

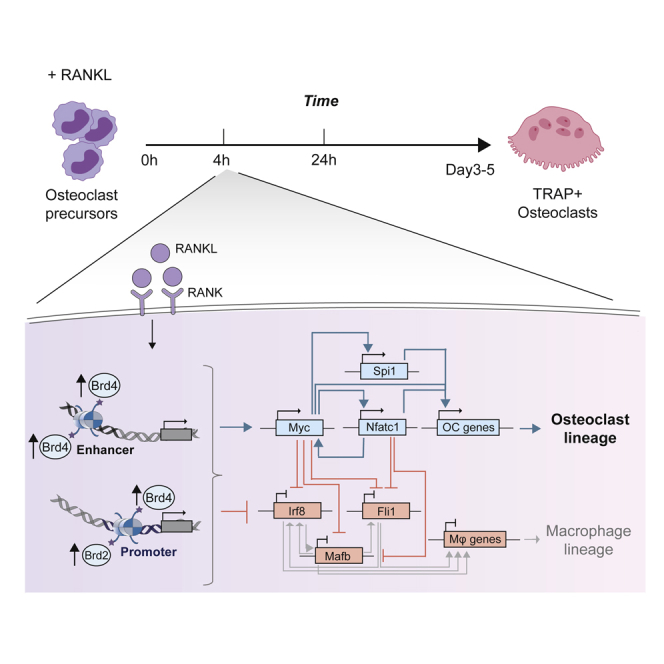
Summary
Osteoclast (OC) development in response to nuclear factor kappa-Β ligand (RANKL) is critical for bone homeostasis in health and in disease. The early and direct chromatin regulatory changes imparted by the BET chromatin readers Brd2-4 and OC-affiliated transcription factors (TFs) during osteoclastogenesis are not known. Here, we demonstrate that in response to RANKL, early OC development entails regulation of two alternative cell fate transcriptional programmes, OC vs macrophage, with repression of the latter following activation of the former. Both programmes are regulated in a non-redundant manner by increased chromatin binding of Brd2 at promoters and of Brd4 at enhancers/super-enhancers. Myc, the top RANKL-induced TF, regulates OC development in co-operation with Brd2/4 and Max and by establishing negative and positive regulatory loops with other lineage-affiliated TFs. These insights into the transcriptional regulation of osteoclastogenesis suggest the clinical potential of selective targeting of Brd2/4 to abrogate pathological OC activation.
Subject areas: Biological Sciences, Developmental Biology, Bioinformatics, Omics, Transcriptomics
Graphical Abstract
Highlights
-
•
Upregulation of osteoclast lineage commitment TFs occurs <24hr after RANKL stimulation
-
•
Enhanced binding of Brd2&4, but not Brd3, is required for osteoclast fate commitment
-
•
Brd2&4 occupancy is associated with super-enhancer reprogramming and TF rewiring
-
•
Myc is the epicenter of the TF regulatory network driving osteoclastogenesis
Biological Sciences; Developmental Biology; Bioinformatics; Omics; Transcriptomics
Introduction
Osteoclasts (OCs) are hematopoietic lineage-derived, bone-resorbing cells that in conjunction with bone-forming osteoblasts regulate bone homeostasis in physiology and in disease (Charles and Aliprantis, 2014; Teitelbaum, 2000). Aberrant hyper-activation of OCs underpins common and serious morbidity in osteoporosis and in cancers such as multiple myeloma, metastatic breast, and prostate cancer (Edwards et al., 2008; Le Pape et al., 2016).
OC lineage commitment requires binding of the lineage defining cytokine receptor activator of nuclear factor kappa-Β ligand (RANKL) to its receptor RANK expressed in myelomonocytic progenitors in the bone marrow. RANKL-RANK interaction activates a cascade of downstream signal transduction pathways that converge to transcriptional programmes that initiate differentiation and the acquisition of OC-specific processes of cell fusion and polylkaryon generation (Kukita et al., 2004; Yang et al., 2008). This is followed by the maturation events of cytoskeleton re-organization and secretion of collagen-degrading enzymes and hydroxyapatite-dissolving proton ions that enable OCs to resorb organic and inorganic bone (Azuma et al., 2000; Soysa et al., 2012; Teitelbaum, 2000).
OC-specific lineage commitment and differentiation requires a compendium of transcription factors (TFs) that are upregulated in response to RANKL, such as the Fos family, Nfkb, NFATc1, Spi1/Pu.1 (Franzoso et al., 1997; Takayanagi et al., 2002; Wagner, 2002). OC commitment also requires a simultaneous repression of existing macrophage/monocytic, inflammatory transcriptional programmes that involve downregulation of TFs such as Irf8, Mafb, and Bcl6 (Kim et al., 2007; Zhao et al., 2009). These TFs in turn are under the repressive regulation of Blimp1 (Prdm1) which is upregulated in response to RANKL (Kurotaki et al., 2020; Miyauchi et al., 2010; Nishikawa et al., 2010).
Similar to other transcriptional processes that define cell lineage commitment, the OC-affiliated TF would be expected to alter gene transcription through changes in the activity of proximal and distal regulatory DNA elements namely enhancers. The latter are often cell type specific, and their activity is regulated by Brd2-4, the bromodomain and extraterminal domain (BET) proteins which act as chromatin readers and transcriptional activators by binding to acetylated histones (Hnisz et al., 2013) (Chiang, 2009; LeRoy et al., 2008; Rahman et al., 2011).
Previous work demonstrated the requirement for Brd4 for OC activation in response to RANKL while BET protein inhibitors inhibit OCs in vivo as well as in vitro and protect from malignancy- or ovariectomy-induced bone disease (Lamoureux et al., 2014; Park-Min et al., 2014). Further, Brd4 binds to the promoters of lineage-defining TFs, such as Nfatc1, cFos, and NF-κB, while pharmacological inhibition by BET inhibitors represses transcription of these TFs (Lamoureux et al., 2014). More recently, the role of Myc in OC differentiation was highlighted and was shown to be required for OC development by regulating transcription of Nfatc1 and Errα (Bae et al., 2017; Lamoureux et al., 2014) (Park-Min et al., 2014).
Despite this progress, how individual BET proteins, singly or in combination, regulate commitment to OC lineage genome-wide at the chromatin level and their interplay with Myc have not been addressed (Baud'huin et al., 2017; Lamoureux et al., 2014; Park-Min et al., 2014). Here, we first identify the initiating regulatory events leading to OC lineage commitment and demonstrate a prominent and non-redundant role of Brd2 and 4 in the chromatin changes underlying these processes. We provide high resolution analysis and integration of the genome-wide binding profile of BET proteins in conjunction with chromatin accessibility maps and early dynamic transcriptome changes in response to RANKL and to BET protein inhibitors. We thus define the combinatorial cross-talk between BET proteins and their interaction with proximal and distant chromatin regulatory areas to define the early stages of OC lineage commitment. Further, we generate the gene regulatory networks (GRNs) driving OC lineage commitment and identify critical TF components defining OC fate determination. We validate the role of Myc as a major hub in these OC-specific GRNs and discover the range of its transcriptional targets and their predicted functions.
Results
A dynamic map of early transcriptome changes in response to RANKL
To model OC lineage commitment in response to RANKL, we used the mouse macrophage cell line RAW264.7 which upon RANKL exposure differentiates to fully bone-resorbing OCs (Collin-Osdoby and Osdoby, 2012; Collin-Osdoby et al., 2003; Raschke et al., 1978). We confirmed that differentiation of RAW264.7 cells, as well as of murine primary bone marrow and purified bone marrow (Lineage−CD115+cKit+) progenitor cells (Hu et al., 2011) toward OCs, was inhibited by the pan-BET protein inhibitor I-BET151 (Figures S1A and S1B) (Baud'huin et al., 2017; Park-Min et al., 2014).
Transcriptome changes brought about 24h after RANKL treatment of RAW264.7 cells correlated highly with those induced by RANKL on OC developing from primary bone marrow (BM) progenitors/precursors (Bae et al., 2017) (Figure S1C), thus attesting further to the suitability of RAW264.7 cells as a model of OC development.
While transcriptome analysis of developing OC has been previously reported at >24h post-RANKL exposure (An et al., 2014; Bae et al., 2017; Davidson et al., 2020; Park-Min et al., 2014), the immediate transcriptional responses triggered by RANKL have not been explored yet. Therefore, to capture the first transcriptional waves during RANKL-driven OC lineage commitment, we performed RNA-seq of RAW264.7 cells at 0, 4, 14, and 24h upon RANKL, I-BET151, and RANKL + I-BET151 treatment (Figure 1A). In addition, we investigated the genome-wide recruitment dynamics of the Brd2-4 proteins that are associated with early transcriptional changes at 4h after treatment (Figure 1A).
Figure 1.

Profiling the dynamics of transcriptional responses underlying early osteoclast development in response to RANKL
(A) Schematic representation of the study outline. RNA-seq analysis performed at 0, 4, 14, and 24 hr after RANKL induction in RAW 264.7 cells. Gene clusters (C1-C12), comprising four main modes of transcriptional response: early repressed; late repressed; early activated; late activated.
(B–D) Heatmap (B) and line graphs (C) illustrate for the transcriptional patterns observed in the 12 clusters; the expression patterns of genes with prominent role in osteoclastogenesis are also highlighted here. Overrepresentation analysis (D) using EnrichR online tool depicts the involvement of key pathways in the four modes of responses.
Upon RANKL treatment, we identified 12 clusters (C1-C12) of differentially expressed genes (DEGs; Figures 1B and 1C) temporally categorized as early (4hr) and late (14 and 24hr). The largest, C6 (late repressed), comprises 780 genes and includes TFs (e.g. Irf8, Mafb, and Bcl6) whose repression is required for extinguishment of the non-permissive macrophage/monocytic transcriptional programmes, thus allowing OC lineage commitment (Figure 1C and Table S1). The early activated C10 contains the OC differentiation master regulators Nfatc1, Fos, and Myc, followed by the upstream repressor of macrophage lineage Blimp1/Prdm1. Using quantitative PCR (qPCR), we confirmed this dynamic, contrasting Myc and Irf8 mRNA expression pattern in murine primary bone-marrow-derived pre-OCs (Figure S1D).
Principal component (PCA) and intesity-ratio (MA) analysis showed a profound impact of I-BET151 in abrogating RANKL-dependent transcriptome changes as early as 4hr after treatment (Figure S1E). These findings confirm the exquisite sensitivity of the RANKL-dependent OC-defining transcriptional programme to BET inhibitors and its dependency on BET proteins. A high-resolution dynamic analysis of Nfatc1 expression further exemplified the dramatic impact of BET inhibitors in the transcriptional regulation of OC lineage commitment (Figure S2A).
Pathway enrichment analysis of early activated clusters C7-C10 was consistent with effects of RANKL-mediated osteoclastogenesis and activation of downstream pathways such as NF-κB, TNFα, and AP-1 (i.e., c-Jun/c-Fos). Of note, among the early activated clusters, and in particular in C7, gene repression at 14 and 24 hr is preceded by increased expression at 4 hr. Pathway enrichment analysis of the 156 C7 genes showed enrichment for putative gene targets of Irf8 in bone marrow macrophages and for genes that define macrophage identity (Figure S2B). This suggests that before definitive repression of the macrophage transcriptional programme occurs, RAW264.7 cells assume a state of transient transcriptional transition to an immature macrophage identity.
Late repressed genes reflected loss of phagocytic capacity and notably decreased expression of genes predicted to be regulated by Irf8 in mouse macrophage cells (Figure 1D).
Notably, genes such as Dcstamp, Ocstamp, and Atp6v0d2 that are required for polykaryon formation, a cellular process that starts at least 48 h after RANKL exposure (Kukita et al., 2004; Wu et al., 2009; Yang et al., 2008), also map to the same early cluster as the master TF (Figure 1B and 1C). In addition, genes such as Ctsk and Mmp9, which are associated with acquisition of OC bone resorptive capacity, a cellular feature present in developing OC after 3 days (Boyce, 2013; Hsu et al., 1999; Sundaram et al., 2007; Troen, 2006), are upregulated at 4–14 hr (Figures 1B and 1C). Therefore, our early post-RANKL transcriptome analysis reveals that transcription of genes required for functions of maturing or mature OC is already active within a few hours following RANKL exposure, a finding that we validated by qPCR Sin primary pre-OCs for Dcstamp and Ocstamp (Figure S1D).
Overall, inspection of all 12 clusters reveals that transcriptome analysis at 14 h after RANKL treatment captures both the early and late transcriptional events that define RANKL-induced OC lineage commitment and differentiation that entails activation of the osteoclastogenic program followed by repression of the macrophage program.
Of note, while double RANKL/I-BET151 treatment reverses the RANKL-induced transcriptional changes, I-BET151 alone represses both alternative cell fate programmes (Figure S2C; OC C10, C12 gene clusters and macrophage C6 cluster) consistent with the role of BET proteins in determining cell fate decisions in development (Huijbregts et al., 2019) (Fernandez-Alonso et al., 2017; Hnisz et al., 2013; Lee et al., 2017). We validated this effect of RANKL/I-BET151 and I-BET151 treatment in primary pre-OCs by measuring expression of genes of interest (Figure S2D). Indeed, while expression of Irf8 increased significantly after 4 hr of treatment with RANKL/I-BET151 and I-BET151, that of Myc, Dcstamp, and Ocstamp was significantly decreased, consistent with the repressive role of BET proteins in the regulation of Irf8 and activating role in the regulation of the other three genes.
Impact of RANKL and I-BET151 on early chromatin recruitment of Brd2-4
To map RANKL- and I-BET151-induced changes in Brd2-4 chromatin binding, we obtained the genome-wide binding profile of the three BET proteins at steady state (S) and at 4h after RANKL (R), RANKL plus I-BET151 (RI), or I-BET151(I) treatment using Chromatin Immunoprecipitation followed by sequencing (ChIP-seq).
By first plotting the average ChIP signal against fold change between R and S conditions (R/S), we identified the differentially enhanced or decreased peaks in R condition for each of the BET proteins. This analysis (Figure 2A and Table S2) demonstrated that RANKL induces mostly increased binding of Brd2 and 4 with minor and nearly equal increase and reduction in binding of Brd3. Overall, 45% of Brd2 and 52% of Brd4 peaks in RANKL-treated cells represented enhanced differentially binding sites (DBSs), while the corresponding fraction for Brd3 was <21% (Figure 2B). However, I-BET151 induced a profound reduction in the binding of all three BET proteins, more pronounced for Brd4 (99%) and less so for Brd2 (75%) and Brd3 (71%) (Figure 2B). Of note, in contrast to Brd3 and 4, a preferential increase in biding of Brd2 was observed in I-BET-only treated cells but not in RANKL+I-BET cells (Figures 2A and 2B). Interestingly, among BET protein genes, expression of Brd2 is the only one that increases in response to I-BET151 treatment (Figures S3A and S3B).
Figure 2.

Early epigenomic alterations in chromatin binding of Brd2,3, and 4 in response to RANKL & I-BET
Differential binding of Brd2, Brd3, and Brd4 in response to RANKL (R), I-BET (I), or a combination of the two (RI) was analyzed 4 h after treatment. S: steady state
(A) Ratio-intensity plots of average ChIP signal and differential binding between conditions (log2 scale) are displayed for each BRD (columns) across treatment contrasts (rows).
(B) Number of differentially bound sites (DBSs) for BET proteins across pairwise treatment comparisons (from A). All DBS are colored by associated condition; non-significantly altered regions are colored gray.
(C) Genomic annotation for all DBS per condition.
(D) Relative enrichment of Brd2,3,4 on Brd2 (top) and Brd4 (bottom) DBSs. Fold change over input in a 20kb window around peak center was plotted for each BRD in S and R conditions, direction of change in Brd2/4 is indicated in lower left corner.
(E) Distribution of DBSs with increased Brd2, Brd4, or dual Brd2,4 enrichment upon RANKL induction.
(F) Average signal profiles for Brd2 and Brd4 across combined differential binding categories, direction of change in Brd2/4 is indicated in top left corner of each group.
(G) Genomic annotation of Brd2/4 binding locations relative to gene features in Brd2/Brd4 common or specific binding.
Genomic feature annotation of the DBS revealed that dynamic changes of Brd4 mostly take place in intronic and intergenic areas, while Brd2 and 3 binding changes are mainly enriched in promoter-overlapping areas (Figure 2C). This distribution chimes with the previously described role of Brd4 as a marker of intergenic and intronic enhancers (Loven et al., 2013; Roe et al., 2015). Gain of Brd2 or Brd4 binding occurs with, but mostly without, concurrent Brd4 and Brd2 increased binding, respectively, and in either case, it does not involve changes in Brd3 binding (Figures 2D–2F). We therefore obtained the union of these Brd2 & 4 peak sets to form four DBS categories for further analysis (i.e., unaltered, Brd2&4 common, Brd2 specific, Brd4 specific; Figure 2F).
Feature annotation demonstrated preferential differential binding of sole Brd4 in intergenic and intronic areas, likely enriched in putative enhancers, while Brd2 DBSs alone and in combination with Brd4 DBSs were enriched preferentially in promoter-overlapping areas (Figure 2G).
Overall, these findings show that RANKL brings about changes in the BET protein chromatin-binding landscape at the very first stages of OC commitment (4h), chiefly detected as enhanced binding of Brd2 and Brd4, and such changes are exquisitely sensitive to pharmacological inhibition.
Enhanced Brd2&4 chromatin occupancy and super-enhancer reprogramming in early osteoclastogenesis
To gain further insights into how dynamic changes in Brd2&4 chromatin binding affect the RANKL-induced transcriptome changes, by annotating DBSs to the nearest DEG, we plotted dynamic transcriptional changes associated with each BET protein DBS categories, i.e., common, Brd2 specific, or Brd4 specific. This analysis revealed that for each group, enhanced binding was associated with the same pattern of transcriptional changes that included up- as well as down-regulated genes (Figure S4A).
Functional annotation of DEGs upon RANKL treatment shows that each of the three DBS categories contributes to the same or unique pathways and TF networks that define OC development (Figure 3A). These included up-regulation of genes involved in Myc, TNF, Nfkb, MAPK, and other OC differentiation pathways, as well as repression of genes involved in commitment to the alternative monocyte-macrophage lineage, e.g. Runx1, Spi1, and Irf8 pathways.
Figure 3.

Differential Brd2 and Brd4 chromatin occupancy is linked to early transcriptional responses and super-enhancer reprogramming in osteoclast differentiation
(A) Top 10 enriched pathways for up- and down-regulated genes (4, 14, and 24hr after RANKL induction) associated with each Brd2/4 binding category (Brd2 specific, Brd4 specific, or shared); color indicates database source for gene set.
(B) Relative motif enrichment per Brd2/4 binding category against either unaltered regions to identify commonly enriched factors (top) or against common regions (middle, bottom) to identify Brd2/4-specific factors. Enriched motifs were grouped by similarity and assigned to putative TFs based on the expression level and differential expression at 4h. Size indicates proportion of regions containing the motif, and color indicates significance of expression change.
(C) Differential expression patterns (RANKL-treated vs untreated) for top 10 TFs predicted to be involved in Brd2/4 regulation of osteoclastogenesis.
(D) Super-enhancer (SE) calling in S, R, RI, and I conditions using H3K27ac ChIPseq.
(E) Epigenomic alterations in genetic loci of the hallmark osteoclastogenesis genes Irf8 and Myc, across treatments.
(F) Relative occupancy (R vs S, RI vs S, and I vs S) of Brd2,3,4 on SE sites (top); time course transcriptional changes associated with differential Brd2,4 binding after RANKL induction (bottom). One-way ANOVA test: ∗∗∗∗,p < 0.0001; n/s, not significant.
To explore how the dynamic changes in Brd2 and 4 binding might interact with the TF in mediating RANKL-induced transcriptome changes for each of the three DBS categories, we correlated changes in TF expression with proportion of DBS enriched in TF motifs (Figure 3B and 3C). This analysis predicted that at 4 hr, sole Brd2 and Brd4 rather than their combined enhanced binding was associated with motif enrichment of TFs that determine alternative cell fates, i.e., Irf8, Fli1, and Mafb, which are downregulated in response to RANKL versus the RANKL-activated, OC-activating TF Fosl1/2, Junb, Myc, Nfkb2, Spi1, and Relb.
Together, these data suggest a non-redundant epigenetic role of Brd2 and Brd4 in OC lineage commitment and differentiation in co-operation with lineage-defining TFs.
Next, by charting the chromatin H3K27ac enrichment load, we sought to identify super-enhancers (SEs), i.e., the regulatory elements that determine cell fate decisions by regulating and being regulated by lineage-affiliated TFs (Figure 3D) often in co-operation with Brd4 (Bae et al., 2017; Loven et al., 2013; Whyte et al., 2013). In total, we identified 728 SEs across different treatments, more than half of which were shared in all four conditions (Figures 3D and Table S3). The majority of SEs which regulate critical TFs (e.g. Irf8, Fos; Figures 3E and S4B) remained unaltered in terms of H3K27ac load across treatments; however, in a few SEs (including the one of Myc), H3K27ac load was reduced upon I-BET151 treatment (Figures 3D and 3E). By contrast, analysis of the BET protein binding load at these SEs confirmed significantly increased binding of both Brd2 and 4 (but not of Brd3) upon RANKL treatment, while I-BET151 treatment resulted in significantly reduced binding of Brd2-4 proteins at SEs (Figures 3E, 3F, and S4B). This effect of I-BET151 was more pronounced for Brd4 and is consistent with its previously described role in SEs (Loven et al., 2013).
Notably, increased binding of Brd2/4 at SEs was associated mostly with transcriptional repression (65% of DEGs) rather than activation (35% of DEGs) of the predicted target genes at 14 and 24 hr (Figure 3F).
Pathway enrichment of DEGs predicted to be upregulated upon differential binding of Brd2&4 to enhancers and SEs confirmed activation of OC differentiation and TNF-α/Nfkb pathways, respectively, while repression of Irf8 targets genes was predicted among down-regulated genes (Figures S4C and S4D)
Thus, in a non-redundant manner, Brd2 and Brd4 via their enhanced binding in response to RANKL and in co-operation of lineage-affiliated TFs determine cell OC lineage commitment and fate by exerting dual, repressive as well activatory transcriptional roles.
Myc is the epicenter of the OC lineage commitment gene regulatory network
By taking advantage of the ability afforded by ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) to identify footprints of DNA binding factors, we obtained and annotated genome-wide footprints of expressed TFs at promoters and enhancers in RANKL-treated RAW264.7 cells. This identified high confidence footprints of several OC-associated TFs (Figure S5A) and allowed construction of a TF-focused GRN.
By displaying the highest expression change than any other TF in response to RANKL (Figure S5B), Myc appears central in the TF-driven GRN of OC lineage commitment (Figure 4A). We confirmed by qPCR that overexpression of Myc in response to RANKL is (a) higher than that of Nfatc1 and (b) it is BET protein dependent, as shown by abrogation of Myc overexpression (Figure S5B) and reduction in Brd2/4 binding at the predicted Myc SE upon I-BET151 treatment (Figure 3E).
Figure 4.

Myc is a central component of the regulatory networks driving osteoclast-lineage commitment
(A) Chromatin accessibility-based construction of gene regulatory network that drives transcriptional changes during OC lineage commitment. Directed TF network diagram: arrow width indicates footprint score for originating TFs at target gene promoter/enhancer, and color indicates direction of expression change for originating TFs (red: upregulated and blue: downregulated). Node size indicates strength of expression change (-log10(padj)∗|log2FC|) and color indicates direction of change at 14 hr after RANKL treatment.
(B) Individual TF modules comprising core components of the overall network.
(C–E) (C) Heatmap representation of ChIP-seq against Myc and Max, along with (D) genomic annotation of significantly enriched areas and (E) thier motif analysis.
(F) Overrepresentation analysis of direct Myc gene targets (as predicted upon integrated cistrome-transcriptome analysis) using EnrichR.
(G) IGV snapshots of Fli1 and Spi1 genetic loci. From top to bottom: showing Brd2/4, Myc, Max and H3K27Ac ChIP-seq, ATAC-seq and Pu.1. and Irf8 ChiP-seq are from (Langlais et al., 2016).
A closer inspection of the GRN reveals that Myc is regulated by Nfatc1, Fosl2, and Nfkb2 while it is predicted to repress Irf8 and Mafb (Figures 4A, 4B, and S5C). Notably, Myc is predicted to establish positive feedback loops with TFs that promote OC differentiation (e.g., Nfatc1, Figure 4B) and a negative feedback loops with TFs that activate the macrophage program (e.g., Irf8 and Mafb, Figure 4B). These findings are consistent with previous low throughput analyses of the role of Brd2/4 proteins in regulating expression of both Myc and Nfatc1 in OC and the Myc requirement for Nfatc1 upregulation in response to RANKL (Lamoureux et al., 2014; Park-Min et al., 2014).
Despite the critical role of Myc in OC development, its genome-wide regulatory landscape in response to RANKL is not known. Herein, we first confirmed that Myc depletion by shRNA or by the Myc/Max dimerization inhibitor 10,058–F4 (Yu et al., 2016) inhibited osteoclastogenesis (Figures S6A and S6B).
Next, we performed ChIP-seq against Myc and its partner Max 4h after RANKL treatment in RAW264.7 cells. Myc binding was equally enriched between putative intergenic and intronic enhancers and proximal regulatory areas. We identified 560 Myc areas also co-bound by Max (Figures 4C and 4D and Table S4); these areas were highly enriched in Fli1, Fosl2, Pu.1/Spi1, Irf8, as well as Myc and Max motifs, supporting a co-operative function of these TFs with Myc in OC development at the genome-wide level (Figure 4E). Over-representation analysis of DEGs predicted to be regulated by Myc/Max binding showed activation of genes involved in ribosome-related pathways, as well as of previously known Myc/Max targets, and repression of Irf8, Runx1, and Spi1/Pu.1 targets (Figure 4F). Moreover, we identified the OC pioneer factor Spi1 and Fli1 as direct targets of Myc, as predicted by the presence of Myc footprints and ChIP-seq peaks at their corresponding SE (Figures 4A and 4G); interestingly, binding of Irf8 and Spi1 on the same SE was previously described to promote macrophage-lineage commitment (Langlais et al., 2016), while the alternative Spi1/Nfact1 partnership favors OC lineage commitment (Carey et al., 2018; Izawa et al., 2019) (Kurotaki et al., 2020).
Integrative epigenomic analysis of the Myc chromatin binding sites revealed significant increase of H3K27ac, Brd2, and Brd4 (but not Brd3) occupancy upon RANKL treatment (Figure S6C). Although no significant change in H3K27ac signal was detected upon RANKL + I-BET151 treatment, Brd2-4 binding signal significantly decreased on the same regions, more prominently for Brd4.
Finally, over-representation analysis of the post-RANKL variable genes predicted to be co-regulated by Myc/Max binding and enhanced binding of BET proteins showed again among upregulated genes the role of Myc in regulating previously identified Myc/Max target genes and in promoting ribosome biology (Figure S6D). Genes predicted to be down-regulated by Myc/Brd2,4 are enriched for Spi1/Pu.1 targets in macrophage cells (Langlais et al., 2016), further supporting a co-operative role of Spi1/Pu.1 with Myc in OC lineage commitment.
Together, these findings show a central role of Myc in OC lineage commitment in co-operation with Brd2/4 and lineage-defining TFs.
Discussion
Here, we dissected the immediate early epigenetic and transcriptional events that are regulated by BET proteins in response to RANKL during OC lineage commitment. Two main insights emerged from the dynamic analysis of the OC transcriptome. First, regulation of the two competing lineage programmes, i.e., repression of macrophage vs activation of the OC program is temporally dissociated, with the former following the latter. This is consistent with the previously described role of Blimp1, a RANKL-activated TF, in repressing Irf8- and Irf8-dependent macrophage-affiliated transcriptional programmes (Miyauchi et al., 2010; Nishikawa et al., 2010). Second, transcriptional activation of genes regulating late functions of OCs, i.e., cell-to-cell fusion and bone resorption, already takes place in the first 24 hr after RANKL exposure, thus providing a paradigm of a cellular differentiation trajectory imprinted in the beginning of the process. Future single-cell-based analyses of developing OCs would provide additional, high-resolution insights into these transcriptional processes.
The non-redundant genetic roles of Brd2 and Brd4 in osteoclastogenesis have been previously shown as has the ability of BET protein inhibitors to abrogate OC development by interfering with transcription of individual critical TF (Baud'huin et al., 2017; Lamoureux et al., 2014; Park-Min et al., 2014). Here, we establish that RANKL induces changes primarily in the binding patterns of Brd2 and Brd4, with the latter mainly focused on enhancers and SEs. While BET protein function has been primarily linked with gene activation (Chiang 2009), our findings clearly show that enhanced Brd2/4 binding in response to RANKL is predicted to mostly regulate repression of the existing macrophage program, highlighting the dual function of Brd2/4 in gene regulation.
Brd2 and Brd4 are predicted to regulate critical pathways for OC development on individual or shared basis, alluding, in line with previous genetic studies (Baud'huin et al., 2017), to a non-redundant functional role in osteoclastogenesis. Therefore, use of selective Brd2 or Brd4 inhibitors would be expected to abrogate pathological, RANKL-dependent OC activation with a more favorable toxicity profile than pan-BET inhibitors such as I-BET151.
Construction of TF-focused GRNs highlighted the role of positive and negative feedback loops of TF cross-regulation with Myc emerging as the most central TF in this process. Indeed, the BET protein-dependent increase in Myc expression is the highest among TFs responding to RANKL. While transcriptome analysis of Myc-deficient OC precursors suggested that Myc regulates metabolic processes such as oxidative phosphorylation in developing OC (Bae et al., 2017) whether relevant genes are directly regulated by Myc binding is not known. Here, we find that Myc is predicted to regulate pro-osteoclastogenic TF such as Nfatc1, Fosl2, and Nfkb2 by establishing positive regulatory loops. In addition, through repression of Irf8 and Mafb and in co-operation with Spi1/Pu.1, Myc participates in the repression of the macrophage transcriptional program required for OC development.
In summary, here we propose a non-redundant role of both Brd2 and 4 which cooperate with Myc to orchestrate early chromatin events and TF regulatory networks that determine OC lineage commitment.
Limitations of the study
Although our study maps the dynamic changes in transcriptional regulation during early osteoclastogenesis in both cell line and primary OC precursor cells, these changes represent average regulatory events at the cell population level and assume synchronous cellular responses upon cytokine stimulation and BET-protein inhibition. Additionally, single-cell epigenomic and transcriptomic analyses could provide further insights into understanding the dynamics of gene expression regulation during OC lineage commitment.
Resource availability
Lead contact
Further requests for resources and materials should be directed to and will be fulfilled by the Lead Contact, Anastasios Karadimitris (a.karadimitris@imperial.ac.uk).
Material availability
This study did not yield new unique reagents.
Data and code availability
Data are available from Gene Expression Omnibus database (Acc. Number: GSE160840).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
V.C., N.T., X.X. and K.P. were supported by the Blood Cancer UK and A.K. was supported by Kay Kendall Leukaemia Fund. We also acknowledge support from Imperial NIHR Biomedical Research Center Genomics Facility, and research funding from GlaxoSmithKline.
Author contributions
V.S.C. and N.T. designed study, performed experiments, and analyzed data. X.X., N.T., and M.R. analyzed data. A.C., K.P., A.K., and H.W.A. provided advice and ideas. A.K. designed study and supervised experiments. V.S.C., N.T., X.X., and A.K. wrote manuscript. All authors contributed to the final draft of the manuscript. N.S. and R.K.P. contributed in provision of compound and related information and reviewed data and the manuscript.
Declaration of interests
The work was in part supported by received research funding from GlaxoSmithKline to A.K. All the other authors declare no conflict of interest.R.K.P. and N.S. are employees and shareholders of GlaxoSmithKline, which is carrying out clinical development of epigenetic inhibitors.
Published: January 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101989.
Supplemental information
References
- An E., Narayanan M., Manes N.P., Nita-Lazar A. Characterization of functional reprogramming during osteoclast development using quantitative proteomics and mRNA profiling. Mol. Cell Proteomics. 2014;13:2687–2704. doi: 10.1074/mcp.M113.034371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma Y., Kaji K., Katogi R., Takeshita S., Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000;275:4858–4864. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- Bae S., Lee M.J., Mun S.H., Giannopoulou E.G., Yong-Gonzalez V., Cross J.R., Murata K., Giguere V., van der Meulen M., Park-Min K.H. MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha. J. Clin. Invest. 2017;127:2555–2568. doi: 10.1172/JCI89935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud'huin M., Lamoureux F., Jacques C., Rodriguez Calleja L., Quillard T., Charrier C., Amiaud J., Berreur M., Brounais-LeRoyer B., Owen R. Inhibition of BET proteins and epigenetic signaling as a potential treatment for osteoporosis. Bone. 2017;94:10–21. doi: 10.1016/j.bone.2016.09.020. [DOI] [PubMed] [Google Scholar]
- Boyce B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013;92:860–867. doi: 10.1177/0022034513500306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey H.A., Hildreth B.E., 3rd, Geisler J.A., Nickel M.C., Cabrera J., Ghosh S., Jiang Y., Yan J., Lee J., Makam S. Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Res. 2018;6:8. doi: 10.1038/s41413-018-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles J.F., Aliprantis A.O. Osteoclasts: more than 'bone eaters'. Trends Mol. Med. 2014;20:449–459. doi: 10.1016/j.molmed.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C.M. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol. Rep. 2009;1:98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin-Osdoby P., Osdoby P. RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Methods Mol. Biol. 2012;816:187–202. doi: 10.1007/978-1-61779-415-5_13. [DOI] [PubMed] [Google Scholar]
- Collin-Osdoby P., Yu X., Zheng H., Osdoby P. RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Methods Mol. Med. 2003;80:153–166. doi: 10.1385/1-59259-366-6:153. [DOI] [PubMed] [Google Scholar]
- Davidson R.K., Himes E.R., Takigawa S., Chen A., Horn M.R., Meijome T., Wallace J.M., Kacena M.A., Yokota H., Nguyen A.V., Li J. The loss of STAT3 in mature osteoclasts has detrimental effects on bone structure. PLoS One. 2020;15:e0236891. doi: 10.1371/journal.pone.0236891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards C.M., Zhuang J., Mundy G.R. The pathogenesis of the bone disease of multiple myeloma. Bone. 2008;42:1007–1013. doi: 10.1016/j.bone.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alonso R., Davidson L., Hukelmann J., Zengerle M., Prescott A.R., Lamond A., Ciulli A., Sapkota G.P., Findlay G.M. Brd4-Brd2 isoform switching coordinates pluripotent exit and Smad2-dependent lineage specification. EMBO Rep. 2017;18:1108–1122. doi: 10.15252/embr.201643534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G., Carlson L., Xing L., Poljak L., Shores E.W., Brown K.D., Leonardi A., Tran T., Boyce B.F., Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H., Lacey D.L., Dunstan C.R., Solovyev I., Colombero A., Timms E., Tan H.L., Elliott G., Kelley M.J., Sarosi I. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. U S A. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M., Bassett J.H., Danks L., Howell P.G., Xu K., Spanoudakis E., Kotsianidis I., Boyde A., Williams G.R., Horwood N. Activated invariant NKT cells regulate osteoclast development and function. J. Immunol. 2011;186:2910–2917. doi: 10.4049/jimmunol.1002353. [DOI] [PubMed] [Google Scholar]
- Huijbregts L., Petersen M.B.K., Berthault C., Hansson M., Aiello V., Rachdi L., Grapin-Botton A., Honore C., Scharfmann R. Bromodomain and extra terminal protein inhibitors promote pancreatic endocrine cell fate. Diabetes. 2019;68:761–773. doi: 10.2337/db18-0224. [DOI] [PubMed] [Google Scholar]
- Izawa N., Kurotaki D., Nomura S., Fujita T., Omata Y., Yasui T., Hirose J., Matsumoto T., Saito T., Kadono Y. Cooperation of PU.1 with IRF8 and NFATc1 defines chromatin landscapes during RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 2019;34:1143–1154. doi: 10.1002/jbmr.3689. [DOI] [PubMed] [Google Scholar]
- Kim K., Kim J.H., Lee J., Jin H.M., Kook H., Kim K.K., Lee S.Y., Kim N. MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood. 2007;109:3253–3259. doi: 10.1182/blood-2006-09-048249. [DOI] [PubMed] [Google Scholar]
- Kukita T., Wada N., Kukita A., Kakimoto T., Sandra F., Toh K., Nagata K., Iijima T., Horiuchi M., Matsusaki H. RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 2004;200:941–946. doi: 10.1084/jem.20040518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki D., Yoshida H., Tamura T. Epigenetic and transcriptional regulation of osteoclast differentiation. Bone. 2020;138:115471. doi: 10.1016/j.bone.2020.115471. [DOI] [PubMed] [Google Scholar]
- Lamoureux F., Baud'huin M., Rodriguez Calleja L., Jacques C., Berreur M., Redini F., Lecanda F., Bradner J.E., Heymann D., Ory B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014;5:3511. doi: 10.1038/ncomms4511. [DOI] [PubMed] [Google Scholar]
- Langlais D., Barreiro L.B., Gros P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med. 2016;213:585–603. doi: 10.1084/jem.20151764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.E., Park Y.K., Park S., Jang Y., Waring N., Dey A., Ozato K., Lai B., Peng W., Ge K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017;8:2217. doi: 10.1038/s41467-017-02403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pape F., Vargas G., Clezardin P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016;5:93–95. doi: 10.1016/j.jbo.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G., Rickards B., Flint S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi Y., Ninomiya K., Miyamoto H., Sakamoto A., Iwasaki R., Hoshi H., Miyamoto K., Hao W., Yoshida S., Morioka H. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J. Exp. Med. 2010;207:751–762. doi: 10.1084/jem.20091957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K., Nakashima T., Hayashi M., Fukunaga T., Kato S., Kodama T., Takahashi S., Calame K., Takayanagi H. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc. Natl. Acad. Sci. U S A. 2010;107:3117–3122. doi: 10.1073/pnas.0912779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park-Min K.H., Lim E., Lee M.J., Park S.H., Giannopoulou E., Yarilina A., van der Meulen M., Zhao B., Smithers N., Witherington J. Inhibition of osteoclastogenesis and inflammatory bone resorption by targeting BET proteins and epigenetic regulation. Nat. Commun. 2014;5:5418. doi: 10.1038/ncomms6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S., Sowa M.E., Ottinger M., Smith J.A., Shi Y., Harper J.W., Howley P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke W.C., Baird S., Ralph P., Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- Roe J.S., Mercan F., Rivera K., Pappin D.J., Vakoc C.R. BET bromodomain inhibition Suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell. 2015;58:1028–1039. doi: 10.1016/j.molcel.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soysa N.S., Alles N., Aoki K., Ohya K. Osteoclast formation and differentiation: an overview. J. Med. Dent. Sci. 2012;59:65–74. [PubMed] [Google Scholar]
- Sundaram K., Nishimura R., Senn J., Youssef R.F., London S.D., Reddy S.V. RANK ligand signaling modulates the matrix metalloproteinase-9 gene expression during osteoclast differentiation. Exp. Cell Res. 2007;313:168–178. doi: 10.1016/j.yexcr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Takayanagi H., Kim S., Koga T., Nishina H., Isshiki M., Yoshida H., Saiura A., Isobe M., Yokochi T., Inoue J. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S.L. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Troen B.R. The regulation of cathepsin K gene expression. Ann. N. Y Acad. Sci. 2006;1068:165–172. doi: 10.1196/annals.1346.018. [DOI] [PubMed] [Google Scholar]
- Wagner E.F. Functions of AP1 (Fos/Jun) in bone development. Ann. Rheum. Dis. 2002;61:40–42. doi: 10.1136/ard.61.suppl_2.ii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Xu G., Li Y.P. Atp6v0d2 is an essential component of the osteoclast-specific proton pump that mediates extracellular acidification in bone resorption. J. Bone Miner. Res. 2009;24:871–885. doi: 10.1359/JBMR.081239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M., Birnbaum M.J., MacKay C.A., Mason-Savas A., Thompson B., Odgren P.R. Osteoclast stimulatory transmembrane protein (OC-STAMP), a novel protein induced by RANKL that promotes osteoclast differentiation. J. Cell Physiol. 2008;215:497–505. doi: 10.1002/jcp.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Niu X., Jin F., Liu Z., Jin C., Lai L. Structure-based inhibitor design for the intrinsically disordered protein c-myc. Sci. Rep. 2016;6:22298. doi: 10.1038/srep22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Takami M., Yamada A., Wang X., Koga T., Hu X., Tamura T., Ozato K., Choi Y., Ivashkiv L.B. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 2009;15:1066–1071. doi: 10.1038/nm.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from Gene Expression Omnibus database (Acc. Number: GSE160840).