

Abstract
Klotho levels decline with age which is an important mechanistic driver of aging. Klotho gene deficiency is associated with hypertension. The purpose of this study is to investigate the potential role of histone 3 lysine (K) 27 trimethylation (H3K27me3) in the regulation of Klotho gene expression and examine the related molecular pathways that may drive kidney cell aging. Kidneys were collected from 6-months-old WT (young WT), 30-months-old wild type (aged WT), and 6-months-old (young) and 20 months (aged) Klotho mutant mice, respectively. We demonstrated that the H3K27me3 level was increased in kidneys of aged WT and Klotho mutant mice vs. young WT mice. Elevation of H3K27me3 levels was likely due to downregulation of the H3K27-specific demethylase JMJD3 in the aged kidneys. Inhibition of Polycomb Repressive Complex C 2 (histone trimethyltransferase) decreased the H3K27me3 levels leading to an increase in the expression of Klotho in cultured primary renal tubule cells assessed by Western blot and Klotho promoter activity assays. The ChIP-qPCR assay revealed that H3K27me3 was physically associated with the Klotho promoter region. Furthermore, aging impaired the SGK-1/FOXO3a signaling leading to upregulation of p53 and p16 (aging markers) in the kidney of aged WT mice. Klotho may regulate the SGK-1/FOXO3 signaling which was decreased due to Klotho deficiency. Thus, aging-associated downregulation of Klotho gene expression may be partly attributed to upregulation of H3K27me3 levels. Downregulation of Klotho may impair the SGK-1/FOXO3 signaling contributing to kidney cell aging.
Keywords: aging, epigenetic, H3K27me3, klotho, SGK-1, FOXO3
Graphical Abstract
Epigenetic Regulation of the Klotho Signaling Pathway in Aging Kidney
Aging-associated downregulation of Klotho gene expression may be partly attributed to upregulation of H3K27me3 levels. Downregulation of Klotho may impair the SGK-1/FOXO3 signaling contributing to kidney cell aging.
INTRODUCTION
Klotho was identified as an anti-aging gene which is primarily expressed in the kidney1. Klotho gene deficiency leads to kidney dysfunction and hypertension1. However, how klotho gene expression is regulated is poorly understood. In the kidney, Klotho serves as a co-receptor of fibroblast growth factor 23 (FGF23) and is coupled with fibroblast growth factor receptor 1 (FGFR1) in regulating phosphate transport in the proximal tubule and stimulating calcium reabsorption in the distal tubule2, as well as mediating the vitamin D homeostasis3. Patients with CKD showed decreased serum soluble Klotho associated with hyperphosphatemia causing cardiac remodeling4. There are similarities between mice lacking Klotho and patients with CKD and Klotho may play an important role in the pathogenesis of CKD, a clinical model of premature aging5. Thus, it is important to understand how Klotho gene expression is regulated.
Epigenetic modification of histone alters gene transcriptional states and regulates various biological processes including development, cell differentiation, diseases, and aging6-9. Histone methylation, specifically histone 3 lysine 27 trimethylation (H3K27me3) is epigenetic modification with close ties to gene regulation and have been directly linked to lifespan and aging in different models10-12. H3K27me3 is primarily a transcriptionally repressive modification catalyzed by the polycomb repressive complex 2 (PRC2) and removed by UTX (also known as KDM6a). PRC2 consists of four subunits: Suz12 (zinc finger), EED, EZH1 or EZH2 (enhancer of zeste homolog 2 gene with histone H3 Lys 27 (H3K27) methyltransferase activity) 13. PRC2 is responsible for establishing and maintaining histone H3K27 methylation during cell differentiation and proliferation. Various animal models have been used for uncovering key pathways linked to aging. However, an opposite effect of histone modification, particularly for H3K27me3, on aging and lifespan, has been observed in different aging models10-12. During C. elegans aging, there is a global decrease in somatic H3K27me3 and an increase in UTX expression10. Loss of function of UTX causes increase in H3K27me3 and extends lifespan via suppression of insulin/IF-1 signaling (IIS) 10. In flies, H3K27me3 appears to regulate lifespan in a opposite manner to that in C. elegans11. Analysis of global occupancy of H3K27me3 between young and aged wild type flies demonstrated that H3K27me3 exhibited genome-wide distributions and there was a dramatic shift in the pattern of H3K27me3 modification during aging11. A similar global upregulation of H3K27me3 was observed in aging killifish12.
Evidence of global changes and imbalance of regional chromatin domains in mammalians was also observed in cultured cells during senescence in vitro and linked to aging-associated tissue deterioration in vivo14. Senescent chromatin is dramatically reorganized in senescent cells through formation of large-scale domains (mesas) of H3K4me3 and H3K27me3 over lamin-associated domains and large losses (canyons) of H3K27me3 outside of LADs results in transcriptional downregulation of lamin B1 in senescence15, 16. Epigenetic changes in aged individuals parallel those seen in patients with chronic kidney disease (CKD) manifested by accelerated renal aging17. Expression of EZH2, a methyltransferase for generation of H3K27me3 is increased in fibrotic kidneys in mice with UUO and in humans with CKD18.
In this study, we aimed to investigate the epigenetic modification of histone H3K27 in the regulation of klotho transcription and expression and examine relevant molecular pathways that may drive kidney cell aging in Klotho mutant (deficiency) and aged mice. We demonstrated that inhibition of PRC2 reduces H3K27me3 recruitment to the Klotho promoter region. These findings provide the first evidence that epigenetic downregulation of Klotho gene expression is due, at least in part, to histone 3 modification in the Klotho promoter region. We further showed that the SGK-1/FOXO3a signaling may contribute to kidney cell aging through upregulation of p53 and p16 genes.
MATERIALS AND METHODS
DATA AVAILABILITY STATEMENT:
The original data including gel blots used to generate the analysis presented in the manuscript is publicly arcived at the following link: https://datadryad.org/stash/share/SIC0Ze7i6Oh1JbU2hdzv7Jyv-W7Yc_VH7IdJdO6qIuo. doi:10.5061/dryad.fn2z34tq0
Animals and isolation of renal tubule cells
All procedures and protocols used in this study were approved by the Institutionla Animal Care and Use Committee of the University of Tennessee Health Science Center (protocal #19-051). Young WT (6 months), aged WT (30 months), as well as young (6 months) and aged (19-21 monhs) Klotho mutant mice were used. Wild type (WT) male mice were fed with regular rodent chow. Klotho mutant male mice were fed with low phosphate diet for 3 months after weaning and switched to regular diet for the rest period of time during the study.
Renal tubule cells were isolated as discrebed previously with some modifications19-21. Briefly, all mice (3 months) were euthanized using a CO2 chamber and kidneys were immediately harvested and washed in sterile ice-cold Hanks’ Balanced Salt Solution (HBSS). Kidneys were decapsulated in HBSS and transferred to Hibernate A medium (BrainBits LLC, USA). Then, kidenys were bisected and the inner medullary was removed. Cortical and outer mudullary tissues were dissected in order to obtain 1mm3 fragments. The tissues were washed twice with Hibernate A medium and then digested in the presence of 1 mg/ml collagenase (type II, Sigma) for 30 min at 37°C. Renal tubues (proximal and distal) were dissected and collected under microscope, and filterred through 70μM and 40μM cell strainer consequently. Renal tubules were washed twice with DMEM/F12 containg 1% penicillin/streptomycin and centrifuged for at 1000 rpm for 5 min. Cells were cultured in 75 cm2 plastic flasks with complete cultue medium (DMEM/F12 medium containng 20% FBS, 15 penicillin/streptomycin, ITS-insulin:transferrin:selenium) at 37°C under 5% CO2 in a humidified incubator. The culture medium was changed after 24h to remove non-adhernet cells.
Promoter analysis
Promoter activity was assessed as we described previously22. Briefly, primary renal tubule cells were plated 18 h before transfection and fed with fresh medium 4 h and pre-treated with EED226 (Axon MechChem, Netherlands ) and/or GSK343 (Sigma) for 2h before transfection. Klotho promoters (pHKP-Sac2 and pHKP-Pml1 ) were constructed into a pGL3 basic reporter gene (Promega, Madison, WI). Klotho reporter plasmid DNAs were introduced into renal tubule cells using cationic liposomes (LipofectAMINE2000, Life technologies, Grand Island, NY). Transfection of plasmid DNAs was carried out for 16–18 h, and then cells were washed twice with phosphate-buffered saline and incubated in fresh medium containing 20% fetal calf serum in the presence of EED226 and GSK343 for another 2 days. To standardize the transfection efficiency, 0.1 μg of pRL-CMV vector (pRL Renilla reniformisluciferase control reporter vector; Promega) was cotransfected in all experiments. Cells were harvested 72 h after transfection and lysed in 50 μl of reporter lysis buffer (Promega). A luciferase assay (20 μl of cell lysed) was performed using a dual luciferase assay kit (Promega), and activity was measured with an Optocomp 1 luminometer (MGM Instruments, Inc., Hamden, CT). Promoter activity (mean ± S.D. of triplicate samples in relative fold changes) is represented by reporter activity normalized to pRL-CMV control. Graphs represent typical results of three-four separate experiments.
Chromatin Immunoprecipitation (ChIP) assays
ChIP assays were performed with a kit from Cell Signaling Technology (Danvers, MA) according to the manufacturer’s instructions with the following modifications as descried previously22, 23. Briefly, approximately 4 X 107 RTC cells in 150 cm2 culture dishes were cross-linked in 1% formaldehyde solution (Fisher Scientific, Pittsburgh, PA) for 10 minutes at room temperature, stopped cross-linking by adding 1M glycine for 5 minutes at room temperature. Cells were washed twice with ice-cold 1× phosphate-buffered saline solution (PBS) (Life Technologies) and harvested in 1-ml of 1× PBS + protease inhibitor cocktail (PIC) + phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO). Cells were then pelleted by centrifugation at 1500 rpm for 5 minutes, 4°C and the pellet was resuspended in 10 ml ice-cold buffer A with DTT, PIC, and PMSF on ice for 10 minutes. Cell nuclei were pelleted by centrifugation at 3000 rpm for 5 minutes at 4°C. Nuclei pellet was then washed in 10 ml ice-cold buffer B with DTT, and resuspened in 1.0 ml buffer B with DTT. Micrococcal nuclease was added to the nuclei and incubated for 20 minutes at 37°C. Digestion was stopped by adding 100μl of 0.5 M EDTA and nuclei was pelleted by centrifugation at 13,000 rpm for 1 minute at 4°C. Nuclear pellet was resuspened in 0.5 ml 1x CHIP buffer with PIC and PMSF for sonication using the VirSonic Ultrasonic cell disrupter 100 (VirTis, Gardiner, NY) to shear the DNA to an average length of 300 to 500 base pairs (six, 15-s bursts on ice). Samples were stored at −80°C before use. For chromatin immunoprecipitation, 200 μl of the cross-linked chromatin preparation were added to 800 μl of ChIP buffer with PIC. Immunoprecipitations were carried out with 2 μg of antibodies (H3K27me3 from Cell Signaling). Histone H3 rabbit mAb (2μg) and normal rabbit IgG (1 μg) were used as positive or negative controls, respectively. After overnight immunoprecipitation on a rotator at 4°C, ChIP samples were then washed and eluted and DNA was un-crosslinked with NaCl and subsequently treated with proteinase K, Tris-Hcl and EDTA. DNA was purified and subjected to PCR amplification of Klotho promoter DNA containing putative binding sites for H3K27me3 using specific primers (Mouse: forward primer 5’-CAG GAT GGA GGC CAC AGG AT-3’, reverse primer 5'- TGA TTA TCC AGA GCA GGC GCC −3').
Western blot analysis
The western blot procedure was described previously24-29. Briefly, samples from mouse kidney and mouse renal tubule cells were prepared in T-per tissue extraction buffer and M-per mammalian extraction buffer (ThermoFisher), respectively. Nuclear and cytoplasmic proteins were isolated by using a NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, Rockford, IL). Protein contents in the samples were quantified and stored at −80°C until use. For electrophoresis, 40μg kidney protein and 20μg renal tubule cell lysates were loaded onto Expressplus Page 4-12% gel (GenScript, USA). Proteins were separated at 120 V for 60 minutes and transferred to nitrocellulose membrane using Trans-Blot Turbo (BioRad). Membranes were blocked with 5% BSA blocking buffer in TBST (Thermo Scientific, Rockford, IL) for 2 hours and then incubated with primary antibody (Klotho, 1:500, R&D; UTX, JMJD3, EZH2, H3K27me3, H3, SGK-1, p-SGK-1, p-mTOR, t-mTOR, p-AKT, t-AKT, p-FOXO3a, t-FOXO3a, p-70S6K, and t-70S6K, 1:1000 from Cell Signaling Technology and p53 1:1000, p21 1:1000, and p16 1:1000 from Abcam #189034) with gentle agitation overnight at 4 °C. After 3 washes with TBST (15 min once and 2x 5 min), membrane was incubated with secondary antibody in 5% BSA blocking buffer at room temperature for 1 hour. Membrane was then washed 4 times (15 min and 2x 5 min) and subjected to ECL (BioRad) and analyzed with BioRad ChemiDoc, MP imaging system. Western bolt using β-actin antibody or Lamin B antibody (Cell Signaling) was used as internal control of protein loadings of cytoplasmic or nuclear, respectively.
Statistical analysis
We evaluated differences between two groups by unpaired t-test and multiple groups by one-way analysis of variance. All values are expressed as means ± SD. All computations were performed using GraphPad Prism5 (GraphPad Software Inc. La Jolla, CA, USA).
RESULTS
Expression of Klotho was decreased in the aging kidney.
To examine the expression and cellular distribution of Klotho in the kidney, we collected the protein samples from whole kidney lysates, nuclear extracts, and cytoplasm of young (6-months-old) and aged (30-months -old) WT mice, and Klotho mutant young (6-months-old) and aged (average 20-months-old) mice. Western blot analysis showed that expression of Klotho was significantly downregulated in the kidney of aged WT mice (Figure 1A) (p<0.05 vs young WT). While, as expected, the expression of Klotho was dramatically decreased in the kidney of Klotho mutant (deficiency) mice (p<0.01 vs young WT, and p<0.05 vs aged WT (Figure 1A). Aging per se did not impact Klotho expression in Klotho mutant mice. Interestingly, we found that Klotho protein was also present in the nuclei as determined by Western blot using the nuclear extracts from the kidney tissue (Figure 1B). Consistently, the level of nucleus Klotho protein expression was higher in the kidney of young WT compared to aged WT mice (Figure 1B) (p<0.05). However, the level of nucleus Klotho protein expression was similar in the kidney of young and aged Klotho-deficient mice (Figure 1B). The level of cytoplasm Klotho was very low in the kidney and was not statistically different among groups (Figure 1C).
Figure 1. Downregulation of Klotho expression in the kidney of aged and Klotho mutant mice.
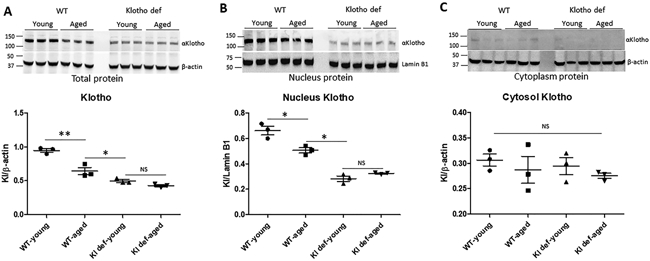
(A) Western blot analysis of Klotho expression using total protein samples isolated from mouse kidneys, and quantitation of αKlotho expression using β-actin as intenal loading control. (B) Nuclear distribution of αKlotho protein ana q uantitation of nucleus αKlotho using lamin B1 as intenal comtrol. (C) Cytosol level of αKlotho protein and quantitatio of cytoplasm distribution of αKlotho using β-actin as loading control. N=4 mice. Data are expressed as the mean ± S.D. * p < 0.05, or ** p<0.01 by unpaired t-test and multiple groups by one-way analysis of variance.
Expression of lysine-specific demethylase was downregulated in the aging kidney
To study the role of epigenetic modification of histone in Klotho gene expression in the aging kidney, we compared the expression of histone 3 lysine-specific demethylase UTX and JMJD3 in the kidney of young WT, aged WT, and Klotho-deficient (young and aged) mice, respectively (Figure 2). We found that expression level of UTX was similar in the kidney of young WT and aged WT mice, suggesting that expression of UTX was not affected by aging in the kidney (Figure 2A). Interestingly, the expression of UTX was significantly lower in the kidney of young and aged Klotho-deficient mice than that of WT controls (Figure 2A) (p<0.05). In contrast, expression of JMJD3, a major demethylase of H3K27me3 in the kidney, was significantly decreased in the aged WT mice (Figure 2B), suggesting that aging downregulates JMJD3 expression in the kidney. Similar to UTX, expression of JMJD3 was also decreased in the kidney of both young and aged Klotho-deficient mice compared to young WT mice (Figure 2B) (p<0.05). The level of JMJD3 was comparable among aged WT and Klotho-deficient mice (Figure 2B). This finding suggests that Klotho deficiency results in downregulation of UTX and JMJD3 demethylase in the kidney.
Figure 2. Epigenetic upregulation of H3K27me3 in aged kidneys.
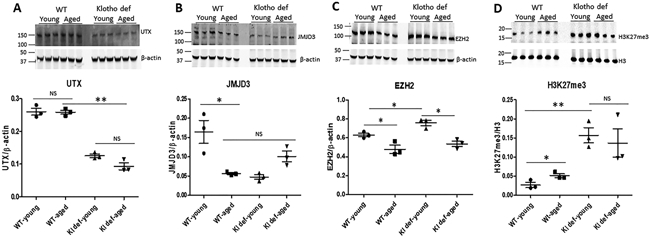
(A) Expression of UTX in mouse kidneys and quantitation of UTX protein using β-actin as internal loading control. (B) Expression of JMJD3 in mouse kidneys and quantitation of JMJD3 protein using β-actin as internal loading control. (C) Expression of EZH2 in mouse kidneys and quantitation of JMJD3 expression using β-actin as internal loading control. (D) Level of H3K27me3 in mouse kidneys and quantitation of H3K27me3 using H3 as loading control. N=4 mice. Data are expressed as the mean ± S.D. * p < 0.05, or ** p<0.01 by unpaired t-test and multiple groups by one-way analysis of variance.
Considering EZH2 is one of the core components of polycomb repressive complex 2 (PRC2) that participates in histone 3 lysine 27 methylation, we measured expression of EZH2 in the kidney. As shown in Figure 2C, expression of EZH2 was significantly higher in the kidney of young mice (WT and Klotho mutant) than that of in aged WT and aged Klotho-deficient mice (p<0.05), suggesting that expression of EZH2 is mainly regulated by aging rather than by Klotho.
To further study if histone methylation is influenced by aging and/or by Klotho deficiency, we examined H3K27me3 levels in mouse kidneys. We found that H3K27me3 levels were significantly increased in kidneys of aged WT mice (Figure 2D), suggesting that aging upregulates H3K27me3 levels. H3K27me3 levels were also increased significantly in kidneys of both young and aged Klotho-deficient mice (Figure 2D, p<0.01), indicating that Klotho deficiency upregulates H3K27me3 levels. The level of H3K27me1, H3K27me2, H3K27AC, and H3K36me was not affected by aging or Klotho deficiency (data not shown).
H3K27me3 directly bound to Klotho promoter and suppressed Klotho gene expression
To study if manipulation of PRC2 activity regulates Klotho gene expression, we isolated and cultured primary renal tubular cells (RTC) in the presence of EED226 (specific inhibitor of PRC2) and GSK343 (specific inhibitor of EZH2). We demonstrated that EED226 decreased expression of H3K27me3 in RTC in a dose-dependent manner (Figure 3A). While, expression of sKlotho (sKL, ~65 kDa) was significantly increased in RTC after EED226 treatment (Figure 3A). GSK343 also decreased H3K27me3 and increased sKL expression in a dose-dependent manner (Figure 3B). Notably, we only detected the short form Klotho (sKL) in primary cultured RTC.
Figure 3. H3K27me3 bound to Klotho promoter and downregulated Klotho gene expression.
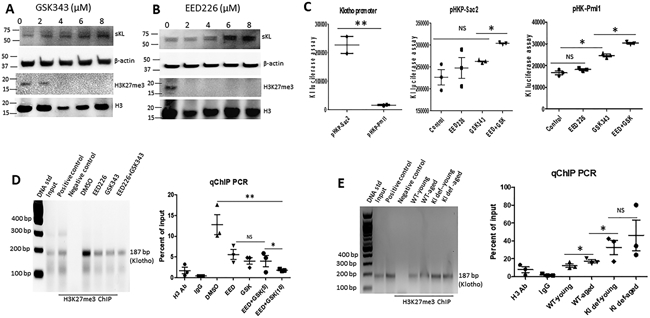
(A) Dose-dependent upregulation of sKL and downregulation of H3K27me3 by GSK343 in primary cultured mouse renal tubule cells. (B) Dose-dependent upregulation of sKL and downregulation of H3K27me3 by EED226 in primary cultured mouse renal tubule cells. (C) Klotho promoter, basal promoter activity of human Klotho gene is controlled by the DNA sequence located between Sac2 and Pml1 site of Klotho promoter region; pHK-Sac2, effect of EED226 and GSK343 on the promoter activity of pHKP-Sca2 construct; pHK-Pml1, upregulation of Klotho promoter activity drived by pHKP-Pml1 construct in the presence of PRC2 inhibitor EED226 and/or GSK343. (D) ChIP assay of H3K27me3 binding to Klotho basal promoter region and ChIP qPCR quantitation of the effect of EED226 and/or GSK343 on H3K27me3 binding to Klotho promoter in vitro. (E) ChIP assay of H3K27me3 binding to Klotho basal promoter region in vivo and ChIP qPCR quantitation of H3K27me3 binding to Klotho promoter in the kidney of WT and Klotho mutant mice in vivo. N=4. Data are expressed as the mean ± S.D. * p < 0.05, or ** p<0.01 by unpaired t-test and multiple groups by one-way analysis of variance.
To test whether H3K27me3 regulates Klotho gene expression at the transcriptional level, we transiently transfected the luciferase construct driven by a human Klotho promoter into RTC cells 22. pHKP-Sac2 (−240 bp KL basal promoter) showed a strong promoter activity, which was 10-fold higher than pHKP-Pml1 (−1270 bp KL promoter) (Figure 3C), suggesting that there is a repressor binding to the Klotho promoter region located between Pml1 and Sac2 sites. To examine if H3K27me3 downregulates Klotho expression through this region, pHKP-Sac2 or pHKP-Pml1 transfected RTC cells were treated with EED226, GSK343, or combination of both for 2 days, then luciferase assay was performed using the cell lysates from these RTC cells. We found that treatment of EED226 and/or GSK343 had little effect on the promoter activity of pHKP-Sac2, but increased promoter activity of pHKP-Pml1 (Figure 3C), indicating that H3K27me3 regulates Klotho expression at the transcriptional level, possibly through binding to the region between Pml1 and Sac2 of Klotho promoter.
To confirm the direct role of H3K27me3 in Klotho expression, we performed a ChIP assay using H3K27me3 antibody and inhibitor treated RTC cells. We demonstrated that H3K27me3 directly bound to Klotho promoter (Figure 3D). Inhibition of EZH2 by GSK343 or PRC2 by EED226 reduced H3K27me3 binding to the Klotho promoter region determined by ChIP-qPCR, respectively (Figure 3D). Furthermore, we first demonstrated that H3K27me3 physically bound to the Klotho promoter in vivo, which was significantly elevated in the kidney of aged WT and Klotho mutant mice compared to that of young WT mice (Figure 3E).
The mTOR signaling pathway was impaired in the aging kidney.
To better understand the pathways, such as mTOR signaling, that may modulate aging and aging-related diseases 30, We ask if reduced Klotho expression in aged WT mice or in Klotho mutant mice per se impairs mTOR signaling in the aging kidney. Consistent with previous studies31, 32, we found that mTOR was hyperphosphorylated in the kidney of aged WT and Klotho mutant mice regardless the levels of Klotho expression (Figure 4A). However, the level of total mTOR (t-mTOR) was similar among each group of mice, suggesting that activation of p-mTOR in the kidney of aged WT or Klotho mutant mice is the result of aging per se rather than downregulation of Klotho. Of note, aging-mediated hyperphosphorylated mTOR failed to activate downstream target P706SK (Figure 4B), indicating that the loss of function of the mTORC1 signaling occurred in the kidney of aged WT and aged Klotho mutant mice. Accordingly, the level of p-AKT was also increased in the kidney of aged WT and aged Klotho mutant mice compared to their young controls, respectively (Figure 4C). Interestingly, the ratio of p-AKT/t-AKT was decreased in young Klotho mutant mice and increased in aged Klotho mutant mice compared to young WT mice (Figure 4C), suggesting that Klotho deficiency differentially regulates AKT activity.
Figure 4. The SGK1 survival signaling was repressed in the aging kidney.
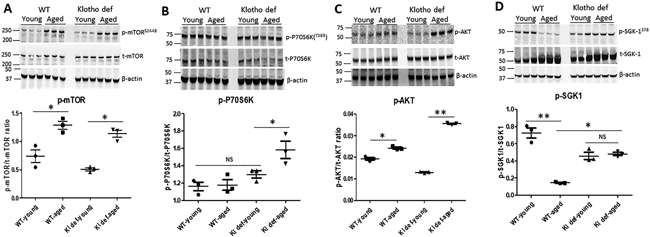
(A) Expression of phosphorylated mTOR and total mTOR in mouse kidneys and ratio of p-mTOR to t-mTOR. (B) Expression of phosphorylated P70S6K and total P70S6K in mouse kidneys and ratio of p-P70S6k to t-P70S6K. (C) Expression of phosphorylated AKT and total AKT in mouse kidneys amd ratio of p-AKT to t-AKT. (D) Expression of phosphorylated p-SGK1 and total SGK1 in each group of mouse kidney, and ratio of p-SGK1 to t-SGK1. N=4. Data are expressed as the mean ± S.D. * p < 0.05, or ** p<0.01 by unpaired t-test and multiple groups by one-way analysis of variance.
To further test if the impaired mTOR signaling affects SGK1 (serum- and glucocorticoid-induced protein kinase 1) activation, we measured phosphorylation of SGK-1 in kidneys of WT and Klotho-deficient mice. Indeed, we found that total SGK1 (t-SGK1) was significantly increased in kidneys of aged WT and in both young and aged Klotho mutant mice (Figure 4D). As a result, the ratio of p-SGK1/t-SGK1 was significantly decreased in the kidney of aged WT and Klotho mutant mice, respectively (p<0.01), suggesting that the SGK1 signaling was suppressed in aging kidneys.
FOXO3a was accumulated in the kidney of aged WT and Klotho mutant mice
FOXO3a belongs to the O subclass of the forkhead family of transcription factors. To study if suppression of SGK-1 signaling causes accumulation of FOXO3a in the aging kidney, we measured the expression of FOXO3a and its downstream target genes p53, p21, and a cell senescence marker p16. We found that expression of FOXO3a (t-FOXO3a) was significantly increased in the kidneys of aged WT and both young and aged Klotho mutant mice (Figure 5A). While, the level of phosphorylated FOXO3a (p-FOXO3a) was similar among each group of mice, leading to a significant decrease in the ratio of p-FOXO3a/t-FOXO3a in the kidneys of aged WT and both young and aged Klotho mutant mice compared to young WT mice (Figure 5A). Expression of p53 and p21 was significantly upregulated in the kidney of aged WT (Figure 5B). Expression of p53 also was significantly increased in the kidney of Klotho mutant (young and aged) mice compared to WT young mice without affecting the expression level of p21 (Figure 5B). Furthermore, expression of p16 was significantly upregulated in parallel with increased FOXO3a in kidneys of aged and Klotho mutant mice, respectively (Figure 5C).
Figure 5. Accumulation of FOXO3a led to upregulation of cell senescence gene p53 and p16 in the aging kidney.
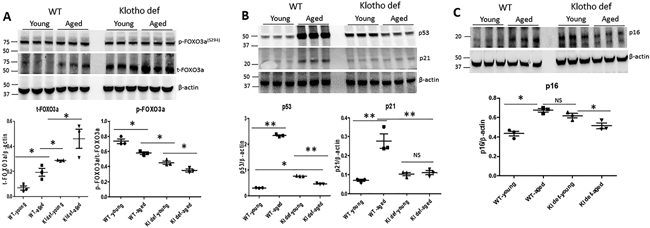
(A) Expression of phosphorylated FOXO3a and total FOXO3a in mouse kidneys, ratio of t-FOXO3a to β-actin and ratio of p-FOXO3a to t-FOXO3a. (B) Expression of p53 and p21 in mouse kidneys and quantitation of p53 and p21 expression in mouse kidneys. (C) Expression of p16 and quantitation of p16 in mouse kidneys. N=4.. Data are expressed as the mean ± S.D. * p < 0.05, or ** p<0.01 by unpaired t-test and multiple groups by one-way analysis of variance.
DISCUSSION
This study generated several interesting findings that may advance our understanding of the regulation of klotho expression in aged kidneys. First, we demonstrated that H3K27me3 is an important histone modification that may downregulate Klotho gene expression in aged kidneys. Using in vitro studies and complementary animal models, we showed that H3K27me3 is enriched in and physically associated with the Klotho promoter region in renal tubule cells and regulates Klotho gene expression directly (Figure 3). We extended this finding by demonstrating that aging-associated upregulation of H3K27me3 is largely due to decreased expression of lysine-specific demethylase (JMJD3) in the aging kidney.
Interestingly, protein expression of UTX and JMJD3 was also downregulated in the kidney of both young and aged Klotho mutant mice (Figure 2). Deficiency of Klotho shortened lifespan in mice accompanied by an array of disorders normally seen with human aging1, 33. Many of these aging-like disorders result from Klotho deficiency-induced hyperphosphatemia1, 34. Of note, Klotho is mainly expressed in the kidney which largely contributes to Klotho-induced antiaging traits1, 26, 35-37. Aging downregulates Klotho expression in the kidney (Figure. 1). However, how aging may regulate Klotho gene expression and how Klotho deficiency may modulate kidney aging remains unclear. Using ChIP assay, we found that H3K27me3 is enriched in the basal promoter region of the Klotho gene. We demonstrated that aging downregulated Klotho expression in the kidney due, at least partially, to increased methylation of H3K27 (H3K27me3). H3K27me3 may directly regulate Klotho gene expression because inhibition of PRC2 (histone trimethyltransferase) decreased H3K27me3 leading to increased Klotho expression in cultured primary renal tubule cells.
In addition to its function as a co-receptor for fibroblast growth factor 23 (FGF23), Klotho may also play a role in the regulation of aging-related gene(s) expression partly through regulation of UTX/JMJD3 and H3K27 methylation in the aged kidney. We showed that high level of αKlotho was detected in the nuclei of kidney cells (Figure. 1). The level of nuclear Klotho was decreased in the kidney of aged WT mice which was accompanied by a reduction of lysine specific demethylase JMJD3. Consistently, both UTX and JMJD3 expression was significantly downregulated in the kidney of young and aged Klotho mutant mice which was associated with an elevated H3K27me3 level. This study cannot determine whether Klotho deficiency downregulated UTX/JMJD3 directly or indirectly through the aging effect resulted from Klotho deficiency.
Epigenetic factors control gene expression and activity in response to environmental arduousness, which lead to changes in the epigenetic landscape that impact kidney function and aging through highly conserved signaling pathways, including the mTOR and insulin/insulin-like growth factor pathways. The mTOR functions as a component of two large complexes, mTORC1 in coordination with mTORC2 play critical role in regulating cell growth and homeostasis38, 39. Several lines of evidence show that the mTOR signaling regulates aging40-44, and inhibition of this pathway extends lifespan in various models44-47. In this study, we showed that activation of the mTOR signaling was age-dependent but Klotho-independent, since the level of p-mTOR in the kidney was similar between young WT and young Klotho mutant mice. This finding suggests that the impaired signaling of mTOR may not be mediated by downregulation of Klotho in the kidney of aged mice. Indeed, we found that elevation of p-mTOR failed to phosphorylate P70S6K, a key downstream target of mTORC1, in the kidney of aged WT mice. The level of p-AKT and p-SGK1, main downstream targets of mTORC2, failed to increase in the kidney of aged WT mice.
Furthermore, we demonstrated that the repressed SGK-1 survival signaling is a common pathway in aged WT and Klotho mutant mice. Although expression of SGK-1 (t-SGK1) was upregulated, SGK-1 activity (p-SGK1) is downregulated in the aged kidney (Figure 4) resulting in a decrease in SGK1-mediated phosphorylation of FOXO3a (p-FOXO3a) and accumulation of FOXO3a (t-FOXO3a) in the nucleus of renal cells of aged kidneys (Figure 5). We further showed that upregulation of FOXO3a was associated with increased expression of p53, p16, and p21 (aging or senescence markers) in aged kidneys in vivo. SGK1, which shares homology with AKT, was also shown to be regulated by mTORC2. In contrast to AKT, which retains a basal activity when mTORC2 is inhibited, SGK1 activity is totally revoked in aged kidneys. Because SGK1 and AKT phosphorylate FOXO3a on common sites, it is possible that the lack of p-SGK1 activity in aging kidney cells reduces the level of phosphorylation of FOXO3a leading to cell growth arrest (aging). By contrast, suppression of the SGK1 signaling observed in the kidney of Klotho mutant mice is likely due to Klotho deficiency because phosphorylation of SGK1 (p-SGK1) by FGF23/FGFRs signaling requires Klotho as a co-receptor in the kidney cells1, 48. Therefore, FGF23/FGFRs/Klotho may constitute an upstream signaling of SGK1 survival pathway.
While many factors may be involved in the complex pathogenesis of aging, the results of this study demonstrated that epigenetic upregulation of H3K27me3 and downregulation of Klotho gene expression may contribute to kidney aging. Although we showed that suppression of SGK1 signaling is a common aging pathway in kidney aging and Klotho deficiency-induced kidney aging, this study did not address whether suppression of the SGK1 signaling is mediated directly by the impaired mTOR pathway or indirectly by decreased Klotho expression in the kidney of aged WT mice.
Klotho gene deficiency causes salt-sensitive hypertension49. Klotho deficiency also contributes to aging-associated hypertension36. This study demonstrates, for the first time, that the aging-associated decline in Klotho expression may be due to epigenetic upregulation of H3K27 methylation mediated by JMJD3. Klotho gene deficiency caused dysregulation of the SGK1 signaling which can lead to upregulation sodium retention through upregulation of epithelial sodium channel (ENaC) and NaCl co-transporter (NCC) in the renal tubular cells50. Importantly, the changes in the SGK1-FOXO3a signaling associated with Klotho gene deficiency may contribute to aging-associated kidney remodeling. Thus, understanding of these regulatory pathways in renal tubule cells may provide new insights into the pathophysiological processes associated with Klotho deficiency, including hypertension and renal disorders in the context of aging and chronic kidney disease (CKD).
Perspectives
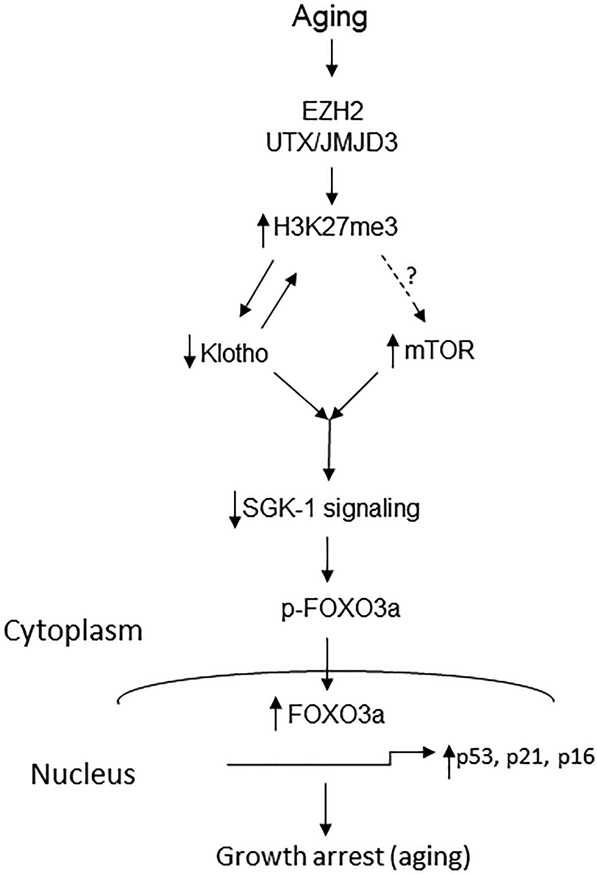
In summary, we found that epigenetic modification of H3K27 regulates Klotho expression in the kidney of aged mice. H3K27me3 is enriched in the Klotho promoter region and downregulates Klotho gene expression in the aged kidney. We propose a new schema (Figure 6), where aging exerts renal effects through upregulation of H3K27me3 and downregulation of Klotho in the renal tubules, and hyperphosphorylation of mTOR which is independent of Klotho status. Normal aging and Klotho-deficiency-mediated aging share a common pathway through the impaired SGK1 survival signaling leading to upregulation of FOXO3a, p53, p21, and p16 in the kidney of aged WT and Klotho mutant mice. This novel epigenetic pathway regulates Klotho-dependent activation of SGK1 in aged kidneys. Thus, hormonal action of Klotho may be an alternative approach for activating SGK1 survival signaling for treating aging-mediated kidney disorders. A new study is warranted for testing the effect of aging on these molecular and epigenetic changes in female mice.
Figure 6. Schematic model of epigenetic regulation of Klotho and mTOR pathway in aging kidney.
Supplementary Material
Novelty and Significance.
- What is new?
- This study reports for the first time that H3K27me3 may regulate Klotho gene expression in aging kidneys.
- This study establishes a new theory that Klotho may regulate the SGK-1/FOXO3 signaling pathway.
- What is relevant?
- There is currently no effective intervention for aging-associated kidney dysfunction and hypertension.
- This study reveals that H3K27-specific demethylase JMJD3 may be a novel therapeutic target for aging-associated kidney disorders and hypertension.
-
Summary
This study provides the first evidence that H3K27me3 may epigenetically regulate Klotho gene expression in aging kidneys. Downregulation of Klotho may impair the SGK-1/FOXO3 signaling pathway contributing to aging-associated kidney disorders and hypertension.
Acknowledgments
Source of Funding
This work was supported by NIH R01 AG049780, AG062375, HL122166, HL118558, and HL116863.
Footnotes
CONFLICTS OF INTEREST
None.
REFERENCE
- 1.Xu Y and Sun Z. Molecular basis of Klotho: from gene to function in aging. Endocr Rev. 2015;36:174–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han X, Yang J, Li L, Huang J, King G and Quarles LD. Conditional Deletion of Fgfr1 in the Proximal and Distal Tubule Identifies Distinct Roles in Phosphate and Calcium Transport. PLoS One. 2016;11:e0147845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuro OM. The Klotho proteins in health and disease. Nat Rev Nephrol. 2019;15:27–44. [DOI] [PubMed] [Google Scholar]
- 4.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, Wolf M, Brand M, Takahashi M, Kuro OM, Hill JA and Moe OW. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stenvinkel P and Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis. 2013;62:339–51. [DOI] [PubMed] [Google Scholar]
- 6.D’Urso A and Brickner JH. Mechanisms of epigenetic memory. Trends Genet. 2014;30:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henikoff S and Greally JM. Epigenetics, cellular memory and gene regulation. Curr Biol. 2016;26:R644–8. [DOI] [PubMed] [Google Scholar]
- 8.Schuettengruber B, Bourbon HM, Di Croce L and Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell. 2017;171:34–57. [DOI] [PubMed] [Google Scholar]
- 9.Sen P, Shah PP, Nativio R and Berger SL. Epigenetic Mechanisms of Longevity and Aging. Cell. 2016;166:822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin C, Li J, Green CD, Yu X, Tang X, Han D, Xian B, Wang D, Huang X, Cao X, Yan Z, Hou L, Liu J, Shukeir N, Khaitovich P, Chen CD, Zhang H, Jenuwein T and Han JD. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011;14:161–72. [DOI] [PubMed] [Google Scholar]
- 11.Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X, Ma H, Yang Y, Tong W, Liu F, Liu Z, Zhang Y, Liu R, Zhu ZJ and Liu N. Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumgart M, Groth M, Priebe S, Savino A, Testa G, Dix A, Ripa R, Spallotta F, Gaetano C, Ori M, Terzibasi Tozzini E, Guthke R, Platzer M and Cellerino A. RNA-seq of the aging brain in the short-lived fish N. furzeri - conserved pathways and novel genes associated with neurogenesis. Aging Cell. 2014;13:965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, Arrowsmith CH and Schapira M. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One. 2013;8:e83737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD and Berger SL. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freund A, Laberge RM, Demaria M and Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012;23:2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiels PG, McGuinness D, Eriksson M, Kooman JP and Stenvinkel P. The role of epigenetics in renal ageing. Nat Rev Nephrol. 2017;13:471–482. [DOI] [PubMed] [Google Scholar]
- 18.Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R, Zhao TC, Liu N, Bayliss G, Dworkin LD and Zhuang S. Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Expression. J Am Soc Nephrol. 2016;27:2092–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gesek FA, Wolff DW and Strandhoy JW. Improved separation method for rat proximal and distal renal tubules. Am J Physiol. 1987;253:F358–65. [DOI] [PubMed] [Google Scholar]
- 20.Kamiyama M, Garner MK, Farragut KM and Kobori H. The establishment of a primary culture system of proximal tubule segments using specific markers from normal mouse kidneys. Int J Mol Sci. 2012;13:5098–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Hauwaert C, Savary G, Gnemmi V, Glowacki F, Pottier N, Bouillez A, Maboudou P, Zini L, Leroy X, Cauffiez C, Perrais M and Aubert S. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS One. 2013;8:e66750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung D, Xu Y and Sun Z. Induction of anti-aging gene klotho with a small chemical compound that demethylates CpG islands. Oncotarget. 2017;8:46745–46755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han X, Xiao Z and Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem. 2015;290:20101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Fan J, Wang S and Sun Z. Secreted Klotho Attenuates Inflammation-Associated Aortic Valve Fibrosis in Senescence-Accelerated Mice P1. Hypertension. 2018;71:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Lin Y and Sun Z. Deficiency in the anti-aging gene Klotho promotes aortic valve fibrosis through AMPKalpha-mediated activation of RUNX2. Aging Cell. 2016;15:853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ullah M and Sun Z. Klotho deficiency accelerates stem cells aging by impairing telomerase activity. J Gerontol A Biol Sci Med Sci. 2019;74:1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Y and Sun Z. In Vivo Pancreatic beta-Cell-Specific Expression of Antiaging Gene Klotho: A Novel Approach for Preserving beta-Cells in Type 2 Diabetes. Diabetes. 2015;64:1444–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao D, Zuo Z, Tian J, Ali Q, Lin Y, Lei H and Sun Z. Activation of SIRT1 Attenuates Klotho Deficiency-Induced Arterial Stiffness and Hypertension by Enhancing AMP-Activated Protein Kinase Activity. Hypertension. 2016;68:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varshney R, Ali Q, Wu C and Sun Z. Monocrotaline-Induced Pulmonary Hypertension Involves Downregulation of Antiaging Protein Klotho and eNOS Activity. Hypertension. 2016;68:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson SC, Rabinovitch PS and Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shavlakadze T, Zhu J, Wang S, Zhou W, Morin B, Egerman MA, Fan L, Wang Y, Iartchouk O, Meyer A, Valdez RA, Mannick JB, Klickstein LB and Glass DJ. Short-term Low-Dose mTORC1 Inhibition in Aged Rats Counter-Regulates Age-Related Gene Changes and Blocks Age-Related Kidney Pathology. J Gerontol A Biol Sci Med Sci. 2018;73:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ and Klickstein LB. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6:268ra179. [DOI] [PubMed] [Google Scholar]
- 33.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R and Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. [DOI] [PubMed] [Google Scholar]
- 34.Kuro-o M Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Current opinion in nephrology and hypertension. 2006;15:437–41. [DOI] [PubMed] [Google Scholar]
- 35.Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Ostman Wernerson A, Lanske B, Olauson H and Larsson TE. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen K and Sun Z. Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell. 2018:e12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen K, Zhou X and Sun Z. Haplodeficiency of Klotho Gene Causes Arterial Stiffening via Upregulation of Scleraxis Expression and Induction of Autophagy. Hypertension. 2015;66:1006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mamane Y, Petroulakis E, LeBacquer O and Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–22. [DOI] [PubMed] [Google Scholar]
- 39.Proud CG. mTORC1 signalling and mRNA translation. Biochem Soc Trans. 2009;37:227–31. [DOI] [PubMed] [Google Scholar]
- 40.Jia K, Chen D and Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–906. [DOI] [PubMed] [Google Scholar]
- 41.Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L and Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. [DOI] [PubMed] [Google Scholar]
- 42.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V and Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S and Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6. [DOI] [PubMed] [Google Scholar]
- 44.Medvedik O, Lamming DW, Kim KD and Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM and Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A and Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E and Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrukhova O, Slavic S, Smorodchenko A, Zeitz U, Shalhoub V, Lanske B, Pohl EE and Erben RG. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol Med. 2014;6:744–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou X, Chen K, Lei H and Sun Z. Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J Am Soc Nephrol. 2015;26:121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faresse N, Lagnaz D, Debonneville A, Ismailji A, Maillard M, Fejes-Toth G, Naray-Fejes-Toth A and Staub O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol. 2012;302:F977–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original data including gel blots used to generate the analysis presented in the manuscript is publicly arcived at the following link: https://datadryad.org/stash/share/SIC0Ze7i6Oh1JbU2hdzv7Jyv-W7Yc_VH7IdJdO6qIuo. doi:10.5061/dryad.fn2z34tq0
Animals and isolation of renal tubule cells
All procedures and protocols used in this study were approved by the Institutionla Animal Care and Use Committee of the University of Tennessee Health Science Center (protocal #19-051). Young WT (6 months), aged WT (30 months), as well as young (6 months) and aged (19-21 monhs) Klotho mutant mice were used. Wild type (WT) male mice were fed with regular rodent chow. Klotho mutant male mice were fed with low phosphate diet for 3 months after weaning and switched to regular diet for the rest period of time during the study.
Renal tubule cells were isolated as discrebed previously with some modifications19-21. Briefly, all mice (3 months) were euthanized using a CO2 chamber and kidneys were immediately harvested and washed in sterile ice-cold Hanks’ Balanced Salt Solution (HBSS). Kidneys were decapsulated in HBSS and transferred to Hibernate A medium (BrainBits LLC, USA). Then, kidenys were bisected and the inner medullary was removed. Cortical and outer mudullary tissues were dissected in order to obtain 1mm3 fragments. The tissues were washed twice with Hibernate A medium and then digested in the presence of 1 mg/ml collagenase (type II, Sigma) for 30 min at 37°C. Renal tubues (proximal and distal) were dissected and collected under microscope, and filterred through 70μM and 40μM cell strainer consequently. Renal tubules were washed twice with DMEM/F12 containg 1% penicillin/streptomycin and centrifuged for at 1000 rpm for 5 min. Cells were cultured in 75 cm2 plastic flasks with complete cultue medium (DMEM/F12 medium containng 20% FBS, 15 penicillin/streptomycin, ITS-insulin:transferrin:selenium) at 37°C under 5% CO2 in a humidified incubator. The culture medium was changed after 24h to remove non-adhernet cells.
Promoter analysis
Promoter activity was assessed as we described previously22. Briefly, primary renal tubule cells were plated 18 h before transfection and fed with fresh medium 4 h and pre-treated with EED226 (Axon MechChem, Netherlands ) and/or GSK343 (Sigma) for 2h before transfection. Klotho promoters (pHKP-Sac2 and pHKP-Pml1 ) were constructed into a pGL3 basic reporter gene (Promega, Madison, WI). Klotho reporter plasmid DNAs were introduced into renal tubule cells using cationic liposomes (LipofectAMINE2000, Life technologies, Grand Island, NY). Transfection of plasmid DNAs was carried out for 16–18 h, and then cells were washed twice with phosphate-buffered saline and incubated in fresh medium containing 20% fetal calf serum in the presence of EED226 and GSK343 for another 2 days. To standardize the transfection efficiency, 0.1 μg of pRL-CMV vector (pRL Renilla reniformisluciferase control reporter vector; Promega) was cotransfected in all experiments. Cells were harvested 72 h after transfection and lysed in 50 μl of reporter lysis buffer (Promega). A luciferase assay (20 μl of cell lysed) was performed using a dual luciferase assay kit (Promega), and activity was measured with an Optocomp 1 luminometer (MGM Instruments, Inc., Hamden, CT). Promoter activity (mean ± S.D. of triplicate samples in relative fold changes) is represented by reporter activity normalized to pRL-CMV control. Graphs represent typical results of three-four separate experiments.
Chromatin Immunoprecipitation (ChIP) assays
ChIP assays were performed with a kit from Cell Signaling Technology (Danvers, MA) according to the manufacturer’s instructions with the following modifications as descried previously22, 23. Briefly, approximately 4 X 107 RTC cells in 150 cm2 culture dishes were cross-linked in 1% formaldehyde solution (Fisher Scientific, Pittsburgh, PA) for 10 minutes at room temperature, stopped cross-linking by adding 1M glycine for 5 minutes at room temperature. Cells were washed twice with ice-cold 1× phosphate-buffered saline solution (PBS) (Life Technologies) and harvested in 1-ml of 1× PBS + protease inhibitor cocktail (PIC) + phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO). Cells were then pelleted by centrifugation at 1500 rpm for 5 minutes, 4°C and the pellet was resuspended in 10 ml ice-cold buffer A with DTT, PIC, and PMSF on ice for 10 minutes. Cell nuclei were pelleted by centrifugation at 3000 rpm for 5 minutes at 4°C. Nuclei pellet was then washed in 10 ml ice-cold buffer B with DTT, and resuspened in 1.0 ml buffer B with DTT. Micrococcal nuclease was added to the nuclei and incubated for 20 minutes at 37°C. Digestion was stopped by adding 100μl of 0.5 M EDTA and nuclei was pelleted by centrifugation at 13,000 rpm for 1 minute at 4°C. Nuclear pellet was resuspened in 0.5 ml 1x CHIP buffer with PIC and PMSF for sonication using the VirSonic Ultrasonic cell disrupter 100 (VirTis, Gardiner, NY) to shear the DNA to an average length of 300 to 500 base pairs (six, 15-s bursts on ice). Samples were stored at −80°C before use. For chromatin immunoprecipitation, 200 μl of the cross-linked chromatin preparation were added to 800 μl of ChIP buffer with PIC. Immunoprecipitations were carried out with 2 μg of antibodies (H3K27me3 from Cell Signaling). Histone H3 rabbit mAb (2μg) and normal rabbit IgG (1 μg) were used as positive or negative controls, respectively. After overnight immunoprecipitation on a rotator at 4°C, ChIP samples were then washed and eluted and DNA was un-crosslinked with NaCl and subsequently treated with proteinase K, Tris-Hcl and EDTA. DNA was purified and subjected to PCR amplification of Klotho promoter DNA containing putative binding sites for H3K27me3 using specific primers (Mouse: forward primer 5’-CAG GAT GGA GGC CAC AGG AT-3’, reverse primer 5'- TGA TTA TCC AGA GCA GGC GCC −3').
Western blot analysis
The western blot procedure was described previously24-29. Briefly, samples from mouse kidney and mouse renal tubule cells were prepared in T-per tissue extraction buffer and M-per mammalian extraction buffer (ThermoFisher), respectively. Nuclear and cytoplasmic proteins were isolated by using a NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific, Rockford, IL). Protein contents in the samples were quantified and stored at −80°C until use. For electrophoresis, 40μg kidney protein and 20μg renal tubule cell lysates were loaded onto Expressplus Page 4-12% gel (GenScript, USA). Proteins were separated at 120 V for 60 minutes and transferred to nitrocellulose membrane using Trans-Blot Turbo (BioRad). Membranes were blocked with 5% BSA blocking buffer in TBST (Thermo Scientific, Rockford, IL) for 2 hours and then incubated with primary antibody (Klotho, 1:500, R&D; UTX, JMJD3, EZH2, H3K27me3, H3, SGK-1, p-SGK-1, p-mTOR, t-mTOR, p-AKT, t-AKT, p-FOXO3a, t-FOXO3a, p-70S6K, and t-70S6K, 1:1000 from Cell Signaling Technology and p53 1:1000, p21 1:1000, and p16 1:1000 from Abcam #189034) with gentle agitation overnight at 4 °C. After 3 washes with TBST (15 min once and 2x 5 min), membrane was incubated with secondary antibody in 5% BSA blocking buffer at room temperature for 1 hour. Membrane was then washed 4 times (15 min and 2x 5 min) and subjected to ECL (BioRad) and analyzed with BioRad ChemiDoc, MP imaging system. Western bolt using β-actin antibody or Lamin B antibody (Cell Signaling) was used as internal control of protein loadings of cytoplasmic or nuclear, respectively.
Statistical analysis
We evaluated differences between two groups by unpaired t-test and multiple groups by one-way analysis of variance. All values are expressed as means ± SD. All computations were performed using GraphPad Prism5 (GraphPad Software Inc. La Jolla, CA, USA).