

Abstract
SARS-CoV-2 is the causative agent of COVID-19, the ongoing global pandemic. It has posed a worldwide challenge to human health as no effective treatment is currently available to combat the disease. Its severity has led to unprecedented collaborative initiatives for therapeutic solutions against COVID-19. Studies resorting to structure-based drug design for COVID-19 are plethoric and show good promise. Structural biology provides key insights into 3D structures, critical residues/mutations in SARS-CoV-2 proteins, implicated in infectivity, molecular recognition and susceptibility to a broad range of host species. The detailed understanding of viral proteins and their complexes with host receptors and candidate epitope/lead compounds is the key to developing a structure-guided therapeutic design.
Since the discovery of SARS-CoV-2, several structures of its proteins have been determined experimentally at an unprecedented speed and deposited in the Protein Data Bank. Further, specialized structural bioinformatics tools and resources have been developed for theoretical models, data on protein dynamics from computer simulations, impact of variants/mutations and molecular therapeutics.
Here, we provide an overview of ongoing efforts on developing structural bioinformatics tools and resources for COVID-19 research. We also discuss the impact of these resources and structure-based studies, to understand various aspects of SARS-CoV-2 infection and therapeutic development. These include (i) understanding differences between SARS-CoV-2 and SARS-CoV, leading to increased infectivity of SARS-CoV-2, (ii) deciphering key residues in the SARS-CoV-2 involved in receptor–antibody recognition, (iii) analysis of variants in host proteins that affect host susceptibility to infection and (iv) analyses facilitating structure-based drug and vaccine design against SARS-CoV-2.
Keywords: SARS-CoV-2, structural bioinformatics, protein 3D structures, structure prediction, mutation/variation, therapeutics
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 19 (COVID-19), which is an ongoing pandemic, causing severe health and socioeconomic burden worldwide. As of 7 September, globally 26 763 217 COVID-19 infection cases, and 876 616 deaths are reported by the WHO [1].
SARS-CoV-2 (previously known as 2019-nCoV), is a single-stranded positive-sense RNA virus, belonging to the genus Betacoronavirus and the family Coronaviridae [2]. SARS-CoV-2 and two related coronaviruses namely SARS-CoV (responsible for an outbreak in 2002–03) and MERS-CoV (Middle East respiratory syndrome coronavirus, the agent of an outbreak in 2012) are known to cause severe disease in humans. The genome of SARS-CoV-2 is 29.9 Kb in length, which encodes for 29 proteins (4 structural proteins, 16 non-structural proteins and 9 accessory proteins) [3–5].
Since the first sequenced genome of SARS-CoV-2 isolated in Wuhan City, China, over 90 000 genome sequences have been deposited in Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/). Three-dimensional (3D) structures were rapidly solved for the key target proteins in SARS-CoV-2 and host proteins, namely the spike protein, RNA-dependent RNA polymerase (RdRp), main protease (Mpro or 3CLpro), Papain-like protease (NSP3 or PLpro) and human angiotensin-converting enzyme 2 (hACE2) [6–11]. As of October 2020, there are over 370 structures deposited in the Protein Data Bank (PDB), corresponding to 21 SARS-CoV-2 proteins [12].
Understanding the functional role of 3D structures is essential to understand viral evolution and transmission, as well as to guide therapeutic research. Structural biology has contributed to the successful development of antiviral therapeutics for other human diseases including HIV and Influenza [13–17]. Kearns [16] has highlighted the important role of structural biology in determining 3D structures of SARS-CoV-2 proteins, in time frames enabling speedy structure-based drug and vaccine development against SARS-CoV-2. In the absence of available vaccines against SARS-CoV-2, structure-guided approaches are being used to identify promising candidates for antigens, particularly from the spike protein which is responsible for mediating infection [18–20]. Several potential drug molecules have been recently proposed for re-purposing based on protein-structure based algorithms (reviewed in Wang et al. [21]). The ongoing efforts by various structural biology groups worldwide are providing valuable data through online resources, as well as insights from analyses of the protein structures. Such insights include the identification of key residues involved in receptor/antibody/drug binding and the impact of variants on viral infectivity/antigenicity and may inform the design of potential drug/vaccine candidates, serving as the foundation for designing the next generation of coronavirus therapeutics.
This manuscript provides (i) an overview of structural bioinformatics tools and resources that provide information on experimentally determined macromolecular structures, theoretically inferred 3D models, descriptions of protein dynamic properties from molecular simulations, structure-based functional annotations, data on variant impacts and therapeutics data for COVID-19 and (ii) a summary of how these tools are informing our understanding of the mechanism of SARS-CoV-2 infection and disease response, and how they are guiding therapeutic strategies.
Experimentally determined 3D structures of SARS-CoV-2 proteins
Experimentally determined structures of macromolecules play an essential role in the effort to discover and develop effective drug molecules against target viral organisms [22, 23]. The worldwide PDB (wwPDB) [24] manages the global archive of macromolecular structures, the PDB [25], with over 165 000 protein and nucleic acid structures and over 30 000 interacting ligand molecules. As of October 2020, 21 of the 29 viral proteins of SARS-CoV-2 have over 300 experimentally determined structures. The overwhelming majority of these structures have canonical amino acid sequences, but a few structures have modified residues, such as PDB 7JR4 (N-Methyl Lysine) or PDB 6XB0 (S-Hydroxycysteine), while a few structures have engineered mutations, such as PDB 6WRH. The majority of these structures focus on the Replicase polyprotein 1ab (over 250 entries covering 10 processed mature proteins to date: Host translation inhibitor, Papain-like proteinase, 3C-like proteinase, RNA-directed RNA polymerase, Helicase, Uridylate-specific endoribonuclease, 2′-O-ribose methyltransferase and non-structural proteins 7, 8 and 9), while most of the non-structural proteins lack experimentally determined structures. As of October 2020, 49% of the sequence of Replicase polyprotein 1ab is covered by experimentally determined structures, 76% of the Spike glycoprotein, 56% of the nucleoproteins, while the ORF proteins have coverages ranging between 39 and 86%. The archived structures are available to the public through the web services of the wwPDB consortium members, namely PDBe [26], RCSB PDB [27] and PDBj [28]. The electrostatic potential maps determined using electron microscopy are archived in the Electron Microscopy Data Bank (EMDB) [29], with the raw EM data available from EMPIAR (Table 1) [30], covering molecular structures from single proteins to organelles and cells (Figure 1).
Table 1.
wwPDB consortium members providing access to experimentally determined macromolecular structures and EMPIAR providing access to raw EM data
| Data resource | Landing page | Example SARS-CoV-2 entry |
|---|---|---|
| Protein Data Bank in Europe (PDBe) | https://pdbe.org | https://pdbe.org/5rgg |
| Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) | https://rcsb.org | https://www.rcsb.org/structure/5rgg |
| Protein Data Bank Japan (PDBj) | https://pdbj.org | https://pdbj.org/mine/summary/5rgg |
| Electron Microscopy Data Bank (EMDB) | https://emdb-empiar.org/ | https://www.ebi.ac.uk/pdbe/entry/emdb/EMD-22126 |
| Electron Microscopy Public Image Archive (EMPIAR) | https://empiar.org/ | https://www.ebi.ac.uk/pdbe/emdb/empiar/entry/10404/ |
Figure 1.
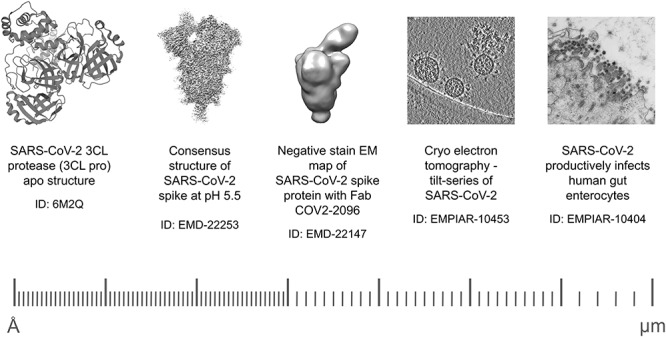
Structural information of SARS-CoV-2 from single proteins to organelles. Structural information on SARS-CoV-2 range from high-resolution single protein structures in PDB to lower resolution EM maps in EMDB and organelles and cells in EMPIAR.
Traditionally, PDB entries correspond to single experiments, covering distinct segments of a protein or nucleic acid sequence. However, to gain valuable biological insights, it is often necessary to collate all the available structural information on the complete length of a protein. The PDB in Europe—Knowledge Base (PDBe-KB) [31] is a community-driven data resource managed by the PDBe team that aims to address this need. PDBe-KB entry pages focus on full-length proteins keyed on their Universal Protein (UniProt) Resource [32] identifiers. These pages provide an aggregated overview of all the available structural information in the PDB in addition to functional and biophysical annotations and cross-references. For example, the aforementioned SARS-CoV-2 protein, Replicase polyprotein 1ab, has over 200 individual PDB entries covering various processed proteins within its sequence (https://www.ebi.ac.uk/pdbe/pdbe-kb/proteins/P0DTD1). Over 160 of these PDB entries correspond to the processed protein 3C-like proteinase, with over 140 distinct small molecules observed to interact with this protein. The strength of an aggregated view is apparent when examining the ligand-binding residues of all these PDB entries side-by-side (Figure 2). The pattern of residues which consistently interact with small molecules underlines their importance and may help to understand the effects of variation at these amino acid positions, in addition to focusing fragment-based drug development efforts on binding pockets defined by the most frequently interacting residues.
Figure 2.
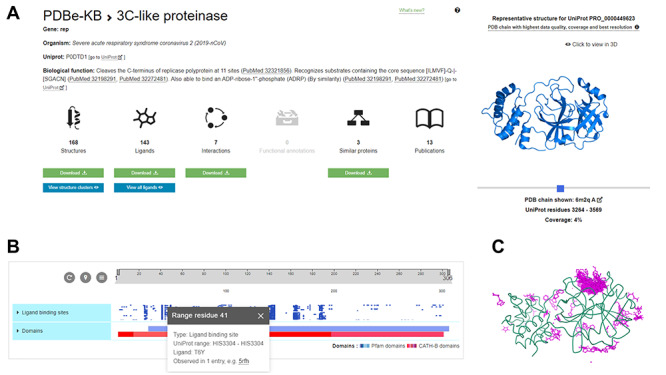
Aggregated overview of structural data for the SARS-CoV-2 3C-like proteinase. Collating all the available structural information on the protein level can yield valuable insights. This is demonstrated in the aggregated view of SARS-CoV-2 3C-like proteinase at PDBe-KB (https://pdbekb.org/proteins/PRO_0000449623) that collates data from over 160 PDB entries (panel A). The residue-level interactions between the protein and over 140 distinct small molecules can be visualized both on a 2D sequence feature viewer (panel B) and using a molecular graphics viewer (panel C), superposing every small molecule and highlighting residues that are consistently involved in binding to various small molecules.
In response to the COVID-19 pandemic, many of the leading data resources for protein structures took the initiative in providing similar overviews of all the available experimentally determined or theoretical structure information. The COVID-19 pages of PDBe and RCSB PDB provide overviews of available PDB entries, while the COVID-19 Data Portal of EMBL-EBI places the known structural information in the context of other data domains such as sequences, predicted structures and genomic data resources (Table 2).
Table 2.
Data resources providing overviews of the available structural information on SARS-CoV-2 proteins
| Service name | Access URL |
|---|---|
| PDBe COVID-19 Portal | https://www.ebi.ac.uk/pdbe/covid-19 |
| RCSB PDB COVID-19 Page | https://rcsb.org/covid19 |
| PDBj COVID-19 Page | https://pdbj.org/featured/covid-19?tab=all |
| BMRB COVID-19 Page | http://www.bmrb.wisc.edu/coronavirus.shtm |
| EMBL-EBI COVID-19 Data Portal | https://www.covid19dataportal.org |
| Swiss-Model COVID-19 Page | https://swissmodel.expasy.org/repository/species/2697049 |
| Coronavirus3D | https://coronavirus3d.org |
| Complex Portal COVID-19 Page | https://www.ebi.ac.uk/complexportal |
| InterPro COVID-19 Page | https://www.ebi.ac.uk/interpro/proteome/uniprot/UP000464024 |
| UniProt COVID-19 Entry Pages | https://covid-19.uniprot.org |
| 3DBioNotes-WS COVID-19 Page | https://3dbionotes.cnb.csic.es/ws/covid19 |
| PDBSum COVID-19 Page | https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/covid-19.html |
| Protepedia COVID-19 Page | http://proteopedia.org/wiki/index.php/Coronavirus_Disease_2019_%28COVID-19%29 |
The SWISS-MODEL [33, 34] web service provides an overview of both experimentally determined and theoretical models on their portal dedicated to COVID-19 as does the Godzik lab at Coronavirus3D [35]. While the Complex Portal [36] collates all the annotated SARS-CoV-2 protein complexes on their pages with a particular focus on the SARS-CoV-2 Spike—human ACE2 receptor complex which provides a key host–virus interaction that allows the virus to enter human cells.
Proteopedia (http://proteopedia.org/) [37], contains pages for PDB structures, which along with a visualization of the structure provide various information such as structural description, functions, mechanisms of actions etc. In addition, the morph tool (http://proteopedia.org/cgi-bin/morph) helps morph large conformational changes in a protein, which can be visualized in the form of a movie. The resource contains pages related to SARS-CoV2 (http://proteopedia.org/wiki/index.php/Coronavirus_Disease_2019_%28COVID-19%29). Along with the visualization of the structures and their description, Proteopedia contains a movie about the closed to open conformation transition of Spike protein.
While having open access to the macromolecular structures of SARS-CoV-2 proteins is essential for understanding the biological mechanisms of COVID-19 infection, it is often insufficient unless these structures are investigated within their biological context [31]. Domain and family annotations of SARS-CoV-2 proteins are key, and data resources such as CATH [38], SUPERFAMILY [39], SCOP and SCOP2 [40, 41], and InterPro [42] provide comprehensive information on these focus areas, such as the COVID-specific pages of InterPro.
There is a rich ecosystem of additional specialist data resources that provide parts of this biological context through functional and biophysical annotations [43–46]. However, the dispersed nature of these data providers hampers FAIR access to annotations, and therefore, it is important to have platforms that collate such information [47]. Data platforms like UniProt-KB [32], PDBe-KB [31] and PDBSum [48] play an important role in making these valuable annotations more accessible.
Resources containing predicted 3D structures
Experimentally determined structures of proteins provide important insights into their functions that can help in the design of therapeutics and in understanding the mechanism of infection. However, the determination of macromolecular structures by experimental methods such as X-ray crystallography, cryo-EM or nuclear magnetic resonance (NMR) spectroscopy is time-consuming, costly and manually intensive. In the absence of experimental structures, theoretical modeling of protein structures can help provide insights into their functions and mechanisms. These theoretical models can be built using homology modeling, threading or ab initio techniques [49].
As described above, various repositories including Coronavirus3D [35], SWISS-Model [34], Aquaria [50] and the Krokin lab repository [51] display both experimentally determined structures and template-based models for SARS-CoV-2 proteins. Other groups have applied tools such as trROSETTA [52], C-I-TASSER [53] and AlphaFold [54] as well as Kiharalab (http://www.kiharalab.org/covid19/index.html) that exploit various machine learning strategies to predict ab initio models for the SARS-CoV-2 proteins and provide their models in an open-access manner. Most of these are exploiting state-of-the-art modeling strategies. For example, AlphaFold was one of the best performing tools in the latest CASP13 experiment [55]. An important caveat is that these models are of unproven and variable quality and it is important to consider the associated model quality scores and perhaps consider further refinements (as discussed below).
SWISS-MODEL has developed a server providing models of the SARS-CoV-2 proteins in addition to models of the associated host interactor proteins, guided by curated associations from IntAct (Figure 3) [56]. Each model is associated with various quality assessment scores such as QMEAN [57], QMEA [33], MolProbilty results [58]. For the host proteins that interact with SARS-CoV-2, information is also provided on natural variants, nucleotide-binding sites, active sites, metal-binding sites and zinc finger motifs. Each protein is associated with links to the corresponding InterPro [42], UniProt [32] and STRING annotation pages [59]. Annotations from sequences similar to the host proteins in other organisms are also provided. In addition to modeling individual viral and host proteins, SWISS-MODEL also contains models of 5 hetero-oligomeric complexes between the viral and host proteins.
Figure 3.
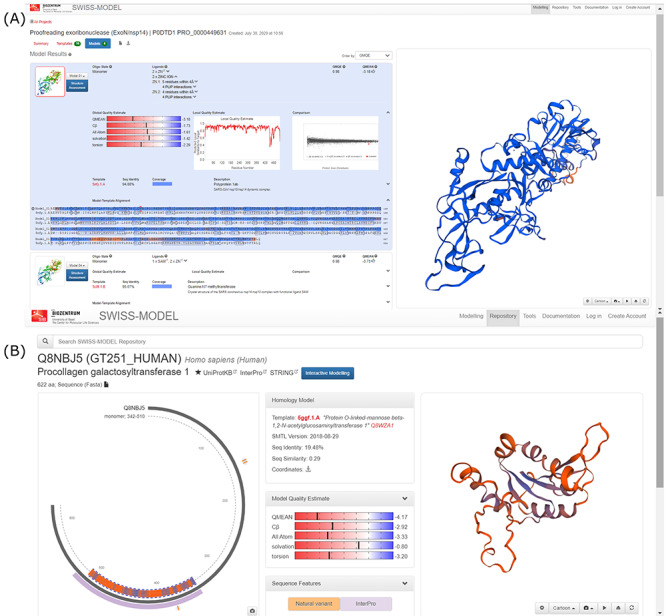
Screenshots from the SWISS-MODEL SARS-CoV-2 web resource (A) model of the viral NSP14 (B) model of the host interactor Procollagen galactosyltransferase. Shown along with the models are the quality estimates, template alignment, etc.
Other resources providing a wide range of functional annotations include Aquaria (Figure 4), which in addition to displaying experimentally determined structures/homology models of the SARS-CoV-2 proteins also displays comprehensive functional information on subcellular localization, functions, related proteins and interactors [60]. These data are collected from a range of resources including CATH (CATH-Superfamily, and functional family (FunFam) annotations), UniProt (active site, metal-binding site, zinc finger region, domain annotations, etc.), SNAP2 (mutational sensitivity, mutation impact score) [61] and PredictProtein (secondary structure, solvent accessibility, conservation, relative B-factor, disordered region) [62]. Aquaria also allows users to add additional custom features to the protein structures.
Figure 4.
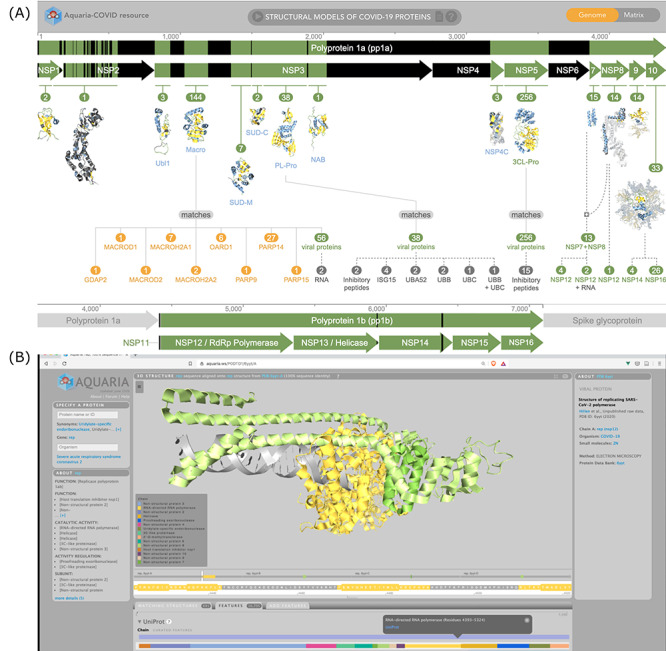
Screenshots from Aquaria web resource (A) Homepage showing all the SARS-CoV-2 proteins (B) RNA polymerase complex colored using UniProt chain features.
Research is facing a unique situation where scientific output has accelerated. Echoing the large amount of information related to COVID-19 made available without review by peers, the vast majority of SARS-CoV-2 or host counterpart proteins are recorded in the PDB without embargo period and not linked to peer-reviewed publication [63]. On 19 August 2020, 269 of the 337 entries tagged ‘COVID-19’ were released in the PDB before publication. Each PDB structure and its associated validation report provides information on the quality of the structure and in some cases the quality of these structures can be improved. With the goal of providing a better structure for the design or discovery of therapies, structure refinement experts have established multiple web resources (COVID-19 bioreproducibility [63], a structural task force at Thorn lab [https://github.com/thorn-lab/coronavirus_structural_task_force], BUSTER [https://www.globalphasing.com/buster/wiki/index.cgi?Covid19] and ISOLDE [64]) to collect the outcome of careful curation and refinement of COVID-related PDB structures. The COVID-19 bioreproducibility web interface provides search filters to select structures with given properties. One filter makes it possible to obtain re-refined structures including small molecule ligands, which constitute the best possible starting point for computer-aided drug design (CADD).
To improve the quality of the theoretical models, Feig lab [65] refined models built using Alphafold, SWISSModel, Baker’s lab (ROSETTA-Gremlin) and trROSETTA using high-resolution molecular dynamics (MD) simulations. This included models of the membrane proteins (M, E, nsp4 and nsp6) which were refined to take into account the membrane environment. The group has also developed a model for the Spike-M protein complex in the endoplasmic reticulum membrane environment. Details on the above-mentioned resources are given in Table 3. As with experimental structures, the models have important applications in MD simulations, structure-based therapeutics design, and in the discovery and study of the effect of mutations (See below).
Table 3.
Details of the various resources containing structures/models of SARS-CoV-2 proteins
| Name, URL and resource leader | Presence of experimental structures | Presence of theoretical structures | Information on human proteins | Type of modeling technique used | Brief description of the modeling technique | Criteria used to decide the model quality | Model refinement technique used | Additional comments |
|---|---|---|---|---|---|---|---|---|
| SWISS-MODEL Repository https://swissmodel.expasy.org/repository/species/2697049 Torsten Schwede |
No | Yes | Yes | Homology modeling | SWISS-MODEL is a fully automated protein structure homology-modeling server, using template-based modeling techniques to model 3-dimensional proteins, as well as homo- and heteromeric complexes. | The model quality estimation tool QMEAN is used to estimate model confidence. | Manually curated a set of 3D homology models and experimental structures for SARS-CoV2 virus proteins and complexes and host proteins. Host proteins have been associated with information from Interpro, STRING, UniProt, variant data, metal-binding site, etc. | |
| Aquaria https://aquaria.ws/covid19 Seán I. O’Donoghue |
Yes | Yes | No | Homology Modeling | Homology models were built by searching sequence homologs of regions of proteins based on a machine learning-based searching method | Contains additional information from CATH, Uniprot, SNAP2, PredictProtein tools. Also contains information about subcellular localization, function, interacting partners, similar proteins, etc. | ||
| Protein Structure Modeling for SARS-CoV-2 at Kiharalab http://www.kiharalab.org/covid19/index.html Daisuke Kihara |
Yes | Yes | No | ab initio modeling, Homology modeling | Inter-residue distances, H-bonds and angles were first predicted with a deep neural network. Then Rosetta was used for modeling the protein structure in ab initio fashion. But when template structures were available, homology modeling was performed. | MD and coarse-grained short simulation | ||
| Coronavirus3d https://coronavirus3D.org Adam Godzik |
Yes | Yes | No | Homology modeling | MODELLER/SWISS-MODEL equivalent | Sequence similarity | Also contains variant data | |
| Structural genomics and interactomics of SARS-COV2 novel coronavirus http://korkinlab.org/wuhan Dmitri Krokin |
Yes | Yes | No | Homology Modeling | MODELLER | Also, contain functional site mapping and a model of the viral interactome. | ||
| CoV3D https://cov3d.ibbr.umd.edu Brian Pierce |
Yes | Yes | No | Glycan modeling on the spike | N-glycan modeling and refinement using Rosetta | |||
| COVID-19 molecular structure and therapeutic hub https://covid.molssi.org/ MOLSSI, BioExcel |
Yes | Yes | Yes | Hub containing the crystal structures, models, docking results, MD studies, therapeutics collated from various groups | ||||
| covid-19.bioreproducibilty https://covid-19.bioreproducibility.org Mariusz Jaskolski, Wladek Minor, Alex Wlodawer |
Yes | No | Yes | Modeling in electron density using Coot, model refinement in Refmac | MolProbity; ligand validation in Twilight; expert evaluation | Maximum Likelihood structure factor refinement | The resource contains refined experimental structures | |
| Coronavirus Structural Taskforce https://github.com/thorn-lab/coronavirus_structural_task_force Andrea Thorn |
Yes | No | No | Modeling in the electron density map | MolProbity based validation | The resource contains refined experimental structures | ||
| GOL COVID-19 https://www.globalphasing.com/buster/wiki/index.cgi?Covid19 Global phasing group |
Yes | No | No | Modeling in the electron density map | Buster | The resource contains refined experimental structures | ||
|
https://drive.google.com/drive/u/0/folders/1S5qJtCnK00NrcbwwBNgImUMewhiBkyPa
Randy Read |
Yes | No | No | Modeling in the electron density map | ISOLDE | The resource contains refined experimental structures | ||
| C-I-TASSER on COVID-19 https://zhanglab.ccmb.med.umich.edu/COVID-19/ Yang Zhang |
No | Yes | No | Deep-learning, threading and ab initio modeling | Integrating contact-maps from deep-learning with I-TASSER fragment assembly simulations | C-score (threading score and convergence of simulation decoys) | Fragment-guided MD simulations | Contains a complex structure of the host-viral complex. |
| Alphafold https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19 Deepmind group |
No | Yes | No | Deep learning | Trained a neural network to make predictions of the distances between pairs of residues. The potential of mean force was constructed to accurately describe the shape of a protein. The resulting potential was optimized by a simple gradient descent algorithm to generate structures. | Best performing tool in the last CASP13 experiment. | ||
| Protein structure models for COVID-19 proteins https://yanglab.nankai.edu.cn/trRosetta/SARS-2-CoV/ Jianyi Yang |
No | Yes | No | Ab initio modeling | trRosetta builds the protein structure based on direct energy minimizations with a restrained Rosetta. The restraints include inter-residue distance and orientation distributions, predicted by a deep residual neural network. | Estimated TM-score | Rosetta fast relax | |
| SARS-CoV2 Protein Structure Models http://feig.bch.msu.edu/web/lab/news/sars-cov-2-protein-structure-models/ Michael Feig |
No | Yes | No | MD-based protein structure refinement | The resource contains refined theoretical structures | |||
| SARS-CoV-2 EVcouplings: mutations, function and structure https://marks.hms.harvard.edu/sars-cov-2/ Nathan Rollins, Debora Marks, Chris Sander |
No | Yes | Yes | Evolutionary couplings contact prediction | Direct interactions were predicted from co-evolution in natural sequences and used to predict direct 3D contacts to solve the 3D fold. The interaction data was also used to predict the effect of mutations on the fitness of the sequence. | 3D structure predictions are compared to experimental structures of SARS-CoV-2 proteins and/or potential homologs. | Contains the prediction of contacts between residues of proteins, and the effect of mutations. They plan to model the proteins and their complexes in the future. Also, contains a comparison between the SARS-CoV2 with SARS-CoV and the nearest coronavirus bat RATG31. | |
| CASP_Commons https://predictioncenter.org/caspcommons/models_consensus2.cgi |
No | Yes | No | Various model accuracy estimates such as ProQ3D, QmeanDISCO, etc | CASP competition for template free modeling of SARS-CoV2 proteins without any template from PDB. Contains structure details, comparisons between models, and scoring of the model by different scoring schemes. | |||
| CAPRI Docking https://www.capri-docking.org/ |
No | Yes | Yes | Various established model accuracy estimates | CAPRI competition to predict the models of COVID complexes. |
CASP commons
The critical assessment of protein structure prediction (CASP) is a worldwide experiment that evaluates structure prediction methods by blind assessment (https://predictioncenter.org/caspcommons/). During the COVID-19 pandemic, CASP initiated the SARS-CoV-2 structure prediction initiative to provide consensus structures of the viral proteins with no experimental structures or known homologs for comparative modeling. CASP identified 10 proteins (nsp2, nsp4, nsp6, PL-PRO, ORF-3a, Membrane protein, ORF-6, ORF-8, ORF-10 and ORF-7b) for the template-free modeling initiative. CASP has created a total of 32 targets from these 10 proteins either to be modeled as a whole protein or domains of the protein.
To date, ~3400 models have been deposited for these and are available in CASP_Commons together with information on their likely accuracy. Each model has been compared pairwise with all other models to provide a per residue score or a global score for the entire model. A global consensus score, which is the average similarity score of a model to all other models is also provided, calculated as the average pairwise local distance difference test (LDDT) [66] and Global distance test total score (GDTTS) [63] scores. The GDTTS score represents the average percentage of residues that are near in two structures optimally superimposed using four selected distance cutoffs. Per residue LDDT and S-score [67] are also provided, and are calculated as the average of all the pairwise residue scores between the models.
In addition to the consensus scores, various model accuracy estimates are provided (such as QmeanDisco [68], ProQ3D [69], VoroCNN [70]) and a predicted estimate of how far away (Cα-Cα distance) a particular residue is from the corresponding target residue position (probable experimentally determined) in their optimal model-target structural superimposition.
CAPRI COVID19 open science initiative
The Critical Assessment of Predicted Interactions (CAPRI) is a community-wide initiative that aims to evaluate tools/groups for the prediction of protein–protein complexes (https://www.ebi.ac.uk/pdbe/complex-pred/capri/; www.capri-docking.org). In April 2020, CAPRI came up with a COVID-19 Open Science initiative to predict the complexes of SARS-CoV-2 with host proteins. The individual component of the target complex was either an experimental structure or good quality theoretical model (built by homology or ab initio). Only those proteins for which reliable biological assembly information could be determined were selected, using the ProtCID server [71]. This resulted in 4 CAPRI targets (from the 332 host–viral interactions detected experimentally by Gordon et al. [72]) which will be open for complex modeling between mid-September and December 2020. A second round is planned in early 2021 with additional target complexes. All the models submitted by the various groups will be made available online immediately. In the absence of the experimental structure for comparison, all the submitted models will be evaluated for their quality, compared and ranked. The initiative also aims to derive consensus models wherever deemed appropriate.
Initiatives like CASP and CAPRI are playing an important role in deriving useful models of individual SARS-CoV-2 proteins and complexes whose structure has not yet been determined experimentally.
Resources containing molecular simulation data
A variety of computer simulation techniques can be used to model the dynamic behavior of molecules and macromolecules, which is intimately associate with their biological function. These techniques, when applied to protein systems, sample to a varying degree the conformational energy landscape of these systems. They may therefore be used to investigate the type of motion a protein undergoes, to estimate the stability of specific conformational states, or the affinity of a protein for binding partners. Various repositories have been developed to store molecular trajectories generated by simulations involving SARS-CoV-2 proteins (Table 4).
Table 4.
Details about repositories for MD simulation data
| Name of resource and group/team | URL | Force field | Description |
|---|---|---|---|
| CHARMM COVID library Wonpil Im |
http://www.charmm-gui.org/?doc=archive&lib=covid19 | CHARMM, NAMD, Gromacs, Amber, GENESIS and OpenMM | Simulation system (CHARMM, NAMD, Gromacs, Amber, GENESIS, and OpenMM) for running MD on COVID proteins. This resource does not contain MD simulation trajectories for the proteins or the complexes. Developed an all-atom model for the SARS-CoV2 spike protein with all the glycans attached along with building a membrane system for the spike protein simulations. |
| DE Shaw group DE Shaw |
https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/ | Variations of Amber on Anton2 supercomputer | Atomistic MD trajectories for SARS-CoV-2 proteins and their complexes (with other viral/host proteins, therapeutics). Simulations of 128 FDA approved drugs with viral targets. |
| The SIRAH-CoV-2 initiative Sergio Pantano |
https://www.cluster.uy/web-covid/ | SIRAH | Coarse-Grained trajectories for all SARS-CoV-2 proteins reported in the PDB |
| COVID-19 molecular structure and therapeutic hub MOLSSI, BioExcel |
https://covid.molssi.org/ | Multiple force fields | Hub containing the crystal structures, models, docking results, MD studies, therapeutics collated from various groups |
| BioExcel COVID19 Bioexcel, Barcelona Supercomputing Center, IRB Barcelona |
https://bioexcel-cv19.bsc.es/#/ | Multiple force fields | Hub containing atomic MD trajectories from different groups |
The CHARMM-GUI Archive-COVID-19 protein library contains inputs for running MD simulations with all atom models implemented in the CHARMM package [73, 74], while the SIRAH-CoV-2 initiative, by the Group of Biomolecular Simulations at Institut Pasteur of Montevideo offers access to trajectories from coarse-grained MD simulations on the SARS-CoV-2 proteins, based on the SIRAH force field [75]. DE-Shaw group has run multiple microseconds long all-atom MD simulations using their Anton-2 supercomputer for SARS-CoV-2 proteins in multiple states (closed, partially open, apo, monomeric or oligomeric forms), or for SARS-CoV-2 proteins interacting with each other, with host proteins, and with various molecular therapeutics. For example, an accelerated weighted ensemble simulation [76, 77] of the binding reaction between the receptor binding domain (RBD) of the viral Spike protein and the host ACE2 protein yielded multiple trajectories, providing important insight into the free energy landscape of the binding process. (https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/).
The BioExcel-COVID-19 portal (https://bioexcel-cv19.bsc.es/#/) is an open-access initiative integrating MD simulation data generated by various groups, including the DE-Shaw group. The portal provides views of the MD trajectory movies and links to all the MD trajectory data. The portal provides data such as the root-mean-square fluctuation of atomic positions, collective motions described by principal component analysis of these fluctuations, root-mean-square deviations from a reference structure or the apparent radius of gyration of the system. The list of available resources for molecular simulation data are given in Table 4.
Tools and resources for predicting protein intrinsic disorder and recognition motifs
In a significant fraction of human proteins and higher vertebrates, the entire polypeptide or segments are devoid of stable secondary or tertiary structures, when in isolation [78]. Such proteins or segments, commonly referred to as intrinsically disordered proteins (IDP) or intrinsically disordered regions (IDR), carry out a range of biological functions, more specifically in signaling and regulatory processes [79], and are also involved in several disease pathologies [80] The experimental characterization of IDPs/IDRs is challenging, considering the range of flexibility properties they tend to display (see [81, 82] for review). On the other hand, it is generally accepted that these properties are encoded by characteristic features of the amino acid sequence, leading to the development of a large number of tools for predicting protein intrinsic disorder from sequence alone (see [83] for review).
A combination of disorder prediction tools has been applied to proteins from SARS-CoV-2 and related viruses [84, 85], showing for example, that with two exceptions (Nsp8 and ORF6) the vast majority of SARS-CoV-2 proteins are predicted to be mostly structured proteins, containing on average a lower fraction of residues in IDRs than typical host proteins. Nonetheless, some IDRs identified in SARS-CoV-2 could be directly related to function, such as those at cleavage sites in its replicase 1ab polyprotein. Using a different set of tools designed to infer molecular recognition features, representing sequence motifs within IDRs involved in the recognition of binding partners, almost all SARS-CoV-2 proteins were found to contain such motifs [84]. A section in the ELM database gathers information on putative short linear motifs in SARS-CoV-2 [86].
More extensive annotations describing flexibility properties and recognition features of SARS-CoV-2 proteins are made available in the resource developed by the Vranken lab at http://bio2byte.be/sars2/. These include predictions of local flexibility properties of the polypeptide, using DynaMine [87, 88], intrinsic disorder predictions with DisoMine [89], early folding regions identified using EFoldMine [90], as well as helix/sheet coil predictions, and regions predicted to interact with other proteins using SerenDip [91].
Structural bioinformatics resources mapping variation data to structures
Discovery of novel variants in SARS-CoV-2 proteins, such as the D614G mutation in the Spike protein identified in Europe, and its significant correlation with the transmission, demonstrates the need for continuous efforts to rapidly analyze and monitor the functional impact of mutations [92]. Structural data are key to interpreting the likely impacts of the variant on the structure and function of the protein. Genome sequencing efforts across the world are being captured in resources like GISAID, enabling the continuous tracking of mutations in SARS-CoV-2 genomes. Various tools have emerged to analyze the variant data, including MicroGMT [93] and CoV-GLUE [94]. CoV-Glue provides an up-to-date database of amino acid variants (concerning the first Wuhan-Hu-1 strain), insertions and deletions. In addition to variation in the viral genomes, several human variation resources such as gnomAD [95] and dbSNP [96] also provide comprehensive annotations on human proteins and their variants for proteins likely to be interacting with SARS-CoV-2.
In parallel, various structure-based resources have been established or extended to explore the structural mapping of these variants and the insights they give on the evolution of the virus or the impacts of mutations on susceptibility to the disease or therapeutics (Table 5). Such resources include Coronavirus3D [35], described above and developed by the Godzik group, Viral Integrated Structural Evolution Dynamic Database (VIStEDD) by Prokop group [97], COVID-3D [98] by the Ascher group and SWISS-MODEL. COVID-3D uses both virus variation (obtained using 45 000 SARS-CoV-2 genomes from GISAID) and population variants data for two human genes (ACE2, B0AT1) available from a wide range of population variation resources from Asia, UK, Europe and the US [98]. Spatial mapping of SARS-CoV-2 variants is displayed in 3D, to aid analysis of potential impacts on the structure and the interactions with other proteins. The molecular impact of mutations on protein–protein interactions was predicted using their in-house developed program called mCSM-PPI2 (http://biosig.unimelb.edu.au/mcsm_ppi2/). mCSM-PPI2 utilizes an integrated computation approach involving graph-based structural signatures, evolutionary information, energetic terms and inter-residue network metrics [99]. This structural information is combined with evolutionary and population variation data to identify sites less likely to undergo mutations in the future, which is invaluable information for the design of therapeutic compounds. These analyses are provided for two major therapeutic targets: the Spike and the Main Protease.
Table 5.
3D structure based resources focusing on variation/mutation data
| Name of resource | URL and reference | Description | Group/team |
|---|---|---|---|
| Coronavirus3D | https://coronavirus3d.org/index.html | The resource maps SARS-CoV-2 genomic variations, from CNCB, using experimental structures as well as models | Adam Godzik |
| COVID-3D | http://biosig.unimelb.edu.au/covid3d/ | The resources utilize both virus (obtained from 45 000 SARS-CoV-2 genomes in GISAID) and population variant data (available from a wide range of population variation resources). The resource utilizes both experimental as well as predicted structures for SAR-CoV-2 proteins, for annotations, analyses and visualization of mutations | David Ascher |
| Viral Integrated Structural Evolution Dynamic Database | https://prokoplab.com/vistedd/ | The resource utilizes predicted models along with information from missense variants (from gnomAD database), MD simulations, and evolutionary mapping. It aims to gain insights into variation and dynamics properties of SARS-CoV-2 proteome | Jeremy Prokop |
| SWISS-MODEL | https://swissmodel.expasy.org/repository/species/2697049 | Provides models for SARS-CoV-2, annotations from UniProt and sites information from dbSNP | Torsten Schwede |
| EVCouplings (SARS-CoV-2: mutations, function and structure) | https://marks.hms.harvard.edu/sars-cov-2/ | For SARS-CoV-2 proteins, the resource provides, in silico deep mutation scans, visualization of mutations on 3D structures, structure prediction from coevolution | Debora Marks and Chris Sander |
| EACoV server | http://cov.lichtargelab.org/ | Provides evolutionary analysis of proteins from SARS-CoV-2 proteome using Evolutionary Trace (ET) method. For every protein, it identifies variants (unique, most frequent and all variants) and epitopes using ET-based approach. These are mapped on corresponding 3D structure. | Olivier Lichtarge |
Coronavirus3D uses SARS-CoV-2 genomic variations, from China National Center for Bioinformation (CNCB) and maps mutations to 3D structures, while the SARS-CoV-2 Dynamicome database, developed by Prokop and co-workers (VIStEDD) provides evolutionary data, structures, dynamics and human variant data (from gnomAD) for 24 proteins from SARS-CoV-2 and 3 human genes (ACE2, TMPRSS2 and SLC6A19). Variant information mapped to protein structures is also provided for SARS-CoV-2 proteins and their interactors in the CATH resource developed by the Orengo group (http://funvar.cathdb.info/). Constituent domains from the proteins are mapped to domain functional families in CATH and variants are visualized on representative structures to establish whether they lie close to known or predicted functional sites and could therefore impact the function of the protein.
Structural bioinformatics tools and resources to support therapeutics discovery
There is an urgent need to develop novel therapeutics to treat COVID-19. Identification of potent drugs through drug-repurposing offers a promising speedy solution. There are various ongoing efforts to compile ligand/small molecule data for SARS-CoV-2, while a few resources such as CoV-AbDAb, provide 3D structure data for SARS-CoV-2 binding antibodies (listed in Table 6).
Table 6.
Structural bioinformatics resources for therapeutics
| Name of resource | URL and reference | Description | Group/team |
|---|---|---|---|
| HADDOCK | https://www.bonvinlab.org/covid/ | A virtual screening platform to guide drug-repurposing: It has performed screening of thousands of chemical compounds against key SARS-CoV-2 proteins | Alexander Bonvin group |
| Diamond Light Resource (COVID-19 moonshot) | https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html | Diamond has initiated a crystallographic fragment screening using the main protease | Diamond Group and collaborators (Credits: https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Credits.html) |
| Postera.ai | https://postera.ai/covid/activity_data | In vitro activity of compounds provided by the COVID Moonshot initiative | PostEra, San Francisco, USA |
| Fragalysis browser | https://fragalysis.diamond.ac.uk/viewer/react/landing/ | 3D visualization and analysis of hit fragments from the XChem initiative | XChem Group, Diamond, UK |
| FOLDING@HOME | https://foldingathome.org/2020/07/28/introducing-covid-moonshot-weekly-sprints-help-us-discover-a-new-therapy/ | Openly distributed computational power for further CADD on compounds obtained by the COVID Moonshot initiative. | FOLDING@HOME consortium (https://foldingathome.org/about/the-foldinghome-consortium/) |
| Exscalate4COV | https://www.exscalate4cov.eu/index.html | High-Performance Computing resources for virtual screening of user-submitted compounds | Exscalate4COV’s Consortium (https://www.exscalate4cov.eu/consortium.html) |
| G2Pdb (Guide to PHARMACOLOGY) | https://www.guidetoimmunopharmacology.org/immuno/index.jsp | Curated data on 64 ligands associated with SARS-CoV-2 | G2Pdb Curation Team (https://www.guidetopharmacology.org/about.jsp#curation) |
| Chemical Checker-based expansion of drugs | https://sbnb.irbbarcelona.org/covid19/ | The group is regularly collecting suggested COVID-19 drugs from literature with different levels of supporting evidence. To date, 307 literature-derived candidates are identified. Chemical Checker tool is used to find small molecules (that exhibit similar bioactivity and chemical features) to these reported drugs | Patric Alloy group |
| FMODB | https://drugdesign.riken.jp/FMODB/covid-19.php | The database is designed to FMO calculations using crystal structures (in PDB) for several SARS-CoV-2 proteins. | The FMODD consortium |
| D3Targets-2019-nCoV | https://www.d3pharma.com/D3Targets-2019-nCoV/index.php | A molecular docking based web server for predicting drug targets and virtual screening against COVID-19 | Zhijian Xu and Weiliang Zhu |
| COVID-19 docking server | https://ncov.schanglab.org.cn/ | A docking server for prediction of the binding modes between the targets and their ligands such as peptides, small molecules, antibodies (The server uses docking tools: CoDockPP and Autodock Vina) | Shan Chang group |
| SARS-CoV-2 data at ChEMBL | https://www.ebi.ac.uk/chembl/ | The page provides a link to the summary of SARS-CoV-2 related data at ChEMBL | ChEMBL |
| Virus Chemogenomics resource [104] | https://www.cbligand.org/g/virus-ckb | Provides data of 3D structures, antiviral drugs, docking utilities, gene and protein annotations | Feng lab |
| CoV-AbDab: The Coronavirus Antibody Database | http://opig.stats.ox.ac.uk/webapps/covabdab/ | This is the first database that compiles antibodies known to bind SARS-CoV-2 and other beta coronaviruses (SARS-CoV-1, MERS-CoV): contains data on >380 patented/published/antibodies and nanobodies that bind to at least one betacoronavirus | The Oxford Protein Informatics Group |
The High Ambiguity Driven protein–protein DOCKing (HADDOCK) web-server [100] has been used for large scale screening of approved drugs against several drug targets in SARS-CoV-2 (main protease, RdRp) and ACE2 receptor. HADDOCK uses a flexible docking approach, which exploits information derived from known (or predicted) protein interfaces and their small molecule ligands [100].
An important initiative called XChem was set up in the Diamond Light Source, UK’s national synchrotron science facility (refer to section Exploiting structural data for drug design). This has already provided a large number of experimental 3D structures of complexes between molecular fragments and the SARS-CoV-2 3CLpro and macrodomain, as a source of inspiration for fragment-based drug design. This initiative led to the creation of the COVID MoonShot project, to experimentally validate compounds obtained by crowdsourcing the drug design effort. The activity of the assayed molecules is made available in collaboration with PostEra on the MoonShot webpage (https://postera.ai/covid/activity_data). Some of the produced molecules achieved noticeable activity, with dissociation constants sometimes lower than 1 μM. X-ray structures were obtained by Diamond XChem for many of these lead compounds in complex with their target, and are viewable through the XChem Fragalysis browser (https://fragalysis.diamond.ac.uk/viewer/react/landing/). These structures can be used as starting points to perform alchemical free energy calculations, and select the most promising lead compound derivatives thanks to the computational power provided by FOLDING@HOME (https://foldingathome.org/2020/07/28/introducing-covid-moonshot-weekly-sprints-help-us-discover-a-new-therapy/).
The EXSCALATE4COV consortium, composed of 18 institutions from 7 European countries, aim to identify new treatments against SARS-CoV-2 (https://www.exscalate4cov.eu/index.html). The EXSCALATE4COV Drugbox allows users to submit structures of compounds they designed. The massive high-performance computing resources of EXSCALATE4COV are then used to perform virtual screening of these compounds against COVID-19 targets to flag the most promising ones as candidates for experimental assays. The results of the latter are planned to be published in open-access databases such as ChEMBL [101]. Some of the target structures were obtained by homology modeling in collaboration with SWISS-MODEL, which also published these models on a dedicated web page (Table 1). In the context of the EXSCALATE4COV consortium, there has also been a comprehensive mapping of druggable cavities on SARS-CoV-2 proteins performed by A. Pedretti and coworkers [102] using the Pockets 2.0 plug-in for the VEGA ZZ suite of programs, whose results were made freely available (https://ds-814.cr.cnaf.infn.it:8443/ex4cov-public/Binding_Sites/).
Many other CADD tools have been created or modified to address the need for COVID-19 treatments. A noticeable example is the application of the deep learning-based Ligand Design tool of Cyclica (https://www.cyclicarx.com/special-perspectives/using-cyclicas-technology-to-identify-repurposed-drug-candidates-for-covid-19) to a collection of approved and developmental drugs to predict their polypharmacology profiles across human proteins associated with SARS-CoV-2 infection and viral drug targets. These profiles were stored in the PolypharmDB resource, and mining this resource yielded promising repurposed drug candidates against COVID-19 that bind host and viral proteins [103].
The ‘COVID-19 Molecular Structure and Therapeutics Hub’ (a collaboration between the Molecular Sciences Software Institute (MolSSI) and BioExcel), is aggregating large and diverse information on data, tools, and results from the numerous other resources. The hub groups information on protein targets, their experimentally determined or modeled 3D structures, known therapeutics, results of molecular modeling simulations and links to useful CADD tools and molecular libraries (https://covid.molssi.org). Similarly, the COVID-19 data portal of the European Bioinformatics Institute (https://www.covid19dataportal.org) and the COVID-19/SARS-CoV-2 Data in PubChem (https://pubchemdocs.ncbi.nlm.nih.gov/covid-19) also collects valuable information for therapeutics.
Other types of analyses, useful as support to drug design, include those reported by FMODB [104] [The database of quantum mechanical data based on the fragment molecular orbital (FMO) method, https://drugdesign.riken.jp/FMODB/covid-19.php]. These involve FMO calculations on PDB structures for several SARS-CoV-2 proteins.
Overview of some of the applications of structural bioinformatics tools and resources to study COVID-19
Structure-guided efforts have made a major contribution to understanding the molecular aspects of SARS-CoV-2 infection by—(i) revealing changes between SARS-CoV and SARS-CoV-2 that promote increased virulence of SARS-CoV-2 (ii) providing information on key sites in the viral proteins, e.g. identification of critical residues involved in receptor and antibody recognition (iii) analysis of variants in host interactors that affect susceptibility to infection, e.g. in human and other hosts and (iv) supporting structure-based drug and vaccine design against COVID-19. These themes are reviewed in more detail below.
Structural insights into the functioning of viral proteins
SARS-CoV-2 has higher infectivity compared to its predecessor SARS-CoV [6–8]. Various MD simulations and binding free-energy calculations point to mutations at the binding interface of the virus as responsible for the higher infectivity [105, 106]. These mutations introduce a larger number of more persistent hydrogen bonds and better stacking interactions at the interface as evident from MD studies [106]. Furthermore, the ACE2-α1 helix of the host makes additional contacts with SARS-CoV-2 spike protein compared to SARS-CoV [106]. Alanine scanning has also shown the importance of individual residues at the spike interface and suggests that all-natural variants of the spike protein have a higher binding affinity to ACE2 compared to SARS-CoV [105].
The spike protein of SARS-CoV-2 is highly glycosylated, which helps in evasion of the immune response [107–110]. The importance of N-glycans at residues N165 and N234 in stabilizing the ACE2 receptor-binding domain have been revealed by MD studies, which also exposed vulnerabilities of the glycan moieties which can be used for vaccine development [110, 111]. MD simulations along with tomographic analyses revealed the contribution of the three hinges in the stalk domain of the spike protein that gives the globular domain freedom to scan the host cell surface [112] and have confirmed the importance of the ACE2 peptidase domain α1 helix for the binding event, insights that can aid inhibitor design [113]. MD simulations combined with evolutionary analyses helped identify two highly dynamic conserved patches on the surface of the N and nsp6 viral proteins that might be regions of protein–protein interaction [114].
The structures also help point toward allosteric sites in the proteins. Protein contact networks, perturbation response scanning, softness-based prediction of intersubunits affinity and binding site prediction tools points toward an allosteric modulation region on the spike protein [115–117]. Elastic network model analysis, binding site predictions followed by MD simulations has been used to predict the allosteric sites on the main protease protein [118, 119]. The identification of allosteric binding sites on the main protease has also been identified by mass spectrometry based studies [120]. The binding of small molecules on these sites is proposed to reduce the rate of substrate processing and dimerization.
Structural insights into variant impacts on phenotypes
Protein structures have been used to understand structural and functional impacts of genetic variations (namely residue mutations) both in SARS-CoV-2 and its associated host proteins in the context of infectivity, host susceptibility, antigenicity and the emergence of antibody escape mutants [121–125]. Early work by Brielle et al. [121] exploited evolutionary and structural data and used MD simulations to obtain important insights into host receptor recognition exerted by distinct pathogenic coronaviruses. The authors compared structures of spike protein: ACE2 complexes in HCoV-NL63 (PDB ID: 3KBH), SARS-CoV (PDB ID:2AJF) and SARS-CoV-2 (based on a predicted model using the template: PDB 3SCI). Although each of these three viruses uses ACE2 as their natural receptor, their work showed that they possess a distinct ACE2 binding interface, featuring a different network of residue–residue contacts. In particular, they found that the SARS-CoV-2: ACE2 complex contains a higher number of contacts, a larger interface area, and decreased interface residue fluctuations relative to the SARS-CoV:ACE2 complex, even though the two viruses have comparable ACE2 binding affinities.
Once the crystal structures of the spike RBD from SARS-CoV-2 in complex with ACE2 became available (Figure 5-I), they provided further insights into key receptor binding residues and the structural basis of receptor recognition and infection (Figure 5-I) [7, 124, 126]. Shang et al. [7] determined the crystal structure of the RBD-ACE2 complex in SARS-CoV-2 (PDB ID: 6VW1) and compared it with those of SARS-CoV and bat coronavirus (RaTG130), showing that SARS-CoV-2 has a more compact conformation of the ACE2-binding ridge, compared to SARS-CoV. Several residue changes in the SARS-CoV-2 RBD were reported to stabilize two virus-binding hotspots at the interface, leading to its increased ACE2-binding affinity.
Figure 5.
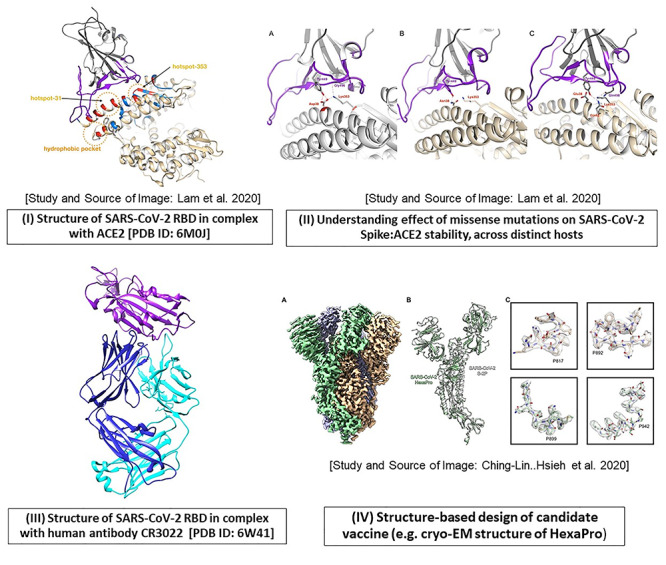
An account of structure-based studies on spike (S) protein of SARS-CoV-2. Some of the applications using spike protein in SARS-CoV-2 are illustrated, as follows. (I) The crystal structure of the SARS-CoV-2 receptor-binding domain (RBD; shown in purple) in complex with human ACE2 receptor (gray) is depicted using Chimera [PDB ID: 6M0J]. The direct contact residues (shown in red while residues in secondary shell are shown in blue) as well as key hotspot positions 31 and 353 (encircled in orange) are studied by various groups [6, 7]. (II) The impact of mutations at hotspot residue 353, on the stability of the RBD-ACE2 complex in various hosts (A. human, B. horseshoe bat, C. cat and dog) are illustrated (Source of images (I) and (II) and more details in Lam et al. [124]). (III) The crystal structure of RBD (purple) in complex with human antibody CR3022 (heavy chain: blue, light chain: cyan) is resolved (PDB ID: 6W41). (IV) Structure-based design of prefusion conformation of spike: design of vaccine candidate namely HexaPro: the high resolution cryo-EM structure is solved by Hsieh et al. [18]; Source of Image: Hsieh et al. [18].
Furthermore, the analysis of mutations in the spike protein of SARS-CoV-2 demonstrated that certain combinations of mutations, confer increased infectivity [92, 127]. The authors use dynamic tracking of SARS-CoV-2 variant across populations world-wide (collecting over 13 000 spike sequences from GISAID) and report the impacts of 80 variants and 26 glycosylation site modifications, on infectivity (using 26 cell lines) and reactivity to a panel of neutralizing monoclonal antibodies and sera from 10 convalescent patients (8 from Wuhan and 2 from Shandong) [127]. They identified the D614G mutation as one of the most prevalent and provide clinical evidence for the higher infectivity of the virus that is carrying this mutant, with however no effect in disease severity. The D614G mutation allows greater availability for receptor binding and reduced antibody interactions [128] and favors the open state of the Spike protein (as compared to the closed state) which is the state that interacts with the ACE2 receptor on the host [129]. In particular, the authors use multiple replicas microsecond all-atom simulations to assess the impact of the D614G mutation on the spike open and closed states. Results show that the mutation influences the interaction energy between the spike protomers, which induces a higher population of (stabilizes) infection-capable (open) states, thereby increasing infectivity.
Some groups have exploited the structural data and information on host variants to predict those likely to destabilize the Spike-ACE2 complex. MacGowan and Barton [130] used mCSM-PPI2 (a platform of programs implementing a machine learning method that uses graph-based structural signatures) to analyze the destabilizing effects of ACE2 missense variants in human proteins reported in the gnomAD database, and validated their approach using binding assays. Some variants cause enhanced spike-ACE2 interaction (G326E). Whereas others inhibit spike binding (E37K, G352V and D355N), and could confer a high degree of resistance to infection. The authors proposed a therapeutic strategy exploiting a mutant ACE2 with a tailored affinity toward the Spike protein.
Structural analyses of Spike-ACE2 complexes have also been used to predict the susceptibility of a broad range of vertebrate hosts to SARS-CoV-2 infection [123, 124, 131]. Lam et al. [124] performed comprehensive structural modeling for 216 vertebrate orthologues of ACE2 and TMPRSS2 and studied impacts of known mutations in ACE2 on the stability of the Spike: ACE2 complex, also using the mCSM-PPI2 tools (Figure 5-II). The available experimental data were used to establish thresholds for likely infection, and when applied to the predictions indicated that many mammals are susceptible to SARS-CoV-2 infection, including ~30 animals in close contact with humans, while most birds, reptiles and fish are not likely to be infected.
Exploiting structural data in vaccine design
Several groups are using structure data to design stable spike protein candidates for vaccines [18, 19]. Hsieh et al. [18] characterized around hundred structure-guided spike designs to obtain a prefusion-stabilized SARS-CoV-2 spike protein. HexaPro was identified as the most promising stable variant (as it exhibits high expression and can withstand temperature stress and storage at room temperature). The cryo-EM structure of HexaPro (at 3.2 Å), confirmed that it retains the prefusion spike conformation [PDB ID:6XKL; EMDB: EMD-22221, EMD-22222] (Figure 5-IV). The Discovery of HexaPro has implications for the accelerated production of stable forms of pre-fusion spikes, and a new generation of coronavirus vaccines [18]. Other studies have used combinatorial computational approaches (structural modeling, MD/molecular docking) to design stable candidates for potential epitope-based vaccines [19, 132–134].
Exploiting structural data for drug design
The severity of the current pandemic has led to unprecedented efforts in collaborative initiatives to discover therapeutic solutions against COVID-19, especially since the development of vaccines and building the necessary level of immunity within the world population to end the pandemic need time. On the other hand, studies resorting to structure-based drug design for COVID-19 are plethoric and show realistically good promise to yield results sooner, mainly by building on the wealth of knowledge from past epidemics, including those caused by CoVs, mainly SARS-CoV, MERS-CoV. Indeed, structure-based design has already demonstrated its value for the discovery of antiviral agents, such as nelfinavir and oseltamivir, to treat HIV and influenza infections, respectively [135]. However, it is too early to report newly discovered molecules validated as effective therapies against COVID-19. Nevertheless, we describe some of the most popular strategies and putative targets arising from on-going efforts. Additionally, a few quick-wins obtained by drug repurposing approaches can be underlined [136]. We briefly review such efforts directed toward the most salient macromolecular host or viral targets for which structural knowledge is available [137]. The very large body of work not yet subjected to peer review, archived in various preprint servers, is not discussed.
Although antiviral pharmacology has proven challenging because of the efficient resistance mechanisms of viruses, therapeutic agents targeting directly the infective moiety are less prone, in principle, to interfere with physiologically important pathways in the host body. This strategy offers a priori a better profile regarding side effects and toxicity. A second strategy consists of modulating host proteins. Although this approach has the advantage of reducing viral resistance to therapy, the risk of negative impacts on important host physiological pathways should be born in mind with regards to unwanted side effects. The great majority of pharmacological strategies targeting host proteins are associated with virus entry mechanisms. Structural data of key targets in their apo and complex form have been made available as discussed above (see Table 1). The characterization of key binding site residues and interactions with drug candidates has been systematically reviewed by Joeng and co-workers, in the context of structure-based drug design [138]. When available, the structure of drug binding sites in the potential therapeutic targets described below can be efficiently analyzed through the PDBe-KB interface (https://www.ebi.ac.uk/pdbe/pdbe-kb/protein).
Viral protein targets
SARS-CoV-2 Spike (S) protein (UniProt ID: P0DTC2), is one of the most studied proteins in CoV, with numerous 3D-structures (54 and 30 resolved by CryoEM and X-ray, respectively to date) yielding insights into the interaction with ACE2 receptor at the atomic level. SARS-CoV-2 is inferred to have increased pathogenicity compared to SARS-CoV due to a better molecular recognition by ACE2 [139]. In particular, two conformations have been precisely defined by Cryo-EM (PDB ID: 6VSB, 6VXX) and lay the basis for a therapeutic strategy aimed at stabilizing the inactive form of protein S, which cannot be cleaved by host proteases (TMPRSS2, furin or cathepsin L) [8]. Recently, a dominant mutation was reported in protein S, which is thought to provide more structural stability and transmission efficacy to SARS-CoV-2 and has emerged as a new target for immunology or drug design approaches [140]. Furthermore, targeting a specific region of protein S (the receptor-binding motif, RBM) with antibodies, peptides or small molecules was shown to reduce recognition by ACE2 and display antiviral potential for SARS-CoV [9]. Since the molecular binding of SARS-CoV-2 to ACE2 is different [6] an interesting strategy consists of designing more effective and selective inhibitors for this receptor [141]. Protein S is also a preferred target for antibody therapy against COVID-19 [142]. To date, 18 X-ray and 12 cryoEM structures of SARS-CoV-2 protein S with Fab can be found in the PDB.
Another structural protein fundamental for delivering CoV RNA into the cell is the nucleocapsid (N) protein (UniProt ID: P0DTC9). The first X-ray structure of the N-terminal domain binding to RNA (PDB ID: 6M3M) [143] and subsequent high-resolution dimeric forms (e.g. PDB IDs: 7C22, 6WZO) enabled the comparison of surface properties of SARS-CoV-2 and other CoVs N proteins. Promising drug discovery approaches for blocking viral replication and transcription rely on the adaptation for SARS-CoV-2 of inhibitors optimized for MERS-CoV [144, 145]. Furthermore, the immunogenicity of protein N was demonstrated as sufficient to consider it as a favorable vaccine target and for the detection of antibodies directed against CoV [146, 147].
Among the well-conserved non-structural proteins in CoVs, one of the most studied is the main protease, or 3CLpro (UniProt ID: P0DTD1), which plays a critical role in virus replication and control of host response. Numerous experimental structures confirm the high level of similarity of 3CLpro in SARS-CoV and SARS-CoV-2 at the atomic levels [148]. This increases the probability that the inhibitors designed for SARS-CoV (and for MERS-CoV) will be efficient against the SARS-CoV-2. Leveraging this antiviral bioactivity knowledge, diverse chemotypes targeting 3CLpro constitute excellent starting points for medicinal chemistry or peptide design programs [149]. Salient examples include vinylsulfone covalent protease inhibitors active at the nanomolar level on SARS-CoV [150] and the structure-based design of sub-micromolar α-ketoamide series involving co-crystallization (PDB entries: 6Y2F and 6Y2G) [148, 151]. Moreover, the output of a virtual screening targeting a high-resolution co-crystal of SARS-CoV-2 3CLpro with an irreversible peptidomimetic inhibitor (PDB ID: 6LU7) suggested disulfiram and carmofur for repurposing among other late-stage drug-candidates. Both drugs demonstrated micromolar activity in vitro [10]. Broader virtual screening campaigns have been undertaken, some involving many hundreds of millions of compounds [152], and also targeting mutations naturally occurring in the binding site of SARS-CoV-2 3CLpro [153]. Potential hit compounds are currently being experimentally confirmed. Furthermore, lopinavir and ritonavir, two antiviral molecules used in multitherapy against other viral infections (mainly HIV), have demonstrated effective inhibition of 3CLpro and are administered to COVID-19 patients [154]. Various fast-track clinical trials are evaluating both inhibitors co-administered with therapeutics having other modes of action (notably, Interferon-β) [155]. Lately, two FDA-approved anti-protease drugs used in chronic hepatitis C, boceprevir and telaprevir, were described as potent inhibitors of SARS-CoV-2 3CLpro in vitro [156]. High-resolution co-crystals involving these new chemotype covalent inhibitors were very recently released (PDB IDs: 6ZRU, 6WNP, 6XQU, 6ZRT, 6XQS, 7C6S, 7COM, 7C7P), as shown in Figure 6. Very recently, a peptide aldehyde compound and its bisulphite prodrug used in veterinary medicine to treat severe CoV peritonitis in cats, have been measured in the nanomolar range as reversible inhibitors of SARS-CoV-2 3CLpro inhibitors [157]; different high-resolution cocrystals are available (PDB entries: 6WTT, 6WTJ, 7C6U, 7C8U, 7CBT, 7BRR).
Figure 6.
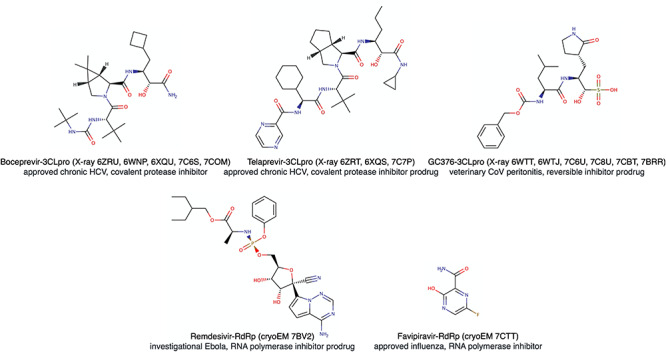
Antiviral drugs repurposed against COVID-19, for which 3D structures of the ligand-protein complex were determined experimentally. Both approved drugs against chronic hepatitis C, boceprevir and telaprevir, inhibit SARS-CoV-2 main protease (3CLpro) and are clinically evaluated in different association. Veterinary molecule against feline CoV infection, GC376, is a prodrug generating an irreversible nanomolar 3CLpro inhibitor and will probably enter clinical phase. Remdesivir, a late development drug against Ebola virus, is a SARS-CoV-2 RNA-Dependent RNA Polymerase (RdRp) strong inhibitor and received emergency use authorization for COVID-19 in Europe and USA. Favipiravir, used in influenza infection, is a RdRp inhibitor investigated against SARS-CoV-2 infection.
3CLpro was chosen as a COVID-19 target for a massive fragment-based crystallographic screening initiative called XChem (https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html) [158] because high-resolution X-ray structures were rapidly obtained. The druggable active site of 3CLpro was soaked with different fragment libraries resulting in 71 co-crystallized active site fragments plus 3 hits at the dimer interface. This rich set of structural information for drug design is available through PDB portals or the XChem website (Figure 7). The initiative has evolved into a crowdsourcing collaboration called COVID MoonShot (https://postera.ai/covid/activity_data), which allows any medicinal chemist to submit rationally designed structures or even ship promising samples to be tested experimentally; more than 500 compounds were subject of in vitro assays on SARS-CoV-2 3CLpro on 11 May 2020, with a handful of sub-micromolar inhibitors.
Figure 7.
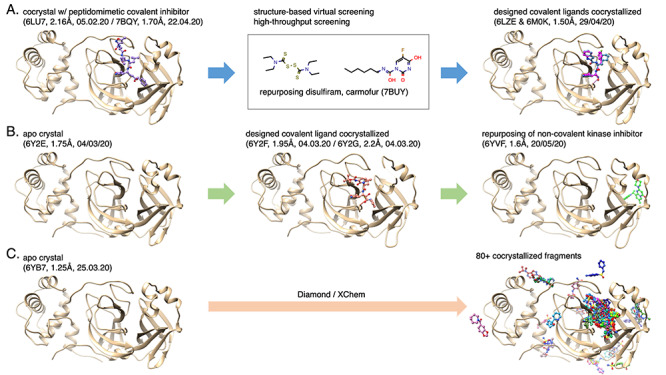
SARS-CoV-2 Main protease (3CLpro): earliest, advanced structure-based drug discovery routes (pdb entry, resolution, release date). The proteins are displayed in beige cartoon and the ligands in colored ball-and-sticks. (A) the first crystal of 3CLpro enabled the discovery of disulfiram and carmofur as potential drugs to repurpose through structure-based virtual and high-throughput screenings [10] as well as the design of peptidomimetic covalent ligands [159]; (B) an apo crystal was subject to structure-based design for covalent ligands [148] and recently brought rational for repurposing a non-covalent small molecule initially developed as a kinase inhibitor; (C) The XChem initiative started with the resolution of an apo structure allowing soaking experiments that led to more than 80 cocrystals with covalent and non-covalent fragments, most are located in the active site.
The main protease has also been a target for many different structure-based studies, including the design of covalent inhibitors guided by X-ray crystallography (PDB IS: 6LZE and 6M0K) [159] and the description of pharmacophore through MD simulations involving His41, Gly143 and Glu166 [160]. Another in-depth description of the molecular interactions between the SARS-CoV-2 3CLpro and its peptide-like inhibitor N3 using FMO (please see section Structural bioinformatics tools and resources to support therapeutics discovery) suggests guidelines for chemical optimization in situ [161]. Furthermore, Altay et al. [162] stress the need to exploit the structural data when determining the risk of drug resistance and for identifying multiple target sites in potential drug candidates.
The papain-like protease or PLpro (Nsp3 protein) is involved in virus replication as well as in the signaling from host infected cells to neighboring healthy cells [163]. Whereas the similarity between different CoVs is slightly lower than for 3CLpro, PLpro remains a viral target of choice to inhibit. This is generally tackled using a strategy of translation from SARS-CoV to SARS-CoV-2 [164]. Antiviral activity and co-crystallized structures (PDB IDs: 6WUU and 6WX4) of PLpro peptide inhibitors are reported as well as zinc-containing advanced compounds for repurposing [165]. Recently, a series of high-quality co-crystals of SARS-CoV-2 PLpro with naphthyl ligands (PDB IDs: 7JN2, 7JNV, 7JIW, 7JIT and 7JIR) was made available by the Center for Structural Genomics of Infectious Diseases, which will help the structure-based design of novel inhibitors (https://csgid.org/deposits/index/title_form:SARS).
Moreover, the macrodomain or X-domain (also part of the multidomain Nsp3 protein) is an interesting target for therapeutics against COVID-19, since macrodomain-deficient CoVs have been observed to be unable to replicate [166]. The macrodomain was crystallized and chosen as a target for an XChem fragment screening initiative. The output is a total of about 80 co-crystallized fragments, among which 54 are inside the active site of the macrodomain. This information is available through PDB portals or the XChem website.
A drug target identified at an early stage of the current outbreak is the RNA-Dependent RNA Polymerase (RdRp). This non-structural enzyme, a central player for viral genome replication and protein synthesis, is well conserved among RNA viruses [167]. The high similarity of RdRp from different viral sources encouraged the rapid application to SARS-CoV-2 of already proven therapeutic approaches. Nucleoside analogs developed for the treatment of other viral diseases were promptly evaluated for COVID-19 repurposing. For example, using a standard molecular docking protocol toward a homology model of SARS-CoV-2 RdRp, remdesivir was rapidly proposed as one of the most promising inhibitors able to force viral transcription to finish prematurely [168]. Although some clinical results have been less positive than expected, this nucleoside prodrug, in late development against Ebola, is still one of the main medications for COVID-19 patients in severe conditions [169, 170].
The 3D-structure of remdesivir with RdRp and two other SARS-CoV-2 important proteins (nsp7 and nsp8) has been solved by electron microscopy (PDB ID: 7BV2, see Figure 6). This, together with more recent cryoEM complexes in different conformations with RNA (PDB IDs: 7BZF and 7C2K), provide excellent opportunities for initiating the design of narrower spectrum inhibitors of this major therapeutic target [171, 172]. Other drugs targeting the RNA polymerase of other viruses are under clinical evaluation. Favipiravir, for instance, approved as a treatment against influenza, is currently being evaluated in COVID-19 patients and a complex cryo-EM structure was released very recently (PDB entry: 7CTT, see Figure 6) [173].
Finally, with a function also important for viral RNA synthesis, the helicase (nsp13) has been targeted in SARS-CoV with diverse small molecule inhibitors obtained by chemical screening [174, 175]. Until very recently, structure-based drug design for COVID-19 required reliable homology models built using 3D-structures of the SARS-CoV helicase (PDB entry: 6JYT), which shows more than 99% sequence identity with the SARS-CoV-2 protein. However, the design of new inhibitors will be boosted by the release of a SARS-CoV-2 helicase crystal structure resolved with a 1.94 Å resolution (PDB entry: 6ZSL).
Host protein targets
As described above, the Angiotensin I Converting Enzyme 2 (ACE2, UniProt ID: Q9BYF1) is the major SARS-CoV-2 receptor tightly bound to protein S and chiefly responsible for viral entry into the cell [176]. This early discovery yielded multiple attempts to modulate ACE2, located in the epithelial cells, essentially in the lung, liver and testis [177]. A supercomputer-based docking effort was achieved by virtually screening 8000 drugs or advanced drug-candidates toward an ensemble of geometries of SARS-CoV-2 S-protein:ACE2 complex generated by molecular dynamic simulations of a homology model [135]. The 77 compounds selected for predicted binding free energies are still under experimental validation to define the molecules to focus on for repurposing in COVID-19. To date, there are 20 unique ligands observed to be bound to the S-protein (https://www.ebi.ac.uk/pdbe/pdbe-kb/proteins/P0DTC2). Subsequently, the determination of co-crystals by X-ray (PDB entries 6M0J and 6LZG) confirmed the molecular recognition of SARS-CoV-2 protein S by ACE2 [6, 178]. Cryo-EM complexes with protein S and transporters provide further supramolecular structural insight that will help in creating targeted inhibitors (PDB ID: 6M17 and 6M18) [126]. Because of the physiological importance and the protective role of ACE2 in respiratory distress syndrome, a therapeutic solution relying on a full blockade of this receptor is questionable. Hence, the design of an engineered host ACE2 that optimizes the binding to Spike protein can also act as a viable inhibitor. Such design by using deep mutagenesis data has shown efficient binding to the spike protein, comparable with neutralizing antibodies [179]. Aside from the already discussed approaches based on viral targets, other host accessory proteins also involved in the viral entry can be modulated. Currently, preferred targets are host proteases that activate or support cell-virus membrane fusion.
The Transmembrane Serine Protease 2 (TMPRSS2, UniProt ID: O15393) expressed primarily in the gastrointestinal, urogenital and respiratory tracts, facilitates the entry of various viruses, including SARS-CoV-2, by priming of protein S for attachment to ACE2 [176]. The prodrug camostat, a protease inhibitor employed in chronic pancreatitis [180], has been demonstrated to lower CoV infection in cells and animal models. To date, there is no experimental structure of TMPRSS2 available to help rationalize the repurposing of camostat or other protease inhibitors. Also, bromhexine, an over-the-counter drug, sold since 1963 as a mucolytic and expectorant medication, has shown in vitro submicromolar inhibition of TMPRSS2 and may therefore be suitable for repurposing in COVID-19 [181, 182]. If validated, in addition to being safe, the molecule would be a good candidate for structural studies since it is chemically very different from known protease inhibitors. Unfortunately, homology modeling has proven difficult with only a few approximate TMPRSS2 modeled structures available.
Another serine protease expressed principally in the lungs is Furin (UniProt ID: P09958). Its cleavage action takes place at a specific site of protein S, not found in other CoVs. This finding partly explains the higher pathogenicity of SARS-CoV-2 and also depicts a potentially more selective therapeutic option for COVID-19 [183]. The design of furin inhibitors (peptide and small molecules) has been already explored in the past, in the context of oncology and infectious diseases, with some issues regarding intrinsic toxicity [183, 184]. The availability of high-resolution X-ray structures including peptide inhibitors (PDB entries: 6EQV, 6EQW, 6EQX) represents an interesting opportunity to speed-up structure-based approaches for the current pandemic [185]. Moreover, molecular analyses of furin bound to protein S of SARS-CoV-2, including MD, docking simulations and functional studies, unveiled the mechanisms of furin binding. It also rationalized the genetic variants, giving important indications for antibody and drug research [186].
Development of molecules targeting Cathepsin L (UniProt: P07711) emerged as an interesting approach as this serine protease is known to activate protein S of SARS-CoV-2 [187], whose penetration in the cell was found to be reduced after application of a nanomolar inhibitor (called SID26681509) [139]. The fact that cathepsin S is the only member of this family of enzymes to be related to viral entry [139] puts this protein at a safe position regarding the side effects of future drugs. On the other hand, selectivity to the target has to be considered at the early steps of the discovery process. For this, a wealth of structural knowledge can be beneficially brought to bear by analyzing the large collection of available crystal structures with different ligands (e.g. PDB 3HWN for a selectivity driven structure). Design of anti-COVID-19 drugs can also leverage the abundant medicinal chemistry aiming to target Cathepsin L in oncology [188–190].
Some kinases are being investigated as putative targets for COVID-19 treatments. Two examples are the Adaptor Protein 2 Associated Kinase 1 (AAK1, UniProt ID: Q2M2I8) and Cyclin G-Associated Kinase (GAK, UniProt ID: O14976). Both these serine–threonine kinases are involved in the entry, assembly and release of RNA viruses (e.g. Ebola, dengue and hepatitis C) [191]. Structure-based inhibitor design will greatly benefit from the co-crystallized structures of AAK1 with different types of ligands, such as an optimized broad-spectrum antiviral compound, nintedanib, an inhibitor used in various pulmonary pathologies (PDB ID: 5L4Q and 5TE0) [192]. Numerous medicinal chemistry efforts to design inhibitors of AAK1 have been reported before the COVID-19 outbreak and novel optimization work led to efficient antiviral small molecules, in particular against dengue [193, 194].
The Phosphatidylinositol 3-Phosphate 5-Kinase (PIKfyve, UniProt ID: Q9Y2I7) is a protein and lipid kinase involved in endocytosis [195]. SARS-CoV-2 entry is significantly reduced by PIKfyve inhibitors, vacuolin-1 or apilimod, a small molecule investigated in Crohn’s disease and psoriasis [196]. To date, no experimental structure of PIKfyve has been made available. However, many kinase templates are suitable to build reliable homology models. Finally, downstream of PIKfyve, the Two-Pore Channel protein (TPC2, UniProt ID: Q8NHX9) is worth mentioning as this voltage-gated channel has been shown to catalyze endocytosis after activation by phospholipids and to control Ebola virus entry in host cells [197]. The elucidation by cryoEM of the substrate-bound and unbound 3D geometries of human TPC2 (PDB IDs: 6NQ0, 6NQ1 and 6NQ2) [198] provides a great opportunity to apply virtual screening, either with existing drugs (including dopamine and estrogen receptors antagonists [199] validated against Ebola) or chemical libraries for repurposing or discovery programs aimed at expanding the therapeutic arsenal against SARS-CoV-2 infections.
In the spite of the many fast-tracks investigated by the urgency of this health crisis, these early stage therapeutic strategies, including drug repurposing, still require considerable clinical work to ensure efficiency and safety for the patient.
Exploiting structure in the design of antibodies
Deep mutation scanning of the RBD in the spike protein, using the crystal structure of the ACE2-bound RBD (PDB ID: 6M0J) and a cryo-EM structure of the full spike ectodomain (ID: 6VXX), is displayed in an interactive visualization of sequence-to-phenotype (https://jbloomlab.github.io/SARS-CoV-2-RBD_DMS/structures/), which reveals multiple mutationally constrained patches on the RBD surface that could be targeted by antibodies [200]. Furthermore, several mutations in the spike protein (N234Q, L452R, A475V, V483A) have been identified as resistant to monoclonal antibodies (mAbs) [92]. These variants are often found at interaction interfaces between the Spike glycoprotein and antibody chains, as is apparent from the aggregated view of this protein at PDBe-KB (https://www.ebi.ac.uk/pdbe/pdbe-kb/proteins/P0DTC2). These findings provide a framework that can inform the design of antibody cocktails for limiting the emergence of viral escape mutants [125, 201, 202]. In this context, the COVID-3D, resource (http://biosig.unimelb.edu.au/covid3d/; Table 5) provides a comprehensive annotation of mutations and visualization of mutation tolerant positions on viral proteins.
There have been several other studies analyzing the structural basis of antibody recognition using electron microscopy [125, 203, 204] and crystallography [142]. Yuan et al. [142] determined the crystal structure of a neutralizing antibody and studied a highly conserved epitope (namely CR3022) and its binding interactions with SARS-CoV-2 and SARS-CoV using structural modeling (Figure 5-III). Pinto et al. [125] indicated that a mixture of a human mAb (namely S309) in combination with other neutralizing mAbs showed enhanced levels of neutralization, thus providing support for developing cocktails of antibodies to limit the emergence of mutants capable of escaping neutralization.
Conclusions
Ongoing efforts by the worldwide structural biology community are providing valuable insights into the structural mechanisms of action between key targets and drug candidates/antibody epitopes. Structural biology can help explain the effect of amino acid variations on interactions with other proteins, leading to changes in the infection rate, associated symptoms etc. This knowledge, when combined with structure-guided efforts to design stable vaccine antigens provides an important foundation for countering the impacts of the disease. These vaccines/drugs should target the regions of the protein which do not mutate fast. Therapeutics against regions with high mutation propensities will make them strain specific. In summary, structure-guided approaches provide an important framework for understanding the increased virulence of this pathogen and for designing therapeutics and will be important for understanding the emergence of drug resistance and antibody-resistant variants of SARS-CoV-2.
Key Points
Since the discovery of the novel coronavirus SARS-CoV-2, experimental structures of key SARS-CoV-2 proteins, along with their complexes have been rapidly solved.
COVID-19 specific structural bioinformatics resources on experimental and predicted structures/models, functional annotations, molecular simulations, variant impact data and vaccine/drug therapeutics data have been developed and are reviewed in this article.
Structure-based tools/resources have also been used for structural mapping of variants which are helpful to gain insights into the evolution of the SARS-CoV-2 and to understand the impacts of mutations in the context of therapeutics.
In the absence of vaccines to combat COVID-19, the roles of structure-based CADD tools and structural bioinformatics in the design of new drugs, and the repurposing of existing ones are discussed in this article.
Conflict of interest: None declared
Vaishali P. Waman holds a Ph.D. in Bioinformatics. She is a postdoctoral Research fellow in the Orengo group, UCL, working on antimicrobial resistance and protein structural domain chopping.
Neeladri Sen holds a Ph.D. in Biology. He is working as a postdoctoral fellow with Prof. Orengo, UCL on improvement of sequence alignment to model proteins and identify functional residues.
Mihaly Varadi joined the PDBe team in 2015 where he worked first as a scientific programmer and more recently as the project leader of the Protein Data Bank in Europe—Knowledge Base.
Antoine Daina holds a Ph.D. in pharmaceutical sciences. He is Senior Scientist in the Molecular Modeling Group at SIB, Swiss Institute of Bioinformatics, in charge of developing and applying computational methods to academic and industrial drug discovery.
Shoshana J. Wodak holds a PhD in Biophysics, Columbia University, USA. She is a computational structural biologist and bioinformatician, specializing in modeling protein interactions. She directed research teams at the Free University of Brussels, EMBL-EBI and University of Toronto.
Vincent Zoete holds a PhD in organic chemistry and is assistant professor in the Department of Fundamental Oncology at the University of Lausanne and Group leader at SIB, where he develops and applies CADD and in silico protein engineering methods.
Sameer Velankar leads the Protein Data Bank in Europe team and is interested in macromolecular structure archiving and in the integration of macromolecular structure data with biomedical data resources.
Christine Orengo is professor of Bioinformatics at University College London. Her group develop algorithms for structure comparison/classification and for protein-function prediction, also for the analysis of functional genomics data.
Contributor Information
Vaishali P Waman, Orengo group, UCL.
Neeladri Sen, Orengo group, UCL.
Mihaly Varadi, Protein Data Bank in Europe—Knowledge Base.
Antoine Daina, Molecular Modeling Group at SIB, Swiss Institute of Bioinformatics.
Shoshana J Wodak, Columbia University, USA.
Vincent Zoete, Department of Fundamental Oncology at the University of Lausanne and Group leader at SIB.
Sameer Velankar, Protein Data Bank in Europe.
Christine Orengo, University College London.
Funding
V.P.W. acknowledges funding from Wellcome Trust. N.S. acknowledges Biotechnology and Biological Sciences Research Council BBSRC (BB/S020144/1). M.V. acknowledges the BBSRC via the FunPDBe (BB/P024351/1). S.V. acknowledges 104948/ Wellcome Trust/United Kingdom.
Data Availability
The online web-links for all the resources/tools, reviewed in this article are provided in main text.
References
- 1. Coronavirus Disease (COVID-19) Situation Reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (7 September 2020, date last accessed).
- 2. Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5:536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan JF-W, Kok K-H, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 2020;9:221–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. COVID-19 protein structures in the PDB . https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=n/a&template=covid19.html (7 September 2020, date last accessed).
- 6. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. [DOI] [PubMed] [Google Scholar]
- 7. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020;581:221–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181(e6):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020;582:289–93. [DOI] [PubMed] [Google Scholar]
- 11. Rut W, Lv Z, Zmudzinski M, et al. Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. biorxiv. 2020. 10.1101/2020.04.29.068890 [DOI] [PMC free article] [PubMed]
- 12. COVID-19 < EMBL-EBI . https://www.ebi.ac.uk/pdbe/covid-19 (28 September 2020, date last accessed)
- 13. Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature 1987;329:351–4. [DOI] [PubMed] [Google Scholar]
- 14. Blundell T, Carney D, Gardner S, et al. Knowledge-based protein modelling and design. Eur J Biochem 1988;172:513–20. [DOI] [PubMed] [Google Scholar]
- 15. Thomas SE, Mendes V, Kim SY, et al. Structural biology and the design of new therapeutics: from HIV and cancer to mycobacterial infections. J Mol Biol 2017;429:2677–93. [DOI] [PubMed] [Google Scholar]
- 16. Kearns S. Structure of the pandemic. Structure 2020;28:874–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Varghese JN. Development of neuraminidase inhibitors as anti-influenza virus drugs. Drug Develop Res 1999;46:176–96. [Google Scholar]
- 18. Hsieh C-L, Goldsmith JA, Schaub JM, et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020;eabd 369(6510):1501–1505. doi: 10.1126/science.abd0826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kar T, Narsaria U, Basak S, et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep 2020;10:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dai L, Zheng T, Xu K, et al. A universal design of betacoronavirus vaccines against COVID-19, MERS, and SARS. Cell 2020;182(e11):722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Guan Y. COVID-19 drug repurposing: a review of computational screening methods, clinical trials, and protein interaction assays. Med Res Rev 2020, med. 21728. doi: 10.1002/med.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Westbrook JD, Burley SK. How structural biologists and the Protein Data Bank contributed to recent FDA new drug approvals. Structure 2019;27:211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bahadur Gurung A, Ajmal Ali M, Lee J, et al. Structure-based virtual screening of phytochemicals and repurposing of FDA approved antiviral drugs unravels lead molecules as potential inhibitors of coronavirus 3C-like protease enzyme. J King Saud Univ Sci 2020;32:2845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol 2003;10:980. [DOI] [PubMed] [Google Scholar]
- 25. ww PDB consortium . Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 2019;47:D520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Armstrong DR, Berrisford JM, Conroy MJ, et al. PDBe: improved findability of macromolecular structure data in the PDB. Nucleic Acids Res 2020;48:D335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burley SK, Berman HM, Bhikadiya C, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 2019;47:D464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kinjo AR, Bekker G-J, Suzuki H, et al. Protein Data Bank Japan (PDBj): updated user interfaces, resource description framework, analysis tools for large structures. Nucleic Acids Res 2017;45:D282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abbott S, Iudin A, Korir PK, et al. EMDB web resources. Curr Protoc Bioinformatics 2018;61:5.10.1–5.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iudin A, Korir PK, Salavert-Torres J, et al. EMPIAR: a public archive for raw electron microscopy image data. Nat Methods 2016;13:387–8. [DOI] [PubMed] [Google Scholar]
- 31. PDBe-KB Consortium . PDBe-KB: a community-driven resource for structural and functional annotations. Nucleic Acids Res 2020;48:D344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The Uni Prot Consortium . Uni Prot: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bienert S, Waterhouse A, Beer TAP, et al. The SWISS-MODEL repository—new features and functionality. Nucleic Acids Res 2017;45:D313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sedova M, Jaroszewski L, Alisoltani A, et al. Coronavirus3D: 3D structural visualization of COVID-19 genomic divergence. Bioinformatics 2020;36(15):4360–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meldal BHM, Bye-A-Jee H, Gajdoš L, et al. Complex portal 2018: extended content and enhanced visualization tools for macromolecular complexes. Nucleic Acids Res 2019;47:D550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hodis E, Prilusky J, Martz E, et al. Proteopedia—a scientific ‘wiki’ bridging the rift between 3D structure and function of biomacromolecules. Genome Biol 2008;9:R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sillitoe I, Dawson N, Lewis TE, et al. CATH: expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res 2019;47:D280–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pandurangan AP, Stahlhacke J, Oates ME, et al. The SUPERFAMILY 2.0 database: a significant proteome update and a new webserver. Nucleic Acids Res 2019;47:D490–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andreeva A, Kulesha E, Gough J, et al. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res 2020;48:D376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andreeva A, Howorth D, Chothia C, et al. SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res 2014;42:D310–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mitchell AL, Attwood TK, Babbitt PC, et al. Inter pro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 2019;47:D351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ribeiro AJM, Holliday GL, Furnham N, et al. Mechanism and catalytic site atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res 2018;46:D618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McSkimming DI, Dastgheib S, Talevich E, et al. ProKinO: a unified resource for mining the cancer kinome. Hum Mutat 2015;36:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Putignano V, Rosato A, Banci L, et al. Metal PDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 2018;46:D459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Levy ED, Pereira-Leal JB, Chothia C, et al. 3D complex: a structural classification of protein complexes. PLoS Comput Biol 2006;2:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henry VJ, Bandrowski AE, Pepin AS, et al. OMICtools: an informative directory for multi-omic data analysis. Database (Oxford) 2014;bau069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res 2001;29:221–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker D. Protein structure prediction and structural genomics. Science 2001;294:93–6. [DOI] [PubMed] [Google Scholar]
- 50. O’Donoghue SI, Sabir KS, Kalemanov M, et al. Aquaria: simplifying discovery and insight from protein structures. Nat Methods 2015;12:98–9. [DOI] [PubMed] [Google Scholar]
- 51. Srinivasan S, Cui H, Gao Z, et al. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020;12:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Improved protein structure prediction using predicted interresidue orientations. PNAS;117:1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng W, Li Y, Zhang C, et al. Deep-learning contact-map guided protein structure prediction in CASP13. Proteins 2019;87(12):1149–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature 2020;577:706–10. [DOI] [PubMed] [Google Scholar]
- 55. AlQuraishi M. Alpha fold at CASP13. Bioinformatics 2019;35:4862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kerrien S, Aranda B, Breuza L, et al. The Int act molecular interaction database in 2012. Nucleic Acids Res 2012;40:D841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011;27:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen VB, Arendall WB, Headd JJ, et al. Mol Probity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47:D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O’Donoghue SI, Schafferhans A, Sikta N, et al. Systematic modeling of SARS-CoV-2 protein structures. bioRxiv 2020; 10.1101/2020.07.16.207308. [DOI]
- 61. Hecht M, Bromberg Y, Rost B. Better prediction of functional effects for sequence variants. BMC Genomics 2015;16(Suppl 8):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yachdav G, Kloppmann E, Kajan L, et al. Predict protein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 2014;42:W337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wlodawer A, Dauter Z, Shabalin IG, et al. Ligand-centered assessment of SARS-CoV-2 drug target models in the Protein Data Bank. FEBS J 2020, 10.1111/febs.15366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Croll TI. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Cryst D 2018;74:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Modeling of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) proteins by machine learning and physics-based refinement. bioRxiv 2020; 10.1101/2020.03.25.008904. [DOI] [Google Scholar]
- 66. Mariani V, Biasini M, Barbato A, et al. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013;29:2722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res 2003;31:3370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Studer G, Rempfer C, Waterhouse AM, et al. QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 2020;36:1765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Uziela K, Menéndez Hurtado D, Shu N, et al. ProQ3D: improved model quality assessments using deep learning. Bioinformatics 2017;33:1578–80. [DOI] [PubMed] [Google Scholar]
- 70. Igashov I, Olechnovic K, Kadukova M, et al. VoroCNN: deep convolutional neural network built on 3D Voronoi tessellation of protein structures. biorXiv 2020; 10.1101/2020.04.27.063586. [DOI] [PubMed]
- 71. Xu Q, Dunbrack RL. ProtCID: a data resource for structural information on protein interactions. Nat Commun 2020;11:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583:459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jo S, Kim T, Iyer VG, et al. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 2008;29:1859–65. [DOI] [PubMed] [Google Scholar]
- 74. Woo H, Park S-J, Choi YK, et al. Developing a fully glycosylated full-length SARS-CoV-2 spike protein model in a viral membrane. J Phys Chem B 2020;124(33):7128–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Machado MR, Barrera EE, Klein F, et al. The SIRAH 2.0 force field: altius, fortius, citius. J Chem Theory Comput 2019;15:2719–33. [DOI] [PubMed] [Google Scholar]
- 76. Abdul-Wahid B, Feng H, Rajan D, et al. AWE-WQ: fast-forwarding molecular dynamics using the accelerated weighted ensemble. J Chem Inf Model 2014;54:3033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Copperman JT, Zuckerman DM. Accelerated estimation of long-timescale kinetics by combining weighted ensemble simulation with Markov model “microstates” using non-Markovian theory. arXiV, 2020; arXiv:1903.04673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dunker AK, Babu MM, Barbar E, et al. What’s in a name? Why these proteins are intrinsically disordered: why these proteins are intrinsically disordered. Intrinsically Disord Proteins 2013;1:e24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Babu MM. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 2016;44:1185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Plitzko JM, Schuler B, Selenko P. Structural biology outside the box—inside the cell. Curr Opin Struct Biol 2017;46:110–21. [DOI] [PubMed] [Google Scholar]
- 82. Tompa P. Unstructural biology coming of age. Curr Opin Struct Biol 2011;21:419–25. [DOI] [PubMed] [Google Scholar]
- 83. Necci M, Piovesan D, Predictors CAID, et al. Critical assessment of protein intrinsic disorder prediction. bioRiv 2020 10.1101/2020.08.11.245852. [DOI]
- 84. Giri R, Bhardwaj T, Shegane M, et al. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell Mol Life Sci 2020;25:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Goh GK-M, Dunker AK, Foster JA, et al. Shell disorder analysis predicts greater resilience of the SARS-CoV-2 (COVID-19) outside the body and in body fluids. Microb Pathog 2020;144:104177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mészáros B, Sámano-Sánchez H, Alvarado-Valverde J, et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. arXiv 2004, 10274 [q-bio] 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cilia E, Pancsa R, Tompa P, et al. The DynaMine webserver: predicting protein dynamics from sequence. Nucleic Acids Res 2014;42:W264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cilia E, Pancsa R, Tompa P, et al. From protein sequence to dynamics and disorder with DynaMine. Nat Commun 2013;4:2741. [DOI] [PubMed] [Google Scholar]
- 89. Orlando G, Raimondi D, Codice F, et al. Prediction of disordered regions in proteins with recurrent neural networks and protein dynamics. bioRxiv 2020 10.1101/2020.05.25.115253. [DOI] [PubMed]
- 90. Pancsa R, Raimondi D, Cilia E, et al. Early folding events, local interactions, and conservation of protein backbone rigidity. Biophys J 2016;110:572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hou Q, De Geest PFG, Griffioen CJ, et al. SeRenDIP: SEquential REmasteriNg to DerIve profiles for fast and accurate predictions of PPI interface positions. Bioinformatics 2019;35:4794–6. [DOI] [PubMed] [Google Scholar]
- 92. Li Q, Wu J, Nie J, et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182(5):1284–1294.e9. doi: 10.1016/j.cell.2020.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Xing Y, Li X, Gao X, et al. MicroGMT: a mutation tracker for SARS-CoV-2 and other microbial genome sequences. Front Microbiol 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Singer J, Gifford R, Cotten M, et al. CoV-GLUE: a web application for tracking SARS-CoV-2 genomic variation. 2020. Preprints.org; 2020. DOI: 10.1126/science.abd5223 [DOI]
- 95. Genome Aggregation Database Consortium, Karczewski KJ, Francioli LC, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sherry ST. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gupta R, Charron J, Stenger CL, et al. SARS-CoV2 (COVID-19) structural/evolution dynamicome: insights into functional evolution and human genomics. bioRxiv 2020 10.1101/2020.05.15.098616. [DOI] [PMC free article] [PubMed]
- 98. Portelli S, Olshansky M, Rodrigues CHM, et al. COVID-3D: an online resource to explore the structural distribution of genetic variation in SARS-CoV-2 and its implication on therapeutic development. Nat Genet, 52, 999–10012020, (2020). 10.1038/s41588-020-0693-3 [DOI] [PubMed] [Google Scholar]
- 99. Rodrigues CHM, Myung Y, Pires DEV, et al. mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res 2019;47:W338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Vries SJ, Dijk M, Bonvin AMJJ. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc 2010;5:883–97. [DOI] [PubMed] [Google Scholar]
- 101. Mendez D, Gaulton A, Bento AP, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 2019;47:D930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gervasoni S, Vistoli G, Talarico C, et al. A comprehensive mapping of the druggable cavities within the SARS-CoV-2 therapeutically relevant proteins by combining pocket and docking searches as implemented in pockets 2.0. Int J Mol Sci 2020;21 5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Redka DS, Mac Kinnon SS, Landon M, et al. PolypharmDB, a deep learning-based resource, quickly identifies repurposed drug candidates for COVID-19. ChemRxiv 2020. http://doi.org/10.26434/chemrxiv.12071271.v1.
- 104. Watanabe C, Watanabe H, Okiyama Y, et al. Development of an automated fragment molecular orbital (FMO) calculation protocol toward construction of quantum mechanical calculation database for large biomolecules. CBIJ 2019;19:5–18. [Google Scholar]
- 105. Ghorbani M, Brooks BR, Klauda JB. Critical sequence hot-spots for binding of nCOV-2019 to ACE2 as evaluated by molecular simulations. bioRxiv 2020, 2020.06.27.175448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Spinello A, Saltalamacchia A, Magistrato A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the Hallmark for its enhanced infectivity? Insights from all-atom simulations. J Phys Chem Lett 2020;11:4785–90. [DOI] [PubMed] [Google Scholar]
- 107. Watanabe Y, Allen JD, Wrapp D, et al. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020;17;369(6501):330–333. doi: 10.1126/science.abb9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Walls AC, Tortorici MA, Frenz B, et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol 2016;23:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yang T-J, Chang Y-C, Ko T-P, et al. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc Natl Acad Sci USA 2020;117:1438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Casalino L, Gaieb Z, Dommer AC, et al. Shielding and beyond: the roles of Glycans in SARS-CoV-2 spike protein. bioRxiv 2020, 2020.06.11.146522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sikora M, Bülow S, FEC B, et al. Map of SARS-CoV-2 spike epitopes not shielded by glycans. bioRxiv 2020, 2020.07.03.186825. [Google Scholar]
- 112. Turoňová B, Sikora M, Schürmann C, et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. 2020;370(6513):203–208. doi: 10.1126/science.abd5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang G, Pomplun S, Loftis AR, et al. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 spike RBD. bioRxiv 2020, 2020.03.19.999318. [Google Scholar]
- 114. Gupta R, Charron J, Stenger CL, et al. SARS-CoV-2 (COVID-19) structural and evolutionary dynamicome: insights into functional evolution and human genomics. J Biol Chem 2020;14;295(33):11742–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Di Paola L, Hadi-Alijanvand H, Song X, et al. The discovery of a putative allosteric site in the SARS-CoV-2 spike protein using an integrated structural/dynamic approach. J Proteome Res 2020, acs.jproteome.0c00273. [DOI] [PubMed] [Google Scholar]
- 116. Di Paola L, Giuliani A. Mapping active allosteric loci SARS-CoV spike proteins by means of protein contact networks. arXiv 2003, 05200 [q-bio, stat] 2020. [Google Scholar]
- 117. Olotu FA, Omolabi KF, Soliman MES. Leaving no stone unturned: allosteric targeting of SARS-CoV-2 spike protein at putative druggable sites disrupts human angiotensin-converting enzyme interactions at the receptor binding domain. Inform Med Unlocked 2020;21:100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Barakat K, Ahmed M, Tabana Y, et al. A “Deep Dive” into the SARS-Cov-2 polymerase assembly: identifying novel allosteric sites and analyzing the hydrogen bond networks and correlated dynamics. bioRxiv 2020 10.1101/2020.06.02.130849. [DOI] [PubMed]
- 119. Dubanevics I, McLeish TCB. Computational analysis of dynamic allostery and control in the SARS-CoV-2 main protease. bioRxiv 2020 10.1101/2020.05.21.105965. [DOI] [PMC free article] [PubMed]
- 120. El-Baba TJ, Lutomski CA, Kantsadi AL, et al. Allosteric inhibition of the SARS-CoV-2 main protease: insights from mass spectrometry based assays. Angew Chem Int Ed 2020. doi: 10.1002/anie.202010316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Brielle ES, Schneidman-Duhovny D, Linial M. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses 2020. 12(5), 497. [DOI] [PMC free article] [PubMed]
- 122. Bojkova D, McGreig JE, McLaughlin K-M, et al. SARS-CoV-2 and SARS-CoV differ in their cell tropism and drug sensitivity profiles. bioRxiv 2020, 2020.04.03.024257. [Google Scholar]
- 123. Damas J, Hughes GM, Keough KC, et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc Natl Acad Sci USA 2020;117(36):22311–22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lam SD, Bordin N, Waman VP, et al. SARS-CoV-2 spike protein predicted to form complexes with host receptor protein orthologues from a broad range of mammals. Sci Rep. 2020;10(1):16471. doi: 10.1038/s41598-020-71936-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Pinto D, Park Y-J, Beltramello M, et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020;583:290–5. [DOI] [PubMed] [Google Scholar]
- 126. Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020;367:1444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020;182:812, e19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Omotuyi IO, Nash O, Ajiboye OB, et al. Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein-enhanced fitness in SARS-COV-2. J Comput Chem 2020;41(24):2158–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mansbach RA, Chakraborty S, Nguyen K, et al. The SARS-CoV-2 spike variant D614G favors an open conformational state. bioRxiv 2020, 2020.07.26.219741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. MacGowan SA, Barton GJ. Missense variants in ACE2 are predicted to encourage and inhibit interaction with SARS-CoV-2 spike and contribute to genetic risk in COVID-19. bioRxiv 2020, 2020.05.03.074781. [Google Scholar]
- 131. Rodrigues JP, Barrera-Vilarmau S, Teixeira JM, et al. Insights on cross-species transmission of SARS-CoV-2 from structural modeling. bioRxiv 2020 10.1101/2020.06.05.136861. [DOI] [PMC free article] [PubMed]
- 132. Ahmad S, Navid A, Farid R, et al. Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur J Pharm Sci 2020;151:105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mukherjee S, Tworowski D, Detroja R, et al. Immunoinformatics and structural analysis for identification of immunodominant epitopes in SARS-CoV-2 as potential vaccine targets. Vaccine 2020;8:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Panda PK, Arul MN, Patel P, et al. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci Adv 2020;6:eabb8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Parks JM, Smith JC. How to discover antiviral drugs quickly. N Engl J Med 2020;382:2261–4. [DOI] [PubMed] [Google Scholar]
- 136. Ciliberto G, Cardone L. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov Today 2020;25:946–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gil C, Ginex T, Maestro I, et al. COVID-19: drug targets and potential treatments. J Med Chem 2020;1723; 12359–12386. [DOI] [PubMed] [Google Scholar]
- 138. Jeong GU, Song H, Yoon GY, et al. Therapeutic strategies against COVID-19 and structural characterization of SARS-CoV-2: a review. Front Microbiol 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 2020;11:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fernández A. Structural impact of mutation D614G in SARS-CoV-2 spike protein: enhanced infectivity and therapeutic opportunity. ACS Med Chem Lett 2020;11(9):1667–1670lett.0c00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Xia S, Liu M, Wang C, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 2020;30:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yuan M, Wu NC, Zhu X, et al. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020;368:630–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Kang S, Yang M, Hong Z, et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B 2020;10:1228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lin S-M, Lin S-C, Hsu J-N, et al. Structure-based stabilization of non-native protein-protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J Med Chem 2020;63:3131–41. [DOI] [PubMed] [Google Scholar]
- 145. Lin S-Y, Liu C-L, Chang Y-M, et al. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J Med Chem 2014;57:2247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020;12 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. McAndrews KM, Dowlatshahi DP, Dai J, et al. Heterogeneous antibodies against SARS-CoV-2 spike receptor binding domain and nucleocapsid with implications on COVID-19 immunity. JCI Insight 2020;5(18):e142386. doi: 10.1172/jci.insight.142386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020;368:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ghosh AK, Brindisi M, Shahabi D, et al. Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem 2020;15:907–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 2015;116:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zhang L, Lin D, Kusov Y, et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem 2020;63:4562–78. [DOI] [PubMed] [Google Scholar]
- 152. Fischer A, Sellner M, Neranjan S, et al. Potential inhibitors for novel coronavirus protease identified by virtual screening of 606 million compounds. Int J Mol Sci 2020;21; 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Gahlawat A, Kumar N, Kumar R, et al. Structure-based virtual screening to discover potential lead molecules for the SARS-CoV-2 main protease. J Chem Inf Model 2020;acs.jcim.0c00546. doi: 10.1021/acs.jcim.0c00546. [DOI] [PubMed] [Google Scholar]
- 154. Nutho B, Mahalapbutr P, Hengphasatporn K, et al. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 2020;59:1769–79. [DOI] [PubMed] [Google Scholar]
- 155. Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol 2020;38:379–81. [DOI] [PubMed] [Google Scholar]
- 156. Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 2020;30:678–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun 2020;11:4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Douangamath A, Fearon D, Gehrtz P, et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat Commun 2020;11:5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dai W, Zhang B, Jiang X-M, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020;368:1331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Yoshino R, Yasuo N, Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci Rep 2020;10:12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hatada R, Okuwaki K, Mochizuki Y, et al. Fragment molecular orbital based interaction analyses on COVID-19 main protease—inhibitor N3 complex (PDB ID: 6LU7). J Chem Inf Model 2020;60:3593–602. [DOI] [PubMed] [Google Scholar]
- 162. Altay O, Mohammadi E, Lam S, et al. Current status of COVID-19 therapies and drug repositioning applications. iScience 2020;23:101303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Morse JS, Lalonde T, Xu S, et al. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 2020;21:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Pillaiyar T, Manickam M, Namasivayam V, et al. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 2016;59:6595–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Maiti BK. Can papain-like protease inhibitors halt SARS-CoV-2 replication? ACS pharmacol. Transl Sci 2020, 3(5):1017–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Fehr AR, Channappanavar R, Jankevicius G, et al. The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. MBio 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. McDonald SM. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip Rev RNA 2013;4:351–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Elfiky AA. Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci 2020;253:117592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020;395:1569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wang Y, Zhou F, Zhang D, et al. Evaluation of the efficacy and safety of intravenous remdesivir in adult patients with severe COVID-19: study protocol for a phase 3 randomized, double-blind, placebo-controlled, multicentre trial. Trials 2020;21:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020;368:1499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wang Q, Wu J, Wang H, et al. Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 2020;182:417–28.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Eloy P, Solas C, Touret F, et al. Dose rationale for favipiravir use in patients infected with SARS-CoV-2. Clin Pharmacol Ther 2020;108:188. [DOI] [PubMed] [Google Scholar]
- 174. Kim MK, Yu M-S, Park HR, et al. 2,6-Bis-arylmethyloxy-5-hydroxychromones with antiviral activity against both hepatitis C virus (HCV) and SARS-associated coronavirus (SCV). Eur J Med Chem 2011;46:5698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Lee C, Lee JM, Lee N-R, et al. Aryl diketoacids (ADK) selectively inhibit duplex DNA-unwinding activity of SARS coronavirus NTPase/helicase. Bioorg Med Chem Lett 2009;19:1636–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271, e8–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Jia Z, Yan L, Ren Z, et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res 2019;47:6538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020;181:894–904.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chan KK, Dorosky D, Sharma P, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020;369:1261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ota S, Hara Y, Kanoh S, et al. Acute eosinophilic pneumonia caused by camostat mesilate: the first case report. Respir Med Case Rep 2016;19:21–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Maggio R, Corsini GU. Repurposing the mucolytic cough suppressant and TMPRSS2 protease inhibitor bromhexine for the prevention and management of SARS-CoV-2 infection. Pharmacol Res 2020;157:104837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Habtemariam S, Nabavi SF, Ghavami S, et al. Possible use of the mucolytic drug, bromhexine hydrochloride, as a prophylactic agent against SARS-CoV-2 infection based on its action on the transmembrane serine protease 2. Pharmacol Res 2020;157:104853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Ivanova T, Hardes K, Kallis S, et al. Optimization of substrate-analogue Furin inhibitors. ChemMedChem 2017;12:1953–68. [DOI] [PubMed] [Google Scholar]
- 184. Dahms SO, Jiao G-S, Than ME. Structural studies revealed active site distortions of human Furin by a small molecule inhibitor. ACS Chem Biol 2017;12:1211–6. [DOI] [PubMed] [Google Scholar]
- 185. Dahms SO, Hardes K, Steinmetzer T, et al. X-ray structures of the proprotein convertase Furin bound with substrate analogue inhibitors reveal substrate specificity determinants beyond the S4 pocket. Biochemistry 2018;57:925–34. [DOI] [PubMed] [Google Scholar]
- 186. Vankadari N. Structure of furin protease binding to SARS-CoV-2 spike glycoprotein and implications for potential targets and virulence. J Phys Chem Lett 2020;11:6655–63. [DOI] [PubMed] [Google Scholar]
- 187. Phillips JM, Gallagher T, Weiss SR. Neurovirulent murine coronavirus JHM.SD uses cellular zinc metalloproteases for virus entry and cell-cell fusion. J Virol 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hardegger LA, Kuhn B, Spinnler B, et al. Halogen bonding at the active sites of human cathepsin L and MEK1 kinase: efficient interactions in different environments. ChemMedChem 2011;6:2048–54. [DOI] [PubMed] [Google Scholar]
- 189. Ehmke V, Winkler E, Banner DW, et al. Optimization of triazine nitriles as rhodesain inhibitors: structure-activity relationships, bioisosteric imidazopyridine nitriles, and X-ray crystal structure analysis with human cathepsin L. ChemMedChem 2013;8:967–75. [DOI] [PubMed] [Google Scholar]
- 190. Sartori GR, Leitão A, Montanari CA, et al. Ligand-induced conformational selection predicts the selectivity of cysteine protease inhibitors. PLoS One 2019;14:e0222055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J Clin Invest 2017;127:1338–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xu J, Berastegui-Cabrera J, Chen H, et al. Structure-activity relationship studies on diversified Salicylamide derivatives as potent inhibitors of human adenovirus infection. J Med Chem 2020;63:3142–60. [DOI] [PubMed] [Google Scholar]
- 193. Xu J, Xie X, Ye N, et al. Design, synthesis, and biological evaluation of substituted 4,6-Dihydrospiro[[1,2,3]triazolo[4,5-b]pyridine-7,3′-indoline]-2′,5(3H)-dione analogues as potent NS4B inhibitors for the treatment of dengue virus infection. J Med Chem 2019;62:7941–60. [DOI] [PubMed] [Google Scholar]
- 194. Saul S, Pu S-Y, Zuercher WJ, et al. Potent antiviral activity of novel multi-substituted 4-anilinoquin(az)olines. Bioorg Med Chem Lett 2020;30:127284. [DOI] [PubMed] [Google Scholar]
- 195. Lartigue J, Polson H, Feldman M, et al. PIKfyve regulation of endosome-linked pathways. Traffic 2009;10:883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Kang Y-L, Chou Y-Y, Rothlauf PW, et al. Inhibition of PIKfyve kinase prevents infection by EBOV and SARS-CoV-2. 2020; Proc Natl Acad Sci U S A.;117(34):20803–20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Sakurai Y, Kolokoltsov AA, Chen C-C, et al. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015;347:995–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. She J, Zeng W, Guo J, et al. Structural mechanisms of phospholipid activation of the human TPC2 channel. Elife 2019;8 e45222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Penny CJ, Vassileva K, Jha A, et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim Biophys Acta Mol Cell Res 1866;2019:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Starr TN, Greaney AJ, Hilton SK, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020182(5);1295–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020;369(6506):1014–1018. doi: 10.1126/science.abd0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020;584:443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Barnes CO, West AP, Huey-Tubman KE, et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 2020;182:828, e16–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Huo J, Zhao Y, Ren J, et al. Neutralization of SARS-CoV-2 by destruction of the Prefusion spike. Cell Host Microbe 2020, 28(3):497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The online web-links for all the resources/tools, reviewed in this article are provided in main text.