

Abstract
Chronic obstructive pulmonary disease (COPD) is a debilitating lung disease associated with cigarette smoking. Alterations in local lung and systemic iron regulation are associated with disease progression and pathogenesis. Hepcidin, an iron regulatory peptide hormone, is altered in subjects with COPD, however, the molecular role of hepcidin in COPD pathogenesis remains to be determined. In this study, utilizing a murine model of smoke-induced COPD, we demonstrate that lung and circulating hepcidin levels are inhibited by cigarette smoke. We show that cigarette smoke exposure increases erythropoietin (EPO) and bone marrow-derived erythroferrone (EFRE) and leads to expanded but inefficient erythropoiesis in murine bone marrow, and an increase in ferroportin on alveolar macrophages (AMs). AMs from smokers and subjects with COPD display increased expression of ferroportin, as well as hepcidin. Notably, murine AMs exposed to smoke fail to increase hepcidin in response to gram-negative or gram-positive infection. Loss of hepcidin in vivo results in blunted functional responses of AMs and exaggerated responses to Streptococcus pneumoniae infection.
Introduction
Chronic obstructive pulmonary disease (COPD) is an incurable, life-limiting inflammatory lung disease associated with smoking and the third leading cause of mortality worldwide (1). While the overwhelming risk factor for the development of COPD is tobacco smoking, genetic susceptibility loci involving iron-associated genes have highlighted the role of abnormal iron metabolism in disease pathogenesis (2–6). Specifically, excessive intracellular iron loading in the lung exacerbates inflammation and the severity of disease progression in murine COPD models, and in patients with COPD (7–11). Iron is mostly localized inside lung tissues with remaining metal associated with bronchoalveolar lavage (BAL) protein (12). In a similar manner to other organs, the lung most likely obtains its iron from the pulmonary vasculature. Such a supply of iron to the lung depends on its systemic plasma availability, which is controlled by the hepcidin/ferroportin regulatory axis (13). Hepcidin (encoded by HAMP), a small peptide hormone chiefly produced in the liver, regulates and is in turn regulated by systemic iron levels (14). Hepcidin expression and release by the liver is induced by increased serum iron, by bone morphogenic protein (BMP) signaling (15) and by a number of pro-inflammatory cytokines, including interleukin-6 (16). In general, up-regulation of hepcidin expression results in the inhibition of cellular iron export and reduces the influx of iron into the plasma from stores, as well as blocking further absorption of dietary iron (17). Ferroportin (encoded by the SLC40A1 gene) is a membrane bound transporter that serves as the major exporter of iron from mammalian cells, including macrophages that recycle iron (18). Hepcidin induces ferroportin endocytosis and subsequent lysosomal degradation (13), which in turn decreases iron efflux from exporting cells (13).
Hepcidin, originally described as a cationic antimicrobial peptide (19, 20), is also found in tissues with no recognized role in systemic iron homeostasis, including the heart (21), the brain (22) and the kidney (23). The function of this extra-hepatic hepcidin remains unknown, but one hypothesis is that it is involved in local iron control (24). Both hepcidin and ferroportin have documented expression in the lung (13, 20, 25) and resistance of ferroportin to hepcidin binding, as well as hepcidin deficiency results in pulmonary iron accumulation (13, 20), suggesting the ferroportin/hepcidin axis plays a role in pulmonary iron regulation. Little is known about the production or role of hepcidin in lung disease, although emerging conflicting evidence suggests that the hepcidin/ferroportin axis may be disrupted in patients with COPD. Specifically, serum hepcidin is increased in patients with mild-moderate disease or during an exacerbation event (increase in respiratory symptoms that require medical intervention) (26, 27), but is decreased in severe end-stage patients with COPD (correlating with hypoxemia) (28). Both non-anemic iron deficiency (NAID) (26, 29) and anemia are highly prevalent in patients with COPD (8, 30, 31). Given hepcidin production can be both modulated by the stimulatory effect of inflammation and the suppressive effect of hypoxia (27), the regulation and role of hepcidin synthesis in COPD is highly complex and requires further investigation. In addition, there are few mechanistic studies linking hepcidin, COPD and the regulation of iron metabolism both in the lung and systemically, particularly at the molecular level.
In this study, we demonstrate that hepcidin expression in the lung is suppressed by exposure to cigarette smoke. Notably, we show that cigarette smoke inhibits the amount of hepcidin produced by alveolar macrophages in response to infection. We propose that such blunted hepcidin production by alveolar macrophages limits the response of these macrophages to produce IL-6 (and TNF-α) in vivo and ex vivo thereby limiting an appropriate immune response to respiratory infections such as Streptococcus pneumoniae. These findings may have direct translational relevance to the pathogenesis of COPD given the progressive course of COPD is accelerated by acute exacerbations predominantly associated with pathogens (32) such as S. pneumoniae (33–35). Understanding determinants of bacterial acquisition and persistence in the lower airways in COPD is important and as yet, poorly understood. Given that iron is essential for bacterial growth, respiration, and metabolism (36, 37), and that bacterial species are thought to be responsible for more than 50% of exacerbation events in COPD, including S. pneumoniae, require iron for survival and growth (38–42), our findings may provide a new investigative avenue to help identify COPD patients at high-risk for infection, as well as provide a novel therapeutic approach to treat them.
Materials and Methods
Mice.
Hamp1−/− mice were generated and provided by Dr. Sophie Vaulont, INSERM (43). Hamp1−/− mice were crossed with C57BL/6J wildtype mice (purchased from The Jackson Laboratory strain # 000664) to generate Hamp1+/− mice to facilitate the production of Hamp1−/− and Hamp1+/+ littermate controls. All animal experiments and procedures were approved by the Institutional Animal Care and Use Committee at Weill Cornell Medicine, and were performed in compliance with all relevant ethical regulations.
Cigarette Smoke exposure.
In vivo experiments involving cigarette smoke (CS) exposure, utilized the inhalation exposure apparatus (TE-10) by Teague Enterprises with 3R4F composition cigarettes (University of Kentucky Center for Tobacco Reference Products). Age, and sex matched mice beginning at 6–12 weeks of age were exposed to cigarette smoke (~150 mg/m3), for a minimum of 3 hours per day, 5 days a week, for 8 or 12 weeks.
Streptococcus pneumoniae infection.
Streptococcus pneumoniae (ATCC 6303, ATCC, Manassas, VA) was grown on 10% sheep blood agar plates (BD Biosciences, 221261) overnight. An inoculating loop of bacteria was cultured in Todd Hewitt broth (THB) containing 2% yeast extract for 4–6 hours (Sigma-Aldrich, T1438). Bacterial cultures were centrifuged at 15,000 × g for 1 minute and resuspended in PBS. Colony forming units (CFU) were quantified by dilution of samples in THB and titers were determined by colony counts from 1×103, 105 or 107 dilution. In vivo S. pneumoniae infection: All mice were anesthetized with isoflurane (5% for induction and 2% for maintenance) prior to intranasal instillation with 1×106 CFU of S. pneumoniae (50 μL volume in PBS). In vitro S. pneumoniae infection: Centrifuged and resuspended bacterial cultures (MOI = 1) were added to wells containing either isolated AMs from BAL or BMDMs for 2–4 hours incubation with cells later collected for RNA extraction, media supernatant collected for ELISA and CFU quantification as described above.
Bronchoalveolar lavage and cell counts.
Mice were euthanized via CO2 asphyxiation, intubated with a 20G × 1” catheter (Terumo, SR-OX2025CA) and the lungs lavaged with ice cold PBS (Gibco, 10010–023) supplemented with 0.5 mM ethylenediaminetetraacetic acid (Quality Biological, 351-027-061) in 0.7 ml increments for a total of 2.1 ml for cell counts, a total of 10 ml for RNA isolation and 15 ml for alveolar macrophage cell culture. The BALF was collected from the first 0.7 ml, after centrifugation at 500 × g for 5 minutes at 4°C and used for ELISA and protein measurement using BCA protein assay kit (Thermo Fisher Scientific, 23225). The cell pellets for cell counts were treated with 1 ml of red blood cell lysis buffer (Sigma-Aldrich, R7757–100ml) incubated for 3 minutes and centrifuged at 6000 × g for 3 minutes at 4°C. The cell pellets were then suspended in 0.5 ml PBS and 20 μl used for cell counting using a hemocytometer. To assess blood count recovery, we collected peripheral blood (50 μl) into EDTA-coated capillary tubes (Thermo Fisher Scientific). Differential blood counts were measured using an automated ADVIA 120 Multispecies Hematology Analyzer (Bayer HealthCare) calibrated for murine blood.
RNA extraction and real time quantitative PCR.
Mice were euthanized, bronchoalveolar lavage performed and necessary organs removed, and flash frozen in liquid nitrogen. BAL cells were centrifuged, re-suspended in 0.35 ml TRIzol reagent (Life Technologies, 15596018) and stored in −80°C. RNA isolation was performed as per the manufacturer’s instructions using TRIzol and clean-up performed using RNeasy Mini Kit (Qiagen, 74104) or RNeasy Micro Kit (Qiagen, 74004). RNA concentration was measured using a NanoDrop (DeNovix, DS-11 Spectrophotometer) and converted into cDNA using the High-Capacity RNA-to-cDNA™ Kit (Thermo Fisher Scientific, 4387406) per the manufacturer’s instructions. Taqman® Gene Expression Master Mix (Thermo Fisher Scientific, 4369016) with the applicable primers for Hamp (Mm04231240_s1 or (Hs00221783_m1) for human studies, Slc40a1 (Mm01254822_m1) or (Hs00205888_m1) for human studies, Fam132b (Mm00557748_m1), Gdf15 (Mm00442228_m1), Tfrc (Mm00441941_m1) and Gapdh (Mm99999915_g1), ActinB (Mm02619580_g1) or Gnb2l1 (Hs00272002_m1) for human studies according to the manufacturer’s instructions.
Human macrophage isolation.
Monocyte-derived macrophages (MDM) were generated from monocytes isolated from peripheral blood mononuclear cells from smokers, subjects with COPD and healthy non-smokers. Subjects provided informed consent, and the study was approved by the NRES London-Chelsea Research Ethics committee (study 09/H0801/ 85). MDMs were isolated using a Percoll gradient and adherence technique as previously described (44). Monocytes were cultured in 2 ng/ml GM-CSF (Cat No. 7954-GM-059/CF, Bio-Techne, Abingdon, UK) for 12 days to generate MDMs. Macrophages were seeded at 0.5×106 cells/well in a 24 well plate and lysed and RNA extracted as described above. Lung tissue macrophages were isolated from lung parenchyma as described previously (45). Lung tissue used in this study was assessed as being non-cancerous by the pathologist and obtained from samples during tissue resection for lung cancer or emphysema.
Perls’ Stain.
BAL cells were collected via bronchoalveolar lavage as previously described, for a total of 10 ml, treated with red blood cell lysis buffer for 3 minutes and following incubation and centrifugation, re-suspended in 200 μl PBS. 100 μl of this suspension was used for cytospin slides which were prepared at a centrifugation of 500 × g for 5 minutes while the rest used for total iron measurement. The slides were fixed using Hema 3™ Stat Pack (Fisherbrand, 122–911) and stained with a 4% w/v of potassium ferrocyanide solution (equal volumes of potassium ferrocyanide (Sigma-Aldrich, P3289–100G) and hydrochloric acid solution (1.2 mmol/l hydrochloric acid in distilled water). The slides were incubated for 30 minutes in the working solution, rinsed in distilled water 3× 5 minutes and stained with 200 μl nuclear fast red solution (Sigma-Aldrich, N3020–100ml) for 4 minutes. The slides were washed 1× 5 minutes in distilled water and dehydrated in a series of washes with ethanol and xylene. Once dried, mounting medium (Vector Laboratories, H-5000) was applied, a coverslip placed and images taken using EVOS FL Auto Imaging System (Life Technologies, AMAFD1000).
ELISA.
Commercial ELISAs were used to measure the following analytes in triplicate in homogenates or BAL of lung samples, in serum, plasma or urine from mice following the manufacturer’s instructions: hepcidin, (Intrinsic LifeSciences, HMC-001), erythropoietin (R&D Systems, MEP00B), IL-6 (Invitrogen, 88–7064) and TNF-alpha (R&D Systems, DY410–05).
Total iron measurement.
Tissue samples that were previously flash frozen were collected, weighed and washed with ice cold PBS and digested with 50% nitric acid in distilled water containing 0.1% digitonin at 1/3 w/v (EMD Millipore, 300410–250MG) for 2 hours at 65°C using iron-free polypropylene tubes after equilibration for 1 hour at room temperature. The samples were cooled and 30% w/w hydrogen peroxide (Sigma-Aldrich, H1009) at a 50% volume was added to the sample mixture. The samples were again digested for 1 hour at 95° and diluted with distilled water for measurement of total iron using the PinAAcle™ 900Z Atomic Absorption Spectrometer (PerkinElmer). BALF was collected as previously described and 60 ul used to digest with 40 ul 50% nitric acid in distilled water containing 0.1% digitonin for 2 hours at 65°C. The samples were allowed to cool, centrifuged at 6000 × g for 2 minutes at room temperature and diluted with 0.2% nitric acid.
Non-heme iron measurement.
Previously frozen tissue samples were weighed and digested with Non-Heme Iron (NHI) acid (10% trichloroacetic acid in 3M hydrochloric acid) at a volume of 100 mg of tissue per 1 ml of acid for 20 hours at 65°C. The samples were cooled to room temperature, vortexed and centrifuged at 1,000–2000 rpm for 15 seconds and the supernatant removed for analysis. Samples and iron standards (0–25 μg/ml) were added to 1 ml cuvettes containing BAT buffer (0.2% thioglycolic acid, 0.02% bathophenanthrolinedisulfonic acid disodium salt hydrate (Sigma-Aldrich 146617) and saturated sodium acetate trihydrate (Sigma-Aldrich, S7670) in distilled water. The absorbance values were then determined using a spectrophotometer at 535 nm and compared against the standard curve.
Immunophenotypic analysis of erythroid precursors by flow cytometry.
Mice were euthanized by CO2 asphyxiation and bones were processed and cell suspensions were filtered through a 40-μm mesh and washed in PEB (2 mM EDTA, 0.2% BSA in PBS, pH 7.4). Cell suspensions were stained with APC-conjugated Ter119 (Biolegend, 116212), APC/Cy7 conjugated CD44 (Biolegend, 103028) Hoechst (Invitrogen, H3570), and in the indicated experiments with a modified lineage cocktail including CD3 (Biolegend, Cat#100304), B220 (Biolegend, 103203), CD11b (Biolegend, 101203), and Gr1 (Biolegend, 108404), stained with streptavidin. Apoptosis in mononuclear cell suspensions was assessed using the Annexin V-FITC Apoptosis Kit (BioLegend, 640914) according to manufacturer’s instructions and analyzed via flow cytometry.
Isolation of AM, BMDM, cell culture and preparation of CSE.
Collected BALF of 15 ml was centrifuged at 500 × g for 5 min at 4°C, and the pellet treated with red blood cell lysis buffer for 3 minutes. The samples were centrifuged at 6000 × g for 5 minutes at 4°C and re-suspended in full RPMI 1640 (Gibco, 11875–093) and added to a 12-well culture dish (1 mouse per well). The cells were left to adhere overnight and treated with 2% cigarette smoke extract (CSE) for 18 hours followed by 100 ng/ml of lipopolysaccharide (Invitrogen, tlrl-eblps) for 6 hours (CSE treatment of 24 hours total). Primary AM media was removed (centrifuged and supernatants stored at −80°C) and remaining cells gently washed with PBS. Primary AMs were removed using a cell lifter with 0.35 ml TRIzol and stored in −80°C for RNA isolation. BMDMs were extracted from the femur and tibia of mice using a 25G needle attached to a 12cc syringe filled with full DMEM in sterile conditions. The cell aggregates were broken up using an 18G needle and afterwards strained through a 70 μM cell strainer. The cells were centrifuged at 1000 rpm for 10 minutes and plated at a concentration of 1 × 106 cells/ml. The medium was changed on the 3rd or 4th day and on the 7th treated with LPS. CSE was prepared using un-supplemented media (either DMEM or RPMI) with a Variable Flow Chemical Transfer Pump (VWR, 23609170) and Reference Cigarettes as previously described (46, 47). Briefly, a peristaltic pump was used to bubble mainstream smoke from five 3R4F cigarettes with filters removed through 50 ml DMEM/RPMI. Each cigarette was smoked within 6 minutes until approximately 17 mm remained. The extract was filter sterilized, stored at −80°C, and used immediately upon thawing. The CSE generated in this fashion was considered 100% strength and was diluted in complete DMEM/RPMI media for cell treatment. Cells pre-treated with CSE (2% for 18 hours, CSE treatment of 24 hours total) were cultured with LPS (100 ng/ml) for 6 hours or S. pneumoniae 0.5 ×106 CFU for 2–4 hours. For experiments using deferoxamine (DFO) (Sigma Aldrich, D9533), cells were pre-treated with 100 μm DFO for 8 hours. For those assessing phagocytosis, the cells were incubated at either 4°C or 37°C, after addition of CSE with shorter CSE treatment times (4 and 8 hours). Experiments using Chelex 100 (Sigma Aldrich, C7901) involved treating media overnight with 10% w/v of the chelating material at 4°C followed by removal of the supernatant for immediate use.
Results
The lung ferroportin/hepcidin axis is altered in a murine model of CS-induced COPD
To characterize the functional role of the hepcidin/ferroportin axis in the pathogenesis of COPD, we utilized a well-established experimental model of cigarette smoke (CS)-induced COPD (48–50). In this model, mice exposed to CS for 6–8 weeks display increased infiltration of AMs, increased markers of lung injury and impairment of mucociliary clearance mechanisms (48–50), indicative of altered inflammation and airway dysfunction. Longer exposure to CS (6 months) results in higher mean alveolar chord length, air space diameter and increased thickness of the small airways; all established indices of experimental emphysema (47, 50–52). In this study, mice exposed to 2–6 months of CS displayed a time-dependent loss of hepcidin transcript in whole lung homogenates, with an increase in ferroportin transcript expression (Figure 1A–B). Consistent with a loss of hepcidin expression and increased ferroportin expression, mice exposed to CS had higher non-heme iron levels in whole lung homogenates, as described previously (49), suggestive of pulmonary iron-overload (Figure 1C).
Figure 1. The lung ferroportin/hepcidin axis is altered in a murine model of CS-induced COPD.
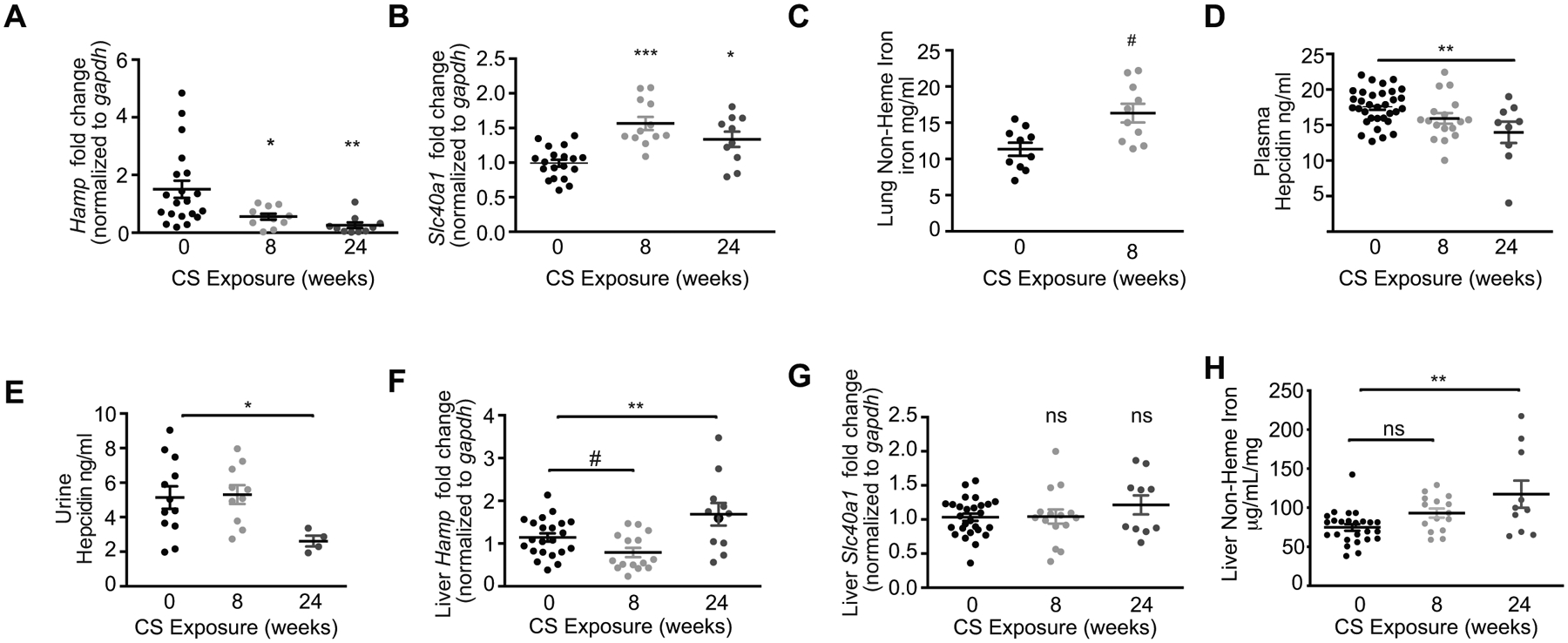
(a) Hamp mRNA (0 weeks n = 20, 8 weeks n = 11, 24 weeks n = 10), (b) Slc40a1 mRNA (0 weeks n = 19, 8 weeks n = 12, and 24 weeks n = 10) in whole lung (including BAL cells) homogenates (post-bronchoalveolar lavaged) of mice exposed to room air (RA) or cigarette smoke (CS) for the indicated times. (c) Non-heme iron (mg/ml) in lung tissue of mice exposed to RA (0 weeks) or CS (8 weeks) (n = 10 per group). (d) Hepcidin expression quantified by ELISA in plasma (0 weeks n = 33, 8 weeks n = 17, 24 weeks n = 9) and (e) urine (0 weeks n = 12, 8 weeks n = 10, 24 weeks n = 4) of mice exposed to RA or CS (8 weeks and 24 weeks). (f) Hamp mRNA (0 weeks n = 22, 8 weeks n = 15, 24 weeks n = 11) and (g) Slc40a1 mRNA (0 weeks, n = 27, 8 weeks n = 15, 24 weeks n = 10) and (h) non-heme iron (0 weeks n = 25, 8 weeks n = 15, 24 weeks n = 10) levels in liver tissue homogenates of mice exposed to RA or CS (8 and 24 weeks). All data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005 by one-way ANOVA with Bonferroni correction. # p<0.05 by unpaired t-test. n.s., not significant.
Systemic and hepatic hepcidin are suppressed in a murine model of CS-induced COPD
To evaluate if changes in hepcidin and ferroportin expression in the lungs of our experimental COPD model was a ubiquitous systemic response to smoke, we assessed hepatic and systemic hepcidin regulation in response to CS exposure. Both plasma and urine levels of hepcidin decreased in response to 8 weeks CS exposure (Figure 1D–E). While plasma and urine hepcidin levels were suppressed after 6 months chronic CS exposure; hepatic hepcidin levels appeared to decrease at the 8-week time point followed by an increase in response to chronic 6-months exposure (Figure 1F). Hepatic ferroportin expression did not change in response to CS (Figure 1G), despite increases in hepatic non-heme iron levels (Figure 1H). Typically, reduced hepcidin expression occurs as a mechanism to enhance iron absorption and recycling to increase body iron stores and availability in the plasma. Consistently, we observed that exposure to 8 weeks cigarette smoke significantly increased hematocrit, hemoglobin, red blood cell counts and plasma iron levels (Supplementary Figure 1A–D).
Mice exposed to cigarette smoke have expanded but iron deficient erythropoietic activity
Hepcidin production is inhibited in physiologic conditions that enhance iron requirements, such as iron deficiency anemia, tissue hypoxia (which increases iron availability for use in erythropoiesis) and/or stress erythropoiesis and dyserythropoiesis (53). To understand if the loss of hepcidin expression in our model involved alterations in erythropoiesis, we evaluated the role of CS on erythroid development in the bone marrow. The cell-surface markers CD71/CD44 and Ter119 were used to characterize erythroblast populations in freshly isolated bone marrow from smoke and room air exposed mice. After 8 weeks of smoke, the number of Ter119 positive cells increased (although not significant) in the marrow with increases across all erythroid progenitors, with particular significant increases in the EIV/EV.44 populations suggestive of expanded erythropoiesis (Figure 2A–B).
Figure 2. Mice exposed to cigarette smoke have expanded but iron deficient erythropoietic activity.
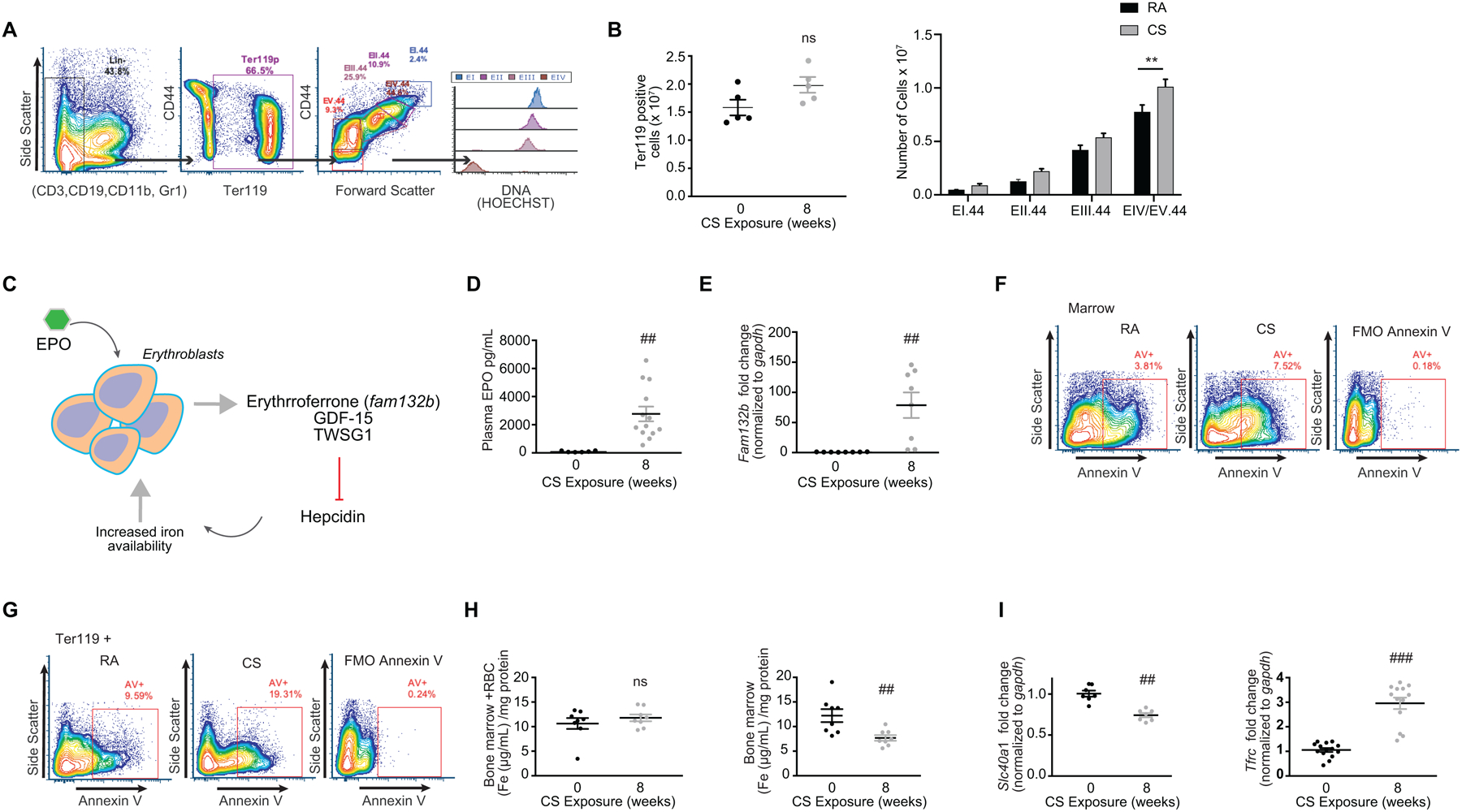
(a) Flow cytometry gating strategy using lineage negative erythroid LinE− (CD3, B220, Gr1, CD11b), CD44, Ter119, Hoechst (DNA) to characterize erythroid maturation with quantification of (b) Ter119 positive cells (left panel) and number of cells per erythropoietic development stage (right panel) in the bone marrow of mice exposed to room air (RA) or cigarette smoke (CS) for 8 weeks. (c) Schematic of erythropoiesis and erythroferrone regulation on hepcidin. (d) EPO levels quantified by ELISA in plasma (0 weeks n = 6, 8 weeks n = 13) and (e) Fam132b mRNA expression (n = 8 per group) in bone marrow of WT mice exposed to RA or CS (8 weeks). (f-g) Representative flow cytometry data showing Annexin V staining in (f) marrow and (g) Ter119+ populations isolated from RA and CS exposed mice (8 weeks). FMO denotes fluorescence minus one. (h) Total iron levels in the bone marrow with red blood cells (left panel) and without red blood cells (right panel) (n = 8 per group). (i) Slc40a1 mRNA (left panel) (n = 7 per group) and Tfrc mRNA (right panel) (0 weeks n = 14, 8 weeks n = 13) levels in the bone marrow of mice exposed to either RA or CS (8 weeks). All data are mean ± s.e.m. **P < 0.01, ****P < 0.001 by one-way ANOVA with Tukeys post-hoc test. ##P < 0.01, ###P < 0.001 by student’s unpaired t-test. n.s., not significant.
Increases in erythropoietic activity suppress hepcidin, at least in part via the erythroblast-secreted factors, erythroferrone (encoded by the gene FAM132b) (53), growth differentiation factor 15 (GDF15), (54) or twisted gastrulation (TWSG1) (55) (Figure 2C). Erythroferrone (ERFE), GDF15 and TWSG1 rapidly suppress hepcidin to allow iron acquisition from absorption and storage sites, thus favoring recovery from anemia secondary to blood loss and inflammation (56). We therefore evaluated the role of CS on the production of erythropoietin (EPO), ERFE, GDF15 and TWSG1 in our murine COPD model. EPO levels dramatically increased in both the plasma and kidneys of mice exposed to CS (8 weeks), whereas hepatic EPO levels did not change in response to CS (Figure 2D and Supplementary Figure 1E–F). ERFE (Fam132b) expression significantly increased in murine bone marrow exposed to CS, whereas GDF15 levels decreased (Figure 2E and Supplementary Figure 1G). We did not observe any change in TWSG1 expression levels upon CS exposure (Supplementary Figure 1H). These data suggest that hepcidin may be chronically suppressed by the exuberant, but ineffective erythropoietic activity (ineffective erythropoiesis) induced by cigarette smoke. Consistently, we observed increased expression of annexin V positive cells in the whole bone marrow fraction, as well as in the Ter119 positive population (Figure 2F–G), suggesting increased activation of apoptosis. To investigate if such changes in erythropoiesis were related to changes in iron handling by erythrocytes, we measured the amount of iron in the bone marrow (with or without red blood cells) of mice exposed to CS. While CS exposure did not change the iron content of the total bone marrow including red blood cells, CS significantly reduced iron levels in the non-RBC population, as well as reducing the expression of ferroportin (Figure 2H–I). This data suggests that there may be a high iron requirement by the bone marrow that is not being fulfilled (iron deficient erythropoiesis), confirmed by increases in transferrin receptor gene expression (Figure 2I).
The ferroportin/hepcidin macrophage axis is altered in smokers and in subjects with COPD
The mononuclear phagocyte system (MPS), which encompasses monocytes and macrophages, is a multifunctional system beyond immunity and tissue repair that also serves to control the body’s metabolic needs for iron. In conditions of expanded but inefficient erythropoiesis such as that observed in our murine smoke model, iron-recycling macrophages may be mobilized to recycle senescent red blood cells, detoxify free heme, and acquire iron for de novo hemoglobin synthesis (57). Alveolar macrophages are the most abundant antigen-presenting cells in the lung regulating immune responses including phagocytosis of particulate matter, secretion of cytokines and enzymes, and control of microbes along with other homeostatic functions (17). During infection, inflammatory signals stimulate the production of hepcidin, resulting in the loss of ferroportin and reduction of extracellular iron, which is thought to be a general defense mechanism against invading pathogens (58, 59). AMs from COPD patients and smokers are dysfunctional with altered inflammatory responses (60–63) and lower phagocytic ability (64). This may have severe implications for AM function in the lung and in the pathogenesis of COPD where recurrent bacterial infections are ubiquitous (65). Previous studies have demonstrated increased prevalence of iron-loaded AMs in smokers and subjects with COPD, however the biological relevance of such observations remains to be elucidated (66, 67). In this study we first assessed if hepcidin and ferroportin are altered in lung tissue-derived and monocyte-derived macrophages (MDM) of non-smokers, smokers and subjects with COPD. Smokers had higher hepcidin expression in tissue-derived macrophages but not in MDMs, when compared to healthy controls (Figure 3A and C). Individuals with COPD trended towards higher hepcidin expression in tissue-derived macrophages and in MDMs, when compared to healthy controls, however these changes were not statistically significant (Figure 3A and C). Smokers also had higher ferroportin expression in tissue derived and MDMs when compared to healthy controls (Figure 3B and D). Individuals with COPD had lower ferroportin expression in both tissue derived and MDMs when compared to smokers, however these changes were not statistically significant (Figure 3B and D). The above results demonstrate that hepcidin and ferroportin are increased by smoke, an effect that may be sustained in tissue resident macrophages and MDMs from individuals with COPD.
Figure 3. The ferroportin/hepcidin macrophage axis is altered in smokers and in subjects with COPD.
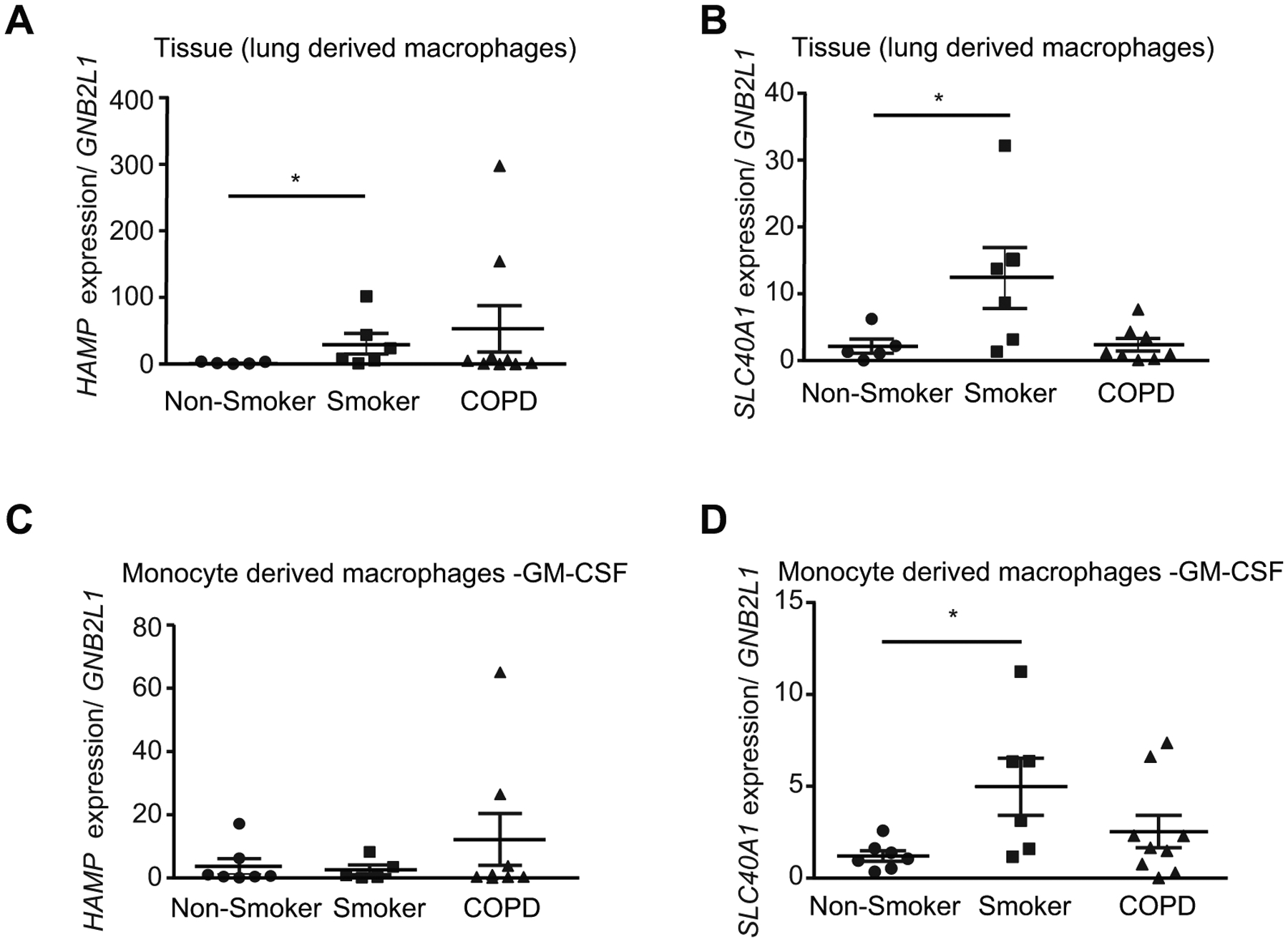
(a) HAMP mRNA (non-smoker n = 5, smoker n = 6, COPD n = 9), (b) SLC40A1 mRNA (non-smoker n = 5, smoker n = 6, COPD n = 8) expression levels in lung-derived macrophages from lung tissue macrophages isolated from lung parenchyma. (c) HAMP mRNA (non-smoker n = 7, smoker n = 5, COPD n = 8) and (d) SLC40A1 mRNA (non-smoker n = 7, smoker n = 6, COPD n = 9) expression levels of monocyte derived macrophages generated from monocytes isolated from peripheral blood mononuclear cells stimulated with granulocyte-macrophage colony stimulating factor (2 ng/ml GM-CSF for 12 days). All data are mean ± s.e.m. *P < 0.05 by Kruskall-Wallis followed by Dunn’s post-hoc test.
Hepcidin production in response to LPS is suppressed by smoke in alveolar macrophages
We next wished to assess if smoke altered the production of hepcidin in response to infection in alveolar macrophages. First, we assessed hepcidin expression in alveolar macrophages isolated from the bronchoalveolar lavage fluid of animals that were exposed to smoke for 8 and 12 weeks. CS exposure alone did not considerably alter hepcidin expression in BAL AMs. Similarly, cigarette smoke extract (CSE, an in vitro model of CS exposure) did not considerably alter hepcidin expression in primary alveolar macrophages or bone marrow derived macrophages (BMDMs) (Figure 4A and Supplementary Figure 2A–C). However, primary AMs exposed to CSE prior to the addition of the gram-negative molecule lipopolysaccharide (LPS) showed blunted Hamp gene expression and blunted Il-6 gene expression and release (Figure 4A and B); BMDMs also had a blunted hepcidin response to LPS with CSE (Supplementary Figure 2C). CS exposure significantly increased ferroportin expression in BAL AMs and in primary AMs or BMDMs treated with CSE (Supplementary Figure 2A–C). LPS treatment reduced ferroportin expression, confirming AMs may try to sequester iron in response to bacterial infection. Treatment of CSE prior to LPS appeared to inhibit the loss of ferroportin in BMDMs but seemed to have little effect in AMs (Figure 4D and Supplementary Figure 2B–C). Notably, the doses and exposure times for CSE used in this study did not alter the viability of AMs or BMDMs (Supplementary Figure 2D). These data suggest that macrophages exposed to cigarette smoke may no longer be able to sequester iron from bacterial pathogens which may lead to increased susceptibility to infection.
Figure 4. The ferroportin/hepcidin macrophage axis is altered in a murine model of CS-induced COPD.
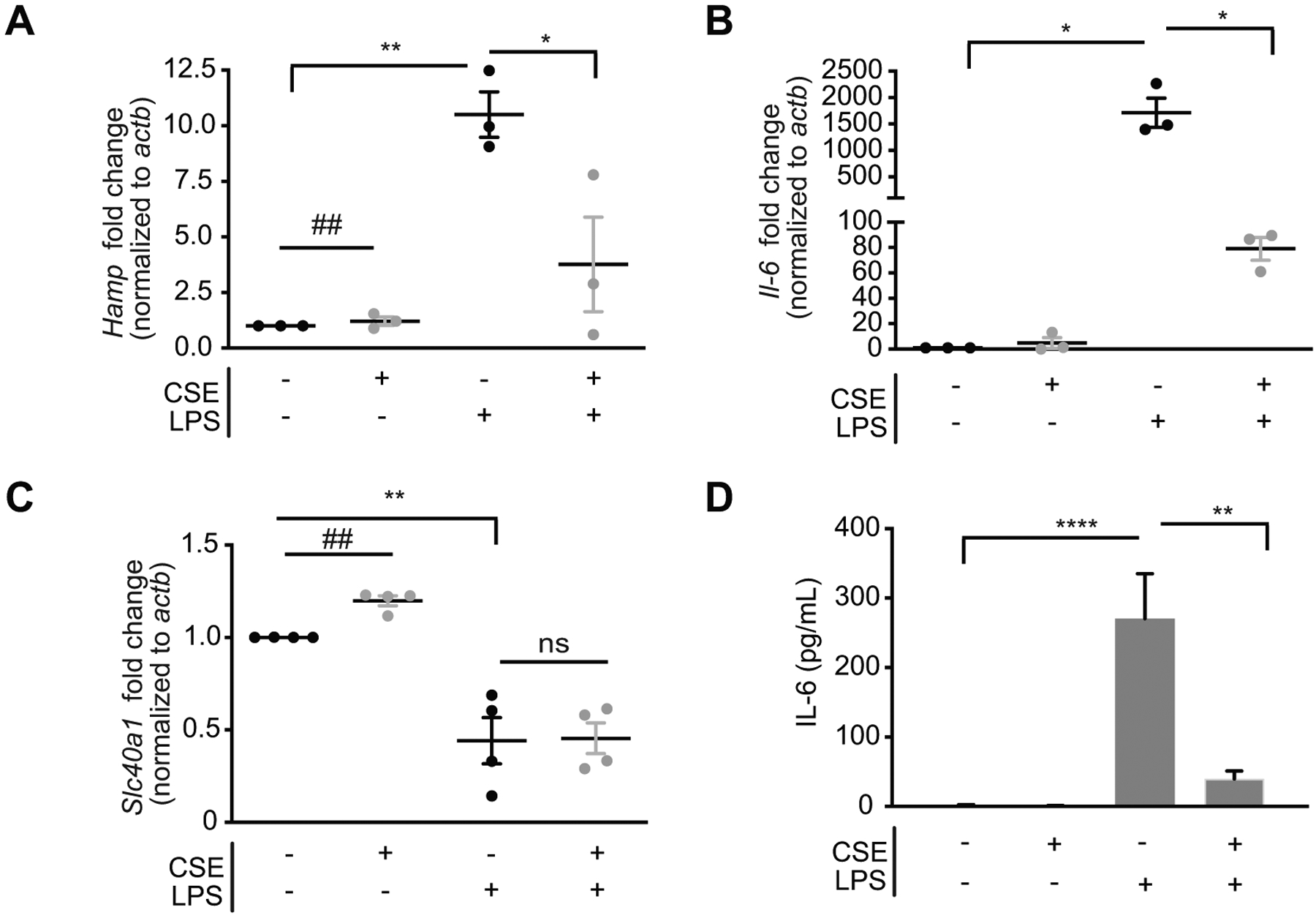
(a) Hamp mRNA, (b) il-6 mRNA (top panel, n = 3 per group) and IL-6 in media supernatant (bottom panel) quantified by ELISA and (c) Slc40a1 mRNA in primary AMs isolated from WT mice, adherence purified and treated with 2% CSE for 18 hours (CSE treatment of 24 hours total) followed by 100 ng/mL LPS (6 hours). Data representative of 3–4 independent experiments. All data are mean ± s.e.m. *P < 0.05, **P < 0.01, ****P < 0.001 by one-way ANOVA with Bonferroni correction. #P < 0.05, ##P < 0.01, ###P < 0.005 by student’s unpaired t-test. n.s., not significant.
Iron uptake by macrophages may occur via phagocytosis of senescent red blood cells or via receptor mediated endocytosis mechanisms involving receptors such as transferrin receptor, divalent metal transporter 1 (DMT1), or zinc transporter ZIP14 (SLC39A14). Both phagocytosis and receptor mediated endocytosis can be non-specifically inhibited by reducing the temperature of cultures to 4°C (68, 69). To assess if phagocytosis or endocytosis alters the expression of hepcidin and ferroportin by smoke, we cultured AMs and BMDMs treated with CSE at 4°C and found that these conditions inhibit the loss of hepcidin and increase in ferroportin expression induced by smoke (+/− LPS), when compared to BMDMs grown at 37°C (Supplementary Figure 2B–C). Removal of iron from CSE using Chelex 100 (a styrene-divinylbenzene co-polymer containing iminodiacetic acid groups that binds to transition metal ions including iron) (Supplementary Figure 2E) had no effect on the expression of hepcidin or ferroportin induced by smoke (Supplementary Figure 2F). Incubating cells with the iron chelator deferoxamine (DFO) trended towards inhibiting the effects of CSE on LPS-induced hepcidin expression and LPS-induced loss of ferroportin expression (Supplementary Figure 2G). These results suggest that cigarette smoke alters the innate response of macrophages to produce hepcidin and lower ferroportin in response to infection, a phenomenon that may involve the regulation of iron via phagocytosis or endocytosis.
Loss of hepcidin in vivo does not alter smoke-induced injury, iron changes or ferroportin expression
To investigate the physiological function of hepcidin in the lung and the physiological response of the lung to smoke-induced injury and inflammation, we utilized a murine model of hepcidin deficiency. As expected, Hamp−/− mice had lower levels of lung and AM Hamp expression and higher levels of lung and AM Slc40A1 expression (Figure 5A–B, Supplementary Figure 2G–H). Hamp−/− mice also had higher non-heme lung iron and higher AM total iron levels as determined by atomic absorption spectroscopy, as well as Perls’ staining (Figure 5C–E). Utilizing a murine model of experimental COPD, exposing Hamp−/− mice to CS for 8 weeks resulted in similar numbers of total BALF cell infiltrates, however, there was a significantly lower number of infiltrating macrophages in the Hamp−/− mice in response to smoke, despite the Hamp−/− mice having higher baseline numbers of macrophages (Figure 5F–G). Interestingly at baseline, Hamp−/− mice had significantly more BAL protein suggestive of increased injury (Figure 5H). In response to smoke Hamp−/− mice appeared to have more BALF protein; however, this was not significant when compared to Hamp+/+ mice exposed to CS (Figure 5H). While iron levels were higher at baseline in the Hamp−/− mice, there was no difference between Hamp+/+ and Hamp−/− mice lung or AM iron content upon smoke exposure (Figure 5C–D). Similarly, ferroportin levels were higher at baseline in the Hamp−/− mice, but there was no difference between ferroportin expression in the Hamp+/+ and Hamp−/− mice upon smoke exposure (Figure 5B). Collectively the above data implies that loss of hepcidin in the lung alters baseline iron and ferroportin levels, but this is not exacerbated by smoke.
Figure 5. Loss of hepcidin in vivo does not alter smoke-induced injury, iron changes or ferroportin expression.
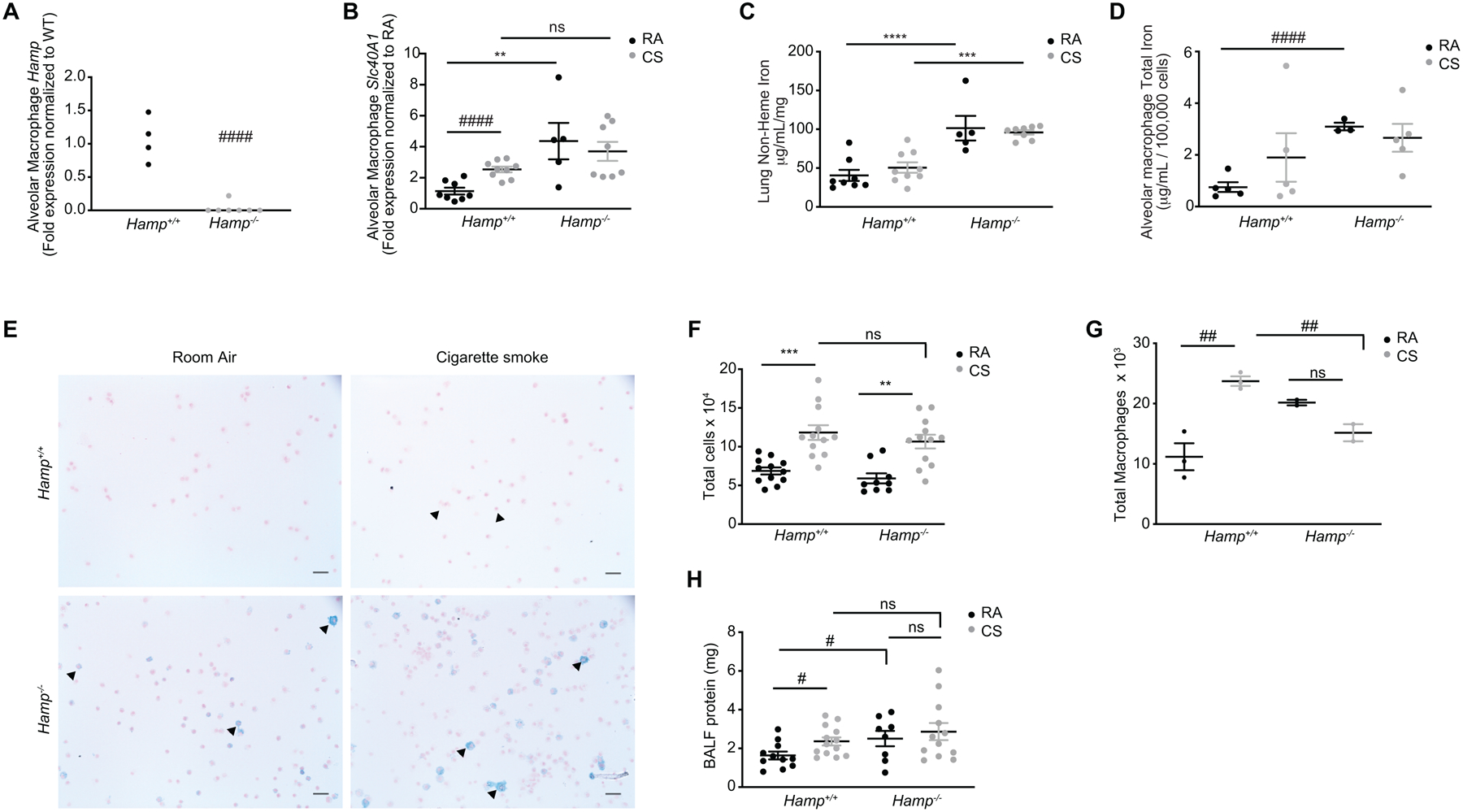
(a) Hamp mRNA expression in BAL AMs of Hamp+/+ (n = 4) and Hamp−/− (n = 7) mice. (b) Slc40a1 mRNA expression in BAL AMs, (c) non-heme iron levels (μg/ml/mg) in whole lung tissue (excluding BAL cells) of Hamp+/+ (RA n = 8, CS n = 9) and Hamp−/− (RA n = 5, CS n = 8) and (d) total iron levels in BAL AMs of Hamp+/+ (RA, CS n = 5) and Hamp−/− (RA n = 3, CS n = 5) mice exposed to RA or CS (8 weeks). (e) Representative Perls’ staining of BAL AMs of Hamp+/+ and Hamp−/− mice exposed to RA or CS (8 weeks) (arrows indicate presence of ferric iron in alveolar macrophages). (f) Total infiltrating leukocyte cell counts of Hamp+/+ (RA, CS n = 12) and Hamp−/− (RA n = 9, CS n = 12) and (g) total macrophage counts calculated from Hematoxylin-Eosin cytospins of BAL cells of Hamp+/+ (n = 3 per group) and Hamp−/− (n = 2 per group) mice exposed to RA and CS (8 weeks). (h) BALF protein (mg) in Hamp+/+ (RA n = 11, CS n = 13) and Hamp−/− (RA n = 8, CS n = 12) of mice exposed to RA or CS (8 weeks). Scale bars, 50 μm. All data are mean ± s.e.m. **P < 0.01, ***P < 0.005, ****P < 0.001 by one-way ANOVA followed by Tukeys post-hoc test. #P < 0.05, ###P < 0.005 ####P < 0.001 by student’s unpaired t-test. n.s., not significant.
Alveolar macrophages and mice deficient in Hamp have altered immune responses to Streptococcus pneumoniae infection
The above results suggest that loss of hepcidin may not alter the injurious response of the lung to cigarette smoke but may alter the number of BALF macrophages infiltrating the lung. To address if loss of hepcidin alters the functional immune response of the lung to smoke, we assessed IL-6 production. Loss of hepcidin resulted in less BALF IL-6 at baseline and in response to smoke (Figure 6A). To assess if hepcidin regulated the functional response of AMs with or without smoke, we isolated primary AMs from Hamp−/− and Hamp+/+ mice and treated them with CSE followed by LPS stimulation. IL-6 was robustly induced by LPS and decreased by CSE in primary Hamp+/+ AMs. AMs isolated from Hamp−/− mice had impaired LPS-induced IL-6 production (Figure 6B–C). Similarly, loss of hepcidin in AMs exacerbated CS-induced inhibition of LPS-associated IL-6 suggesting hepcidin may play a role in the mechanism by which CS inhibits IL-6 responses by LPS and by CS (Figure 6B–C).
Figure 6. Alveolar macrophages and mice deficient in Hamp have altered immune responses to Streptococcus pneumoniae infection.
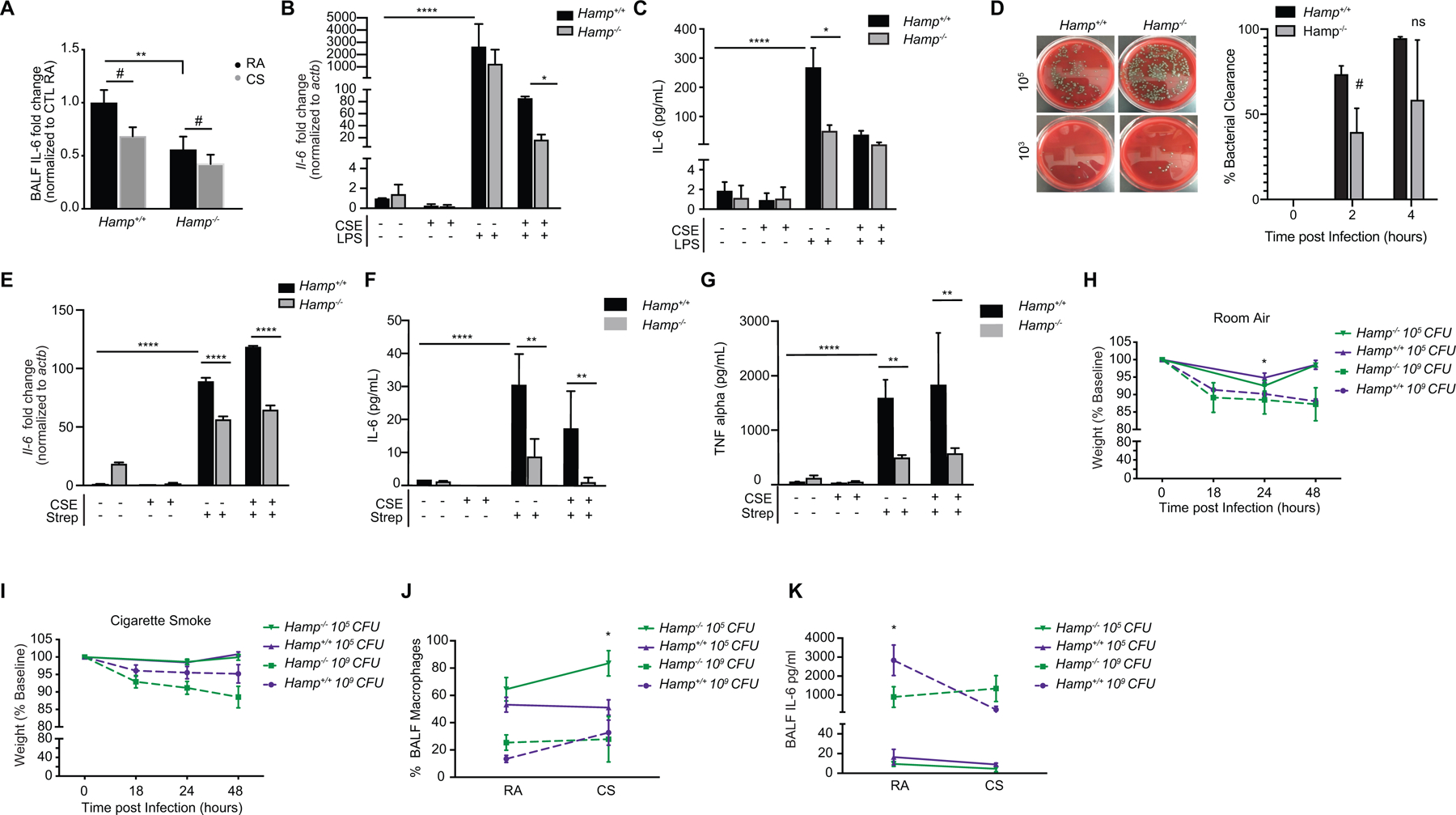
(a) Fold change in IL-6 levels in the bronchoalveolar lavage fluid (BALF) of Hamp+/+ (RA n = 8, CS n = 9) and Hamp−/− (RA n = 5, CS n = 8) mice exposed to RA and CS (8 weeks) measured by ELISA. (b) Il-6 mRNA and (c) IL-6 levels (pg/ml by ELISA) in the media of primary AMs isolated from Hamp+/+ and Hamp−/− mice treated with 2% CSE for 18 hours (CSE treatment of 24 hours total) followed by 100 ng/mL LPS (6 hours). Data representative of 3–4 independent experiments. (d) Bacterial titer (left panel) at 103 and 105 dilutions with the number of colony forming units (CFUs) quantified (right panel) in the media supernatants of Hamp+/+and Hamp−/− primary AMs treated with 0.5 ×106 CFU S. pneumoniae for 2 or 4 hours. Data representative of n=4–6 independent experiments. (e) Il-6 mRNA expression (f) IL-6 levels (pg/ml ELISA) and (g) TNF-α levels (pg/ml ELISA) in primary AMs and corresponding media supernatants from Hamp+/+ and Hamp−/− mice treated with 2% CSE (24 hours) followed by S. pneumoniae (4 hours). Data representative of n=2 independent experiments. (h-i) Relative weights, (j) percentage of macrophages in BALF by Hematoxylin-Eosin staining and (k) BALF IL-6 levels (pg/ml ELISA) in Hamp+/+and Hamp−/− (n = 5 per group) mice at 0, 18, 24 and 48 hours post-infection of 105 and 109 (CFU) of S. pneumoniae after exposure to either (h) RA or (i) CS (8 weeks). All data are mean ± s.e.m. *P < 0.05, **P < 0.01, ****P < 0.001 by one-way ANOVA followed by Tukeys post-hoc test. #P < 0.05 by student’s unpaired t-test.
One of the most common organisms cultured from sputum and bronchoscopic samples of patients with COPD is S. pneumoniae (35). Both smokers and individuals with COPD have an increased susceptibility to S. pneumoniae infection (33, 34), and bacterial colonization and infection by S. pneumoniae typically occurs early in the course of COPD (35). To assess if loss of hepcidin alters the response of AMs to S. pneumoniae, primary AMs and BMDMs isolated from Hamp+/+ and Hamp−/− mice were cultured and treated with live S. pneumoniae (ATTC strain 6303). AMs deficient in Hamp were less able to clear S. pneumoniae with a greater burden of colony forming units (CFU) remaining in the supernatants 2–4 hours post infection (Figure 6D). Similarly, AMs isolated from Hamp−/− mice had impaired S. pneumoniae -induced IL-6 and TNF-α production at baseline and in the presence of CSE (Figure 6E–G). In vivo, Hamp−/− mice infected with S. pneumoniae displayed signs of a higher bacterial burden reflected by more significant loss in weight, higher numbers of BALF macrophage infiltrates and lower AM Il-6 levels (Figure 6H–K and Supplementary Figure 2I–J). Hamp−/− mice exposed to smoke, followed by S. pneumoniae infection also had more severe loss in body weight and heightened BALF macrophage infiltrates (Figure 6I–J).
Discussion
Changes in pulmonary and systemic iron regulation are associated with the progression and pathogenesis of COPD. In this study, we show that the master iron hormone hepcidin is lower in whole lung homogenates, plasma and urine of animals exposed to smoke. We show that a loss in hepcidin expression may be associated with increased levels of EPO, EFRE and expanded, but inefficient erythropoiesis in the bone marrow. We hypothesize that this may in turn lead to changes in iron uptake and release in macrophages of the lung. Consistently, murine AMs exposed to smoke have higher ferroportin and higher iron levels. Upon exposure to smoke, AMs fail to increase hepcidin in response to gram-negative or gram-positive infection and loss of hepcidin in vivo results in blunted functional responses of AMs and altered responses to Streptococcus pneumoniae infection.
These results support a theory whereby a rise in hepcidin expression is an essential bacteriostatic response of alveolar macrophages, consistent with previous reports that inflammatory signals stimulate the production of hepcidin (58). Our observed increases in hepcidin in macrophages derived from healthy smokers and subjects with COPD is also consistent with hepcidin activation in patients with mild-moderate disease or during an exacerbation event (commonly associated with infection (70, 71)), reflective of the pro-inflammatory phenotype of alveolar macrophages in smokers and individuals with COPD (26, 27). Similarly, our observations that whole lung and systemic hepcidin levels decline with chronic smoking is consistent with the loss of hepcidin expression in severe end-stage patients with COPD, most likely correlating with hypoxemia (28). These findings further highlight that hepcidin production can be both modulated by the stimulatory effect of inflammation and the suppressive effect of hypoxia (27).
While a rise in hepcidin in macrophages appears to be a protective response to infection, whether or not this rise in hepcidin is directly related to the effect of hepcidin on ferroportin expression and in turn iron export, requires further extensive investigation. Notably, we were unable to validate the transcript levels of ferroportin on alveolar macrophages observed in this study by immunoblotting due to our inability to validate commercially available murine ferroportin antibodies for immunoblotting (data not shown). Preliminary data generated by this study suggests that the effect of smoking on ferroportin expression and/or iron changes may be independent to hepcidin expression. In addition, loss of hepcidin expression in the lung does not alter the injurious response of the lung to smoke. Irrespective, cigarette smoking impairs innate hepcidin induction in macrophages, which reduces their ability to respond to infection. AMs are plastic cells with many important roles in the lung including clearance of inhaled particulates and microbes and modulation of inflammation and tissue repair (72, 73). Defects in the ability of these cells to adapt their intracellular metabolism may alter their ability to produce inflammatory cytokines and reactive oxygen species (ROS), and to perform other defense and repair functions (74). The observed impairments in AM responses may lead to an inefficient adaptive response to environmental cues such as the presence of bacteria resulting in an increased microbial burden in COPD, enhancing the risk of recurrent infections. Bacterial colonization and impaired clearance are characteristic of COPD and are associated with disease progression (65). Increased rates of recurrent infection in COPD may manifest as chronic bronchitis and/or excessive mucus production and are associated with reduced quality of life, more rapid decline in lung function and increased mortality (70, 71).
Our results also highlight the important systemic effects cigarette smoking has on erythropoiesis and systemic iron regulation. We demonstrate that exposure to smoke results in expanded but inefficient erythropoietic activity, most likely due to the decline in iron levels in erythropoietic precursors. While there is some evidence to suggest that patients with COPD have altered responses to hypoxia and may have higher EPO levels (75) and that chronic cigarette smoking increases the size of the mitotic and post-mitotic pools in the marrow (76), little is known about the direct effects of CS on erythropoietic function or EPO production. These findings suggest that under acute hypoxic conditions dietary iron absorption by enterocytes and release by macrophages cannot match the increased requirements for erythropoiesis. Defects in iron reutilization, ineffective erythropoiesis, and impaired marrow response to normal EPO levels have all been incriminated in the pathogenesis of anemia of which there is an increased prevalence in subjects with severe COPD (8, 30, 31). Further studies investigating the effects of cigarette smoke on the production of hematopoietic precursors supplying the mononuclear phagocyte system and the role of iron in this process will be of great interest to understand macrophage defects in subjects with COPD. In addition, more extensive studies utilizing pharmacological inhibitors of phagocytosis and endocytosis are required to fully understand the role that iron-associated phagocytosis has in this process.
This work provides an early foundation for further studies to understand the role of hepcidin in lung biology and disease as well as the interplay between the bone marrow, erythropoiesis and the mononuclear phagocyte system in the lung. Mechanistic studies assessing the ferroportin-independent role of hepcidin in the lung in response to infection and varying environmental conditions are required and are the subject of further investigation. Our findings suggest that a loss of hepcidin systemically and at the level of the alveolar macrophage (in response to infection) may play an important role in the pathogenesis and progression of COPD. In particular, given that iron is essential to bacterial growth, respiration, and metabolism (36, 37) and the bacterial species thought to be responsible for more than 50% of exacerbation events in COPD, including S. pneumoniae require iron for survival and growth (38–42); understanding the regulation of iron metabolism in the lung microenvironment and interaction with the immune system is of great importance. To conclude, if replicated, our findings that hepcidin repression in the lung upon smoke exposure may alter the bactericidal response of the lung macrophages provides a new investigative avenue to help identify, understand and treat COPD patients at high-risk for infection.
Supplementary Material
Key Points.
Cigarette Smoke reduces hepcidin levels in the lung, plasma and urine of mice
Cigarette smoke inhibits hepcidin production by murine alveolar macrophages
Alveolar macrophages from smokers and individuals with COPD have higher ferroportin
Acknowledgments
The authors wish to thank Dr. Gregory Sonnenberg, Dr. William Zhang and Dr. Augustine M.K. Choi for critical insight and discussion.
This work is supported by the US National Institute of Health grants R00-HL125899 (S.M.C.), by R01AG052530 (H.S.D.) and R01AG056699 (H.S.D.), by Science Foundation Ireland grant FRL4862 (S.M.C) and by a European Respiratory Society, short-term research fellowship (J.R.B). This work was also supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
References
- 1.2016. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (London, England) 388: 1545–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A, Bueno R, Pillai SG, Lomas DA, Sparrow D, Shapiro SD, Criner GJ, Kim HP, Chen Z, Choi AM, Reilly J, and Silverman EK. 2009. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. American journal of human genetics 85: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chappell SL, Daly L, Lotya J, Alsaegh A, Guetta-Baranes T, Roca J, Rabinovich R, Morgan K, Millar AB, Donnelly SC, Keatings V, MacNee W, Stolk J, Hiemstra PS, Miniati M, Monti S, O’Connor CM, and Kalsheker N. 2011. The role of IREB2 and transforming growth factor beta-1 genetic variants in COPD: a replication case-control study. BMC medical genetics 12: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du Y, Xue Y, and Xiao W. 2016. Association of IREB2 Gene rs2568494 Polymorphism with Risk of Chronic Obstructive Pulmonary Disease: A Meta-Analysis. Medical science monitor : international medical journal of experimental and clinical research 22: 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardin M, Zielinski J, Wan ES, Hersh CP, Castaldi PJ, Schwinder E, Hawrylkiewicz I, Sliwinski P, Cho MH, and Silverman EK. 2012. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. American journal of respiratory cell and molecular biology 47: 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y, Yang D, Xun X, Wang Z, Sun P, Xu D, He P, Niu H, and Jin T. 2015. Association of genetic polymorphisms with chronic obstructive pulmonary disease in the Hainan population: a case-control study. International journal of chronic obstructive pulmonary disease 10: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turi JL, Yang F, Garrick MD, Piantadosi CA, and Ghio AJ. 2004. The iron cycle and oxidative stress in the lung. Free Radic Biol Med 36: 850–857. [DOI] [PubMed] [Google Scholar]
- 8.Portillo K, Martinez-Rivera C, and Ruiz-Manzano J. 2013. Anaemia in chronic obstructive pulmonary disease. Does it really matter? International journal of clinical practice 67: 558–565. [DOI] [PubMed] [Google Scholar]
- 9.Guo J, Zheng C, Xiao Q, Gong S, Zhao Q, Wang L, He J, Yang W, Shi X, Sun X, and Liu J. 2015. Impact of anaemia on lung function and exercise capacity in patients with stable severe chronic obstructive pulmonary disease. BMJ open 5: e008295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasquez A, and Logomarsino JV. 2016. Anemia in Chronic Obstructive Pulmonary Disease and the Potential Role of Iron Deficiency. Copd 13: 100–109. [DOI] [PubMed] [Google Scholar]
- 11.Zhang WZ, Oromendia C, Kikkers SA, Butler JJ, O’Beirne S, Kim K, O’Neal WK, Freeman CM, Christenson SA, Peters SP, Wells JM, Doerschuk C, Putcha N, Barjaktarevic I, Woodruff PG, Cooper CB, Bowler RP, Comellas AP, Criner GJ, Paine R 3rd, Hansel NN, Han MK, Crystal RG, Kaner RJ, Ballman KV, Curtis JL, Martinez FJ, and Cloonan SM. 2020. Increased airway iron parameters and risk for exacerbation in COPD: an analysis from SPIROMICS. Scientific reports 10: 10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heilig EA, Thompson KJ, Molina RM, Ivanov AR, Brain JD, and Wessling-Resnick M. 2006. Manganese and iron transport across pulmonary epithelium. American journal of physiology. Lung cellular and molecular physiology 290: L1247–1259. [DOI] [PubMed] [Google Scholar]
- 13.Neves J, Leitz D, Kraut S, Brandenberger C, Agrawal R, Weissmann N, Muhlfeld C, Mall MA, Altamura S, and Muckenthaler MU. 2017. Disruption of the Hepcidin/Ferroportin Regulatory System Causes Pulmonary Iron Overload and Restrictive Lung Disease. EBioMedicine 20: 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arezes J, and Nemeth E. 2015. Hepcidin and iron disorders: new biology and clinical approaches. International journal of laboratory hematology 37 Suppl 1: 92–98. [DOI] [PubMed] [Google Scholar]
- 15.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, and Lin HY. 2006. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nature genetics 38: 531–539. [DOI] [PubMed] [Google Scholar]
- 16.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, and Ganz T. 2004. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. The Journal of clinical investigation 113: 1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganz T, and Nemeth E. 2015. Iron homeostasis in host defence and inflammation. Nature reviews. Immunology 15: 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang F, Haile DJ, Wang X, Dailey LA, Stonehuerner JG, and Ghio AJ. 2005. Apical location of ferroportin 1 in airway epithelia and its role in iron detoxification in the lung. American journal of physiology. Lung cellular and molecular physiology 289: L14–23. [DOI] [PubMed] [Google Scholar]
- 19.Houamel D, Ducrot N, Lefebvre T, Daher R, Moulouel B, Sari MA, Letteron P, Lyoumi S, Millot S, Tourret J, Bouvet O, Vaulont S, Vandewalle A, Denamur E, Puy H, Beaumont C, Gouya L, and Karim Z. 2016. Hepcidin as a Major Component of Renal Antibacterial Defenses against Uropathogenic Escherichia coli. Journal of the American Society of Nephrology : JASN 27: 835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deschemin JC, Mathieu JRR, Zumerle S, Peyssonnaux C, and Vaulont S. 2017. Pulmonary Iron Homeostasis in Hepcidin Knockout Mice. Frontiers in physiology 8: 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merle U, Fein E, Gehrke SG, Stremmel W, and Kulaksiz H. 2007. The iron regulatory peptide hepcidin is expressed in the heart and regulated by hypoxia and inflammation. Endocrinology 148: 2663–2668. [DOI] [PubMed] [Google Scholar]
- 22.McCarthy RC, and Kosman DJ. 2014. Glial cell ceruloplasmin and hepcidin differentially regulate iron efflux from brain microvascular endothelial cells. PloS one 9: e89003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulaksiz H, Theilig F, Bachmann S, Gehrke SG, Rost D, Janetzko A, Cetin Y, and Stremmel W. 2005. The iron-regulatory peptide hormone hepcidin: expression and cellular localization in the mammalian kidney. The Journal of endocrinology 184: 361–370. [DOI] [PubMed] [Google Scholar]
- 24.Lakhal-Littleton S, Wolna M, Chung YJ, Christian HC, Heather LC, Brescia M, Ball V, Diaz R, Santos A, Biggs D, Clarke K, Davies B, and Robbins PA. 2016. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen NB, Callaghan KD, Ghio AJ, Haile DJ, and Yang F. 2006. Hepcidin expression and iron transport in alveolar macrophages. American journal of physiology. Lung cellular and molecular physiology 291: L417–425. [DOI] [PubMed] [Google Scholar]
- 26.Nickol AH, Frise MC, Cheng HY, McGahey A, McFadyen BM, Harris-Wright T, Bart NK, Curtis MK, Khandwala S, O’Neill DP, Pollard KA, Hardinge FM, Rahman NM, Armitage AE, Dorrington KL, Drakesmith H, Ratcliffe PJ, and Robbins PA. 2015. A cross-sectional study of the prevalence and associations of iron deficiency in a cohort of patients with chronic obstructive pulmonary disease. BMJ open 5: e007911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tandara L, Grubisic TZ, Ivan G, Jurisic Z, Tandara M, Gugo K, Mladinov S, and Salamunic I. 2015. Systemic inflammation up-regulates serum hepcidin in exacerbations and stabile chronic obstructive pulmonary disease. Clin Biochem 48: 1252–1257. [DOI] [PubMed] [Google Scholar]
- 28.Duru S, Bilgin E, and Ardic S. 2012. Hepcidin: A useful marker in chronic obstructive pulmonary disease. Annals of thoracic medicine 7: 31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silverberg DS, Mor R, Weu MT, Schwartz D, Schwartz IF, and Chernin G. 2014. Anemia and iron deficiency in COPD patients: prevalence and the effects of correction of the anemia with erythropoiesis stimulating agents and intravenous iron. BMC pulmonary medicine 14: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Rivera C, Portillo K, Munoz-Ferrer A, Martinez-Ortiz ML, Molins E, Serra P, Ruiz-Manzano J, and Morera J. 2012. Anemia is a mortality predictor in hospitalized patients for COPD exacerbation. Copd 9: 243–250. [DOI] [PubMed] [Google Scholar]
- 31.Boutou AK, Karrar S, Hopkinson NS, and Polkey MI. 2013. Anemia and Survival in Chronic Obstructive Pulmonary Disease: A Dichotomous rather than a Continuous Predictor. Respiration; international review of thoracic diseases 85: 126–131. [DOI] [PubMed] [Google Scholar]
- 32.Qureshi H, Sharafkhaneh A, and Hanania NA. 2014. Chronic obstructive pulmonary disease exacerbations: latest evidence and clinical implications. Therapeutic Advances in Chronic Disease 5: 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuorti JP, Butler JC, Farley MM, Harrison LH, McGeer A, Kolczak MS, and Breiman RF. 2000. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. The New England journal of medicine 342: 681–689. [DOI] [PubMed] [Google Scholar]
- 34.Grau I, Ardanuy C, Calatayud L, Schulze MH, Linares J, and Pallares R. 2014. Smoking and alcohol abuse are the most preventable risk factors for invasive pneumonia and other pneumococcal infections. International journal of infectious diseases : IJID : official publication of the International Society for Infectious Diseases 25: 59–64. [DOI] [PubMed] [Google Scholar]
- 35.Sethi S, Evans N, Grant BJ, and Murphy TF. 2002. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. The New England journal of medicine 347: 465–471. [DOI] [PubMed] [Google Scholar]
- 36.Skaar EP 2010. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS pathogens 6: e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cloonan SM, Mumby S, Adcock IM, Choi AMK, Chung KF, and Quinlan GJ. 2017. The IRONy of Iron-overload and Iron-deficiency in Chronic Obstructive Pulmonary Disease. American journal of respiratory and critical care medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honsa E, Johnson M, and Rosch J. 2013. The roles of transition metals in the physiology and pathogenesis of Streptococcus pneumoniae. Frontiers in Cellular and Infection Microbiology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitby PW, VanWagoner TM, Seale TW, Morton DJ, and Stull TL. 2006. Transcriptional Profile of Haemophilus influenzae: Effects of Iron and Heme. Journal of Bacteriology 188: 5640–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, Facchini M, Pasquali P, Bragonzi A, and Visca P. 2016. Role of Iron Uptake Systems in Pseudomonas aeruginosa Virulence and Airway Infection. Infection and Immunity 84: 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Vries SPW, Burghout P, Langereis JD, Zomer A, Hermans PWM, and Bootsma HJ. 2013. Genetic requirements for Moraxella catarrhalis growth under iron-limiting conditions. Molecular Microbiology 87: 14–29. [DOI] [PubMed] [Google Scholar]
- 42.Hoyer J, Bartel J, Gómez-Mejia A, Rohde M, Hirschfeld C, Heß N, Sura T, Maaß S, Hammerschmidt S, and Becher D. 2018. Proteomic response of Streptococcus pneumoniae to iron limitation. International Journal of Medical Microbiology 308: 713–721. [DOI] [PubMed] [Google Scholar]
- 43.Lesbordes-Brion JC, Viatte L, Bennoun M, Lou DQ, Ramey G, Houbron C, Hamard G, Kahn A, and Vaulont S. 2006. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108: 1402–1405. [DOI] [PubMed] [Google Scholar]
- 44.Taylor AE, Finney-Hayward TK, Quint JK, Thomas CM, Tudhope SJ, Wedzicha JA, Barnes PJ, and Donnelly LE. 2010. Defective macrophage phagocytosis of bacteria in COPD. The European respiratory journal 35: 1039–1047. [DOI] [PubMed] [Google Scholar]
- 45.Chana KK, Fenwick PS, Nicholson AG, Barnes PJ, and Donnelly LE. 2014. Identification of a distinct glucocorticosteroid-insensitive pulmonary macrophage phenotype in patients with chronic obstructive pulmonary disease. The Journal of allergy and clinical immunology 133: 207–216 e201–211. [DOI] [PubMed] [Google Scholar]
- 46.Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, Nakahira K, Pilewski JM, Lee JS, Zhang Y, Ryter SW, and Choi AM. 2008. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PloS one 3: e3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen ZH, Lam HC, Jin Y, Kim HP, Cao J, Lee SJ, Ifedigbo E, Parameswaran H, Ryter SW, and Choi AM. 2010. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A 107: 18880–18885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, Cervo M, Yao H, Chung AL, Mizumura K, An CH, Shan B, Franks JM, Haley KJ, Owen CA, Tesfaigzi Y, Washko GR, Quackenbush J, Silverman EK, Rahman I, Kim HP, Mahmood A, Biswal SS, Ryter SW, and Choi AM. 2013. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. The Journal of clinical investigation 123: 5212–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA, Konrad C, Polverino F, Siempos II, Perez E, Mizumura K, Ghosh MC, Parameswaran H, Williams NC, Rooney KT, Chen ZH, Goldklang MP, Yuan GC, Moore SC, Demeo DL, Rouault TA, D’Armiento JM, Schon EA, Manfredi G, Quackenbush J, Mahmood A, Silverman EK, Owen CA, and Choi AM. 2016. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, and Choi AM. 2014. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. The Journal of clinical investigation 124: 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, and Pare PD. 2004. The nature of small-airway obstruction in chronic obstructive pulmonary disease. The New England journal of medicine 350: 2645–2653. [DOI] [PubMed] [Google Scholar]
- 52.Laucho-Contreras ME, Taylor KL, Mahadeva R, Boukedes SS, and Owen CA. 2015. Automated Measurement of Pulmonary Emphysema and Small Airway Remodeling in Cigarette Smoke-exposed Mice. LID - 10.3791/52236 [doi]. J Vis Exp. 16 (95). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasricha SR, Lim PJ, Duarte TL, Casu C, Oosterhuis D, Mleczko-Sanecka K, Suciu M, Da Silva AR, Al-Hourani K, Arezes J, McHugh K, Gooding S, Frost JN, Wray K, Santos A, Porto G, Repapi E, Gray N, Draper SJ, Ashley N, Soilleux E, Olinga P, Muckenthaler MU, Hughes JR, Rivella S, Milne TA, Armitage AE, and Drakesmith H. 2017. Hepcidin is regulated by promoter-associated histone acetylation and HDAC3. Nature communications 8: 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, and Miller JL. 2007. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 13: 1096–1101. [DOI] [PubMed] [Google Scholar]
- 55.Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, Bhupatiraju A, Lee YT, Goodnough JB, Harandi O, Ganz T, Paulson RF, and Miller JL. 2009. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 114: 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camaschella C, Pagani A, Nai A, and Silvestri L. 2016. The mutual control of iron and erythropoiesis. International journal of laboratory hematology 38 Suppl 1: 20–26. [DOI] [PubMed] [Google Scholar]
- 57.Nairz M, Theurl I, Swirski FK, and Weiss G. 2017. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Archiv : European journal of physiology 469: 397–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stefanova D, Raychev A, Arezes J, Ruchala P, Gabayan V, Skurnik M, Dillon BJ, Horwitz MA, Ganz T, Bulut Y, and Nemeth E. 2017. Endogenous hepcidin and its agonist mediate resistance to selected infections by clearing non-transferrin-bound iron. Blood 130: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Agoro R, Taleb M, Quesniaux VFJ, and Mura C. 2018. Cell iron status influences macrophage polarization. PloS one 13: e0196921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCrea KA, Ensor JE, Nall K, Bleecker ER, and Hasday JD. 1994. Altered cytokine regulation in the lungs of cigarette smokers. American journal of respiratory and critical care medicine 150: 696–703. [DOI] [PubMed] [Google Scholar]
- 61.Vlahos R, and Bozinovski S. 2014. Role of alveolar macrophages in chronic obstructive pulmonary disease. Frontiers in immunology 5: 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O’Connor TP, and Crystal RG. 2009. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. Journal of immunology (Baltimore, Md. : 1950) 183: 2867–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV, Kaner RJ, Crystal RG, and Cloonan SM. 2020. Alveolar Macrophage Immunometabolism and Lung Function Impairment in Smoking and Chronic Obstructive Pulmonary Disease. American journal of respiratory and critical care medicine 201: 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, and Reynolds PN. 2007. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. American journal of respiratory cell and molecular biology 37: 748–755. [DOI] [PubMed] [Google Scholar]
- 65.Berenson CS, Kruzel RL, Wrona CT, Mammen MJ, and Sethi S. 2015. Impaired Innate COPD Alveolar Macrophage Responses and Toll-Like Receptor-9 Polymorphisms. PloS one 10: e0134209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wesselius LJ, Nelson ME, and Skikne BS. 1994. Increased release of ferritin and iron by iron-loaded alveolar macrophages in cigarette smokers. American journal of respiratory and critical care medicine 150: 690–695. [DOI] [PubMed] [Google Scholar]
- 67.Philippot Q, Deslee G, Adair-Kirk TL, Woods JC, Byers D, Conradi S, Dury S, Perotin JM, Lebargy F, Cassan C, Le Naour R, Holtzman MJ, and Pierce RA. 2014. Increased iron sequestration in alveolar macrophages in chronic obstructive pulmonary disease. PloS one 9: e96285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hemmaplardh D, and Morgan EH. 1977. The role of endocytosis in transferrin uptake by reticulocytes and bone marrow cells. Br J Haematol 36: 85–96. [DOI] [PubMed] [Google Scholar]
- 69.Peterson PK, Verhoef J, and Quie PG. 1977. Influence of temperature on opsonization and phagocytosis of staphylococci. Infect Immun 15: 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han MK, Quibrera PM, Carretta EE, Barr RG, Bleecker ER, Bowler RP, Cooper CB, Comellas A, Couper DJ, Curtis JL, Criner G, Dransfield MT, Hansel NN, Hoffman EA, Kanner RE, Krishnan JA, Martinez CH, Pirozzi CB, O’Neal WK, Rennard S, Tashkin DP, Wedzicha JA, Woodruff P, Paine R 3rd, and Martinez FJ. 2017. Frequency of exacerbations in patients with chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. The Lancet. Respiratory medicine 5: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, and Ochando R. 2005. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 60: 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marriott HM, and Dockrell DH. 2007. The role of the macrophage in lung disease mediated by bacteria. Experimental lung research 33: 493–505. [DOI] [PubMed] [Google Scholar]
- 73.Stuart LM, and Ezekowitz RA. 2005. Phagocytosis: elegant complexity. Immunity 22: 539–550. [DOI] [PubMed] [Google Scholar]
- 74.Van den Bossche J, O’Neill LA, and Menon D. 2017. Macrophage Immunometabolism: Where Are We (Going)? Trends in immunology 38: 395–406. [DOI] [PubMed] [Google Scholar]
- 75.Sharma RK, and Chakrabarti S. 2016. Anaemia secondary to erythropoietin resistance: important predictor of adverse outcomes in chronic obstructive pulmonary disease. Postgraduate medical journal. [DOI] [PubMed] [Google Scholar]
- 76.Terashima T, Wiggs B, English D, Hogg JC, and van Eeden SF. 1997. The effect of cigarette smoking on the bone marrow. American journal of respiratory and critical care medicine 155: 1021–1026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.