

Abstract
Transmission of arthropod-borne viruses (arboviruses) involves infection and replication in both arthropod vectors and vertebrate hosts. Nearly all arboviruses are RNA viruses with high mutation frequencies, which leaves them vulnerable to genetic drift and fitness losses owing to population bottlenecks during vector infection, dissemination from the midgut to the salivary glands and transmission to the vertebrate host. However, despite these bottlenecks, they seem to avoid fitness declines that can result from Muller’s ratchet. In addition, founder effects that occur during the geographic introductions of human-amplified arboviruses, including chikungunya virus and Zika virus, can affect epidemic and endemic circulation, as well as virulence. In this Review, we discuss the role of genetic drift following population bottlenecks and founder effects in arboviral evolution and spread, and the emergence of human disease.
Subject terms: Viral evolution, Evolution, Viral vectors, Viral transmission
In this Review, Weaver and colleagues discuss the role of genetic drift following population bottlenecks and founder effects in mosquito-borne arboviral evolution and spread, and the emergence of human disease, focusing on chikungunya virus and Zika virus.
Introduction
Arthropod-borne viruses (arboviruses) are transmitted to vertebrates by arthropod vectors (mainly mosquitoes and ticks)1 (Fig. 1). Most of the more than 500 known arboviruses are zoonotic agents that use wild animals as amplification and/or reservoir hosts (Box 1). A small fraction of these (about 100) are human-pathogenic and cause disease via ‘spillover’ infections (that is, enzootic or bridge vectors bite humans following infection from an enzootic vertebrate host). Notable examples include West Nile virus (WNV) and Japanese encephalitis virus, which use avian hosts but can be highly virulent when humans are infected by Culex spp. mosquito vectors. However, four arboviruses with non-human primate enzootic hosts have initiated human-amplified transmission cycles that last from months to centuries, which increases their ability to infect humans and to cause human disease. These include dengue virus (DENV), which infects nearly 400 million humans annually throughout many tropical and subtropical regions and causes disease in nearly 100 million individuals, thus making DENV the most important arbovirus2; and yellow fever virus (YFV), which despite the availability of an effective vaccine for nearly a century still infects tens of thousands of humans annually, with recent epidemics in Brazil3, Nigeria4 and Angola5. In addition, the past 15 years have brought unexpected outbreaks caused by two other human-amplified arboviruses, namely chikungunya virus (CHIKV) and Zika virus (ZIKV). Like DENV and YFV, these viruses are transmitted by the anthropophilic mosquitoesAedes (Stegomyia) aegypti and sometimes Aedes (Stegomyia) albopictus. The expansion of these mosquitoes from Africa (A. aegypti) or Asia (A. albopictus) increased the disease burden throughout much of the tropics and permitted transmission in temperate regions (A. albopictus)6. However, our understanding of the emergence mechanisms remains incomplete. For example, why was CHIKV relatively inactive in the urban setting between the 1960s and 2005, and why were no ZIKV outbreaks documented before 2007? The ability of all arboviruses to survive and circulate despite adverse weather conditions (for example, droughts or winters when vectors and sometimes vertebrate hosts are inactive or limited in number) remains largely enigmatic, although current research is beginning to shed light on overwintering mechanisms7–9.
Fig. 1. Infection and transmission of arthropod-borne viruses by mosquitoes and bottlenecks.

Arthropod-borne viruses (arboviruses) undergo repeated bottlenecks during vector infection, dissemination from the midgut to the salivary glands and transmission to the vertebrate host. All of these population bottlenecks can present fitness challenges for the virus and may result in drift related to founder effects at each step outlined below. Infection of the mosquito midgut occurs upon ingestion of an infectious bloodmeal (steps 1, 2). Depending on the virus and the mosquito, the number of epithelial cells infected varies greatly (typically between 10 and most of the cells in the single layer of midgut epithelial cells)119. The severity of the midgut entry bottleneck seems to primarily depend on the titre of the initial infection and the genetic diversity of the virus population, as reductions in titre and diversity both result in severe reduction in the population to as little as 1–10 virus particles that found the next population following dissemination. Following infection and replication in the midgut cells, the arbovirus disseminates into the haemocoel (or open body cavity) (step 3). Then, access is gained to diverse secondary target organs and tissues such as the fat body, neurons and muscles, which support replication and may enhance infection of the salivary glands. Dissemination into the haemocoel is considered the major bottleneck during mosquito infection. The mechanism remains obscure, but generally the virus infects the midgut epithelium and then inefficiently disseminates into the haemocoel, either through microperforations in the basal lamina or during basal lamina remodelling after blood feeding. Infection of the salivary glands is essential for transmission, and evidence suggests that there is a small bottleneck at this stage, probably because the arbovirus must pass through a basal lamina surrounding this organ (step 4). However, the impact and severity of this bottleneck remains to be determined. Following replication in the salivary gland acinar cells and shedding into the apical cavities, transmission can occur during a subsequent bloodmeal when saliva is injected during mosquito probing and feeding on blood vessels (step 5). The severity of this virus population bottleneck has been estimated to average between approximately 10 infectious units for some alphaviruses and over 104 infectious units for West Nile virus. However, most of these estimates come from in vitro saliva collection, and few have been confirmed experimentally during in vivo transmission. Both dissemination from the midgut and infection of the salivary glands necessitate passage through basal laminae, predominantly consisting of collagen IV and laminin. The midgut basal lamina seems to become permissive for virus passage during bloodmeal digestion120 and stretching during a subsequent bloodmeal can create microperforations that seem to enhance dissemination46, but passage through the salivary gland basal lamina remains poorly understood.
Nearly all arboviruses have RNA genomes that replicate in the absence of error correction mechanisms, and thus they exhibit high mutation frequencies in the range of 10−3 to 10−5 per nucleotide replicated10. Populations of RNA viruses exist as mutant swarms (Fig. 2), often selected at the population level as quasispecies. These swarms comprise a range of mutants (representing intra-host variation) that are dominated by a master sequence, which is typically characterized by high fitness in the current replicative environment. These mutant distributions are thought to enable RNA viruses to rapidly adapt to changing environments, which sometimes results in the emergence of epidemic and/or epizootic disease. Despite their potential for rapid evolution, many arboviruses exhibit high degrees of consensus genome sequence stability in nature, which may reflect, in part, the requirement to maintain fitness for infection of divergent vertebrate and arthropod hosts, and may involve fitness trade-offs11. However, studies of some arboviruses such as WNV fail to support this trade-off hypothesis12. Alternative views include that the different host replicative environments are sufficiently similar to avoid a ‘trade-off’ or that one host may dominate as the driving force in arbovirus evolution13.
Fig. 2. The impacts of founder effects and population bottlenecks on the evolution of human-amplified arthropod-borne viruses.

Schematic depicting diverse viral populations (mutant swarms) that circulate. These swarms comprise a range of mutants that are dominated by a master sequence, which is typically characterized by high fitness in the current replicative environment. An infected human traveller initiates a founder effect by introducing a virus into a new geographic region, which results in a new population with randomly derived genetic variants, some of which may have reduced fitness depending on whether the master sequence is eliminated stochastically. Following population expansion during viraemia in the human host, further bottlenecks occur during infection of the vector and dissemination in the vector, as well as during transmission to another human host. Founder effects and other bottlenecks lead to the loss of genetic variation and fitness declines that can result from Muller’s ratchet, as random mutations become sequentially fixed in the population in the absence of efficient recombination to restore master sequences without infrequent direct reversions18.
One critical challenge to arbovirus survival is represented by sequential anatomic barriers, especially in the vector (Fig. 1). These anatomic barriers to viral dissemination result in repeated population bottlenecks, which strongly reduce the number (to as little as one) of virus particles available to maintain infection and permit transmission14.
Population bottlenecks during the transmission cycle combined with the high mutation frequencies suggest that arboviruses undergo major challenges to maintain their fitness. Efficient natural selection (positive; or, more typically, purifying selection to remove random and potentially deleterious mutations) usually dominates virus evolution when populations are large. By contrast, reductions in virus populations reduce the efficiency of selection, which can result in stochastic changes in mutant frequencies and loss of genetic variation (genetic drift). Such population reductions can occur when arboviruses orally infect or disseminate inefficiently within the vector prior to transmission, or during transmission of small populations to the vertebrate host (Fig. 1).
Reductions in virus populations can also manifest at the level of infected hosts when viruses are transported by a single vector or amplifying host (the latter seems to be much more common for urban arboviruses) to initiate point-source transmission. This scenario, where a founder population is established by a small number of individuals from a larger, ancestral population, can result in a loss in genetic variation and even the fixation of random mutations, termed a founder effect15–17 (Fig. 2). Therefore, founder effects typically represent a population bottleneck, although some definitions of the latter focus exclusively on the random extermination of most of a population. Along with the loss of genetic variation that accompanies a founder effect comes a stochastic sampling of the founder genomes that establish the new population, many of which may, by chance, include deleterious or neutral mutations that become fixed in the new population in the absence of strong natural selection. Founder effects, therefore, are a form of genetic drift, whereby the frequency of a given genotype in a population changes due to stochastic sampling rather than due to selection. Each individual vector infection event and transmission to a vertebrate host can be thought of as a founder event. However, in the case of many parallel chains of transmission, as is typical of endemic–enzootic or epidemic–epizootic transmission, these events probably have far less influence compared with a new geographic introduction that begins with a single transmission chain.
Despite the apparent benefits for RNA viruses to respond to changing fitness landscapes, high mutation frequencies may leave arboviruses vulnerable to drift and fitness declines following repeated population bottlenecks due to the phenomenon termed Muller’s ratchet18 (Fig. 2). Without efficient recombination or re-assortment mechanisms, repeated bottlenecks may result in the stepwise accumulation of deleterious mutations in a ratchet-like manner. Each of these mutations has a low probability of direct reversion unless the population size remains above ~104 (the reciprocal of the mutation frequency for a given nucleotide). Muller’s ratchet has been demonstrated to occur experimentally with several arboviruses19,20, which underscores the risk they face if they are subjected to repeated bottlenecks during their transmission cycle (requiring replication and spread in divergent hosts)21.
In this Review, we discuss viral population genetic factors that have recently been shown to be involved in arboviral spread and emergence of human disease. Adaptive evolution has been the focus of several recent, well-publicized examples of arboviral emergence22–25. However, genetic drift has been far less investigated and has received less attention despite its potential to have an equally strong impact on arboviral spread and disease emergence. Therefore, we discuss new evidence — best exemplified by recent studies of CHIKV and ZIKV — that genetic drift following founder effects accompanying geographic introductions, and other population bottlenecks (Fig. 2), can also have dramatic influences on arboviral epidemics and disease. Although we focus on horizontal transmission, many arboviruses also undergo vertical, trans-ovarial transmission, which may also influence their evolution (but this is not discussed in this Review).
Box 1 Arbovirus transmission cycles and emergence.
Arbovirus transmission involves two ecologically and genetically distinct cycles: the enzootic cycle (often called the sylvatic cycle) and the human-amplified cycle (often called the urban cycle) (see the figure). Zoonotic viruses, such as West Nile virus (WNV) and western equine encephalitis virus (WEEV) (see the figure, part a), use various wild animals as enzootic hosts, with spillover infections affecting humans. Some viruses, such as Venezuelan equine encephalitis virus (VEEV) (see the figure, part b), undergo secondary amplification involving domesticated animals, which increases spillover to humans in agricultural settings. Several of the most medically important viruses, including dengue virus (DENV), chikungunya virus (CHIKV), yellow fever virus (YFV) and Zika virus (ZIKV), use humans as direct amplification hosts (see the figure, part c), resulting in endemic and/or epidemic transmission and sometimes severe outbreaks.
For mosquito-borne viruses such as ZIKV, DENV and CHIKV, extant, ancestral virus strains cycle between various arboreal Aedes mosquito species and non-human primates (NHPs)6, and remain active in several parts of Africa and Southeast Asia (Asian enzootic transmission demonstrated only for DENV). During the human cycle, viruses circulate between humans (amplification hosts), and peridomestic Aedes albopictus and domestic Aedes aegypti mosquitoes (see the figure, part c). Other Aedes mosquito species, mostly belonging to the subgenus Stegomyia, may vector these viruses in niche geographic regions within this endemic and/or epidemic cycle.
In rural areas of Africa and Asia, also known as the ‘zone of emergence’, these viruses can transferred between NHPs and humans through direct spillover, whereby an enzootic or a bridge vector transmits the virus from an enzootic host to a human. Under appropriate conditions, this can result in a transition from the enzootic cycle to a human–mosquito–human cycle (see the figure, part c).
Arboviruses with this potential for human-amplified transmission are among the most important for public health, such as ZIKV and CHIKV. They are likely to have emerged from the forest, associated with clearing of forests and development of human settlements. On the basis of phylogenetic analyses and detection in NHPs and A. aegypti mosquitoes, ZIKV spread to Asia at least decades ago99,110,122. By contrast, CHIKV is believed to have spread to Asia and the Americas centuries ago through the sailing ship trade6. The first ZIKV outbreak outside Africa was detected in Micronesia in 2007, followed by another outbreak in French Polynesia in 2013, before being introduced into South America and reaching pandemic levels (reviewed in ref.123).
The historical emergence of CHIKV from Africa (from the East/Central/South African (ECSA) enzootic lineage) has been traced through phylogenetics to South and Southeast Asia, where the emergent Asian strain continues to circulate and was the source of a recent introduction into the Americas124. Currently, a distinct ECSA strain introduced directly into Brazil co-circulates with Asian lineage strains in South America125. A second recent emergence from Africa (Indian Ocean lineage (IOL)) affected the islands of the Indian Ocean, India, Southeast Asia and Europe. Spread of all epidemic strains involved transmission mainly by A. aegypti, except for IOL strains that in some locations have adapted through a series of envelope glycoprotein substitutions for efficient transmission by A. albopictus126.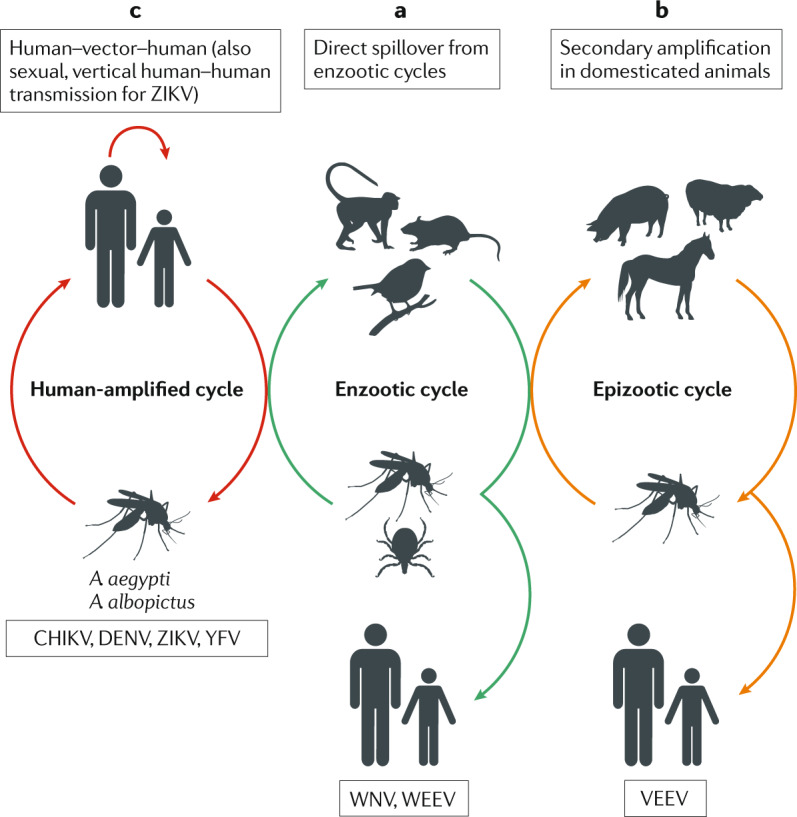
Bottlenecks during transmission
Infection of and dissemination in the vector
Infection of the mosquito vector is a driving force in arbovirus evolution, given the persistent nature of the infection as well as the length of time it takes for the virus to disseminate from the site of primary infection to the salivary glands. It has long been understood that the anatomy of the mosquito, in particular the basal laminae that surround the midgut, salivary glands and other organs, imposes bottlenecks on arboviral populations (Fig. 1). Thus, there has been great interest in the response of arboviruses to these bottlenecks and how they overcome them for transmission. We discuss three critical stages of infection with regard to the bottlenecks: infection of the mosquito midgut; dissemination into the haemocoel; and infection of the salivary glands.
Infection of the midgut represents the first bottleneck encountered by arboviruses (Fig. 1). This bottleneck was first thought to represent an anatomical barrier, a notion supported by early work on the transmission of WNV by Culex pipiens quinquefasciatus26 and Venezuelan equine encephalitis virus (VEEV) by Aedes (Ochlerotatus) taeniorhynchus27, showing infection of only a subset of epithelial cells. Whether this small number of infected cells resulted from differences or changes in cell susceptibility, even though most are absorptive/digestive enterocytes with common functions, or inefficient entry into the small number of these cells where infection is initiated, remains to be determined. However, recent work with the enzootic subtype IE VEEV vector Culex taeniopus suggests the latter. When infection of this highly susceptible vector occurs by a high-titre bloodmeal, most cells become infected, which is a substantial departure from previous work28. However, these findings may reflect the long-term co-evolution of enzootic subtype IE VEEV–C. taeniopus compared with more recent WNV–vector interactions in North America, and may therefore represent an outlier for human pathogenic arboviruses. Patterns and efficiency of vector infection by different arbovirus taxa (for example, alphaviruses compared with flaviviruses) may also differ. Further work examining multiple arboviruses including co-evolved virus–vector systems is needed to clarify these potential differences. Similar studies have not been carried out with DENV, ZIKV or CHIKV, but the few studies of midgut infections suggest that there is great variation among the virus–vector pairings as to the number of cells initially infected29,30. Studies investigating the midgut infection barrier using reporter-marked viral clones show that changes in the titre of the bloodmeal have a major impact on the severity of the bottleneck14. A limitation of all these studies is that these infections are generally initiated using high oral doses; yet, although the susceptibility of the midgut is important, the lower titres typical of natural infection are likely to reduce the number of infected cells during a typical infection, thus substantially increasing the bottleneck in the midgut during initial infection. This view is supported by studies examining infections of mosquito vectors with VEEV, showing that reducing the bloodmeal titre from 106 to 105 pfu ml–1 reduces the number of virions that infect the C. taeniopus midgut by 100-fold14. Similar studies with DENV have estimated that 5–100 virions initiate infection31,32. As natural vertebrate viraemia levels reach titres from 102 to 106 pfu ml–1, it is likely that the bottleneck associated with natural midgut infection is much greater than we observe in laboratory infections.
Another major factor that influences midgut infection and transmission competence is the mosquito microbiome, which could enhance the impact of bottlenecks by altering the immune status of the vector and susceptibility to oral infection (reviewed previously33); however, a detailed discussion is beyond the scope of this Review. Moreover, recent studies on insect-specific viruses and endogenous viral elements, which are naturally found in various mosquito species, suggest they may also have a role in vector competence, warranting further investigation34–38.
In addition, the virus itself represents a bottleneck during midgut infection. Arboviruses exhibit high mutation rates during replication, but most of the mutations do not become fixed within the population. Studies of viral diversity during infection have shown that arboviruses generate diverse populations during infection of the midgut31,39. Sampling viral diversity during early infection revealed that the amount of diversity observed during infection days 1–4 is lower than that observed through days 12–19 (refs39,40). However, for DENV, no reduction in viral diversity was observed but substantial bottlenecks were detected at each stage of dissemination within the vector32, which suggests that sequence diversity was restored following each bottleneck41. Moreover, mosquitoes with disseminated infections tend to have higher viral diversity than those without dissemination from the midgut. This suggests that viral diversity has a role in overcoming the midgut infection barrier39 as well as the midgut escape barrier discussed below, as any alteration in viral polymerase fidelity and mutational frequency tends to reduce the ability of the virus to overcome the bottleneck42.
The mechanism of arbovirus dissemination from the midgut into the haemocoel (Fig. 1) remains poorly understood. In addition to periodic basal lamina remodelling, which may permit virions to penetrate the otherwise exclusionary structure, the bloodmeal stretches the midgut basal lamina due to the approximately 20-fold increase in luminal volume. This results in basal lamina tears near the surrounding circular and longitudinal muscles43–45. When midguts of CHIKV-infected mosquitoes were studied using transmission electron microscopy, virions were present in the basal lamina 24 h after feeding, and by 48 h the virus had disseminated from the midgut43. Structural alterations and microperforations provide a likely escape mechanism for the virus from the midgut, especially in A. aegypti mosquitoes that feed more frequently than most other species. Recent studies examining the effect of repeated feeding suggest that a second, non-infectious bloodmeal expedites dissemination from the midgut of arboviruses infecting via an initial bloodmeal, probably due to similar mechanisms of microperforations46.
Despite these physical mechanisms that promote arbovirus dissemination from the midgut, the dissemination barrier still presents a population bottleneck as demonstrated for CHIKV47, ZIKV48, VEEV14, WNV49 and DENV32. Depending on the virus–vector pairing, this bottleneck can be more or less stringent. For example, in subtype IE VEEV–C. taeniopus, the bottleneck has been estimated to reduce the population to approximately 2–50 virions, depending on the titre of the bloodmeal. However, 95% of infected mosquitoes support dissemination into the haemocoel. For WNV and CHIKV, the number of mosquitoes that develop disseminated infections is generally between 50 and 80%, again depending on the strains of mosquito and virus. Regardless of the severity of the bottleneck, arboviruses seem to recover genetic diversity quickly; for example, VEEV in approximately 8 days39 and DENV even more quickly32. The ability of arboviruses to restore diversity following a bottleneck probably enables them to complete dissemination and, ultimately, transmission.
Prior to horizontal transmission, arboviruses must infect the salivary glands50 (Fig. 1). Regardless of how the virus passes through the surrounding basal lamina to infect the salivary glands, a population bottleneck occurs. Using intrathoracic infections that bypass the midgut bottleneck, several studies have demonstrated that a salivary gland infection bottleneck nevertheless persists14,49,51. These founder effects have not been demonstrated to the same degree for alphaviruses as for WNV because the former are generally transmitted in much smaller amounts, which results in technical challenges to assessing the alphavirus populations in saliva.
For completion of the transmission cycle, an arbovirus must be present in the saliva of the vector during a bloodmeal subsequent to the infectious meal (Fig. 1). Some mosquitoes, especially A. aegypti, take frequent, partial bloodmeals and others only take one per reproductive cycle, which lasts approximately 1 week. The extrinsic incubation period, the minimum time between infection and transmission potential, can be as little as 2 days for CHIKV and a few other viruses52. Given the difficulties in establishing experimental transmission by infected mosquitoes in the laboratory, where in vitro collection of saliva can overestimate the amount of virus transmitted53, it is unclear how stringent this transmission bottleneck is. This is especially true due to the artificial methods of expectoration typically used to assess virus titres in mosquito saliva. Flaviviruses such as YFV or DENV can transmit as large populations with titres up to 105 infectious units54,55, whereas alphaviruses such as CHIKV52 and VEEV53 are present in saliva at much lower titres, which suggests a more stringent transmission bottleneck.
Replication and viraemia in the vertebrate host
The early stages of arboviral replication and resulting viraemia in the vertebrate host are not well understood, in part because of limitations in the ability of animal models to reproduce natural hosts. For many arboviruses, critical sites of initial infection are immune cells near the site of the vector bite that precede dissemination to the lymphoid system and then transition to the circulatory system for viraemia. Studies with WNV suggest that the initial sites of replication are keratinocytes56, which may represent a staging point from which the virus then spreads to the blood and other tissues and organs. Of the few arboviral population genetic studies in vertebrates, one showed that there was little to no bottleneck involved in generating viraemia for VEEV42. A second study with ZIKV indicated few bottlenecks during the first 7 days of non-human primate infection57, which suggests that vectors represent a more important bottleneck challenge than vertebrate hosts. This conclusion is also supported by studies with WNV that suggest limited bottlenecks and a strong purifying selection in birds58. Thus, it is likely that the level of viraemia and the degree of viral diversity within the viraemia population are the major factors in transmission from vertebrates to arthropods, if only because this influences the degree to which mosquitoes are infected.
Arbovirus evolution and emergence
Overall, the above reviewed findings suggest that arboviruses regularly undergo major population bottlenecks at several stages of their transmission cycle. Therefore, they may face a major challenge of avoiding fitness declines due to drift when population sizes become too small for effective natural selection. Because total mutation frequencies of arboviruses are typically about 10−4 per nucleotide replicated, resulting in a per-genome frequency of about 1, direct reversion of the same mutation fixed in a population by drift occurs less frequently (once in about 104 genomes), and repeated bottlenecks can rapidly reduce the fitness of a population due to the accumulation of random mutations that are typically deleterious (Fig. 2). This phenomenon, Muller’s ratchet, occurs in the absence of efficient recombination, which can quickly and at higher frequency restore the wild-type sequence if two different genomes with deleterious mutations in different genome regions recombine. Some arboviruses have segmented genomes that may facilitate recombination via segment re-assortment, but most do not. Experimental studies of arboviruses including vesicular stomatitis virus19 and eastern equine encephalitis virus20 have shown that Muller’s ratchet reduces fitness when repeated plaque cloning is used to introduce bottlenecks to in vitro evolution systems.
However, considerable evidence suggests that many arboviruses avoid major fitness losses and continue to circulate efficiently in the face of both genetic and environmental challenges. In fact, their consensus sequences can be remarkably stable in nature59, which suggests that they can avoid major drift and its potentially deleterious effects. One of the few exceptions of an arbovirus that stably circulates in nature is western equine encephalitis virus, an avian virus that during the mid nineteenth century caused tens of thousands of annual spillover infections of equids and humans in North America, with high rates of mortality. However, western equine encephalitis virus underwent a dramatic reduction in its enzootic circulation, as well as human and equine disease, during the late twentieth century for unknown reasons60,61. Recent reverse genetic studies provided no evidence of overall fitness declines, which suggests that ecological factors might have had a role in the decline of western equine encephalitis virus62.
One potential explanation for the ability of arboviruses to avoid Muller’s ratchet is that, following bottlenecks during various stages of mosquito infection and transmission (Fig. 1), the mutant swarm is quickly restored via rapid, low-fidelity replication. This hypothesis is supported by several population-level sequencing studies, most recently involving deep sequencing21,31,39. Population-level sequencing of specific mosquito tissues has shown that arboviruses generate specific minority variants that may increase the likelihood of dissemination and transmission39. There are at least two hypotheses for this: the first is that, following initial infection, there is ‘leakage’ from the midgut, which restores viral diversity, including the most fit variants. Alternatively, it is possible that arboviruses have optimized their genome structure and mutation frequencies such that specific mutations, which confer some benefits during mosquito infection and dissemination, are consistently produced42,63,64. If these hypotheses are true, this may be yet another way arboviruses overcome the deleterious effects of the numerous bottlenecks during transmission cycles.
Although recombination has seldom been detected in nature for most unsegmented arboviruses, studies of recombination involving the formation of defective interfering viral RNA genomes suggest that template switching, thought to be the major mechanism of recombination, occurs regularly65. However, the frequency at which appropriate recombinants, capable of restoring a master sequence, occur is unknown. To confirm that natural recombination leading to the transmission of the recombinant strain indeed occurs, the following prerequisites should be met: the recombinant crossover should be demonstrated in a single PCR amplicon following cloning to ensure that it occurs in a single cDNA molecule; the recombination event should be demonstrated repeatedly in clonal populations of viable virus (for example, a plaque-purified or limited end point dilutions); and the recombinant should maintain adequate sequence conservation during post-recombination evolution66. These conditions have rarely been met in descriptions of arbovirus recombinants in the literature67, although recombinants between closely related genomes would be technically challenging to detect.
Although drift would be frequently expected immediately following major bottlenecks regardless of mutant swarm restoration, its impacts may be minimized across multiple arboviral lineages. Discrete viral lineages within individual transmission chains may indeed undergo fitness declines in nature due to drift. However, very large numbers of individual transmission chains circulate in an active focus or region. This large number of lineages may ensure that, by chance, high-fitness genomes with master sequences remain propagated in nature to outcompete those that are affected by bottleneck-associated drift. Therefore, the ability of arboviruses to sustain huge numbers of transmission chains during efficient circulation in endemic or enzootic locations may enable them to avoid deleterious effects on their fitness, and thus permit them to persist with the ability to emerge via positive selection under favourable conditions.
Geographic spread and founder effects
In addition to the virus population bottlenecks encountered during the transmission cycle as described above, arboviruses frequently undergo bottlenecks during spread into new geographic regions or during reintroductions following dry seasons or winters that are not conducive to continuous circulation or persistence. In recent years, WNV68, ZIKV69 and CHIKV70 spread for the first time in the modern scientific era to the New World. Although other arboviruses, especially DENV, also undergo frequent geographic introductions, their more widespread distribution for many decades may render the associated fitness effects less dramatic. Although the mechanism for the introduction of WNV into New York in 1999 remains enigmatic, modelling and phylogenetic studies suggest that most introductions of human-amplified ZIKV, CHIKV and DENV occurred via infected air travellers or individuals travelling by ship71–73 rather than through mosquitoes that have been inadvertently transported74 (Fig. 2). Although nearly all expansions of the geographic ranges of these arboviruses seem to represent point-source introductions68,75–78, detailed studies of ZIKV in Florida in 2016 demonstrated multiple independent introductions, with only a few of these initiating prolonged transmission of weeks to a few months73. For a stable geographic introduction, the arrival of an infected traveller must be followed by exposure to a competent vector (or vectors) that survives extrinsic incubation and then bites a susceptible person to initiate a transmission chain. This is presumably rare, considering the large numbers of infected travellers diagnosed with CHIKV, ZIKV and DENV in permissive regions such as the southern United States, and almost always without evidence of ensuing vector-borne circulation. However, when an infected traveller upon arrival is exposed to large populations of vectors such as A. aegypti, a major epidemic may occasionally ensue, as detected in multiple locations for all of these viruses.
The above geographic spread scenario includes several bottlenecks that provide the potential for founder effects (Fig. 2) to result in suboptimal transmission efficiency and altered amplification and/or virulence in humans. Based on the experimental evidence discussed above, in the case of many arboviruses, including CHIKV52 and ZIKV79,80, the infected traveller is likely to be inoculated with a small number of infectious virions present in mosquito saliva (Fig. 1), with the potential for this population to contain random, sometimes fixed, deleterious mutations. When the infected traveller is subsequently bitten by a susceptible vector, the mosquito is likely to be infected by a small number of virions owing to the poor susceptibility of A. aegypti to these viruses and the midgut bottleneck described above6,80. Thus, point-source introduction of these viruses by infected travellers probably involves at least two major arboviral bottlenecks during traveller infection and initial vector infection. Random mutations are far more likely by chance to be deleterious because non-synonymous mutations outnumber synonymous mutations, and random amino acid changes usually reduce protein function. Therefore, random mutations often result in less efficient vector transmission and/or host viraemia (sometimes with an accompanying reduction in virulence) than beneficial effects that increase transmission efficiency. Thus, these bottlenecks may influence emergence and human disease. Because large numbers of transmission chains, which may overcome drift as described above, do not initially exist following a new introduction, founder effects may have more dramatic impacts than the bottlenecks that all arboviruses undergo during the transmission cycle. Below, we discuss the evidence that these bottlenecks result in substantial fitness changes that affect the potential for arboviral outbreaks, further spread and disease severity.
Chikungunya virus
In the modern scientific era, several geographic CHIKV introductions have resulted in ongoing and amplified human–mosquito–human transmission chains that resulted in major disease outbreaks. In 2004, an epidemic was initiated by an East/Central/South African (ECSA) (Fig. 3) enzootic (sylvatic mosquito–non-human primate cycle) (Box 1) strain in coastal Kenya81, followed by its spread into islands in the Indian Ocean82 and also, independently, from Kenya into South Asia83, and subsequently to Southeast Asia84. Spread of this ECSA-derived Indian Ocean lineage (IOL) was characterized by severe arthralgia with high rates of chronic disease, and the virus exhibited a series of envelope glycoprotein E2/E3 and E1 mutations that adapted some IOL strains for more efficient transmission by the recently invasive mosquito, A. albopictus25,85,86 (Fig. 3). Compared with all other ECSA strains examined, descendant IOL strains exhibit similar mosquito infection phenotypes and virulence in a mouse model87, with no indication of fitness declines following the emergence in 2004. Thus, any founder effects and bottlenecks associated with the 2004–2005 CHIKV emergence did not reduce fitness, possibly because of multiple introductions from coastal Kenya. Although IOL strains were exported from Asia via infected travellers to initiate small outbreaks in Italy and France between 2007 and 2017, they did not initiate detectable transmission in the Americas despite thousands of introductions88.
Fig. 3. Phylogenetic tree of CHIKV.

Phylogenetic tree based on 45 representative complete open-reading frame nucleotide sequences of chikungunya virus (CHIKV). Evolutionary distances were computed using the maximum composite likelihood method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test are shown next to the branches (1,000 replicates). Evolutionary analyses were performed in MEGA X121 using the neighbour-joining method. CHIKV strains are labelled by GenBank Accession number, followed by country and year of collection. Coloured arrows show the branches that include mutations known to affect CHIKV fitness. The duplication mutation in the 3′ untranslated region (UTR; green), which is associated with increased fitness upon introduction of an Asian lineage CHIKV strain into the Caribbean, may represent a rare beneficial founder effect. The deletion mutation in the 3′ UTR (red) was associated with the introduction into Asia of an East/Central/South African (ECSA) African strain prior to 1959. Other mutations (indicated in blue), including the convergent (evolved at least four times) envelope glycoprotein 1 (E1)-A226V substitution and several E2/E3 substitutions, led to an increase in fitness for infection of Aedes albopictus mosquitoes. Red asterisk indicates the epistatic founder mutation (E1-A98T) and blue asterisks indicate ECSA strains with E2-211I; both of these amino acids decrease the ability of CHIKV to efficiently transmit via A. albopictus in Asia and the Americas (that is, decreasing the beneficial effect conferred by the E1-A226V mutation).
Prior to the emergence of the IOL in 2004, the first phylogenetically delineated spread from Africa resulted from the earlier introduction of an ECSA enzootic strain in South and Southeast Asia approximately 60–100 years ago78,89. This endemic and/or epidemic strain, called the Asian lineage (Fig. 3), has continued to circulate in Southeast Asia ever since the first outbreaks were reported in the 1950s. These outbreaks in Asia were generally less severe than those caused by the IOL beginning in 2005, and it was initially unclear whether this reflected higher levels of human herd immunity during the 1950s compared with 2005, lower transmission efficiency during the earlier outbreaks or lower virulence of Asian lineage strains. Compared with African and IOL CHIKV strains, there is little or no difference in infectivity for A. aegypti, the principal urban CHIKV vector in most locations. However, based on epidemiologic90–92 and animal model87 studies, the Asian lineage strains seem to be less virulent in terms of apparent to inapparent human infection ratios and rates of chronic arthralgia. In 2013, this Asian CHIKV lineage was introduced into the Caribbean island of Saint Martin, followed by the extensive spread through much of the New World tropics and subtropics6.
In contrast to the IOL that underwent adaptive evolution for more efficient mosquito transmission, evidence indicates that the Asian CHIKV lineage underwent founder effects and fitness losses following its introduction from Africa many decades ago. Asian strains possess a unique, low-fitness (in all hosts examined) 3′ untranslated region (UTR) that is much longer than that of African or IOL CHIKV strains, which strongly suggests a founder effect. The determinants of the lower virulence of the Asian lineage have not been thoroughly investigated, but phylogenetic reconstructions of the 3′ UTR indicate that the progenitor arrived from Africa with a major deletion in two direct sequence repeats, which reduced its fitness for replication in both mosquito and vertebrate cells93. This fitness loss of the Asian lineage was partially restored over time by a series of point mutations and repeated sequence duplications. However, in every CHIKV sequence studied so far, the 3′ UTR of the Asian lineage remains debilitated (it results in reduced fitness for all hosts examined when placed using reverse genetics into the backbone of any major CHIKV lineage) compared with the ancestral ECSA and IOL 3′ UTR sequences93. Furthermore, the 3′ UTR was also implicated in a probable founder effect following the 2013 introduction of the Asian lineage into the Americas94. All Asian/American CHIKV strains contain a unique 177-nucleotide duplication in the repeated sequence region of the 3′ UTR (Fig. 3). This insertion increases fitness for replication in mosquito cells94, which suggests that CHIKV founder effects can also confer fitness advantages (as expected in a minority of introductions based on the majority of random mutations expected to be deleterious).
In addition to the direct 3′ UTR-mediated fitness effects resulting from the introductions of the Asian CHIKV lineage into Asia and the Americas, other founder effects have limited the ability of this strain to adapt for more efficient urban transmission. The detection of convergent evolution of the initial envelope glycoprotein 1 (E1)-A226V substitution in IOL strains as they spread into the Indian Ocean basin and South Asia suggested a strong selective advantage of this mutation for epidemic transmission82,95. This hypothesis was supported by the major fitness advantage conferred by this mutation for infection of A. albopictus85,86, which had spread from its ancestral range in Asia to many regions of Africa, southern Europe and the Americas since 1985 (ref.96). However, the lack of Asian lineage CHIKV strains with the E1-226V residue seemed paradoxical because this lineage had circulated in Asia, native territory for A. albopictus, for many decades without evidence of the same adaptive mutation. This conundrum was resolved when subsequent reverse genetic studies demonstrated that the E1-A226V substitution does not appreciably increase infectivity of the Asian lineage CHIKV for A. albopictus due to epistasis97. The generation of chimeric Asian–IOL viruses followed by mutagenesis of an Asian lineage clone showed that a single epistatic E1 glycoprotein threonine residue at position 98 is responsible for the inability of the Asian strain to benefit from the E1-A226V mutation (Fig. 3). Furthermore, the presence of the implicated E1-98T in all Asian strains and the lack of this residue in any other CHIKV strain, including those collected in Africa (all have E1-98A), along with the lack of a detectable phenotype for this mutation alone, strongly suggest that the threonine at position 98 in the Asian lineage resulted from a founder effect that accompanied the 3′ UTR mutation upon introduction into Asia many decades ago.
In summary, major founder effects that accompanied the introduction of an ECSA CHIKV strain into Asia many decades ago probably limited its ability to cause major outbreaks due to 3′ UTR-mediated fitness reductions that were difficult to restore, along with an amino acid substitution in the E1 envelope glycoprotein that prevented adaptation for transmission by A. albopictus. The latter epistatic constraint was implicated in limiting the spread of the Asian and/or American CHIKV strains into temperate regions, a notion that is supported by the lack of the E1-226V residue in any sequenced CHIKV strain from the Americas, as well as by a lack of evidence for transmission in the Americas by A. albopictus98.
Zika virus
The pathway and timing of the recent ZIKV introduction into the Americas is remarkably similar to that of CHIKV (Fig. 4). Following its evolution in sub-Saharan Africa in enzootic cycles involving arboreal mosquitoes and non-human primates, ZIKV was introduced many decades ago into Asia99. Following many decades without evidence of causing severe human disease, the first recognized outbreaks occurred during 2007 in Gabon100 and Yap Island101. Then, in 2013, ZIKV spread into the South Pacific followed by Brazil, before causing a major epidemic that spread throughout the American tropics, subtropics and beyond76,102.
Fig. 4. Phylogenetic tree of ZIKV.

Phylogenetic tree based on 73 representative, complete open-reading frame nucleotide sequences of Zika virus (ZIKV). Evolutionary distances were computed using the maximum likelihood method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test are shown next to the branches (1,000 replicates). Evolutionary analyses were performed in MEGA X121. ZIKV strains are labelled by GenBank Accession number, followed by strain and country and year of collection. Coloured arrows indicate the substitutions in the capsid (C-A106T), precursor membrane (PrM-A1V), non-structural protein 1 (NS1-V188A) and non-structural protein 5 (NS5-V872M) that occurred during the early circulation of ZIKV in Asia, presumably due to founder effects or another form of genetic drift during early circulation. These same substitutions reverted just prior to its introduction into the South Pacific and the Americas, which resulted in increased fitness for transmission by Aedes aegypti mosquitoes among humans103.
The reasons for the sudden emergence of ZIKV accompanied by Guillain–Barré syndrome and congenital Zika syndrome remain at least partially unresolved. Studies examining a mutation that occurred just prior to the spread of ZIKV into the South Pacific suggest that the virus adapted for more efficient urban transmission, similar to what was observed for the CHIKV IOL. An amino acid in the non-structural protein 1 (NS1-188) (Fig. 4) differs among ancestral African, some Asian and American ZIKV strains, with all African and American strains including valine and early Asian strains alanine. When the valine residue was engineered into a pre-epidemic 2010 Cambodian ZIKV strain, the resultant virus generated slightly increased infectivity for A. aegypti mosquitoes103, the principal urban ZIKV vector104,105. Furthermore, phylogenetic reconstruction of the evolution of this NS1-188 residue (Fig. 4) reveals that the pre-epidemic A188V substitution actually reflects the direct reversion of an earlier V188A substitution that occurred following the introduction of ZIKV into Asia, or soon thereafter. This strongly suggests that the V188A substitution confers reduced fitness for infection of A. aegypti. This substitution therefore likely resulted from a founder effect, much like the CHIKV 3′ UTR and E1-A98V mutations that accompanied the same geographic introduction from Africa to Asia. In addition, three additional substitutions in the capsid (C-A106T), precursor membrane (PrM-A1V) and non-structural protein 5 (NS5-V872M), also representing direct reversions of earlier mutations, confer enhanced transmission by A. aegypti and/or enhanced viraemia in human cell or mouse models of human viraemia106. However, comparisons of ancestral African strains with Asian and American ZIKV strains suggest that the ability to infect A. aegypti has not been completely restored by these four mutations, as indicated by the greater transmissibility of African strains by A. aegypti80,107,108 as well as higher virulence in models of human viraemia when compared with American strains with the same residues108,109. This lack of complete fitness restoration despite decades of presumed urban transmission by A. aegypti, as suggested by a 1966 isolation from this mosquito species in Malaysia110, is surprising and underscores potential evolutionary constraints of the ability of arboviruses to adapt to new transmission opportunities due to their complex transmission cycles111. Recently, another transmission-adaptive mutation that occurred shortly before the American outbreaks was identified. A substitution of methionine for valine in the envelope glycoprotein (E-V473M) enhances viraemia in mice and cynomolgus macaques, which suggests that it increased the epidemic potential via greater human viraemia112. However, unlike the four revertant substitutions, E-V473M did not involve reversion of a prior mutation, which suggests classic Darwinian evolution.
The similarity in the geographic spread and fitness histories of ZIKV and CHIKV, with founder effects followed by direct reversions (ZIKV) or pseudo-reversions (CHIKV 3′ UTR) to only partially restore fitness, underscores the importance of founder effects in determining the emergence and epidemic potential of arboviruses. Both viruses reached the Americas only after many decades of circulation in Asia, with only partial restoration of the ancestral fitness exhibited by African strains. In the case of CHIKV, this limited vector usage (not by A. albopictus) and virulence in the Americas, whereas there was no direct evidence that ZIKV virulence was affected by founder effects or drift (NS1-A188V does not affect this phenotype). Thus, founder effects may limit both the timing and magnitude of emergence events, and their stochastic nature is likely to limit our ability to predict future epidemics.
Conclusions
There is compelling evidence that arboviruses face major population bottlenecks during several steps of their transmission cycles: infection of the mosquito midgut, dissemination from the midgut into the haemocoel, infection of the salivary glands and transmission to the vertebrate host. These bottlenecks can result in the stochastic fixation of random mutations that result from the inherently error-prone RNA virus replication, a form of genetic drift. Despite these bottlenecks and their potential for fitness losses via Muller’s ratchet, arboviruses are typically genetically stable in nature with no evidence of frequent fitness losses. These ubiquitous bottlenecks may be overcome through the rapid regeneration of the genetically diverse population following subsequent replication, along with large numbers of viral lineages in independent transmission chains from which lineages with master or high-fitness sequences can be selected. Founder effects, also a form of a bottleneck that can fix stochastic mutations via drift, can affect emergence trajectories in particularly unpredictable ways. They seem to have negatively affected the fitness of CHIKV and ZIKV following their introduction from Africa to Asia, possibly accompanied by drift from initially inefficient urban circulation in Asia. Although only one of several CHIKV lineages introduced from Africa to Asia or the Americas shows evidence for such a founder effect-related fitness decline, it is likely that this resulted in constraints on its vector usage and probably also affected its virulence. The emergence of ZIKV into the Americas followed the partial restoration of its fitness following founder effect-mediated drift and fitness loss. The lack of detected outbreaks caused by the ancestral African ZIKV lineage seems counter-intuitive, and contrasts with the observations of African CHIKV epidemics. However, the low symptomatic to asymptomatic ratio of ZIKV infections, compared with CHIKV, may result in greater under-reporting of the former virus. Furthermore, the greater experimental evidence of epidemic potential (A. aegypti infection and viraemia in animal models) of African ZIKV strains compared with Asian and American strains could also reflect limitations of the animal models.
It is important to recognize that predicting most arboviral emergence events remains nearly impossible due to the random nature of the mutations that accompany founder effects and bottleneck-mediated drift. Neither the emergence of the Asian lineage of CHIKV nor the emergence of ZIKV into the Americas could have been predicted without the retrospectively acquired information reviewed above. Nonetheless, some important inferences for the potential of a pathogen to emerge can be deduced through comprehensive surveillance, modelling and carefully controlled experimental studies11,79,80,113–115.
Finally, drift and founder effects are only a few of many evolutionary mechanisms known to affect arboviral emergence. Other major drivers include changes in land use, increased international commerce and travel, all of which have been key for the spread of urban arboviruses116. These factors have not been shown to directly impact bottlenecks that accompany arbovirus circulation, and more research is required. During past centuries, DENV, YFV and CHIKV (and possibly ZIKV) have spread regularly but more slowly from Africa to Asia and the Americas on ships, with only seasonal persistence in many temperate locations. However, the widespread dissemination of A. aegypti centuries ago, and that of A. albopictus since 1985 resulting from used tyre and other trades, along with urban population growth, especially in the tropics, has greatly accelerated global arbovirus spread117. Huge populations of uncontrolled A. aegypti in large cities in the tropics have only compounded the risk for efficient transmission, along with resistance to insecticides by this and other arbovirus vectors. Finally, adaptation to vectors and hosts can also have substantial effects on arboviral spread, as underscored by both ZIKV103,112 and CHIKV118. All of these emergence drivers require concerted, sustained study, and overcoming their effect on human health will necessitate innovative combinations of control approaches.
Acknowledgements
Research in the authors’ laboratories is supported by National Institutes of Health (NIH) grants AI120942 and AI121452 (to S.C.W.) and AI145918 (to N.V.).
Glossary
- Zoonotic
Infections that spill over from non-human animals to humans.
- Enzootic
Ancestral, often continuous, transmission cycles of zoonotic arboviruses involving wild animals serving as amplification and/or reservoir hosts.
- Anthropophilic mosquitoes
Mosquitoes with a preference for and typically a tendency to bite humans.
- Mutant swarms
A population of RNA viruses with numerous, randomly derived mutations resulting from low-fidelity RNA replication (lack of proofreading). Mutant swarms are also termed quasispecies when they are selected and evolve as populations rather than as individual genetic variants.
- Quasispecies
A diverse RNA (virus) population that contains many mutants closely related to a master sequence (usually the most abundant) and selected as a population, rather than as individuals, during its evolution.
- Population bottlenecks
Major reductions in the population size of organisms, often used in the context of near-extinction events. Population bottlenecks result in a loss in genetic diversity and can also fix mutations at random. Population bottlenecks also represent a form of genetic drift.
- Genetic drift
Random changes in the genetic make-up of a population due to chance, random sampling. Drift can dominate the evolution of a virus when population sizes remain small, reducing the efficiency of selection and genetic diversity.
- Founder effects
The loss of genetic variation, sometimes resulting in the fixation of random mutations. Founder effects occur when a new population is established by a small number of individuals (founder population) randomly derived from a larger ancestral population. Founder effects represent a form of genetic drift.
- Muller’s ratchet
The step-wise fixation following population bottlenecks of random mutations, which are typically deleterious, resulting in a decline in fitness that is difficult to restore in the absence of efficient recombination.
- Vector competence
The intrinsic ability of an arthropod to become infected and transmit a pathogen.
- Epistasis
Interactions of genes or mutations such that one can suppress the effect of another on a phenotype.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Microbiology thanks D. Brackney, G. Ebel and S. Lequime for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Karabatsos, N. International Catalog of Arboviruses Including Certain Other Viruses of Vertebrates 4th edn (American Society of Tropical Medicine and Hygiene, 1985).
- 2.Bhatt S, et al. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paules CI, Fauci AS. Yellow fever — once again on the radar screen in the Americas. N. Engl. J. Med. 2017;376:1397–1399. doi: 10.1056/NEJMp1702172. [DOI] [PubMed] [Google Scholar]
- 4.Nwachukwu WE, et al. The response to re-emergence of yellow fever in Nigeria, 2017. Int. J. Infect. Dis. 2020;92:189–196. doi: 10.1016/j.ijid.2019.12.034. [DOI] [PubMed] [Google Scholar]
- 5.Kraemer MUG, et al. Spread of yellow fever virus outbreak in Angola and the Democratic Republic of the Congo 2015–16: a modelling study. Lancet Infect. Dis. 2017;17:330–338. doi: 10.1016/S1473-3099(16)30513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weaver SC, Charlier C, Vasilakis N, Lecuit M. Zika, chikungunya, and other emerging vector-borne viral diseases. Annu. Rev. Med. 2018;69:395–408. doi: 10.1146/annurev-med-050715-105122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kramer IM, et al. Does winter cold really limit the dengue vector Aedes aegypti in Europe? Parasites Vectors. 2020;13:178. doi: 10.1186/s13071-020-04054-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tippelt L, Werner D, Kampen H. Tolerance of three Aedes albopictus strains (Diptera: Culicidae) from different geographical origins towards winter temperatures under field conditions in northern Germany. PLoS ONE. 2019;14:e0219553. doi: 10.1371/journal.pone.0219553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ko HY, et al. Inter- and intra-host sequence diversity reveal the emergence of viral variants during an overwintering epidemic caused by dengue virus serotype 2 in southern Taiwan. PLoS Negl. Trop. Dis. 2018;12:e0006827. doi: 10.1371/journal.pntd.0006827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domingo E, Holland JJ. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 11.Coffey LL, et al. Arbovirus evolution in vivo is constrained by host alternation. Proc. Natl Acad. Sci. USA. 2008;105:6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deardorff ER, et al. West Nile virus experimental evolution in vivo and the trade-off hypothesis. PLoS Pathog. 2011;7:e1002335. doi: 10.1371/journal.ppat.1002335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novella IS, Presloid JB, Smith SD, Wilke CO. Specific and nonspecific host adaptation during arboviral experimental evolution. J. Mol. Microbiol. Biotechnol. 2011;21:71–81. doi: 10.1159/000332752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forrester NL, Guerbois M, Seymour RL, Spratt H, Weaver SC. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog. 2012;8:e1002897. doi: 10.1371/journal.ppat.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayr, E. Systematics and the Origin of Species (Columbia Univ. Press, 1942).
- 16.Templeton AR. The reality and importance of founder speciation in evolution. Bioessays. 2008;30:470–479. doi: 10.1002/bies.20745. [DOI] [PubMed] [Google Scholar]
- 17.Mayr, E. in Evolution as a Process (eds Huxley, J., Hardy, A. C., & Ford E. B.) 157–180 (Princeton Univ. Press, 1954).
- 18.Muller HJ. The relation of recombination to mutational advance. Mutat. Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 19.Duarte E, Clarke D, Moya A, Domingo E, Holland J. Rapid fitness losses in mammalian RNA virus clones due to Muller’s ratchet. Proc. Natl Acad. Sci. USA. 1992;89:6015–6019. doi: 10.1073/pnas.89.13.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weaver SC, Brault AC, Kang W, Holland JJ. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 1999;73:4316–4326. doi: 10.1128/JVI.73.5.4316-4326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forrester NL, Coffey LL, Weaver SC. Arboviral bottlenecks and challenges to maintaining diversity and fitness during mosquito transmission. Viruses. 2014;6:3991–4004. doi: 10.3390/v6103991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moudy RM, Meola MA, Morin LL, Ebel GD, Kramer LD. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am. J. Trop. Med. Hyg. 2007;77:365–370. doi: 10.4269/ajtmh.2007.77.365. [DOI] [PubMed] [Google Scholar]
- 23.Brault AC, et al. Venezuelan equine encephalitis emergence: enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc. Natl Acad. Sci. USA. 2004;101:11344–11349. doi: 10.1073/pnas.0402905101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anishchenko M, et al. Venezuelan encephalitis emergence mediated by a phylogenetically predicted viral mutation. Proc. Natl Acad. Sci. USA. 2006;103:4994–4999. doi: 10.1073/pnas.0509961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsetsarkin KA, et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 2014;5:4084. doi: 10.1038/ncomms5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholle F, Girard YA, Zhao Q, Higgs S, Mason PW. trans-Packaged West Nile virus-like particles: infectious properties in vitro and in infected mosquito vectors. J. Virol. 2004;78:11605–11614. doi: 10.1128/JVI.78.21.11605-11614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith DR, Adams AP, Kenney JL, Wang E, Weaver SC. Venezuelan equine encephalitis virus in the mosquito vector Aedes taeniorhynchus: infection initiated by a small number of susceptible epithelial cells and a population bottleneck. Virology. 2008;372:176–186. doi: 10.1016/j.virol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenney JL, Adams AP, Gorchakov R, Leal G, Weaver SC. Genetic and anatomic determinants of enzootic Venezuelan equine encephalitis virus infection of Culex (Melanoconion) taeniopus. PLoS Negl. Trop. Dis. 2012;6:e1606. doi: 10.1371/journal.pntd.0001606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuckols JT, et al. Infection of Aedes albopictus with chikungunya virus rectally administered by enema. Vector Borne Zoonotic Dis. 2013;13:103–110. doi: 10.1089/vbz.2012.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Coupanec A, et al. Co-infection of mosquitoes with chikungunya and dengue viruses reveals modulation of the replication of both viruses in midguts and salivary glands of Aedes aegypti mosquitoes. Int. J. Mol. Sci. 2017;18:1708. doi: 10.3390/ijms18081708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lequime S, Fontaine A, Ar Gouilh M, Moltini-Conclois I, Lambrechts L. Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. PLoS Genet. 2016;12:e1006111. doi: 10.1371/journal.pgen.1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim S, et al. Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 2015;9:e0004052. doi: 10.1371/journal.pntd.0004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jupatanakul N, Sim S, Dimopoulos G. The insect microbiome modulates vector competence for arboviruses. Viruses. 2014;6:4294–4313. doi: 10.3390/v6114294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colmant AMG, et al. The recently identified flavivirus Bamaga virus is transmitted horizontally by Culex mosquitoes and interferes with West Nile virus replication in vitro and transmission in vivo. PLoS Negl. Trop. Dis. 2018;12:e0006886. doi: 10.1371/journal.pntd.0006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki Y, et al. Non-retroviral endogenous viral element limits cognate virus replication in Aedes aegypti ovaries. Curr. Biol. 2020;30:3495–3506.e6. doi: 10.1016/j.cub.2020.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romo H, Kenney JL, Blitvich BJ, Brault AC. Restriction of Zika virus infection and transmission in Aedes aegypti mediated by an insect-specific flavivirus. Emerg. Microbes Infect. 2018;7:181. doi: 10.1038/s41426-018-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goenaga S, et al. Potential for co-infection of a mosquito-specific flavivirus, Nhumirim virus, to block West Nile virus transmission in mosquitoes. Viruses. 2015;7:5801–5812. doi: 10.3390/v7112911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roundy CM, et al. Viruses: a historical overview and recent developments. Adv. Virus Res. 2017;98:119–146. doi: 10.1016/bs.aivir.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 39.Patterson EI, et al. Mosquito bottlenecks alter viral mutant swarm in a tissue and time-dependent manner with contraction and expansion of variant positions and diversity. Virus Evol. 2018;4:vey001. doi: 10.1093/ve/vey001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karna AK, et al. Colonized Sabethes cyaneus, a sylvatic New World mosquito species, shows a low vector competence for Zika virus relative to Aedes aegypti. Viruses. 2018;10:434. doi: 10.3390/v10080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lequime S, Richard V, Cao-Lormeau VM, Lambrechts L. Full-genome dengue virus sequencing in mosquito saliva shows lack of convergent positive selection during transmission by Aedes aegypti. Virus Evol. 2017;3:vex031. doi: 10.1093/ve/vex031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warmbrod KL, et al. Viral RNA-dependent RNA polymerase mutants display an altered mutation spectrum resulting in attenuation in both mosquito and vertebrate hosts. PLoS Pathog. 2019;15:e1007610. doi: 10.1371/journal.ppat.1007610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kantor AM, Grant DG, Balaraman V, White TA, Franz AWE. Ultrastructural analysis of chikungunya virus dissemination from the midgut of the yellow fever mosquito, Aedes aegypti. Viruses. 2018;10:571. doi: 10.3390/v10100571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weaver SC, Scott TW, Lorenz LH, Lerdthusnee K, Romoser WS. Togavirus-associated pathologic changes in the midgut of a natural mosquito vector. J. Virol. 1988;62:2083–2090. doi: 10.1128/jvi.62.6.2083-2090.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weaver SC, Scott TW. Ultrastructural changes in the abdominal midgut of the mosquito, Culiseta melanura, during the gonotrophic cycle. Tissue Cell. 1990;22:895–909. doi: 10.1016/0040-8166(90)90051-A. [DOI] [PubMed] [Google Scholar]
- 46.Armstrong PM, et al. Successive blood meals enhance virus dissemination within mosquitoes and increase transmission potential. Nat. Microbiol. 2020;5:239–247. doi: 10.1038/s41564-019-0619-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stapleford KA, et al. Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microbe. 2014;15:706–716. doi: 10.1016/j.chom.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Weger-Lucarelli J, et al. Using barcoded Zika virus to assess virus population structure in vitro and in Aedes aegypti mosquitoes. Virology. 2018;521:138–148. doi: 10.1016/j.virol.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grubaugh ND, et al. Genetic drift during systemic arbovirus infection of mosquito vectors leads to decreased relative fitness during host switching. Cell Host Microbe. 2016;19:481–492. doi: 10.1016/j.chom.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franz AW, Kantor AM, Passarelli AL, Clem RJ. Tissue barriers to arbovirus infection in mosquitoes. Viruses. 2015;7:3741–3767. doi: 10.3390/v7072795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ciota AT, et al. Quantification of intrahost bottlenecks of West Nile virus in Culex pipiens mosquitoes using an artificial mutant swarm. Infect. Genet. Evol. 2012;12:557–564. doi: 10.1016/j.meegid.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dubrulle M, Mousson L, Moutailler S, Vazeille M, Failloux AB. Chikungunya virus and Aedes mosquitoes: saliva is infectious as soon as two days after oral infection. PLoS ONE. 2009;4:e5895. doi: 10.1371/journal.pone.0005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith DR, et al. Venezuelan equine encephalitis virus transmission and effect on pathogenesis. Emerg. Infect. Dis. 2006;12:1190–1196. doi: 10.3201/eid1708.050841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vancini R, Kramer LD, Ribeiro M, Hernandez R, Brown D. Flavivirus infection from mosquitoes in vitro reveals cell entry at the plasma membrane. Virology. 2013;435:406–414. doi: 10.1016/j.virol.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Hanley KA, Azar SR, Campos RK, Vasilakis N, Rossi SL. Support for the transmission-clearance trade-off hypothesis from a study of Zika virus delivered by mosquito bite to mice. Viruses. 2019;11:1072. doi: 10.3390/v11111072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boylan BT, Moreira FR, Carlson TW, Bernard KA. Mosquito cell-derived West Nile virus replicon particles mimic arbovirus inoculum and have reduced spread in mice. PLoS Negl. Trop. Dis. 2017;11:e0005394. doi: 10.1371/journal.pntd.0005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aliota MT, et al. Molecularly barcoded Zika virus libraries to probe in vivo evolutionary dynamics. PLoS Pathog. 2018;14:e1006964. doi: 10.1371/journal.ppat.1006964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grubaugh ND, et al. Mosquitoes transmit unique west nile virus populations during each feeding episode. Cell Rep. 2017;19:709–718. doi: 10.1016/j.celrep.2017.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weaver SC, Rico-Hesse R, Scott TW. Genetic diversity and slow rates of evolution in New World alphaviruses. Curr. Top. Microbiol. Immunol. 1992;176:99–117. doi: 10.1007/978-3-642-77011-1_7. [DOI] [PubMed] [Google Scholar]
- 60.Reisen WK, Fang Y, Brault AC. Limited interdecadal variation in mosquito (Diptera: Culicidae) and avian host competence for western equine encephalomyelitis virus (Togaviridae: Alphavirus) Am. J. Trop. Med. Hyg. 2008;78:681–686. doi: 10.4269/ajtmh.2008.78.681. [DOI] [PubMed] [Google Scholar]
- 61.Bergren NA, et al. Western equine encephalitis virus: evolutionary analysis of a declining alphavirus based on complete genome sequences. J. Virol. 2014;88:9260–9267. doi: 10.1128/JVI.01463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergren NA, et al. “Submergence” of western equine encephalitis virus: evidence of positive selection argues against genetic drift and fitness reductions. PLoS Pathog. 2020;16:e1008102. doi: 10.1371/journal.ppat.1008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moratorio G, Vignuzzi M. Monitoring and redirecting virus evolution. PLoS Pathog. 2018;14:e1006979. doi: 10.1371/journal.ppat.1006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kutchko KM, et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res. 2018;46:3657–3670. doi: 10.1093/nar/gky012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaworski E, Routh A. Parallel ClickSeq and nanopore sequencing elucidates the rapid evolution of defective-interfering RNAs in Flock House virus. PLoS Pathog. 2017;13:e1006365. doi: 10.1371/journal.ppat.1006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weaver SC, Vasilakis N. Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 2009;9:523–540. doi: 10.1016/j.meegid.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weaver SC, et al. Recombinational history and molecular evolution of western equine encephalomyelitis complex alphaviruses. J. Virol. 1997;71:613–623. doi: 10.1128/jvi.71.1.613-623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lanciotti RS, et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333–2337. doi: 10.1126/science.286.5448.2333. [DOI] [PubMed] [Google Scholar]
- 69.Cardoso CW, et al. Outbreak of exanthematous illness associated with Zika, chikungunya, and dengue viruses, Salvador, Brazil. Emerg. Infect. Dis. 2015;21:2274–2276. doi: 10.3201/eid2112.151167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. Chikungunya in the Americas. Lancet. 2014;383:514. doi: 10.1016/S0140-6736(14)60185-9. [DOI] [PubMed] [Google Scholar]
- 71.Rezza G, et al. Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet. 2007;370:1840–1846. doi: 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- 72.Venturi, G. et al. Detection of a chikungunya outbreak in central Italy, August to September 2017. Euro Surveill.22, 17-00646 (2017). [DOI] [PMC free article] [PubMed]
- 73.Grubaugh ND, et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature. 2017;546:401–405. doi: 10.1038/nature22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mier YT-RL, Tatem AJ, Johansson MA. Mosquitoes on a plane: disinsection will not stop the spread of vector-borne pathogens, a simulation study. PLoS Negl. Trop. Dis. 2017;11:e0005683. doi: 10.1371/journal.pntd.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Faria NR, et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature. 2017;546:406–410. doi: 10.1038/nature22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faria, N. R. et al. Zika virus in the Americas: early epidemiological and genetic findings. Science (2016). [DOI] [PMC free article] [PubMed]
- 77.Nunes MR, et al. Emergence and potential for spread of chikungunya virus in Brazil. BMC Med. 2015;13:102. doi: 10.1186/s12916-015-0348-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen R, et al. Comprehensive genome scale phylogenetic study provides new insights on the global expansion of chikungunya virus. J. Virol. 2016;90:10600–10611. doi: 10.1128/JVI.01166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Azar SR, et al. Differential vector competency of Aedes albopictus populations from the Americas for Zika virus. Am. J. Trop. Med. Hyg. 2017;97:330–339. doi: 10.4269/ajtmh.16-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roundy CM, et al. Variation in Aedes aegypti mosquito competence for Zika virus transmission. Emerg. Infect. Dis. 2017;23:625–632. doi: 10.3201/eid2304.161484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chretien JP, et al. Drought-associated chikungunya emergence along coastal East Africa. Am. J. Trop. Med. Hyg. 2007;76:405–407. doi: 10.4269/ajtmh.2007.76.405. [DOI] [PubMed] [Google Scholar]
- 82.Schuffenecker I, et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006;3:e263. doi: 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yergolkar PN, et al. Chikungunya outbreaks caused by African genotype, India. Emerg. Infect. Dis. 2006;12:1580–1583. doi: 10.3201/eid1210.060529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Soon YY, et al. Chikungunya virus of central/east african genotype detected in Malaysia. Med. J. Malaysia. 2007;62:214–217. [PubMed] [Google Scholar]
- 85.Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007;3:e201. doi: 10.1371/journal.ppat.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vazeille M, et al. Two chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS ONE. 2007;2:e1168. doi: 10.1371/journal.pone.0001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Langsjoen RM, et al. Chikungunya virus strains show lineage-specific variations in virulence and cross-protective ability in murine and nonhuman primate models. mBio. 2018;9:e02449-17. doi: 10.1128/mBio.02449-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rezza G, Weaver SC. Chikungunya as a paradigm for emerging viral diseases: evaluating disease impact and hurdles to vaccine development. PLoS Negl. Trop. Dis. 2019;13:e0006919. doi: 10.1371/journal.pntd.0006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Powers AM, Brault AC, Tesh RB, Weaver SC. Re-emergence of chikungunya and O’nyong-nyong viruses: evidence for distinct geographical lineages and distant evolutionary relationships. J. Gen. Virol. 2000;81:471–479. doi: 10.1099/0022-1317-81-2-471. [DOI] [PubMed] [Google Scholar]
- 90.Yoon IK, et al. High rate of subclinical chikungunya virus infection and association of neutralizing antibody with protection in a prospective cohort in the Philippines. PLoS Negl. Trop. Dis. 2015;9:e0003764. doi: 10.1371/journal.pntd.0003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Langsjoen RM, et al. Molecular virologic and clinical characteristics of a chikungunya fever outbreak in La Romana, Dominican Republic, 2014. PLoS Negl. Trop. Dis. 2016;10:e0005189. doi: 10.1371/journal.pntd.0005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bustos Carrillo F, et al. Epidemiological evidence for lineage-specific differences in the risk of inapparent chikungunya virus infection. J. Virol. 2019;93:e01622-18. doi: 10.1128/JVI.01622-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen R, Wang E, Tsetsarkin KA, Weaver SC. Chikungunya virus 3′ untranslated region: adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog. 2013;9:e1003591. doi: 10.1371/journal.ppat.1003591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stapleford KA, et al. Whole-genome sequencing analysis from the chikungunya virus Caribbean outbreak reveals novel evolutionary genomic elements. PLoS Negl. Trop. Dis. 2016;10:e0004402. doi: 10.1371/journal.pntd.0004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Lamballerie X, et al. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: a sign of things to come? Virol. J. 2008;5:33. doi: 10.1186/1743-422X-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kraemer MUG, et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 2019;4:854–863. doi: 10.1038/s41564-019-0376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tsetsarkin KA, et al. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl Acad. Sci. USA. 2011;108:7872–7877. doi: 10.1073/pnas.1018344108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Diaz-Gonzalez EE, et al. First report of Aedes aegypti transmission of chikungunya virus in the Americas. Am. J. Trop. Med. Hyg. 2015;93:1325–1329. doi: 10.4269/ajtmh.15-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haddow AD, et al. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012;6:e1477. doi: 10.1371/journal.pntd.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grard G, et al. Zika virus in Gabon (Central Africa)—2007: a new threat from Aedes albopictus? PLoS Negl. Trop. Dis. 2014;8:e2681. doi: 10.1371/journal.pntd.0002681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lanciotti RS, et al. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008;14:1232–1239. doi: 10.3201/eid1408.080287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Musso D, Gubler DJ. Zika virus. Clin. Microbiol. Rev. 2016;29:487–524. doi: 10.1128/CMR.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu Y, et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature. 2017;545:482–486. doi: 10.1038/nature22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Azar SR, Diaz-Gonzalez EE, Danis-Lonzano R, Fernandez-Salas I, Weaver SC. Naturally infected Aedes aegypti collected during a Zika virus outbreak have viral titres consistent with transmission. Emerg. Microbes Infect. 2019;8:242–244. doi: 10.1080/22221751.2018.1561157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Azar SR, Weaver SC. Vector competence: what has Zika virus taught us? Viruses. 2019;11:867. doi: 10.3390/v11090867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu, J. et al. Role of mutational reversions and fitness restoration in Zika virus spread to the Americas. Nat. Commun.10.1038/s41467-020-20747-3 (2021). [DOI] [PMC free article] [PubMed]
- 107.Weger-Lucarelli J, et al. Vector competence of American mosquitoes for three strains of Zika virus. PLoS Negl. Trop. Dis. 2016;10:e0005101. doi: 10.1371/journal.pntd.0005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith DR, et al. African and Asian Zika virus isolates display phenotypic differences both in vitro and in vivo. Am. J. Trop. Med. Hyg. 2018;98:432–444. doi: 10.4269/ajtmh.17-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duggal NK, et al. Differential neurovirulence of African and Asian genotype Zika virus isolates in outbred immunocompetent mice. Am. J. Trop. Med. Hyg. 2017;97:1410–1417. doi: 10.4269/ajtmh.17-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marchette NJ, Garcia R, Rudnick A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am. J. Trop. Med. Hyg. 1969;18:411–415. doi: 10.4269/ajtmh.1969.18.411. [DOI] [PubMed] [Google Scholar]
- 111.Weaver SC. Host range, amplification and arboviral disease emergence. Arch. Virol. Suppl. 2005;19:33–44. doi: 10.1007/3-211-29981-5_4. [DOI] [PubMed] [Google Scholar]
- 112.Shan C, et al. A Zika virus envelope mutation preceding the 2015 epidemic enhances virulence and fitness for transmission. Proc. Natl Acad. Sci. USA. 2020;117:20190–20197. doi: 10.1073/pnas.2005722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vasilakis N, et al. Potential of ancestral sylvatic dengue-2 viruses to re-emerge. Virology. 2007;358:402–412. doi: 10.1016/j.virol.2006.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Althouse BM, et al. Potential for Zika virus to establish a sylvatic transmission cycle in the Americas. PLoS Negl. Trop. Dis. 2016;10:e0005055. doi: 10.1371/journal.pntd.0005055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Young KI, et al. Abundance and distribution of sylvatic dengue virus vectors in three different land cover types in Sarawak, Malaysian Borneo. Parasit. Vectors. 2017;10:406. doi: 10.1186/s13071-017-2341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Swei A, Couper LI, Coffey LL, Kapan D, Bennett S. Patterns, drivers, and challenges of vector-borne disease emergence. Vector Borne Zoonotic Dis. 2020;20:159–170. doi: 10.1089/vbz.2018.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guzman MG, et al. Dengue: a continuing global threat. Nat. Rev. Microbiol. 2010;8:S7–S16. doi: 10.1038/nrmicro2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weaver SC, Chen R, Diallo M. Chikungunya virus: role of vectors in emergence from Enzootic cycles. Annu. Rev. Entomol. 2019;65:313–332. doi: 10.1146/annurev-ento-011019-025207. [DOI] [PubMed] [Google Scholar]
- 119.Conway MJ, Colpitts TM, Fikrig E. Role of the vector in arbovirus transmission. Annu. Rev. Virol. 2014;1:71–88. doi: 10.1146/annurev-virology-031413-085513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dong S, et al. Chikungunya virus dissemination from the midgut of Aedes aegypti is associated with temporal basal lamina degradation during bloodmeal digestion. PLoS Negl. Trop. Dis. 2017;11:e0005976. doi: 10.1371/journal.pntd.0005976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smithburn KC. Neutralizing antibodies against arthropod-borne viruses in the sera of long-time residents of Malaya and Borneo. Am. J. Hyg. 1954;59:157–163. doi: 10.1093/oxfordjournals.aje.a119630. [DOI] [PubMed] [Google Scholar]
- 123.Aliota MT, et al. Zika in the Americas, year 2: what have we learned? What gaps remain? A report from the Global Virus Network. Antivir. Res. 2017;144:223–246. doi: 10.1016/j.antiviral.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cassadou S, et al. Emergence of chikungunya fever on the French side of Saint Martin island, October to December 2013. Euro Surveill. 2014;19:20752. doi: 10.2807/1560-7917.ES2014.19.13.20752. [DOI] [PubMed] [Google Scholar]
- 125.Machado LC, et al. Genome sequencing reveals coinfection by multiple chikungunya virus genotypes in a recent outbreak in Brazil. PLoS Negl. Trop. Dis. 2019;13:e0007332. doi: 10.1371/journal.pntd.0007332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsetsarkin KA, Chen R, Weaver SC. Interspecies transmission and chikungunya virus emergence. Curr. Opin. Virol. 2016;16:143–150. doi: 10.1016/j.coviro.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]