

Abstract
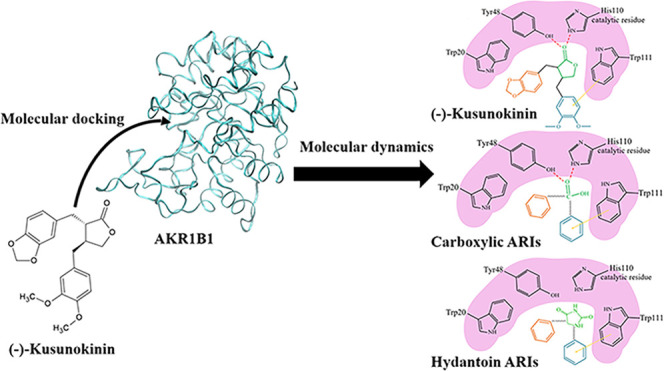
(−)-Kusunokinin performed its anticancer potency through CFS1R and AKT pathways. Its ambiguous binding target has, however, hindered the next development phase. Our study thus applied molecular docking and molecular dynamics simulation to predict the protein target from the pathways. Among various candidates, aldo-keto reductase family 1 member B1 (AKR1B1) was finally identified as a (−)-kusunokinin receptor. The predicted binding affinity of (−)-kusunokinin was better than the selected aldose reductase inhibitors (ARIs) and substrates. The compound also had no significant effect on AKR1B1 conformation. An intriguing AKR1B1 efficacy, with respect to the known inhibitors (epalrestat, zenarestat, and minalrestat) and substrates (UVI2008 and prostaglandin H2), as well as a similar interactive insight of the enzyme pocket, pinpointed an ARI equivalence of (−)-kusunokinin. An aromatic ring and a γ-butyrolactone ring shared a role with structural counterparts in known inhibitors. The modeling explained that the aromatic constituent contributed to π–π attraction with Trp111. In addition, the γ-butyrolactone ring bound the catalytic His110 using hydrogen bonds, which could lead to enzymatic inhibition as a consequence of substrate competitiveness. Our computer-based findings suggested that the potential of (−)-kusunokinin could be furthered by in vitro and/or in vivo experiments to consolidate (−)-kusunokinin as a new AKR1B1 antagonist in the future.
1. Introduction
Chemotherapy is commonly used in the treatment of cancer. Chemotherapeutic agent development is ongoing, so as to increase treatment efficacy and reduce adverse effects.1 The major source of synthetic chemotherapeutic agents is active plant-derived chemicals. Various types of natural products have been successfully developed into practical chemotherapeutic drugs.2
Lignans, a dimeric phenylpropanoid from natural products, provide a wide range of biological functions. In particular, flaxseed lignans have been revealed to have anticancer effects by multiple targets of the hallmarks of cancer.3 Well-known cases include lignan-based anticancer drugs, namely, etoposide and other podophyllotoxin semisynthetic derivatives.4,5 Another reported lignan, arctigenin and its glycoside, arctiin, hindered the cancer cell growth.6,7 Arctigenin prevented the osteoarthritis progression via targeting of the PI3K/AKT pathway.8 Arctiin possessed antiangiogenic effects on human myeloma cells via inhibition of STAT3 phosphorylation.9 In addition, arctigenin and arctiin also exhibited anti-inflammatory effects through the inhibition of reactive oxygen species (ROS) production and COX-2.10
(−)-Kusunokinin, one of the reported lignan compounds, exhibited anticancer activity on various human cancer cell lines, including breast cancer, colon cancer, and cholangiocarcinoma.11 This compound inhibited tumor growth with no side effects and exhibited the normal complete blood count parameters and clinical chemistry of the renal and liver function test in the breast cancer rat model.12 The anticancer mechanism of (−)-kusunokinin could occur through the partial binding with CSF1R, resulting in the suppression of AKT and its downstream proteins, including c-myc, cyclinB1, and cyclinD1.12,13
Computational modeling of (−)-kusunokinin was carried out to investigate its protein target based on a similar case of bursehernin,13,14 a structural isomer of (−)-kusunokinin. Colony stimulating factor 1 receptor (CSF1R) was reported as a possible target of these compounds, in which the stabilized π–π interaction with aromatic amino acids played a crucial role. This observation suggested that the selectivity of (−)-kusunokinin depended on the π–π stacking with aromatic amino acids at the binding pocket, such as tyrosine, phenylalanine, or tryptophan. In addition, the (−)-kusunokinin structure could perform interactions via various hydrogen-bond acceptor counterparts, namely, an oxygen atom(s) of the methoxy group (−OCH3) or/and an oxygen atom at the ketone group on the γ-butyrolactone ring.13,14
As we had already noticed some clues in the interaction and specificity of (−)-kusunokinin, we thus speculated that another protein containing an aromatic-rich domain could behave with (−)-kusunokinin in the same manner. Moreover, due to the partial binding with CSF1R, the downstream molecules of the CSF1R and AKT signaling pathways were selected for screening for the possibility of (−)-kusunokinin target proteins in this study. Herein, we used the molecular docking technique to predict an additional target protein of (−)-kusunokinin. The time-dependent conformational behavior of the (−)-kusunokinin–target complex was elucidated using molecular dynamics simulation. Then, the binding manner of (−)-kusunokinin was also evaluated and compared with the reported inhibitors. Finally, our present work provided more information regarding the target protein of (−)-kusunokinin with aldo-keto reductase family 1 member B1 (AKR1B1) and the downstream molecules of CSF1R and AKT. (−)-Kusunokinin bound this protein at the anion binding pocket. These findings could become constructive for furthering the (−)-kusunokinin development phase as a specifically targeted anticancer inhibitor.
2. Methods
2.1. Protein Structure Preparation
Structures of 41 proteins associated with the CSF1R and AKT pathways were retrieved from the Protein Data Bank (https://www.rcsb.org/) in a PDB format file. All atoms were selected based on the highest occupancy. Crystallographic waters were removed and polar hydrogen atoms were added using AutoDockTools 1.5.6 (ADT).15 The protein structures were written into a PDBQT format file.
2.2. Ligand Preparation
The structure files of (−)-kusunokinin and the other ligands used were obtained from the PubChem database. (−)-Kusunokinin was taken from a structure with CID 384876. The crystal-bound (native) ligands were directly separated from the crystallography structure using the Visual Molecular Dynamics (VMD) package.16 All ligands used in this study are summarized in Table S1. The structure was then converted into a PDB file using the Biovia Discovery Studio 2019 Client.17 Hydrogen atoms were added to all ligands. Gasteiger charges and default atom parameters were automatically assigned from ADT. Finally, the structure was written into the PDBQT file format using ADT.
2.3. Molecular Docking Parameters
Molecular docking studies were performed using the AutoDock4 program. During the process, the protein structure was set as a rigid molecule with a flexible ligand. The docking site on the protein target was defined by a grid box of 120 × 120 × 120 cubic angstrom (Å3), with the grid spacing at the center of the protein structure. Other parameters followed the default values in ADT. Fifty genetic algorithm (GA) runs with a population size of 200 were executed for conformational sampling. The conformation with the lowest binding energy was considered as the best pose. The interaction of the best docked pose was compared with the native ligands in the crystallographic structure.
2.4. Molecular Docking Study and Binding Energy Calculation
The reproducibility of the AutoDock4 program was validated via a self-redocking experiment of the native reference ligand. The all-atom root-mean-square deviation (RMSD) cutoff of less than 2 Å between docked and native crystal structures was set as the criterion for a successful procedure. The AutoDock4 program score was recorded from the binding free energy between the ligand and the protein.18,19
2.5. Molecular Dynamics Simulation
The AKR1B1 structure selected was the PDB structure code 1US0 (a resolution of 0.66 Å). All water and other solvents were removed. Only AKR1B1 with the cofactor dihydro-nicotinamide-adenine-dinucleotide phosphate (NADPH) was retained. All polar hydrogen in the best ligand docked pose was eliminated. AMBER16 force field was exploited to model the drug–AKR1B1 complex, similar to previous reports of AKR1B1 molecular dynamics study.20−23 The hydrogen atom was instead replaced via the leap module implemented in the AMBER16 package.24 An ionizable side chain of the amino acid was considered at pH 7. The side chain in lysine or arginine contained +1e charge, while the glutamate and aspartate side chains contained −1e charge. The imidazole ring in histidine was neutral. No disulfide was observed in the AKR1B1 structure. All ligand molecules, namely, (−)-kusunokinin, AKR1B1 inhibitors, and a substrate (PGH2), were parameterized using the Gaussian16 package,25 based on calculations of restrained electrostatic atomic potential (RESP). NADPH parameters were adopted from a previous report.26
The structure of ligand-free AKR1B1 or AKR1B1 with the best docked conformer was solvated by TIP3P water with a distance of 14 Å from the surface, leading to approximately 12000 water molecules. Sodium ion (Na+) was used to neutralize the system, and 18 NaCl pairs were included to yield a 0.1 M NaCl solution. The aforementioned AKR1B1–ligand complex was used as an initial coordinate for the molecular dynamics process. The AKR1B1 solution was first equilibrated in a canonical (NVT) ensemble using Langevin dynamics at 310 K (37 °C). The harmonic restraint potential was applied to the positions of AKR1B1, NADPH, and the ligand, with a force constant of 200, 100, 50, 20, and 10 kcal/mol-Å2, respectively. Each restrained NVT ensemble lasted for 200 ps using a time step of 1 fs. The system was switched into an NPT ensemble via the Berendsen algorithm at 310 K and 1.013 bar.
The molecular dynamics (MD) simulation was run for 150 ns with a time step of 2 fs. An MD trajectory with 2500 equidistant snapshots was obtained. The last 60-ns-MD conformation (1500 snapshots) was taken for conformation and binding analysis. Root-mean-square deviation (RMSD) calculation and structure visualization were conducted using the VMD package. Root-mean-square fluctuation (RMSF) was computed, and the pattern of distance geometry was acquired using the cpptraj module.
2.6. Binding Free Energy Evaluation
Average binding free energy (ΔG) was calculated using the molecular mechanics/generalized Born-surface area (MM/GBSA)27−30 method from MD trajectories. Average molecular mechanics energy was obtained from the AMBER functional formula.18,28,31
3. Results and Discussion
3.1. (−)-Kusunokinin Target Screening
From the selected 41 CSF1R and AKT associated proteins, the top 10 for the binding energy of (−)-kusunokinin are shown in Figure 1. All binding energies of (−)-kusunokinin and the native ligands with 41 selected proteins are also summarized in Table S2. (−)-Kusunokinin exhibited the best predicted binding potential toward aldo-keto reductase family 1 member B1 (AKR1B1), with a binding energy of −11.11 kcal/mol. The other candidate was the dual specificity mitogen-activated protein kinase kinase2 (MEK2), with a binding energy of −10.14 kcal/mol.
Figure 1.

Binding energy of (−)-kusunokinin with protein candidates in the CSF1R and AKR1B1 pathways. A number denotes the binding energy in kcal/mol.
In spite of the closeness in the predicted binding energy between AKR1B1 and MEK2, AKR1B1 piqued our interest not only for its lower binding energy but also due to some supporting data regarding its lignan-based inhibitor. AKR1B1, an NADPH-dependent enzyme, is a target for diabetic complication treatment through the regulation of the polyol metabolic glucose pathway.32 Recently, AKR1B1 was found in various human cancers,33 and it could be the novel target for cancer aggressiveness reduction.34−36 Arctigenin and arctiin exhibited potency in the aldose reductase (AKR1B1) inhibition.37−39 Since the lignans share a feature for the aromatic ring structure, the π–π stacking with the aromatic binding site of AKR1B1 would then be expected to be like CSF1R.13,14
3.2. Preliminary Atomistic View of (−)-Kusunokinin on AKR1B1 Binding
(−)-Kusunokinin was suitably inserted in the AKR1B1 active pocket cleft, which was surrounded by several hydrophobic and aromatic residues (Figure 2). The site was categorized into three apparent pockets: an anion binding pocket (Tyr48, His110, and Trp111), a small hydrophobic pocket (Trp20, Val47, and Tyr48), and a large hydrophobic specific pocket (Trp79, Trp111, Phe115, Phe122, Trp219, Cys298, Leu300, Cys303, and Tyr309). These active pockets resembled previously reported AKR1B1 binding with β-aminophenylpropanoic acid derivatives.23
Figure 2.

(−)-Kusunokinin binding site on AKR1B1. The site is composed of anion binding pocket (orange), small hydrophobic pocket (green), and large hydrophobic pocket (yellow).
Later, a molecular docking study of (−)-kusunokinin against AKR1B1 was performed and compared with the other available AKR1B1 substrates,40,41 commercial carboxylic acid aldose reductase inhibitors (ARIs), commercial hydantoin ARIs, and some suggested potential ARIs, respectively.42−44 The result is summarized in Table 1. Interestingly, the binding energy of (−)-kusunokinin (−11.11 kcal/mol) was in a similar range to the reported ARIs, from −6.92 to −11.54 kcal/mol. Moreover, (−)-kusunokinin provided a better predicted energy than substrates (PGH2 and retinol), substrate analogs (UVI2008 and PGA1), and some reported potential ARIs. (−)-Kusunokinin was likewise inserted in the active pocket with a similar position and orientation to the well-known ARIs (Figure 3).
Table 1. Predicted Binding Energy of Docked Ligands with AKR1B1.
| ligands | ΔG (kcal/mol) | ligands | ΔG (kcal/mol) |
|---|---|---|---|
| (−)-kusunokinin | –11.11 | suggested potential ARIs | |
| carboxylic ARIs | oleanolic acid | –10.90 | |
| zenarestat | –11.54 | AD5467 | –10.84 |
| zopolrestat | –11.21 | γ-mangostin | –9.63 |
| sulindac | –11.19 | arctiin | –9.85 |
| lidorestat | –10.92 | arctigenin | –9.55 |
| IDD1219 | –10.86 | 10C | –9.32 |
| epalrestat | –10.57 | caffeic acid phenethyl ester | –9.08 |
| IDD740 | –10.52 | substrates | |
| IDD594 | –9.83 | Retinol | –9.91 |
| IDD552 | –9.53 | PGH2 | –9.71 |
| tolrestat | –9.38 | substrate analogs | |
| tolmetin | –9.22 | UVI2008 | –10.71 |
| alrestatin | –8.92 | PGA1 | –10.01 |
| hydantoin ARIs | |||
| minalrestat | –10.88 | ||
| ranirestat | –10.41 | ||
| fidarestat | –7.50 | ||
| sorbinil | –6.92 |
Figure 3.
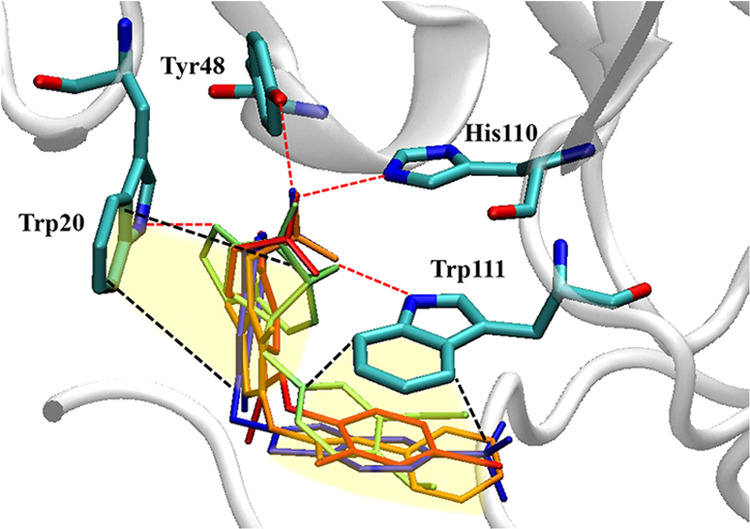
(−)-Kusunokinin and ARI interaction with AKR1B1. Interaction of ARIs include epalrestat (orange), zenarestat (blue), minalrestat (red), and (−)-kusunokinin (lime green). Tyr48 and His110 bound ligands using hydrogen bonds, while Trp20 and Trp111 were mainly responsible for π–π interactions. The π–π interactions are shown as black dashed lines, and the hydrogen bonds are shown as red dashed lines.
Trp20 and Trp111 sustained ARI conformation by forming π–π stacking with the aromatic rings of ARIs. Furthermore, Tyr48 and His110 also formed hydrogen bonds with the AKR1B1-bound ligand. These interactions, found in most ARIs, are concordant with previous reports that Trp20, Tyr48, His110, and Trp111 are present in the anion binding pocket.32 The results gave a hint that the aromatic ring-containing compounds could be the potential ARIs due to π–π stacking formation at the active pocket.
Since molecular docking was performed on the basis of rigid proteins with a flexible ligand, the study of molecular dynamics simulation was later carried out to investigate the time-dependent characteristics of AKR1B1 and its bound compounds.
3.3. Molecular Dynamics Trajectory Analysis
Molecular dynamics simulation was performed to reflect the more realistic dynamic behaviors of the drug-free AKR1B1 or drug-AKR1B1 complex. All of the MD trajectories of all systems became stable after 90 ns (Figure 4a). The flexibility pattern of ligand–AKR1B1 complexes was similar mostly throughout the structure. A distinctive exception was observed in the AKR1B1 residue 210 to 230 in PGH2-AKR1B1 and UVI2008-AKR1B1, which was distinctively more flexible (Figure 4b).
Figure 4.
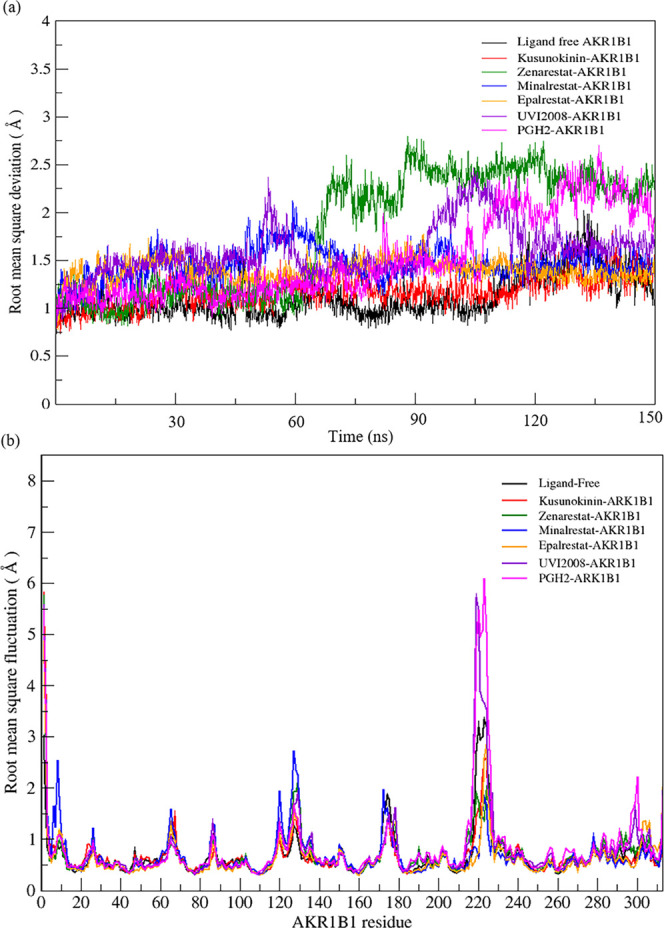
MSD and RMSF of MD trajectory from the AKR1B1 simulation. The interesting simulations were the AKR1B1 and AKR1B1 structures with six docked compounds. RMSD was plotted from 150 ns simulations (a). RMSF was computed from a 90 to 150 MD trajectory (b).
The next point was to investigate whether the bound ligand can affect the AKR1B1 conformation. The distance between a center of geometry in each amino acid and an origin point was plotted to give the pattern of distance. The reference structure was ligand-free AKR1B1 MD simulation. The same distance pattern indicated a similar conformation of the assigned amino acid. However, a different distance pattern pinpointed a different conformation from the AKR1B1. Herein, similar patterns among the seven AKR1B1 simulations were observed (Figure 5). The conclusive finding was that (−)-kusunokinin and other ARIs did not affect the AKR1B1 conformation.
Figure 5.
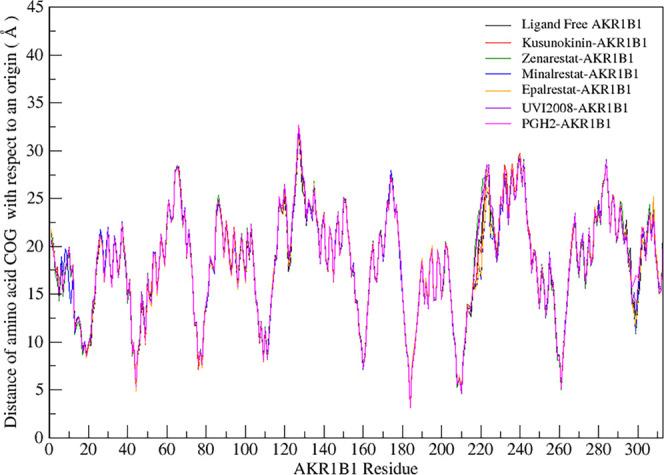
Distance pattern among ligand-free AKR1B1 and drug-AKR1B1 MD simulations. The distance pattern of all simulations was similar to the ligand-free AKR1B1, giving a hint that a bound ligand showed no significant effect on AKR1B1 conformation.
AKR1B1 binding affinity of (−)-kusunokinin was considered from an average MM/GBSA binding free energy with respect to other experimentally known ARIs, which were zenarestat, minalrestat, and epalrestat (Table 2). MM/GBSA calculations revealed that the binding free energy of (−)-kusunokinin (−42.23 kcal/mol) was comparable to other selected ARIs, ranging from −29.63 to −38.96 kcal/mol, and suggested that (−)-kusunokinin would bind with AKR1B1 like other inhibitors.
Table 2. MM/GBSA Binding Energy of (−)-Kusunokinin and Selective ARIs toward AKR1B1.
| ligand | MM/GBSA binding energy (kcal/mol) |
|---|---|
| (−)-kusunokinin | –42.23 ± 0.09 |
| zenarestat | –29.63 ± 0.10 |
| minalrestat | –36.55 ± 0.07 |
| epalrestat | –38.96 ± 0.08 |
An oxygen atom at the ketone group on the γ-butyrolactone ring of (−)-kusunokinin interacted with His110 via hydrogen bonding. Meanwhile, the aromatic ring on 1,2-dimethoxybenzene was stabilized by π–π stacking with Trp111 throughout the MD progression (Figure 6a,b). The γ-butyrolactone ring acted equivalently to the carboxylic acid group of epalrestat and zenarestat to access into a key catalytic His110 in an AKR1B1 anion binding site. The hydantoin ring of minalrestat, however, formed temporary hydrogen bonds with His110. This observed finding is concordant with the reported role of His110 with carboxylic acid or hydantoin ARIs.45
Figure 6.

Binding interaction of (−)-kusunokinin and other inhibitors/substrates with AKR1B1. Six compounds were chosen for MD simulation: (−)-kusunokinin (a), zenarestat (b), minalrestat (c), epalrestat (d), UVI2008 (e), and prostaglandin H2 (PGH2) (f). The black, red, and green dotted lines represent hydrogen bonds, π–π interaction, and π–alkyl interaction, respectively.
Furthermore, Trp111 reportedly acted as a key residue in ARI specificity.32,46 Our study showed that an aromatic ring of (−)-kusunokinin was stabilized by π–π stacking with Trp111 (Figure 6a), similar to selected ARIs (Figure 6b–d) and UVI2008 (an AKR1B1 substrate analog) (Figure 6e). Although no aromatic ring was found in the PGH2 structure, Trp111 was also responsible in PGH2 binding, using π–alkyl interaction with the PGH2 hydrocarbon chain (Figure 6f). Besides, Tyr48 could temporarily form hydrogen bonds with (−)-kusunokinin and ARIs.
3.4. Comparative Behavior of (−)-Kusunokinin with ARIs and AKR1B1 Substrate
AKR1B1 requires NADPH cofactor to catalyze the various substrates, including prostaglandin H2 (PGH2). PGH2 is converted into PGF2α and relays the signals in the NF-κB pathway to regulate the epithelial–mesenchymal transition (EMT) process on various cancers. The catalytic imidazole ring in His110 acts as a proton donor for the dioxabicycloheptane ring of PGH2 to produce PGF2α,4040 as shown in Figure 7a.
Figure 7.

His110 catalytic role in PGH2 conversion. PGH2 conversion to PGF2α using proton transfer from His110 (a). Position alignment between (−)-kusunokinin (red) and dioxabicycloheptane ring of PGH2 (blue) at the ARK1B1 catalytic site (His110) (b).
The molecular dynamics simulations showed that the PGH2 dioxabicycloheptane ring made contact with a transferable proton of NADPH and His110, as expected. The stable hydrogen bond formation with a catalytic His110 due to (−)-kusunokinin, as well as epalrestat, directly prevented PGH2 access into the catalytic site (Figure 7b). However, no stable hydrogen bond with His110 was noticed in the other ARIs. Instead, aromatic residue Trp111 was found to be the principal stabilizer of (−)-kusunokinin, carboxylic and hydantoin ARIs. Mutual interaction with Trp111 would be a rather major contributor to AKR1B1 inhibition because the ligand hindered PGH2 access into the catalytic site, no matter whether or not the compound could form a hydrogen bond with His110 (Figure 8).
Figure 8.

Proposed binding model of ARI in AKR1B1 anion binding pocket. (−)-Kusunokinin (a), carboxylate ARIs (b), hydantoin ARIs (c); red dashed lines: hydrogen bond; orange arrow: π–π interaction.
The binding behaviors of (−)-kusunokinin were closer to carboxylic ARI than hydantoin ARIs. In Figure 8, (−)-kusunokinin could bind AKR1B1 via its carbonyl oxygen in the γ-butyrolactone ring and aromatic ring (Figure 8a). The γ-butyrolactone ring resembled a carboxylic group carboxylic ARI. Carboxylic ARIs have exerted carboxylate groups into anion binding pockets,47 including His110, similar to the carbonyl oxygen of (−)-kusunokinin (Figure 8a,b). This structural information could also be supported by ARI-reported lignans (arctigenin and arctiin)37−39 because their core structures contain the γ-butyrolactone ring and aromatic entity (Figure 9).
Figure 9.

(−)-Kusunokinin, arctigenin, and arctiin structures.
Although many ARIs were synthesized, several ARIs were withdrawn in consequence of the severe adverse effects of hepatotoxicity48−50 and hypersensitivity.51,52 Only commercial epalrestat is used in Asian countries for diabetic neuropathy treatment53 and metastatic triple-negative breast cancer in clinical trials.54 However, its poor efficiency and adverse effects remain a problem in other countries.55 Hence, the search for new, efficient ARIs with low toxicity is important.
4. Conclusions
Related to (−)-kusunokinin action in CSF1R and AKT pathways, AKR1B1 was found as a tentative (−)-kusunokinin target. Predicted binding affinity from (−)-kusunokinin was investigated and compared with other enzyme inhibitors and substrates. Interestingly, (−)-kusunokinin exhibited distinctive binding affinity toward AKR1B1 with respect to the reported AKR1B1 inhibitors (epalrestat, zenarestat, and minalrestat) and substrates (UVI2008 and prostaglandin H2). No conformational alteration in AKR1B1 was found due to compound binding. Interchangeable structural features in (−)-kusunokinin reflected an equivalency to AKR1B1 inhibitors. An aromatic ring acted as an AKR1B1 binding counterpart through π–π stacking with Trp11. An obstruction in AKR1B1 function was also expected because of the hydrogen bond formation of the γ-butyrolactone ring with a crucial catalytic His110. Due to persuasive in silico information, further experiments are encouraged to determine the AKR1B1 inhibitory effect of (−)-kusunokinin.
Acknowledgments
This research project was supported financially by The Royal Golden Jubilee Ph.D. Program (PHD/0186/2561), the Thailand Research Fund (TRF), Thailand, the National Research Council of Thailand (NRCT), and Thailand and Agricultural Research Development Agency (Public Organization) (ARDA), Thailand (CRP6205031700).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05102.
Table S1 summarizes all of the compounds used in the molecular docking along with PubChem CID. Table S2 reports all of the binding energies of (−)-kusunokinin compared with the known inhibitors from the 41 proteins. PDB identification code and structure resolution were included in each protein candidate (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Schirrmacher V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. 10.3892/ijo.2018.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg G. M.; Newman D. J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. 10.1016/j.jep.2005.05.011. [DOI] [PubMed] [Google Scholar]
- De Silva S. F.; Alcorn J. Flaxseed lignans as important dietary polyphenols for cancer prevention and treatment: Chemistry, pharmacokinetics, and molecular targets. Pharmaceuticals 2019, 12, 68 10.3390/ph12020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordaliza M.; Garcia P. A.; del Corral J. M.; Castra M. A.; Gomez-Zurita M. A. Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. 10.1016/j.toxicon.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Ardalani H.; Avan A.; Ghayour-Mobarhan M. Podophyllotoxin: a novel potential natural anticancer agent. Avicenna J. Phytomed. 2017, 7, 285–294. [PMC free article] [PubMed] [Google Scholar]
- Hao Q.; Diaz T.; del Rio Verduzco A.; Magyar C. E.; Zhong J.; Elshimali Y.; Rettig M. B.; Henning S. M.; Vadgama J. V.; Wang P. Arctigenin inhibits prostate tumor growth in high-fat diet fed mice through dual actions on adipose tissue and tumor. Sci. Rep. 2020, 10, 1403 10.1038/s41598-020-58354-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki Y.; Koyama M.; Hitomi T.; Yokota T.; Kawanaka M.; Nishikawa A.; Germain D.; Sakai T. Arctiin induces cell growth inhibition through the down-regulation of cyclin D1 expression. Oncol. Rep. 2008, 19, 721–727. 10.3892/or.19.3.721. [DOI] [PubMed] [Google Scholar]
- Tang S.; Zhou W.; Zhong X.; Xu J.; Huang H.; Zheng X.; Zhang J.; Yang S.; Shang P.; Tang Q.; Liu H. Arctigenin prevents the progression of osteoarthritis by targeting PI3K/Akt/NF-κB axis: In vitro and in vivo studies. J. Cell. Mol. Med. 2020, 24, 4183–4193. 10.1111/jcmm.15079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Kim C.; Lee J.; Um J-Y.; Sethi G.; Ahn K. S. Arctiin is a pharmacological inhibitor STAT3 phosphorylation at tyrosine 705 residue and potentiates bortezomib-induced apoptotic and anti-angiogenic effects in human multiple myeloma cells. Phytomedicine 2019, 55, 282–292. 10.1016/j.phymed.2018.06.038. [DOI] [PubMed] [Google Scholar]
- Gao Q.; Yang M.; Zuo Z. Overview of the anti-inflammatory effects, pharmacokinetic properties and clinical efficacies of arctgenin and arctiin from Arctium lappa L.. Acta Pharmacol. Sin. 2018, 39, 787–801. 10.1038/aps.2018.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattanaburee T.; Thongpanchang T.; Wongma K.; Tedasen A.; Sukpondma Y.; Graidist P. Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomed. Pharmacother. 2019, 117, 109115 10.1016/j.biopha.2019.109115. [DOI] [PubMed] [Google Scholar]
- Tedasen A.; Dokduang S.; Sukpondma Y.; Lailerd N.; Madla S.; Sriwiriyajan S.; Rattanaburee T.; Tipmanee V.; Graidist P. (-)-kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. Eur. J. Pharmacol. 2020, 882, 173311 10.1016/j.ejphar.2020.173311. [DOI] [PubMed] [Google Scholar]
- Rattanaburee T.; Tipmanee V.; Tedasen A.; Thongpanchang T.; Graidist P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. 2020, 129, 110361 10.1016/j.biopha.2020.110361. [DOI] [PubMed] [Google Scholar]
- Tedasen A.; Choomwattana S.; Graidist P.; Tipmanee V. Structure-guided cancer blockade between bioactive bursehernin and proteins: Molecular docking and molecular dynamics study. J. Mol. Graphics Model. 2017, 74, 215–224. 10.1016/j.jmgm.2017.04.021. [DOI] [PubMed] [Google Scholar]
- Morris G.; Huey R.; Linkstrom W.; Sanner M.; Belew R.; Goodsell D.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- BIOVIA . Discovery Studio Modeling Environment, release 2017; Dassault Systèmes: San Diego, 2016.
- Jewboonchu J.; Saetang J.; Saeloh D.; Siriyong T.; Rungrotmongkol T.; Voravuthikunchai S. P.; Tipmanee V. Atomistic insight and modeled elucidation of conessine towards Pseudomonas aeruginos efflux pump. J. Biomol. Struct. Dynamics 2020, 1–10. 10.1080/07391102.2020.1828169. [DOI] [PubMed] [Google Scholar]
- Huey R.; M. Morris G.; Olson A. J.; Goodsell D. S. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J. Comput. Chem. 2007, 28, 1145–1152. 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- Gallego O. F.; Ruiz X.; Ardèvo A.; Domínguez M.; Alvarez R.; de Lera A. R.; Rovira C.; Farrés J.; Fita I.; Parés X. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20764–20769. 10.1073/pnas.0705659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez-Dacal B.; Sánchez-Gómez F. J.; Sánchez-Murcia P. A.; Milackova I.; Zimmerman T.; Ballekova J.; García-Martín E.; Agúndez J. A.; Gharbi S.; Gago F.; Stefek M.; Pérez-Sala D. Molecular Interactions and Implications of Aldose Reductase Inhibition by PGA1 and Clinically Used Prostaglandins. Mol. Pharmacol. 2016, 89, 42–52. 10.1124/mol.115.100693. [DOI] [PubMed] [Google Scholar]
- Zhan J.-Y.; Ma K.; Zheng Q.-C.; Yang G.-H.; Zhang H.-X. Exploring the Interactional Details between Aldose Reductase (AKR1B1) and 3-Mercapto-5H-1,2,4-triazino[5,6-b]indole-5-acetic Acid through Molecular Dynamics Simulations. J. Biomol. Struct. Dynamics 2019, 37, 1724–1735. 10.1080/07391102.2018.1465851. [DOI] [PubMed] [Google Scholar]
- Wang L.; Gu Q.; Zheng X.; Ye J.; Liu Z.; Li J.; Hu X.; Hagler A.; Xu J. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J. Chem. Inf. Model. 2013, 53, 2409–2422. 10.1021/ci400322j. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Botello-Smith W.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; York D. M.; Kollman P. A.. Amber 16; University of California: San Francisco: 2016.
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.. Gaussian 16, revision C.01; Gaussian, Inc., 2016.
- Ryde U. Molecular dynamic simulations of alcohol dehydrogenase with varying coordination number of the catalytic zinc ion. Proteins 1995, 21, 40–50. 10.1002/prot.340210106. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Kollman P. Free energy calculations: applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. 10.1021/cr00023a004. [DOI] [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram B.; Sprous D.; Young M. A.; Beveridge D. L. 1998. Free energy analysis of the conformational preferences of A and B forms of DNA in solution. J. Am. Chem. Soc. 1998, 120, 10629–10633. 10.1021/ja981307p. [DOI] [Google Scholar]
- Nutho B.; Mahalapbutr P.; Hengphasatporn K.; Pattaranggoon N. C.; Simanon N.; Shigeta Y.; Hannongbua S.; Rungrotmongkol T. Why are lopinavir and ritonavir effective against the newly emerged Coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 2020, 59, 1769–1779. 10.1021/acs.biochem.0c00160. [DOI] [PubMed] [Google Scholar]
- Zhu C.Aldose Reductase Inhibitors as Potential Therapeutic Drugs of Diabetic Complications; IntechOpen, 2013; Vol. 23, pp 19–43. [Google Scholar]
- Laffin B.; Petrash J. M. Expression of the aldo-ketoreductases AKR1B1 and AKR1B10 in human cancers. Front. Pharmacol. 2012, 3, 104 10.3389/fphar.2012.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A.; Siddiqui A.; Vazakidou M. E.; Napoli F.; Bottcher M.; Menchicchi B.; Raza U.; Saatci O.; Krebs A. M.; Ferrazzi F.; Rapa I.; Dettmer-Wilde K.; Waldner M. J.; Ekici A. B.; Rasheed S.A.K.; Mougiakakos D.; Oefner P. J.; Sahin O.; Volante M.; Greten F. R.; Brabletz T.; Ceppi P. Polyol pathway links glucose metabolism to the aggressiveness of cancer cells. Cancer Res. 2018, 78, 1604–1618. 10.1158/0008-5472.CAN-17-2834. [DOI] [PubMed] [Google Scholar]
- Wu X.; Li X.; Fu Q.; Cao Q.; Chen X.; Wang M.; Yu J.; Long J.; Yao J.; Liu H.; Wang D.; Liao R.; Dong C. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J. Exp. Med. 2017, 214, 1065–1079. 10.1084/jem.20160903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana K. V.; Tammali R.; Srivastava S. K. Inhibition of aldose reductase prevents growth factor-induced G1-S phase transition through the AKT/phosphoinositide 3-kinase/E2F-1 pathway in human colon cancer cells. Mol. Cancer Ther. 2010, 9, 813–824. 10.1158/1535-7163.MCT-09-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.; Wang J. J.; Yan J.; Cui C. F.; Wu W. H.; Li L.; Wang Z. S.; Yu M.; Gao N.; Liu L.; Ouyang D. S. Effects of lignans extracted from Eucommia ulmoides and aldose reductase inhibitor epalrestat on hypertensive vascular remodeling. J. Ethnopharmacol. 2011, 133, 6–13. 10.1016/j.jep.2010.08.055. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yang H.; Zhou M.; Feng Y.; Jia W. Inhibitory effect of total lignan from Fructus arctii on aldose reductase. Phytother. Res. 2010, 24, 472–473. 10.1002/ptr.2828. [DOI] [PubMed] [Google Scholar]
- Xie H.; Wang T.; Maatsuda H.; Morikawa T.; Yoshikawa M.; Tani T. Bioacitve Constituents from Chinese Natural Medicines. XV. Inhibitory Effect on Aldose Reductase and Structures of Saussureosides A and B from Saussurea medusa. Chem. Pharm. Bull. 2005, 53, 1416–1422. 10.1248/cpb.53.1416. [DOI] [PubMed] [Google Scholar]
- Nagata N.; Kusakari Y.; Fukunishi Y.; Inoue T.; Urade Y. Catalytic mechanism of the primary human prostaglandin F2α synthase, aldo-keto reductase 1B1-Prostaglandin D2 synthase activity in the absence of NADP(H). FEBS J. 2011, 278, 1288–1298. 10.1111/j.1742-4658.2011.08049.x. [DOI] [PubMed] [Google Scholar]
- Crosas B.; Hyndman D. J.; Gallego O.; Martras S.; Parés X.; Flynn T. G.; Farrés J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: Consequences for retinoid metabolism. Biochem. J. 2003, 373, 973–979. 10.1042/bj20021818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M.; Endo S.; Matsunaga T.; Zhao H. T.; El-Kabbani O.; Iinuma M.; Yamamura K.; Hara A. Inhibition of human aldose reductase-like protein (AKR1B10) by α- and γ-mangostins, major components of pericarps of mangosteen. Biol. Pharm. Bull. 2012, 35, 2075–2080. 10.1248/bpb.b12-00538. [DOI] [PubMed] [Google Scholar]
- Soda M.; Hu D.; Endo S.; Takemura M.; Li J.; Wada R.; Ifuku S.; Zhao H. T.; El-Kabbani O.; Ohta S.; Yamamura K.; Toyooka N.; Hara A.; Matsunaga T. Design, synthesis and evaluation of caffeic acid phenethyl ester-based inhibitors targeting a selectivity pocket in the active site of human aldo-keto reductase 1B10. Eur. J. Med. Chem. 2012, 48, 321–329. 10.1016/j.ejmech.2011.12.034. [DOI] [PubMed] [Google Scholar]
- Takemura M.; Endo S.; Matsunaga T.; Soda M.; Zhao H. T.; El-Kabbani O.; Tajima K.; Iinuma M.; Hara A. Selective inhibition of the tumor marker aldo-keto reductase family member 1B10 by oleanolic acid. J. Nat. Prod. 2011, 74, 1201–1206. 10.1021/np200118q. [DOI] [PubMed] [Google Scholar]
- Lee Y. S.; Chen Z.; Kador P. F. Molecular modeling studies of the binding modes of aldose reductase inhibitors at the active site of human aldose reductase. Bioorg. Med. Chem. 1998, 6, 1811–1819. 10.1016/S0968-0896(98)00139-4. [DOI] [PubMed] [Google Scholar]
- Khan S.; Somvanshi P.; Singh A.; Khan M.; Mandal R. K.; Dar S. A.; Wahid M.; Jawed A.; Mishra B. N.; Haque S. Potency of inhibitors depends upon the accessibility of their aromatic rings within the hydrophobic specificity pocket: a novel avenue for future aldose reductase inhibitor design. J. Biomol. Struct. Dynamics 2020, 1–7. 10.1080/07391102.2020.1733090. [DOI] [PubMed] [Google Scholar]
- Chatzopoulou M.; Alexiou P.; Kotsampasakou E.; Demopoulos V. J. Novel aldose reductase inhibitors: A patent survey (2006–present). Expert Opin. Ther. Pat. 2012, 22, 1303–1323. 10.1517/13543776.2012.726615. [DOI] [PubMed] [Google Scholar]
- van Gerven J. M. A.; Lemkes H. H. P. J.; van Dijk J. G. Long-term effects of tolrestat on symptomatic diabetic sensory polyneuropathy. J. Diabetes Complications 1992, 6, 45–48. 10.1016/1056-8727(92)90048-P. [DOI] [PubMed] [Google Scholar]
- Ao S.; Shingu Y.; Kikuchi C.; Takano Y.; Nomura K.; Fujiwara T.; Ohkubo Y.; Notsu Y.; Yamaguchi I. Characterization of a novel aldose reductase inhibitor, FR74366, and its effects on diabetic cataract and neuropathy in the rat. Metabolism 1991, 40, 77–87. 10.1016/0026-0495(91)90196-4. [DOI] [PubMed] [Google Scholar]
- Mylari B. L.; Larson E. R.; Beyer T. A.; Zembrowski W. J.; Aldinger C. E.; Dee M. F.; Siegel T. W.; Singleton D. H. Novel, potent aldose reductase inhibitors: 3,4-Dihydro-4-oxo-3-[[5-(trifluoromethyl)-2-benzothiazolyl] methyl]-1-phthalazineacetic acid (zopolrestat) and congeners. J. Med. Chem. 1991, 34, 108–122. 10.1021/jm00105a018. [DOI] [PubMed] [Google Scholar]
- Sarges R.; Schnur R. C.; Belletire J. L.; Peterson M. J. spiro hydantoin aldose reductase inhibitor. J. Med. Chem. 1988, 31, 230–243. 10.1021/jm00396a037. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Aldinger C. E.; Beyer T. A.; Bordner J.; Burdi D. F.; Bussolotti D. L.; Inskeep P. B.; Siegel T. W. Hydantoin bioisosteres. in vivo active spiro hydroxy acetic acid aldose reductase inhibitors. J. Med. Chem. 1992, 35, 2169–2177. 10.1021/jm00090a004. [DOI] [PubMed] [Google Scholar]
- Hotta N.; Kawamori R.; Atsumi Y.; Baba M.; Kishikawa H.; Nakamura J.; Oikawa S.; Yamada N.; Yasuda H.; Shigeta Y. Stratified analyses for selecting appropriate target patients with diabetic peripheral neuropathy for long-term treatment with an aldose reductase inhibitor, epalrestat. Diabetic Med. 2008, 25, 818–825. 10.1111/j.1464-5491.2008.02490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaled N.; Bidet Y. New insights into the implication of epigenetic alterations in the EMT of triple negative breast cancer. Cancers 2019, 11, 559–580. 10.3390/cancers11040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schemmel K. E.; Padiyara R. S.; D’Souza J. J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. J. Diabetes Complications 2010, 24, 354–360. 10.1016/j.jdiacomp.2009.07.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.