genomic Instability Score (GIS). GIS is an algorithmic measurement of loss of heterozygosity, telomeric allelic imbalance, and large‐scale state transitions. A GIS ≥42 is considered positive for the HRD status, whereas a GIS <42 (biomarker negative) suggested that the homologous recombination pathway is not defective.
Assessment Aid
Both submissions were reviewed using the Assessment Aid (AAid), initially launched as an FDA Oncology Center of Excellence pilot in 2018 [15, 16]. The AAid is a voluntary submission from the applicant. Use of the AAid facilitates a streamlined review process and focuses the FDA's efforts on critical thinking and evaluation of the data while ensuring clear delineation of the results, the position of the applicant, and the assessment of the FDA.
Prior to submitting the application, the applicant receives a structured AAid template modeled after the FDA's Multidisciplinary Review. Each section of the AAid is separated into three parts—(a) the data, (b) the applicant's position, and (c) the FDA's assessment—and allows for objective presentation of the results (in the data part) and interpretation of these results by the applicant and the FDA. For marketing applications, the applicant completes the data and the applicant's position parts of each section in the template. During the review, the FDA evaluates the information provided by the applicant and, if indicated, conducts its own independent in‐depth analysis of efficacy and safety, including analysis of the provided patient‐level datasets, as is customary for FDA reviews. The FDA's analyses and assessments are then included in the FDA's assessment part of the AAid template, and any areas of agreement or disagreement with the applicant are discussed in detail.
Results
Patient and Treatment Characteristics
SOLO‐1 randomized 391 women from 15 countries (28% from the U.S.). Most patients had received six cycles of platinum‐based chemotherapy prior to randomization; carboplatin/paclitaxel was the most commonly reported previous chemotherapy regimens in both arms. Demographic and disease characteristics were balanced between the two arms. Most patients had no evidence of disease on imaging at study entry (n = 290), as well as a good performance status (Eastern Cooperative Oncology Group performance status 0; n = 305) in both arms. Of all patients, 82% were in complete response to their most recent platinum‐based regimen. The majority of patients in SOLO‐1 had germline BRCA mutation (n = 389), whereas two patients had somatic BRCA mutation, which was confirmed retrospectively using a next‐generation sequencing–based F1CDx test.
PAOLA‐1 randomized 806 women from 11 countries (no clinical sites in the U.S.). Demographic and baseline disease characteristics were balanced and comparable between both arms. All patients had received first‐line platinum‐based therapy and bevacizumab. First‐line treatment outcomes at screening indicated that 32% of patients in both arms had NED with complete macroscopic resection at initial debulking surgery, 31% of patients in both arms had NED/CR with complete macroscopic resection at interval debulking surgery, 15% of patients in both arms had NED/CR with either incomplete resection (at initial or interval debulking surgery) or no debulking surgery, and 22% of patients in both arms had a partial response. Thirty percent of patients in both arms had a deleterious mutation. Patients were not restricted by the surgical outcome, with 65% having complete cytoreduction at initial or interval debulking surgery and 35% having residual macroscopic disease. Of the 806 patients, 48% (n = 387) had HRD‐positive tumors, including 29% (n = 235) that were tBRCAm; 34% (n = 277) were HRD negative; and 18% (n = 142) had unknown HRD status.
Efficacy Results
Results from SOLO‐1 and PAOLA‐1 demonstrated that BRCAm patients treated with olaparib monotherapy and HRD‐positive patients treated with O+B, respectively, had a clinically meaningful improvement in investigator‐assessed PFS (Table 3).
Table 3.
Efficacy results from SOLO‐1 and PAOLA‐1 trials
| Trial name | Patient population | Maintenance treatment arms | PFS in indicated population | Companion diagnostic |
|---|---|---|---|---|
| SOLO‐1 | BRCAm newly diagnosed advanced ovarian cancer | Olaparib vs. placebo |
BRCAm population PFS NR vs. 13.8 months HR: 0.30 (0.23, 0.41) |
BRACAnalysis CDx and FoundationOne CDx |
| PAOLA‐1 | Newly diagnosed advanced ovarian cancer | O+B vs. P+B |
HRD‐positive population PFS 37.2 vs. 17.7 months HR: 0.33 (0.25, 0.45) |
Myriad myChoice CDx |
Abbreviations: BRCAm, BRCA mutated; CDx, companion diagnostic; HR, hazard ratio; HRD, homologous recombination deficient; NR, not reached; O+B, olaparib plus bevacizumab; P+B, placebo plus bevacizumab; PFS, progression‐free survival.
For SOLO‐1, the estimated hazard ratio equaled 0.30 (95% CI, 0.23–0.41; p<.0001) with a median PFS not reached in patients randomized to the olaparib arm and 13.8 months in patients randomized to the placebo arm (Fig. 1). At the time of the primary PFS analysis, OS data were immature with approximately 21% of patients having died. The data did not show any detrimental effect on OS. For this trial, clinical validation of the Myriad BRACAnalysis CDx test was established by comparing the mutation results and the associated clinical outcomes for the overall study population to the subset of 383 patients with confirmed germline BRCA status upon prospective or retrospective testing. The effectiveness of the FMI F1CDx test was based on a subset of 313 patients with ovarian cancer whose tumor tissue was confirmed to carry deleterious BRCA mutation (may suggest presence of germline or somatic BRCA mutation) status. The PFS results in patients with tumor BRCA mutations confirmed by FMI F1CDx and in patients with germline BRCA mutation confirmed Myriad BRACAnalysis CDx were consistent with the results of the overall study population.
Figure 1.
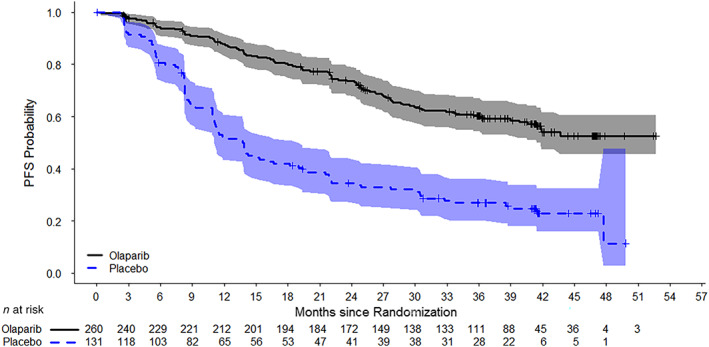
Kaplan‐Meier curves of investigator‐assessed progression‐free survival in SOLO‐1.Abbreviation: PFS, progression‐free survival.
The PAOLA‐1 trial enrolled a total of 806 patients (intention‐to‐treat [ITT] population). Although the ITT population demonstrated a statistical improvement in PFS, this improvement was primarily driven by the HRD‐positive subgroup (inclusive of all tBRCAm patients). In the HRD‐positive subgroup, the hazard ratio for PFS was 0.33 (95% CI, 0.25–0.45), with a median PFS of 37.2 months in the O+B arm compared with 17.7 months in the P+B arm (Fig. 2). At the time of the primary PFS analysis, OS data were immature with approximately 16% of patients having died. The data did not show any detrimental effect on OS among HRD‐positive patients. No benefit of PFS and OS was observed in the HRD‐negative subgroup.
Figure 2.
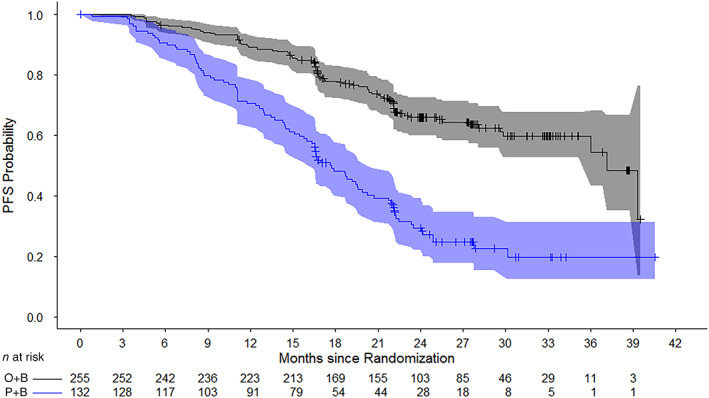
Kaplan‐Meier Curves of investigator‐assessed progression‐free survival in PAOLA‐1 (homologous recombination deficient–positive population).Abbreviations: O+B, olaparib + bevacizumab; P+B, placebo + bevacizumab; PFS, progression‐free survival.
Based on these results, the HRD‐positive population supported a favorable risk‐benefit profile for approval. FDA also approved the Myriad myChoice CDx (Myriad Genetic Laboratories, Inc.) as a companion diagnostic for O+B.
Safety Results
The primary safety population for SOLO‐1 consisted of all patients who received at least one dose of trial drug, olaparib or placebo (olaparib, n = 260; placebo, n = 130). The median total duration of exposure to olaparib was approximately two times longer than exposure to placebo (24.6 months vs. 13.9 months), consistent with the 2‐year treatment cap for olaparib‐treated patients and PFS for placebo‐treated patients (Table 4). Serious adverse events (AEs) occurred in 21% of the patients in the olaparib group and 12% of the patients in the placebo group. Dose interruptions because of AEs were reported in 52% and dose reductions were reported in 29% of patients treated with olaparib, compared with 17% and 3% of patients on placebo, respectively. Hematological AEs were the most common AEs leading to treatment interruptions or reductions. Treatment discontinuation because of AEs occurred in 12% of patients treated with olaparib compared with 2% of patients on placebo. The most common AEs leading to treatment discontinuation included fatigue, anemia, nausea, and vomiting. The most common grade 1–4 and grade 3–4 AEs in patients receiving olaparib included fatigue, anemia, and gastrointestinal disorders.
Table 4.
Safety results from SOLO‐1 and PAOLA‐1 trials
| Trial name | Patient population | Maintenance treatment arms | Median time on therapy (months) | Discontinuations because of AE | All grade AEs | Grade 3–4 AEs | On treatment deaths |
|---|---|---|---|---|---|---|---|
| SOLO‐1 (n = 391) | BRCAm newly diagnosed advanced ovarian cancer | Olaparib vs. placebo |
O: 24.6 P: 13.9 |
O: 12% P: 2% |
O: 99% P: 92% |
O: 30% P: 5% |
No deaths because of AEs while on study drug or within 30 days after last dose in either arm |
| PAOLA‐1 (n = 806) | Newly diagnosed advanced ovarian cancer | O+B vs. P+B |
O+B: 17.3 P+B: 15.6 |
O+B: 20% P+B: 6% |
O+B: 99% P+B: 96% |
O+B: 57% P+B: 50% |
O+B: 1 death because of aplastic anemia/ pneumonia P+B: 4 deaths (2 myocardial infarction, 1 intestinal perforation, 1 cardiovascular failure) |
Abbreviations: AE, adverse event; B, bevacizumab; BRCAm, BRCA mutated; O, olaparib; O+B, olaparib plus bevacizumab; P, placebo; P+B, placebo plus bevacizumab.
The primary safety population for PAOLA‐1 was based on 535 patients in the O+B arm and 267 patients in the P+B arm who received at least one dose of study treatment (Table 4). The median total treatment duration in the O+B arm (17.3 months) was longer than the median total treatment duration in the P+B arm (15.6 months), with the median duration of exposure to bevacizumab 11.0 months versus 10.4 months, respectively. A similar percentage of patients in the O+B arm experienced serious AEs compared with the P+B arm (31%, both arms). AEs leading to treatment discontinuation occurred in 20% of patients in the O+B arm compared with 6% on the P+B arm. In the O+B arm, 54% of patients required treatment interruptions because of AEs compared with 24% in the P+B arm, and dose reductions were 41% versus 7.5% in each arm, respectively. Hypertension was reported in a lower percentage of patients in the O+B arm (46%) compared with the P+B arm (60%). Similarly, grade 3–4 AEs of hypertension were lower in the O+B arm versus the P+B arm (19% vs. 30%, respectively). There were no sequelae of hypertension such as stroke or myocardial infarction that were more prevalent in the O+B arm. Venous thromboembolism was observed more commonly in patients receiving O+B versus those receiving P+B (5% vs. 1.9%), although this difference was minimal after adjusting for exposure. It is unclear at this time whether there is a mechanistic reason for this finding, and these results should be interpreted with caution given their exploratory nature.
Myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and pneumonitis have been identified as key adverse events of interest with olaparib [17]. Incidence of MDS/AML and pneumonitis from clinical trials enrolling single agent olaparib has been identified (by pooling olaparib monotherapy data) as 1.2% and 0.9%, respectively. In the SOLO‐1 trial, the incidence of MDS/AML was 1.2%, as three new cases of MDS/AML were identified in patients treated with olaparib (after discontinuation of therapy), and all cases resulted in a fatal outcome. In the PAOLA‐1 trial, the incidence of MDS/AML was 0.7%, as three new cases of AML (two of the cases had fatal outcome) and one case of MDS were identified in O+B arm, which is slightly lower than the overall percentage observed in pooled olaparib monotherapy studies (1.2%); however, PAOLA‐1 had shorter follow‐up compared with data from monotherapy studies. Pneumonitis/ interstitial lung disease (ILD) is also a risk associated with olaparib therapy, and the incidence of pneumonitis was 1.9%, as five patients developed pneumonitis/ILD on the olaparib arm of the SOLO‐1 trial. The incidence of pneumonitis was 1.1%, with six new cases in PAOLA‐1, and did not differ substantially from the pooled incidence of 0.9% reported in olaparib monotherapy studies.
Discussion
Olaparib was the first PARP inhibitor to receive FDA approval for the maintenance treatment, alone or in combination with bevacizumab, for patients with newly diagnosed advanced ovarian cancer, and represents a major change to the standard of care for adult patients undergoing initial therapy for ovarian cancer.
Although the SOLO‐1 trial was designed to enroll patients with both germline and somatic BRCAm tumors, only two patients with somatic BRCA mutations were accrued to the trial based on a local tissue test result, and the remainder of the patients had germline BRCA mutations (n = 389). Despite the small number of enrolled patients with somatic BRCA mutations, there is a biologic rationale that an underlying BRCA mutation results in HRR deficiency and confers sensitivity to PARP inhibition, irrespective of the mutation type (germline or somatic BRCA mutation) [18, 19]. In accordance with the FDA Guidance for Industry Developing Targeted Therapies in Low‐Frequency Molecular Subsets of a Disease, the SOLO‐1 trial grouped the patients with different molecular alterations (germline and somatic BRCA mutations) with a reasonable expectation that the grouped patients would have similar pharmacological responses based on a strong scientific rationale [20]. Because the results from the clinical trial demonstrated a favorable benefit‐risk profile, the FDA approved the drug for patients with BRCA‐mutated tumors regardless of somatic or germline origin. However, a postmarketing commitment was agreed upon to gather further evidence of clinical efficacy in patients who harbor a tBRCAm status from the PAOLA‐1 trial.
Compared with SOLO‐1, PAOLA‐1 included a wider representation of patients with advanced ovarian cancer, as selection was not based on BRCAm status. The trial demonstrated a clinically meaningful improvement in investigator‐assessed PFS for patients with HRD‐positive tumors (including all tBRCAm) randomized to O+B (median PFS, 37.2 months) versus P+B (median PFS, 17.7 months), with a hazard ratio of 0.33 (95% CI, 0.25–0.45). At the time of the primary PFS analysis, OS data were immature with approximately 16% of patients having died. The data did not show any detrimental effect on OS among HRD‐positive patients. No benefit of PFS and OS was observed in the HRD‐negative subgroup. Because the HRD unknown subgroup constitutes an unobserved mixture of HRD‐positive and ‐negative patients instead of a biologically distinct population, the HRD‐unknown population is not included in the indicated population. The differential efficacy between the HRD‐positive and HRD‐negative populations is biologically plausible based on the mechanism of action of olaparib and was not thought to be due to chance. Thus, the indication was restricted to only the HRD‐positive population, defined by presence of a deleterious or suspected deleterious BRCA mutation and/or genomic instability.
The PFS benefit observed with O+B in patients with tBRCAm tumors in the PAOLA‐1 appears consistent with the results reported in the SOLO‐1 and supported the clinical benefit of olaparib in BRCA‐mutated tumors regardless of somatic or germline origin. The toxicity profile of olaparib is well understood, with rare but significant risk for MDS/AML and pneumonitis/ILD events. The safety profile for O+B was generally consistent with that observed in SOLO‐1 and other monotherapy trials of olaparib, with the notable exception of hypertension, which is a known AE of bevacizumab.
Although the PFS benefit observed in SOLO‐1 and PAOLA‐1 represents a clinically meaningful improvement for maintenance therapy and is a major shift from the prevailing standard, the impact of this change on the natural history of ovarian cancer and responsiveness to later‐line therapies remain to be seen. Although mature overall survival analyses are postmarketing commitments for these two approvals, postprogression therapies that will likely include PARP inhibitors may affect the interpretation of these results.
For these applications, we employed a new regulatory tool, the AAid, to streamline the review while supporting our standard in‐depth analyses. Performing the review under the AAid pilot allowed the FDA to focus the written portion of the review on critical thinking and improved review efficiency by means of a shared document that outlined the applicant's positions as well as the in‐depth independent analyses performed by our review team. The use of the AAid streamlined the written product and contributed to a more efficient review process, and FDA was able to grant both these approvals on an expedited review clock.
Conclusion
Olaparib monotherapy and the combination of O+B represent a new treatment paradigm for patients with patients with newly diagnosed advanced ovarian cancer (Table 5). Although the SOLO‐1 trial primarily included patients carrying a germline BRCA mutation, there is a biologic rationale that an underlying BRCA mutation (germline or somatic) results in HRR deficiency and confers sensitivity to PARP inhibition, irrespective of the mutation type; therefore, the indication was granted for patients with both germline and somatic BRCA mutations. The BRACAnalysis CDx and the F1CDx tests were designated as companion diagnostic devices for the safe and effective use of olaparib monotherapy in these patient populations.
Table 5.
SOLO‐1 and PAOLA‐1 Food and Drug Administration benefit‐risk analysis
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
Ovarian cancer is the fifth most common cause of cancer death in U.S. women. Seventy‐five percent of patients present with advanced disease at diagnosis (stage III or IV), and most patients die from their disease, with 5‐year survival rates of only 29% for advanced stages. Patients with advanced ovarian cancer undergo surgical cytoreduction followed by platinum‐based doublet chemotherapy, but a majority of patients relapse within the first 3 years. Maintenance treatment after completion of platinum‐based chemotherapy may delay the need for subsequent treatment and may reduce cumulative toxicity. |
Advanced ovarian cancer is a serious and life‐threatening disease with a significant unmet medical need for more effective therapies. |
| Current treatment options a |
Cytoreductive surgery and platinum‐based chemotherapy are treatments of choice for newly diagnosed advanced ovarian cancer. Most patients with newly diagnosed advanced ovarian cancer achieve CR after first‐line treatment; however, 70% relapse within the first 3 years of diagnosis, and their disease becomes largely incurable. Bevacizumab in combination with carboplatin and paclitaxel followed by bevacizumab as maintenance is the first biological treatment to be approved in the first‐line ovarian cancer treatment setting. |
There is an unmet medical need to improve the outcomes of newly diagnosed patients with advanced ovarian cancer after first‐line treatment. |
| Benefit |
SOLO‐1 The clinical data from the randomized, double‐blind, placebo‐controlled SOLO‐1 trial, conducted in patients with advanced ovarian cancer with BRCA mutation who had responded to first‐line platinum‐based chemotherapy (complete or partial response), demonstrated a 70% reduction in the risk of disease progression or death (HR, 0.30; 95% CI, 0.23–0.41) for olaparib compared with placebo. The estimated median PFS in the olaparib arm was not reached compared with 13.8 months for the placebo arm. PAOLA‐1 The efficacy of olaparib in combination with bevacizumab as first‐line maintenance treatment of advanced ovarian cancer was evaluated in the randomized, double‐blind, placebo‐controlled PAOLA‐1 study, which compared the efficacy of olaparib in combination with bevacizumab vs. placebo plus bevacizumab. There was a PFS improvement in the ITT population of all‐comers (HR, 0.59; 95% CI, 0.49–0.72) The HR for PFS was 0.33 (95% CI, 0.25–0.45) in the HRD‐positive subpopulation, a prespecified subgroup consisting of 387 patients with HRD‐positive tumors identified after randomization using the Myriad myChoice HRD Plus assay. |
SOLO‐1 Evidence of effectiveness was supported by a clinically meaningful improvement in PFS. PAOLA‐1 Evidence of effectiveness in patients with HRD‐positive tumors was demonstrated for olaparib in combination with bevacizumab by a clinically meaningful improvement in PFS compared with bevacizumab monotherapy in patients receiving first‐line maintenance treatment for advanced ovarian cancer. There was no evidence of benefit and some evidence for potential harm in patients who were HRD‐negative or had unknown HRD status. Olaparib in combination with bevacizumab will not be indicated for these patients. |
| Risk and risk management |
SOLO‐1 The most common adverse reactions experienced by at least 20% of patients in the SOLO‐1 study included anemia, abdominal pain, dizziness, nausea, fatigue (including asthenia), vomiting, neutropenia, leukopenia, nasopharyngitis/upper respiratory tract infection/influenza, respiratory tract infection, diarrhea, arthralgia/myalgia, dysgeusia, headache, dyspepsia, decreased appetite, constipation, and stomatitis. PAOLA‐1 The most common adverse events experienced by at least 20% of patients in PAOLA‐1 included fatigue, nausea, anemia, lymphopenia, and vomiting. Pneumonitis and MDS/AML occurred at similar incidences to that seen in the monotherapy population. Venous thromboembolism occurred more commonly in the olaparib/bevacizumab arm than the placebo/bevacizumab arm; however, the exposure‐adjusted incidences were similar. Labeling details dose interruption, reduction, or discontinuation for SOLO‐1 and PAOLA‐1. MDS/AML and pneumonitis are described in the Warnings and Precautions section of the label. Laboratory and vital sign monitoring are recommended before and during treatment. |
The safety profile of olaparib is acceptable for the intended population. Venous thromboembolism is described in the Adverse Reactions section of the label. MDS/AML and pneumonitis are the adverse reactions described in the Warnings and Precautions sections of the label. The safe use of olaparib can be managed through accurate labeling and routine oncology care. |
Olaparib monotherapy was approved as maintenance therapy for patients with advanced BRCA‐mutated ovarian cancer in complete or partial response to platinum‐based chemotherapy based on the SOLO‐1 trial at the time of PAOLA‐1 review.
Abbreviations: AML, acute myeloid leukemia; CI, confidence interval; CR, complete response; HR, hazard ratio; HRD, homologous recombination deficient; ITT, intention‐to‐treat; MDS, myelodysplastic syndrome; PFS, progression‐free survival.
Given the results observed in PAOLA‐1 as well as the biologically plausibility of effect only in HRD‐positive subgroup, O+B was granted the indication for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first‐line platinum‐based chemotherapy and whose cancer is associated with HRD‐positive status defined via presence of a deleterious or suspected deleterious BRCA mutation and/or genomic instability. Myriad myChoice CDx was designated as the companion diagnostic device for the safe and effective use of O+B combination therapy.
Olaparib alone and in combination with bevacizumab each have an acceptable safety profile, and safe use can be managed through accurate labeling and routine oncology care.
Author Contributions
Conception/design: Shaily Arora, Sanjeeve Balasubramaniam
Collection and/or assembly of data: Shaily Arora, Sanjeeve Balasubramaniam, Hui Zhang, Tara Berman, Preeti Narayan, Daniel Suzman, Erik Bloomquist, Shenghui Tang, Yutao Gong, Francisca Reyes Turcu, Deb Chatterjee, Banu Saritas‐Yildirim, Soma Ghosh
Data analysis and interpretation: Shaily Arora, Sanjeeve Balasubramaniam, Hui Zhang, Tara Berman, Preeti Narayan, Daniel Suzman, Erik Bloomquist, Shenghui Tang, Yutao Gong, Rajeshwari Sridhara, Francisca Reyes Turcu, Deb Chatterjee, Banu Saritas‐Yildirim, Soma Ghosh, Reena Philip, Anand Pathak, Jennifer J. Gao, Laleh Amiri‐Kordestani, Richard Pazdur, Julia A. Beaver
Manuscript writing: Shaily Arora, Sanjeeve Balasubramaniam, Hui Zhang, Tara Berman, Preeti Narayan, Daniel Suzman, Erik Bloomquist, Shenghui Tang, Yutao Gong, Rajeshwari Sridhara, Francisca Reyes Turcu, Deb Chatterjee, Banu Saritas‐Yildirim, Soma Ghosh, Reena Philip, Anand Pathak, Jennifer J. Gao, Laleh Amiri‐Kordestani, Richard Pazdur, Julia A. Beaver
Final approval of manuscript: Shaily Arora, Sanjeeve Balasubramaniam, Hui Zhang, Tara Berman, Preeti Narayan, Daniel Suzman, Erik Bloomquist, Shenghui Tang, Yutao Gong, Rajeshwari Sridhara, Francisca Reyes Turcu, Deb Chatterjee, Banu Saritas‐Yildirim, Soma Ghosh, Reena Philip, Anand Pathak, Jennifer J. Gao, Laleh Amiri‐Kordestani, Richard Pazdur, Julia A. Beaver
Disclosures
The authors indicated no financial relationships.
Acknowledgments
Dr. Sanjeeve Balasubramaniam completed this work while employed at the FDA and is currently affiliated with BeiGene.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Cancer Statistics Center . American Cancer Society Web site. Updated 2020. Available at https://cancerstatisticscenter.cancer.org/. Accessed May 11, 2020.
- 2. Hennessy BT, Coleman RL, Markman M. Ovarian cancer. Lancet 2009;374:1371–1382. [DOI] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 4. Miki Y, Swensen J, Shattuck‐Eidens D et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- 5. Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol 2016;27(suppl 1):i40–i44. [DOI] [PubMed] [Google Scholar]
- 6. Kuchenbaecker KB, Hopper JL, Barnes DR et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017;317:2402–2416. [DOI] [PubMed] [Google Scholar]
- 7. Pennington KP, Walsh T, Harrell MI et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 2014;20:764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Polterauer S, Vergote I, Concin N et al. Prognostic value of residual tumor size in patients with epithelial ovarian cancer FIGO stages IIA‐IV: Analysis of the OVCAD data. Int J Gynecol Oncol 2012;22:380–385. [DOI] [PubMed] [Google Scholar]
- 10. National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology: Ovarian Cancer, Including Fallopian Tube Cancer and Primary Peritoneal Cancer. Version 1.2020. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2020. Available at https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf. Accessed May 11, 2020.
- 11. Burger RA, Brady MF, Bookman MA et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 2011;365:2473–2483. [DOI] [PubMed] [Google Scholar]
- 12. Avastin [package insert] . South San Francisco, CA: Genentech, Inc., June 2019.
- 13. Ledermann JA, Raja FA, Fotopoulou C et al.; ESMO Guidelines Working Group . Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2013;24(suppl 6):vi24–vi32. [DOI] [PubMed] [Google Scholar]
- 14. Lynparza [package insert] . Wilmington, DE: AstraZeneca Pharmaceuticals LP, May 2020.
- 15. Oncology Center of Excellence : Assessment Aid. U.S. Food and Drug Administration Web site. Available at https://www.fda.gov/about-fda/oncology-center-excellence/assessment-aid-pilot-project. Accessed June 8, 2019.
- 16. Blumenthal G, Cheng J, Couvillon K et al. Real‐Time Oncology Review and the Assessment Aid: Increasing Review Efficiency Through Standardization and Earlier Data Access. White paper. Washington, DC: Friends of Cancer Research, August 2018. Available at https://www.focr.org/sites/default/files/pdf/ROTR%20White%20Paper%201.pdf. Accessed June 8, 2019.
- 17. Kim G, Ison G, McKee AE et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA‐mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 2015;21:4257–4261. [DOI] [PubMed] [Google Scholar]
- 18. Dougherty BA, Lai Z, Hodgson DR et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high‐grade serous ovarian cancers in the maintenance setting. Oncotarget 2017;8:43653–43661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hennessy BT, Timms KM, Carey MS et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP Ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol 2010;28:3570–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. U.S. Food and Drug Administration . Developing Targeted Therapies in Low‐Frequency Molecular Subsets of a Disease: Guidance for Industry. Rockville, MD: U.S. Food and Drug Administration, October 2018. Available at https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/developing‐targeted‐therapies‐low‐frequency‐molecular‐subsets‐disease. Accessed June 8, 2019. [Google Scholar]