

Summary
In the face of ineffective vaccines, increasing antibiotic resistance and the decline in new antibacterial drugs in the pipeline, tuberculosis (TB) still remains pandemic. Exposure to Mycobacterium tuberculosis (Mtb), which causes TB, results in either direct elimination of the pathogen, most likely by the innate immune system, or infection and containment that requires both innate and adaptive immunity to form the granuloma. Host defence strategies against infectious diseases are comprised of both host resistance, which is the ability of the host to prevent invasion or to eliminate the pathogen, and disease tolerance, which is defined by limiting the collateral tissue damage. In this review, we aim to examine the metabolic demands of the immune cells involved in both host resistance and disease tolerance, chiefly the macrophage and T‐lymphocyte. We will further discuss how baseline metabolic heterogeneity and inflammation‐driven metabolic reprogramming during infection are linked to their key immune functions containing mycobacterial growth and instructing protective immunity. Targeting key players in immune cellular metabolism may provide a novel opportunity for treatments at different stages of TB disease.
Keywords: Mycobacterium tuberculosis, macrophage, lymphocyte, alveolar macrophage, T‐cell, disease tolerance, host resistance, immunometabolism, glycolysis, oxidative phosphorylation, fatty acid oxidation, ATP, metabolism, Trained immunity, vaccine
The host defence against infectious diseases is comprised of both host resistance and disease tolerance. Resistance is the ability of the host to eliminate the pathogen, while disease tolerance is defined by limiting the collateral tissue damage caused by the pathogen and/or the immune response and activating tissue repair mechanisms. We need a better understanding of the entire spectrum of immunity against complex diseases such as TB.
Abbreviations
- TB
Tuberculosis
- Mtb
Mycobacterium tuberculosis
- BCG
bacille Calmette–Guérin
- AM
alveolar macrophage
- IMMs
interstitial monocyte‐derived macrophages
- DCs
dendritic cells
- AEC
alveolar epithelial cell
- BMDMs
bone marrow‐derived macrophages
- MDMs
monocyte‐derived macrophages
- HSCs
haematopoietic stem cells
- TCA
tricarboxylic acid cycle
- OXPHOS
oxidative phosphorylation
- FAO
fatty acid oxidation
- NO
nitric oxide
- T2D
type 2 diabetes
- TST
tuberculin skin test
- PD1
programmed cell death
CELLULAR RESPONSES TO Mtb
1.5 million people died because of infection with Mycobacterium tuberculosis (Mtb) in 2018 1 , mainly in sub‐Saharan Africa and South‐East Asia, although tuberculosis (TB) cases remain a global pandemic with 1 in 4 people worldwide estimated to be infected with the disease‐causing pathogen, Mtb. Moreover, when this old TB pandemic collides with a novel threats (e.g. HIV or COVID‐19), this silent killer will only get worse as a result of syndemic and the reduced accessibility to health care 2 , 3 . In spite of staggering numbers in infection rate, illness and mortality, we face significant challenges, both politically and scientifically, which we must overcome to curb the development of this global health problem.
The heterogeneity in infection outcomes ranging from elimination, inactive TB or active disease manifests as a result of the complex evolution and interaction between Mtb and the human immune system 4 . This involves multiple cell types including resident pulmonary and recruited immune cells, each of which display plasticity and heterogeneity in their ability to eliminate or contain infection 5 . Recent scientific advances have allowed us to more precisely profile the multiple cell types at various stages of infection 6 , ranging from initial host–pathogen interaction, subsequent immune cell recruitment and granuloma formation. However, our understanding of the inherent functional differences in these cells and the systemic factors in the host that impinge upon their function is very limited.
One of the most sophisticated strategies of Mtb is that the bacteria have adapted to specifically infect the lung‐resident alveolar macrophage (AM), which plays a dual role as both the effector against and reservoir for TB infection 7 . Following internalization of the bacteria via various phagocytic receptors 8 , 9 , pattern recognition receptors trigger cytokine and chemokine production in the alveolar and interstitial space 10 , causing the recruitment of blood‐borne immune cells via underlying blood vessels. These include monocyte‐derived macrophages classified as interstitial monocyte‐derived macrophages (IMMs) 11 and multinucleated granular neutrophils 12 . Both classes of phagocytic myeloid cells play key roles containing bacteria through various intracellular mechanisms, as well as orchestrating the inflammatory response. However, similar to the AM, they also can act as intracellular niches for Mtb replication and the balance between these processes impacts the outcome of infection and the extent of bacterial dissemination. Following pulmonary infection, Mtb‐infected dendritic cells (DCs) or monocyte‐derived macrophages migrate to the thoracic draining lymph nodes where they present antigens to naïve T cells, which leads effector T cells to traffic to the lung 4 . Interestingly, Mtb hijacks this process by delaying it up to 3 weeks post‐infection 13 . B cells also play a role in TB immunity by providing Mtb‐specific antibodies to promote opsonization, as well as producing cytokines to modulate macrophage and T‐cell function 14 , 15 . Where complete sterilization fails, the extracellular inflammatory milieu and recruited immune cells form a granuloma, with infected macrophages at the centre, surrounded by layers of uninfected macrophages of various phenotypes and ultimately T cells with some follicular B cells and fibroblasts to limit infection and prevent subsequent dissemination.
Although the induction of inflammatory mediators within a granuloma is required for preventing Mtb dissemination, overly intense pro‐inflammatory responses lead to reduced disease tolerance, including the destruction of granulomas via necrosis, enhanced lung parenchymal damage, lung cavitation and transmission that results in the onset of active disease 4 , 16 , 17 . Indeed, targeting TNF in immunodeficient patients with advanced inflammatory mycobacterial disease, including tuberculosis, actually improved outcome 18 . Furthermore, studies in animal models of TB and in humans have elegantly demonstrated that inflammatory signalling is highly organized within the granuloma as pro‐inflammatory signalling is mainly found at the core of the granuloma, while anti‐inflammatory signalling predominates in the periphery 19 . This spatial compartmentalization of pro‐ and anti‐inflammatory signalling determines the granuloma's function in controlling both disease tolerance and host resistance to Mtb (Figure 1). Thus, the optimal host response may be a balance of inflammatory and anti‐inflammatory signalling that leads to the regulation of inflammation within and around the granuloma and reduced frequency of active disease 20 .
Figure 1.
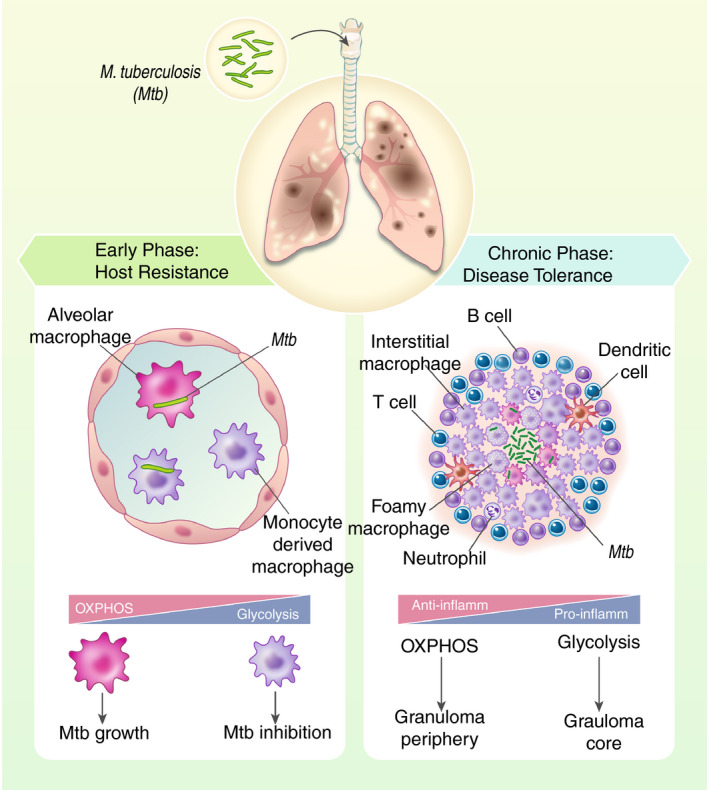
Immune cell metabolism in host defence against Mtb. The early phase of host defence against Mtb is dominated by macrophage attempts to resist the infection. However, resident alveolar macrophages that rely on fatty acid oxidation (FAO) in the lipid‐rich lung microenvironment to fuel oxidative phosphorylation (OXPHOS) provide a niche for Mtb replication. Recruited interstitial monocyte‐derived macrophage (IMM) up‐regulate glycolytic activities to promote pro‐inflammatory activities to eliminate the bacteria. If the bacteria persist by migrating into the lung interstitial tissue that leads to the granuloma formation, the host defence mechanism switches to disease tolerance to containing infection and limiting tissue damage. This is driven mainly by activated T cells, B cells, phagocytic cells such as DC, and interstitial macrophages. As the granuloma develops, pro‐inflammatory glycolytic macrophages attempt to restrict mycobacterial growth in the centre, while anti‐inflammatory interstitial macrophages balance the inflammatory response and limit bacterial dissemination. Foamy macrophages in which lipid metabolism is remobilized have been also associated with both bacterial growth and pro‐inflammatory antimicrobial activities. While the centre of the granuloma appears to be pro‐inflammatory, the periphery is anti‐inflammatory. This spatial compartmentalization of pro‐ and anti‐inflammatory signalling that regulates by cellular metabolism determines the capacity of the granuloma to prevent bacterial dissemination
Considering both the pro‐ and anti‐inflammatory phenotypes of immune cells are linked to their cellular metabolism, it is imperative to delineate the molecular mechanisms that dictate their metabolism. The granuloma itself is a complex metabolic environment with areas of hypoxia, lipid accumulation and necrosis and is completely dependent on cytokine instruction to maintain integrity 21 . With obesity now recognized as a metabolic disease that alters homeostatic immune function 22 and TB–diabetes now an increasing comorbidity observed in South‐East Asia 23 , changes in whole‐body metabolic signalling impact immune cells’ ability to contain Mtb. Thus, metabolic disease should be considered a key factor that determines susceptibility to active TB. In this review, we will dissect the metabolic phenotypes observed in two hallmark cell types of the immune response to Mtb – macrophages and T lymphocytes – and overlay how recent knowledge on immunometabolism in these cells may dictate host resistance or disease tolerance in TB.
RESIDENT ALVEOLAR MACROPHAGES
The AM response to Mtb
It is now well recognized that AMs, like many tissue‐resident macrophages, is not originated from blood‐borne monocytes but rather from embryologically distinct progenitors (yolk sac), which can proliferate locally 24 . AMs were first described as ‘dust cells’, due to the large amounts of darkly staining particulate matter indicative of their key phagocytic function surveying the external environment and maintaining homeostasis 25 . These features however predispose these cells to Mtb infection, as they express a wide array of phagocytic scavenger receptors (SRs) used by Mtb to enter phagocytes, 4 , 10 . The immunotolerant environment of the lung also favours Mtb infection, promoted by both AM and alveolar epithelial cell (AEC)‐derived TGF‐β and vitamin A metabolite all‐trans retinoic acid driving anti‐inflammatory 26 and regulatory T‐cell signalling 27 . Pulmonary surfactant, the lipoprotein mixture that regulates lung compliance 28 , also affects AM phenotype 29 . This combination of lung features promotes disease tolerance, and thus, AMs possess a phenotype to maintain the lung immunotolerant environment at the steady state. However, AMs express many surface and endosomal TLRs, which recognize mycobacterial epitopes and pro‐inflammatory activation 30 . Mtb evolved exquisite strategies to evade and inhibit this, the most crucial of which is inhibiting phagolysosomal fusion to escape destruction and allow an intracellular niche for survival 9 . Although this responsiveness of AMs is seemingly dictated by their expression of surface receptors and interactions with the immunotolerant pulmonary environment, investigators are now examining the metabolic activity of AM, which may explain their failure to efficiently contain Mtb during the initial phase of infection.
Macrophage metabolism
We now know that the flux, fate and substrate preference of central C‐metabolism pathway can be altered to support T‐cell energy and biosynthetic demands, in response to changes in the tissue microenvironment, which uniquely affects migrating and patrolling immune cells with varying functional demands 31 . The predominant view in macrophage biology has been that macrophages exist on 2 metabolic spectrums, in line with broad in vitro classifications used to distinguish opposing functional programmes 32 . Classically activated (M1) macrophages, generated in vitro by treating naïve human blood or mouse bone marrow‐derived macrophages (hMDM or BMDM) with the TLR ligand LPS and IFN‐γ, adopt a glycolytic phenotype with impaired tricarboxylic acid cycle (TCA) dynamics, even in the presence of oxygen 33 , 34 . Through the up‐regulation of glycolytic rates, this metabolic reprogramming provides rapid ATP from cellular glycolysis without the need for mitochondrial oxidative phosphorylation (OXPHOS). TCA metabolites and glycolytic intermediates are syphoned into alternative pathways including citrate‐derived fatty acid synthesis (FAS) and increased pentose–phosphate pathway (PPP) activity to support the biosynthetic demands of activation 33 , 35 , 36 , 37 , which in cancer cells that adopt similar Warburg metabolism or in activated T cells, is easily explained by the proliferative demand 12 . It has been hypothesized that in macrophages this anabolic metabolism supports the antimicrobial oxidative burst, as well as the increased transcriptional and translational activity required for cytokine production 38 , 39 . In contrast, alternatively activated (M2) macrophages, generated in vitro through anti‐inflammatory cytokine stimulation, chiefly IL‐4, maintain high levels of OXPHOS with an intact TCA cycle 33 . While M1 and M2 are the example of two extreme spectrum of cellular metabolism, nonetheless these are useful benchmark to define the differences observed in macrophage metabolism and phenotype during the course of Mtb infection.
AM immunometabolism
At baseline, AMs represent M2‐like macrophages, showing similar levels of oxidative metabolism as IL‐4‐treated human MDMs 40 . This aligns with their phagocytic function, which is an energetically demanding activity. Although rapid cellular proliferation requires anabolic metabolism, long‐lived self‐renewal cells such as AM rely heavily on OXPHOS to maintain viability 41 . The lung is a lipid‐rich environment due to surfactant, and therefore, a diversity of fuels is available for AM. While glucose is the preferential fuel, its availability is often limited in microenvironments 42 , and therefore, immune cells have adapted to undertake fatty acid metabolism. Indeed, lung glucose levels are thought to be lower than serum levels 43 , which may force alternative metabolism in immune cells engaged in the granuloma as an adaptation to the new environment. Increased fatty acid oxidation (FAO) is a feature of M2 macrophages 33 . Similar up‐regulation of FAO has also been observed in mouse AMs ex vivo 44 . Up‐regulation of CD36 expression by IL‐4 drives FA uptake and lipolysis to feed FAO in M2 macrophages and is linked to M2 marker up‐regulation and anti‐inflammatory cytokine production 45 . Thus, it is possible that a similar switch occurs in the lung with surfactant lipoprotein SP‐A known to drive CD36 expression and activity 46 , which could skew towards an M2‐like AM phenotype forcing both FAO and anti‐inflammatory gene expression. Although Mtb can utilize glucose, multiple studies suggest FA as the preferred C‐source for Mtb during infection 44 , while Mtb does not possess enzymatic machinery for cholesterol biosynthesis 43 . However, it has been postulated that the lipid‐rich environment of the AM favours Mtb by providing triacylglycerols, cholesterol and FA as a C‐source for mycobacterial growth. CD36‐mediated uptake of surfactant lipoproteins by AMs may in fact provide a double hit for the bacteria, limiting inflammation and providing access to nutrients for growth. Indeed, ex vivo studies suggest that mouse AM represent a more favourable niche for Mtb replication than observed in BMDM or recruited lung IMs 44 , 47 , in a manner completely dependent upon FAO 44 .
A major question that remains to be answered is the functional and metabolic plasticity of AMs during Mtb infection. While in vitro infection of both human and mouse AMs with attenuated or inactivated forms of Mtb allows glycolysis, live virulent Mtb abolishes the glycolytic capacity and activity of these cells 44 , 48 . More extensive bioenergetic analysis reveals a strong dependence on OXPHOS for glucose metabolism and ATP production in LPS‐treated AM ex vivo 49 . These findings are supported by in vivo analysis of influenza infection and profiling of lung populations 49 . Extensive transcriptomic profiling of both uninfected and infected AMs reveals increased expression of FAO and lipid metabolism genes 44 , 50 . Although glycolysis could be driven in AMs through the stabilization of HIF‐1α 49 , this does not significantly contribute to LPS responses observed. Impaired IL‐4 responses in AM in the lung were also found to be due to environmental suppression of glycolysis and were restored to similar levels as peritoneal macrophages when AMs were treated ex vivo 50 . AMs therefore have adapted their immunometabolic pathways to circumvent the low availability of glucose and lipid‐rich environment in the lung. This would suggest that following Mtb infection, the threshold for AM activation via glycolytic metabolism should be higher than IMMs. Thus, identifying the regulatory mechanism involved in AM metabolism can map out targeted therapy for switching AM metabolism from OXPHOS towards the glycolytic pathway and therefore halting the initial phase of Mtb infection.
At the interface of the external environment, AMs are uniquely susceptible to factors that can impinge upon immune function. Smoking increases the risk of developing TB disease by twofold 51 . Recent evidence suggests smokers’ AMs are defective at multiple cellular compartments, with increased expression of oxidative stress genes 52 , mitochondrial impairment leading to defective autophagy 53 and lysosomal storage disorders 54 . These intriguing findings link mitochondrial reprogramming in these cells with antimicrobial mechanisms and could explain the observed increased bacillary burden in smokers’ AMs 51 . In our model of ex vivo AM activation with non‐viable Mtb or LPS treatment, AMs from smokers were unable to shift towards glycolytic metabolism after stimulation, although basal glycolytic levels were higher 40 . This was also noted in a separate study examining smokers’ AM and AM from COPD patients against healthy AM 52 . Together, these data suggest cigarette smoke exposure triggers basal reprogramming of AM metabolism, impairing normal oxidative phosphorylation required for AM homeostatic activities, which triggers compensatory glycolysis. This low‐level induction of glycolysis exhausts the AM‐limited capacity for glycolysis, such that appropriate immunometabolic‐mediated inflammatory activation to pathogens is lost.
RECRUITED MACROPHAGES
Metabolic reprogramming supports Mtb host defence
Most studies to date examining the immunometabolic effects of Mtb infection have been performed in macrophages, which model naïve, newly recruited macrophages at sites of inflammation (hMDM, BMDM and THP1 cells), and for the most part, the results are consistent with what is observed in M1 macrophage models, with a profound up‐regulation of glycolytic activity 55 , 56 , 57 , 58 , 59 . Encouragingly, these cells contain Mtb growth in a superior fashion and their ability to do so is completely dependent upon the up‐regulation of glycolysis 44 , 57 . The landmark study by Huang et al characterized the recruited IMMs in mouse models of Mtb infection, and targeting glycolysis in these mouse models completely abolished the ability to contain infection 44 , a feature also observed in vitro when glycolysis is poisoned in mouse BMDM or hMDM 44 , 57 , 60 or in BMDM from mice that lack a key regulator of the glycolytic response, HIF‐1α 58 , 61 . While multiple groups have now reported the up‐regulation of glycolysis in Mtb‐infected macrophages, a recent extensive metabolic screen in hMDM and THP1 cells infected with various forms and strains of Mtb found virulent Mtb can actually decelerate flux through this pathway 55 , supporting the notion that during successful infection, this pathway is attenuated to promote immune evasion. This, coupled with the finding that one of the chief functions of Th1‐derived IFN‐γ is to drive glycolysis in Mtb‐infected macrophages 48 , 58 , supports an important pro‐inflammatory role for the glycolytic response in host defence.
Although initial work suggested a mechanism whereby maintenance of glycolysis supports pro‐inflammatory IL‐1β to regulate eicosanoid PGE2 production and bacterial containment 57 , 62 , the existence of other glycolysis‐linked pathways to host defence has emerged. We and others have observed production of nitric oxide (NO) linked to increased macrophage glycolysis 48 , 58 , 61 . This could not only occur as a consequence of increased IL‐1β signalling, but may also result from NADP generation through glycolysis‐linked PPP 37 . This glycolysis‐associated NO also acts as an important immunomodulatory signal by feeding back and enhancing glycolysis via HIF‐1α activation, while simultaneously promoting inflammatory resolution through inhibition of NF‐kB 61 . Although NO production is undoubtedly an important component of the host response to Mtb, not all models support a direct role of NO as antimycobacterial. In fact, recent studies suggest that NO production not only does represent an important immunomodulatory signal impacting on magnitude of inflammation, but may also regulate subsequent immunometabolic responses 63 , 64 . It has been elegantly demonstrated that nitric oxide (NO) inhibits NLRP3 inflammasome‐mediated IL‐1β production to prevent neutrophil‐dependent pulmonary tissue damage 65 . A recent study has shown that the role of NO in host resistance to Mtb acts via the recruitment of neutrophils, which are permissive to Mtb growth 63 . Importantly, this immunoregulatory function of NO is co‐ordinated with the initial recruitment of IFN‐γ‐producing T cells into the lung, which leads to granuloma formation and perhaps the transition from host resistance to disease tolerance. In a mouse model of influenza viral infection, accumulation of IMM in the lung tissue compromises disease tolerance that leads to pulmonary dysfunction. Interestingly, the production of IFN‐α via leukotriene B4 signalling was required to minimize IMM‐mediated lung tissue damage 66 . Much of our understanding of the role of NO comes from both targeting and measuring its production in mouse models. Human macrophages, including AM, however produce limited amounts of NO in comparison 67 , and therefore, it is important to confirm its role in the immunometabolic response in these systems. However, these studies collectively indicate that the regulation of IMM by various pro‐inflammatory mediators is required for maintaining disease tolerance in lung tissue.
Regulation of IMM metabolism
While examining the potential of antibiotic‐resistant strains of Mtb for the modulation of macrophage metabolism that limits IL‐1β responses 68 , Howard et al identified mycobacterial lipids, which when mutated in MDR‐TB strains were unable to drive glycolysis in BMDM. This aligns with the work of Lachmandas and colleagues who identified the bacterial lipoprotein sensor TLR2 and subsequent PI3/AKT kinase signalling as a key axis through which recognition of Mtb drives metabolic reprogramming 56 , 69 . This intersects with IFN‐γ signalling to activate pro‐glycolytic HIF‐1α 58 . The up‐regulation of glycolytic genes by HIF‐1α is a key step supporting enhanced glycolytic flux during Mtb infection 59 . Intriguingly, tracing studies suggests that only a small amount of glucose‐derived carbon makes it to the pyruvate and lactate steps of later glycolysis in Mtb‐infected cells 55 . This metabolic consequence of increased glycolytic flux in Mtb‐infected macrophages may do more than promote a pro‐inflammatory and antimicrobial environment through the regulation of inflammation, but, in itself, can deprive replicating Mtb of essential nutrients required for intracellular growth. Osaka‐Oka et al, 2019, demonstrate that IFN‐γ treatment restores LdhA expression and activity in Mtb‐infected macrophages and LdhA‐deficient BMDMs allow uncontained intracellular mycobacterial growth 70 . De Carvalho et al recently demonstrated that pyruvate and lactate are in fact superior carbon sources for Mtb over glucose and fatty acids but only when oxygen is plentiful 71 , which may not be entirely reflective of the situation in a developing TB granuloma.
Although a feature of classically studied LPS‐activated macrophages 33 , 34 , 72 , impaired TCA cycle activity has not been well documented in Mtb‐infected macrophages. This is consistent with TLR2‐mediated metabolic reprogramming, which preserves oxidative metabolism to maintain phagocytic capacity 69 . Although less virulent mycobacterial strains drive glycolysis without significantly altering TCA activity, the thorough analysis performed by Cumming et al in hMDM revealed that virulent Mtb reduces total bioenergetic activity, including both glycolytic metabolism and oxidative metabolism 55 . Another key observation is that although oxidative metabolism remains intact in Mtb‐infected macrophages, anaplerotic pathways feed TCA cycling and infected macrophages display a profound fuel preference for fatty acids as their carbon source 55 . This fits with the long‐held observation of foamy macrophage appearance following Mtb infection both in vitro and in vivo 73 , 74 , discussed in the following section.
FOAMY MACROPHAGES
Lipid droplet‐laden foam cell macrophages are hallmarks of the developing TB granuloma 83 . These differ from those observed in systemic lipid disorders such as obesity and atherosclerosis 22 both quantitatively (numbers of lipid droplets per cell and foamy cells per granuloma) and quantitatively (lipid composition). Additionally, the source of lipids fuelling this remains unclear. It has been shown, however, that Mtb can use host cholesterol as a carbon source in long‐term murine models of infection 75 . Thus, it is plausible that the lipid‐rich pulmonary environment provided by surfactant is a source of exogenous lipids, both FA and cholesterol, which are taken up by unregulated SR activity in Mtb‐infected macrophages to drive foamy macrophage appearance, although this has not been formally examined. While atherosclerotic foam cells are cholesterol‐dominated, Mtb granulomas are triglyceride‐rich 76 , with mycobacterial ligand signalling through macrophage receptors to alter cellular triglyceride content 77 . This indicates a dynamic remobilization of intracellular lipids in Mtb‐infected macrophages that can favour bacterial growth 78 . In fact, using FA‐loaded macrophages, Ouimet et al observed strong colocalization of Mtb with lipid droplets in a pathway regulated by microRNA‐33‐mediated control of the autophagy machinery 79 .
Although hyperlipidaemia often accompanies type 2 diabetes (T2D) – an increasing risk factor for TB 23 – it is unclear whether changes in systemic lipoprotein levels affect intracellular lipids in recruited or alveolar macrophages to predispose to Mtb infection. Recently, oxLDL, the modified form of LDL cholesterol that accumulates subendothelially in atherosclerotic plaques and that drives unregulated SR uptake and pro‐inflammatory activation 80 , was found to both accumulate in guinea‐pig granuloma models 81 and circulate in serum from T2D patients 82 . Vrieling et al demonstrated that oxLDL‐loaded hMDMs display higher mycobacterial burden consistent with lysosomal cholesterol accumulation and subsequent dysfunction 82 .
How Mtb infection drives lipid droplet formation in macrophages remains unclear, although a role for the lipid‐responsive nuclear receptor PPARγ has been demonstrated 83 , 84 , 85 . While this process may serve as another immunometabolic evasion mechanism to co‐opt the intracellular environment towards bacterial growth, recent evidence suggests that the appearance of a foamy macrophage phenotype may in fact serve as a key host defence mechanism. Knight et al profiled the lipid content of Mtb‐infected macrophages with and without IFN‐γ treatment. Remarkably, while both treatments induced the appearance of lipid droplets, the lipid species in IFN‐γ‐treated macrophages were qualitatively different from those measured in Mtb foamy macrophages with more apparent triglycerides and cholesterol esters 86 . They demonstrate that IFN‐γ drives metabolic reprogramming through HIF‐1α, which alters the macrophage lipidome to promote the production of host bioactive lipids prostaglandin E2 and leukotriene B4, which are protective in Mtb infection 87 , and reprogramming lipid metabolism away from species that would favour Mtb.
Finally, it is must be noted that lipids themselves represent a key feature of the TB granuloma, with an area of extracellular lipid deposition apparent in developed granuloma, termed the caseum 88 . This is thought to originate from cell death of infected and bystander foamy macrophages, suggesting that although these cells may play a role containing Mtb, over time the metabolic stresses caused by exhausted glycolysis, extensive FAO and mitochondrial repurposing may eventually become too much for the cell and loaded with the added burden of pathogen growth, eventually commits to cell death. The form of cell death and the way in which surrounding phagocytes recognize and respond to this can determine the fate of the granuloma and the response to TB 89 . Regulated apoptosis driven by functional healthy mitochondria can lead to efferocytosis and successful degradation of its infectious cargo. Unregulated macrophage necrosis indicative of dysfunctional, repurposed mitochondria will release the cell metabolic contents alongside extracellular Mtb and eventually lead to granuloma breakdown and dissemination. Although the impact of various Mtb strains and macrophage backgrounds on cell death pathways has been well documented 90 , how they overlay with the metabolic stresses triggered by Mtb‐induced metabolic reprogramming and impinge upon mitochondria function may in fact determine the outcome of infection as shown in Figure 2, and therefore is a fruitful area for further investigation.
Figure 2.
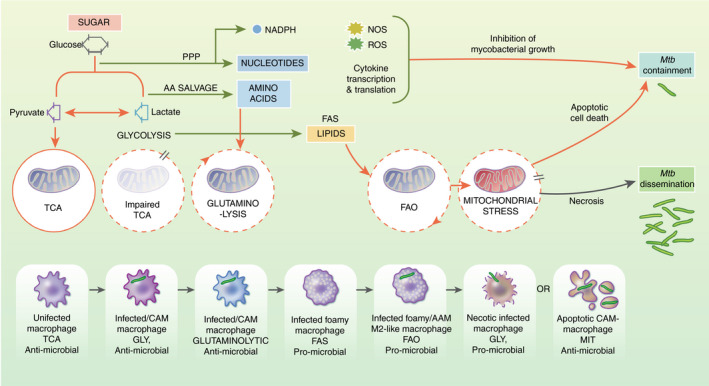
Metabolic reprogramming in the macrophage response to Mtb. 6‐C Sugar is metabolized via cellular glycolysis to 3‐C pyruvate, reduced to 2‐C acetyl‐CoA to feed mitochondrial TCA and associated oxidative phosphorylation (OXPHOS). After infection or classical activation (CAM), macrophages adopt glycolysis and up‐regulate 3‐C lactate, which attenuates TCA cycling but supports pro‐inflammatory and antimicrobial mechanisms. In Mtb‐infected macrophages, AA derived from glycolytic salvage pathways or endogenously can support TCA via glutaminolysis and support pro‐inflammatory activities. TCA repurposing supports fatty acid synthesis (FAS) and is associated with foamy macrophage appearance after Mtb infection. De novo fatty acids can generate acetyl‐coA via b‐oxidation to support TCA (FAO), as observed in altenatively‐activated macrophages. Distinct foamy macrophage phenotypes are associated with host defence or Mtb growth. Finally, if infection is not contained by the induction of pro‐inflammatory cytokines and antimicrobial species (ROS and NO), macrophages commit to cell death. Metabolic reprogramming and associated glycolysis and defective TCA could lead to mitochondrial repurposing and induction of apoptosis, which supports host defence through restricting niches for bacillary replication. However, if other metabolic signals support mitochondrial activities, the bacteria can preferentially drive host T‐cell lysis and necrosis, leading to enhanced bacterial dissemination. Thus, the metabolic output of the macrophage has a major effect on host cell fate and bacterial containment
T‐CELL RESPONSES IN TB
T‐cell and Mtb containment
During a nascent Mtb infection, a robust T‐cell response can be detected by 6–8 weeks after infection. Epidemiological observations indicate that this immune response is not associated with clearance of the pathogen. Rather, this response is used in clinical medicine as a proxy for ongoing infection, with tuberculin skin test (TST)‐positive individuals offered antibiotic therapy, precisely because they are thought to harbour live Mtb. In the minority of infected contacts (5%–10%) who progress to active pulmonary TB disease, the size of their TST response is indistinguishable from infected contacts without disease 91 , again providing evidence that this immune response does not correlate with pathogen killing. In fact, in studies that have enumerated interferon‐gamma‐producing cells that respond to Mtb‐specific antigens, a stronger T‐cell response if anything correlates with disease, rather than protection 92 , 93 . Among patients with TB disease, a significantly lower proportion of individuals have a measurable TST response in disseminated forms of TB, such as miliary TB and meningeal TB 94 . Consistent with this, in CD4 T‐cell‐deficient HIV‐positive individuals who have been exposed to Mtb, extrapulmonary TB is more frequent 95 . Thus, while there is epidemiologic evidence supporting an association between T‐cell responses and containment of infection, there is no evidence that these responses are associated with Mtb elimination.
Mtb has coevolved with humans to achieve an evolutionary trade‐off that infrequently compromises host fitness for survival. Usually, rapid changes in the genome of a pathogen (e.g. influenza virus) provide an evolutionary strategy to outpace host immunity. However, the mutation rate in Mtb is low 96 and human T‐cell epitopes of Mtb reveal no more sequence variation than genes essential for pathogen survival, indicating that Mtb genomes are not being selected for evasion of T‐cell recognition 97 . T cells have in fact been proposed to contribute to TB transmission by participating in the induction of cavitary lung disease 98 , 99 . Furthermore, a mouse strain (C3HeB/FeJ) that generates robust T‐cell responses rapidly succumbs to experimental Mtb infection due to necrotic granulomas 100 , 101 . In addition to the studies above, increasing T‐cell responses above the normal generation of natural immunity may not provide enhanced protection. A TB vaccine candidate, called MVA85A (recombinant vaccinia Ankara‐expressing Ag85A), generates enhanced T‐cell mediated immunity, yet, in a phase IIb infant clinical trial, there was no demonstrable protection against TB disease 102 . Similarly, a large randomized adult clinical trial of comparison of BCG revaccination with H4:IC31 (a candidate subunit vaccine contains both Ag85B and TB10.4 Mtb antigens) showed a significant protection in the BCG group 103 . However, genetic investigations have revealed that prominent T‐cell antigens of Mtb (e.g. ESAT‐6, CFP‐10, MPT64, MPT70 and MPT83) are missing from BCG strains used in clinical trials 104 , consistent with the clinical observation that in BCG trials, there was no correlation between the proportion of subjects that converted their TST and subsequent protection against TB 105 , 106 , 107 . Furthermore, in human studies that monitored T‐cell responses, while it was shown that BCG induced increased T‐cell responses, there were no data linking these responses to protection 108 , 109 .
Although the basis for using adjuvants in non‐live vaccines (e.g. M72 in TB) is their ability to promote effector T‐ and B‐cell differentiation, the direct impact of adjuvants on innate immune cells via pattern recognition receptors (PRR) and innate immunity has so far been neglected. Indeed, adjuvants containing diverse PRR agonists have significantly improved the efficacy of several human vaccines. For instance, the ASO1 adjuvant is common to the current M72 TB vaccine with 54% efficacy among individuals with prior Mtb infection 110 , the shingles vaccine (Shingrix) with more than 95% efficacy among older adults (>50 years old) 111 , and the RTS,S malaria vaccine with 50% efficacy 112 . AS01 is a liposome‐based adjuvant containing MPLA (TLR4 agonist) and the purified saponin fraction QS‐21 (NLRP3 agonist) 113 , 114 . Thus, in M72‐TB vaccine, the direct impact of ASO1 in boosting innate immunity and protection versus Mtb antigen‐specific T‐cell‐mediated protection remains to be determined. It is also important to indicate that the preclinical trials of subunit vaccines with Mtb antigens provide modest protection against Mtb 115 , 116 , 117 . In addition, people previously diagnosed and treated for TB retain strong T‐cell responses, yet remain at high risk for developing disease again 118 . Similarly, in mice cured of primary pulmonary Mtb infection, North and colleagues demonstrated that the secondary T‐cell responses to subsequent Mtb challenge do not prevent and resolve the infection 119 . This argues that although the primary infection has elicited a robust Mtb antigen‐specific T‐cell responses, which helps to contain the infection, this was not sufficient to prevent additional episodes of disease. Whether the generation of enhanced T‐cell‐mediated responses is detrimental, or merely not beneficial, remains to be determined.
For a long time, immunologists have studied host resistance to infections, which focuses on detection and destruction of pathogens. However, it appears that tolerating an infection, as well as controlling tissue damage, is also critical for host defence against pathogens 120 . While the concept of disease tolerance is well established in plants 121 , its contribution to mammalian host defence has only recently been appreciated. We have also recently shown (unpublished data) that a mitochondrial protein known as cyclophilin D is a key molecular mediator of T‐cell‐mediated host tolerance to TB 122 , 123 . Thus, one possibility is that T cells contribute to containing Mtb and tolerating its presence, to mitigate immunopathology and maximize host fitness (Figure 1). As T cells have not been shown to generate sterilizing immunity, efforts to generate a TB vaccine based on T cells might exceed their intrinsic functional capacity, which is potentially containment of Mtb.
Contribution of T cells to disease tolerance
The identification of mutations in the IL‐12/IFN‐γ/STAT1 axis that lead to disseminated mycobacterial infections, termed Mendelian susceptibility to mycobacterial disease (MSMD), along with the susceptibility of T‐cell‐deficient hosts to mycobacterial infections established the dogma that IFN‐γ‐producing T cells play a crucial role in host resistance against TB. However, there is no direct evidence of T cells/IFN‐γ in protection against Mtb, but rather contains infection 92 , 93 , 95 , via regulation of the inflammatory response. For instance, extrapulmonary TB is associated with individuals having lower measurable tuberculin skin test (TST) responses 94 , as well as with HIV‐positive individuals with very low CD4+ T‐cell counts 95 . In addition, IFN‐γ has been shown to inhibit pulmonary neutrophilic inflammation to prevent lung tissue damage during the chronic phase of Mtb infection 124 , 125 . High levels of neutrophils generate a strong inflammatory response that results in increased pulmonary pathology and mortality. Importantly, neutrophil depletion in IFN‐γR−/− mice prolonged their survival 124 . The contribution of neutrophils to immunopathology during Mtb infection has been well established in mice 126 , NHP 127 , 128 and humans 129 . These studies collectively indicate that the IFN pathway is critical in the regulation of inflammatory signals and disease tolerance rather than host resistance.
Furthermore, dysregulated T‐cell responses appear to be detrimental for the host by inducing overt immunopathology. It has been well documented that during chronic viral infection, constant exposure of T cells to antigens and inflammatory cytokines leads to loss of T‐cell function, a process termed ‘T‐cell exhaustion' 130 . One of the well‐defined pathways in T‐cell exhaustion is programmed cell death (PD1). The interaction between PD1, which is expressed on antigen‐experienced T cells, and its ligands PDL‐1 and PDL‐2 prevents T‐cell proliferation and cytokine production. Thus, it was thought that the inhibition of PD1 signalling should promote protection via ‘reviving’ T‐cell‐mediated immunity to chronic Mtb infection. However, while disruption of PD1 signalling through either genetically or neutralizing antibodies significantly enhanced T‐cell‐mediated immunity to Mtb infection, this was associated with increased bacterial growth, massive pulmonary immunopathology and reduced survival 131 , 132 . Thus, the regulatory mechanisms involved in the expansion and contraction of T‐cell responses become a critical determinant of the outcome of TB infection. While the surface expression of some of these markers (e.g. PD1 or KLRG) on T cells appears to be critical for dictating their functional role during infection, the intrinsic immunoregulatory mechanisms of T cells are poorly understood.
T‐cell immunometabolism
To meet the metabolic demands of active cells, mitochondria rapidly switch from a state of catabolism to anabolism to provide the biosynthetic intermediates that are particularly important for lymphocyte function. Naïve T cells have a low rate of metabolic activity, characterized by minimal nutrient uptake and biosynthesis. These cells procure ATP from the energetically efficient processes (OXPHOS) and fatty acid oxidation (FAO) 133 . Upon TCR activation however, dramatic metabolic reprogramming occurs to generate the increased energy needed for T‐cell proliferation, differentiation and cytokine production. To ensure adequate metabolic resources are available, activated T cells increase nutrient uptake and switch from OXPHOS and FAO to aerobic glycolysis 133 . While energetically inefficient, glycolysis enables the cells to rapidly produce ATP and other biosynthetic precursors essential for cell growth and proliferation. This switch from predominantly OXPHOS to aerobic glycolysis, despite the presence of abundant oxygen, is known as the ‘Warburg effect'. Metabolic shift from OXPHOS to glycolysis or vice versa is also highly associated with the inflammatory and anti‐inflammatory role of immune cells 134 .
The metabolic condition also controls the cell fate determination 135 . Th17 cell differentiation relies on glycolysis, whereas blocking glycolysis inhibits Th17 development and promotes regulatory T‐cell (Treg) differentiation. Th17 cells are important in host resistance to Mtb, but uncontrolled production of IL‐17 induces inflammation via recruitment of neutrophils and increases the mortality of Mtb‐infected mice 124 . Higher susceptibility of TLR‐2‐KO mice to Mtb has been linked to reduced accumulation of Treg cells and concomitant increased inflammation 136 . Thus, a balance is required for Th17/Treg development in TB. These findings suggest that the metabolic state determines the fate of immune cells, which is critical in promoting or dampening inflammation.
An equally important function of mitochondria is their role in the cell death programme. Cyclophilin D (CypD), a member of the cyclophilin protein family, is a conserved protein located in the mitochondrial matrix 137 . It has been previously shown that CypD plays a key role in necrosis by regulating the mitochondrial permeability transition pore (MPTP), which allows the passage of solutes and water from the cytoplasm into the mitochondria 138 , 139 . Necrosis of macrophages represents an exit mechanism for Mtb 13 , 140 , 141 , 142 . Remold and colleagues initially demonstrated that the pharmacological inhibition of CypD in human macrophages leads to the inhibition of necrosis and reduction of Mtb growth in vitro 143 . This observation has been recently extended to the zebrafish and mouse models of tuberculosis where the genetic blockade of CypD prevented macrophage necrosis and enhanced their antimycobacterial capacity 144 , 145 . Based on the role of CypD in macrophage immunity to Mtb infection, we initially hypothesized that CypD‐deficient mice (CypD−/−) are resistant to Mtb infection. Surprisingly, CypD−/− mice were highly susceptible to Mtb infection compared with control animals, despite similar numbers of bacteria in both groups. We further identified that this susceptibility was related to an enhanced T‐cell response that promoted lung immunopathology independent of host resistance. We could show that CypD intrinsically regulates T‐cell metabolism and critically regulates disease tolerance in TB 146 . Similarly, the C3HeB/FeJ mouse strain that generates a profound T‐cell response to Mtb infection quickly succumbs to death due to the overgrowth of necrotic granulomas 147 . Although it is still unclear why the functional role of CypD is different in macrophages versus T cells, we envision that as T cells are intrinsically programmed to proliferate, the functional role of CypD in these cells may be wired to regulate the metabolism and proliferation rather than cell death. Collectively, these data indicate that, similar to granulomas, T cells are a double‐edged sword: while they are crucial to initiate granuloma formation during the early phase of Mtb infection and prevent the dissemination of disease, they also play an important role in transmission of Mtb by promoting granuloma necrosis during the active phase of the disease 2 . Thus, the function and location of these effector immune cells are critical determinants of host resistance and disease tolerance in TB.
TARGETING INNATE IMMUNITY AND CELLULAR METABOLISM AGAINST MTB
In TB, while there is evidence to support a role for T cells in disease tolerance during infection, the evidence that they can contribute to host resistance is limited. We envision that the translocation of Mtb from the airways into the lung interstitial tissue signals the transition of host defence mechanisms from resistance to tolerance 148 . However, the remarkable efficacy of innate immunity to eliminate a significant numbers of pathogens from plant to invertebrates and vertebrates, as well as their memory‐like capacity (trained immunity), indicates that the power of innate immune cells, particularly macrophages, can be harnessed against pathogens. The finding that BCG vaccination provides protection against C. albicans in SCID mice lacking both B and T cells, which depends on macrophages 149 , along with human studies indicating that BCG protection in volunteers challenged with yellow fever or malaria was associated with inflammatory monocytes 150 , 151 supports the concept of trained immunity, which can be used to promote host resistance against TB. A variety of live vaccine (e.g. BCG or oral polio vaccine) and adjuvants (e.g. β‐glucan or TLR agonists) have shown to induce trained immunity, which drive metabolic and epigenetic changes in target cells, including innate immune cells (e.g. monocytes, macrophages, NK cells) or stromal cells (e.g. epithelial cells). While direct training of fully differentiated innate immune cells has a limited efficacy for generating long‐term memory response, bone marrow haematopoietic stem cells (HSCs) with self‐renewing capacity can overcome this limitation 152 . For instance, we have recently demonstrated that exposure of HSCs to bacille Calmette–Guérin (BCG) or β‐glucan in the BM results in their reprogramming to promote myelopoiesis and protective trained immunity in macrophages against Mtb infection in type II IFN‐ or IL‐1‐dependent manner, respectively 153 , 154 . Activation and proliferation of HSCs by β‐glucan appears to be dependent on IL‐1β and glycolytic metabolism 155 . Up‐regulation of glycolysis alongside a broken TCA cycle provides acetyl and methyl groups required as both cofactors and substrates for histone remodelling at inflammatory genes 156 . In addition to up‐regulated glycolytic metabolism, increased lipid biosynthesis is also a feature of Training 154 , 155 , 157 . Mevalonate is a key metabolic intermediate in the cholesterol biosynthesis pathway, and mevalonate treatment can drive trained immunity 158 . Finally, both BCG and β‐glucan‐trained macrophages differ from classically primed macrophages as not only they increase glycolytic metabolism but also they manage to retain some oxidative capacity through anaplerotic feeding of TCA, particularly using amino acids as alternative carbon sources, chiefly through glutaminolysis 156 , 159 . This preserves the viability of trained cells and allows the replenishment of TCA mediators including α‐keto‐glutarate production, to provide the substrates and cofactors for epigenetic remodelling. Collectively, these studies suggest that targeting trained immunity can be a novel approach for developing vaccine against TB. Additionally, the use of trained immunity inducers to drive metabolic reprogramming in immune progenitor cells can be a useful strategy to promote disease resistance (Table 1).
Table 1.
Select examples of targeting immunometabolism for improved TB host defence
| Strategy | Metabolite | Immune Pathway | Outcome | Ref |
|---|---|---|---|---|
| Micronutrient supplementation | Vitamin D | (AMPS) cathelicidins | Promote resistance | 163, 164, 165 |
| Vitamin A | Autophagy, lipid metabolism | |||
| Macronutrients supplementation | Glutamine | Metabolic reprogramming, | Promote resistance | 166, 167 |
| Arginine | NO production | Modulate tolerance | ||
| Macronutrient depletion | Tryptophan | Mycobacterial metabolism, granuloma formation | Promote tolerance | 168 |
| Microbial metabolites | SCFA | Anti‐inflammatory | Modulate tolerance | 173, 174 |
| IPA | Promote resistance | |||
| Repurposing small molecule | Rapamycin analogues (everolimus) | mTOR/AKT1 glycolytic reprogramming | Modulate tolerance | 56, 169 |
| Targeting whole‐body metabolism | Metformin | Modulate hyperglycaemia, autophagy, cytokine production | Modulate tolerance | 170, 171, 172 |
| Statins | Macrophage foam cells | Promote resistance | ||
| Promote immune training | BCG | Promote bone marrow myeloid expansion | Increased resistance | 153, 154 |
| Beta‐glucans |
Abbreviations: AMPK, AMP protein kinase; AMPs, antimicrobial peptides; BCG, bacille Calmette–Guérin vaccine; HMGCR, HMG‐CoA reductase; IPA, indole‐3‐propionic acid; NO, nitric oxide; SCFA, short‐chain fatty acids.
CONCLUDING REMARKS
Mtb has coevolved with humans for ~70,000 years 160 , 161 and achieved an evolutionary trade‐off that infrequently compromises host survival. This trade‐off has been conventionally considered to be dependent on host resistance for limiting the growth of Mtb. This ultimately led to the discovery of extensive cellular and molecular mechanisms that were thought to be only engaged in host resistance to TB. However, recent studies indicate an equally important arm of immunity to TB is disease tolerance that limits the collateral tissue damage caused by Mtb or immune responses maintaining the physiological function of the lung. Thus, understanding the entire landscape of both innate and adaptive immunity and their contributions to host resistance and disease tolerance against Mtb is required for developing novel vaccine or therapy. In TB, while there is evidence to support a role for T cells in disease tolerance during infection, the evidence that they can contribute to host resistance is limited. We envision that the translocation of Mtb from the airways into to the lung interstitial tissue, which leads to granuloma formation, signals the transition of host defence mechanisms from resistance to tolerance 148 . Therefore, it becomes essential to dissect the functional capacity of T cells, which is regulated by their cellular metabolism and dictated by the complex microenvironment of the granuloma, preventing dysregulated T‐cell responses and overt immunopathology.
Although we know a great deal about the immune response to chronic pulmonary Mtb infection, our understanding of Mtb‐Mφ interactions and its contribution to the chronicity of disease is extremely limited. Throughout the tug of war between macrophages and Mtb that includes lysosomal function and ultimately the mitochondria metabolism, the fate of the macrophages is decided to either become a permissive host for Mtb and die of necrosis promoting infection or induce apoptosis terminating the niche of infection 13 , 89 , 140 , 162 . Thus, strategies targeting trained Immunity and cellular metabolism (Table 1) are novel approaches to enhance host defence against TB.
TB was known as ‘consumption’ as this chronic infection causes dramatic cachexia (wasting). Interestingly, a study examining Mycobacterium marinum infection in the fruit fly Drosophila melanogaster showed that the increased mortality is independent of bacterial load and is mediated by altered host metabolism and increased body wasting. Thus, the fitness of the host during infection can not only be evaluated by the ability to eliminate the pathogen, but also by the ability to survive chronic infection. Unravelling the cellular and molecular mechanisms involved in the regulation of metabolism in different population of Mφ and T cells is essential for a complete understanding of the pathogenesis of TB.
CONFLICT OF INTEREST
The authors declare no competing interests.
ACKNOWLEDGMENTS
M.D. is supported by the Canadian Institute of Health Research (CIHR) Operating Grants (168884 and 168885) Bill & Melinda Gates Foundation. M.D. holds a Fonds de la Recherche du Quebec‐Sante (FRQS) Award and the Strauss Chair in Respiratory Diseases. FJS is supported by Science Foundation Ireland Frontiers for the Future Award (19/FFP/6625) and Enterprise Ireland Innovation Partnerships (EI/2018/0710 & EI/2019/0880). The authors acknowledge Servier for the artwork used in the figures of this manuscript. Servier Medical ART by Servier is licensed under a Creative Commons Attribution 3.0 Unported Licence; https://smart.servier.com.
Frederick J. Sheedy and Maziar Divangahi contributed equally to this study.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Sweet talk: Metabolic conversations between host and microbe during infection. Immunology 2021, 162: 121‐122.
Schistosome and intestinal helminth modulation of macrophage immunometabolism. Immunology 2021, 162: 123‐134.
Targeting metabolism to reverse T‐cell exhaustion in chronic viral infections. Immunology 2021, 162: 135‐144.
Contributor Information
Frederick J. Sheedy, Email: sheedyf@tcd.ie.
Maziar Divangahi, Email: maziar.divangahi@mcgill.ca.
DATA AVAILABILITY STATEMENT
Not applicable.
References
REFERENCES
- 1. Global Tuberculosis Programme W . Global Tuberculosis Report. Geneva, Switzerland: World Health Organization; 2019. Report No.: 978–92‐4‐156571‐4. [Google Scholar]
- 2. Kwan CK, Ernst JD. HIV and tuberculosis: a deadly human syndemic. Clin Microbiol Rev. 2011;24(2):351‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manyazewal T, Woldeamanuel Y, Blumberg HM, Fekadu A, Marconi VC. The fight to end tuberculosis must not be forgotten in the COVID‐19 outbreak. Nat Med. 2020;26(6):811‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12(8):581‐91. [DOI] [PubMed] [Google Scholar]
- 5. Mayer‐Barber KD, Barber DL. Innate and adaptive cellular immune responses to Mycobacterium tuberculosis infection. Cold Spring Harb Perspect Med. 2015;5(12):1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38(1):79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, et al. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol. 2001;166(7):4604‐11. [DOI] [PubMed] [Google Scholar]
- 8. Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annu Rev Pathol. 2012;7:353‐84. [DOI] [PubMed] [Google Scholar]
- 9. Queval CJ, Brosch R, Simeone R. The macrophage: a disputed fortress in the battle against Mycobacterium tuberculosis . Front Microbiol. 2017;8:2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. Innate immune recognition of Mycobacterium tuberculosis . Clin Dev Immunol. 2011;2011:405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skold M, Behar SM. Tuberculosis triggers a tissue‐dependent program of differentiation and acquisition of effector functions by circulating monocytes. J Immunol. 2008;181(9):6349‐60. [DOI] [PubMed] [Google Scholar]
- 12. Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, et al. Neutrophil‐mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117(7):1988‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Divangahi M, Desjardins D, Nunes‐Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis . Nat Immunol. 2010;11(8):751‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lugo‐Villarino G, Hudrisier D, Benard A, Neyrolles O. Emerging trends in the formation and function of tuberculosis granulomas. Front Immunol 2013;3(405):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benard A, Sakwa I, Schierloh P, Colom A, Mercier I, Tailleux L, et al. B Cells producing type I IFN modulate macrophage polarization in tuberculosis. Am J Respir Crit Care Med. 2018;197(6):801‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coleman MT, Maiello P, Tomko J, Frye LJ, Fillmore D, Janssen C, et al. Early Changes by (18)Fluorodeoxyglucose positron emission tomography coregistered with computed tomography predict outcome after Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. 2014;82(6):2400‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun. 2003;71(12):7099‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu DC, Faldetta KF, Pei L, Sheikh V, Utay NS, Roby G, et al. A paradoxical treatment for a paradoxical condition: infliximab use in three cases of mycobacterial IRIS. Clin Infect Dis. 2015;62(2):258‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med. 2016;22(5):531‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin PL, Maiello P, Gideon HP, Coleman MT, Cadena AM, Rodgers MA, et al. PET CT identifies reactivation risk in cynomolgus macaques with latent M. tuberculosis . PLoS Pathog. 2016;12(7):e1005739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qualls JE, Murray PJ. Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin Immunopathol. 2016;38(2):139‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18(3):363‐74. [DOI] [PubMed] [Google Scholar]
- 23. Martinez N, Kornfeld H. Diabetes and immunity to tuberculosis. Eur J Immunol. 2014;44(3):617‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long‐lived cells in the first week of life via GM‐CSF. J Exp Med. 2013;210(10):1977‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robertson O. Phagocytosis of foreign material in the lung. Physiol Rev. 1941;21(1):112‐39. [Google Scholar]
- 26. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319‐28. [DOI] [PubMed] [Google Scholar]
- 27. Coleman MM, Ruane D, Moran B, Dunne PJ, Keane J, Mills KH. Alveolar macrophages contribute to respiratory tolerance by inducing FoxP3 expression in naive T cells. Am J Respir Cell Mol Biol. 2013;48(6):773‐80. [DOI] [PubMed] [Google Scholar]
- 28. Goerke J. Pulmonary surfactant: functions and molecular composition. Biochim Biophys Acta. 1998;1408(2–3):79‐89. [DOI] [PubMed] [Google Scholar]
- 29. Nguyen HA, Rajaram MV, Meyer DA, Schlesinger LS. Pulmonary surfactant protein A and surfactant lipids upregulate IRAK‐M, a negative regulator of TLR‐mediated inflammation in human macrophages. Am J Physiol‐Lung Cell Mol Physiol. 2012;303(7):L608‐L616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Juarez E, Nunez C, Sada E, Ellner JJ, Schwander SK, Torres M. Differential expression of Toll‐like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir Res. 2010;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez‐Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin‐Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185(1):605‐14. [DOI] [PubMed] [Google Scholar]
- 32. Xue J, Schmidt Susanne V, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014;40(2):274‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015;42(3):419‐30. [DOI] [PubMed] [Google Scholar]
- 34. Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL‐1beta through HIF‐1alpha. Nature 2013;496(7444):238‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodriguez AE, Ducker GS, Billingham LK, Martinez CA, Mainolfi N, Suri V, et al. Serine metabolism supports macrophage IL‐1beta production. Cell Metab. 2019;29(4):1003‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15(6):813‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. 2015;6:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP‐citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. 2013;440(1):105‐11. [DOI] [PubMed] [Google Scholar]
- 40. Gleeson LE, O'Leary SM, Ryan D, McLaughlin AM, Sheedy FJ, Keane J. Cigarette smoking impairs the bioenergetic immune response to Mycobacterium tuberculosis infection. Am J Respir Cell Mol Biol. 2018;59(5):572‐9. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013;38(4):792‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lawless SJ, Kedia‐Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, et al. Glucose represses dendritic cell‐induced T cell responses. Nat Commun. 2017;8(1):15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baker EH, Clark N, Brennan AL, Fisher DA, Gyi KM, Hodson ME, et al. Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol 2007;102(5):1969‐75. [DOI] [PubMed] [Google Scholar]
- 44. Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med. 2018;215(4):1135‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang SC‐C, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell‐intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014;15(9):846‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dodd CE, Pyle CJ, Glowinski R, Rajaram MV, Schlesinger LS. CD36‐mediated uptake of surfactant lipids by human macrophages promotes intracellular growth of Mycobacterium tuberculosis . J Immunol. 2016;197(12):4727‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, et al. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe. 2018;24(3):439‐46e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hackett EE, Charles‐Messance H, O'Leary SM, Gleeson LE, Munoz‐Wolf N, Case S, et al. Mycobacterium tuberculosis limits host glycolysis and IL‐1beta by restriction of PFK‐M via MicroRNA‐21. Cell Rep. 2020;30(1):124‐36e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Woods PS, Kimmig LM, Meliton AY, Sun KA, Tian Y, O'Leary EM, et al. Tissue‐resident alveolar macrophages do not rely on glycolysis for LPS‐induced inflammation. Am J Respir Cell Mol Biol. 2020;62(2):243‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svedberg FR, Brown SL, Krauss MZ, Campbell L, Sharpe C, Clausen M, et al. The lung environment controls alveolar macrophage metabolism and responsiveness in type 2 inflammation. Nat Immunol. 2019;20(5):571‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette smoking impairs human pulmonary immunity to Mycobacterium tuberculosis . Am J Respir Crit Care Med. 2014;190(12):1430‐6. [DOI] [PubMed] [Google Scholar]
- 52. O'Beirne SL, Kikkers SA, Oromendia C, Salit J, Rostmai MR, Ballman KV, et al. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2020;201(6):735‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, et al. Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol. 2010;185(9):5425‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berg RD, Levitte S, O'Sullivan MP, O'Leary SM, Cambier CJ, Cameron J, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell 2016;165(1):139‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cumming BM, Addicott KW, Adamson JH, Steyn AJ. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife. 2018;7:1‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lachmandas E, Beigier‐Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, et al. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur J Immunol. 2016; 46(11):2574‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gleeson LE, Sheedy FJ, Palsson‐McDermott EM, Triglia D, O'Leary SM, O'Sullivan MP, et al. Cutting Edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol. 2016;196(6):2444‐9. [DOI] [PubMed] [Google Scholar]
- 58. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF‐1alpha Is an essential mediator of IFN‐gamma‐dependent immunity to Mycobacterium tuberculosis . J Immunol. 2016;197(4):1287‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci Rep. 2015;5:18176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palsson‐McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif‐1alpha activity and IL‐1beta induction and is a critical determinant of the warburg effect in LPS‐activated macrophages. Cell Metab. 2015;21(1):65‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Braverman J, Stanley SA. Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF‐1alpha and repression of NF‐kappaB. J Immunol. 2017;199(5):1805‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mayer‐Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host‐directed therapy of tuberculosis based on interleukin‐1 and type I interferon crosstalk. Nature 2014;511(7507):99‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, et al. Nitric oxide prevents a pathogen‐permissive granulocytic inflammation during tuberculosis. Nat Microbiol. 2017;2:17072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Palmieri EM, Gonzalez‐Cotto M, Baseler WA, Davies LC, Ghesquiere B, Maio N, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun. 2020;11(1):698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome‐dependent processing of IL‐1beta. Nat Immunol. 2013;14(1):52‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pernet E, Downey J, Vinh DC, Powell WS, Divangahi M. Leukotriene B4–type I interferon axis regulates macrophage‐mediated disease tolerance to influenza infection. Nature Microbiol. 2019;4(8):1389‐400. 10.1038/s41564-019-0444-3 [DOI] [PubMed] [Google Scholar]
- 67. Rich EA, Torres M, Sada E, Finegan CK, Hamilton BD, Toossi Z. Mycobacterium tuberculosis (MTB)‐stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tuber Lung Dis. 1997;78(5–6):247‐55. [DOI] [PubMed] [Google Scholar]
- 68. Howard NC, Marin ND, Ahmed M, Rosa BA, Martin J, Bambouskova M, et al. Mycobacterium tuberculosis carrying a rifampicin drug resistance mutation reprograms macrophage metabolism through cell wall lipid changes. Nat Microbiol. 2018;3(10):1099‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, et al. Microbial stimulation of different Toll‐like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. 2016;2:16246. [DOI] [PubMed] [Google Scholar]
- 70. Osada‐Oka M, Goda N, Saiga H, Yamamoto M, Takeda K, Ozeki Y, et al. Metabolic adaptation to glycolysis is a basic defense mechanism of macrophages for Mycobacterium tuberculosis infection. Int Immunol. 2019;31(12):781‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Serafini A, Tan L, Horswell S, Howell S, Greenwood DJ, Hunt DM, et al. Mycobacterium tuberculosis requires glyoxylate shunt and reverse methylcitrate cycle for lactate and pyruvate metabolism. Mol Microbiol. 2019;112(4):1284‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO‐producing inflammatory dendritic cells. Blood 2012;120(7):1422‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. D'Avila H, Melo RC, Parreira GG, Werneck‐Barroso E, Castro‐Faria‐Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette‐Guerin induces TLR2‐mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol. 2006;176(5):3087‐97. [DOI] [PubMed] [Google Scholar]
- 74. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient‐rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4(11):e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105(11):4376‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guerrini V, Prideaux B, Blanc L, Bruiners N, Arrigucci R, Singh S, et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease‐specific. PLoS Pathog. 2018;14(8):e1007223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dkhar HK, Nanduri R, Mahajan S, Dave S, Saini A, Somavarapu AK, et al. Mycobacterium tuberculosis keto‐mycolic acid and macrophage nuclear receptor TR4 modulate foamy biogenesis in granulomas: a case of a heterologous and noncanonical ligand‐receptor pair. J Immunol. 2014;193(1):295‐305. [DOI] [PubMed] [Google Scholar]
- 78. Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis‐driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. 2012;12(5):669‐81. [DOI] [PubMed] [Google Scholar]
- 79. Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium tuberculosis induces the miR‐33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17(6):677‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14(8):812‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martens GW, Arikan MC, Lee J, Ren F, Vallerskog T, Kornfeld H. Hypercholesterolemia impairs immunity to tuberculosis. Infect Immun. 2008;76(8):3464‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vrieling F, Wilson L, Rensen PCN, Walzl G, Ottenhoff THM, Joosten SA. Oxidized low‐density lipoprotein (oxLDL) supports Mycobacterium tuberculosis survival in macrophages by inducing lysosomal dysfunction. PLoS Pathog. 2019;15(4):e1007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Almeida PE, Silva AR, Maya‐Monteiro CM, Torocsik D, D'Avila H, Dezso B, et al. Mycobacterium bovis bacillus Calmette‐Guerin infection induces TLR2‐dependent peroxisome proliferator‐activated receptor gamma expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. J Immunol. 2009;183(2):1337‐45. [DOI] [PubMed] [Google Scholar]
- 84. Rajaram MV, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator‐activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J Immunol. 2010;185(2):929‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guirado E, Rajaram MV, Chawla A, Daigle J, La Perle KM, Arnett E, et al. Deletion of PPARgamma in lung macrophages provides an immunoprotective response against M. tuberculosis infection in mice. Tuberculosis 2018;111:170‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN‐gamma/HIF‐1alpha signaling and supports host defense. PLoS Pathog. 2018;14(1):e1006874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mayer‐Barber KD, Sher A. Cytokine and lipid mediator networks in tuberculosis. Immunol Rev. 2015;264(1):264‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2(7):258‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage affects immunity to tuberculosis. Adv Exp Med Biol. 2013;783:103‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Spira A, Carroll JD, Liu G, Aziz Z, Shah V, Kornfeld H, et al. Apoptosis genes in human alveolar macrophages infected with virulent or attenuated Mycobacterium tuberculosis: a pivotal role for tumor necrosis factor. Am J Respir Cell Mol Biol. 2003;29(5):545‐51. [DOI] [PubMed] [Google Scholar]
- 91. Al Zahrani K, Al Jahdali H, Menzies D. Does size matter? Utility of size of tuberculin reactions for the diagnosis of mycobacterial disease. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1419‐22. [DOI] [PubMed] [Google Scholar]
- 92. Hinks TS, Dosanjh DP, Innes JA, Pasvol G, Hackforth S, Varia H, et al. Frequencies of region of difference 1 antigen‐specific but not purified protein derivative‐specific gamma interferon‐secreting T cells correlate with the presence of tuberculosis disease but do not distinguish recent from remote latent infections. Infect Immun. 2009;77(12):5486‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Diel R, Loddenkemper R, Niemann S, Meywald‐Walter K, Nienhaus A. Negative and positive predictive value of a whole‐blood interferon‐gamma release assay for developing active tuberculosis: an update. Am J Respir Crit Care Med. 2011;183(1):88‐95. [DOI] [PubMed] [Google Scholar]
- 94. Yaramis A, Gurkan F, Elevli M, Soker M, Haspolat K, Kirbas G, et al. Central nervous system tuberculosis in children: a review of 214 cases. Pediatrics 1998;102(5):E49. [DOI] [PubMed] [Google Scholar]
- 95. Tornheim JA, Dooley KE. Tuberculosis associated with HIV infection. Microbiol Spectr. 2017;5(1):1‐16. [DOI] [PubMed] [Google Scholar]
- 96. Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug‐resistant tuberculosis. Nat Genet. 2013;45(7):784‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. 2010;42(6):498‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mukadi Y, Perriens JH, St Louis ME, Brown C, Prignot J, Willame JC, et al. Spectrum of immunodeficiency in HIV‐1‐infected patients with pulmonary tuberculosis in Zaire. Lancet 1993;342(8864):143‐6. [DOI] [PubMed] [Google Scholar]
- 99. Rodrigo T, Cayla JA, Garcia de Olalla P, Galdos‐Tanguis H, Jansa JM, Miranda P, et al. Characteristics of tuberculosis patients who generate secondary cases. Int J Tuberc Lung Dis. 1997;1(4):352‐7. [PubMed] [Google Scholar]
- 100. Kamath AB, Alt J, Debbabi H, Behar SM. Toll‐like receptor 4‐defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis . Infect Immun. 2003;71(76):4112‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kamath AB, Alt J, Debbabi H, Taylor C, Behar SM. The major histocompatibility complex haplotype affects T‐cell recognition of mycobacterial antigens but not resistance to Mycobacterium tuberculosis in C3H mice. Infect Immun. 2004;72(12):6790‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo‐controlled phase 2b trial. Lancet 2013;381(9871):1021‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, et al. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N Engl J Med. 2018;379(2):138‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Behr MA. BCG–different strains, different vaccines? Lancet Infect Dis. 2002;2(2):86‐92. [DOI] [PubMed] [Google Scholar]
- 105. Comstock GW. Does the protective effect of neonatal BCG vaccination correlate with vaccine‐induced tuberculin reactions? Am J Respir Crit Care Med. 1996;154(1):263‐4. [DOI] [PubMed] [Google Scholar]
- 106. Kagina BM, Abel B, Scriba TJ, Hughes EJ, Keyser A, Soares A, et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette‐Guerin vaccination of newborns. Am J Respir Crit Care Med. 2010;182(8):1073‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mittrucker HW, Steinhoff U, Kohler A, Krause M, Lazar D, Mex P, et al. Poor correlation between BCG vaccination‐induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci U S A. 2007;104(30):12434‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Murray RA, Mansoor N, Harbacheuski R, Soler J, Davids V, Soares A, et al. Bacillus Calmette Guerin vaccination of human newborns induces a specific, functional CD8+ T cell response. J Immunol. 2006;177(8):5647‐51. [DOI] [PubMed] [Google Scholar]
- 109. Soares AP, Scriba TJ, Joseph S, Harbacheuski R, Murray RA, Gelderbloem SJ, et al. Bacillus Calmette‐Guerin vaccination of human newborns induces T cells with complex cytokine and phenotypic profiles. J Immunol. 2008;180(5):3569‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tait DR, Hatherill M, Van Der Meeren O, Ginsberg AM, Van Brakel E, Salaun B, et al. Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. N Engl J Med. 2019;381(25):2429‐39. [DOI] [PubMed] [Google Scholar]
- 111. Maltz F, Fidler B. Shingrix: a new herpes zoster vaccine. P T. 2019;44(7):406‐33. [PMC free article] [PubMed] [Google Scholar]
- 112. Kazmin D, Nakaya HI, Lee EK, Johnson MJ, van der Most R, van den Berg RA, et al. Systems analysis of protective immune responses to RTS, S malaria vaccination in humans. Proc Natl Acad Sci U S A. 2017;114(9):2425‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vandepapeliere P, Horsmans Y, Moris P, Van Mechelen M, Janssens M, Koutsoukos M, et al. Vaccine adjuvant systems containing monophosphoryl lipid A and QS21 induce strong and persistent humoral and T cell responses against hepatitis B surface antigen in healthy adult volunteers. Vaccine 2008;26(10):1375‐86. [DOI] [PubMed] [Google Scholar]
- 114. Welsby I, Detienne S, N'Kuli F, Thomas S, Wouters S, Bechtold V, et al. Lysosome‐dependent activation of human dendritic cells by the vaccine adjuvant QS‐21. Front Immunol. 2016;7:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Reed SG, Coler RN, Dalemans W, Tan EV, DeLa Cruz EC, Basaraba RJ, et al. Defined tuberculosis vaccine, Mtb72F/AS02A, evidence of protection in cynomolgus monkeys. Proc Natl Acad Sci U S A. 2009;106(7):2301‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Brandt L, Skeiky YA, Alderson MR, Lobet Y, Dalemans W, Turner OC, et al. The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72‐kilodalton fusion polyprotein Mtb72F in M. tuberculosis‐infected guinea pigs. Infect Immun. 2004;72(11):6622‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Darrah PA, DiFazio RM, Maiello P, Gideon HP, Myers AJ, Rodgers MA, et al. Boosting BCG with proteins or rAd5 does not enhance protection against tuberculosis in rhesus macaques. NPJ Vaccines. 2019;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Verver S, Warren RM, Beyers N, Richardson M, van der Spuy GD, Borgdorff MW, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am J Respir Crit Care Med. 2005;171(12):1430‐5. [DOI] [PubMed] [Google Scholar]
- 119. Jung YJ, Ryan L, LaCourse R, North RJ. Properties and protective value of the secondary versus primary T helper type 1 response to airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2005;201(12):1915‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science 2012;335(6071):936‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schafer JF. Tolerance to plant disease. Annu Rev Phytopathol. 1971;9:235‐52. [Google Scholar]
- 122. Gillard J, Tzelepis F, Jaworska J, Divangahi M. Mitochondrial cyclophilin D is a critical regulator of T cell mediated immunity to Mycobacterium tuberculosis . Am J Respir Crit Care Med. 2013;187:A6028. [Google Scholar]
- 123. Divangahi M. How to Win Against TB: Host Resistance Versus Host Tolerance Keystone Symposia: New Development in Our Basic Understanding of Tuberculosis. 2017.
- 124. Nandi B, Behar SM. Regulation of neutrophils by interferon‐gamma limits lung inflammation during tuberculosis infection. J Exp Med. 2011;208(11):2251‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Desvignes L, Ernst JD. Interferon‐gamma‐responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis . Immunity 2009;31(6):974‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73(3):1744‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Flynn JL, Gideon HP, Mattila JT, Lin PL. Immunology studies in non‐human primate models of tuberculosis. Immunol Rev. 2015;264(1):60‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mattila JT, Maiello P, Sun T, Via LE, Flynn JL. Granzyme B‐expressing neutrophils correlate with bacterial load in granulomas from Mycobacterium tuberculosis‐infected cynomolgus macaques. Cell Microbiol. 2015;17(8):1085‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon‐inducible neutrophil‐driven blood transcriptional signature in human tuberculosis. Nature 2010;466(7309):973‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Barber DL, Mayer‐Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD‐1‐mediated inhibition. J Immunol. 2011;186(3):1598‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lazar‐Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death‐1 (PD‐1)‐deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A. 2010;107(30):13402‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science 2013;342(6155):1242454.24115444 [Google Scholar]
- 134. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo‐starvation. Nature 2013;493(7432):346‐55. [DOI] [PubMed] [Google Scholar]
- 135. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. McBride A, Konowich J, Salgame P. Host defense and recruitment of Foxp3(+) T regulatory cells to the lungs in chronic Mycobacterium tuberculosis infection requires toll‐like receptor 2. PLoS Pathog. 2013;9(6):e1003397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621‐32. [DOI] [PubMed] [Google Scholar]
- 138. Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005;434(7033):652‐8. [DOI] [PubMed] [Google Scholar]
- 139. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434(7033):658‐62. [DOI] [PubMed] [Google Scholar]
- 140. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10(8):899‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tzelepis F, Verway M, Daoud J, Gillard J, Hassani‐Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross‐presentation during Mycobacterium tuberculosis infection. J Clin Invest. 2015;125(2):752‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8(9):668‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gan H, He X, Duan L, Mirabile‐Levens E, Kornfeld H, Remold HG. Enhancement of antimycobacterial activity of macrophages by stabilization of inner mitochondrial membrane potential. J Infect Dis. 2005;191(8):1292‐300. [DOI] [PubMed] [Google Scholar]
- 144. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013;153(3):521‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zhao X, Khan N, Gan H, Tzelepis F, Nishimura T, Park SY, et al. Bcl‐xL mediates RIPK3‐dependent necrosis in M. tuberculosis‐infected macrophages. Mucosal Immunol. 2017;10(6):1553‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Tzelepis F, Blagih J, Khan N, Gillard J, Mendonca L, Roy DG, et al. Mitochondrial cyclophilin D regulates T cell metabolic responses and disease tolerance to tuberculosis. Sci Immunol. 2018;3(23). [DOI] [PubMed] [Google Scholar]
- 147. Irwin SM, Driver E, Lyon E, Schrupp C, Ryan G, Gonzalez‐Juarrero M, et al. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis . Dis Model Mech. 2015;8(6):591‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Meunier I, Kaufmann E, Downey J, Divangahi M. Unravelling the networks dictating host resistance versus tolerance during pulmonary infections. Cell Tissue Res. 2017;367(3):525‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette‐Guerin induces NOD2‐dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA 2012;109(43):17537‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang S‐Y, Oosting M, et al. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018;23(1):89‐100.e5. [DOI] [PubMed] [Google Scholar]
- 151. Walk J, de Bree LCJ, Graumans W, Stoter R, van Gemert G‐J, van de Vegte‐Bolmer M, et al. Outcomes of controlled human malaria infection after BCG vaccination. Nat Commun. 2019;10(1):1‐8. 10.1038/s41467-019-08659-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Netea MG, Dominguez‐Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol 2020;20(6):375‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 2018;172(1–2):176‐90 e19. [DOI] [PubMed] [Google Scholar]
- 154. Moorlag S, Khan N, Novakovic B, Kaufmann E, Jansen T, van Crevel R, et al. beta‐Glucan induces protective trained immunity against Mycobacterium tuberculosis infection: a key role for IL‐1. Cell Rep. 2020;31(7):107634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 2018;172(1–2):147‐61e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24(6):807‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3‐dependent innate immune reprogramming. Cell 2018;172(1–2):162‐75 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell 2018;172(1–2):135‐46e9. [DOI] [PubMed] [Google Scholar]
- 159. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG‐induced trained immunity. Cell Rep. 2016;17(10):2562‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gutierrez MC, Brisse S, Brosch R, Fabre M, Omais B, Marmiesse M, et al. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis . PLoS Pathog. 2005;1(1):e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato‐Maeda M, et al. Out‐of‐Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Keane J, Balcewicz‐Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. 1997;65(1):298‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: vitamin D‐mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol. 2007;179(4):2060‐3. [DOI] [PubMed] [Google Scholar]
- 164. Coleman MM, Basdeo SA, Coleman AM, Cheallaigh CN, Peral de Castro C, McLaughlin AM, et al. All‐trans retinoic acid augments autophagy during intracellular bacterial infection. Am J Respir Cell Mol Biol. 2018;59(5):548‐56. [DOI] [PubMed] [Google Scholar]
- 165. Wheelwright M, Kim EW, Inkeles MS, De Leon A, Pellegrini M, Krutzik SR, et al. All‐trans retinoic acid‐triggered antimicrobial activity against Mycobacterium tuberculosis is dependent on NPC2. J Immunol. 2014;192(5):2280‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Koeken V, Lachmandas E, Riza A, Matzaraki V, Li Y, Kumar V, et al. Role of glutamine metabolism in host defense against Mycobacterium tuberculosis infection. J Infect Dis. 2019;219(10):1662‐70. [DOI] [PubMed] [Google Scholar]
- 167. Rapovy SM, Zhao J, Bricker RL, Schmidt SM, Setchell KD, Qualls JE. Differential requirements for L‐citrulline and L‐arginine during antimycobacterial macrophage activity. J Immunol. 2015;195(7):3293‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, et al. Tryptophan biosynthesis protects mycobacteria from CD4 T‐cell‐mediated killing. Cell 2013;155(6):1296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Ashley D, Hernandez J, Cao R, To K, Yegiazaryan A, Abrahem R, et al. Antimycobacterial effects of everolimus in a human granuloma model. J Clin Med. 2020;9(7):2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159. [DOI] [PubMed] [Google Scholar]
- 171. Lachmandas E, Eckold C, Bohme J, Koeken V, Marzuki MB, Blok B, et al. Metformin alters human host responses to Mycobacterium tuberculosis in healthy subjects. J Infect Dis. 2019;220(1):139‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014;209(5):754‐63. [DOI] [PubMed] [Google Scholar]
- 173. Lachmandas E, van den Heuvel CNAM, Damen MSMA, Cleophas MCP, Netea MG, van Crevel R. Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short‐Chain Fatty Acids. J Diabetes Res. 2016. 10.1155/2016/6014631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Negatu DA, Yamada Y, Xi Y, Go ML, Zimmerman M, Ganapathy U, et al. Gut Microbiota Metabolite Indole Propionic Acid Targets Tryptophan Biosynthesis in Mycobacterium tuberculosis. mBio. 2019;10 10.1128/mBio.02781-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.