

Abstract
Self-nanoemulsifying drug delivery systems (SNEDDS) were used to enhance the dissolution rate of furosemide as a model for class IV drugs and the system was solidified into liquisolid tablets. SNEDDS of furosemide contained 10% Castor oil, 60% Cremophor EL, and 30% PEG 400. The mean droplets size was 17.9 ± 4.5 nm. The theoretical model was used to calculate the amounts of the carrier (Avicel PH101) and coating materials (Aerosil 200) to prepare liquisolid powder. Carrier/coating materials ratio of 5/1 was used and Ludipress was added to the solid system, thus tablets with hardness of 45 ± 2 N were obtained. Liquisolid tablets showed 2-folds increase in drug release as compared to the generic tablets after 60 min in HCl 0.1 N using USP apparatus-II. Furosemide loaded SNEDDS tablets have great prospects for further in vivo studies, and the theoretical model is useful for calculating the adequate amounts of adsorbents required to solidify these systems.
Subject terms: Drug development, Nanomedicine
Introduction
Class IV drugs of the Biopharmaceutical Classification System (BCS) present a great challenge in oral formulations due to their low solubility and permeability since solubility enhancement approaches alone may not be sufficient to enhance oral bioavailability of these drugs (e.g., amphotericin B, furosemide, acetazolamide, ritonavir, paclitaxel)1.
Self-emulsifying drug delivery systems (SEDDS) present a useful mean to enhance both solubility and permeability of both class II and IV drugs2,3. The drug is dissolved in a mixture of oil, surfactant and co-solvent which forms oil-in-water (o/w) micro- or nano-emulsion with the gastrointestinal aqueous fluids after oral administration under gentle agitation by the digestive system4–7. SEDDS promote the intestinal lymphatic transport of the drug and inhibit its enzymatic degradation and gut wall efflux, which increase the intracellular concentration of the drug, and reduce the variability in rate and extent of absorption8–12.
Liquisolid systems (LS) were also developed to enhance drugs solubility. The drug solution in non-volatile solvent is adsorbed on a carrier material, and then mixed with a coating material with high surface area to prepare a dry powder13–15.
Liquid SEDDS are usually filled in soft gelatine capsules which limited their widespread16–18. This led to the development of solid SEDDS by several methods into powder to be either filled in hard capsules or compressed into tablets19,20. However, the preparation of tablets is highly beneficial since hard capsules may only be filled up to about 400 mg due to the low bulk density of the carriers used (e.g. silicates)21.
Furosemide (FUR), a potent loop diuretic, is a class IV drug due to its low solubility in water (5–25 µg/ml) and low permeability22,23. The oral bioavailability of FUR is highly variable and the response to treatment is unpredictable with a large degree of differences within and between patients24.
The aim of this study was to optimize a hybrid liquisolid/self-emulsifying system of FUR as a model for class IV drugs and to prepare liquisolid tablets with acceptable properties.
Materials and methods
Materials
FUR was provided as a gift sample from UNIPHARMA pharmaceutical industries (AMRI, Aurangabad, India). Tween 80 was obtained from Riedel-De häen (Seelze-Hannover, Germany). Cremophor EL and Cremophor RH40 were a gift sample from Dar Al Dawa' pharmaceutical Co. Ltd (Amman, Jordan). Castor oil, Oleic acid, Glycerol, Propylene glycol (PG) and Polyethylene glycol 400 (PEG 400) were purchased from Panreac Co. Ltd (Barcelona, Spain). Sesame oil, Sunflower oil, and Soybean oil were purchased from the local market. Avicel PH101 was obtained from FMC Co. Ltd (Philadelphia, USA). Aerosil 200 was from Evonik industries (Rheinfelden, Germany). Ludipress was purchased from BASF (Ludwigshafen, Germany).
Formulation of FUR loaded self-nanoemulsifying drug delivery system (FUR-SNEDDS)
Equilibrium solubility of FUR in excipients
In order to prepare FUR-SNEDDS, the required dose of FUR (20 mg) should dissolve in a small amount of the system. The solubility of FUR in various oils (Sesame oil, Castor oil, Sunflower oil, Soybean oil, Oleic acid), surfactants (Tween 80, Cremophor El, Cremophor RH40), and co-solvents (Glycerol, PG, PEG 400) was determined by shake flask method25. An excess amount of FUR was added to each capped tube containing 5 ml of each of the selected vehicles and was shaken at 25 °C for 48 h to reach equilibrium. Each tube was centrifuged at 6000 rpm (HERMLE Z200A, Germany) for 15 min. The supernatant was then filtered and diluted with methanol as necessary. The amount of soluble FUR was determined using UV-spectrophotometer (Scinco S-3100, Korea) at 274 nm, with methanol as a blank. All measurements were done in triplicates.
Pseudo-ternary phase diagram
The concentration of each component of the system that produced clear emulsions under mild stirring at room temperature (25 °C) was then determined using water titration method26. Three mixtures of surfactant/co-solvent (S/coS) were prepared in 3 ratios: A-mix (1/1), B-mix (2/1), and C-mix (1/2), then each mixture was mixed with the oil phase in 9 ratios (9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 2:8, 1:9). Water was added drop-wise under mild agitation by a magnetic stirrer for 15 min. The resulted mixtures were inspected visually for the formation of a clear/translucent emulsion that is associated with the formation of microemulsions26. The ratios that produced optically clear emulsions were plotted on a pseudo-ternary phase diagram using Tri-plot v1-2–4. No attempt was made to evaluate other phases when a white emulsion or gel-like phase was formed.
Preparation of FUR-SNEDDS
FUR-SNEDDS was prepared by dissolving FUR in sufficient amount of the self-emulsifying system (20 mg FUR/100 mg SNEDDS) and was sonicated for 30 min at 40 °C to facilitate solubilization. The droplets size and polydispersibility index (PdI) of the prepared system were measured using Zetasizer (Nano S, Malvern Instruments Ltd, UK) after diluting 1 ml with 100 ml of distilled water under gentle mixing by a magnetic stirrer.
Formulation of LS powders
Hybrid LS compacts were prepared using the theoretical model14 to calculate the required amounts of the carrier and coating materials and produce flowable powders of FUR-SNEDDS as the liquid vehicle. Avicel PH101 and Aerosil 200 were used as carrier and coating materials, respectively.
The angle of slide (θ) was measured as described by Karmarkar et al.27, where uniform FUR-SNEDDS/powder admixtures that contained either Avicel PH101 or Aerosil 200 with increasing quantities of FUR-SNEDDS were prepared. The liquid/solid ratio that corresponded to an angle of slide of 33° was considered the flowable liquid-retention potential (Φ-value). Φ-value is the maximum amount of liquid that can be retained inside powder bulk (w/w) while maintaining acceptable flowability27.
Φ-value for SNEDDS of Avicel PH101 (ΦCa) and Aerosil 200 (ΦCo) were used to calculate liquid loading factor (Lf) with variant R-values (5, 10, 15, 20) using Eq. (1). The amount of carrier (Q) and coating (q) materials can be calculated by rearranging Eqs. (2) and (3) once the amount of liquid medication (W), and (R) values were determined28.
| 1 |
| 2 |
| 3 |
Flowability of each LS compact with different R-ratios was evaluated using Carr’s compressibility index29.
Reconstitution test
The ability of the hybrid LS compacts to re-emulsify spontaneously was tested by diluting an amount of LS powder that contained 1 ml of SNEDDS 100 times with water under mild agitation to form a clear emulsion and was then filtered to remove solids. The droplets size of the formed emulsion was measured using Zetasizer (Nano S, Malvern Instruments Ltd, UK).
Preparation and characterization of hybrid LS tablets
The calculated amounts of Avicel PH101 and FUR-SNEDDS were blended using a mortar and pestle. The coating material (Aerosil 200) was then added to the mixture to obtain a dry LS powder. Ludipress was added to the LS powder as an adjuvant. A schematic representation of the preparation of the hybrid LS tablets (LST) is illustrated in Fig. 130. Conventional directly compressed tablets (DCT) of FUR mixed with the powder components without the self-emulsifying system were prepared to evaluate the effect of self-emulsifying on drug release. The compositions of LST and DCT are shown in Table 1.
Figure 1.

Schematic representation of the formulation of hybrid LS powder30.
Table 1.
The compositions of LST and DCT for 1 tablet.
| Ingredients | LST | DCT |
|---|---|---|
| Furosemide | 20 mg | 20 mg |
| SNEDDS | 100 mg | – |
| Avicel PH101 | 260 mg | 260 mg |
| Aerosil 200 | 65 mg | 65 mg |
| Ludipress | 300 mg | 300 mg |
Single punch tablet machine (Erweka AR 402, Germany) with flat-faced punch was used for both tablets (LST and DCT). The hardness and friability of LST were evaluated using hardness (Erweka TBH 300, Germany) and friability testers (Erweka TAR 120, Germany).
Differential Scanning Calorimeter (DSC)
The physical state of FUR in SNEDDS and LS powder was studied by DSC (Mettler Toledo TG50, Germany). Changes in melting enthalpy, glass transition temperature, and percentage of crystallinity due to any interactions with excipients were evaluated. Accurately weighed samples (2–10 mg) were placed in standard aluminium pans and the sample cell was purged with dry nitrogen at a flow rate of 100 ml/min. All samples were scanned from 25 to 400 °C at a heating rate of 10 °C/min.
X-ray diffraction (XRD)
XRD patterns for FUR in LS powder and its pure components were recorded at room temperature (Stadi-P diffractometer, STOE, Germany). The samples were subjected to 40 kV voltage and 30 mA current conditions. The patterns were scanned over the angular range 3–90° (2θ) with 0.05° intervals and a counting time of 60 s per step.
Scanning electron microscopy (SEM)
SEM imaging was utilized to examine the surface morphological characters of LS powder. The samples were mounted on a slab of metal with a double-sided adhesive carbon tape and were examined (VEGA-II, Tescan, Czech Republic).
In-vitro dissolution studies
FUR release from LST in comparison to DCT and generic FUR tablets (GT) was determined by USP apparatus-II (DIS 8000, Copley, UK). The dissolution medium was 900 ml of either phosphate buffer pH 5.8 or HCl 0.1 N at 37 ± 0.5 °C, stirred at 100 rpm for 60 min. At the predetermined intervals (5, 10, 20, 30, 45, 60 min), 5 ml aliquots were withdrawn, filtered through a 0.45 µm membrane filter and assayed spectrophotometrically (Scinco S-3100, Korea) at 274 nm. Measurements were done in triplicates. Two-tailed student's t-test was performed to evaluate the significant differences in FUR release from LST and DCT or GT. The difference was considered statistically significant at p-value < 0.05.
Results and discussion
The solubility of FUR in each phase is displayed in Table 2. The highest solubility of FUR was in Castor oil (1.23 ± 0.045 mg/g) and Oleic acid (1.1 ± 0.057 mg/g) as oily phase, and in Cremophor EL (47.62 ± 2.227 mg/g) and PEG 400 (158.48 ± 6.379 mg/g) as surfactant and co-solvent, respectively. Therefore, the pseudo-ternary phase diagrams consisted of Cremophor El/PEG400 (S/coS) in different ratios: A-mix (1:1), B-mix (2:1), and C-mix (1:2) with Castor oil. The concentrations at which a clear/translucent emulsion was formed are represented as (X) in the diagrams in Fig. 2. Oleic acid was excluded from the results since its mixture with (S/coS) did not form a clear emulsion at any ratios.
Table 2.
The solubility of FUR in various oils, surfactants, and co-solvents (mean ± SD, n = 3).
| Vehicle | Solubility (mg/1 g) ± SD |
|---|---|
| Soybean oil | 0.06 ± 0.00 |
| Sunflower oil | 0.06 ± 0.00 |
| Castor oil | 1.23 ± 0.04 |
| Oleic acid | 1.10 ± 0.06 |
| Sesame oil | 0.23 ± 0.02 |
| Tween 80 | 31.4 ± 0.93 |
| Cremophor RH40 | 30.71 ± 1.17 |
| Cremophor El | 47.62 ± 2.23 |
| Glycerol | 0.87 ± 0.01 |
| PG | 13.17 ± 0.55 |
| PEG-400 | 158.48 ± 6.38 |
Figure 2.

Pseudo-ternary phase diagrams of Cremophor El /PEG400:Castor oil:water with different Cremophor El/PEG400 ratios: (A)1:1, (B)2:1 and (C)1:2. (X) Represents the concentrations at which a clear/translucent emulsion was formed. (diamond) Gel-like phase. (triangle) White emulsion.
The diagrams show that the system consisting of 90% B-mix with 10% Castor oil formed a clear emulsion even after the addition of water up to 90% (Fig. 2B). On the other hand, for A-mix and C-mix the emulsion turned white after water addition up to 50% and 40%, respectively (Fig. 2A,C). This may be due to the higher concentration of Cremophor El (60%) as surfactant in B-mix, which formed a nano-emulsion upon water dilution with mean droplet size of (17.9 ± 4.5 nm), and PdI was 0.064. PdI below 0.3 indicates a good droplets size distribution. The droplets size was reported to have an impact on drug absorption since the smaller is the droplets size the larger is the interfacial surface for absorption31.
The solubility of FUR in the SNEDDS was (210.8 ± 26.6 mg/ml, n = 3), which is mainly attributed to the use of PEG 400 in the system as co-solvent.
Preparation of FUR-SNEDDS LS powders
The measured angle of slide of each admixture was plotted against Φ-value of either Avicel PH101 or Aerosil 200 with SNEDDS as the liquid vehicle in Fig. 3. Φ-values that corresponded to the angle of slide of 33° were 0.25 and 0.75 for Avicel PH101 (ΦCa) and Aerosil 200 (ΦCo), respectively. These values were used to calculate Lf using the Eqs. (1–3), where the amount of liquid FUR-SNEDDS (W) was considered (120 mg) in a dosage unit, as each unit contained 20 mg FUR dissolved in 100 mg SNEDDS. Several admixtures with variant R-values (5, 10, 15, 20) were prepared and their flowability and compressibility were evaluated using Carr's compressibility index as demonstrated in Table 3. LS-1 powder with the highest Aerosil 200 amount displayed the best flowability with Carr's index of 15.38% (Table 3). Increasing the amount of Aerosil 200 enhanced the flow properties of LS powder. This may be due the large surface area of Aerosil 200 that covers the surface of carrier particles and works as a lubricant and enhance powder flowability.
Figure 3.
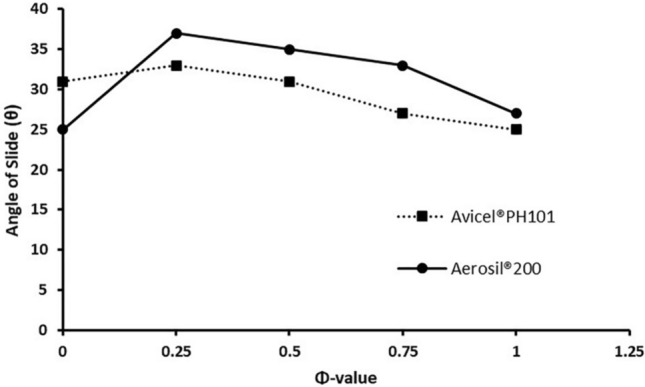
Angle of slide for admixtures of Avicel PH101 and Aerosil 200 SNEDDS as the liquid vehicle in different ratios.
Table 3.
The compositions and compressibility indexes of LS compacts.
| LS compact | R value | Lf | Avicel PH101 (mg) |
Aerosil 200 (mg) |
CI* (%) |
|---|---|---|---|---|---|
| LS-1 | 5 | 0.4 | 300 | 60 | 15.38 |
| LS-2 | 10 | 0.325 | 369.23 | 36.92 | 27.27 |
| LS-3 | 15 | 0.3 | 400 | 26.67 | 27.91 |
| LS-4 | 20 | 0.2875 | 417.39 | 20.87 | 26.67 |
*CI Carr's compressibility index.
Reconstitution test
The mean droplets size of the reconstituted hybrid LS system was (19.82 ± 2.71 nm). The hybrid LS system maintained its ability to form a nanoemulsion after adsorption onto the solid carrier. PdI increased to (0.137) compared to the liquid system after water dilution, but still indicates a good uniformity in droplets size (PdI < 0.3)31.
Characterization of LST
The adsorbents used to solidify SNEDDS have large surface area and consequently have low bulk density which presents a challenge upon filling the required quantities in hard gelatine capsules. However, tablets preparation holds some challenges due to the high compressibility index of silicates and squeezing out of liquids under compression that would lead to fragile tablets; especially at high liquid loads17. Decreasing drug load by increasing the powder quantity would significantly increase tablets size and weight to more than 1 g which makes it harder to swallow.
In the present study, Avicel PH101 and Aerosil 200 were employed as carrier and coating materials, respectively. Avicel PH101 is a commonly used carrier in liquisolid tablets formulation due to its good compactability32. Microcrystalline cellulose (Avicel) exhibits plastic deformation under compression, its microcrystalline nature and the hydrogen bonds between cellulose molecules gives tablets cohesion and strength15. On the other hand, very fine silica derivatives powders like Aerosil 200 with high adsorption properties are employed as coating materials in liquisolid compacts. The coating material (e.g. Aerosil 200) covers the wet carrier particles saturated with liquid to produce dry flowable powder33. Increasing the amount of Aerosil 200 in LS-1 increased Lf, which lowered the amount of powders needed to solidify the emulsifying system. To overcome the compressibility issues of Aerosil 200, an adjuvant was added. Kollidon K30 and Methocel K4M were respectively added to the LS powder in 1:1 ratio with no significant changes in powder compressibility (data not shown).
Ludipress (a mixture of Lactose monohydrate, Povidone, and Crospovidone) was added in increased ratios and tested. When Ludipress was added in 1:1 ratio to LS powder (see Table 2), tablets hardness and friability were 45 ± 2 N and 0.53%, respectively. The mean weight of LST unit was (745.62 ± 11.67 mg, n = 10) and none of the tablets showed a percentage deviation from the mean weight more than ± 5%. The prepared LST fulfilled the pharmacopeial standards of USP-38. Drug content was (99.81%) and within the accepted limits of 95.0–105.0%.
DSC
The thermogram of FUR in Fig. 4D showed a big sharp exothermal peak at 226.94 °C due to its decomposition at its melting temperature34–37. Aerosil 200 is an amorphous material, the thermogram only showed a broad shallow feature due to the evaporation of adsorbed water (Fig. 4B). Similar observation was noticed for Avicel PH101 (Fig. 4C) at 50° to 120 °C before its melting at about 285 °C. The thermogram of LS powder (Fig. 4A) lacked the distinguishing exothermal peak of FUR with broadened endothermic peak of Avicel PH101. The disappearance of drug features in LS formulation indicates the formation of an amorphous solid solution as the drug is molecularly dispersed in LS system38,39.
Figure 4.
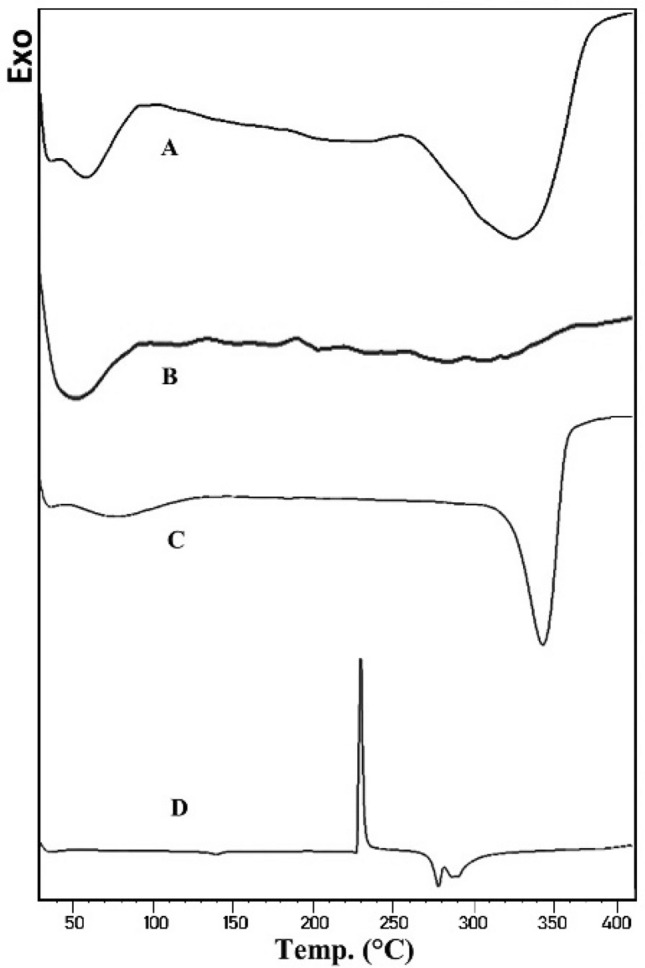
DSC thermograms of: (A) LS powder, (B) Aerosil 200, (C) Avicel PH101, (D) FUR.
XRD
XRD patterns demonstrate the crystalline nature of FUR and the solid components of LS powder (Fig. 5). The diffraction pattern of FUR (Fig. 5D) exhibited its crystalline nature with sharp and well-defined peaks. Avicel PH101 showed a typical microcrystalline pattern with broad peaks due to the small crystallites (Fig. 5C). Aerosil 200 is amorphous, thus the pattern has no defined features that indicate a crystalline module (Fig. 5B). The diffraction pattern of LS powder in (Fig. 5A) showed the broad peaks of Avicel PH101 with a small decline in its intensity. The disappearance of FUR features could be due to its conversion from crystalline to amorphous state. These results are in agreement with the previously mentioned DSC results and implicate that FUR is solubilized in the emulsifying system40.
Figure 5.
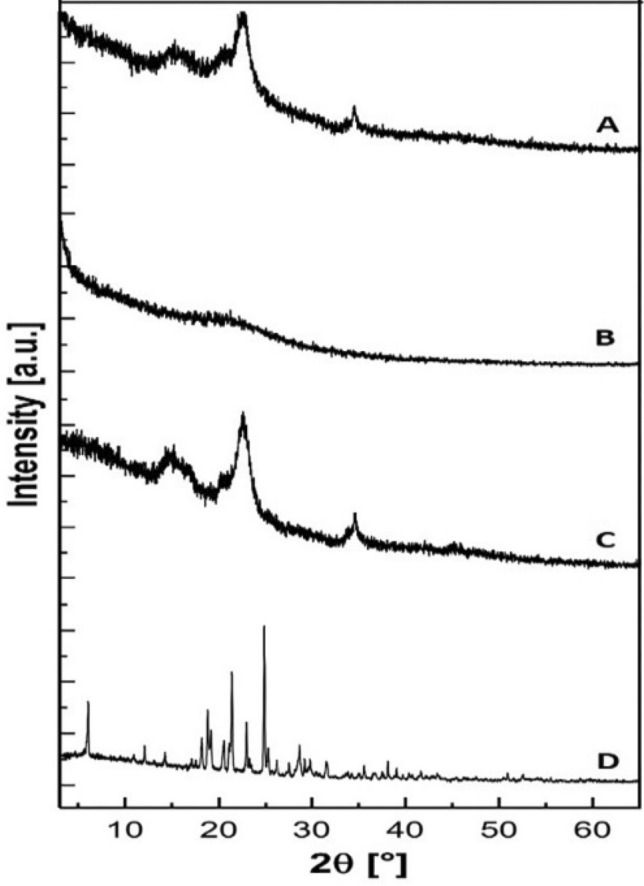
XRD patterns of (A) LS powder, (B) Aerosil 200, (C) Avicel PH101, (D) FUR.
SEM
SEM images in Fig. 6C,D showed dry LS powder. This implicates; according to the liquisolid theory30, that the liquid system is adsorbed onto Avicel PH101 (the carrier material shown in Fig. 6B) and Aerosil 200 (the coating material shown in Fig. 6A) is adsorbed on Avicel particles that appears as a coating layer to form dry surface (see Fig. 1). This would interpret the compressibility index of LS-1 powder as Aerosil 200 acted as a lubricant and enhanced the powder's flowability.
Figure 6.

SEM images of (A) Aerosil 200, 1000x. (B) Avicel PH101, 1000x. (C) LS powder, 1000x, (D) LS powder, 2000x.
In-vitro dissolution studies
The percentages of FUR released from LST, GT and DCT in different mediums are illustrated in Fig. 7. Both LST and GT released about 89% of FUR after 60 min with similar release profiles (P > 0.05, n = 3) in phosphate buffer pH 5.8. This may be attributed to the solubility of FUR in the dissolution medium due to its weak acidic properties (pKa ~ 3.9)24. This would lead to rapid absorption of FUR in the stomach, and the absorption is slowed down along the rising pH in the gastrointestinal tract from 3 to 541.
Figure 7.
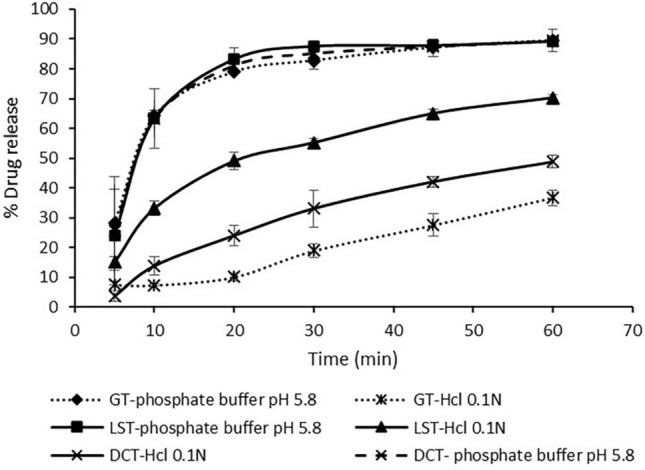
Percentage drug release of FUR from various tablets against time (min) in different mediums. Data are expressed as mean ± SD (n = 3).
Therefore, FUR release was studied in HCl 0.1 N as a dissolution medium. The release profile of LST was significantly different (P < 0.05, n = 3) from both GT and DCT release profiles. LST released 70.3% of FUR after 60 min, and 50% of FUR was released in 20 min. GT and DCT released about 36.7% and 48.8% of FUR after 60 min, respectively.
The self-emulsifying technique enhanced the solubility of FUR in the acidic medium up to 2-folds. The formation of nano-sized droplets upon mixing with the dissolution medium provided a large interfacial surface area, and presented FUR in a dissolved form9. Moreover, FUR is in the more soluble amorphous state in the LS compacts, which increased its solubility in the dissolution medium.
Conclusion
LST demonstrated 2-folds increment in FUR release in the acidic medium (HCl 0.1 N), thus, have great potentials for further in vivo studies because of its higher dissolution in the medium where FUR is mostly absorbed. LS theoretical model is useful in calculating the adequate amounts of adsorbents required to solidify these systems, and the addition of Ludipress in tablets' formulation helped to overcome the poor compressibility characters of Aerosil 200.
Author contributions
All authors have contributed to the work and reviewed the manuscript. Specifically, L.D. conducted the experiments, analysed the results and wrote the main manuscript text. A.W.A and H.EZ supervised the project and the interpretation of the results.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ghadi R, Dand N. BCS class IV drugs: highly notorious candidates for formulation development. J. Control. Release. 2017;248:71–95. doi: 10.1016/j.jconrel.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Murtaza G, Khan SA, Najam-ul-haq M, Hussain I. Comparative evaluation of various solubility enhancement strategies for furosemide. Pak. J. Pharm. Sci. 2014;27:963–973. [PubMed] [Google Scholar]
- 3.Mohsin K, et al. Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug. Int. J. Nanomed. 2016;11:2829. doi: 10.2147/IJN.S104187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohsin K, Pouton CW. The influence of the ratio of lipid to surfactant and the presence of cosolvent on phase behaviour during aqueous dilution of lipid-based drug delivery systems. J. Drug Deliv. Sci. Technol. 2012;22:531–540. doi: 10.1016/S1773-2247(12)50092-4. [DOI] [Google Scholar]
- 5.Alqahtani S, Alayoubi A, Nazzal S, Sylvester PW, Kaddoumi A. Enhanced solubility and oral bioavailability of Y-tocotrienol using a self-emulsifying drug delivery system (SEDDS) Lipids. 2014;49:819–829. doi: 10.1007/s11745-014-3923-6. [DOI] [PubMed] [Google Scholar]
- 6.Khan F, Islam MS, Roni MA, Jalil RU. Systematic development of self-emulsifying drug delivery systems of atorvastatin with improved bioavailability potential. Sci. Pharm. 2012;80:1027–1043. doi: 10.3797/scipharm.1201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov. Today. 2010;15:958–965. doi: 10.1016/j.drudis.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 9.Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int. J. Pharm. 2014;472:40–47. doi: 10.1016/j.ijpharm.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 10.Mohsin K, Long MA, Pouton CW. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: precipitation of drug after dispersion of formulations in aqueous solution. J. Pharm. Sci. 2009;98:3582–3595. doi: 10.1002/jps.21659. [DOI] [PubMed] [Google Scholar]
- 11.Patel D, Sawant KK. Oral bioavailability enhancement of acyclovir by self-microemulsifying drug delivery systems (SMEDDS) Drug Dev. Ind. Pharm. 2007;33:1318–1326. doi: 10.1080/03639040701385527. [DOI] [PubMed] [Google Scholar]
- 12.Mohsin K. Design of lipid-based formulations for oral administration of poorly water-soluble drug fenofibrate: effects of digestion. AAPS PharmSciTech. 2012;13:637–646. doi: 10.1208/s12249-012-9787-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spireas SS, Jarowski CI, Rohera BD. Powdered solution technology: principles and mechanism. Pharm. Res. 1992;9:1351–1358. doi: 10.1023/A:1015877905988. [DOI] [PubMed] [Google Scholar]
- 14.Spireas S, Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int. J. Pharm. 1998;166:177–188. doi: 10.1016/S0378-5173(98)00046-5. [DOI] [Google Scholar]
- 15.Tiong N, Elkordy AA. Effects of liquisolid formulations on dissolution of naproxen. Eur. J. Pharm. Biopharm. 2009;73:373–384. doi: 10.1016/j.ejpb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997;25:47–58. doi: 10.1016/S0169-409X(96)00490-5. [DOI] [Google Scholar]
- 17.Sander C, Holm P. Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech. 2009;10:1388–1395. doi: 10.1208/s12249-009-9340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gumaste SG, Dalrymple DM, Serajuddin ATM, Development of solid SEDDS, V Compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto neusilin US2. Pharm. Res. 2013;30:3186–3199. doi: 10.1007/s11095-013-1106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan A, Rao S, Prestidge CA. Transforming lipid-based oral drug delivery systems into solid dosage forms: an overview of solid carriers, physicochemical properties, and biopharmaceutical performance. Pharm. Res. 2013;30:2993–3017. doi: 10.1007/s11095-013-1107-3. [DOI] [PubMed] [Google Scholar]
- 20.Nikolakakis I, Malamataris S. Self-emulsifying pellets: Relations between kinetic parameters of drug release and emulsion reconstitution—influence of formulation variables. J. Pharm. Sci. 2014;103:1453–1465. doi: 10.1002/jps.23919. [DOI] [PubMed] [Google Scholar]
- 21.Gumaste SG, et al. Development of solid SEDDS, IV: Effect of adsorbed lipid and surfactant on tableting properties and surface structures of different silicates. Pharm. Res. 2013;30:3170–3185. doi: 10.1007/s11095-013-1114-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perioli L, D’Alba G, Pagano C. New oral solid dosage form for furosemide oral administration. Eur. J. Pharm. Biopharm. 2012;80:621–629. doi: 10.1016/j.ejpb.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Van Wart SA, Shoaf SE, Mallikaarjun S, Mager DE. Population-based meta-analysis of furosemide pharmacokinetics. Biopharm. Drug Dispos. 2014;35:119–133. doi: 10.1002/bdd.1874. [DOI] [PubMed] [Google Scholar]
- 24.Pacifici GM. Clinical pharmacology of furosemide in neonates: a review. Pharmaceuticals. 2013;6:1094–1129. doi: 10.3390/ph6091094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prajapat MD, Patel NJ, Bariya A, Patel SS, Butani SB. Formulation and evaluation of self-emulsifying drug delivery system for nimodipine, a BCS class II drug. J. Drug Deliv. Sci. Technol. 2017;39:59–68. doi: 10.1016/j.jddst.2017.02.002. [DOI] [Google Scholar]
- 26.Syed HK, Peh KK. Identification of phases of various oil, surfactant / co-surfactants and water system by ternary phase diagram. Acta Pol. Pharm. Drug Res. 2014;71:301–309. [PubMed] [Google Scholar]
- 27.Karmarkar AB, Gonjari ID, Hosmani AH. Liquisolid technology for dissolution rate enhancement or sustained release. Expert Opin. Drug Deliv. 2010;7:1227–1234. doi: 10.1517/17425247.2010.511173. [DOI] [PubMed] [Google Scholar]
- 28.Elkordy AA, Essa EA, Dhuppad S, Jammigumpula P. Liquisolid technique to enhance and to sustain griseofulvin dissolution: effect of choice of non-volatile liquid vehicles. Int. J. Pharm. 2012;434:122–132. doi: 10.1016/j.ijpharm.2012.05.072. [DOI] [PubMed] [Google Scholar]
- 29.Carr RL. Evaluating flow properties of solids. Chem. eng. 1965;72:163–168. [Google Scholar]
- 30.Hentzschel CM, Sakmann A, Leopold CS. Suitability of various excipients as carrier and coating materials for liquisolid compacts. Drug Dev. Ind. Pharm. 2011;37:1200–1207. doi: 10.3109/03639045.2011.564184. [DOI] [PubMed] [Google Scholar]
- 31.Bandi S, Sanka K, Bakshi V. Enhanced oral delivery of risperidone through a novel self-nanoemulsifying powder (SNEP) formulations: in-vitro and ex-vivo assessment. J. Microencapsul. 2016;33:544–553. doi: 10.1080/02652048.2016.1223200. [DOI] [PubMed] [Google Scholar]
- 32.Elkordy AA, Tan XN, Essa EA. Spironolactone release from liquisolid formulations prepared with Capryol 90, Solutol HS-15 and Kollicoat SR 30 D as non-volatile liquid vehicles. Eur. J. Pharm. Biopharm. 2013;83:203–223. doi: 10.1016/j.ejpb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Kulkarni AS, Aloorkar NH, Mane MS, Gaja JB. LiquisolidSystems : a Review ABSTRACT. Int. J. Pharm. Sci. Nanotechnol. 2010;3:795–802. [Google Scholar]
- 34.Babu NJ, Cherukuvada S, Thakuria R, Nangia A. Conformational and synthon polymorphism in furosemide (Lasix) Cryst. Growth Des. 2010;10:1979–1989. doi: 10.1021/cg100098z. [DOI] [Google Scholar]
- 35.Darandale SS, Vavia PR. Design of a gastroretentivemucoadhesive dosage form of furosemide for controlled release. Acta Pharm. Sin. B. 2012;2:509–517. doi: 10.1016/j.apsb.2012.05.004. [DOI] [Google Scholar]
- 36.Spamer E, Müller DG, Wessels PL, Venter JP. Characterization of the complexes of furosemide with 2-hydroxypropyl-β-cyclodextrin and sulfobutyl ether-7-β-cyclodextrin. Eur. J. Pharm. Sci. 2002;16:247–253. doi: 10.1016/S0928-0987(02)00107-0. [DOI] [PubMed] [Google Scholar]
- 37.Yang W, de Villiers MM. Aqueous solubilization of furosemide by supramolecularcomplexation with 4-sulphonic calix[n]arenes. J. Pharm. Pharmacol. 2004;56:703–708. doi: 10.1211/0022357023439. [DOI] [PubMed] [Google Scholar]
- 38.Fahmy RH, Kassem MA. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2008;69:993–1003. doi: 10.1016/j.ejpb.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Parmar K, Patel J, Sheth N. Formulation and development of embelinliquisolid systems using quality by design approach. J. Pharm. Investig. 2016;46:547–556. doi: 10.1007/s40005-016-0239-y. [DOI] [Google Scholar]
- 40.Parmar K, Patel J, Sheth N. Fabrication and characterization of liquisolid compacts of Embelin for dissolution enhancement. J. Pharm. Investig. 2014;44:391–398. doi: 10.1007/s40005-014-0134-3. [DOI] [Google Scholar]
- 41.Boles Ponto LL, Schoenwald RD. Furosemide (Frusemide): a pharmacokinetic/pharmacodynamic review (part 1) Clin. Pharmacokinet. 1990;18:381–408. doi: 10.2165/00003088-199018050-00004. [DOI] [PubMed] [Google Scholar]