

SUMMARY
In this review, I summarize historical and recent features of the classical pathways activated by growth hormone (GH) through the cell surface GH receptor (GHR). GHR is a cytokine receptor superfamily member that signals by activating the non-receptor tyrosine kinase, JAK2, and members of the Src family kinases. Activation of the GHR engages STATs, PI3K, and ERK pathways, among others, and details of these now-classical pathways are presented. Modulating elements, including the SOCS proteins, phosphatases, and regulated GHR metalloproteolysis, are discussed. In addition, a novel physical and functional interaction of GHR with IGF-1R is summarized and discussed in terms of its mechanisms, consequences, and physiological and therapeutic implications.
Keywords: growth hormone receptor, signaling, IGF-1 receptor
1. Molecules involved in initial GH signal generation
Nearly a century ago, Evans and Long published their fundamental observation that rats injected intraperitoneally with extracts of cow anterior (but not posterior) pituitary glands “…grew invariably much heavier than their litter mate sisters… Increase in weight results to a great extent from a storage of fat, but is not solely due to this, the skeleton being invariably somewhat larger and heavier, and, as would be expected, the heart, lung, alimentary canal and kidney are heavier” (Evans and Long 1922). This suggested the existence of pituitary-derived growth hormone (GH). Using a five step extraction and purification procedure, Li and Evans two decades later first isolated GH from ox pituitaries (Li and Evans 1944).
It was another 35 years later that molecular cloning and recombinant technology allowed discovery of the gene encoding GH (Martial, Hallewell et al. 1979) and eventually the high level production of human GH for therapeutic use. The GH gene in humans and most other vertebrates is found on chromosome 17; in humans, GH is one of five similar genes in a 48 kilobase cluster (Chen, Liao et al. 1989). The GH gene encodes a 22 kDa circulating protein hormone that emanates from the pituitary in many species, signaling the profound anabolic and metabolic actions noted by Evans and Long and others (Isaksson, Eden et al. 1985, Waters, Hoang et al. 2006, Moller and Jorgensen 2009). Elucidation of the three-dimensional structures of both porcine and human proteins in the late 1980s-early 1990s (Abdel-Meguid, Shieh et al. 1987, de Vos, Ultsch et al. 1992) revealed GH to be composed of four antiparallel α-helices connected by loops of varying length; its overall structure was similar to what at the time was an emerging group of cytokines, hormones, interleukins, and interferons.
Binding sites for GH were shown to be present in multiple tissues, being notably enriched in liver; indeed, the first isolation of cDNAs for the rabbit and human GH receptors (GHRs) in 1987 employed liver tissue as the starting material (Leung, Spencer et al. 1987). In short order, GHR cDNAs encoding mouse, rat, cow, sheep, pig, and chicken were cloned between 1989-1991 (Baumbach, Horner et al. 1989, Smith, Kuniyoshi et al. 1989, Adams, Baker et al. 1990, Cioffi, Wang et al. 1990, Hauser, McGrath et al. 1990, Burnside, Liou et al. 1991). As anticipated, the GHR proteins encoded in these species, while varying in amino acid sequence, shared similar overall topology. All predicted single membrane-spanning type 1 proteins (620 residues in length in human) with a ligand-binding extracellular domain (246 residues in human) that is N-glycosylated and a substantial (350 residues in human) intracellular domain that can be ubiquitinated (Leung, Spencer et al. 1987). As discussed in greater detail by Brooks, et al elsewhere in this volume, deVos and colleagues (de Vos, Ultsch et al. 1992), in a tour de force co-crystallization study in 1992, determined the three dimensional structure of human GH bound to the human GHR extracellular domain; they found a novel 1:2 GH:GHR stoichiometry that supports the notion that the signaling unit of GHR is a dimer, whether it is either constitutively or inducibly formed (Cunningham, Ultsch et al. 1991, Frank 2002, Gent, van Kerkhof et al. 2002, Liu, Berry et al. 2014, Wilmes, Hafer et al. 2020). Unlike what we now know is likely an intrinsically disordered structure in the intracellular domain (Haxholm, Nikolajsen et al. 2015), the GHR extracellular domain was found to be comprised of a set of modular elements. Each GHR monomer’s extracellular domain has two subdomains (1 and 2 with 1 N-terminal to 2), each comprised of seven β-strands arranged into two antiparallel β-sheets; subdomain 1 harbors most of the GH contact points and subdomain 2 facilitates dimerization of the monomers (Leung, Spencer et al. 1987). The fact that each GH molecule in the GH/GHR assemblage has two asymmetric binding sites (one for each GHR monomer in the assemblage) was a noteworthy finding that laid the groundwork for development of therapeutically useful GH antagonists (discussed in detail elsewhere (Fuh, Cunningham et al. 1992, Kopchick, Parkinson et al. 2002)).
Validation that the cloned GHR was indeed a physiological receptor for GH came in 1997 with the generation by the Kopchick group of the GHR knockout mouse (Zhou, Xu et al. 1997) (discussed in greater detail elsewhere in this volume). Thirty years prior (Laron, Pertzelan et al. 1966), Laron had described the first cases of human GH resistance (Laron syndrome). These patients had a phenotype similar to those with GH deficiency, except that circulating GH was present and often elevated. We now know that this syndrome is explained by a variety of defects in GHR that alter receptor expression, structure, or cell surface targeting and function. (Human GHR defects leading to Laron syndrome are discussed in this volume by Guevara-Aguirre.) The GHR knockout mouse (the “Laron mouse”) phenocopied human Laron syndrome in displaying substantial growth retardation and proportionate dwarfism with raised levels of GH in the serum.
By virtue of structural similarities, it was rapidly appreciated that GHR fell within the large family of so-called cytokine (or hematopoietin) receptors that included receptors for various interleukins, interferons, cytokines, and hormones (Bazan 1990, Argetsinger and Carter-Su 1996, Bole-Feysot, Goffin et al. 1998, Frank and O’Shea 1999), including the related prolactin receptor PRLR. Indeed, human GH can bind and activate human PRLR (but not vice-versa (Hughes and Friesen 1985, Cunningham, Bass et al. 1990, Fu, Arkins et al. 1992, Somers, Ultsch et al. 1994)); although beyond the scope of this review, such GH-induced PRLR signaling may prove quite physiologically and pathophysiologically relevant. Further (also beyond the scope of this review), recent studies suggest important physical and functional interactions between GHR and PRLR by which each may impact the trafficking and signaling capacities of the other (Xu, Zhang et al. 2011, Xu, Sun et al. 2013, Liu, Zhang et al. 2016, Liu, Jiang et al. 2017).
2. GH-induced JAK2 kinase activation.
Unlike other cell surface receptor proteins whose primary sequences encoded enzymatic activities (e.g., tyrosine or serine/threonine kinases), the GHR’s intracellular domain sequence did not reveal activity to suggest a signaling mechanism. Yet, Carter-Su and colleagues made the seminal observations in 1988-89 that GH treatment of mouse preadipocytes acutely and robustly caused tyrosine phosphorylation of cellular proteins, including GHR itself (Foster, Shafer et al. 1988, Carter-Su, Stubbart et al. 1989), suggesting the existence of a tightly GHR-associated GH-regulated tyrosine kinase. Indeed, that tyrosine kinase was identified in 1993 by the Carter-Su group (Argetsinger, Campbell et al. 1993) as the non-receptor cytoplasmic tyrosine kinase, JAK2, one of four members of the Janus kinase family (JAKs 1-3 and TYK2) (Firmbach-Kraft, Byers et al. 1990, Wilks, Harpur et al. 1991, Harpur, Andres et al. 1992, Silvennoinen, Witthuhn et al. 1993, Witthuhn, Quelle et al. 1993, Johnston, Kawamura et al. 1994) that each possess a C-terminal kinase domain, a kinase-like domain, and an N-terminal (FERM) domain that interacts with particular hematopoeitin receptor family members. Subsequent studies in the 1990s and early 2000s by several labs identified the regions within GHR (a proline-rich proximal cytoplasmic domain element) and JAK2 (the N-terminus) that mediate physical and functional interaction between the two proteins (Frank, Gilliland et al. 1994, Sotiropoulos, Perrot-Applanat et al. 1994, Vanderkuur, Wang et al. 1994, Frank, Yi et al. 1995, Tanner, Chen et al. 1995, He, Wang et al. 2003). The mechanism(s) by which GH binding to the dimerized GHR causes activation of the GHR-associated JAK2 have recently been intensely investigated (Jiang, Wang et al. 2004, Brown, Adams et al. 2005, Rowlinson, Yoshizato et al. 2008, Jiang, Wan et al. 2011, Brooks, Dai et al. 2014, Liu, Berry et al. 2014, Sedek, van der Velden et al. 2014, Liu, Jiang et al. 2017) and are reviewed by Brooks and colleagues in this volume. Despite these advances, many aspects of this activation process remain enigmatic and are worthy of continued research. The reader is directed to Figure 1 for a diagram of the pathways activated by the GH-engaged GHR, which are discussed below.
Figure 1. GHR signaling pathways.
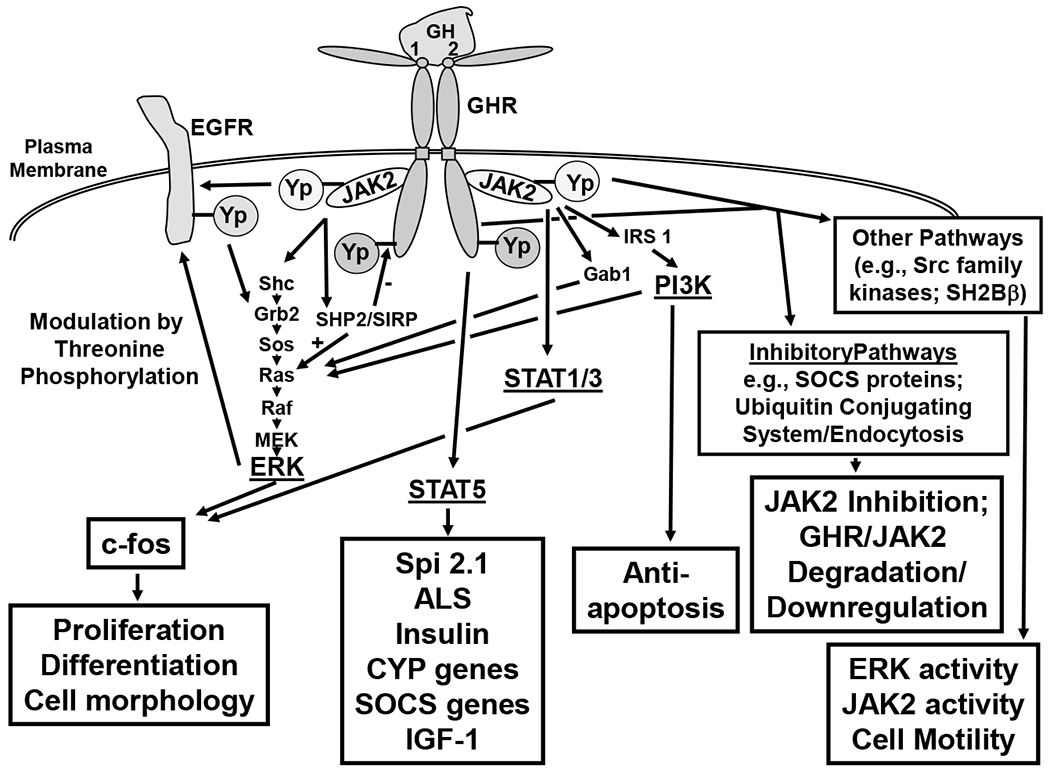
This figure summarizes the GHR signaling pathways discussed in the text. Major signaling systems engaged downstream of the GH-engaged GHR-JAK2 complex are shown. These include the STATs, ERKs, and PI3K pathways. Intermediate molecules on these pathways are shown, as are loci of crosstalk with EGFR. Yp indicates tyrosine phosphorylation. Other pathways referred to in the text are indicated in boxes, as are cell biological and biochemical outcomes of activated pathways.
These studies clearly linked JAK2 structurally and biochemically to GH signaling. In terms of function among the JAK family members, JAK3 and TYK2 are largely involved in immune signaling pathways (Velazquez, Fellous et al. 1992, Johnston, Kawamura et al. 1994, Stahl, Boulton et al. 1994, Lin, Migone et al. 1995, Nosaka, van Deursen et al. 1995), while JAK2 and JAK1 mediate important signals both within and outside of the immune system (Witthuhn, Quelle et al. 1993, Lebrun, Ali et al. 1994, Smit, Meyer et al. 1996, Tian, Tapley et al. 1996, Drachman and Kaushansky 1997, Neubauer, Cumano et al. 1998, Rodig, Meraz et al. 1998). Notably, developmental knockout of JAK2 manifests not in deficient GH action, but rather as embryonic lethality resulting from ineffective hematopoiesis associated with lack of erythropoietin action (Neubauer, Cumano et al. 1998). This finding was consistent with the notion that JAK2 couples to a variety of cytokine receptors. Confirmatory in vivo evidence that JAK2 couples to GHR action has emerged from the elegant studies of Weiss and colleagues (Sos, Harris et al. 2011, Corbit, Camporez et al. 2017, Corbit, Wilson et al. 2019). In those studies, hepatocyte-specific deletion of JAK2 phenocopied aspects of previously-described mice with hepatocyte-specific deletion of either GHR (Fan, Menon et al. 2009) or the key GH effector, insulin-like growth factor-1 (IGF-1) (Yakar, Liu et al. 1999) (more below) in having raised circulating GH levels and markedly reduced circulating IGF-1.
3. GH-induced STAT5 activation.
STATs (signal transducers and activators of transcription) are a family of latent cytoplasmic transcription factors that translocate to the nucleus in response to various cytokines, growth factors, and hormones that effect target gene transcription by interacting with specific DNA enhancer sequences (Stark and Darnell 2012, O’Shea, Holland et al. 2013). The family has seven members (STATs 1,2,3,4,5A,5B, and 6), the first of which were discovered in the early 1990s; each intimately interacts functionally with specific cytokine receptor/JAK assemblages to initiate rapid gene expression signaling cascades. Studies in the 1990s revealed that only STATs 1,3, and 5 are activated by GH (Gronowski and Rotwein 1994, Meyer, Campbell et al. 1994, Campbell, Meyer et al. 1995, Gouilleux, Pallard et al. 1995, Gronowski, Zhong et al. 1995, Wood, Sliva et al. 1995, Han, Leaman et al. 1996, Ram, Park et al. 1996, Silva, Lu et al. 1996, Smit, Vanderkuur et al. 1997). STATs 1 and 3 were first shown to be critical in interferon signaling and antiviral immunity (STAT1) and interleukin-6-family cytokine signaling and the acute phase response (STAT3) (Darnell Jr, Kerr et al. 1994). Indeed, GH treatment in cell culture model systems causes formation of tyrosine phosphorylation-mediated dimers (homo- and hetero-) comprised of STAT1 and STAT3 on the promoter of the c-fos gene that corresponds to expression of reporter genes relating to GH’s known regulation of c-fos gene expression (Doglio, Dani et al. 1989, Gurland, Ashcom et al. 1990, Gronowski and Rotwein 1994, Meyer, Campbell et al. 1994, Campbell, Meyer et al. 1995, Frank, Yi et al. 1995). However, disruption of neither the STAT1 nor STAT3 genes in mice results in a phenotype clearly indicative of impaired GH action (Durbin, Hackenmiller et al. 1996, Meraz, White et al. 1996, Takeda, Noguchi et al. 1997); it remains unknown the degree to which either of these two STATs contributes to the physiological actions of GH.
In contrast, STAT5 has emerged as a key GH signal transducer with clearly-understood impact on GH action. As the topic is extensively reviewed in this volume by Rotwein, it will only briefly be discussed here. STAT5 was originally not distinguished as either the A or B isoform (highly homologous, but not identical gene products (Ambrosio, Fimiani et al. 2002, Hwa 2016)) and was first found as a prolactin-responsive transcription factor in sheep (Wakao, Gouilleux et al. 1994). We now know that various cytokines and growth factors activate STAT5 and that it has pro-proliferative and immunoregulatory roles in coupling to the receptors for these factors. With regards to GH action, studies in the mid-1990s implicated STAT5 in governing GH-dependent expression of genes encoding serine protease inhibitor 2.1 and a cytochrome P450 activity (Bergad, Shih et al. 1995, Subramanian, Teixeira et al. 1995). Landmark studies by Waxman and colleagues in rats and mice have shown that STAT5B, in particular, is critical in mediating sexually-dimorphic effects of GH on liver gene expression that rely on the pulsatility of GH in the circulation (Waxman and O’Connor 2006). Further, STAT5B is a key mediator of GH-induced expression in liver and other tissues of IGF-1 (Woelfle, Chia et al. 2003, Woelfle and Rotwein 2004). Indeed, targeted deletion of STAT5B in mice results in loss of sexually-dimorphic growth and hepatic gene expression, reduced serum IGF-1 levels, and elevated GH levels (Udy, Towers et al. 1997, Teglund, McKay et al. 1998), in several ways phenocopying the GHR knockout mouse and strongly implicating STAT5B in GH action. Even further evidence of this is found in humans with an autosomal recessive pattern of mutation of STAT5B (reviewed in (Hwa 2016)), who manifest short stature, low IGF-1, non-suppressed GH levels, and relative GH resistance. These human STAT5B mutations and their relationship to Laron syndrome are discussed in this volume by Hwa.
Mechanistically, STAT5B also differs from other GH-induced STATs in that its activation requires phosphorylation of at least one of several tyrosine residues of the GHR cytoplasmic tail, as well as activation of JAK2 (Hansen, Wang et al. 1996, Moriggl, Gouilleux-Gruart et al. 1996, Smit, Meyer et al. 1996, Wang, Darus et al. 1996, Yi, Kim et al. 1996, Smit, Vanderkuur et al. 1997, Rowland, Kerr et al. 2005, Rowland, Lichanska et al. 2005), consistent with the notion that transient recruitment of STAT5B to the GHR via the STAT SH2 domain allows processive tyrosine phosphorylation of STAT5B by GH-activated JAK2, followed by SH2-phosphotyrosine-mediated STAT5B dimerization and translocation to the nucleus. Indeed, genetic knock-in of GHR truncation and tyrosine mutants in the setting of the GHR knockout mouse suggest an important correlation between the GHR tail’s ability to support STAT5B activation with IGF-1 expression and linear growth (Rowland, Lichanska et al. 2005).
Notably, very recent studies using this GHR knock-in system highlight the metabolic effects of uncoupling GHR activation from STAT5B signaling in that inability of GH to activate STAT5B results in both enhanced insulin sensitivity in multiple tissues and diminished hepatic gluconeogenesis (Chhabra, Nelson et al. 2019). Interestingly, liver GHR knockout in adult mice results in an augmentation of de novo lipogenesis, also suggesting important metabolic regulatory roles for GH in that model system (Cordoba-Chacon, Majumdar et al. 2015). Very likely, further comprehensive approaches using multi-omics (akin to recent studies of GHR-deficient pigs (Riedel, Hinrichs et al. 2020)) will better elucidate GH-dependent metabolic effects that are or are not dependent on GH’s ability to couple to STAT5B activation.
4. GH-induced activation of other pathways.
4.1. Src family kinases
Like JAKs, the Src family kinases are cytoplasmic non-receptor tyrosine kinases that can couple to cytokine and growth factor receptors (Roskoski 2015, Espada and Martin-Perez 2017); in addition to Src, the family includes eight other members (Yes, Fyn, Lyn, Fgr, Hck, Lck, Yrk, and Blk), each roughly half the size of and structurally quite dissimilar from JAKs, except in the kinase domain. Src, Yes, Fyn, and Yrk are widely expressed. The others are more restricted in their tissue distribution. Collectively, they are involved in multiple cellular functions, including migration, proliferation, survival, and differentiation. Early evidence that GH acutely activated Src and Fyn came from the Lobie group, working in GHR-reconstituted CHO tissue culture cells (Zhu, Goh et al. 1998). Further work from the same group implicated a role for Src in promoting GH-dependent, JAK2-independent ERK pathway activation (more below) via small GTPases and phospholipase D activation (Zhu, Ling et al. 2002, Ling, Zhu et al. 2003). Waters and colleagues, in cell culture models, implicated Lyn and phospholipase Cγ in mediating GH-dependent ERK activation (Rowlinson, Yoshizato et al. 2008) and, in knock-in mice with GHR mutations incapable of coupling to JAK2, identified a GH-induced Src-mediated ERK activation pathway (Barclay, Kerr et al. 2010). However, GH-induced Src family kinase activation has not been universally detected (Jin, Lanning et al. 2008). Also, it is of note that knock-in mice with a GHR incapable of coupling to JAK2 do not differ from GHR knockout mice with regard to growth (Barclay, Kerr et al. 2010), suggesting the primacy of JAK2 for coupling to this aspect of GH action. Whether there are other facets of GH’s effects that are specifically mediated by Src family kinases is yet unknown.
4.2. ERKs and other interrelated pathways
ERK1 and ERK2 (extracellular signal regulated kinases -1 and -2) are cytosolic serine-threonine kinases that are part of the Ras-Raf-MEK-ERK signaling cascade (Roskoski 2012). ERKs are enzymatically activated by tyrosine and threonine phosphorylation downstream of many growth factor and other receptors. ERKs themselves catalyze proline-directed serine-threonine phosphorylation of important substrates to effect cellular processes including proliferation, cell cycle progression, migration, survival, and metabolism. ERK1/2 were among the earliest identified substrates for GH-induced tyrosine phosphorylation (Anderson 1992, Campbell, Pang et al. 1992, Moller, Hansson et al. 1992, Winston and Bertics 1992). Because other important serine-threonine kinases activated by GH (e.g., p70rsk, p90rsk, and S6 kinase) (Anderson 1993) were known to be ERK substrates, the ERK pathway was early on seen as a link between multiple GH-activated signals. As above, there are varying conclusions as to whether JAK2 activation (versus Src-family kinase activation) is necessary for ERK activation; indeed, this may vary depending on cellular context. Early mapping studies indicated that the proximal GHR intracellular domain required for JAK2 activation was sufficient for GH-induced ERK activation (Moller, Hansson et al. 1992, Sotiropoulos, Perrot-Applanat et al. 1994) and that a GHR incapable of associating with or activating JAK2 was unable to allow GH-induced ERK activation (Vanderkuur, Wang et al. 1994). Likewise, JAK2 mutants rendered either catalytically inactive or unable to couple to GHR failed to foster GH-induced ERK tyrosine phosphorylation (Frank, Yi et al. 1995). However, in these early studies, the activation state of Src-family kinases was not specifically assessed.
Independent of whether JAK2 is the only kinase required for ERK activation, several pathway components are known to be upstream of ERK activation by GH (for a more detailed discussion, the reader is directed to (Carter-Su, Schwartz et al. 2016, Bergan-Roller and Sheridan 2018, Dehkhoda, Lee et al. 2018)). Coupling of each of these pathways (and, indeed, GH-induced ERK activation itself) may be cell type- and context-specific (Love, Whatmore et al. 1998, Yang, Huang et al. 2004), much like the primacy of JAK2 vs. Src-family kinases. Substantial evidence exists for involvement of the classical SHC-Grb2-SOS-Ras-Raf-MEK1-ERK cascade in mediating GH-induced ERK activity via direct interaction of SHC with tyrosine phosphorylated JAK2 (VanderKuur, Allevato et al. 1995, Vanderkuur, Butch et al. 1997), although SHC has also been shown to bind the tyrosine phosphorylated GHR tail in an in vitro assay (Moutoussamy, Renaudie et al. 1998).
However, other molecules have been found to at least partially contribute to GH-induced ERK activation. GH causes activation of phosphoinositide-3 kinase (PI3K) (Kilgour, Gout et al. 1996, Hodge, Liao et al. 1998, MacKenzie, Yarwood et al. 1998, Costoya, Finidori et al. 1999, Liang, Zhou et al. 1999, Liang, Jiang et al. 2000, Jeay, Sonenshein et al. 2001) and pretreatment with chemical inhibitors of PI3K can block GH’s ability to activate ERKs (Kilgour, Gout et al. 1996, Liang, Zhou et al. 1999, Liang, Jiang et al. 2000), placing PI3K upstream of ERKs in these cellular systems. Consistent with its ability to activate PI3K, GH has been amply demonstrated to cause tyrosine phosphorylation of the large adaptor protein, insulin receptor substrate-1 (IRS-1), which often associates with PI3K activity (Ridderstrale and Tornqvist 1994, Souza, Frick et al. 1994, Argetsinger, Hsu et al. 1995, Kilgour, Gout et al. 1996), and IRS-1 has been shown in mutagenesis and coimmunoprecipitation experiments to interact with JAK2 (Argetsinger, Hsu et al. 1995, Liang, Jiang et al. 2000). Further, both cellular reconstitution of IRS-1-deficient (murine promonocytic) cells with IRS-1 (Liang, Jiang et al. 2000) and shRNA-mediated knockdown of endogenous IRS-1 (in murine preadipocytes) (Wang, Yang et al. 2009) revealed that IRS-1 selectively augments GH-induced PI3K and ERK pathway signaling without affecting GHR, JAK2, or STAT5 abundance or activatability. Interestingly, GH-induced SHC phosphorylation was also lessened with IRS-1 silencing, suggesting that SHC may in part reside downstream of IRS-1 in this pathway (Wang, Yang et al. 2009). Work from our own laboratory also indicates that another large docking protein in the IRS family, Gab1 (Kim, Loesch et al. 2002), and the protein tyrosine phosphatase, SHP-2 (Kim, Jiang et al. 1998), participate in the GH-induced selective activation of ERKs, although the exact nature of inclusion of each in the GHR’s signaling complex remains uncertain. Intriguingly, for SHP-2, activating mutations associated with Noonan Syndrome (which includes short stature) cause the expected ERK hyperactivation, but also lead to reduced GH-induced IGF-1 generation in cell systems; detailed mechanisms of this uncoupling remain to be elucidated (De Rocca Serra-Nedelec, Edouard et al. 2012).
In addition to docking molecules and enzymes, another receptor has been implicated both as a link to the GH-induced ERK activation pathway and as being affected by this pathway. Epidermal growth factor receptor (EGFR; ErbB1) is a tyrosine kinase growth factor receptor involved in multiple cellular processes in health and disease. Yamauchi et al (Yamauchi, Ueki et al. 1997) demonstrated in cell culture systems that GH caused JAK2-dependent tyrosine phosphorylation of EGFR’s cytoplasmic domain independent of EGFR kinase activity and that this allowed Grb2 to bind EGFR and activate the ERK cascade. Thus, EGFR can function as a docking protein for the GH-induced ERK activation pathway. The physiological significance of this is as yet unknown, but it is also interesting that GH induces ERK to phosphorylate EGFR at a cytoplasmic domain threonine residue, the net effect of which is to dampen subsequent EGF-induced EGFR downregulation and allow GH to cooperatively sustain EGF signaling (Huang, Kim et al. 2003, Li, Huang et al. 2008). One implication of these findings is that that GH-induced ERK activation may exert indirect effects by virtue of the propensity of GHR to interact physically or functionally with other receptor signaling systems (more below).
As mentioned above, GH’s ability to activate ERKs may be uncoupled from its triggering of STAT5 signaling (Love, Whatmore et al. 1998). Mechanisms for this pathway selectivity are incompletely known, but there are indications that subcellular localization based on local plasma membrane architecture may play a role. In subcellular fractionation experiments in which plasma membranes were separated to isolate cholesterol-rich lipid rafts (LR) vs. non-LR fractions, GHR was highly LR-enriched in both murine preadipocytes (in which GH promotes both STAT5 and ERK activation) as well as in human IM-9 lymphoblasts (in which GH promotes STAT5, but not ERK activation); ERKs and Grb-2 were also LR-enriched in preadipocytes, but although amply expressed, ERKs and Grb-2 were not associated with LR in IM-9 cells and, in preadipocytes, GH caused further LR accumulation of ERK pathway components (Yang, Huang et al. 2004). STAT5 (activated in both cells by GH) was similarly non-membranous in both cells. Further, in preadipocytes, GH-induced ERK activation was highly LR-enriched, but markedly less so in LR when IRS-1 was silenced (Wang, Yang et al. 2009). These data raise the intriguing possibility that there is spatial compartmentalization of pathway utilization downstream of GHR.
5. Modulation of GH signaling.
What follows is a brief discussion of examples of mechanisms by which the strength of GH signaling may be modulated either by external factors or as a result of GHR activation itself.
5.1. Modulation of cell surface GHR abundance and GH sensitivity by constitutive and inducible GHR metalloproteolysis
One important determinant of cellular GH sensitivity is cell surface GHR abundance, which, not surprisingly, is regulated by a number of mechanisms (Baumann and Frank 2002, Deng, He et al. 2007, Frank and Fuchs 2008). A series of studies beginning in the late 1990s have demonstrated that the GHR in multiple species is a target for constitutive and inducible proteolysis in its extracellular domain membrane-proximal stem region (Alele, Jiang et al. 1998, Wang, He et al. 2002, Wang, He et al. 2003). This proteolysis results in loss of the full-length GHR with concomitant generation of a cell-associated cytoplasmic domain-containing GHR remnant and shedding of a soluble GHR extracellular domain (also known as GH binding protein or GHBP) (Alele, Jiang et al. 1998, Wang, He et al. 2002, Wang, He et al. 2003). The transmembrane enzyme that primarily cleaves GHR is TACE (tumor necrosis factor-α converting enzyme; also known as ADAM17 (Black, Rauch et al. 1997, Moss, Jin et al. 1997)) (Zhang, Jiang et al. 2000), which contains a zinc-dependent protease in its extracellular domain and can be activated by growth factors and stimuli via ERK and protein kinase C (PKC) pathways. Indeed, treatment of cells with serum, PDGF, or PKC activators rapidly induces GHR proteolysis and GHBP shedding (Zhang, Jiang et al. 2000, Guan, Zhang et al. 2001). The net effect is to rapidly downregulate cell surface GHR and desensitize the cell to subsequent acute treatment with GH. Of note, acute inflammatory states, as modeled by administration in rodents of lipopolysaccharide (LPS), have been shown to be associated with diminished hepatic GH sensitivity by various putative mechanisms (Defalque, Brandt et al. 1999, Mao, Ling et al. 1999, Bergad, Schwarzenberg et al. 2000, Wang, Li et al. 2002, Denson, Held et al. 2003, Hong-Brown, Brown et al. 2003, Chen, Sun et al. 2007). Interestingly, in mice, LPS treatment rapidly caused loss of hepatic GHR protein without altering GHR mRNA levels and this GHR loss was associated with diminished subsequent acute GH-induced hepatic STAT5 signaling (i.e., desensitization to GH) (Wang, Jiang et al. 2008). In this model, LPS also caused rapid proteolytic shedding of the extracellular domain (the GHBP) of the hepatically-expressed GHR. Thus, LPS-induced modulation of GHR abundance by proteolytic cleavage corresponds to desensitization to GH in the liver. Recent studies also revealed that TIMP3 (tissue inhibitor of metalloprotease3), an extracellular matrix binding protein (Blenis and Hawkes 1984, Staskus, Masiarz et al. 1991, Yu, Yu et al. 2000) and a natural inhibitor of TACE (Amour, Slocombe et al. 1998, Mohammed, Smookler et al. 2004), reversed the desensitizing effects of TACE on GHR abundance and cellular GH action (Zhang, Wang et al. 2016). These findings support the notion that modulation of GHR availability by regulation of the balance of TACE/TIMP3 activity by factors (such as inflammation) in the cell’s environment may meaningfully modulate aspects of GH sensitivity at a posttranscriptional level. Given GH’s influence on metabolism and energy economy, this is a topic worthy of further investigation.
5.2. Involvement of SOCS proteins
The SOCS (suppressor of cytokine signaling) proteins are a family of molecules generally rapidly translated upon activation of a number of cytokine receptors that function as a negative feedback loop to dampen cytokine signaling (Linossi, Babon et al. 2013, Dehkhoda, Lee et al. 2018). There are eight family members (SOCS1-7 and CIS), all of which are cytosolic modular proteins that each contain a central phosphotyrosine-binding SH2 domain and a conserved C-terminal 40-residue motif called the SOCS box. This motif mediates interaction with several proteins that collectively form an active E3 ubiquitin ligase complex that facilitates proteasomal degradation of molecules targeted by the particular SOCS protein. In addition to this degradative function, SOCS proteins can variably interfere with formation of cytokine receptor-JAK-STAT complexes and diminish JAK kinase activity, all of which accomplish negative regulation of cytokine action. In terms of GH signaling, four SOCS proteins (SOCS1-3 and CIS) have been implicated to varying degrees (Adams, Hansen et al. 1998, Hansen, Lindberg et al. 1999, Ram and Waxman 1999, Tollet-Egnell, Flores-Morales et al. 1999, Paul, Seiliez et al. 2000). The most compelling in vivo evidence suggests particular physiological regulation of GH action is contributed by SOCS2. SOCS2 knockout mice exhibit postnatal gigantism akin to acromegaly, but unaccompanied by excess pituitary GH secretion (Metcalf, Greenhalgh et al. 2000, Greenhalgh, Bertolino et al. 2002); these studies implicated prolonged GH-induced STAT5 activation as mechanistically important in the setting of SOCS2 deficiency. Recent studies of a GHR mutant incapable of interacting with SOCS2 in the setting of lung cancer suggest that SOCS2 principally regulates GH sensitivity via degradation of GHR (Chhabra, Wong et al. 2018).
5.3. Other modulators
Although a detailed treatment is beyond the scope of this review, there have emerged other molecules, often with multiple other functions, that modulate GH signaling. The reader is referred to recent excellent reviews for more complete discussion of some of these (Carter-Su, Schwartz et al. 2016, Dehkhoda, Lee et al. 2018).
Protein tyrosine phosphatases (PTPs) catalyze removal of phosphate from tyrosine residues in their substrates and are thus often thought of as negative regulators of tyrosine kinase-induced signaling. Several PTPs (including SHP1, SHP2, PTP-H1, and PTP-1B) have been implicated as modulators of GH-induced STAT5 activity (Hackett, Wang et al. 1997, Ram and Waxman 1997, Hodge, Liao et al. 1998, Kim, Jiang et al. 1998, Stofega, Herrington et al. 2000, Gu, Dube et al. 2003, Pasquali, Curchod et al. 2003, Choi, Kim et al. 2006, Pilecka, Patrignani et al. 2007, De Rocca Serra-Nedelec, Edouard et al. 2012, Gan, Zhang et al. 2013). In most cases, PTP activity likely negatively regulates GH signaling by dephosphorylating proximal GHR signaling elements; however, as mentioned above (Kim, Jiang et al. 1998), some findings suggest a positive regulatory effect of SHP2 on GH-induced ERK activation.
The examples of modulation discussed above (inducible GHR metalloproteolysis, GH-induced SOCS expression, and GH-induced PTP involvement) generally act as brakes or negative regulators of GH signaling (either heterologous in the case of GHR metalloproteolysis or homologous in the cases of SOCS proteins and most PTPs). One type of positive modulator of GH signaling was first identified by the Carter-Su laboratory in 1997. The SH2B family of adaptor proteins (reviewed in (Maures, Kurzer et al. 2007)) were first found to be important in immune cell activation. In an attempt to define JAK2-interacting proteins, Rui, et al (Rui, Mathews et al. 1997) demonstrated that GH induced an SH2B isoform called SH2Bβ to associate with tyrosine phosphorylated JAK2 (at Tyr-813) and itself become tyrosine phosphorylated by JAK2. This group later demonstrated that SH2Bβ is a strong activator of JAK2 in response to GH (Rui and Carter-Su 1999); this may be based on SH2Bβ’s ability to enhance JAK2 dimerization to encourage activation and/or its ability to preserve JAK2’s activation status. Functionally, subsequent studies have identified important roles for SH2Bβ in enhancing GH-induced cell motility by interacting with the actin cytoskeleton, as well as other non-GH signaling effects on energy homeostasis and obesity (Maures, Kurzer et al. 2007).
6. GHR and IGF-1R – a novel signaling relationship.
In this section, we discuss a new set of findings derived from cellular signaling studies that suggest a heretofore unappreciated relationship between GHR and IGF-1R and that broaden our conception of how the two receptors may collaborate. We first briefly review the essence of the somatomedin hypothesis of GH action and then review findings on the physical and functional consequences of GHR-IGF-1R interaction for GH signaling.
6.1. Somatomedin hypothesis of GH action
Although Evans and Long implicated pituitary GH in longitudinal growth and enhancement of muscle mass in the 1920s, as described above, understanding of the physiological role(s) of GH in these somatogenic actions was greatly advanced by critical experimental observations in the 1950s. Their groundbreaking work led Salmon and Daughaday to propose the “somatomedin hypothesis” of GH action in 1957 (Salmon and Daughaday 1957, Daughaday 2000) (Figure 2A). As originally suggested, this hypothesis held that circulating GH induced the liver to secrete a circulating factor (mediator of growth) known as somatomedin-C (later defined as the ~6-kD peptide, IGF-1), which then acted as an endocrine hormone to promote growth of bone, muscle, and some other tissues. IGF-1 was found to exert its effects by binding to the type 1 IGF-1 receptor (IGF-1R), which is a disulfide-linked heterotetramer comprised of two extracellular α-chains and two transmembrane β-chains, the latter of which encode intrinsic tyrosine kinase activity in their intracellular domains (Ullrich, Gray et al. 1986, LeRoith 2000, Nakae, Kido et al. 2001). Notably, liver normally has little to no IGF-1R expression (see below) (Pivonello, De Martino et al. 2014).
Figure 2. Role of IGF-1R-GHR interaction in augmenting GH signaling in the context of the somatomedin hypothesis.
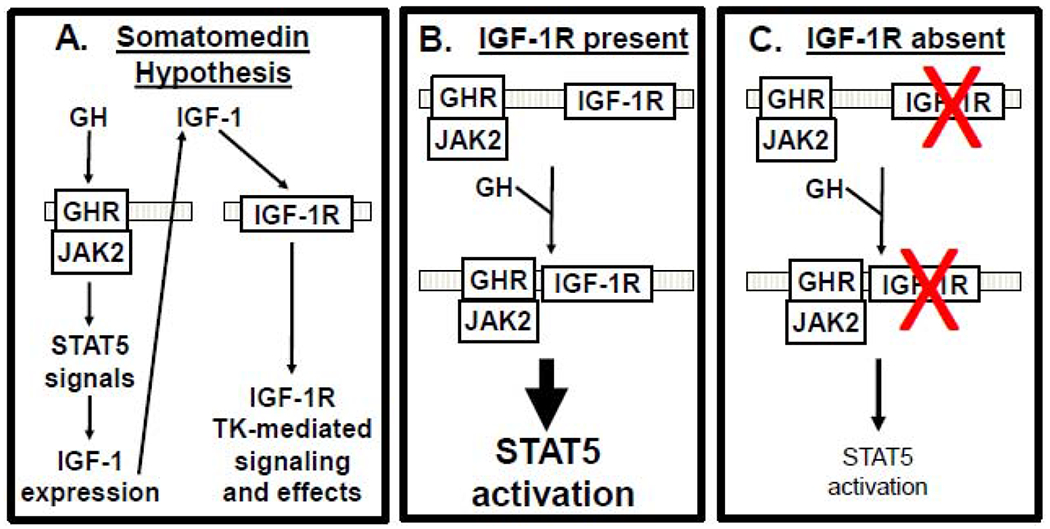
A) Somatomedin hypothesis. GH binds GHR, activating JAK2/STAT5 and IGF-1 production. IGF-1 then acts in an endocrine fashion, stimulating IGF-1R signaling. B,C) GH promotes GHR-IGF-1R association. IGF-1R acts as a component of GH signaling (B). In IGF-1R’s absence (or reduction) (red X)(C), GH action is reduced. See the text for discussion.
The somatomedin hypothesis elegantly addressed major issues in endocrinology and the physiology of GH and IGF-1 actions. Indeed, global genetic knockouts of IGF-1 and IGF-1R in mice were characterized by intrauterine growth retardation and, if viable, postnatal growth defects (Liu, Baker et al. 1993, Powell-Braxton, Hollingshead et al. 1993, Liu and LeRoith 1999, Wang, Zhou et al. 1999); the latter effect was consistent with the notion that IGF-1 functions “downstream” of GH. Notably, liver-specific deletion of IGF-1 (Sjogren, Liu et al. 1999, Yakar, Liu et al. 1999) resulted in ~75% reduction in circulating IGF-1 and substantial rise in GH, but did not diminish growth; however a further 15% reduction of IGF-1, accomplished by combined deletion of the liver acid-labile subunit of the IGF binding complex (Yakar, Rosen et al. 2002), did result in postnatal growth retardation. These and other findings suggest that either non-hepatically-derived IGF-1 and/or a minimum circulating threshold of liver-derived IGF-1 are important to facilitate GH action (at least as it pertains to growth). Yet another dual gene deletion mouse, a combined GHR and IGF-1 global knockout (Lupu, Terwilliger et al. 2001), presented yet another wrinkle. This mouse exhibited postnatal growth retardation greater than either the GHR knockout or the IGF-1 knockout, calling into some question the primacy of the “linear” GH ► GHR ► IGF-1 ► IGF-1R pathway predicted by the somatomedin hypothesis and perhaps allowing for collaboration of GH and IGF-1 (as in (Ashcom, Gurland et al. 1992, Edmondson, Russo et al. 1999, Huang, Kim et al. 2004)) or that each contributes in independent, but overlapping, fashion.
6.2. GHR-IGF-1R physical and functional interaction
In a series of studies using several cell culture model systems, we in our own laboratory have pursued a potential novel variation on the theme outlined above for the signaling relationships between GHR and IGF-1R. As summarized below, these findings suggest that, in addition to delivering the signaling payload of the GH ► GHR ► IGF-1 ► IGF-1R pathway, IGF-1R may serve as a component of the GH signaling pathway, modulating GHR’s signaling strength and allowing for more local heterogeneity of GH/IGF-1 actions.
GH-induced association of GHR with IGF-1R
GHR/IGF-1R signaling interactions were originally explored in mouse preadipocytes – 3T3-F442A and 3T3-L1 cells – that endogenously express GHR, IGF-1R, and JAK2 and respond to both GH and IGF-1 (Huang, Kim et al. 2004). Interestingly, acute GH stimulation of these cells allowed specific coimmunoprecipitation of GHR with IGF-1R. Acute formation of this novel GHR-IGF-1R complex (also including JAK2) in response to physiologic GH concentrations did not depend on IGF-1R or JAK2 kinase activity or phosphorylation of any partner, as it proceeded even with pretreatment with a chemical kinase inhibitor, and did not depend on IGF-1 treatment. GHR-IGF-1R complex formation was also observed in several GH-responsive rodent β-cell lines (Ma, Wei et al. 2011), as well as in human prostate cancer (LNCaP) cells (Gan, Buckels et al. 2014). Notably, complex formation was inhibited by pretreatment with a GHR extracellular domain antagonistic monoclonal antibody known to prevent GH-induced GHR conformational changes, but not to block GH binding (Jiang, Wan et al. 2011, Gan, Buckels et al. 2014), suggesting that GH-induced changes in GHR conformation may allow IGF-1R to associate with GHR.
IGF-1R participates in acute GH signaling
New model systems were developed to probe the question of whether the physical association of IGF-1R with GHR might have functional impact on GH signaling. The first of these systems was mouse primary calvarial osteoblasts, which acutely respond to both GH and IGF-1 (DiGirolamo, Mukherjee et al. 2007). Osteoblasts from newborns bearing lox-P-flanked IGF-1R alleles (Zhang, Xuan et al. 2002, DiGirolamo, Mukherjee et al. 2007) were infected in vitro with Ad-Cre (adenovirus driving Cre recombinase) or a control adenovirus and Ad-Cre treatment specifically silenced IGF-1R protein, but did not affect GHR expression or abundance (Gan, Zhang et al. 2010). Yet, notably, IGF-1R deletion resulted in marked reduction of acute GH-induced STAT5 phosphorylation and GH-dependent endogenous IGF-1 mRNA, suggesting signaling impact downstream of STAT5 (Figure 2B,C). Two other model cell line systems (mouse MIN6 β-cells (Ma, Wei et al. 2011) and human LNCaP cells (Gan, Zhang et al. 2013, Gan, Buckels et al. 2014)) were developed in which stable shRNA transfection knocked down IGF-1R substantially without affecting GHR, JAK2, or STAT5 levels. In both systems, GH-induced STAT5 phosphorylation was markedly reduced in cells with low IGF-1R. Further, careful time course analyses in the LNCaP system revealed that lowering IGF-1R similarly led to reduced GH-induced JAK2 and GHR tyrosine phosphorylation. Because acute GH signaling was affected, these findings are not explained by altered GH-induced IGF-1 expression (somatomedin hypothesis); rather, they suggest that IGF-1R can serve as a proximal component of acute GH signaling and that full GH-induced activation is realized when IGF-1R is present.
IGF-1R determinants for its participation in acute GH signaling
If IGF-1R, by GH-regulated interaction with GHR, is a relevant and potentially modulatable participant in GH action in cells expressing both receptors, what are the IGF-1R determinants that allow this physical and functional interaction? This was explored in the osteoblast system, in which re-expression of wild-type IGF-1R in Ad-Cre-mediated endogenous IGF-1R-deleted cells rescued GH-induced STAT5 activation and IGF-1 gene expression (Gan, Zhang et al. 2010). Similarly, an IGF-1R mutant truncated just below the transmembrane domain to lack the beta chain’s intracellular domain (including its kinase) also rescued GH signaling, These findings suggested that IGF-IR’s presence, but not its kinase activity, augments GH signaling and that extracellular domain and/or transmembrane domain elements mediate its effect.
Examination of extracellular domain elements involved was furthered by the finding that reconstitution with the structurally-related insulin receptor (IR), unlike the IGF-1R, was unable to rescue GH signaling in osteoblasts with silenced endogenous IGF-1R (Gan, Paterson et al. 2014). In addition to emphasizing the specificity of GHR-IGF-1R collaboration, this finding allowed a platform to assess IGF-1R extracellular domain determinants involved by exploiting the interchangeability of extracellular domain modules in IGF-1R/IR chimeric constructs. Utilizing this “domain swapping” approach in rescue experiments, it was determined that an N-terminal IGF-1R α-chain extracellular domain region(s), likely within the CR and/or L2 subdomains, is critical to allow the IGF-1R to specifically augment acute GH signaling (Gan, Paterson et al. 2014). Mechanistically, it is hypothesized that GH promotes IGF-1R’s interaction via its extracellular domain with another (likely transmembrane) protein, the intracellular domain of which effectively augments GH action. This protein, cartooned as “X” (see (Gan, Zhang et al. 2013)) in Figure 3, is envisioned as either a single protein or a group of proteins that enhance GH-induced JAK2 activation, delay GH-induced downregulation of activated GHR, or prevent negative regulators of GHR/JAK2 (e.g., PTP1B) from acting on targets (Gan, Zhang et al. 2013, Gan, Buckels et al. 2014).
Figure 3. Putative IGF-1R-associated accessory protein(s) augment GH signaling.
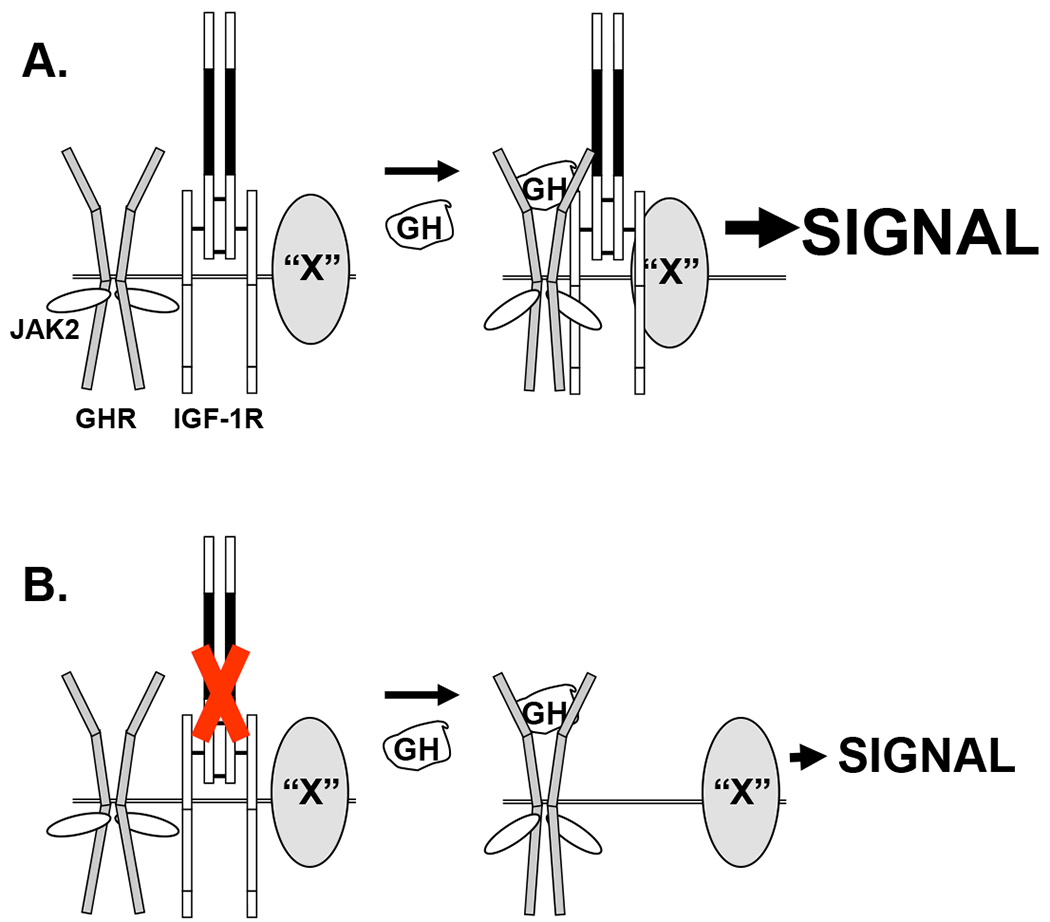
A) GH enhances association of GHR/JAK2 with IGF-1R and an IGF-1R-associated protein(s), “X”, augmenting GH signaling. “X” is cartooned as a transmembrane protein to suggest likely association with IGF-1R ECD and actions exerted intracellularly, but “X” could be a protein complex that either augments signaling or lessens down-regulation of signaling. B) In IGF-1R’s absence, GH does not induce “X” to join the GHR/JAK2 signaling complex and signaling is relatively diminished. CR-L2 region of IGF-1R ECD is shown in solid black and is envisioned as the region of IGF-1R that interacts with GHR. See the text for discussion.
The ability of a soluble IGF-1R-α N-terminal L1-CR-L2 fragment, when expressed alone, to affect GH signaling produced interesting and important results (Huang, Kim et al. 2004, Gan, Zhang et al. 2010, Gan, Paterson et al. 2014). This fragment (termed soluble IGF-1R or sol IGF-1R) and its analogous insulin receptor fragment (sol IR; used as a control) fold normally and do not bind ligands (Garrett, McKern et al. 1998, Menting, Whittaker et al. 2013) and sol IGF-1R, when expressed in IGF-1R-deleted cells, did not rescue diminished GH signaling (Gan, Paterson et al. 2014). However, conditioned medium of cells programmed to express sol IGF-1R, but not sol IR, strongly inhibited acute GH-induced GHR/STAT5 signaling and downstream IGF-1 gene expression of cells bearing the normal IGF-1R and GHR (Gan, Buckels et al. 2014), indicating that sol IGF-1R affects (inhibits) very proximal GH signaling. In principle, sol IGF-1R could inhibit GH signaling by interaction with GH, IGF-1R, or GHR. Only the latter (sol IGF-1R/GHR) interaction has been detected and it is enhanced by GH treatment (Gan, Buckels et al. 2014). The sol IGF-1R/GHR functional interaction was shown to be specific for GHR and not PRLR in that only GH-induced signaling mediated by GHR, and not PRLR, is inhibited by sol IGF-1R (Zhang, Gc et al. 2019). Thus, it is hypothesized that sol IGF-1R, by a dominant-negative effect, specifically inhibits GH signaling in cells expressing IGF-1R by competing with IGF-1R for GHR association and that IGF-1R augments GH signaling, while sol IGF-1R is inhibitory (Figure 4).
Figure 4. Putative effects of sol IGF-1R on GH signaling.
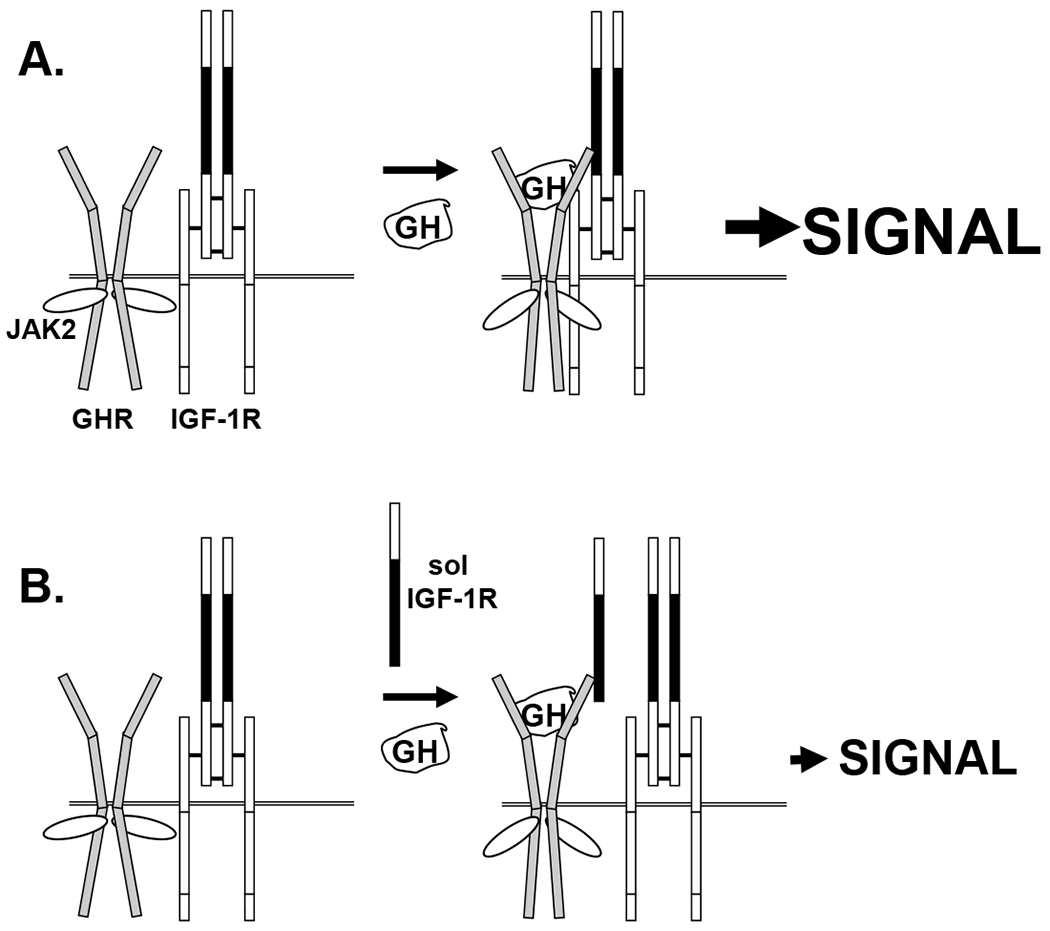
A) GH enhances association of GHR/JAK2 with IGF-1R, augmenting GH signaling, as in Figure 3A. Strong GH-induced signaling is indicated by “SIGNAL”. B) GH induces GHR/sol IGF-1R association and dampened signaling ensues. Sol IGF-1R contains the L1-CR-L2 region (CR-L2 region in black). Sol IGF-1R allows GH-induced GHR and JAK2 conformational changes and signaling, but competes with IGF-1 R for GHR association. GH signaling is similar in strength as with IGF-1R silencing seen in Figure 3B. See the text for discussion.
Implications of these findings
These findings suggest a non-canonical role for IGF-1R as a contributor to GH action. While the classical somatomedin hypothesis clearly identifies IGF-1 as a GH effector, it is now clear that: GH can have direct effects; IGF-1 itself has endocrine, paracrine, and autocrine effects; and GH and IGF-1 can have overlapping, counteracting, and/or collaborative effects (Ashcom, Gurland et al. 1992, Edmondson, Russo et al. 1999, Le Roith, Bondy et al. 2001, Lupu, Terwilliger et al. 2001, Huang, Kim et al. 2004, Kaplan and Cohen 2007, Wu, Sun et al. 2013). Similarly, it is proposed that the novel physical and functional interaction between IGF-1R and GHR allows IGF-1R, in addition to mediating its own ligand’s effects, to contribute proximally to efficient GH signaling. Why would a role for IGF-1R in proximal GH signaling evolve? One possibility is that IGF-1 action in GH-responsive tissues could be locally enhanced/modulated by virtue of IGF-1R sensitizing cells to GH, allowing enhanced local IGF-1 production and therefore local IGF-1 action, without necessarily affecting circulating IGF-1 and thus GH levels. Thus, the dynamic range of GH target tissue response could be even further enhanced by regulation of IGF-1R expression levels – via both proximal GH sensitivity and IGF-1 signaling (Figure 5). As liver normally has little to no IGF-1R expression, this could be most relevant in extra-hepatic tissues and/or in cancers, including hepatocellular carcinomas, in which aberrant IGF-1R expression is likely etiologically important (Wu and Zhu 2011, Pivonello, De Martino et al. 2014). Further, manipulation of IGF-1R’s availability or its capacity for GHR interaction is a therapeutically-exploitable implication of these findings, as is further development of sol IGF-1R as a GHR antagonist. Thus, they present a more enriched paradigm for interplay between GH/GHR and IGF-1/IGF-1R that may complement and synergize with the somatomedin hypothesis.
Figure 5. Potential selective modulation of local GH action in IGF-1R-expression GH target tissues.
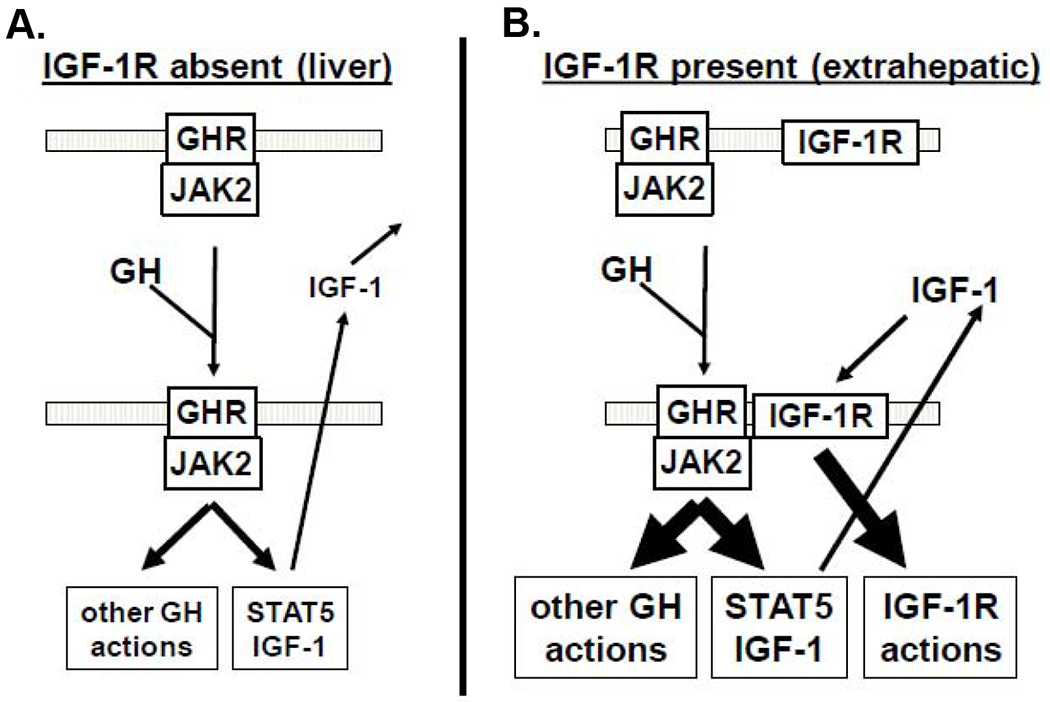
A) In IGF-1R-negative tissues (such as liver), GH induces STAT5 signaling and other GH actions, culminating with IGF-1 secretion and endocrine actions, but not local IGF-1 effects. B) In IGF-1R-positive tissues (designated as extrahepatic) the presence of IGF-1R augments GH signaling (as above) and local IGF-1 production, which exerts further local effects via the IGF-1R, thereby augmenting the dynamic range of GH/IGF-1 action without necessarily changing the circulating GH or IGF-1 levels. See the text for discussion.
Acknowledgements
The author appreciates the helpful conversations with members of the Frank laboratory. This work was supported by a Veterans Affairs Merit Review Award, NIH grants DK107441, and DK58259 (to S.J.F.).
This work was supported by NIH grants DK107441 and a VA Merit Review grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT: The author has nothing to disclose.
References
- Abdel-Meguid SS, Shieh HS, Smith WW, Dayringer HE, Violand BN and Bentle LA (1987). “Three-dimensional structure of a genetically engineered variant of porcine growth hormone.” Proc Natl Acad Sci U S A 84(18): 6434–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams TE, Baker L, Fiddes RJ and Brandon MR (1990). “The sheep growth hormone receptor: molecular cloning and ontogeny of mRNA expression in the liver.” Mol Cell Endocrinol 73(2-3): 135–145. [DOI] [PubMed] [Google Scholar]
- Adams TE, Hansen JA, Starr R, Nicola NA, Hilton DJ and Billestrup N (1998). “Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling.” J Biol Chem 273(3): 1285–1287. [DOI] [PubMed] [Google Scholar]
- Alele J, Jiang J, Goldsmith JF, Yang X, Maheshwari HG, Black RA, Baumann G and Frank SJ (1998). “Blockade of growth hormone receptor shedding by a metalloprotease inhibitor.” Endocrinology 139(4): 1927–1935. [DOI] [PubMed] [Google Scholar]
- Ambrosio R, Fimiani G, Monfregola J, Sanzari E, De Felice N, Salerno MC, Pignata C, D’Urso M and Ursini MV (2002). “The structure of human STAT5A and B genes reveals two regions of nearly identical sequence and an alternative tissue specific STAT5B promoter.” Gene 285(1-2): 311–318. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ and Murphy G (1998). “TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3.” FEBS Lett 435(1): 39–44. [DOI] [PubMed] [Google Scholar]
- Anderson NG (1992). “Growth hormone activates mitogen-activated protein kinase and S6 kinase and promotes intracellular tyrosine phosphorylation in 3T3-F442A preadipocytes.” Biochem J 284(Pt 3): 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NG (1993). “Simultaneous activation of p90rsk and p70s6k S6 kinases by growth hormone in 3T3-F442A preadipocytes.” Biochem Biophys Res Commun 193(1): 284–290. [DOI] [PubMed] [Google Scholar]
- Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN and Carter-Su C (1993). “Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase.” Cell 74(2): 237–244. [DOI] [PubMed] [Google Scholar]
- Argetsinger LS and Carter-Su C (1996). “Mechanism of signaling by growth hormone receptor.” Physiol Rev 76(*): 1089–1107. [DOI] [PubMed] [Google Scholar]
- Argetsinger LS, Hsu GW, Myers MGJ, Billestrup N, White MF and Carter-Su C (1995). “Growth hormone, interferon-gamma, and leukemia inhibitory factor promoted tyrosyl phosphorylation of insulin receptor substrate-1.” J Biol Chem 270(*): 14685–14692. [DOI] [PubMed] [Google Scholar]
- Ashcom G, Gurland G and Schwartz J (1992). “Growth hormone synergizes with serum growth factors in inducing c-fos transcription in 3T3-F442A cells.” Endocrinology 131(4): 1915–1921. [DOI] [PubMed] [Google Scholar]
- Barclay JL, Kerr LM, Arthur L, Rowland JE, Nelson CN, Ishikawa M, d’Aniello EM, White M, Noakes PG and Waters MJ (2010). “In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2.” Molecular endocrinology 24(1): 204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann G and Frank SJ (2002). “Metalloproteinases and the modulation of GH signaling.” J Endocrinol 174(3): 361–368. [DOI] [PubMed] [Google Scholar]
- Baumbach WR, Horner DL and Logan JS (1989). “The growth hormone-binding protein in rat serum is an alternatively spliced form of the rat growth hormone receptor.” Genes Dev 3(8): 1199–1205. [DOI] [PubMed] [Google Scholar]
- Bazan JF (1990). “Structural design and molecular evolution of a cytokine receptor superfamily.” Proc Natl Acad Sci U S A 87(18): 6934–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergad PL, Schwarzenberg SJ, Humbert JT, Morrison M, Amarasinghe S, Towle HC and Berry SA (2000). “Inhibition of growth hormone action in models of inflammation.” Am J Physiol Cell Physiol 279(6): C1906–1917. [DOI] [PubMed] [Google Scholar]
- Bergad PL, Shih HM, Towle HC, Schwarzenberg SJ and Berry SA (1995). “Growth hormone induction of hepatic serine protease inhibitor 2.1 transcription is mediated by a Stat5-related factor binding synergistically to two gamma-activated sites.” J Biol Chem 270(42): 24903–24910. [DOI] [PubMed] [Google Scholar]
- Bergan-Roller HE and Sheridan MA (2018). “The growth hormone signaling system: Insights into coordinating the anabolic and catabolic actions of growth hormone.” Gen Comp Endocrinol 258: 119–133. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ and Cerretti DP (1997). “A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells.” Nature 385(6618): 729–733. [DOI] [PubMed] [Google Scholar]
- Blenis J and Hawkes SP (1984). “Characterization of a transformation-sensitive protein in the extracellular matrix of chicken embryo fibroblasts.” J Biol Chem 259(18): 11563–11570. [PubMed] [Google Scholar]
- Bole-Feysot C, Goffin V, Edery M, Binart N and Kelly PA (1998). “Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice [In Process Citation].” Endocr Rev 19(3): 225–268. [DOI] [PubMed] [Google Scholar]
- Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, Parker MW, Sierecki E, Gambin Y, Gomez GA, Alexandrov K, Wilson IA, Doxastakis M, Mark AE and Waters MJ (2014). “Mechanism of activation of protein kinase JAK2 by the growth hormone receptor.” Science 344(6185): 1249783. [DOI] [PubMed] [Google Scholar]
- Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW and Waters MJ (2005). “Model for growth hormone receptor activation based on subunit rotation within a receptor dimer.” Nat Struct Mol Biol 12(9): 814–821. [DOI] [PubMed] [Google Scholar]
- Burnside J, Liou SS and Cogburn LA (1991). “Molecular cloning of the chicken growth hormone receptor complementary deoxyribonucleic acid: mutation of the gene in sex-linked dwarf chickens.” Endocrinology 128(6): 3183–3192. [DOI] [PubMed] [Google Scholar]
- Campbell GS, Meyer DJ, Raz R, Levy DE, Schwartz J and Carter-Su C (1995). “Activation of acute phase response factor (APRF)/Stat 3 transcription factor by growth hormone.” J Biol Chem 270(*): 3974–3979. [DOI] [PubMed] [Google Scholar]
- Campbell GS, Pang L, Miyasaka T, Saltiel AR and Carter-Su C (1992). “Stimulation by growth hormone of MAP kinase activity in 3T3-F442A fibroblasts.” J Biol Chem 267(9): 6074–6080. [PubMed] [Google Scholar]
- Carter-Su C, Schwartz J and Argetsinger LS (2016). “Growth hormone signaling pathways.” Growth Horm IGF Res 28: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Su C, Stubbart JR, Wang XY, Stred SE, Argetsinger LS and Shafer JA (1989). “Phosphorylation of highly purified growth hormone receptors by a growth hormone receptor-associated tyrosine kinase.” J Biol Chem 264(31): 18654–18661. [PubMed] [Google Scholar]
- Chen EY, Liao YC, Smith DH, Barrera-Saldana HA, Gelinas RE and Seeburg PH (1989). “The human growth hormone locus: nucleotide sequence, biology, and evolution.” Genomics 4(4): 479–497. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sun D, Krishnamurthy VM and Rabkin R (2007). “Endotoxin attenuates growth hormone-induced hepatic insulin-like growth factor I expression by inhibiting JAK2/STAT5 signal transduction and STAT5b DNA binding.” Am J Physiol Endocrinol Metab 292(6): E1856–1862. [DOI] [PubMed] [Google Scholar]
- Chhabra Y, Nelson CN, Plescher M, Barclay JL, Smith AG, Andrikopoulos S, Mangiafico S, Waxman DJ, Brooks AJ and Waters MJ (2019). “Loss of growth hormone-mediated signal transducer and activator of transcription 5 (STAT5) signaling in mice results in insulin sensitivity with obesity.” FASEB J 33(5): 6412–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra Y, Wong HY, Nikolajsen LF, Steinocher H, Papadopulos A, Tunny KA, Meunier FA, Smith AG, Kragelund BB, Brooks AJ and Waters MJ (2018). “A growth hormone receptor SNP promotes lung cancer by impairment of SOCS2-mediated degradation.” Oncogene 37(4): 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Kim HS, Kim SH, Yang YR, Bae YS, Chang JS, Kwon HM, Ryu SH and Suh PG (2006). “Phospholipase Cgamma1 negatively regulates growth hormone signalling by forming a ternary complex with Jak2 and protein tyrosine phosphatase-1B.” Nat Cell Biol 8(12): 1389–1397. [DOI] [PubMed] [Google Scholar]
- Cioffi JA, Wang X and Kopchick JJ (1990). “Porcine growth hormone receptor cDNA sequence.” Nucleic Acids Res 18(21): 6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Camporez JPG, Tran JL, Wilson CG, Lowe DA, Nordstrom SM, Ganeshan K, Perry RJ, Shulman GI, Jurczak MJ and Weiss EJ (2017). “Adipocyte JAK2 mediates growth hormone-induced hepatic insulin resistance.” JCI Insight 2(3): e91001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Wilson CG, Lowe D, Tran JL, Vera NB, Clasquin M, Mattis AN and Weiss EJ (2019). “Adipocyte JAK2 mediates spontaneous metabolic liver disease and hepatocellular carcinoma.” JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordoba-Chacon J, Majumdar N, List EO, Diaz-Ruiz A, Frank SJ, Manzano A, Bartrons R, Puchowicz M, Kopchick JJ and Kineman RD (2015). “Growth Hormone Inhibits Hepatic De Novo Lipogenesis in Adult Mice.” Diabetes 64(9): 3093–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costoya JA, Finidori J, Moutoussamy S, Searis R, Devesa J and Arce VM (1999). “Activation of growth hormone receptor delivers an antiapoptotic signal: evidence for a role of Akt in this pathway.” Endocrinology 140(12): 5937–5943. [DOI] [PubMed] [Google Scholar]
- Cunningham BC, Bass S, Fuh G and Wells JA (1990). “Zinc mediation of the binding of human growth hormone to the human prolactin receptor.” Science 250(4988): 1709–1712. [DOI] [PubMed] [Google Scholar]
- Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR and Wells JA (1991). “Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule.” Science 254(5033): 821–825. [DOI] [PubMed] [Google Scholar]
- Darnell J Jr, Kerr IM and Stark GR (1994). “Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins.” Science 264(5164): 1415–1421. [DOI] [PubMed] [Google Scholar]
- Daughaday WH (2000). “Growth hormone axis overview--somatomedin hypothesis.” Pediatr Nephrol 14(7): 537–540. [DOI] [PubMed] [Google Scholar]
- De Rocca Serra-Nedelec A, Edouard T, Treguer K, Tajan M, Araki T, Dance M, Mus M, Montagner A, Tauber M, Salles JP, Valet P, Neel BG, Raynal P and Yart A (2012). “Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature.” Proc Natl Acad Sci U S A 109(11): 4257–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos AM, Ultsch M and Kossiakoff AA (1992). “Human growth hormone and extracellular domain of its receptor: crystal structure of the complex.” Science 255(5042): 306–312. [DOI] [PubMed] [Google Scholar]
- Defalque D, Brandt N, Ketelslegers JM and Thissen JP (1999). “GH insensitivity induced by endotoxin injection is associated with decreased liver GH receptors.” Am J Physiol 276(3 Pt 1): E565–572. [DOI] [PubMed] [Google Scholar]
- Dehkhoda F, Lee CMM, Medina J and Brooks AJ (2018). “The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects.” Front Endocrinol (Lausanne) 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, He K, Wang X, Yang N, Thangavel C, Jiang J, Fuchs SY and Frank SJ (2007). “Determinants of growth hormone receptor down-regulation.” Mol Endocrinol 21(7): 1537–1551. [DOI] [PubMed] [Google Scholar]
- Denson LA, Held MA, Menon RK, Frank SJ, Parlow AF and Arnold DL (2003). “Interleukin-6 inhibits hepatic growth hormone signaling via upregulation of Cis and Socs-3.” Am J Physiol Gastrointest Liver Physiol 284(4): G646–654. [DOI] [PubMed] [Google Scholar]
- DiGirolamo DJ, Mukherjee A, Fulzele K, Gan Y, Cao X, Frank SJ and Clemens TL (2007). “Mode of growth hormone action in osteoblasts.” J Biol Chem 282(43): 31666–31674. [DOI] [PubMed] [Google Scholar]
- Doglio A, Dani C, Grimaldi P and Ailhaud G (1989). “Growth hormone stimulates c-fos gene expression by means of protein kinase C without increasing inositol lipid turnover.” Proc Natn Acad Sci USA 86(*): 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman JG and Kaushansky K (1997). “Dissecting the thrombopoietin receptor: functional elements of the Mpl cytoplasmic domain.” Proc Natl Acad Sci U S A 94(6): 2350–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC and Levy DE (1996). “Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease.” Cell 84(3): 443–450. [DOI] [PubMed] [Google Scholar]
- Edmondson SR, Russo VC, McFarlane AC, Wraight CJ and Werther GA (1999). “Interactions between growth hormone, insulin-like growth factor I, and basic fibroblast growth factor in melanocyte growth.” J Clin Endocrinol Metab 84(5): 1638–1644. [DOI] [PubMed] [Google Scholar]
- Espada J and Martin-Perez J (2017). “An Update on Src Family of Nonreceptor Tyrosine Kinases Biology.” Int Rev Cell Mol Biol 331: 83–122. [DOI] [PubMed] [Google Scholar]
- Evans HM and Long JA (1922). “Characteristic Effects upon Growth, Oestrus and Ovulation Induced by the Intraperitoneal Administration of Fresh Anterior Hypophyseal Substance.” Proc Natl Acad Sci U S A 8(3): 38–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, Le Roith D, Trucco M and Sperling MA (2009). “Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism.” J Biol Chem 284(30): 19937–19944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R and Krolewski JJ (1990). “tyk2, prototype of a novel class of non-receptor tyrosine kinase genes.” Oncogene 5(9): 1329–1336. [PubMed] [Google Scholar]
- Foster CM, Shafer JA, Rozsa FW, Wang X, Lewis SD, Renken DA, Natale JE, Schwartz J and Carter-Su C (1988). “Growth hormone promoted tyrosyl phosphorylation of growth hormone receptors in murine 3T3-F442A fibroblasts and adipocytes.” Biochemistry 27(*): 326–334. [DOI] [PubMed] [Google Scholar]
- Frank SJ (2002). “Receptor dimerization in GH and erythropoietin action--it takes two to tango, but how?” Endocrinology 143(1): 2–10. [DOI] [PubMed] [Google Scholar]
- Frank SJ and Fuchs SY (2008). “Modulation of growth hormone receptor abundance and function: roles for the ubiquitin-proteasome system.” Biochim Biophys Acta 1782(12): 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SJ, Gilliland G, Kraft AS and Arnold CS (1994). “Interaction of the growth hormone receptor cytoplasmic domain with the JAK2 tyrosine kinase.” Endocrinology 135(5): 2228–2239. [DOI] [PubMed] [Google Scholar]
- Frank SJ and O’Shea JJ, Eds. (1999). Recent advances in cytokine signal transduction: lessons from growth hormone and other cytokines Advances in Molecular and Cellular Endocrinology. Greenwich, CT. [Google Scholar]
- Frank SJ, Yi W, Zhao Y, Goldsmith JF, Gilliland G, Jiang J, Sakai I and Kraft AS (1995). “Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor.” J Biol Chem 270(24): 14776–14785. [DOI] [PubMed] [Google Scholar]
- Fu YK, Arkins S, Fuh G, Cunningham BC, Wells JA, Fong S, Cronin MJ, Dantzer R and Kelley KW (1992). “Growth hormone augments superoxide anion secretion of human neutrophils by binding to the prolactin receptor.” J Clin Invest 89(2): 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV and Wells JA (1992). “Rational design of potent antagonists to the human growth hormone receptor.” Science 256(5064): 1677–1680. [DOI] [PubMed] [Google Scholar]
- Gan Y, Buckels A, Liu Y, Zhang Y, Paterson AJ, Jiang J, Zinn KR and Frank SJ (2014). “Human GH receptor-IGF-1 receptor interaction: implications for GH signaling.” Mol Endocrinol 28(11): 1841–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Paterson AJ, Zhang Y, Jiang J and Frank SJ (2014). “Functional collaboration of insulin-like growth factor-1 receptor (IGF-1R), but not insulin receptor (IR), with acute GH signaling in mouse calvarial cells.” Endocrinology 155(3): 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Zhang Y, Buckels A, Paterson AJ, Jiang J, Clemens TL, Zhang ZY, Du K, Chang Y and Frank SJ (2013). “IGF-1R modulation of acute GH-induced STAT5 signaling: role of protein tyrosine phosphatase activity.” Mol Endocrinol 27(11): 1969–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Zhang Y, Digirolamo DJ, Jiang J, Wang X, Cao X, Zinn KR, Carbone DP, Clemens TL and Frank SJ (2010). “Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling.” Mol Endocrinol 24(3): 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Frenkel MJ, Bentley JD, Lovrecz GO, Elleman TC, Cosgrove LJ and Ward CW (1998). “Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor.” Nature 394(6691): 395–399. [DOI] [PubMed] [Google Scholar]
- Gent J, van Kerkhof P, Roza M, Bu G and Strous GJ (2002). “Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis.” Proc Natl Acad Sci U S A 99(15): 9858–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouilleux F, Pallard C, Dusanter-Fourt I, Wakao H, Haldosen LA, Norstedt G, Levy D and Groner B (1995). “Prolactin, growth hormone, erythropoietin and granulocyte-macrophage colony stimulating factor induce MGF-Stat5 DNA binding activity.” Embo J 14(9): 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh CJ, Bertolino P, Asa SL, Metcalf D, Corbin JE, Adams TE, Davey HW, Nicola NA, Hilton DJ and Alexander WS (2002). “Growth enhancement in suppressor of cytokine signaling 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b).” Mol Endocrinol 16(6): 1394–1406. [DOI] [PubMed] [Google Scholar]
- Gronowski AM and Rotwein P (1994). “Rapid changes in nuclear protein tyrosine phosphorylation after growth hormone treatment in vivo. Identification of phosphorylated mitogen-activated protein kinase and STAT91.” J Biol Chem 269(11): 7874–7878. [PubMed] [Google Scholar]
- Gronowski AM, Zhong Z, Wen Z, Thomas MJ, Darnell JE Jr. and Rotwein P (1995). “In vivo growth hormone treatment rapidly stimulates the tyrosine phosphorylation and activation of Stat3.” Mol Endocrinol 9(2): 171–177. [DOI] [PubMed] [Google Scholar]
- Gu F, Dube N, Kim JW, Cheng A, Ibarra-Sanchez Mde J, Tremblay ML and Boisclair YR (2003). “Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling.” Mol Cell Biol 23(11): 3753–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Zhang Y, Jiang J, Baumann CA, Black RA, Baumann G and Frank SJ (2001). “Phorbol ester- and growth factor-induced growth hormone (GH) receptor proteolysis and GH-binding protein shedding: relationship to GH receptor down-regulation.” Endocrinology 142(3): 1137–1147. [DOI] [PubMed] [Google Scholar]
- Gurland G, Ashcom G, Cochran BH and Schwartz J (1990). “Rapid events in growth hormone action. Induction of c-fos and c-jun transcription in 3T3-F442A preadipocytes.” Endocrinology 127(*): 3187–3195. [DOI] [PubMed] [Google Scholar]
- Hackett RH, Wang YD, Sweitzer S, Feldman G, Wood WI and Larner AC (1997). “Mapping of a cytoplasmic domain of the human growth hormone receptor that regulates rates of inactivation of Jak2 and Stat proteins.” J Biol Chem 272(17): 11128–11132. [DOI] [PubMed] [Google Scholar]
- Han Y, Leaman DW, Watling D, Rogers NC, Groner B, Kerr IM, Wood WI and Stark GR (1996). “Participation of JAK and STAT proteins in growth hormone-induced signaling.” J Biol Chem 271(10): 5947–5952. [DOI] [PubMed] [Google Scholar]
- Hansen JA, Lindberg K, Hilton DJ, Nielsen JH and Billestrup N (1999). “Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins.” Mol Endocrinol 13(11): 1832–1843. [DOI] [PubMed] [Google Scholar]
- Hansen LH, Wang X, Kopchick JJ, Bouchelouche P, Nielsen JH, Galsgaard ED and Billestrup N (1996). “Identification of tyrosine residues in the intracellular domain of the growth hormone receptor required for transcriptional signaling and Stat5 activation.” J Biol Chem 271: 12669–12673. [DOI] [PubMed] [Google Scholar]
- Harpur AG, Andres AC, Ziemiecki A, Aston RR and Wilks AF (1992). “JAK2, a third member of the JAK family of protein tyrosine kinases.” Oncogene 7(7): 1347–1353. [PubMed] [Google Scholar]
- Hauser SD, McGrath MF, Collier RJ and Krivi GG (1990). “Cloning and in vivo expression of bovine growth hormone receptor mRNA.” Mol Cell Endocrinol 72(3): 187–200. [DOI] [PubMed] [Google Scholar]
- Haxholm GW, Nikolajsen LF, Olsen JG, Fredsted J, Larsen FH, Goffin V, Pedersen SF, Brooks AJ, Waters MJ and Kragelund BB (2015). “Intrinsically disordered cytoplasmic domains of two cytokine receptors mediate conserved interactions with membranes.” Biochem J 468(3): 495–506. [DOI] [PubMed] [Google Scholar]
- He K, Wang X, Jiang J, Guan R, Bernstein KE, Sayeski PP and Frank SJ (2003). “Janus kinase 2 determinants for growth hormone receptor association, surface assembly, and signaling.” Mol Endocrinol 17(11): 2211–2227. [DOI] [PubMed] [Google Scholar]
- Hodge C, Liao J, Stofega M, Guan K, Carter-Su C and Schwartz J (1998). “Growth hormone stimulates phosphorylation and activation of elk-1 and expression of c-fos, egr-1, and junB through activation of extracellular signal-regulated kinases 1 and 2.” J Biol Chem 273(47): 31327–31336. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Cooney RN, Frost RA and Lang CH (2003). “Sepsis-induced muscle growth hormone resistance occurs independently of STAT5 phosphorylation.” Am J Physiol Endocrinol Metab 285(1): E63–72. [DOI] [PubMed] [Google Scholar]
- Huang Y, Kim S-O, Yang N, Jiang J and Frank SJ (2004). “Physical and functional interaction of GH and IGF-1 signaling elements.” Mol Endocrinol. 18(6): 1471–1485. [DOI] [PubMed] [Google Scholar]
- Huang Y, Kim SO, Jiang J and Frank SJ (2003). “Growth hormone-induced phosphorylation of epidermal growth factor (EGF) receptor in 3T3-F442A cells. Modulation of EGF-induced trafficking and signaling.” J Biol Chem 278(21): 18902–18913. [DOI] [PubMed] [Google Scholar]
- Hughes JP and Friesen HG (1985). “The nature and regulation of the receptors for pituitary growth hormone.” Annu Rev Physiol 47: 469–482. [DOI] [PubMed] [Google Scholar]
- Hwa V (2016). “STAT5B deficiency: Impacts on human growth and immunity.” Growth Horm IGF Res 28: 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson OG, Eden S and Jansson JO (1985). “Mode of action of pituitary growth hormone on target cells.” Annu Rev Physiol 47(1): 483–499. [DOI] [PubMed] [Google Scholar]
- Jeay S, Sonenshein GE, Kelly PA, Postel-Vinay MC and Baixeras E (2001). “Growth hormone exerts antiapoptotic and proliferative effects through two different pathways involving nuclear factor-kappaB and phosphatidylinositol 3-kinase.” Endocrinology 142(1): 147–156. [DOI] [PubMed] [Google Scholar]
- Jiang J, Wan Y, Wang X, Xu J, Harris JM, Lobie PE, Zhang Y, Zinn KR, Waters MJ and Frank SJ (2011). “Inhibitory GH receptor extracellular domain monoclonal antibodies: three-dimensional epitope mapping.” Endocrinology 152(12): 4777–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Wang X, He K, Li X, Chen C, Sayeski PP, Waters MJ and Frank SJ (2004). "A conformationally sensitive GHR [growth hormone (GH) receptor] antibody: impact on GH signaling and GHR proteolysis.” Mol Endocrinol 18(12): 2981–2996. [DOI] [PubMed] [Google Scholar]
- Jin H, Lanning NJ and Carter-Su C (2008). “JAK2, but not Src family kinases, is required for STAT, ERK, and Akt signaling in response to growth hormone in preadipocytes and hepatoma cells.” Mol Endocrinol 22(8): 1825–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW and O’Shea JJ (1994). “Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2.” Nature 370(6485): 151–153. [DOI] [PubMed] [Google Scholar]
- Kaplan SA and Cohen P (2007). "The somatomedin hypothesis 2007: 50 years later.” J Clin Endocrinol Metab 92(12): 4529–4535. [DOI] [PubMed] [Google Scholar]
- Kilgour E, Gout I and Anderson NG (1996). “Requirement for phosphoinositide 3-OH kinase in growth hormone signalling to the mitogen-activated protein kinase and p70s6k pathways.” Biochem J 315(Pt 2): 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SO, Jiang J, Yi W, Feng GS and Frank SJ (1998). “Involvement of the Src homology 2-containing tyrosine phosphatase SHP-2 in growth hormone signaling.” J Biol Chem 273(4): 2344–2354. [DOI] [PubMed] [Google Scholar]
- Kim SO, Loesch K, Wang X, Jiang J, Mei L, Cunnick JM, Wu J and Frank SJ (2002). “A role for Grb2-associated binder-1 in growth hormone signaling.” Endocrinology 143(12): 4856–4867. [DOI] [PubMed] [Google Scholar]
- Kopchick JJ, Parkinson C, Stevens EC and Trainer PJ (2002). “Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly.” Endocr Rev 23(5): 623–646. [DOI] [PubMed] [Google Scholar]
- Laron Z, Pertzelan A and Mannheimer S (1966). “Genetic pituitary dwarfism with high serum concentation of growth hormone--a new inborn error of metabolism?” Isr J Med Sci 2(2): 152–155. [PubMed] [Google Scholar]
- Le Roith D, Bondy C, Yakar S, Liu JL and Butler A (2001). “The somatomedin hypothesis: 2001.” Endocr Rev 22(1): 53–74. [DOI] [PubMed] [Google Scholar]
- Lebrun JJ, Ali S, Sofer L, Ullrich A and Kelly PA (1994). “Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2.” J Biol Chem 269(19): 14021–14026. [PubMed] [Google Scholar]
- LeRoith D (2000). “Insulin-like growth factor I receptor signaling--overlapping or redundant pathways?” Endocrinology 141(4): 1287–1288. [DOI] [PubMed] [Google Scholar]
- Leung DW, Spencer SA, Cachianes G, Hammonds RG, Collins C, Henzel WJ, Barnard R, Waters MJ and Wood WI (1987). “Growth hormone receptor and serum binding protein: purification, cloning and expression.” Nature 330(*): 537–543. [DOI] [PubMed] [Google Scholar]
- Li CH and Evans HM (1944). “The Isolation of Pituitary Growth Hormone.” Science 99(2566): 183–184. [DOI] [PubMed] [Google Scholar]
- Li X, Huang Y, Jiang J and Frank SJ (2008). “ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling.” Cell Signal 20(11): 2145–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Jiang J and Frank SJ (2000). “Insulin receptor substrate-1-mediated enhancement of growth hormone-induced mitogen-activated protein kinase activation.” Endocrinology 141(9): 3328–3336. [DOI] [PubMed] [Google Scholar]
- Liang L, Zhou T, Jiang J, Pierce JH, Gustafson TA and Frank SJ (1999). “Insulin receptor substrate-1 enhances growth hormone-induced proliferation.” Endocrinology 140(5): 1972–1983. [DOI] [PubMed] [Google Scholar]
- Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, Yamauchi A, Bloom ET, Mietz J, John S and et al. (1995). “The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15.” Immunity 2(4): 331–339. [DOI] [PubMed] [Google Scholar]
- Ling L, Zhu T and Lobie PE (2003). “Src-CrkII-C3G-dependent activation of Rap1 switches growth hormone-stimulated p44/42 MAP kinase and JNK/SAPK activities.” J Biol Chem 278(29): 27301–27311. [DOI] [PubMed] [Google Scholar]
- Linossi EM, Babon JJ, Hilton DJ and Nicholson SE (2013). “Suppression of cytokine signaling: the SOCS perspective.” Cytokine Growth Factor Rev 24(3): 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JL and LeRoith D (1999). “Insulin-like growth factor I is essential for postnatal growth in response to growth hormone.” Endocrinology 140(11): 5178–5184. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ and Efstratiadis A (1993). “Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r).” Cell 75(1): 59–72. [PubMed] [Google Scholar]
- Liu Y, Berry PA, Zhang Y, Jiang J, Lobie PE, Paulmurugan R, Langenheim JF, Chen WY, Zinn KR and Frank SJ (2014). “Dynamic analysis of GH receptor conformational changes by split luciferase complementation.” Mol Endocrinol 28(11): 1807–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jiang J, Lepik B, Zhang Y, Zinn KR and Frank SJ (2017). “Subdomain 2, Not the Transmembrane Domain, Determines the Dimerization Partner of Growth Hormone Receptor and Prolactin Receptor.” Endocrinology 158(10): 3235–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang Y, Jiang J, Lobie PE, Paulmurugan R, Langenheim JF, Chen WY, Zinn KR and Frank SJ (2016). “GHR/PRLR Heteromultimer Is Composed of GHR Homodimers and PRLR Homodimers.” Mol Endocrinol 30(5): 504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DW, Whatmore AJ, Clayton PE and Silva CM (1998). “Growth hormone stimulation of the mitogen-activated protein kinase pathway is cell type specific.” Endocrinology 139(4): 1965–1971. [DOI] [PubMed] [Google Scholar]
- Lupu F, Terwilliger JD, Lee K, Segre GV and Efstratiadis A (2001). “Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth.” Dev Biol 229(1): 141–162. [DOI] [PubMed] [Google Scholar]
- Ma F, Wei Z, Shi C, Gan Y, Lu J, Frank SJ, Balducci J and Huang Y (2011). “Signaling cross talk between growth hormone (GH) and insulin-like growth factor-I (IGF-I) in pancreatic islet beta-cells.” Mol Endocrinol 25(12): 2119–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie SJ, Yarwood SJ, Peden AH, Bolger GB, Vernon RG and Houslay MD (1998). “Stimulation of p70S6 kinase via a growth hormone-controlled phosphatidylinositol 3-kinase pathway leads to the activation of a PDE4A cyclic AMP-specific phosphodiesterase in 3T3-F442A preadipocytes.” Proc Natl Acad Sci US A 95(7): 3549–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ling PR, Fitzgibbons TP, McCowen KC, Frick GP, Bistrian BR and Smith RJ (1999). “Endotoxin-induced inhibition of growth hormone receptor signaling in rat liver in vivo.” Endocrinology 140(12): 5505–5515. [DOI] [PubMed] [Google Scholar]
- Martial JA, Hallewell RA, Baxter JD and Goodman HM (1979). “Human growth hormone: complementary DNA cloning and expression in bacteria.” Science 205(4406): 602–607. [DOI] [PubMed] [Google Scholar]
- Maures TJ, Kurzer JH and Carter-Su C (2007). “SH2B1 (SH2-B) and JAK2: a multifunctional adaptor protein and kinase made for each other.” Trends Endocrinol Metab 18(1): 38–45. [DOI] [PubMed] [Google Scholar]
- Menting JG, Whittaker J, Margetts MB, Whittaker LJ, Kong GK, Smith BJ, Watson CJ, Zakova L, Kletvikova E, Jiracek J, Chan SJ, Steiner DF, Dodson GG, Brzozowski AM, Weiss MA, Ward CW and Lawrence MC (2013). “How insulin engages its primary binding site on the insulin receptor.” Nature 493(7431): 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, Du Bois RN, Clark R, Aguet M and Schreiber RD (1996). “Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway.” Cell 84(3): 431–442. [DOI] [PubMed] [Google Scholar]
- Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ and Alexander WS (2000). “Gigantism in mice lacking suppressor of cytokine signalling-2.” Nature 405(6790): 1069–1073. [DOI] [PubMed] [Google Scholar]
- Meyer DJ, Campbell GS, Cochran BH, Argetsinger LS, Larner AC, Finbloom DS, Carter-Su C and Schwartz J (1994). “Growth hormone induces a DNA binding factor related to the interferon-stimulated 91-kDa transcription factor.” J Biol Chem 269(*): 4701–4704. [PubMed] [Google Scholar]
- Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC and Khokha R (2004). “Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration.” Nat Genet 36(9): 969–977. [DOI] [PubMed] [Google Scholar]
- Moller C, Hansson A, Enberg B, Lobie PE and Norstedt G (1992). “Growth hormone (GH) induction of tyrosine phosphorylation and activation of mitogen-activated protein kinases in cells transfected with rat GH receptor cDNA.” J Biol Chem 267(32): 23403–23408. [PubMed] [Google Scholar]
- Moller N and Jorgensen JO (2009). “Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects.” Endocr Rev 30(2): 152–177. [DOI] [PubMed] [Google Scholar]
- Moriggl R, Gouilleux-Gruart V, Jahne R, Berchtold S, Gartmann C, Liu X, Hennighausen L, Sotiropoulos A, Groner B and Gouilleux F (1996). “Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype.” Mol Cell Biol 16(10): 5691–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Warner J, Willard D and Becherer JD (1997). “Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha.” Nature 385(6618): 733–736. [DOI] [PubMed] [Google Scholar]
- Moutoussamy S, Renaudie F, Lago F, Kelly PA and Finidori J (1998). “Grb10 identified as a potential regulator of growth hormone (GH) signaling by cloning of GH receptor target proteins.” J Biol Chem 273(26): 15906–15912. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kido Y and Accili D (2001). “Distinct and overlapping functions of insulin and IGF-I receptors.” Endocr Rev 22(6): 818–835. [DOI] [PubMed] [Google Scholar]
- Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U and Pfeffer K (1998). “Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis.” Cell 93(3): 397–409. [DOI] [PubMed] [Google Scholar]
- Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC and Ihle JN (1995). “Defective lymphoid development in mice lacking Jak3.” Science 270(5237): 800–802. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Holland SM and Staudt LM (2013). “JAKs and STATs in immunity, immunodeficiency, and cancer.” N Engl J Med 368(2): 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali C, Curchod ML, Walchli S, Espanel X, Guerrier M, Arigoni F, Strous G and Van Huijsduijnen RH (2003). “Identification of protein tyrosine phosphatases with specificity for the ligand-activated growth hormone receptor.” Mol Endocrinol 17(11): 2228–2239. [DOI] [PubMed] [Google Scholar]
- Paul C, Seiliez I, Thissen JP and Le Cam A (2000). “Regulation of expression of the rat SOCS-3 gene in hepatocytes by growth hormone, interleukin-6 and glucocorticoids mRNA analysis and promoter characterization.” Eur J Biochem 267(19): 5849–5857. [DOI] [PubMed] [Google Scholar]
- Pilecka I, Patrignani C, Pescini R, Curchod ML, Perrin D, Xue Y, Yasenchak J, Clark A, Magnone MC, Zaratin P, Valenzuela D, Rommel C and Hooft van Huijsduijnen R (2007). “Protein-tyrosine phosphatase H1 controls growth hormone receptor signaling and systemic growth.” J Biol Chem 282(48): 35405–35415. [DOI] [PubMed] [Google Scholar]
- Pivonello C, De Martino MC, Negri M, Cuomo G, Cariati F, Izzo F, Colao A and Pivonello R (2014). “The GH-IGF-SST system in hepatocellular carcinoma: biological and molecular pathogenetic mechanisms and therapeutic targets.” Infect Agent Cancer 9: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N and Stewart TA (1993). “IGF-I is required for normal embryonic growth in mice.” Genes Dev 7(12B): 2609–2617. [DOI] [PubMed] [Google Scholar]
- Ram PA, Park SH, Choi HK and Waxman DJ (1996). “Growth hormone activation of Stat 1, Stat 3, and Stat 5 in rat liver. Differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phosphorylation.” J Biol Chem 271(*): 5929–5940. [DOI] [PubMed] [Google Scholar]
- Ram PA and Waxman DJ (1997). “Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase.” J Biol Chem 272(28): 17694–17702. [DOI] [PubMed] [Google Scholar]
- Ram PA and Waxman DJ (1999). “SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms.” J Biol Chem 274(50): 35553–35561. [DOI] [PubMed] [Google Scholar]
- Ridderstrale M and Tornqvist H (1994). “PI-3-kinase inhibitor Wortmannin blocks the insulin-like effects of growth hormone in isolated rat adipocytes.” Biochem Biophys Res Commun 203(1): 306–310. [DOI] [PubMed] [Google Scholar]
- Riedel EO, Hinrichs A, Kemter E, Dahlhoff M, Backman M, Rathkolb B, Prehn C, Adamski J, Renner S, Blutke A, de Angelis MH, Bidlingmaier M, Schopohl J, Arnold GJ, Frohlich T and Wolf E (2020). “Functional changes of the liver in the absence of growth hormone (GH) action - Proteomic and metabolomic insights from a GH receptor deficient pig model.” Mol Metab 36: 100978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson EM Jr. and Schreiber RD (1998). “Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses.” Cell 93(3): 373–383. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2012). “ERK1/2 MAP kinases: structure, function, and regulation.” Pharmacol Res 66(2): 105–143. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2015). “Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors.” Pharmacol Res 94: 9–25. [DOI] [PubMed] [Google Scholar]
- Rowland JE, Kerr LM, White M, Noakes PG and Waters MJ (2005). “Heterozygote effects in mice with partial truncations in the growth hormone receptor cytoplasmic domain: assessment of growth parameters and phenotype.” Endocrinology 146(12): 5278–5286. [DOI] [PubMed] [Google Scholar]
- Rowland JE, Lichanska AM, Kerr LM, White M, d’Aniello EM, Maher SL, Brown R, Teasdale RD, Noakes PG and Waters MJ (2005). “In vivo analysis of growth hormone receptor signaling domains and their associated transcripts.” Mol Cell Biol 25(1): 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlinson SW, Yoshizato H, Barclay JL, Brooks AJ, Behncken SN, Kerr LM, Millard K, Palethorpe K, Nielsen K, Clyde-Smith J, Hancock JF and Waters MJ (2008). “An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway.” Nat Cell Biol 10(6): 740–747. [DOI] [PubMed] [Google Scholar]
- Rui L and Carter-Su C (1999). “Identification of SH2-bbeta as a potent cytoplasmic activator of the tyrosine kinase Janus kinase 2.” Proc Natl Acad Sci U S A 96(13): 7172–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Mathews LS, Hotta K, Gustafson TA and Carter-Su C (1997). “Identification of SH2-Bbeta as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling.” Mol Cell Biol 17(11): 6633–6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon WD and Daughaday WH (1957). “A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro.” J. Lab. Clin. Med 49: 825–836. [PubMed] [Google Scholar]
- Sedek M, van der Velden LM and Strous GJ (2014). “Multimeric growth hormone receptor complexes serve as signaling platforms.” J Biol Chem 289(1): 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva CM, Lu H and Day RN (1996). “Characterization and cloning of STAT5 from IM-9 cells and its activation by growth hormone.” Mol Endocrinol 10(5): 508–518. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi T and Ihle JN (1993). “Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction.” Proc Natl Acad Sci USA 90(*): 8429–8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO and Ohlsson C (1999). “Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice.” Proc Natl Acad Sci U S A 96(12): 7088–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit LS, Meyer DJ, Billestrup N, Norstedt G, Schwartz J and Carter-Su C (1996). “The role of the growth hormone (GH) receptor and JAK1 and JAK2 kinases in the activation of Stats 1, 3, and 5 by GH.” Mol Endocrinol 10(5): 519–533. [DOI] [PubMed] [Google Scholar]
- Smit LS, Vanderkuur JA, Stimage A, Han Y, Luo G, Yu-Lee LY, Schwartz J and Carter-Su C (1997). “Growth hormone-induced tyrosyl phosphorylation and deoxyribonucleic acid binding activity of Stat5A and Stat5B.” Endocrinology 138(8): 3426–3434. [DOI] [PubMed] [Google Scholar]
- Smith WC, Kuniyoshi J and Talamantes F (1989). “Mouse serum growth hormone (GH) binding protein has GH receptor extracellular and substituted transmembrane domains.” Mol Endocrinol 3(6): 984–990. [DOI] [PubMed] [Google Scholar]
- Somers W, Ultsch M, De Vos AM and Kossiakoff AA (1994). “The X-ray structure of a growth hormone-prolactin receptor complex.” Nature 372(6505): 478–481. [DOI] [PubMed] [Google Scholar]
- Sos BC, Harris C, Nordstrom SM, Tran JL, Balazs M, Caplazi P, Febbraio M, Applegate MA, Wagner KU and Weiss EJ (2011). “Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2.” J Clin Invest 121(4): 1412–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos A, Perrot-Applanat M, Dinerstein H, Pallier A, Postel-Vinay MC, Finidori J and Kelly PA (1994). “Distinct cytoplasmic regions of the growth hormone receptor are required for activation of JAK2, mitogen-activated protein kinase, and transcription.” Endocrinology 135(4): 1292–1298. [DOI] [PubMed] [Google Scholar]
- Souza SC, Frick GP, Yip R, Lobo RB, Tai LR and Goodman HM (1994). “Growth hormone stimulates tyrosine phosphorylation of insulin receptor substrate-1.” J Biol Chem 269(48): 30085–30088. [PubMed] [Google Scholar]
- Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G, Pellegrini S and et al. (1994). “Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components.” Science 263(5143): 92–95. [DOI] [PubMed] [Google Scholar]
- Stark GR and Darnell JE Jr. (2012). “The JAK-STAT pathway at twenty.” Immunity 36(4): 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staskus PW, Masiarz FR, Pallanck LJ and Hawkes SP (1991). “The 21-kDa protein is a transformation-sensitive metalloproteinase inhibitor of chicken fibroblasts.” J Biol Chem 266(1): 449–454. [PubMed] [Google Scholar]
- Stofega MR, Herrington J, Billestrup N and Carter-Su C (2000). “Mutation of the SHP-2 binding site in growth hormone (GH) receptor prolongs GH-promoted tyrosyl phosphorylation of GH receptor, JAK2, and STAT5B.” Mol Endocrinol 14(9): 1338–1350. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Teixeira J, Wang J and Gil G (1995). “A STAT factor mediates the sexually dimorphic regulation of hepatic cytochrome P450 3A10/lithocholic acid 6 beta-hydroxylase gene expression by growth hormone.” Mol Cell Biol 15(9): 4672–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T and Akira S (1997). “Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality.” Proc Natl Acad Sci U S A 94(8): 3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner JW, Chen W, Young RL, Longmore GD and Shaw AS (1995). “The conserved box 1 motif of cytokine receptors is required for association with JAK kinases.” J Biol Chem 270(12): 6523–6530. [DOI] [PubMed] [Google Scholar]
- Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G and Ihle JN (1998). “Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses.” Cell 93(5): 841–850. [DOI] [PubMed] [Google Scholar]
- Tian SS, Tapley P, Sincich C, Stein RB, Rosen J and Lamb P (1996). “Multiple signaling pathways induced by granulocyte colony-stimulating factor involving activation of JAKs, STAT5, and/or STAT3 are required for regulation of three distinct classes of immediate early genes.” Blood 88(12): 4435–4444. [PubMed] [Google Scholar]
- Tollet-Egnell P, Flores-Morales A, Stavreus-Evers A, Sahlin L and Norstedt G (1999). “Growth hormone regulation of SOCS-2, SOCS-3, and CIS messenger ribonucleic acid expression in the rat.” Endocrinology 140(8): 3693–3704. [DOI] [PubMed] [Google Scholar]
- Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, Waxman DJ and Davey HW (1997). “Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression.” Proc Natl Acad Sci U S A 94(14): 7239–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E and et al. (1986). “Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity.” Embo J 5(10): 2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderKuur J, Allevato G, Billestrup N, Norstedt G and Carter-Su C (1995). “Growth hormone-promoted tyrosyl phosphorylation of SHC proteins and SHC association with Grb2.” J Biol Chem 13(*): 7587–7593. [DOI] [PubMed] [Google Scholar]
- Vanderkuur JA, Butch ER, Waters SB, Pessin JE, Guan KL and Carter-Su C (1997). “Signaling molecules involved in coupling growth hormone receptor to mitogen-activated protein kinase activation.” Endocrinology 138(10): 4301–4307. [DOI] [PubMed] [Google Scholar]
- Vanderkuur JA, Wang X, Zhang L, Campbell GS, Allevato G, Billestrup N, Norstedt G and Carter-Su C (1994). “Domains of the growth hormone receptor required for association and activation of JAK2 tyrosine kinase.” J Biol Chem 269(*): 21709–21717. [PubMed] [Google Scholar]
- Velazquez L, Fellous M, Stark GR and Pellegrini S (1992). “A protein tyrosine kinase in the interferon alpha/beta signaling pathway.” Cell 70(2): 313–322. [DOI] [PubMed] [Google Scholar]
- Wakao H, Gouilleux F and Groner B (1994). “Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response.” The EMBO J 13(9): 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhou J, Powell-Braxton L and Bondy C (1999). “Effects of Igf1 gene deletion on postnatal growth patterns.” Endocrinology 140(7): 3391–3394. [DOI] [PubMed] [Google Scholar]
- Wang P, Li N, Li JS and Li WQ (2002). “The role of endotoxin, TNF-alpha, and IL-6 in inducing the state of growth hormone insensitivity.” World J Gastroenterol 8(3): 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Darus CJ, Xu BC and Kopchick JJ (1996). “Identification of growth hormone receptor (GHR) tyrosine residues required for GHR phosphorylation and JAK2 and STAT5 activation.” Mol Endocrinol 10(10): 1249–1260. [DOI] [PubMed] [Google Scholar]
- Wang X, He K, Gerhart M, Huang Y, Jiang J, Paxton RJ, Yang S, Lu C, Menon RK, Black RA, Baumann G and Frank SJ (2002). “Metalloprotease-mediated GH receptor proteolysis and GHBP shedding. Determination of extracellular domain stem region cleavage site.” J Biol Chem 277(52): 50510–50519. [DOI] [PubMed] [Google Scholar]
- Wang X, He K, Gerhart M, Jiang J, Paxton RJ, Menon RK, Black RA, Baumann G and Frank SJ (2003). “Reduced proteolysis of rabbit growth hormone (GH) receptor substituted with mouse GH receptor cleavage site.” Mol Endocrinol 17(10): 1931–1943. [DOI] [PubMed] [Google Scholar]
- Wang X, Jiang J, Warram J, Baumann G, Gan Y, Menon RK, Denson LA, Zinn KR and Frank SJ (2008). “Endotoxin-induced proteolytic reduction in hepatic growth hormone (GH) receptor: a novel mechanism for GH insensitivity.” Mol Endocrinol 22(6): 1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang N, Deng L, Li X, Jiang J, Gan Y and Frank SJ (2009). “Interruption of growth hormone signaling via SHC and ERK in 3T3-F442A preadipocytes upon knockdown of insulin receptor substrate-1.” Mol Endocrinol 23(4): 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA and Brown RJ (2006). “New insights into growth hormone action.” J Mol Endocrinol 36(1): 1–7. [DOI] [PubMed] [Google Scholar]
- Waxman DJ and O’Connor C (2006). “Growth hormone regulation of sex-dependent liver gene expression.” Mol Endocrinol 20(11): 2613–2629. [DOI] [PubMed] [Google Scholar]
- Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G and Ziemiecki A (1991). “Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase.” Mol Cell Biol 11(4): 2057–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmes S, Hafer M, Vuorio J, Tucker JA, Winkelmann H, Lochte S, Stanly TA, Pulgar Prieto KD, Poojari C, Sharma V, Richter CP, Kurre R, Hubbard SR, Garcia KC, Moraga I, Vattulainen I, Hitchcock IS and Piehler J (2020). “Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations.” Science 367(6478): 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston LA and Bertics PJ (1992). “Growth hormone stimulates the tyrosine phosphorylation of 42- and 45-kDa ERK-related proteins.” J Biol Chem 267(*): 4747–4751. [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O and Ihle JN (1993). “JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin.” Cell 74(2): 227–236. [DOI] [PubMed] [Google Scholar]
- Woelfle J, Chia DJ and Rotwein P (2003). “Mechanisms of growth hormone (GH) action. Identification of conserved Stat5 binding sites that mediate GH-induced insulin-like growth factor-I gene activation.” J Biol Chem 278(51): 51261–51266. [DOI] [PubMed] [Google Scholar]
- Woelfle J and Rotwein P (2004). “In vivo regulation of growth hormone-stimulated gene transcription by STAT5b.” Am J Physiol Endocrinol Metab 286(3): E393–401. [DOI] [PubMed] [Google Scholar]
- Wood TJ, Sliva D, Lobie PE, Pircher TJ, Gouilleux F, Wakao H, Gustafsson JA, Groner B, Norstedt G and Haldosen LA (1995). “Mediation of growth hormone-dependent transcriptional activation by mammary gland factor/Stat 5.” J Biol Chem 270(16): 9448–9453. [DOI] [PubMed] [Google Scholar]
- Wu J and Zhu AX (2011). “Targeting insulin-like growth factor axis in hepatocellular carcinoma.” J Hematol Oncol 4: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Sun H, Basta-Pljakic J, Cardoso L, Kennedy OD, Jasper H, Domene H, Karabatas L, Guida C, Schaffler MB, Rosen CJ and Yakar S (2013). “Serum IGF-1 Is Insufficient to Restore Skeletal Size in the Total Absence of the Growth Hormone Receptor.” J Bone Miner Res 28(7): 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sun D, Jiang J, Deng L, Zhang Y, Yu H, Bahl D, Langenheim JF, Chen WY, Fuchs SY and Frank SJ (2013). “The role of prolactin receptor in GH signaling in breast cancer cells.” Mol Endocrinol 27(2): 266–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang Y, Berry PA, Jiang J, Lobie PE, Langenheim JF, Chen WY and Frank SJ (2011). “Growth hormone signaling in human T47D breast cancer cells: potential role for a growth hormone receptor-prolactin receptor complex.” Mol Endocrinol 25(4): 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B and LeRoith D (1999). “Normal growth and development in the absence of hepatic insulin-like growth factor I.” Proc Natl Acad Sci U S A 96(13): 7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR and LeRoith D (2002). “Circulating levels of IGF-1 directly regulate bone growth and density.” J Clin Invest 110(6): 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Ueki K, Tobe K, Tamemoto H, Sekine N, Wada M, Honjo M, Takahashi M, Takahashi T, Hirai H, Tushima T, Akanuma Y, Fujita T, Komuro I, Yazaki Y and Kadowaki T (1997). “Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone.” Nature 390(6655): 91–96. [DOI] [PubMed] [Google Scholar]
- Yang N, Huang Y, Jiang J and Frank SJ (2004). “Caveolar and lipid raft localization of GH receptor and its signaling elements: impact on GH signaling.” J Biol Chem 279(20): 20898–20905. [DOI] [PubMed] [Google Scholar]
- Yi W, Kim SO, Jiang J, Park SH, Kraft AS, Waxman DJ and Frank SJ (1996). “Growth hormone receptor cytoplasmic domain differentially promotes tyrosine phosphorylation of signal transducers and activators of transcription 5b and 3 by activated JAK2 kinase.” Mol Endocrinol 10(11): 1425–1443. [DOI] [PubMed] [Google Scholar]
- Yu WH, Yu S, Meng Q, Brew K and Woessner JF Jr. (2000). “TIMP-3 binds to sulfated glycosaminoglycans of the extracellular matrix.” J Biol Chem 275(40): 31226–31232. [DOI] [PubMed] [Google Scholar]
- Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A and Clemens TL (2002). “Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization.” J Biol Chem 277(46): 44005–44012. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gc S, Patel SB, Liu Y, Paterson AJ, Kappes JC, Jiang J and Frank SJ (2019). “Growth hormone (GH) receptor (GHR)-specific inhibition of GH-Induced signaling by soluble IGF-1 receptor (sol IGF-1R).” Mol Cell Endocrinol 492: 110445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Jiang J, Black RA, Baumann G and Frank SJ (2000). “TACE is a growth hormone binding protein sheddase: the metalloprotease TACE/ADAM-17 is critical for (PMA-induced) growth hormone receptor proteolysis and GHBP generation.” Endocrinology 141: 4324–4348. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang X, Loesch K, May LA, Davis GE, Jiang J and Frank SJ (2016). “TIMP3 Modulates GHR Abundance and GH Sensitivity.” Mol Endocrinol 30(6): 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, Baumann G and Kopchick JJ (1997). "A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse).” Proc Natl Acad Sci U S A 94(24): 13215–13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Goh EL, LeRoith D and Lobie PE (1998). “Growth hormone stimulates the formation of a multiprotein signaling complex involving p130(Cas) and CrkII. Resultant activation of c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK).” J Biol Chem 273(50): 33864–33875. [DOI] [PubMed] [Google Scholar]
- Zhu T, Ling L and Lobie PE (2002). “Identification of a JAK2-independent pathway regulating growth hormone (GH)-stimulated p44/42 mitogen-activated protein kinase activity. GH activation of Ral and phospholipase D is Src-dependent.” J Biol Chem 277(47): 45592–45603. [DOI] [PubMed] [Google Scholar]