

Abstract

Current chemotherapeutics for leishmaniasis have multiple deficiencies, and there is a need for new safe, efficacious, and affordable medicines. This study describes a successful drug repurposing approach that identifies the over-the-counter antihistamine, clemastine fumarate, as a potential antileishmanial drug candidate. The screening for inhibitors of the sphingolipid synthase (inositol phosphorylceramide synthase, IPCS) afforded, following secondary screening against Leishmania major (Lmj) promastigotes, 16 active compounds. Further refinement through the dose response against LmjIPCS and intramacrophage L. major amastigotes identified clemastine fumarate with good activity and selectivity with respect to the host macrophage. On target engagement was supported by diminished sensitivity in a sphingolipid-deficient L. major mutant (ΔLmjLCB2) and altered phospholipid and sphingolipid profiles upon treatment with clemastine fumarate. The drug also induced an enhanced host cell response to infection indicative of polypharmacology. The activity was sustained across a panel of Old and New World Leishmania species, displaying an in vivo activity equivalent to the currently used drug, glucantime, in a mouse model of L. amazonensis infection. Overall, these data validate IPCS as an antileishmanial drug target and indicate that clemastine fumarate is a candidate for repurposing for the treatment of leishmaniasis.
Keywords: Leishmania, ceramide, sphingolipids, repurposing, IPCS, clemastine fumarate
Infecting over 1 billion people,1,2 neglected tropical diseases (NTDs) are a group of 20 diseases that are prevalent in poorer populations of the world. These diseases are classified as “neglected” due to both this predominance in areas of poverty and their relatively low priority on national and international health agendas.3 Causing an estimated 3.32 million disability adjusted life years (DALYs) and accounting for 13% of all NTD DALYs,4,5 the leishmaniases are one of the most widespread and serious of the NTDs.1,5−7
Caused by insect vector-borne kinetoplastid parasites of the genus Leishmania, the leishmaniases are endemic in 98 countries, placing an estimated 310 million people at risk. There are 1.3 million new cases recorded annually, and the disease claims in excess of 30,000 lives each year.6 Leishmaniasis encompasses a range of clinical manifestations that affect both humans and animals. There are three main human disease states that are caused by 21 of the 30 Leishmania species that infect mammals.8 Cutaneous leishmaniasis (CL), commonly caused by L. major, presents as ulcerations that may heal spontaneously. In more extreme cases, as with recurrent and diffuse cutaneous leishmaniasis, these lesions are chronic and difficult to treat.2 Mucocutaneous leishmaniasis (MCL) is the most disfiguring form involving the destruction of buccopharyngeal tissue. While they are nonfatal, CL and MCL can be extremely debilitating and can lead to long-term stigma for those afflicted.9 Visceral leishmaniasis (VL), the most severe form of leishmaniasis caused mainly by L. donovani, manifests with fever, hepatosplenomegaly, and pancytopenia and is fatal if untreated. Treated VL patients can relapse and suffer from postkala-azar dermal leishmaniasis (PKDL), which is a reservoir for the emergence of drug resistance.2,10
There is currently no approved vaccine against Leishmania infections in humans;5 thus, the control of the disease relies on a limited number of drug treatments that all suffer from a number of inadequacies including cost, long-term and painful modes of administration, toxicity, and the emergence of relapse and resistance.11−13 When one considers this, there is an urgent need for novel antileishmanial treatments that are inexpensive and free of side effects.
New treatments require new modes of actions, and the differences in essential sphingolipid synthesis offer opportunities for selective intervention in parasite metabolism. In contrast to mammalian cells, where the predominant complex sphingolipid is sphingomyelin (SM), Leishmania, as well as fungi, plants, and some other protozoa, synthesize inositol phosphorylceramide (IPC).14−18 The final step in the synthesis, catalyzed by sphingolipid synthases (SLS), involves the conversion of ceramides (phytoceramides in plants and fungi) to phosphosphingolipids via the addition of a headgroup that varies according to the substrate used. Mammals predominantly incorporate phosphocholine from the lipid phosphatidylcholine (PC) to generate SM species through the action of sphingomyelin synthase (SMS) while plants, fungi, and kinetoplastid parasites mainly use phosphatidylinositol (PI) in a reaction catalyzed by inositol phosphorylceramide synthase (IPCS). This divergence enabled the identification and validation of the fungal IPC synthase (AUR1p) as a drug target for pathogenic fungi.19−23 Subsequently, work in our group identified and isolated the orthologous Leishmania enzyme from L. major (LmjIPCS).24−26 Using a microsomal preparation derived from a LmjIPCS-complemented Saccharomyces cerevisiae lacking the fungal AUR1p, we were able to develop a microtiter plate-compatible assay of enzyme activity and use this to confirm the mechanism of action and identify critical substrate parameters.27 With this resource in hand, we sought to identify potential inhibitors of the enzyme.
In this report, we describe the further application of this assay to screen a set of pharmacologically active compounds and to use the most active and selective of the hits, clemastine fumarate 1, to chemically validate IPCS as an antileishmanial drug target both in vitro and in vivo.
Results
Using the previously reported microtiter plate assay,26 a diverse set of 1040 pharmacologically active compounds (National Institute of Neurological Disorders and Stroke [NINDS] (NINDS set))28 were screened initially at 20 μM for activity against LmjIPCS (Figures 1 and S1 and Table S1). Fifty-seven compounds were found to exhibit ≥70% inhibition of the enzyme; these were tested for efficacy against L. major proliferative, insect stage promastigotes at 10 μM using a resazurin-based cell-viability assay29,30 (Figure S2). While the majority of the compounds inhibited parasite growth at 24 h (data not shown), the extension of the assay duration to 72 h revealed a smaller set of 16 active compounds (Figures 1, 2, and S2 and Table S1).
Figure 1.

Schematic flowchart of the high-throughput screening workflow. LmjIPCS-inositol phosphorylceramide synthase from L. major.
Figure 2.
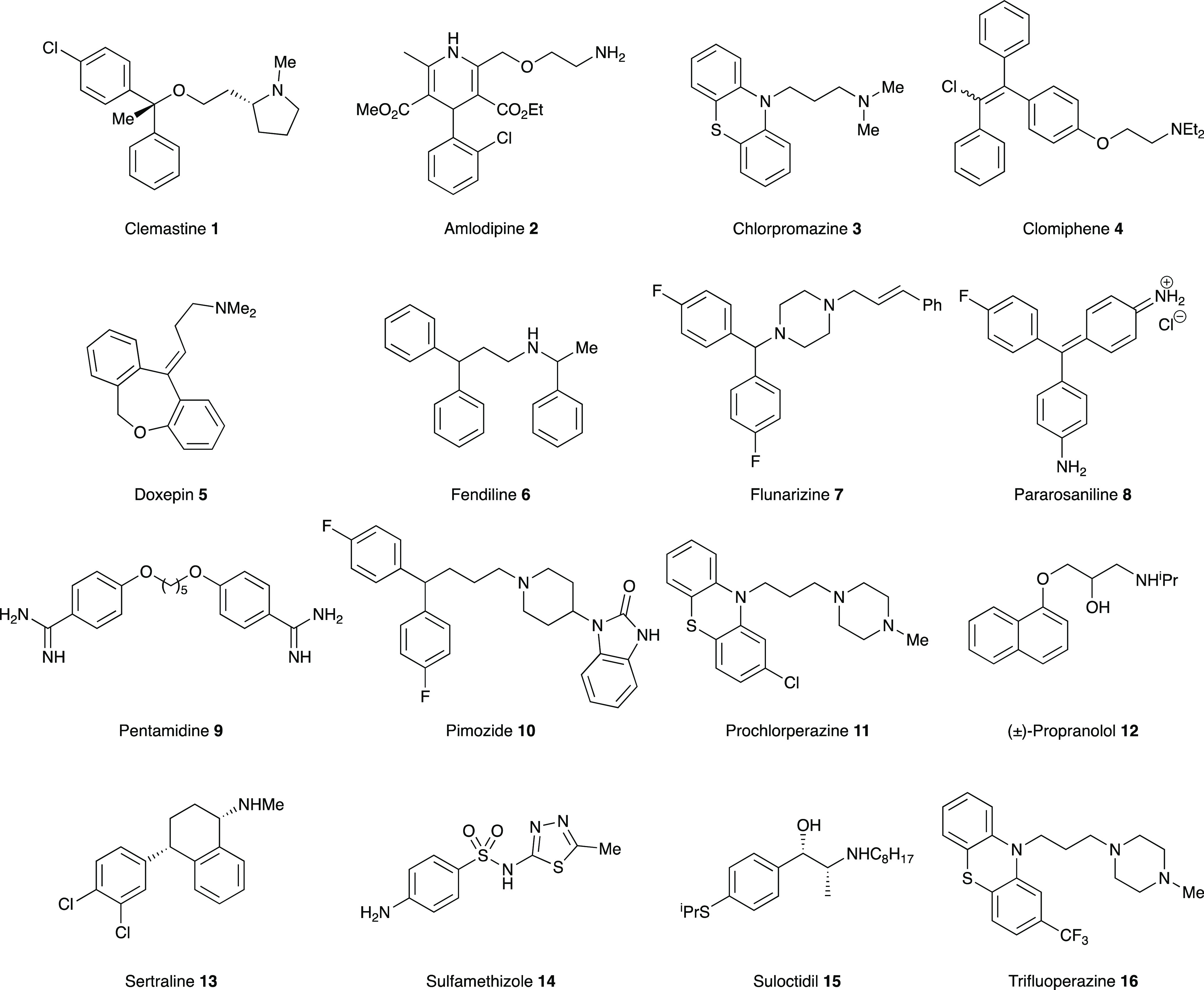
NINDS library hit compounds selected for further study.
Using commercially sourced samples, secondary dose response assays for these 16 compounds were then undertaken against L. major (FV1) promastigotes and LmjIPCS (Figures 1, 2, and S2 and Table S1). While LmjIPCS inhibition was confirmed for amlodipine 2, chlorpromazine 3, clomiphene 4, doxepin 5, fendiline 6, flunarizine 7, pimozide 10, prochlorperazine edisylate 11, propranolol 12, sertraline 13, suloctidil 15, and trifluoperazine 16, these compounds displayed low to moderate (EC50 > 1 μM) activity against the L. major promastigotes and were not further explored. Pentamidine 9 is a clinically utilized antileishmanial, and this study revealed an unexpected alternative mode-of-action to that previously reported.31,32 IC50 values against LmjIPCS could not be determined for pararosaniline 8 and sulfamethizole 14, which also exhibited poor antileishmanial activity, and these were also discarded.
This left clemastine fumarate 1, which demonstrated potent activity against LmjIPCS (IC50 = 2.90 μM) and submicromolar inhibition of promastigote proliferation (EC50 = 0.18 μM). In a subsequent experiment utilizing L. major (JISH118) infected bone marrow derived macrophages (BMDMs), this compound demonstrated activity against intramacrophage amastigote L. major (EC50 = 3.16 μM) and a selectivity index of 6.05 (BMDM CC50 = 19.13 μM) (Figure 3A and Table S1). The fact that the compound demonstrated higher activity against the axenic L. major (EC50 = 0.18 μM) than the IPCS target (IC50 = 2.90 μM) initially appeared counterintuitive. However, the enzyme material extracted from yeast is highly concentrated and, assuming cellular equivalence, estimated to be ∼100-fold greater than that present in the promastigote cell assay. Therefore, while other modes of action were not discounted at this stage, clemastine fumarate 1 was selected as the most interesting hit from the NINDS library screen and advanced to further explore the inhibition of IPCS as a viable antileishmanial drug strategy.
Figure 3.

Clemastine fumarate broad range antileishmanial activity. Dose–response curves of clemastine fumarate plotting the percentage of macrophage, intracellular amastigote, and promastigote growth inhibition for L. major (A) and L. amazonensis (B). Dose–response curves of clemastine fumarate against L. donovani (C) and L. infantum (D) promastigotes; each curve shows a representative experiment of at least three biological replicates. For each species, amphotericin B was used as a positive control (EC50 = 0.027 ± 0.003 μM). EC50 and CC50 values are the mean ±95% CI from at least three experiments. SI, selectivity index (macrophage CC50/amastigote EC50).
Clemastine Fumarate Shows Activity against a Wide Range of Leishmania Species
The results presented thus far revealed the potential of clemastine fumarate 1 as an antileishmanial drug with a novel mode-of-action (LmjIPCS inhibition). L. major is classified as an Old World species, prevalent in the Eastern hemisphere, and manifests as CL, the most common form of the disease.33 However, given the global reach of leishmaniasis, the ideal drug is one that is active against all pathogenic species of Leishmania and therefore has the potential to treat all forms of the disease. To explore this possibility, clemastine fumarate was also tested against promastigote forms of L. amazonensis, which causes New World CL in the Western hemisphere,34 and L. infantum and L. donovani, which both manifest as VL, the most fatal form of the disease.33
Clemastine fumarate showed significant levels of activity against all species of Leishmania promastigote forms tested. EC50 [μM] values are as follows: L. major (JISH118), 0.179 ± 0.047; L. amazonensis (Josefa), 0.028 ± 0.012; L. donovani (LV1), 0.019 ± 0.043; L. infantum (MAP263), 0.708 ± 0.043 (Figure 3B–D). Selecting L. amazonensis due to its sensitivity to clemastine fumarate and relevance to many cases of CL and MCL in South America, we then confirmed intramacrophage amastigote activity. This assay revealed that clemastine fumarate exhibits submicromolar inhibition of L. amazonensis amastigote intramacrophage growth (EC50 = 0.462 ± 0.100 μM) and a promising selectivity index of 39.49 (CC50 BMDM = 18.25 ± 0.05 μM) (Figure 3B).
Clemastine Fumarate Activity Is Correlated with Inositol Phosphorylceramide Synthase Inhibition
Coupled with the well-documented in vivo safety profile of clemastine fumarate 1(35,36) [oral LD50 values of 730 mg.kg–1 (mouse) and 3550 mg.kg–1 (rat), low toxicity against human liver HepG2 cells (CC50 > 50 μM), and low cardiotoxicity], the observed potency against both promastigote and, the more clinically relevant, intramacrophage amastigote life stages suggested that clemastine fumarate 1 had significant potential to be employed as an antileishmanial chemotherapeutic. In order to establish whether the antileishmanial action of clemastine fumarate 1 occurred via its activity as an inhibitor of the Leishmania IPCS, experiments were conducted with the sphingolipid-free L. major mutant created through the targeted deletion of the catalytic subunit (LmjLCB2) of the first enzyme in the pathway, serine palmitoyltransferase.37 While these parasites (ΔLmjLCB2) contain a functional IPCS, the lack of de novo synthesized ceramide renders this activity redundant.37 Notably, it has been demonstrated that L. major ΔLmjLCB2 parasites maintain high levels of IPCS activity during animal infection and, furthermore, it is known that LCB2 and serine palmitoyltransferase activity is downregulated in pathogenic amastigotes forms, which scavenge host ceramide to enable IPC synthesis.38,39 In these experiments, as a control, we used an “add-back” cell line in which the ΔLmjLCB2 mutant was “rescued” by the expression of LmjLCB2 from a plasmid [pXNEO LmLCB2] to generate a line in which ceramide and IPC synthesis is restored.37 Dose–response assays for clemastine fumarate 1 against L. major wild type promastigotes, ΔLmjLCB2 mutant, and add-back (AB) lines revealed that the EC50 of clemastine fumarate 1 against ΔLmjLCB2 parasites was 0.45 ± 0.07 μM, a significantly lower efficacy to that observed for wild type L. major in this assay (0.16 ± 0.02 μM; Figure 4). Furthermore, the restoration of sphingolipid biosynthesis in the add-back line restored the sensitivity of the mutant parasites to clemastine fumarate 1 (EC50 = 0.08 ± 0.01 μM). The reasons for the increased sensitivity of the AB lines remain unclear, although the overexpression of LmjLCB2 in these mutant parasites37 may lead to other changes in sphingolipid biosynthesis. Importantly, the positive control, cycloheximide, which acts by a different mode-of-action, maintained an EC50 value of 0.02 μM against all three parasite lines tested. Collectively, these results supported the hypothesis that the inhibition of IPCS is a key component of the antileishmanial activity of clemastine fumarate 1.
Figure 4.

Clemastine fumarate antileishmanial activity is correlated with IPCS inhibition. Promastigotes of L. major wild type (WT), sphingolipid-free mutant (ΔLCB2), or the add-back (AB) line were incubated with serial dilutions of clemastine fumarate for 48 h. Then, parasite viability was assessed fluorometrically by the addition of resazurin solution to the culture. Clemastine fumarate EC50 values are the mean of at least three biological replicates with standard error (bars). Statistically significant values in relation to WT (one-way ANOVA, P < 0.05, 95% confidence interval) are shown with stars: **P ≤ 0.01; ***P ≤ 0.001.
To further verify this effect, a study of the effect of clemastine fumarate 1 on the sphingolipid pathway was initiated. Initially, we examined the phospholipid components of the reaction catalyzed by IPCS. Targeted phospholipid analysis by direct infusion ESI-MS/MS provided the initial indication that parasites treated with nonlethal levels of clemastine fumarate compound had a decreased ratio of IPC relative to PI compared to the untreated control samples (Figure S4). This was confirmed by GC-MS inositol quantification of these two phospholipid pools (IPC and PI). Encouraged by these data, we then undertook a broader investigation of the effects of clemastine fumarate 1 (10 μM for 12 h) on the sphingolipid biosynthetic pathway in L. major promastigotes using untargeted LC-MS metabolomics and lipidomics. Consistent with inhibition of IPCS, these analyses confirmed a remarkable increase in the relative abundance of ceramide, alongside other intermediates linked to the pathway (1.15- to 2.17-fold, P < 0.05), demonstrating significant disruption of this metabolic pathway (Figure 5).
Figure 5.

Relative abundance of metabolites (LCMS) in wild type L. major promastigotes treated with clemastine fumarate (10 μM) for 12 h. Treated (WTTx) and untreated parasites (WTCx) (x-axis) were analyzed in four biological replicates. Multivariate data analysis was performed with PiMP pipeline.40 The Benjamini-Hochberg procedure adjusted raw P-values (q-values) < 0.05 for ANOVA. Differences among samples (WTTx vs WTCx) were evaluated using one-way ANOVA with the Dunnett’s multiple comparison test P-value < 0.05 with the Prism software version 8. Statistically significant values (P < 0.05, 95% confidence interval) are shown with stars: ns, nonsignificant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). Changes in other ceramide species are provided in Table S2.
Selection for Clemastine Fumarate Resistance and Cross Resistance against Other Antileishmanials
Two individual clones of the L. major promastigote form of the parasites (cl.C and cl.D) selected in vitro for resistance to clemastine fumarate 1 showed EC50 values 2.5- to 3-fold greater than the parental wild type (Figures 6 and S3). Importantly, no evidence for cross-resistance against the front-line antileishmanials (pentamidine, amphotericin B, miltefosine, paromomycin, and the antimonials) was observed in these parasite populations (Figure 6), indicating a different mode-of-action or mode-of-resistance. Interestingly, the selection for resistance to clemastine fumarate 1in vitro generated mutants with variants (SNPs) within the coding sequence (CDS) and the untranslated region (UTRs) in eight genes of the sphingolipid pathway in L. major (Table 1 and Figure S5), consistent with the changes observed in the abundance of intermediates involved in sphingolipid metabolism after exposure to clemastine fumarate 1 (Figure 6). Clone cl.C showed the higher number of SNPs (18 compared to 12 in cl.D), including the only four found within the CDS of the genes (Figure 6). The majority of mutations were found in the UTRs (14 and 12, respectively), perhaps indicating that these affect regulatory elements such as noncoding RNAs present in these regions in Leishmania.41 Notably, in Leishmania and other kinetoplastids, UTRs are involved in transcription and gene expression;42,43 therefore, these mutations could influence enzyme content. The total number of variants identified is provided (Table S3), and the high number of SNPs found across the genomes of the selected clones (cl.C and cl.D compared with parental wild type) complicates our interpretation of these polymorphisms. However, changes in copy number (CNVs) were also observed in a number of chromosomes from cl.C and cl.D. Of those encoding proteins involved in sphingolipid biosynthesis, the copy numbers of chromosomes 31 and 34 were increased in clone cl.D, while a reduction of chromosome 8 was observed for both clones (Table 1 and Figure S6). Table S4 provides the CNV values in all chromosomes in both clones. Chromosomes 31 and 34 harbor the genes encoding ceramide synthase (CerS) and serine palmitoyltransferase (SPT) subunit 2 (LCB2), respectively, with amplification perhaps influencing the expression level, sphingolipid content, and sensitivity to clemastine fumarate 1. Chromosome 8, whose decrease in copy number was common to both clones, harbors inositol phosphosphingolipid phospholipase C (ISCL). Interestingly, the loss of ISCL has previously been demonstrated to lead to the accumulation of IPC, indicating that a reduction in copy number may be directly linked to clemastine fumarate 1 resistance in both selected clones.39
Figure 6.

Cross resistance (EC50) against known antileishmanials of cloned clemastine fumarate-resistant L. major promastigotes. CLE, clemastine fumarate; PENT, pentamidine; AmB, amphotericin B; MF, miltefosine; PAR, paromomycin; PAT, potassium antimony tartrate. EC50 values are the mean of at least three biological replicates with standard error (bars). Statistically significant values (one-way ANOVAs with the Dunnett’s multiple comparison test, P-value < 0.05, were processed with Prism software version 8, P < 0.05, 95% confidence interval) are shown with stars: ns, nonsignificant; *P ≤ 0.05; **P ≤ 0.01.
Table 1. Genomic Changes Identified in Genes Encoding Sphingolipid Biosynthetic Enzymes in the Clemastine Fumarate Resistance (CleR) to the Leishmania major Promastigote Form of the Parasitesa.
| genes
encoding sphingolipid synthetic enzymes in L. major |
single
nucleotide polymorphisms (SNPs) |
copy
number variations (CNVs) |
||||||
|---|---|---|---|---|---|---|---|---|
| num. | gene ID | abbreviation | cl.C | cl.D | total | Chr. | cl.C | cl.D |
| 1 | LmjF.34.3740 | SPT | 1 | 3 | 4 | 34 | increase | |
| 2 | LmjF.35.0320 | SPT-like | 3 | 3 | 35 | |||
| 3 | LmjF.35.0330 | 3-KSR | 1 | 1 | 35 | |||
| 4 | LmjF.31.1780 | CerS | 31 | increase | ||||
| 5 | LmjF.26.1670/80 | CerD | 1 | 1 | 2 | 26 | ||
| 6 | Nd | CerAse | ||||||
| 7 | LmjF.35.4990 | IPCS | 2 | 2 | 35 | |||
| 8 | LmjF.08.0200 | ISCL | 2 | 2 | 8 | decrease | decrease | |
| 9 | Nd | CerK | ||||||
| 10 | Nd | CerP | ||||||
| 11 | LmjF.26.0710 | SK | (2*) | 2 | 4 | 26 | ||
| 12 | LmjF.32.2290 | S1PAse | 2 (1*) | 3 | 6 | 32 | ||
| 13 | LmjF.30.2350 | S1PLY | 30 | |||||
| 14 | LmjF.18.0440 | PAF | 3 (1*) | 2 | 6 | 18 | ||
| total | 18 | 12 | 30 | |||||
| Chrπ | ||||||||
| 33 | increase | |||||||
| 11 | increase | |||||||
Missense (nonsynonymous) single nucleotide polymorphisms (SNPs) are indicated with a star (*); other SNPs correspond to intergenic region variants or UTRs. Genes IDs are from the TritrypDB (https://tritrypdb.org/). SPT: serine palmitoyltransferase; 3-KSR: 3-dehydrosphinganine reductase; CerS: ceramide synthase; CerD: sphingolipid 4-desaturase; CerA: ceramidase; IPCS: inositol phosphorylceramide synthase; ISCL: inositol phosphosphingolipid phospholipase C; CerK: ceramide kinase; CerP: ceramide phosphatase; SK: sphingosine kinase; S1PAse: sphingosine-1-phosphate phosphatase; S1PLY: sphingosine 1-phosphate lyase; PAF: phosphatidic acid phosphatase. Nd: nondetermined. Copy number variations (CNVs) are indicated. πChromosomes are those not known to contain genes encoding sphingolipid biosynthetic enzymes.
Clemastine Fumarate Stimulates a Macrophage Response to Leishmania Infection
The host response to Leishmania infection is driven by macrophage activity that proceeds via two major mechanisms involving either the generation of reactive oxygen and nitrogen species (ROS/RNS), which combine with nitric oxide (NO) to generate peroxynitrites, powerful oxidants capable of killing microorganisms,44 or the formation of the acidified phagolysosome, which rapidly kills and degrades the pathogen.45Leishmania spp. amastigotes have adapted to survive in macrophage phagolysosomes by disrupting the generation of ROS. However, in order to differentiate into amastigotes, the promastigotes must first survive antimicrobial activities of the macrophages. A mechanism used to achieve this involves the inhibition of phagolysosome biogenesis.46 This leads to a decrease in NADPH oxidase complex activity and reduces exposure to ROS. In addition, the inhibition of phagosome maturation excludes the vesicular proto-ATPase (v-ATPase), required for phagosomal acidification and, consequently, microbicidal properties of the phagosomes are impaired. Clemastine fumarate has been reported to affect host immune responses, and it was therefore of interest to see if these effects were contributing to the observed antiparasitic effect.47
It was postulated that clemastine fumarate 1 could increase NO synthesis and, consequently, peroxynitrite levels, leading to enhanced parasite death,48 so uninfected macrophages were treated with serial dilutions of clemastine fumarate 1 and incubated for 48 h. Subsequently, supernatants were treated with the Griess reagent, and the absorbance at 570 nm was measured to quantify nitrite formation. However, no significant effect was observed, suggesting that the enhancement of NO levels is not a contributing factor to the observed antiparasitic activity of clemastine fumarate 1 (Figure S7).
Clemastine fumarate 1 has been shown to stimulate the P2X7 receptor (P2X7R) which, upon activation by extracellular ATP, leads to the secretion of pro-inflammatory cytokines and the production of ROS/RNS.49 Significantly, macrophages infected with L. amazonensis display an enhanced expression of P2X7R and are significantly more responsive to extracellular ATP.50,51 Collectively, this suggested that the enhancement of P2X7R, resulting from the treatment with clemastine fumarate 1, could contribute to the elimination of the parasites.52 To explore this idea, antiamastigote assays were performed with macrophages from C57BL/6 wild type mice and from C57BL/6 mice in which P2X7R had been knocked out (P2X7R KO) (Figure 7). This study revealed that clemastine fumarate 1 is >4 times more active against infected wild type cells when a low concentration of extracellular ATP (100 μM) is added. In contrast, a similar experiment with P2X7R KO macrophages did not show any effect on the addition of extracellular ATP. This suggested that wild type infected cells treated with clemastine fumarate 1 have an enhanced P2X7R activity, which is responsive to low concentrations of extracellular ATP with a downstream impact on intracellular parasite viability.
Figure 7.

Host cell antileishmanial activity modulation by clemastine fumarate. BMDM from C57BL/6 mice wild type (WT) or knockout for P2X7 receptor (KO) were infected with promastigotes of L. amazonensis. After 24 h of infection, cells were treated with serial dilutions of clemastine fumarate in the presence or absence of ATP (100 μM) for 48 h at 37 °C. Cells were stained with Giemsa stain, and the number of intracellular amastigotes in 200 macrophages was quantified by light microscopy. Clemastine fumarate EC50 values are the mean of one biological replicate with standard error (bars). Statistically significant values in relation to WT (one-way ANOVA, P < 0.05, 95% confidence interval) are shown with stars: ns, nonsignificant; ***P ≤ 0.001).
Clemastine Fumarate Is an Effective Antileishmanial Agent In Vivo
Once we demonstrated that clemastine fumarate 1 was effective against Leishmania promastigotes and intramacrophage amastigotes, with a good safety profile against host cell macrophages, it was necessary to confirm the activity in an animal model. On the basis of the reported safety profile for clemastine fumarate in a mouse and the high activity against L. amazonensis, we opted to explore in vivo efficacy using BALB/c mice infected with L. amazonensis expressing green fluorescent protein (GFP). For this CL model, using the classical antimonial drug glucantime as a reference, animals were divided into five groups comprising clemastine fumarate 1 (oral, 134 mg kg–1, five times a week); clemastine fumarate 1 (intraperitoneal [IP], 11.65 mg kg–1, twice a week); clemastine fumarate 1 (intralesional [IL], 1.17 mg kg–1, twice a week); glucantime (IL, 1.30 g kg–1, twice a week); untreated. Mice were treated for 28 days, and lesion size was measured regularly to monitor the progression of the disease. On day 41, the animals were euthanized and parasite load was quantified using the limiting dilution assay (LDA). There was a significant reduction in lesion size in all clemastine fumarate-treated mice at day 35 postinfection when compared with the untreated control group (Figure 8A,C). All clemastine fumarate 1 treatments led to a statistically significant reduction in parasite burden as determined by LDA (Figure 8B). Pleasingly, clemastine fumarate administered via IP decreased the parasite burden to a greater extent than IL glucantime, even using lower concentrations of active compound (5.83 mM clemastine fumarate versus 819 mM glucantime [221 mM pentavalent antimonial]).
Figure 8.

Efficacy of clemastine fumarate in L. amazonensis-infected mice. BALB/c mice were infected with 2 × 106L. amazonensis GFP promastigotes in the right ear. Seven days after the infection, mice were treated for 28 days with clemastine fumarate by the oral route (134 mg kg–1, five times a week), the intraperitoneal route (IP; 11.65 mg kg–1, twice a week), or intralesional injections (IL; 1.17 mg kg–1, twice a week); glucantime by the intraperitoneal route (IP; 1.30 g kg–1, twice a week); or left without treatment (UNT). Mice were euthanized seven days after the end of treatment. (A) Lesion thickness throughout treatment. (B) Ear parasite loads on day 41 post-infection. (C) Representative photographs of the infected ear for each group. Data are the mean of two biological replicates (n = 5) with the standard deviation. Statistically significant values in relation to the untreated group (one-way ANOVA or two-way ANOVA, P < 0.05, 95% confidence interval) are shown with stars: *P < 0.05, **P < 0.01, and ***P < 0.001.
Discussion
Given the cost for discovering and developing a NCE drug is estimated at ∼$1 billion (US) and development can take up to 20 years,53 it is not surprising that the repurposing of existing medications to treat NTDs has become a popular strategy.54−56 Indeed, many of the currently used antileishmanials were initially developed for other applications; e.g., amphotericin B, a repurposed antifungal treatment;57 paromomycin, a broad spectrum antibiotic;58 miltefosine, a failed antineoplastic agent.59 However, all of the current therapies have difficulties in cost, efficacy, resistance, or the mode-of-administration, and there is a pressing need for new therapies that address these issues and are amenable for the application against the diverse variety of species that cause leishmaniasis.
In this respect, the identification of clemastine fumarate as an effective antileishmanial in an in vivo mouse model of infection is a significant starting point. Clemastine fumarate is an orally available antihistamine used for the treatment of allergic rhinitis, marketed under the brand name Tavegil, which functions as an antagonist for the histamine H1 receptor.60 This over-the-counter drug has a well-established safety profile,36 with the major side effects being sedation, dizziness, disturbed coordination, epigastric distress, and dry mouth.61 As a licensed medicine, clemastine fumarate has proven pharmacokinetic exposure and the high volume of distribution (9.5 ± 3.8 L kg–1) could be contributing to the enhanced perfusion of parasite infected cells. As such, it is perhaps not surprising that clemastine fumarate has been identified in other repurposing studies for NTDs,62−68 including those looking for new antileishmanial applications.69 However, significant antiparasitic activity has not been reported nor has a putative mode-of-action been identified. The discovery of antileishmanial efficacy as a selective inhibitor of the parasite enzyme (IPCS) that catalyzes the formation of the primary complex sphingolipid in Leishmania spp. is exciting given that this activity is not found in the host. Sphingolipid biosynthesis is generally highly conserved across the Eukaryota and has, therefore, been rarely explored in the search for new therapeutic targets.17,18 A number of studies have explored the various enzymes in the pathway with most effort concentrated on the initial enzyme, serine palmitoyltransferase (SPT), as well as the kinases and lyases that generate and degrade the key signaling molecules, sphingosine-1-phosphate and ceramide-1-phosphate.16 However, possibly reflecting the fact that being membrane bound they are more difficult to study, many of the intermediate enzymes in the pathway remain to be fully explored.
The validation of the fungal IPCS (AUR1p) as a drug target by workers at Upjohn was an important landmark that has been largely overlooked by the pharmaceutical industry.21,70−72 Since then, studies have shown that other orthologues may be targeted in mammals (sphingomyelin synthase; SMS), plants, and protozoa.17,18 However, a viable drug acting in this fashion has yet to be identified. The higher efficacy observed for clemastine fumarate 1 against wild type (WT) L. major when compared to the ΔLmjLCB2 mutant, where LmjIPCS is redundant, is consistent with this mode-of-action, an observation further supported by metabolomic and lipidomic data, which showed that the addition of the compound led to enhanced levels of ceramide and its biosynthetic precursors. These data provided chemical validation of IPCS as an antileishmanial target.
In this study, clemastine fumarate was identified from a screen of a diverse set of bioactive compounds (National Institute of Neurological Disorders and Stroke [NINDS] of 1040 compounds) for inhibitors of LmjIPCS. While we identified 14 with micromolar activity against the enzyme, which also had activity against L. major promastigote parasites, the structural diversity within the set challenged the establishment of any significant SAR. However, consistent with a common therapeutic target, it is interesting to note that many of these compounds have some structural similarity, in that they contain both an aryl-rich portion and an amine moiety, connected via an aliphatic linker. Significantly, the polypharmacology apparent in clemastine fumarate, is not unexpected for such basic and relatively lipophilic compounds. One of the 14 compounds was the known antileishmanial drug pentamidine 9, with the data shedding light on a possible new mode-of-action, and four exhibited greater antiparasitic efficacy than this. Interestingly, these four compounds were found to have higher potency against the parasite than the LmjIPCS enzyme in our assays. This may reflect pleiotropism, a possibility supported by the fact that pentamidine 9 also displayed a lower EC50 value against L. major promastigotes than its IC50 value against LmjIPCS and has additional known modes of action including the inhibition of polyamine biosynthesis and uptake73 and DNA binding.74 Importantly, although parasite populations showing low levels of resistance to clemastine fumarate could be generated, no evidence for cross resistance against the front-line antileishmanials (pentamidine, amphotericin B, miltefosine, paromomycin, and the antimonials) was observed. Again, while the low level of resistance could, in part, be attributed to the effects of polypharmacology exhibited by clemastine fumarate, the genomic analyses of these resistant lines showed strong support for changes in sphingolipid metabolism. Interestingly, both clemastine fumarate-resistant clones tested demonstrated statistically significant hypersensitivity to pentamidine 9 (Figure 6). While this suggested that the antileishmanial mode-of-action for clemastine fumarate 1 and pentamidine 9 is not the same, these data are consistent with a common biosynthetic pathway (e.g., sphingolipid metabolism) being affected.
Importantly, given the broad spectrum of Leishmania spp. associated with the disease and the challenges in diagnosing the specific species causing a given infection, the ideal drug will act against multiple species, both Old and New World. As such, clemastine fumarate 1 was tested against the insect stage promastigote form of L. amazonesis, L. donovani, L. infantum, and L. major, with the greatest activity observed against Old World L. donovani and New World L. amazonesis (EC50 against promastigote forms of <20 nM). The subsequent in vivo assessment using a mouse model of L. amazonesis CL demonstrated that IP delivered clemastine fumarate 1 outperformed the currently used antimonial glucantime. Surprisingly, the IL dose of clemastine fumarate was less effective than the IP dose. The reasons for this are not obvious, but potentially, the kinetics of dosing, with IP delivery providing a longer, slower delivery of the active agent, may have accounted for this observation. As a repurposed drug, it is not surprising that clemastine fumarate 1 exhibits other effects, with the IP treated mice showing evidence of drowsiness, a common and known side effect of clemastine fumarate 1. The beneficial immunostimulatory effect observed on P2X7 receptors is further evidence that the in vivo antileishmanial activity observed with clemastine fumarate arises from multiple pathways. Whether this effect is independent or linked to disruption of the IPCS activity and sphingolipid signaling remains to be established. While no significant effect on NO levels could be observed, the possibility for clemastine fumarate to directly interact with other ROS or RNS species cannot be disregarded.
In contrast to IP treatment, the oral treatment was only partially effective in reducing the parasite load and promoting somnolence (Figure S8), although it is pertinent to note that the dose used is approximately 3× lower than the equivalent recommended human oral dose.75 Improved formulation or dosing regimens to increase absorption and achieve blood concentrations equivalent to that found in humans will possibly demonstrate the usefulness of clemastine in oral leishmaniasis treatment.
In conclusion, this study has chemically validated IPCS as a viable target for antileishmanial chemotherapy and identified a safe over-the-counter drug, clemastine fumarate, as a potential candidate for drug repurposing. IPCS inhibition therefore represents a much needed new antileishmanial drug strategy that exploits a distinct difference between the parasite and host and enables the development of better drugs for this important neglected disease. Although complete clearance of parasitaemia was not observed with clemastine fumarate, such a sterile cure in the BALB/C mouse model is seldom achieved,76,77 and the results obtained with this marketed drug are exciting. Current efforts are focused on enhancing the formulation and exploring combination therapies to provide a higher level of in vivo efficacy to enable future human trials.
Methods
Materials
Biological grade materials, solvents, reagents, and media components were purchased from commercial suppliers and used as provided. l-α-Phosphatidylinositol (bovine liver PI; predominant species 1-stearoyl-2-arachidonoyl-sn-glycero-3-phospho-1-myo-inositol sodium salt) was from Avanti Polar Lipids. NBD-C6-ceramide and BODIPY FL C5-ceramide complexed to BSA were from Invitrogen. AG 4-X4 ion-exchange resin was obtained from Bio-Rad. Yeast nitrogen base was from Invitrogen. Protease inhibitor, Complete EDTA-free Protease Inhibitor Cocktail Tablets were from Roche Applied Science. Amino acids drop-out packages were from Clontech. Acid-washed glass beads (212–300 μm) were obtained from Sigma-Aldrich. Clemastine fumarate, amphotericin B, and compounds 2–16 were purchased from Sigma-Aldrich and used as supplied. Glucantime solution (meglumine antimoniate, 300 mg mL–1) was a gift from Sanofi Aventis. The protein assay kit was from BioRad using Coomassie Brilliant Blue G-250.
Reactions and media were prepared using high purity distilled, deionized water. All other solvents used were of the highest purity available commercially. The solutions of the test compounds were made up in DMSO, unless otherwise stated.
Animals and Ethics Statement
All mice used in the experiments were maintained under a controlled temperature, filtered air and water, autoclave bedding, and commercial food at the animal facilities at Federal University of Rio de Janeiro. The animal protocols for this study were approved by the Federal University of Rio de Janeiro Institutional Animal Care and Use Committee under the number 030/17. The research was conducted in compliance with the principles stated in the Guide for the Care and Use of Laboratory Animals (NIH).78 Mice were euthanized by an overdose of isoflurane inhalation followed by cervical dislocation.
Preparation of the LmjIPCS Enriched Microsomal Membrane Fraction
Auxotrophic AUR1 mutant S. cerevisiae was complemented by the expression of the L. major IPCS to create YPH499-HIS-GAL-AUR1 pRS246 LmjIPCS. All the steps required to obtain yeast extraction and enrich the fraction containing microsomal membranes were performed as described by Mina et al.26 These steps were carried out under nonsterile conditions.
Determination of Protein Activity in Units (U)
To standardize the assay and remove variability from the sample preparation, microsome samples were normalized with respect to active enzyme content. Enzymatic activity was measured in enzyme units (U), where 1 U of enzyme is defined as that which converts 1 pmol of substrate per minute under the conditions described (i.e., 1 U = 1 pmol(product) min–1).
A stock solution of NBD-C6-ceramide at a concentration of 100 μM was used to create a standard curve ranging from 0.2 to 80 pmol. The volumes were adjusted to 200 μL with 1 M potassium formate in MeOH, and the fluorescence was measured (Ex460/Em540).
Samples of the washed microsomal membranes were incubated with NBD-C6-ceramide and phosphatidylinositol (PI) under assay conditions, and the product fluorescence was measured. Correlation with the standard curve allowed the activity of the microsome preparation to be calculated in U μL–1. The membranes were adjusted to 1.5 U μL–1 with storage buffer and stored in LoBind Eppendorf tubes at −80 °C.
HPTLC NBD-C6-Ceramide Fluorescence Assay26
Ten mM PI in CHCl3 (1 μL) was dried into a LoBind Eppendorf tube using a vacuum concentrator (Eppendorf Concentrator 5301). To the dried PI, Tris/EDTA/BSA buffer (20 μL) was added, and the solution was mixed by vortexing. The volume was adjusted to 48 μL with distilled H2O, followed by the addition of test compound (0.5 μL) and 100 μM NBD-C6-ceramide in DMSO (1 μL). The reaction was started by the addition of microsomal membranes (1.5 U·μL–1, 0.5 μL), and the mixture was incubated at 30 °C for 25 min. After quenching with CHCl3/MeOH/H2O (10:10:3, 150 μL), the mixture was centrifuged (14 400g, RT, 5 min) to separate phases and the organic layer was removed. The aqueous phase was then re-extracted with 50 μL of chloroform. The combined organic extracts were dried in a rotavapor (Eppendorf Concentrator 5301) and resuspended in 20 μL of chloroform/methanol:/water (10:10:3). 5–10 μL of the reaction products was loaded onto an HTPLC plate, and the lipid components were separated using the solvent system CHCl3/MeOH/0.25% KCl(aq.) (55:45:10). The Rf values for the substrate NBD-C6-ceramide and the product NBD-C6-IPC were 0.96 and 0.57, respectively. Product quantification was carried out using a fluorescence plate reader (Fujifilm, FLA3000) (Ex473/Em520) and AIDA Image Analyzer software (version 3.52).
96-Well Plate NBD-C6-Ceramide Fluorescence Assay
For each 96-well assay plate, in a 10 mL vial, 48 μL of PI (10 mM in CHCl3) was dried using rotary evaporator at RT for 20 min. To the dried PI, 120 μL of NBD-C6-ceramide (200 μM in DMSO), 480 μL of CHAPS (3 mM), and 1680 μL of assay buffer (PO4 buffer 71.9 mM) were added. The mixture was vortexed briefly, and 19 μL was added to each well of a 96-well V-bottom assay plate followed by the addition of 0.8 μL control/test compound (in DMSO); the assay plate was kept at 4 °C.
To another 10 mL vial, 1680 μL of assay buffer, 480 μL of CHAPS, 192 μL of storage buffer, and 48 μL of microsomal material (0.5–0.6 U CHAPS-washed membranes) were mixed and kept on ice. Twenty μL/well of this mixture was added to the assay plates.
The assay was started by moving to an incubator, and the assay plate was incubated at 30 °C for 25 min. The reaction was quenched with 200 μL of MeOH (HPLC grade).
The filter plates were prepared briefly by suspending 10 mg of AG4-X4 resin in 50 mL of EtOH (100%) in a 50 mL falcon tube and were added to the filter plates as 2× additions of 100 μL/well. The filter plates were centrifuged to remove the solvent; 50 μL of formic acid was added to each well, and the wells were incubated for 5 min and then centrifuged to remove the liquid phase. The plates were washed with 100 μL of dH2O and centrifuged to remove the liquid phase.
230 μL of assay reaction mixture was loaded onto the filter plates, and the plates were centrifuged to remove the liquid phase followed by washing 5× with 200 μL of MeOH. The collection plate was removed, and a new plate was used. The product, NBD-C6-IPC, was eluted 4× with 50 μL of MeOH. The collection plate was read at Ex480/Em540 using a fluorescent plate reader (Synergy HT BioTek). The assays were carried out in triplicate; data were analyzed and IC50 values were calculated using sigmoidal regression analysis (GraphPad Prism).
Leishmania Culture
L. major (MHOM/IL/81/Friedlin; FV1 strain [wild type, WT; add-back, AB; serine palmitoyltransferase mutant, ΔLmLCB2]), L. major (MHOM/SA/85/JISH118), L. amazonensis (MHOM/BR/75/Josefa; WT and GFP),79L. infantum (MHOM/MA67ITMAP263), and L. donovani (MHOM/ET/67/HU3/LV9) promastigotes were maintained at 26 °C in Schneider’s insect medium (pH 7) or M199 medium, supplemented with 15% heat-inactivated fetal bovine serum (FBS), 100 μg mL–1 streptomycin, and 100 IU mL–1 penicillin. L. major (FV1) AB and L. amazonensis-GFP promastigotes were cultivated under antibiotic pressure G418 (Gibco) at 40 or 1000 μg mL–1, respectively.
Antipromastigote Assay
L. major WT, AB, ΔLmLCB2 (4 × 105 or 1 × 106 mL–1), L. amazonensis (5 × 105 mL–1), L. donovani (2 × 106 mL–1), and L. infantum (2 × 106 mL–1) promastigotes were incubated in sterile 96-well plates with compounds in triplicate (amphotericin B was used as a positive control and untreated parasites with DMSO, as a negative control) at 26 °C during 48 h for L. major (FV1) and 72 h for L. major (JISH118) and all other Leishmania species. Resazurin solution (10 μL) was then added and the plate, incubated at 26 °C for 4 h prior to measurement using a fluorescence plate reader (Ex555/Em585). EC50 values were calculated using sigmoidal regression analysis (GraphPad Prism).
Isolation of Bone Marrow Derived Macrophages
Bone marrow derived macrophages (BMDMs) were differentiated from bone marrow of BALB/c, C57BL/6, and knock out C57BL/6 mice using L929-cell conditioned medium (LCCM) as a source of macrophage colony-stimulating factor (M-CSF) as described by Marim et al.80
Antiamastigote Intramacrophage Assay
BMDM from BALB/c mice were diluted in RPMI 1640 medium (supplemented with 200 mM l-glutamine, 16.5 mM NaHCO3, and 10% FBS) to a concentration of 4 × 105 mL–1 in a 24-well plate with round coverslips and incubated for 24 h at 37 °C and 5% CO2. Then, macrophages were infected with L. major (JISH118) (7:1) or L. amazonensis (10:1) promastigotes at 34 °C for 4 h, and noninternalized parasites were removed by washing. After 24 h of infection, serial dilutions of the test compounds in medium were added and the slides were incubated at 37 °C and 5% CO2 for 5 days (L. major) or 2 days (L. amazonensis). Amphotericin B and miltefosine were used as controls. At the end, cells were fixed with methanol and stained with Giemsa solution, and the intracelular amastigotes were counted using a light microscope. Results were expressed as the percentage reduction of intracellular amastigotes (compared with untreated control wells). EC50 values were calculated using sigmoidal regression analysis (GraphPad Prism).
Macrophage Cytotoxicity Assay
BMDMs (1 × 106 mL–1) were seeded in sterile 96-well plates in RPMI 1640 medium and incubated for 24 h at 37 °C and 5% CO2. Following removal of the media, serial dilutions of the test compounds in fresh medium (200 μL) were added and the plate was incubated for 24 or 48 h at 37 °C and 5% CO2. Resazurin solution (10 μL) was added to the wells, and cell-viability measurements were carried out using a fluorescence plate reader (Ex555/Em585). Podophyllotoxin or Triton X-100 were used as a reference compound. CC50 values were calculated using sigmoidal regression analysis (GraphPad Prism).
Nitric Oxide Assay
BMDM uninfected macrophages (1 × 106 mL–1, 100 μL well–1) in 96-well plates were incubated for 24 h at 37 °C and 5% CO2. Following removal of the media, serial dilutions of the test compounds in fresh RPMI medium (100 μL) were added and the plate was incubated for 48 h at 37 °C and 5% CO2. The release of nitric oxide was measured in the culture supernatant by the Griess method, as described previously.81 The absorbance was measured at 570 nm, and the nitrite concentration was determined using a standard curve of sodium nitrite (0 to 50 μM). The positive control was macrophages incubated with 1 μg mL–1 LPS (Sigma-Aldrich, Brazil) and 10% conditioned medium of lymphocytes as a source of IFN. Negative controls were cells treated with DMSO and untreated cells.
Antiamastigote Activity in P2X7R–/– Macrophages
BMDMs from wild type (WT) and P2X7R–/– C57BL/6 mice (4 × 105 mL–1) were plated in 24-well plates with round coverslips (BMDM) and incubated for 24 h at 37 °C and 5% CO2. Then, macrophages were infected with L. amazonensis (10:1) promastigotes at 34 °C for 4 h. After 24 h of infection, cells were incubated with serial dilutions of clemastine in the presence or absence of 100 μM ATP in RPMI medium for 48 h at 37 °C and 5% CO2. At the end, cells were fixed with methanol and stained with Giemsa solution, and the intracelular amastigotes were counted using a light microscope. IC50 values were calculated using sigmoidal regression analysis (GraphPad Prism).
Phospholipid ESI-MS/MS Analysis
Leishmania major FV1 cells, both treated with clemastine (3× biological replicates) and untreated controls (3× biological replicates), were extracted in chloroform/methanol/water (2:2:1; V/V)82 and, following phase separation, the organic layer was subjected to ESI-MS/MS analysis. Organic phases were suspended in a 1:1 ratio of 2:1 methanol/chloroform and 6:7:2 acetonitrile/isopropanol/water for direct infusion using an Advion TriVersa NanoMate interface (∼125 nL/min) to deliver samples to an AB Sciex 4000 QTRAP. Samples were analyzed in negative ion mode using a capillary voltage of 1.25 kV. MS/MS scanning (precursors of m/z 241 for inositol-phosphate containing lipids) was performed using nitrogen as the collision gas, with collision energies between 35 and 55 V. Each spectrum encompasses at least 30 repetitive scans. Phospholipid species annotations were determined in reference to previous assignments83 and the LIPID MAPS database.
GC-MS Quantitative Inositol Phospholipid Analysis
Relative amounts of myo-inositol containing lipids (IPC and PI) were quantified, and the means of three separate analyses were determined for IPC and PI inositol quantification. Briefly, lipid samples underwent base hydrolysis: treatment with 500 μL of 50% concentrated ammonia and 50% propan-1-ol (1:1), followed by incubation for at least 5 h at 50 °C. Upon drying under nitrogen and removal of traces of ammonia with 2 rounds of H2O/MeOH evaporation, samples were suspended in chloroform/methanol/water (2:2:1; V/V) in order to separate (organic phase) IPC from inositol-phospho-glycerol derived from hydrolyzed PI species (aqueous phase). These two phases were dried and processed for GC-MS inositol content determination as described previously.83
Selection for Clemastine Resistance
Two independent lines of L. major (FV1) were selected for resistance against clemastine over 5 months by increasing the concentration of drug in the culture medium in a stepwise manner. The parental wild type was also cultured in parallel in the absence of drug. Individual clones were then selected from each resistant line by limiting dilution.
Whole Genome Sequencing
WGS was performed for the two clones to investigate if resistance to clemastine fumarate was associated with genomic changes. Reads of 2 × 75 bp paired-end sequencing were obtained at Glasgow Polyomics using an Illumina NextSeq500 sequencer. The reference genomes and gene annotation of L. major strain Friedlin was obtained from TriTrypDB release 46 (http://tritrypdb.org). Reads were mapped to the reference genomes using BWA-MEM.84 PCR duplicates were removed using GATK (version 4.1.4.1).85 Variants that exist in resistant strains but not in parental wild type strains were identified using MuTect (version 1.1.7)86 and MuTect287 with the default settings. The effects of the variants were predicted using snpEff software.88 Copy ratio alterations were detected using GATK (version 4.1.8.1). Reference genomes were divided into equally sized bins of 1000 base pairs using PreprocessIntervals tool. Read counts in each bin were collected from alignment data using CollectReadCounts tool. Copy ratios of a resistant sample over a matched non-resistant sample were obtained from read counts using CreateReadCountPanelOfNormals and DenoiseReadCounts tools. Genome contigs were segmented using ModelSegments tool from copy ratios of resistant and non-resistant samples. Amplified and deleted segments were identified using CallCopyRatioSegments tool with the default settings. Plots of denoised and segmented copy-ratios were generated using PlotModeledSegments tool. Sequencing raw data was deposited at the European Nucleotide Archive (ENA) under project number PRJNA665266.
LC/MS Metabolomics and Lipidomics Analysis
Metabolite Extraction
Time-to-kill and dose-to-kill were determined (Table S5), and L. major (FV1) promastigotes were treated with high concentrations of clemastine for 12 h or left untreated as the control condition. At the 12 h exposure time point, metabolites were extracted (1 × 107 cells per sample, 4× biological replicates per condition) using an MTBE/methanol/water procedure for biphasic extraction.89 The aqueous phase from each sample was subjected to metabolomics analysis and the organic phase, to lipidomics analysis.
Metabolomic Analysis
Samples were analyzed at Glasgow Polyomics using liquid chromatography–mass spectrometry (LC-MS) as previously described.90 Briefly, hydrophilic interaction liquid chromatography (HILIC) was performed using a Dionex UltiMate 3000 RSLC system (Thermo Fisher Scientific, Hemel Hempstead, UK) using a ZIC-pHILIC column (150 mm × 4.6 mm, 5 μm column, Merck Sequant). The column was maintained at 30 °C, and samples were eluted with a linear gradient using the solvent system: (A) 20 mM ammonium carbonate in water and (B) acetonitrile over 26 min (flow rate of 0.3 mL/min). Samples were injected (10 μL) and were maintained at 5 °C prior to injection. MS analysis was performed using a Thermo Orbitrap Fusion (Thermo Fisher Scientific) operated in polarity switching mode (resolution = 120 000; AGC = 2 × 105; m/z = 70–1000; sheath gas = 40; auxiliary gas = 5; sweep gas = 1; probe temperature = 150 °C; capillary temperature = 325 °C). For positive mode ionization, the source voltage was +4.3 kV; for negative mode ionization, the source voltage was −3.2 kV, and S-lens RF level was 60%. Pooled samples were employed for quality control purposes to assess the reproducibility of the instrument, being analyzed at 5-sample intervals throughout the run.
Lipidomics Analysis
Lipidomics sample analysis at Glasgow Polyomics was conducted in a similar way to metabolomics analysis with adjustments made to coincide with previously described HILIC lipidomics methodology.91 Adjustments were as follows: an ACE unbonded silica column (150 mm × 3.0 mm, 3 μm column, Advanced Chromatography Technologies) was used; samples were eluted using the solvent system: (A) 20% isopropanol and 80% acetonitrile and (B) 20% isopropanol with 20 mM ammonium formate in water over 40 min.
Data Analysis
Both metabolomics and lipidomics data was analyzed using the PiMP analysis pipeline for data filtering and metabolite annotation.40 Except in instances where metabolite retention times matched those of the authentic standards (Level 1 assignments), annotated metabolite identities are assigned at Level 2 in accordance with Metabolomics Standards Initiative directives.92
Antileishmanial Efficacy In Vivo
BALB/c female mice were infected in the right ear with 2 × 106 promastigotes of L. amazonensis-GFP. Seven days after the infection, animals were randomly distributed into 5 groups and treated for 28 days with clemastine by oral the route (134 mg kg–1, five times a week), by the intraperitoneal route (11.65 mg kg–1, twice a week), or by subcutaneous injections (1.17 mg kg–1, twice a week). OraPlus (Perrigo) was used as a vehicle for clemastine oral formulation while PBS was the vehicle for the other routes. Glucantime was given by the intraperitoneal route (1.30 g kg–1, twice a week), as a reference drug, and the untreated group as the negative control. Infected ear thicknesses were measured once or twice a week with a calliper gauge, and the lesion sizes were expressed as the difference between the thickness of the infected and noninfected ear. On day 41 post-infection, the animals were euthanized and the parasite loads in the individual lesions were quantified using a limiting dilution assay (LDA).93 Pictures of each lesion were taken at the end of the treatment.
Behavioral Analyses
BALB/c female mice received a single dose of clemastine by oral (134 mg kg–1) and intraperitoneal (11.65 mg kg–1) routes or each respective vehicle, OraPlus or PBS. Five minutes after treatment, mice exploratory capacity was carried out in an open field arena measuring 0.3 × 0.3 × 0.45 m3, as described previously.94 In each box, one animal was allowed to freely move for 30 min, and the total distance was recorded and quantified using the ANY-maze software (Stoelting Co.). The experiment was performed twice using two animals per group.
Statistical Analysis
Data were analyzed by one-way or two-way ANOVA followed by Dunnett’s multiple comparison test. Data were expressed as arithmetic mean ± standard deviation (SD) values, and samples were considered significantly different when P ≤ 0.05 in a series of at least three independent experiments.
Acknowledgments
We thank CNPq (PVE Grant: 400894/2014-9 to B.R.-B. and P.G.S.), The Royal Society (The Royal Society International Collaboration Awards for Research Professors 2016: IC160044 to B.R.-B. and P.G.S.), MRC (iCASE studentship for C.B.; Grant MR/Mo20118/1 to T.K.S.; MRC Confidence in Concept MC-PC-17157), UKRI Grand Challenges Research Fund (“A Global Network for Neglected Tropical Diseases” grant number MR/P027989/1), Wellcome Trust (104111/Z/14/Z “Wellcome Centre for Integrative Parasitology”), and FAPERJ (Grant: E-26/010.002985/2014 to R.C.-S.) for financial support; Glasgow Polyomics for data acquisition; Professor Simon Croft and Dr. Vanessa Yardley for help with the intramacrophage antiamastigote assays.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00546.
Figure S1, percentage inhibition frequency distribution of LmjIPCS by NINDS set at 20 μM; Figure S2, L. major promastigote growth inhibition of compounds from the NINDS set; Figure S3, selection for clemastine fumarate resistance and EC50 values in L. major promastigotes; Figure S4, phospholipid analysis of L. major in the absence and presence of clemastine fumarate; Figure S5, SNPs identified in SLS genes and changes in the abundance of sphingolipid intermediates in four CleR-lines in L. major (LmjF) promastigotes; Figure S6, copy number variations (CNVs) found in two individual clones of Leishmania major promastigotes; Figure S7, nitric oxide production by macrophage after clemastine fumarate treatment; Figure S8, clemastine fumarate intraperitoneally reduces mice mobility; Table S2, relative abundance of metabolites (LCMS) in wild type L. major promastigotes treated with clemastine fumarate (10 μM) for 12 h; Table S3, polymorphisms identified in two individual clones of Leishmania major promastigotes; Table S5, wild type Leishmania major promastigotes treated with clemastine fumarate (PDF)
Table S1, NINDs library screening data (XLSX)
Table S4, copy number variation (ploidy) in whole genome sequencing data of clemastine-resistant clones of Leishmania major (XLSX)
Author Present Address
¶ J.G.M.M.: The National Horizons Centre, School of Health and Life Sciences, Teesside University, Darlington, DL1 1HG, United Kingdom.
Author Contributions
# J.G.M.M., R.L.C., E.A.-S., and D.O.E. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO (accessed 2020-05-10) http://www.who.int/neglected_diseases/en/.
- WHO (2010) Working to overcome the global impact of neglected tropical diseases: First WHO report on neglected tropical diseases, WHO, Geneva. [Google Scholar]
- Pedrique B.; Strub-Wourgaft N.; Some C.; Olliaro P.; Trouiller P.; Ford N.; Pécoul B.; Bradol J.-H. (2013) The drug and vaccine landscape for neglected diseases (2000–11): a systematic assessment. Lancet Glob. Health 1, e371–379. 10.1016/S2214-109X(13)70078-0. [DOI] [PubMed] [Google Scholar]
- Hotez P. J.; Alvarado M.; Basáñez M.-G.; Bolliger I.; Bourne R.; Boussinesq M.; Brooker S. J.; Brown A. S.; Buckle G.; Budke C. M.; Carabin H.; Coffeng L. E.; Fèvre E. M.; Fürst T.; Halasa Y. A.; Jasrasaria R.; Johns N. E.; Keiser J.; King C. H.; Lozano R.; Murdoch M. E.; O’Hanlon S.; Pion S. D. S.; Pullan R. L.; Ramaiah K. D.; Roberts T.; Shepard D. S.; Smith J. L.; Stolk W. A.; Undurraga E. A.; Utzinger J.; Wang M.; Murray C. J. L.; Naghavi M. (2014) The Global Burden of Disease Study 2010: Interpretation and Implications for the Neglected Tropical Diseases. PLoS Neglected Trop. Dis. 8, e2865. 10.1371/journal.pntd.0002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez P. J.; Bottazzi M. E.; Strych U. (2016) New Vaccines for the World’s Poorest People. Annu. Rev. Med. 67, 405–417. 10.1146/annurev-med-051214-024241. [DOI] [PubMed] [Google Scholar]
- Alvar J.; Vélez I. D.; Bern C.; Herrero M.; Desjeux P.; Cano J.; Jannin J.; Boer M. (2012) Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS One 7, e35671. 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjeux P. (2004) Leishmaniasis: current situation and new perspectives. Comp. Immun. Microbiol. Infect. Dis. 27, 305–318. 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- WHO (accessed 2020-05-10) https://www.who.int/leishmaniasis/disease/clinical_forms_leishmaniases/en/.
- Bailey F.; Eaton J.; Jidda M.; van Brakel W. H.; Addiss D. G.; Molyneux D. H. (2019) Neglected Tropical Diseases and Mental Health: Progress, Partnerships, and Integration. Trends Parasitol. 35, 23–31. 10.1016/j.pt.2018.11.001. [DOI] [PubMed] [Google Scholar]
- Zijlstra E. E.; Alves F.; Rijal S.; Arana B.; Alvar J. (2017) Post-kala-azar dermal leishmaniasis in the Indian subcontinent: A threat to the South-East Asia Region Kala-azar Elimination Programme. PLoS Neglected Trop. Dis. 11, e0005877. 10.1371/journal.pntd.0005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer M.; Argaw D.; Jannin J.; Alvar J. (2011) Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 17, 1471–1477. 10.1111/j.1469-0691.2011.03635.x. [DOI] [PubMed] [Google Scholar]
- Sangshetti J. N.; Kalam Khan F. A.; Kulkarni A. A.; Arote R.; Patil R. H. (2015) Antileishmanial drug discovery: comprehensive review of the last 10 years. RSC Adv. 5, 32376–32415. 10.1039/C5RA02669E. [DOI] [Google Scholar]
- Croft S. L.; Sundar S.; Fairlamb A. H. (2006) Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19, 111–126. 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H.; Riezman H. (2005) The ins and outs of sphingolipid synthesis. Trends Cell Biol. 15, 312–318. 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Gault C. R., Obeid L. M., and Hannun Y. A. (2010) An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. In Sphingolipids as Signaling and Regulatory Molecules. Advances in Experimental Medicine and Biology (Chalfant C., and DelPoeta M., Eds.) Vol. 688, pp 1–23, Springer, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P. J.; Dunn T. M.; Campopiano D. J. (2018) Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep. 35, 921–954. 10.1039/C8NP00019K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina J. G. M.; Denny P. W. (2018) Everybody needs sphingolipids, right! Mining for new drug targets in protozoan sphingolipid biosynthesis. Parasitology 145, 134–147. 10.1017/S0031182017001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S. A.; Mina J. G.; Denny P. W.; Smith T. K. (2012) Sphingolipid and ceramide homeostasis: potential therapeutic targets. Biochem. Res. Int. 2012, 248135. 10.1155/2012/248135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidler S. A.; Radding J. A. (1995) The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 39, 2765–2769. 10.1128/AAC.39.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashida-Okado T.; Ogawa A.; Endo M.; Yasumoto R.; Takesako K.; Kato I. (1996) AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 251, 236–244. 10.1007/BF02172923. [DOI] [PubMed] [Google Scholar]
- Nagiec M. M.; Nagiec E. E.; Baltisberger J. A.; Wells G. B.; Lester R. L.; Dickson R. C. (1997) Sphingolipid synthesis as a target for antifungal drugs - Complementation of the inositol phosphorylceramide synthase defect in strain of Saccharomyces cerevisiae by the AUR1 gene. J. Biol. Chem. 272, 9809–9817. 10.1074/jbc.272.15.9809. [DOI] [PubMed] [Google Scholar]
- Heidler S. A.; Radding J. A. (2000) Inositol phosphoryl transferases from human pathogenic fungi. Biochim. Biophys. Acta, Mol. Basis Dis. 1500, 147–152. 10.1016/S0925-4439(99)00097-6. [DOI] [PubMed] [Google Scholar]
- Georgopapadakou N. H. (2000) Antifungals targeted to sphingolipid synthesis: focus on inositol phosphorylceramide synthase. Expert Opin. Invest. Drugs 9, 1787–1796. 10.1517/13543784.9.8.1787. [DOI] [PubMed] [Google Scholar]
- Denny P. W.; Shams-Eldin H.; Price H. P.; Smith D. F.; Schwarz R. T. (2006) The protozoan inositol phosphorylceramide synthase: a novel drug target that defines a new class of sphingolipid synthase. J. Biol. Chem. 281, 28200–28209. 10.1074/jbc.M600796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina J. G.; Mosely J. A.; Ali H. Z.; Denny P. W.; Steel P. G. (2011) Exploring Leishmania major inositol phosphorylceramide synthase (LmjIPCS): insights into the ceramide binding domain. Org. Biomol. Chem. 9, 1823–1830. 10.1039/c0ob00871k. [DOI] [PubMed] [Google Scholar]
- Mina J. G.; Mosely J. A.; Ali H. Z.; Shams-Eldin H.; Schwarz R. T.; Steel P. G.; Denny P. W. (2010) A plate-based assay system for analyses and screening of the Leishmania major inositol phosphorylceramide synthase. Int. J. Biochem. Cell Biol. 42, 1553–1561. 10.1016/j.biocel.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Norcliffe J. L.; Mina J. G.; Alvarez E.; Cantizani J.; de Dios-Anton F.; Colmenarejo G.; Valle S. G.-D.; Marco M.; Fiandor J. M.; Martin J. J.; Steel P. G.; Denny P. W. (2018) Identifying inhibitors of the Leishmania inositol phosphorylceramide synthase with antiprotozoal activity using a yeast-based assay and ultra-high throughput screening platform. Sci. Rep. 8, 3938. 10.1038/s41598-018-22063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NINDS Custom Collection 2, https://iccb.med.harvard.edu/ninds-custom-collection-2.
- Mikus J.; Steverding D. (2000) A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue(R). Parasitol. Int. 48, 265–269. 10.1016/S1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
- Chadbourne F. L.; Raleigh C.; Ali H. Z.; Denny P. W.; Cobb S. L. (2011) Studies on the antileishmanial properties of the antimicrobial peptides temporin A, B and 1Sa. J. Pept. Sci. 17, 751–755. 10.1002/psc.1398. [DOI] [PubMed] [Google Scholar]
- Waller D. G., and Sampson A. P. (2018) Chemotherapy of Infections. In Medical Pharmacology and Therapeutics, 5th ed., pp 581–629, Elsevier, Oxford. [Google Scholar]
- Sands M.; Kron M. A.; Brown R. B. (1985) PENTAMIDINE - A REVIEW. Clin. Infect. Dis. 7, 625–634. 10.1093/clinids/7.5.625. [DOI] [PubMed] [Google Scholar]
- Lindoso J. A. L.; Costa J. M. L.; Queiroz I. T.; Goto H. (2012) Review of the current treatments for leishmaniases. Res. Rep. Trop Med. 3, 69–77. 10.2147/RRTM.S24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato V. S.; Tuon F. F.; Bacha H. A.; Neto V. A.; Nicodemo A. C. (2008) Mucosal leishmaniasis. Current scenario and prospects for treatment. Acta Trop. 105, 1–9. 10.1016/j.actatropica.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Budavari S., Ed. (1989) Clemastine animal LD50. In The Merck Index - Encyclopedia of Chemicals, Drugs and Biologicals, p 366, Merck and Co. Inc., Rahway, NJ. [Google Scholar]
- McEvoy G. K., Ed. (1992) Clemastine animal toxicity. In American Hospital Formulary Service - Drug Information 92, p 15, American Society of Hospital Pharmacists Inc., Bethesda, MD. [Google Scholar]
- Denny P. W.; Goulding D.; Ferguson M. A. J.; Smith D. F. (2004) Sphingolipid-free Leishmania are defective in membrane trafficking, differentiation and infectivity. Mol. Microbiol. 52, 313–327. 10.1111/j.1365-2958.2003.03975.x. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Hsu F. F.; Scott D. A.; Docampo R.; Turk J.; Beverley S. M. (2005) Leishmania salvage and remodelling of host sphingolipids in amastigote survival and acidocalcisome biogenesis. Mol. Microbiol. 55, 1566–1578. 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang O.; Wilson M. C.; Xu W.; Hsu F.-F.; Turk J.; Kuhlmann F. M.; Wang Y.; Soong L.; Key P.; Beverley S. M.; Zhang K. (2009) Degradation of Host Sphingomyelin Is Essential for Leishmania Virulence. PLoS Pathog. 5, e1000692. 10.1371/journal.ppat.1000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloaguen Y.; Morton F.; Daly R.; Gurden R.; Rogers S.; Wandy J.; Wilson D.; Barrett M.; Burgess K. (2017) PiMP my metabolome: an integrated, web-based tool for LC-MS metabolomics data. Bioinformatics 33, 4007–4009. 10.1093/bioinformatics/btx499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas Castro F.; Ruy P. C.; Nogueira Zeviani K.; Freitas Santos R.; Simoes Toledo J.; Kaysel Cruz A. (2017) Evidence of putative non-coding RNAs from Leishmania untranslated regions. Mol. Biochem. Parasitol. 214, 69–74. 10.1016/j.molbiopara.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Campbell D. A.; Sturm N. R. (2007) The untranslated regions of genes from Trypanosoma cruzi: perspectives for functional characterization of strains and isolates. Memorias Do Instituto Oswaldo Cruz 102, 125–126. 10.1590/S0074-02762007000100022. [DOI] [PubMed] [Google Scholar]
- Saxena A.; Lahav T.; Holland N.; Aggarwal G.; Anupama A.; Huang Y.; Volpin H.; Myler P. J.; Zilberstein D. (2007) Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol. Biochem. Parasitol. 152, 53–65. 10.1016/j.molbiopara.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolo C.; Alvarez M. N.; Radi R. (2014) Peroxynitrite, a potent macrophage- derived oxidizing cytotoxin to combat invading pathogens. BioFactors 40, 215–225. 10.1002/biof.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slauch J. M. (2011) How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 80, 580–583. 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradin N.; Descoteaux A. (2012) Leishmania promastigotes: building a safe niche within macrophages. Front. Cell. Infect. Microbiol. 2, 121. 10.3389/fcimb.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen P.; Weiss A.; Bunter A.; Waeckerle-Men Y.; Fettelschoss A.; Odermatt B.; Kundig T. M. (2011) Clemastine causes immune suppression through inhibition of extracellular signal-regulated kinase-dependent proinflammatory cytokines. J. Allergy Clin. Immunol. 128, 1286–1294. 10.1016/j.jaci.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Coleman J. W. (2001) Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 1, 1397–1406. 10.1016/S1567-5769(01)00086-8. [DOI] [PubMed] [Google Scholar]
- Norenberg W.; Hempel C.; Urban N.; Sobottka H.; Illes P.; Schaefer M. (2011) Clemastine Potentiates the Human P2 × 7 Receptor by Sensitizing It to Lower ATP Concentrations. J. Biol. Chem. 286, 11067–11081. 10.1074/jbc.M110.198879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves S. P.; Torres-Santos E. C.; Marques C.; Figliuolo V. R.; Persechini P. M.; Coutinho-Silva R.; Rossi-Bergmann B. (2009) Modulation of P2X(7) purinergic receptor in macrophages by Leishmania amazonensis and its role in parasite elimination. Microbes Infect. 11, 842–849. 10.1016/j.micinf.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Figliuolo V. R.; Chaves S. P.; Savio L. E. B.; Thorstenberg M. L. P.; Salles E. M.; Takiya C. M.; D’Imperio-Lima M. R.; Guedes H. L. D.; Rossi-Bergmann B.; Coutinho-Silva R. (2017) The role of the P2 × 7 receptor in murine cutaneous leishmaniasis: aspects of inflammation and parasite control. Purinergic Signalling 13, 143–152. 10.1007/s11302-016-9544-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio L. E. B.; Coutinho-Silva R. (2019) Immunomodulatory effects of P2 × 7 receptor in intracellular parasite infections. Curr. Opin. Pharmacol. 47, 53–58. 10.1016/j.coph.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Wouters O. J.; McKee M.; Luyten J. (2020) Research and Development Costs of New Drugs. JAMA, J. Am. Med. Assoc. 324, 518. 10.1001/jama.2020.8651. [DOI] [PubMed] [Google Scholar]
- Andrews K. T.; Fisher G.; Skinner-Adams T. S. (2014) Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol.: Drugs Drug Resist. 4, 95–111. 10.1016/j.ijpddr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubé J. (2012) Drug repurposing and the medicinal chemist. ACS Med. Chem. Lett. 3, 442–444. 10.1021/ml300114c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton R. L.; Rossi-Bergmann B.; Denny P. W.; Steel P. G. (2018) Repurposing as a strategy for the discovery of new anti-leishmanials: the-state-of-the-art. Parasitology 145, 219–236. 10.1017/S0031182017000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halde C.; Newcomer V. D.; Wright E. T.; Sternberg T. H. (1957) An Evaluation of Amphotericin B In Vitro and In Vivo in Mice Against Coccidioides Immitis and Candida Albicans, and Preliminary Observations Concerning the Administration of Amphotericin B to Man. J. Invest. Dermatol. 28, 217–232. 10.1038/jid.1957.26. [DOI] [PubMed] [Google Scholar]
- Vicens Q.; Westhof E. (2001) Crystal Structure of Paromomycin Docked into the Eubacterial Ribosomal Decoding A Site. Structure 9, 647–658. 10.1016/S0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- Dorlo T. P. C.; Balasegaram M.; Beijnen J. H.; de Vries P. J. (2012) Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 67, 2576–2597. 10.1093/jac/dks275. [DOI] [PubMed] [Google Scholar]
- Thomson N. C.; Kerr J. W. (1980) Thorax 35, 428–434. 10.1136/thx.35.6.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant T. G. (2010) Bicyclic modulators of H1 receptors, US20100160271A1.
- Duffy S.; Sykes M. L.; Jones A. J.; Shelper T. B.; Simpson M.; Lang R.; Poulsen S. A.; Sleebs B. E.; Avery V. M. (2017) Screening the Medicines for Malaria Venture Pathogen Box across Multiple Pathogens Reclassifies Starting Points for Open-Source Drug Discovery. Antimicrob. Agents Chemother. 61, e00379-17. 10.1128/AAC.00379-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planer J. D.; Hulverson M. A.; Arif J. A.; Ranade R. M.; Don R.; Buckner F. S. (2014) Synergy Testing of FDA-Approved Drugs Identifies Potent Drug Combinations against Trypanosoma cruzi. PLoS Neglected Trop. Dis. 8, e2977. 10.1371/journal.pntd.0002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. (2015) Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol.: Drugs Drug Resist. 5, 215–228. 10.1016/j.ijpddr.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. (2018) 3-pyridyl inhibitors with novel activity against Trypanosoma cruzi reveal in vitro profiles can aid prediction of putative cytochrome P450 inhibition. Sci. Rep. 8, 4901. 10.1038/s41598-018-22043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M. E.; Naudzius E. M.; Diaz S. J.; Wismar T. W.; Martchenko S. M.; Schulz D. (2020) Identification of clinically approved small molecules that inhibit growth and affect transcript levels of developmentally regulated genes in the African trypanosome. PLoS Neglected Trop. Dis. 14, e0007790. 10.1371/journal.pntd.0007790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G. S.; Lee N.; Ioset J. R.; No J. H. (2017) Evaluation of Parameters Impacting Drug Susceptibility in Intracellular Trypanosoma cruzi Assay Protocols. Slas Discovery 22, 125–134. 10.1177/1087057116673796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M.; Thomas J.; Riley J.; Brough S. J.; Miles T. J.; Gray D. W. (2016) Identification of Trypanocidal Activity for Known Clinical Compounds Using a New Trypanosoma cruzi Hit-Discovery Screening Cascade. PLoS Neglected Trop. Dis. 10, e0004584. 10.1371/journal.pntd.0004584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadele M.; Abay S. M.; Makonnen E.; Hailu A. (2020) Leishmania donovani Growth Inhibitors from Pathogen Box Compounds of Medicine for Malaria Venture. Drug Des. Devel. Ther. 14, 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagiec M. M.; Young C. L.; Zaworski P. G.; Kobayashi S. D. (2003) Yeast sphingolipid bypass mutants as indicators of antifungal agents selectively targeting sphingolipid synthesis. Biochem. Biophys. Res. Commun. 307, 369–374. 10.1016/S0006-291X(03)01164-1. [DOI] [PubMed] [Google Scholar]
- Aeed P. A.; Sperry A. E.; Young C. L.; Nagiec M. M.; Elhammer A. P. (2004) Effect of membrane perturbants on the activity and phase distribution of inositol phosphorylceramide synthase; Development of a novel assay. Biochemistry 43, 8483–8493. 10.1021/bi049141u. [DOI] [PubMed] [Google Scholar]
- Aeed P. A.; Young C. L.; Nagiec M. M.; Elhammer A. P. (2009) Inhibition of Inositol Phosphorylceramide Synthase by the Cyclic Peptide Aureobasidin A. Antimicrob. Agents Chemother. 53, 496–504. 10.1128/AAC.00633-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basselin M.; Badet-Denisot M. A.; Lawrence F.; Robert-Gero M. (1997) Effects of pentamidine on polyamine level and biosynthesis in wild-type, pentamidine-treated, and pentamidine-resistant Leishmania. Exp. Parasitol. 85, 274–282. 10.1006/expr.1996.4131. [DOI] [PubMed] [Google Scholar]
- Cruz A.; de Toledo J.; Falade M.; Terrao M.; Kamchonwongpaisan S.; Kyle D.; Uthaipibull C. (2009) Current treatment and drug discovery against Leishmania spp. and Plasmodium spp.: a review. Curr. Drug Targets 10, 178–192. 10.2174/138945009787581177. [DOI] [PubMed] [Google Scholar]
- Nair A. B.; Jacob S. (2016) A simple practice guide for dose conversion between animals and human. J. Basic. Clin, Pharm. 7, 27–31. 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]; The recommended human oral dose (33 mg kg-21/day, Tavegil tablets), when adjusted using a standard mouse/human dose conversion, equates to a dosing of 2429 mg kg-2011/day in mice. Consequently, the mouse oral dose used here (2134 mg kg-2011/day), although maximized for a 2010.2011 mL intake, is ∼2013× smaller than the standard human dose.
- Valenzuela J. G.; Belkaid Y.; Garfield M. K.; Mendez S.; Kamhawi S.; Rowton E. D.; Sacks D. L.; Ribeiro J. M. C. (2001) Toward a defined anti-Leishmania vaccine targeting vector antigens: Characterization of a protective salivary protein. J. Exp. Med. 194, 331–342. 10.1084/jem.194.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks D.; Noben-Trauth N. (2002) The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2, 845–858. 10.1038/nri933. [DOI] [PubMed] [Google Scholar]
- National Research Council (US) (2011) Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Guide for the Care and Use of Laboratory Animals, 8th ed., National Academies Press (US), Washington, DC. [Google Scholar]
- Costa S. d. S.; de Assis Golim M.; Rossi-Bergmann B.; Costa F. T. M.; Giorgio S. (2011) Use of In Vivo and In Vitro Systems to Select Leishmania amazonensis Expressing Green Fluorescent Protein. Korean J. Parasitol. 49, 357–364. 10.3347/kjp.2011.49.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marim F. M.; Silveira T. N.; Lima D. S.; Zamboni D. S. (2010) A Method for Generation of Bone Marrow-Derived Macrophages from Cryopreserved Mouse Bone Marrow Cells. PLoS One 5, e15263. 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger D. L.; Taintor R. R.; Boockvar K. S.; Hibbs J. B. (1996) Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol. 268, 142–151. 10.1016/S0076-6879(96)68016-1. [DOI] [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. (1959) A rapid method for total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- Fernandez-Prada C.; Vincent I. M.; Brotherton M. C.; Roberts M.; Roy G.; Rivas L.; Leprohon P.; Smith T. K.; Ouellette M. (2016) Different Mutations in a P-type ATPase Transporter in Leishmania Parasites are Associated with Cross-resistance to Two Leading Drugs by Distinct Mechanisms. PLoS Neglected Trop. Dis. 10, e0005171. 10.1371/journal.pntd.0005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv.org, arXiv:1303.3997, https://arxiv.org/abs/1303.3997. [Google Scholar]
- McKenna A.; Hanna M.; Banks E.; Sivachenko A.; Cibulskis K.; Kernytsky A.; Garimella K.; Altshuler D.; Gabriel S.; Daly M.; DePristo M. A. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K.; Lawrence M. S.; Carter S. L.; Sivachenko A.; Jaffe D.; Sougnez C.; Gabriel S.; Meyerson M.; Lander E. S.; Getz G. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D., Sato T., Cibulskis K., Getz G., Stewart C., and Lichtenstein L. (2019) Calling Somatic SNVs and Indels with Mutect2. bioRxiv, https://doi.org/10.1101/861054. [Google Scholar]
- Cingolani P.; Platts A.; Wang L. L.; Coon M.; Nguyen T.; Wang L.; Land S. J.; Lu X. Y.; Ruden D. M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly 6, 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyash V.; Liebisch G.; Kurzchalia T. V.; Shevchenko A.; Schwudke D. (2008) Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 49, 1137–1146. 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamleh A.; Barrett M. P.; Wildridge D.; Burchmore R. J. S.; Scheltema R. A.; Watson D. G. (2008) Metabolomic profiling using Orbitrap Fourier transform mass spectrometry with hydrophilic interaction chromatography: a method with wide applicability to analysis of biomolecules. Rapid Commun. Mass Spectrom. 22, 1912–1918. 10.1002/rcm.3564. [DOI] [PubMed] [Google Scholar]
- Reis A.; Rudnitskaya A.; Blackburn G. J.; Fauzi N. M.; Pitt A. R.; Spickett C. M. (2013) A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J. Lipid Res. 54, 1812–1824. 10.1194/jlr.M034330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner L. W.; Amberg A.; Barrett D.; Beale M. H.; Beger R.; Daykin C. A.; Fan T. W. M.; Fiehn O.; Goodacre R.; Griffin J. L.; Hankemeier T.; Hardy N.; Harnly J.; Higashi R.; Kopka J.; Lane A. N.; Lindon J. C.; Marriott P.; Nicholls A. W.; Reily M. D.; Thaden J. J.; Viant M. R. (2007) Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221. 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima H. C.; Bleyenberg J. A.; Titus R. G. (1997) A simple method for quantifying Leishmania in tissues of infected animals. Parasitol. Today 13, 80–82. 10.1016/S0169-4758(96)40010-2. [DOI] [PubMed] [Google Scholar]
- Figueiredo C. P.; Barros-Aragao F. G. Q.; Neris R. L. S.; Frost P. S.; Soares C.; Souza I. N. O.; Zeidler J. D.; Zamberlan D. C.; de Sousa V. L.; Souza A. S.; Guimaraes A. L. A.; Bellio M.; de Souza J. M.; Alves-Leon S. V.; Neves G. A.; Paula-Neto H. A.; Castro N. G.; De Felice F. G.; Assuncao-Miranda I.; Clarke J. R.; Da Poian A. T.; Ferreira S. T. (2019) Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 10, 3890. 10.1038/s41467-019-11866-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.