

ABSTRACT
Immune checkpoint therapy has resulted in minimal clinical response in many pediatric cancers. We sought to understand the influence of immune checkpoint inhibition using anti-PD-1 and anti-CTLA-4 antibodies individually, in combination, and after chemotherapy on immune responses in minimal and established murine neuroblastoma models. We also sought to understand the role of the tumor microenvironment (TME) and PD-L1 expression and their alteration post-chemotherapy in our models and human tissues. PD-L1 expression was enriched in human tumor-associated macrophages and up-regulated after chemotherapy. In a murine minimal disease model, single and dual immune checkpoint blockade promoted tumor rejection, improved survival, and established immune memory with long-term anti-tumor immunity against re-challenge. In an established tumor model, only dual immune checkpoint blockade showed efficacy. Interestingly, dual immune checkpoint therapy distinctly influenced adaptive and innate immune responses, with significant increase in CD8+CD28+PD-1+ T cells and inflammatory macrophages (CD11bhiCD11c−F4/80+Ly6Chi) in tumor-draining lymph nodes. Adding chemotherapy before immunotherapy provided significant survival benefit for mice with established tumors receiving anti-PD-1 or dual immune checkpoint blockade. Our findings demonstrate anti-PD-1 and anti-CTLA-4 therapy induces a novel subset of effector T cells, and support administration of induction chemotherapy immediately prior to immune checkpoint blockade in children with high-risk neuroblastoma.
Keywords: neuroblastoma, immune checkpoint therapy, tumor microenvironment, dual immune checkpoint therapy, tumor-associated macrophages
Introduction
Monoclonal antibodies directed against the immune checkpoint receptors Cytotoxic T-Lymphocyte Associated Protein 4 (CTLA-4) and Programmed Cell Death 1 (PD-1/PDCD1/CD279) have been at the forefront of cancer immunotherapies. Although signaling through these receptors induces unresponsiveness in effector immune cells, their mechanisms of action are not redundant.1,2 CTLA-4 has been shown to inhibit T cell activation by competing with the co-stimulatory receptor CD28 for B7-1 and B7-2 ligands (CD80 and CD86, respectively) on antigen-presenting cells during early immune responses, particularly in lymph nodes (LNs). In contrast, PD-1 (CD279) regulates the effector functions of T cells in various peripheral tissues.3–5 CTLA-4 and PD-1 are up-regulated on activated T cells and are considered markers of activation. In the absence of signaling through CD28, primary effector T cells can be generated via T cell receptor signaling but the memory T cells generated without co-stimulation through CD28 are anergic and fail to respond to secondary antigen exposure. Single-agent immune checkpoint inhibition strategies targeting CTLA-4 (CD152) or PD-1 have demonstrated efficacy for treatment of several types of cancers including metastatic melanoma, lung cancers, and others, 6 and their combination was found to lead to superior survival in such patients.7–9 However, the success of immune checkpoint therapies has not been replicated in pediatric malignancies including high-risk neuroblastomas, which account for 15% of childhood cancer-related mortality.10 Identification of successful combination therapies with immune checkpoint inhibitors and a deeper understanding of mechanisms of their synergy for pediatric cancers are needed.
In the neuroblastoma tumor microenvironment (TME), the importance of inflammation and tumor-associated macrophages (TAM) and their prognostic relevance has been well established.11–14 PD-L1 expression on as-yet unidentified cells has been described in neuroblastoma tumors.15 Since immune checkpoint pathways utilized by Tregs and TAMs can suppress CTLs, we hypothesized that combination immune checkpoint blockade using anti-CTLA-4 and anti-PD-1 antibodies in an immunocompetent murine model of neuroblastoma may enhance in vivo efficacy and enable insight into mechanisms of synergy. We also hypothesized that combination of neoadjuvant chemotherapy with immune checkpoint blockade may further enhance survival, since chemotherapy can attenuate regulatory immune cells and stimulate immune effector cells in addition to its direct tumoricidal effects.16 In a cohort of human specimens, we examined PD-L1 to identify cells responsible for its expression and to assess the effect of chemotherapy on its expression.
Materials and methods
Animals
All experiments and procedures were completed in accordance with the CHLA Institutional Animal Care and Use Committee. Transgenic mice carrying the SV40 large T-antigen gene (NB-Tag) on a C57BL6 background were previously characterized to represent MYCN-non-amplified neuroblastoma.17,18 The NBT3L cell line represents a luciferase-expressing line derived from NB-Tag mice in our laboratory. The transplantable model (NB-SQ) was established by injecting 4 × 106 NBT3L cells subcutaneously over the shoulder of syngeneic four-week-old C57BL/6 male mice. To generate an intra-renal model of neuroblastoma, 1 × 106 NBT3L cells were injected into the renal capsule. Treatment for animals with established tumors was started when bioluminescence signal of at least >100 photons/second (log10 scale) was achieved (minimum of 21 days after tumor inoculation and minimum of 100 mm3 tumor volume in subcutaneous model). Animals were randomized to various groups prior to initiation of any therapy.
Immunohistochemistry (IHC) and flow cytometry
Tumor samples were obtained at diagnosis and post five cycles of chemotherapy and stained for PD-L1 (clone-SP142), PHOX2B (clone-EPR14423), CD163 (clone-10D6) on the same slide using a de-staining and re-staining protocol.19 In between each staining, the slides were imaged using an Aperio scanner (Leica Biosystems). All slides were reviewed by a neuroblastoma pathology expert. Manual scoring involved a three-category system: <1% expression, 1–5% expression, and >5% expression. Automatic scoring was performed using a custom pipeline in ImageJ after image alignment of all IHC images. Briefly, the pipeline included assessment of nucleated cells using a nuclear counterstain in PD-L1 images and definition of cell boundaries by dilation of a predefined nuclei algorithm. PHOX2B+ tumor cells, CD163+ macrophages, and PD-L1+ cells were enumerated by their presence within the identified cells.
Tumor, tumor-draining LNs, and spleen single-cell suspensions, were prepared by mechanical dissociation then passed through a 70 µm filter. Cells were surface-stained, then permeabilized followed by intracellular staining using designated antibodies (Supplemental Table S1) for flow cytometry on a BD LSRII cytometer.
Treatment of mice
Monoclonal antibodies used for treatment were purchased from BioXCell. Anti-CTLA-4 (clone 9D9) or corresponding isotype control was administered intraperitoneally to mice at 100 μg/day. Anti-PD-1 (clone RMP1-14) or corresponding isotype control was administered intraperitoneally at 250 μg/day. Both antibodies were administered twice weekly on days 1 and 3 owing to the relatively rapid clearance of antibodies from mice, for a duration of 6 weeks. Tumor size in treated and control groups was assessed weekly by bioluminescence imaging on an IVIS Spectrum platform (Xenogen). Tumor size in treated or isotype-treated groups of NB-Tag mice was measured every other week by MRI. Mice were sacrificed when tumor volume exceeded 500 mm3.
For depletion of T cell subsets, anti-CD4 (clone GK1.5), anti-CD8a (clone 2.43) or corresponding isotype control antibody (clone LTF-2) was administered intraperitoneally at 500 µg daily for 3 days prior to injection of tumor cells and maintained with twice weekly injections (Mondays and Fridays) for the duration of the experiment. For re-challenge experiments, NB-SQ mice previously treated with monoclonal antibody therapy were injected subcutaneously above the shoulder with 4 × 106 NBT3L cells 5–14 weeks after the date of final therapeutic antibody injection to ensure sufficient time for complete clearance of antibodies. For chemotherapy treatment, doses of cyclophosphamide (110 mg/kg/day) and topotecan (0.4 mg/kg/day) were chosen after determining maximum tolerated doses for C57BL/6 J; they were injected intraperitoneally for 5 consecutive days to model induction therapies given to children with high-risk neuroblastoma.
Gene expression studies
Tumors from NB-Tag mice and control tissue (adrenal glands) from syngeneic wild-type mice harvested at 4, 8, 12 and 14 weeks of age and RNA prepared using STAT-60 (Tel-Test) and RNeasy Mini Kit (Qiagen) per manufacturer instructions. Quantitative real-time PCR (qRT-PCR) for murine immune genes was performed using a TaqMan Low-Density Array (TLDA) mouse immune panel (Applied Biosystems). Cycle thresholds (Ct) for genes of interest were standardized by subtraction of the geometric mean of four housekeeping genes (18S, Actb, Gapdh, and Hprt1) and the resultant ΔCt values were normalized to baseline from 4-week-old syngeneic control tissue (ΔΔCt). All age and species groups were repeated in at least three mice per group.
Affymetrix human exon arrays were used to analyze RNA expression in 17 neuroblastoma cell lines and in primary tumor specimens of all stages collected at diagnosis prior to therapy from 249 patients with neuroblastoma. Tumor specimens were evaluated as part of the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative (https://target-data.nci.nih.gov/Public/NBL/clinical/harmonized/; See file named “ClinicalData_Discovery”), supported by NCI U10-CA98543.
Statistical analysis
All statistical analyses were performed using R version-3. Differences in means were determined with Student’s t-test or one-way analysis of variance (ANOVA), not assuming equal variances; the Wilcoxon rank test was substituted for ANOVA where indicated for non-normal data. Differences in survival were determined using the log-rank test.
Results
Elevated expression of PD-L1 in human MYCN-non-amplified tumors and cell lines
Expression of the PD-L1 gene was evaluated in neuroblastoma tumors isolated from a cohort of 249 patients and 17 human neuroblastoma cell lines (Figure 1A-C & Suppl. Fig. S1A). Gene expression analysis revealed significantly increased expression of the PD-L1 gene in MYCN-non-amplified (NA) cell lines compared to MYCN-amplified (A) cell lines (p = .033, Figure 1A). MYCN-NA tumors (n = 181) from patients had notably higher expression of PD-L1 compared to MYCN-A tumors (n = 30) (p < .0001). This over-expression of PD-L1 was also higher than in cell lines (MYCN-A and -NA, combined) (p = .0001; Figure 1B), suggesting cells within the TME as a possible source of PD-L1 expression in MYCN-non-amplified tumors. Interestingly, expression of PD-L1 among patients with MYCN-non-amplified disease was highest in tumors of children with high-risk neuroblastoma (n = 151) compared to low-risk MYCN-non-amplified neuroblastoma (n = 68) (Figure 1C) or to MYCN-amplified (n = 30)(Figure 1A,B). There was no association between PD-L1 gene expression and overall survival or disease severity (Suppl. Fig. S1B). These gene expression data reveal higher expression of PD-L1 in MYCN-non-amplified neuroblastomas and suggest non-tumor cells in the TME as a major source of PD-L1 expression.
Figure 1.
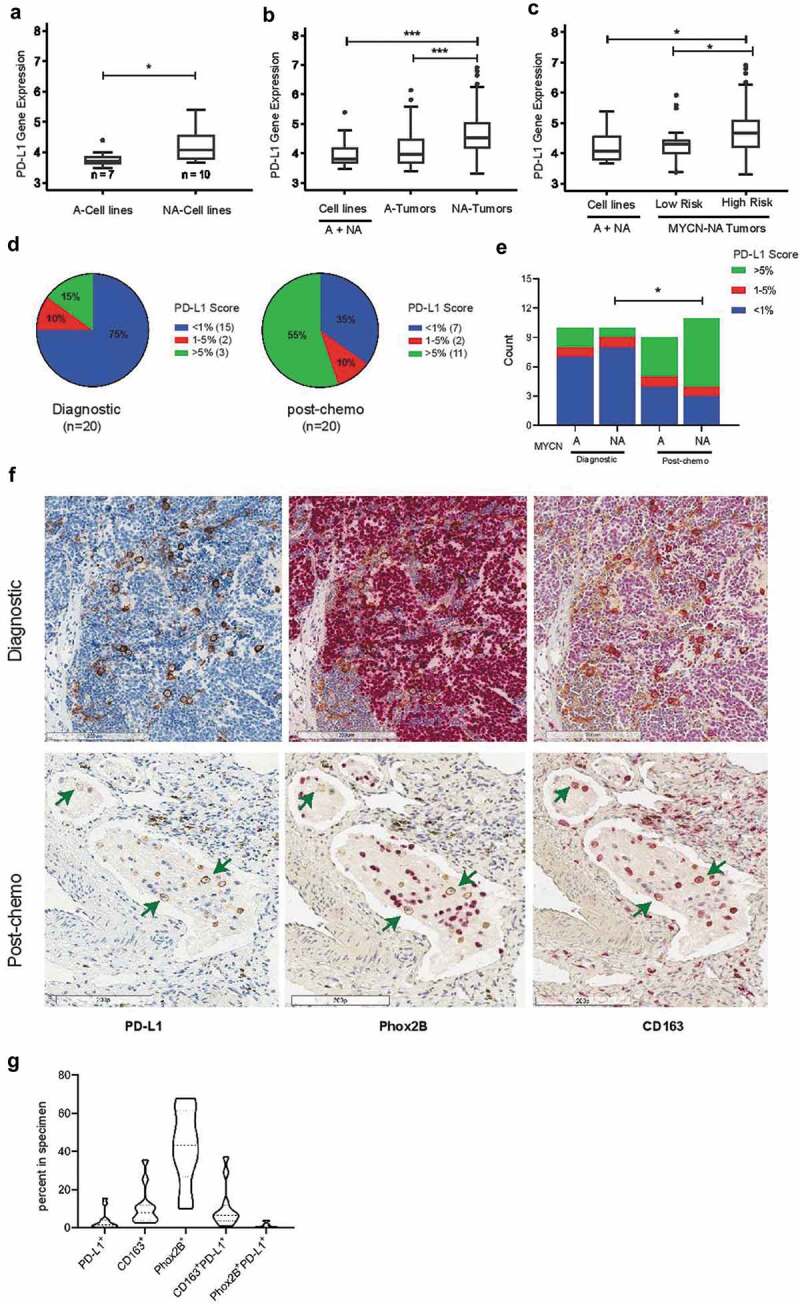
PD-L1 (CD274) gene expression and immunohistochemical analysis in human neuroblastoma samples. (A) Gene expression analysis (Affymetrix Human Exon Arrays) of 17 cell lines demonstrate significantly higher expression of PD-L1 in MYCN-non-amplified (NA) cell lines (n=10) compared to MYCN-amplified (A) cell lines (n=7, p=0.033); (B) Comparison of 249 neuroblastoma tumors and 17 cell lines revealed significantly higher gene expression of PD-L1 in MYCN -non-amplified (NA) tumors (n=181) compared to MYCN-amplified (A) tumors (n=68, p<0.0001) or to all (A+NA) cell lines (n=17, p=0.0001); (C) Among MYCN-NA tumor samples, those from high-risk patients (n=151) showed significantly increased PD-L1 expression compared to low-risk tumors (n=30, p=0.005) or to all (A+NA) cell lines (n=17, p=0.02); (D) Scoring of PD-L1 protein expression on 40 neuroblastoma tissues (20 at diagnosis and 20 post-chemotherapy) stained by immunohistochemistry and classified into three groups based on the expression of PD-L1: negative or lower than 1% (< 1%), 1 to 5% (1-5%) and higher than 5% (> 5%). (E) PD-L1 protein expression in 40 neuroblastoma tissue sections categorized by their MYCN status at diagnosis or post-chemotherapy; (F) Representative images of neuroblastoma sections serially stained for PD-L1 protein (membrane staining, brown, arrows pointing at some PD-L1+ cells), followed by staining for PHOX2B (nuclear staining, red, arrows pointing at couple of PHOX2B+ cells negative for PD-L1) and for CD163 (membrane staining, red, arrows pointing at CD163+ cells co-expressing PD-L1). (G) Violin plot distribution of PD-L1+ cells along with CD163+ TAMs and PHOX2B+ tumor cells and their subsets expressing PD-L1 in neuroblastoma samples at diagnosis of PD-L1+ cells along with CD163+ Tas, and PHOX2B+ tumor cells (n=12 samples analyzed)
PD-L1 protein expressed predominately on TAMs at diagnosis and up-regulated after chemotherapy
We have previously elucidated the presence and contribution of TAMs in maintaining an inflammatory microenvironmental niche for metastatic neuroblastoma17 and have shown that low risk neuroblastomas have a significantly lower number of CD163+ macrophages, with a direct correlation between presence of macrophages and stage of disease.11 To assess the contribution of TAMs as the source of PD-L1 expression, human neuroblastoma tissues (n = 40) were stained and imaged by serial immunohistochemical analyses for PD-L1, PHOX2B (tumor marker), and CD163 (TAM marker) proteins and analyzed both manually and by using an image processing pipeline. Manual scoring of PD-L1 indicated expression of PD-L1 on 25% (5/20) of neuroblastomas at diagnosis, with a marked increase to 65% (13/20, p = .011) in tumors collected after 5-cycles of chemotherapy (Figure 1D). At diagnosis, NA-MYCN tumors did not exhibit more PD-L1 than MYCN-A tumors (Figure 1E); this may at least partially reflect the finding that half of samples in the MYCN-NA subgroup were from low-/intermediate-risk group as low-risk NA-MYCN tumors had less PD-L1 expression than high-risk NA-MYCN tumors Figure 1C. There were not significant differences in PD-L1 expression between MYCN-A versus NA tumors in diagnostic or post-chemotherapy specimens; however, there was a significant increase in PD-L1 expression in MYCN-NA tumors post-chemotherapy compared to those obtained at diagnosis (Mann-Whitney U-test p = .02) (Figure 1E), suggesting that chemotherapy selected for cells with high PD-L1 expression. Notably, histologic analysis revealed PD-L1+ expression on CD163+ TAMs both at diagnosis and post-chemotherapy, whereas expression of PD-L1 on PHOX2B+ tumor cells was detectable at very low frequency (Figure 1F). Automated, quantitative analysis of diagnostic tissues indicated that PD-L1+ TAMs comprised an average of about 12% (median 8%) of all cells within the 12 neuroblastomas analyzed, and confirmed qualitative, manual immunohistochemical findings by showing that an average of 10% (median 7%) of cells in neuroblastomas were PD-L1+ CD163+ TAMs whereas only 1% (median 1%) were PD-L1+PHOX2B+ tumor cells (Figure 1G). Automated analysis of post-chemotherapy tissues was not possible due to induction of hemosiderin deposits and necrosis. Together, these findings indicate that CD163+ TAMs are a dominant source of PD-L1 positivity within the neuroblastoma TME.
An immunosuppressive TME in the NB-Tag and NB-SQ murine models
Prior to evaluating the efficacy of immune checkpoints and combination therapies, we assessed immune cell composition of the neuroblastoma TME in transgenic spontaneous tumor (NB-Tag)17,18 and syngeneic subcutaneous injection (NB-SQ) tumor models. This aggressive MYCN-non-amplified neuroblastoma model shows a consistent age-dependent growth of tumors that allows detailed analysis of gene expression and immunomodulatory cytokines. Significantly higher expression of the TGFB1 gene was observed in NB-Tag tumors at different stages of disease progression at both < 12 weeks (p = .03) and ≥ 12 weeks (p = .005) old mice. We also observed higher expression of CD4 and IL10 genes in NB-Tag tumors of mice ≥ 12 weeks (p = .004 and p = .03, respectively) (Figure 2A). Flow-cytometric analysis of NB-Tag tumors revealed presence approximately one-fifth of T cells in the TME comprised Tregs (CD3+CD4+CD25+Foxp3+), with their frequency remaining relatively unchanged at all stages of disease progression (Figure 2B and Suppl. Fig. S2). In contrast, the frequency of myeloid-derived suppressor cells (MDSCs) identified by the CD11b+CD11c−Ly6Gbright phenotype in the TME continuously increased (p = .0003) while the frequency of NK cells (NK1.1+) decreased over time (p = .02) (Figure 2C). PD-L1 expression in a subset of CD45− cells (Figure 2D) and TAMs (Figure 2E) increased from 14 to 18 weeks of age (p < .05) with peak expression observed at 16 weeks of age. Analysis of the expression of immune checkpoint proteins PD-1 and CTLA-4 on tumor-infiltrating T cells also showed a statistically significant increase in their frequency in the CD4+ and CD8+ T cell subsets over time, mirroring the timing of alterations observed in PD-L1+ TAMs (Figure 2F). Overall, these observations reveal a dynamically evolving immunosuppressive microenvironment during the tumor growth phase of NB-Tag mice (Figure 2A-F & Suppl. Fig.S2).
Figure 2.
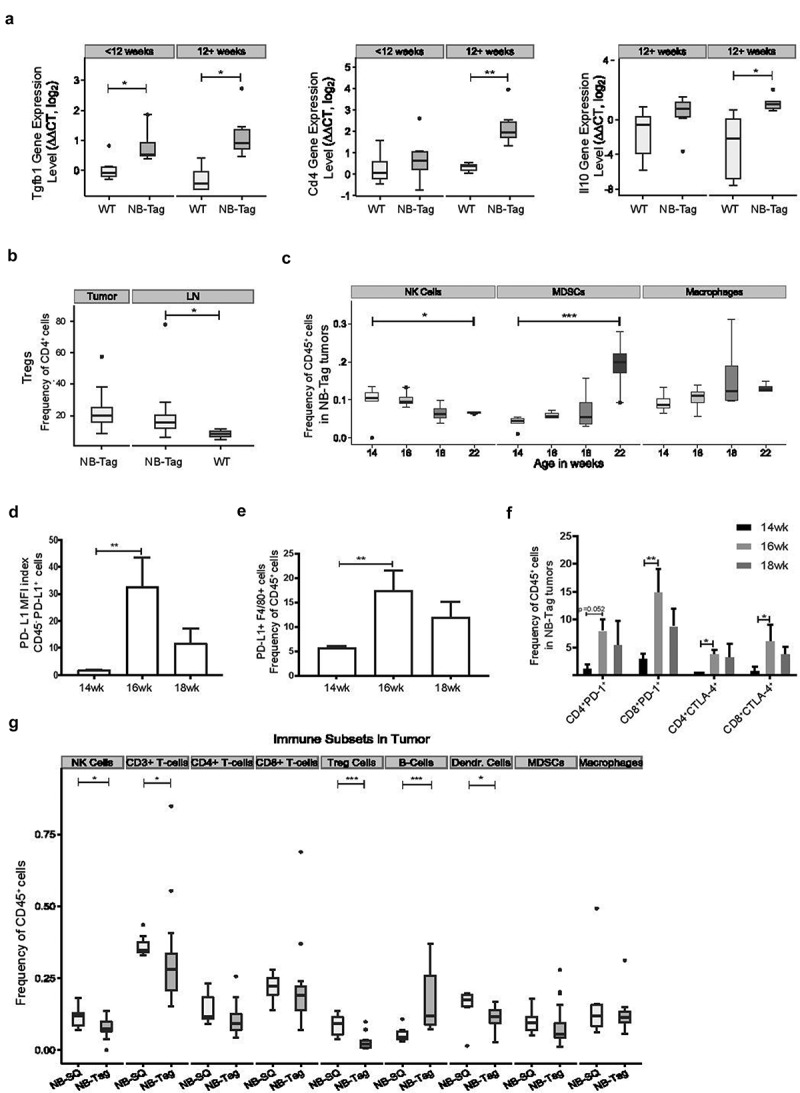
The immunosuppressive microenvironment of transgenic and syngeneic murine neuroblastomas and association with PD-L1 mRNA and protein expression. (A) Boxplot showing intensity of mRNA expression (Log2) of inflammation related genes TGFB1, CD4 and IL-10 in tissue lysates from the adrenal gland and NB-Tag tumors of wild-type C57BL/6 and NB-Tag mice, respectively, before and after 12 weeks of age by qRT-PCR (n = 6 per group); (B) Flow cytometric analysis of the Treg population (mean = 22.6% of CD4+ T-cells, n = 20) in tumors and draining para-aortic lymph nodes of transgenic NB-Tag mice and the para-aortic nodes of wild-type mice (mean 20.2% vs 8.5%, p=0.03, n = 3-15/group); (C) Summary of changes in population frequencies observed in the TME population of MDSCs [CD11b+CD11c−Ly6G(Gr-1)bright], NK (NK1.1+) cells, TAMs (CD11b+CD11c−Ly6G−F4/80high) over time between 14-22 weeks of age as measured by flow cytometry. Boxplots suggest a temporal decrease in NK cells and increase in MDSCs over time. A trend towards increase in TAMs was not significant (p = 0.07). P-values reflect testing for a linear trend over time by regression analysis; n = 3-5 mice per age group; (D) Mean fluorescence intensity (MFI) index of PD-L1 protein expression on tumor cells (CD45−) and (E) frequency of PD-L1+ TAMs (F4/80+) in tumors from NB-Tag mice between 14-18 weeks of age. (F) PD-1 and CTLA-4 expression on tumor-infiltrating CD4+ and CD8+ T-cells over time by flow cytometry. (G) Comparison of the immune landscape in the TME of the transgenic NB-Tag model versusthe subcutaneous (NB-SQ) syngeneic murine model by flow cytometry (average of weeks 14-22). Significance: *, < 0.05; **, < 0.005; ***, < 0.0005
We extended our investigations to blood, bone marrow and draining lymph nodes (LNs) of NB-Tag and wild-type mice to document changes in the frequencies of immune subsets over time. In draining para-aortic lymph nodes, a significantly larger portion of CD4+ T cells was identified as Tregs in NB-Tag mice compared to wild-type mice (mean 20.2% versus 8.5%, p = .03) (Figure 2B), while no significant differences in Treg frequency were observed in blood or bone marrow (data not shown). The data clearly indicate that the majority of alterations in the distribution of immune cell types in the transgenic NB-Tag mouse model occur predominantly within the TME or draining LNs.
We next compared the TME of the NB-Tag model with syngeneic tumor models derived from NB-Tag derived cell line (NBT3L) used in our immunotherapy experiments. The NBT3L model has an engraftment rate of 85% in either subcutaneous or orthotopic injection into C57BL/6 mice (Suppl. Fig. S3). Flow-cytometric analysis of tumor and tumor-infiltrating cells in subcutaneous tumors indicated an immunosuppressive microenvironment, similar to that observed in the NB-Tag model (Figure 2G). Barring a few incongruities such as a notably higher frequency of Tregs (p = .0003) and fewer B-cells (p = .0004) in subcutaneous versus NB-Tag tumor milieus (Figure 2G), the distribution of the majority of CD45+ cells including macrophages, MDSCs, CD8+ T cells, and CD4+ T cells was comparable in the two models (all p > .10). These results indicate remarkable similarity in the TMEs of the syngeneic tumor model and transgenic mouse model, and suggest that these models can be used to reproducibly study the targeting of tumor immune regulation with immune checkpoint blockade.
Combination of high-dose chemotherapy and immune checkpoint inhibition regresses established tumors and improves survival
Twenty-four hours after subcutaneous injection with NBT3L cells, mice were treated with anti-CTLA-4, anti-PD-1, or in combination, for a total of 6 weeks (see schema in Figure 3A). Significant rejection of transplanted tumor cells was observed upon early intervention with any form of immune checkpoint therapy, with an overall survival of 86.7%, 80%, or 88.9%, respectively, versus 23.8% in isotype control treated C56BL/6 mice (p = .0005) (Figure 3B-C). We did not observe any additive or synergistic advantage for combination immune checkpoint therapy compared to monotherapies in this setting.
Figure 3.
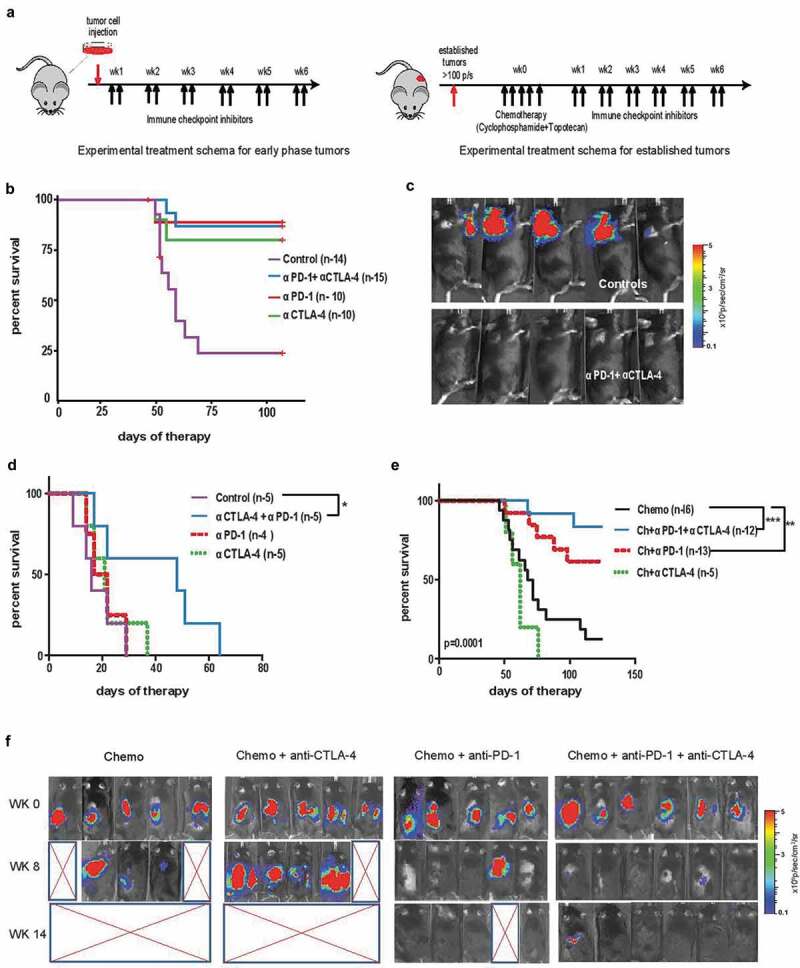
Efficacy of single and dual immune checkpoint blockade during minimal and established disease, and post-chemotherapy in established neuroblastoma tumors . (A) Schematic representation of immune checkpoint blockade as an early intervention in the subcutaneous (NB-SQ) murine model and late intervention in established tumors with or without chemotherapy in the orthotopic model of neuroblastoma. Mice were administered intraperitoneally twice a week with anti-CTLA-4 and anti-PD-1 antibodies or their combination for 6 weeks starting 24 hours after tumor cell injection (4x106 cells) in the NB-SQ model (minimal disease model, left schematic In the established tumor model (right schematic), 1x106 tumor cells were injected orthotopically during surgery to induce renal tumors. Once tumors achieved a bioluminescence signal > 100 photons/second (p/s; log10 scale) (minimum of 21 days after tumor cell injection), chemotherapy consisting of cyclophosphamide plus topotecan was administered consecutively for 5 days followed by 6 weeks of either single or dual immune checkpoint therapy as described above. (B) Kaplan−Meier survival curve of mice from the NB-SQ model receiving early intervention with immune checkpoint blockade. (C) Bioluminescence imaging for luciferase-expressing tumors in NB-SQ mice during early intervention with combination immune checkpoint treatment compared to isotype-treated controls. Panels show 5 representative mice for each treatment group at 6 weeks after checkpoint blockade. (D, E) Kaplan-Meier survival curve of mice with established renal tumors receiving delayed intervention with single or combination immune checkpoint antibodies either (D) without, or (E) with chemotherapy. (F) Bioluminescence imaging for luciferase-expressing tumors of mice treated with the indicated immune checkpoint blockade following chemotherapy starting at week 0 when tumors reached a bioluminescence signal > 100 p/s. Panels include images of (5-6) representative mice for each treatment group at 0, 8 and 14 weeks post chemotherapy. Significance: *, < 0.05;, **,: < 0.005;, ***, < 0.0005
Next, mice with established tumors were evaluated for the combination of immune checkpoint inhibitors alone and after a course of high-dose chemotherapy with cyclophosphamide (110 mg/kg/day) and topotecan (0.4 mg/kg/day), simulating the 5-day chemotherapy induction protocol used for treatment of children with high-risk neuroblastoma. Unlike the responses obtained with immune checkpoint therapies administered soon after tumor inoculation, all mice with established tumors receiving the same therapy had to be euthanized by day 65 because of large tumors. Within these treatment groups, combination immune checkpoint therapy (anti-PD-1 plus anti-CTLA-4 antibodies) showed a statistically significant (p = .04) delay in tumor growth and an increase in survival compared to immune checkpoint monotherapy or isotype control groups (Figure 3D). Encouragingly, treatment of established tumors with chemotherapy prior to immune checkpoint inhibition dramatically improved outcome. Compared to isotype controls, a large increase in survival was observed for mice with established tumors receiving anti-PD-1 monotherapy (p < .005) or anti-PD-1 plus anti-CTLA-4 together (p < .0005) when administered after chemotherapy, and a trend toward significance was observed between these two groups (p = .09) (Figure 3E,F, Figure S6). These findings demonstrate that anti-PD-1 and its combination with anti-CTLA-4 enhance anti-tumor immunity post-chemotherapy in the bulky murine neuroblastoma model, suggesting two potential therapeutic approaches capable of producing long-lasting, durable clinical responses in children with high-risk neuroblastomas.
Tumor rejection and long-term anti-tumor memory response occurs in a CD8+ T cell-dependent manner in the NB-SQ model of neuroblastoma
The above findings indicate that immune checkpoint inhibition can render different anti-tumor responses depending on the stage of tumor development and degree of TME establishment. We first demonstrated that effective depletion of CD8+ T cells but not CD4+ T cells led to complete ablation of efficacy of immune checkpoints, as expected. In the CD8+ T cell-depleted cohort, all engrafted mice developed tumors with accelerated growth compared to non-depleted controls (p < .005) (Figure 4A,B). We then sought to examine the long-term immune memory response in our NB-SQ murine model following early immune checkpoint therapy and tumor rejection. Mice that had successfully rejected tumors following immune checkpoint therapy were re-challenged with the same dose of NBT3L cells at one month (Figure 4C,E), and three months (Figure 4D) following completion of the last dose of treatment. A brief increase in the bioluminescence signal after re-challenge in mice treated with immune checkpoint inhibitors was observed, indicative of tumor growth. However, unlike the control group, the signal intensity of mice previously treated with immune checkpoint therapy showed diminished signal after one week and none of these animals developed tumors within the next 6 weeks of monitoring (Figure 4C). Rejection of repeated tumor cell inoculations and long-term immune protection was observed in all treatment groups irrespective of whether the mice had received single or combination immune checkpoint therapy (Figure 4E). These observations clearly indicate that inhibition of tumor growth by immune checkpoint blockade is mediated by CD8+ T cells and induces a long-term memory response.
Figure 4.
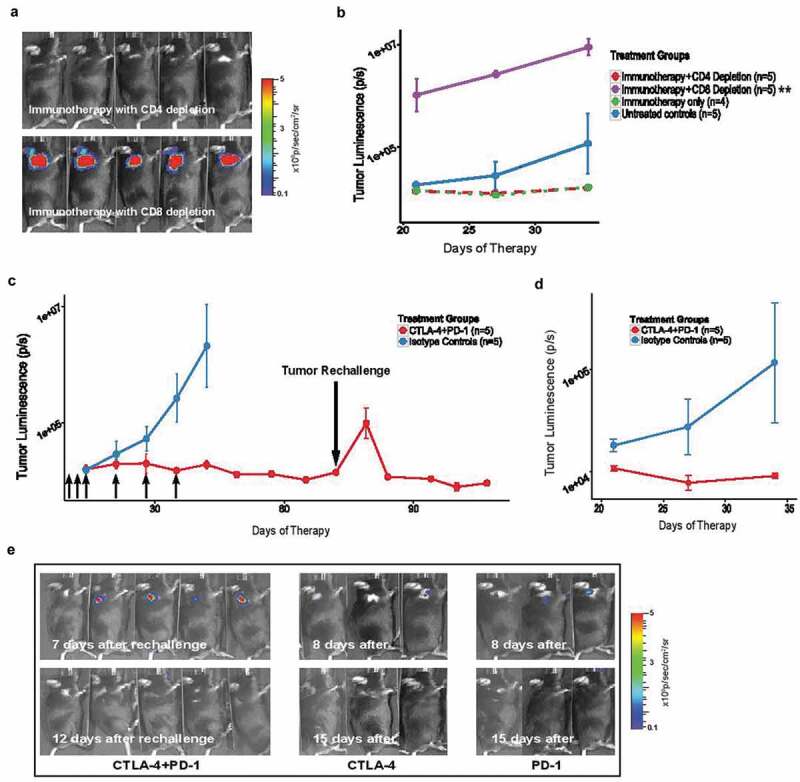
Tumor rejection by early immune checkpoint treatment induces long-term anti-tumor immunity in a CD8+ T-cell dependent manner. (A) Bioluminescence imaging for luciferase expressing tumors in five representative mice depleted of either CD4+ or CD8+ T cells prior to treatment with a combination of anti-CTLA-4 and anti-PD-1 antibodies 24 hours after tumor injection in the NB-SQ model.; (B) Tumor growth curve in NB-SQ mice with and without immunodepletion of CD4+ and CD8+ T cells (monitored by bioluminescence image analysis). Treatment with combination immunotherapy and isotype controls served as positive and negative controls in the experiment. P-values were calculated using the difference of logs of D11 and D34. (p=0.001); (C-D) Mice treated twice weekly for 6 weeks with the combination of anti-CTLA-4 and anti-PD-1 antibodies or isotype controls (small black arrows) were re-challenged with tumor cells (4x106) approximately 1 month (C) and 3 months (D) after completion of antibody therapy. Mice treated with isotype control and tumor-naïve mice served as controls for tumor rejection and subsequent re-challenge, respectively (n = 5 per group). (E) Bioluminescence imaging of representative mice for luciferase expression demonstrates induction of long-term immune memory with either single or dual immune checkpoint treatment. *, < 0.05; **, < 0.005; ***, < 0.0005
Immune checkpoint inhibitors modulate the immune system via distinct mechanisms
As chemotherapy significantly depletes lymphocytes in circulation and eviscerates draining lymph nodes, we attempted to gain insight into the mechanisms of tumor rejection by immune checkpoint inhibitors by analyzing subsets of immune cells in secondary lymphoid organs and blood starting five days after tumor cell inoculation and following either monotherapy or combination immune checkpoint therapy. A significant increase in the number of CD3+, CD4+, CD8+ T cells and their CD28+ and PD-1+ subsets were observed in tumor-draining LNs, spleens and blood in mice treated with either monotherapy or combination of immune checkpoint inhibitors compared to isotype controls with the greatest effect observed in draining LNs and with combination of immune checkpoint inhibitors (Figure 5 and Fig. S4). However, the pattern of alterations varied between monotherapies and combination therapies, with increase in the CD8+CD28+ T cell population only seen in anti-CTLA-4 and the combination of anti-CTLA-4 plus anti-PD-1 immunotherapy in draining LNs (Figure 5A). Meanwhile, the CD8+ CD28+PD-1+ subset significantly increased only in tumor-draining LNs of animals treated with the combination of anti-CTLA-4 plus anti-PD-1 therapy (Figure 5B). The combination therapy also increased CD8+ CD28+PD-1+ in spleen but not blood (Figure 5C-D) indicating that the likely benefit of combination therapy is enhancement of the number and frequency of CD28+PD-1+ effector T cells, particularly in draining LNs. We also noted an unexpected and significant increase in CD4+CD28+PD-1+ cells in draining LNs and blood of mice treated with anti-PD-1 or anti-CTLA-4 plus anti-PD-1 (Figure 5B,D). Among the innate immune cells, an increase in number of F4/80+ macrophages in the LNs after combination immune checkpoint treatment was predominantly observed in the C D11bhiCD11c−F4/80+Ly6Chi subset, a phenotype characteristic of the inflammatory subgroup of macrophages (Figure 5E,G). In the spleen, the total macrophage population did not increase in frequency after single or combined immune checkpoint inhibition (Figure 5F), but the frequency of the inflammatory subgroup of macrophages was significantly higher in mice receiving anti-PD-1 monotherapy compared to other treatment groups (Figure 5H). Circulating blood neutrophils were also observed to increase in response to anti-PD-1 (suppl. Fig. S5A), while the NK cell population significantly increased in draining LNs following anti-CTLA-4 and combination treatment (suppl. Fig. S5B), possibly owing to a treatment-induced inflammation-associated milieu of cytokines and chemokines in the TME, such as IL-23 known to recruit neutrophils and CXCL9 and CXCL11 known to recruit NK cells. Overall, these data indicate that treatment with anti-PD-1, anti-CTLA-4, or their combination affects the innate and adaptive immune system distinctly in different tissues, with combinatorial immune checkpoint therapy generating discernable anti-tumor CD8+CD28+PD-1+ adaptive immunity in the draining lymph nodes of the NB-Tag murine neuroblastoma model.
Figure 5.
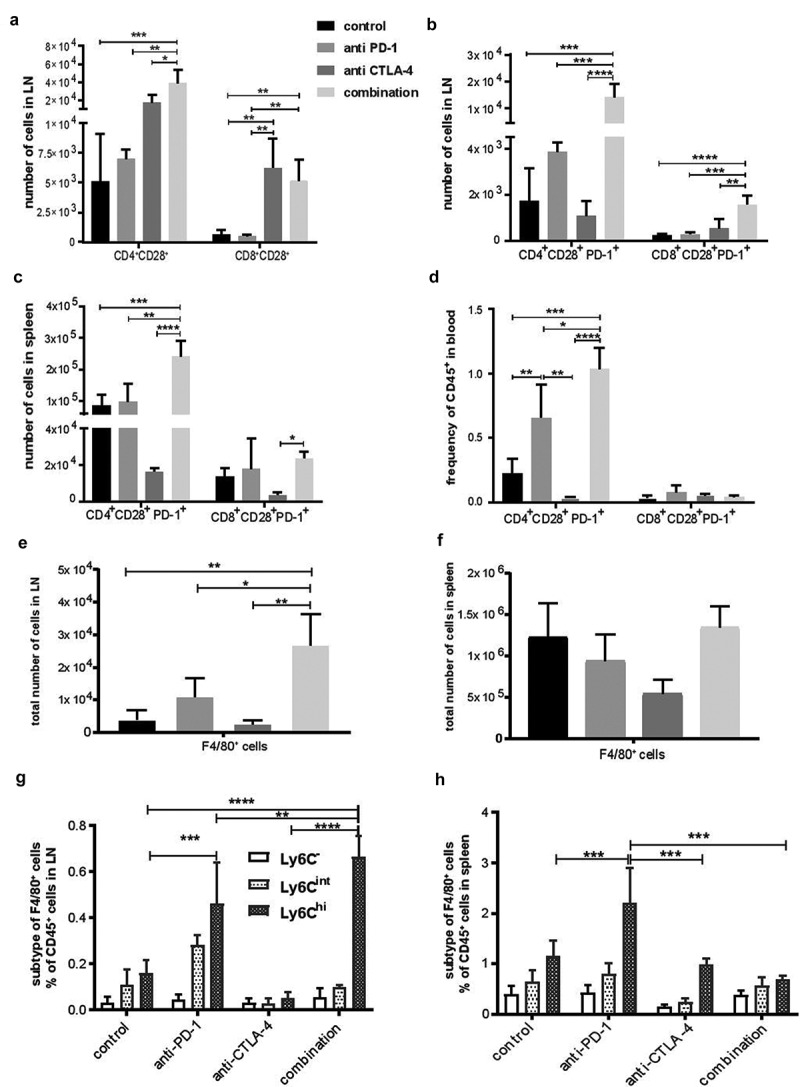
Induction of CD8+CD28+ PD-1 cells and innate immune cells in response to dual immune checkpoint blockade in secondary lymphoid organs and peripheral blood. Flow cytometric analysis of tumor draining LNs, spleens, and blood for different T cells population (A-D) and macrophages (E-H). (A) Average number (+SD) of CD4+CD28+ and CD8+CD28+ cells in tumor draining LNs. (B) Average number (+SD) of CD4+CD28+PD-1+ and CD8+CD28+PD-1+ cells in tumor draining LNs and (C), spleens (D), and peripheral blood. (E) Average number (+SD) of F4/80+ macrophages in draining lymph nodes (F) and spleens. (G) Average percentage (+SD) of various F4/80+ macrophage subtypes based on Ly6C expression in tumor draining lymph nodes and (H) spleen. (n=4-5 mice per group). *, < 0.05; **, < 0.005; ***, < 0.0005
Discussion
Use of chemotherapy regimens, having both cytotoxic and immune modulatory effects, together with immune checkpoint blockade has potential to stimulate an effective immune response to control residual tumor cells.16 However, results of early phase clinical trials with immune checkpoint therapies in childhood cancers have been disappointing, and new therapies and drug combinations that achieve effective and durable treatment responses are needed. In the iMATRIX phase I/II trial of the anti-PD-L1 antibody atezolizumab as a single agent in previously treated patients with neuroblastoma and other malignancies, atezolizumab was well tolerated; however, while 4 of the 11 patients with neuroblastoma exhibited stable disease, 0 of the 11 exhibited a partial or complete response.20 In a clinical trial of the anti-CTLA-4 antibody ipilimumab as a single agent in 21 pediatric patients including one patient with neuroblastoma, ipilimumab was well tolerated but no objective tumor regression was detected, although improved overall survival was observed for patients who exhibited immune-related toxicities compared to those who did not.21 These studies are consistent with results from our pre-clinical murine model of established neuroblastoma in the absence of chemotherapy, in which we observed no response to anti-PD-1 or anti-CTLA-4 antibodies as single agents. However, when combined, anti-PD-1 with anti-CTLA-4 induced significant systemic and local immune responses in the murine model, with alteration of myeloid cells to an inflammatory phenotype, marked increase in CD28+ PD-1+ effector T cells in draining lymph nodes, and establishment of immune memory protective against tumor re-challenge. Interestingly, in tumors of children with high-risk neuroblastoma, we demonstrate that chemotherapy significantly and specifically increased expression of PD-L1 in TAMs. Most importantly, in the murine model, preceding immune checkpoint blockade with neoadjuvant chemotherapy in mice with established tumors significantly improved long-term survival and led to cures.
Our present analysis of the TME in the transgenic and syngeneic NB-Tag model of MYCN-non-amplified neuroblastoma17 revealed an immune suppressive TME where TAM infiltration is associated with an increase in PD-L1 and TGFB1 gene expression and decrease in frequency of cytotoxic lymphocytes. This data is in line with our serial IHC analysis of primary human neuroblastoma specimens revealing expression of PD-L1 on infiltrating TAMs. These results support the concept of dynamic PD-L1 expression on both tumor cells and tumor-infiltrating immune cells during tumor evolution and post chemotherapy. Relevant to these dynamics, PD-L1 gene regulation has been shown to be IFN-γ-dependent. For example, PD-L1 protein is expressed on some human neuroblastoma cell lines and can be further induced by treatment with IFN-γ; interestingly, constitutive PD-L1 expression can be lost when neuroblastoma cells are injected subcutaneously into mice, but PD-L1 expression can be restored by bolus injection of IFN-γ directly into the tumor.22 Similarly, PD-L1 is produced in an IFN-γ-dependent manner in melanoma cells and ovarian cancer cells.23,24 Neuroblastoma samples that we obtained post-chemotherapy were collected from patients that had undergone a minimum of five cycles of combination chemotherapies including combinations of alkylator, platinum, topoisomerase inhibitor, and anthracycline-based chemotherapies. It has been proposed that combinations of these chemotherapies are responsible for an immunogenic cell death leading to type I interferon response and alteration in the TME with subsequent PD-L1 expression, thus providing the rationale for combining chemotherapy with anti-PD-1.25 Future studies using multiplex imaging techniques of human neuroblastoma samples at various timepoints of therapy including recurrence should shed light to the dynamic expression of PD-L1 and additional cells that may express this immune checkpoint. Taken together, our data demonstrate an immunosuppressive microenvironment in the NB-Tag murine model of neuroblastoma, and increased PD-L1 protein expression on TAMs in both the murine model and in post-chemotherapy tumor tissues from patients.
Our gene expression data demonstrated higher PD-L1 expression in human MYCN-non-amplified neuroblastomas at diagnosis, with significant increase in frequency of PD-L1 positive tumors noted post-chemotherapy. TAMs play an important role in maintaining an inflammatory microenvironmental niche for metastatic neuroblastoma lacking MYCN amplification. In turn, MYCN-non-amplified neuroblastomas actively promote monocyte/TAM infiltration via secretion of CCL2, a well-known and potent attractant of macrophages. We and others have previously shown using an anti-CCL2 neutralizing antibody that blockade of CCL2 (which was secreted from neuroblastoma cells in vitro) inhibits monocyte migration toward the neuroblastoma cells, and relatedly, that MYCN activity in MYCN-amplified neuroblastomas suppresses production of CCL2, as demonstrated by an inverse correlation between MYCN amplification and CCL2 gene expression, increase in CCL2 expression upon knock-down of MYCN, decrease in CCL2 protein expression upon enforced expression of MYCN, and direct binding of MYCN to the CCL2 promoter.26,27 The increase in PD-L1+ macrophage recruitment by MYCN-non-amplified tumor cells along with dynamic regulation of PD-L1 expression provides an explanation for the increase in PD-L1 positivity in MYCN-non-amplified tumors post chemotherapy.
Minimal clinical response has been observed in pediatric clinical trials of immune checkpoint therapies.21 These results are consistent with our findings of failure of immune checkpoint monotherapies in mice with established disease. However, we did find significant prolongation of survival in mice with established tumors treated with the combination of anti-PD-1 and anti-CTLA-4, suggesting a synergistic effect and consistent with these agents working through distinct mechanisms. Importantly, in mice with established neuroblastomas, our data provide evidence for efficacy of chemotherapy with cyclophosphamide and topotecan followed by anti-PD-1 or anti-PD-1 plus anti-CTLA-4 therapy. Cyclophosphamide is known to deplete immunosuppressive cells, release antigens from tumor cells, and stimulate T cells, and topotecan up-regulates major histocompatibility class I molecules and Fas expression, thereby rendering tumor cells more sensitive to T cell-mediated cytotoxicity.28 These findings add to recent literature describing the success of immune checkpoint blockade in combination with various therapeutic strategies for achieving synergistic and durable clinical responses. Combining chemotherapies with immune checkpoint blockade has also shown encouraging results in retrospective and meta-analysis of adult clinical studies, including non-small cell lung cancer and Hodgkin lymphoma.29,30 Dual blockade with anti-PD-1 and anti-CTLA-4 antibodies in combination with tumor vaccines has led to increased levels of tumor rejection in murine models for melanoma, colon and ovarian cancer.31,32 Similarly, combination of immune checkpoint inhibition with local radiation therapy has been shown to activate the adaptive immune system and enhance abscopal effects.33,34
We also demonstrate a novel mechanism for the success of dual immune checkpoint therapy. Expansion of a CD28+ subset of T cells exhibiting a transitional memory phenotype has been observed in patients receiving either anti-CTLA-4 or combination anti-CTLA-4 plus anti-PD-1 therapy.35 CD28 is a pivotal positive costimulatory molecule that defines T cell motility and formation of immune synapse between T cells and antigen-presenting cells (APCs) leading to activation of effector T cells and induction of an anti-tumor response.36 Proliferating CD8+ T effector cells expressing CD28 and high levels of PD-1 with co-expression of CTLA-4 in peripheral blood were observed after PD-1-targeted therapy in lung cancer patients and associated with clinical benefits.37 The CD28 receptor on T cells has been shown to be a primary target for PD-1 mediated inhibition.38 Expression of CD28 by T cells is critical for PD-1 signaling and for expansion of effector T cells, as it has been shown that CD28 deficiency or blocking the CD28/B7 pathway abrogates the anti-tumor effects of anti-PD-1 treatment in a colon carcinoma murine model.38,39 Expression of CD28-PD-1 fusion proteins in CD8 + T cells also overcomes the PD-1:PDL-1 immunosuppressive axis and improves cytolytic activity.40 The increase in CD4+CD28+PD-1+ T cell subset likely improves the anti-tumor effect of their CD8+ counterparts as the CD28:PD-1 fusion protein expressed in CD4 + T cells has been shown to improve the cytotoxic efficacy of CD8+CD28+PD-1+ T cells in models of pancreatic carcinoma and non-Hodgkin’s lymphoma.41 While increase in the number of CD4+CD28+ T cell subsets likely enhances CD8+CD28+ effector functions in our model, we believe the increase in CD8+CD28+ effector cells most predominantly seen in tumor-draining LN of the mice treated with combination of immune checkpoint therapies was the crucial component of the anti-tumor response. This is supported by our finding that depletion of CD8+ but not CD4+ cells abrogates the effects of immune checkpoint blockers. Additional alterations in innate immune cell profiles likely contributed to the efficacy of combination immune checkpoint therapy, such as the observed increases in neutrophil counts and a subset of inflammatory macrophages in draining LN and spleen of mice receiving anti-PD-1 or combined anti-PD-1 and anti-CTLA-4 therapies.
It has previously been shown that the efficacy of anti-PD-1 can be increased by anti-GD2 antibody in the NXS2-HGW model of murine neuroblastoma42 and that the efficacy of anti-PD-1 or anti-PD-L1 can be increased by anti-CD4 antibody in the Neuro2a and NXS2 models of murine neuroblastoma.43 Our results complement these earlier reports by showing that treatment with anti-PD-1 antibody or with anti-PD-1 plus anti-CTLA-4 can provide increased efficacy when administered soon after standard induction chemotherapy. If tested in the clinic, pediatric patients could be managed with standard tools available for treating patients for chemotherapy-induced neutropenia and for side effects related to immune checkpoint immune checkpoint blockade such as endocrinopathies, colitis, and pneumonitis using therapies such as steroids, non-steroidal anti-inflammatory drugs, or in some cases anti-TNFα monoclonal antibody (infliximab) or anti-integrin α4β7 antibody (vedolizumab).44
While our study supports the administration of anti-PD-1 or anti-PD-1 plus anti-CTLA-4 after induction chemotherapy, our experimental models had limitations. First, it was not feasible for us to examine tumor-infiltrating cells by IHC after chemotherapy, owing to deposits of hemosiderin and severe intratumor necrosis induced by cyclophosphamide and topotecan in our mouse model. Second, it should be noted that the subcutaneous and orthotopic models used in this study are of localized disease and may not faithfully represent all cases of metastatic disease in which some sites of disease may be more resistant to immune cell-mediated cytotoxicity than others.
In summary, our data demonstrate that dual immune checkpoint blockade using anti-PD-1 and anti-CTLA-4 antibodies activates CD8+CD28+PD-1+ T cells and inflammatory TAMs in a potentially synergistic manner leading to enhanced tumor control. The prolonged survival observed after sequential treatment with induction chemotherapy followed by either anti-PD-1 or anti-PD-1 plus anti-CTLA-4 immunotherapy in the syngeneic murine NB model suggests opportunities for application in children with recurrent neuroblastoma.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr. Y. Maitani and associates at the Institute of Medicinal Chemistry in Hoshi University of Tokyo, Japan for providing us with the NB-Tag model, Prof. Omid Akbari and Dr. Hadi Maazi for their assistance and protocols, Tsen-Yin Lin in CHLA’s flow cytometry core, Meenakshi Upreti for manuscript review, Dr. Martine Torres for editorial assistance, and Drs. Yves DeClerck, Robert C. Seeger, Eleanora Heisterkamp and Chintan Parekh for their valuable input. The authors would like to acknowledge the Children’s Oncology Group (U10–CA98543) for providing tumor specimens from patients with neuroblastoma from which microarray data were generated.
Funding Statement
This work was supported in part by grant CA170257P1 from the Department of Defense (SA); by a T32 CA009659 training grant (RYC) from the National Cancer Institute (PI Y.A. DeClerck); by R01 CA215753 and R01 CA219024 from the National Cancer Institute (PI MF), and in part by the 5 P01 CA81403-14 National Cancer Institute (co-I SA, PI R.C. Seeger, J Maris), T.J. Martell Foundation, V Foundation, and Nautica Malibu Triathlon (SA).
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Soheila Shirinbak, Randall Y. Chan, Shilpa Shihani, Sakunthala Muthugounder, Rebekah Kennedy, Long T. Hung, G. Esteban Fernandez, Michael D. Hajidaniel, Muller Fabbri, Babak Moghimi, Michael A. Sheard, Alan L. Epstein, and Hiroyuki Shimada. The first draft of the manuscript was written by Soheila Shirinbak, Randall Y. Chan, Shilpa Shahani, Michael A. Sheard, and Shahab Asgharzadeh and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Data availability statement
Microarray data are accessible through the TARGET Data Matrix (http://target.nci.nih.gov/dataMatrix/TARGET_DataMatrix.html).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. (Children’s Hospital Los Angeles IRB: CCI-06-00198)
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW.. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272:1170–13. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98:13866–13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. 2017;5:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson RAM, Evans TRJ, Fraser AR, Nibbs RJB. Immune checkpoint inhibitors: new strategies to checkmate cancer. Clin Exp Immunol. 2018;191:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Errico A. Melanoma: checkMate 067–frontline nivolumab improves PFS alone or in combination with ipilimumab, Nature reviews. Clin Oncol. 2015;12:435. [DOI] [PubMed] [Google Scholar]
- 8.Schadendorf D, Larkin J, Wolchok J, Hodi FS, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao C, et al. Health-related quality of life results from the phase III CheckMate 067 study. Eur J Cancer. 2017;82:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kabir TF, Chauhan A, Anthony L, Hildebrandt GC. Immune checkpoint inhibitors in pediatric solid tumors: status in 2018. Ochsner J. 2018;18:370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, Hadjidaniel M, Liu CW, Metelitsa LS, Pique-Regi R, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol. 2012;30:3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsson K, Kock A, Idborg H, Arsenian Henriksson M, Martinsson T, Johnsen JI, Korotkova M, Kogner P, Jakobsson PJ. COX/mPGES-1/PGE2 pathway depicts an inflammatory-dependent high-risk neuroblastoma subset. Proc Natl Acad Sci U S A. 2015;112:8070–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang P, Wu X, Basu M, Dong C, Zheng P, Liu Y, Sandler AD. MYCN amplification is associated with repressed cellular immunity in neuroblastoma: an in silico immunological analysis of TARGET database. Front Immunol. 2017;8:1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borriello L, DeClerck YA. [Tumor microenvironment and therapeutic resistance process]. Med Sci. 2014;30:445–451. [DOI] [PubMed] [Google Scholar]
- 15.Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, Santi M, Merchant MS, Geoerger B, Hezam I, Marty V, et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. 2017;123:3807–3815. [DOI] [PubMed] [Google Scholar]
- 16.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. [DOI] [PubMed] [Google Scholar]
- 17.Hadjidaniel MD, Muthugounder S, Hung LT, Sheard MA, Shirinbak S, Chan RY, Nakata R, Borriello L, Malvar J, Kennedy RJ, et al. Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget. 2017;8:91516–91529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwakura H, Ariyasu H, Kanamoto N, Hosoda K, Nakao K, Kangawa K, Akamizu T. Establishment of a novel neuroblastoma mouse model. Int J Oncol. 2008;33:1195–1199. [PubMed] [Google Scholar]
- 19.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017;19:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geoerger B, Zwaan CM, Marshall LV, Michon J, Bourdeaut F, Casanova M, Corradini N, Rossato G, Farid-Kapadia M, Shemesh CS, et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): a multicentre phase 1-2 study. Lancet Oncol. 2020;21:134–144. [DOI] [PubMed] [Google Scholar]
- 21.Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, Delbrook C, Lodish M, Bishop R, Wolchok JD, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res. 2016;22:1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dondero A, Pastorino F, Della Chiesa M, Corrias MV, Morandi F, Pistoia V, Olive D, Bellora F, Locatelli F, Castellano A, et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology. 2016;5:e1064578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112:1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y-J, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018;5:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, Seeger RC. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J.Exp.Med. 2004;199:1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song L, Ara T, Wu HW, Woo CW, Reynolds CP, Seeger RC, DeClerck YA, Thiele CJ, Sposto R, Metelitsa LS. Oncogene MYCN regulates localization of NKT cells to the site of disease in neuroblastoma. J Clin Invest. 2007;117:2702–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30:219–235. [DOI] [PubMed] [Google Scholar]
- 29.Zhou Y, Chen C, Zhang X, Fu S, Xue C, Ma Y, Fang W, Yang Y, Hou X, Huang Y, et al. Immune-checkpoint inhibitor plus chemotherapy versus conventional chemotherapy for first-line treatment in advanced non-small cell lung carcinoma: a systematic review and meta-analysis. J Immunother Cancer. 2018;6:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossi C, Gilhodes J, Maerevoet M, Herbaux C, Morschhauser F, Brice P, Garciaz S, Borel C, Ysebaert L, Oberic L, et al. Efficacy of chemotherapy or chemo-anti-PD-1 combination after failed anti-PD-1 therapy for relapsed and refractory Hodgkin lymphoma: A series from Lysa centers. Am J Hematol. 2018;93:1042-1049. [DOI] [PubMed] [Google Scholar]
- 31.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sindoni A, Minutoli F, Ascenti G, Pergolizzi S. Combination of immune checkpoint inhibitors and radiotherapy: review of the literature. Crit Rev Oncol Hematol. 2017;113:63–70. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, Dong Y, Kong L, Shi F, Zhu H, Yu J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J Hematol Oncol. 2018;11:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dilek N, Poirier N, Hulin P, Coulon F, Mary C, Ville S, Vie H, Clemenceau B, Blancho G, Vanhove B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PloS One. 2013;8:e83139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114:4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobold S, Grassmann S, Chaloupka M, Lampert C, Wenk S, Kraus F, Rapp M, Duwell P, Zeng Y, Schmollinger JC, et al. Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst. 2015;107:djv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rataj F, Kraus FBT, Chaloupka M, Grassmann S, Heise C, Cadilha BL, Duewell P, Endres S, Kobold S. PD1-CD28 fusion protein enables CD4+ T cell help for adoptive T cell therapy in models of pancreatic cancer and non-hodgkin Lymphoma. Front Immunol. 2018;9:1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siebert N, Zumpe M, Juttner M, Troschke-Meurer S, Lode HN. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology. 2017;6:e1343775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rigo V, Emionite L, Daga A, Astigiano S, Corrias MV, Quintarelli C, Locatelli F, Ferrini S, Croce M. Combined immunotherapy with anti-PDL-1/PD-1 and anti-CD4 antibodies cures syngeneic disseminated neuroblastoma. Sci Rep. 2017;7:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winer A, Bodor JN, Borghaei H. Identifying and managing the adverse effects of immune checkpoint blockade. J Thorac Dis. 2018;10:S480–S489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data are accessible through the TARGET Data Matrix (http://target.nci.nih.gov/dataMatrix/TARGET_DataMatrix.html).