

Abstract
Objective
Erythropoietin (EPO), the cytokine required for erythropoiesis, contributes to metabolic regulation of fat mass and glycemic control. EPO treatment in mice on high-fat diets (HFD) improved glucose tolerance and decreased body weight gain via reduced fat mass in males and ovariectomized females. The decreased fat accumulation with EPO treatment during HFD in ovariectomized females was abrogated with estradiol supplementation, providing evidence for estrogen-related gender-specific EPO action in metabolic regulation. In this study, we examined the cross-talk between estrogen mediated through estrogen receptor α (ERα) and EPO for the regulation of glucose metabolism and fat mass accumulation.
Methods
Wild-type (WT) mice and mouse models with ERα knockout (ERα−/−) and targeted deletion of ERα in adipose tissue (ERαadipoKO) were used to examine EPO treatment during high-fat diet feeding and after diet-induced obesity.
Results
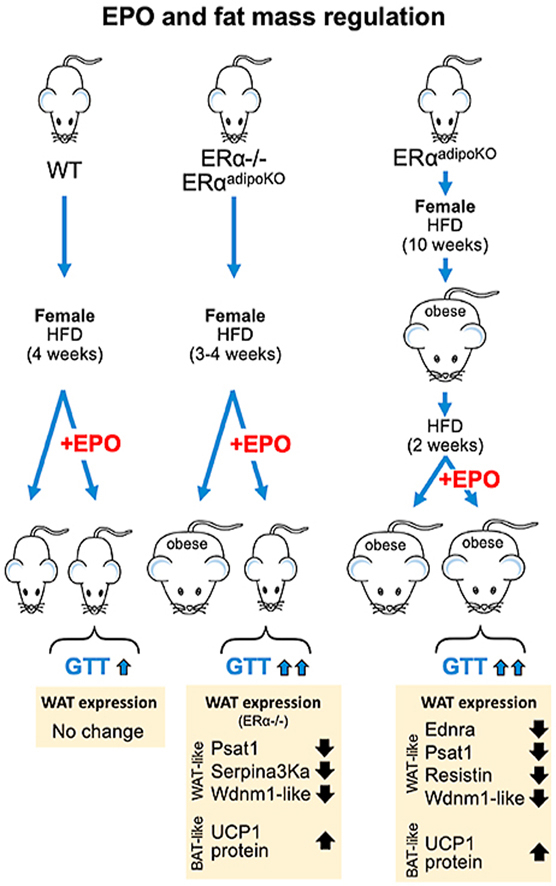
ERα−/− mice on HFD exhibited increased fat mass and glucose intolerance. EPO treatment on HFD reduced fat accumulation in male WT and ERα−/− mice and female ERα−/− mice but not female WT mice. EPO reduced HFD increase in adipocyte size in WT mice but not in mice with deletion of ERα independent of EPO-stimulated reduction in fat mass. EPO treatment also improved glucose and insulin tolerance significantly greater in female ERα−/− mice and female ERαadipoKO compared with WT controls. Increased metabolic activity by EPO was associated with browning of white adipocytes as shown by reductions in white fat-associated genes and induction of brown fat-specific uncoupling protein 1 (UCP1).
Conclusions
This study clearly identified the role of estrogen signaling in modifying EPO regulation of glucose metabolism and the sex-differential EPO effect on fat mass regulation. Cross-talk between EPO and estrogen was implicated for metabolic homeostasis and regulation of body mass in female mice.
Keywords: Erythropoietin, Estrogen receptor α, Fat mass, Obesity, Gender-specific
Graphical abstract
Highlights
-
•
Erythropoietin regulates fat mass in male but not female mice on high-fat diets.
-
•
Female estrogen receptor alpha deletion restores erythropoietin fat mass regulation.
-
•
Estrogen receptor alpha deletion increases erythropoietin regulation of glucose tolerance.
-
•
Erythropoietin reduced white fat-associated genes and increased uncoupling protein 1.
-
•
Erythropoietin and estrogen cross-talk is implicated for metabolic homeostasis.
1. Introduction
Obesity is a worldwide epidemic and a high-risk factor for serious health conditions including type 2 diabetes, heart disease, cardiovascular disease, and cancer due to the strong relationship between body mass index and diabetes and insulin resistance. Several factors, including genetics, diet, hormones, or various cytokines, are documented to regulate adiposity and glucose metabolism, but how these factors affect obesity and its associated metabolic syndromes are unknown. Estrogens are known to play a role in energy metabolism, control of food intake, insulin sensitivity, and lipid metabolism [1]. Men and postmenopausal women accumulate more fat in the intra-abdominal depot than premenopausal women, suggesting that premenopausal women have a reduced risk of developing metabolic complications associated with obesity and insulin resistance [2,3]. The incidence of obesity and type 2 diabetes in women gradually increases after menopause [4,5], and hormonal replacement therapy was reported to reduce the incidence of type 2 diabetes in postmenopausal women [6,7]. In ovariectomized mice, estrogens protect against high-fat diet-induced insulin resistance [8]. These estrogens’ effects in fat regulation and improved glucose metabolism can be explained by estrogen receptors (ER), particularly ERα-mediated response [9,10]. Deletion of ERα in mice leaving ERβ activity intact increased adipose tissue, fatty liver, insulin resistance, and susceptibility to diet-induced obesity [9,[11], [12], [13]], and activity of the AF-1 transcriptional activation domain of ERα prevented fat accumulation by inducing energy expenditure [14].
Erythropoietin (EPO) is a glycoprotein secreted in the adult kidney that targets erythroid progenitor cells in the bone marrow to stimulate red blood cell production. Animal studies showed that non-erythroid EPO response contributes to metabolic regulation. EPO is protective in cardiac ischemia/reperfusion injury through the production of nitric oxide, a regulator of vascular tone, as an acute response to increase oxygen delivery [15,16]. EPO also exhibits neuroprotective activity against ischemia and traumatic injury [17,18]. Transgenic mouse models with disrupted EPO signaling in non-erythroid tissues developed obesity and insulin resistance while overexpression of a human EPO transgene in mice reduced body weight and improved glucose tolerance [19,20]. Interestingly, female mice with EPO receptor restricted to erythroid tissue developed an obese phenotype and insulin resistance earlier than male mice [19]. In full-heritage Pima Indians with a high prevalence of obesity and type 2 diabetes, endogenous EPO levels were associated with percentage weight change per year in a gender-specific manner with increased EPO levels associated with decreased weight change per year in males only [21]. EPO treatment also attenuated body weight gain in male mice including young mice on normal chow, high-fat diet-induced obese mice, and ob/ob mice but not female young or obese mice [19,22]. In ovariectomized female mice on high-fat diet, EPO decreased fat mass gain, but the effect was abrogated after estradiol supplementation [22]. These results provided evidence for estrogen-related gender-specific EPO action in metabolic regulation beyond erythropoiesis.
In this study, we determined the contribution of estrogen response in EPO metabolic regulation including food intake, body composition, and glucose homeostasis by assessing EPO treatment in estrogen receptor α knockout (ERα−/−) mice and mice with ERα deleted in adipose tissue (ERαadipoKO). Accompanying increased hematocrit with EPO treatment in mice during high-fat diet (HFD) feeding is a reduction in fat mass in male wild-type (WT) and ERα−/− mice and female ERα−/− mice, but no change in fat mass in female WT mice. Among non-hematopoietic tissue, EPO receptor is expressed at high levels in white adipose tissue; deletion of EPO receptor in adipose tissue in mice decreased glucose tolerance and insulin sensitivity and increased susceptibility to HFD-induced obesity [19,23]. Using ERαadipoKO, we found that EPO treatment also reduced fat mass in female ERαadipoKO mice. We previously showed that obese male WT mice treated with EPO for 2 weeks was sufficient to improve glucose tolerance and insulin sensitivity and shift inflammation in white adipose tissue toward an anti-inflammatory state [24]. We found that obese female ERαadipoKO mice but not WT female mice treated with EPO for 2 weeks was sufficient to improve fasting glucose levels and insulin sensitivity with reduction in inflammatory markers in white adipose tissue without changes in fat mass. These data suggest that cross-talk between EPO and estrogen-signaling pathways, particularly in female mice, affects regulation of body mass and metabolic homeostasis, and direct ERα response in adipose tissue blunts EPO-stimulated improvement in glycemic control.
2. Methods
2.1. Animals
Animal procedures were approved by the National Institute of Diabetes and Digestive and Kidney Diseases Animal Care and Use Committee and carried out in accordance with the National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals. Mice were maintained under controlled temperature (23 °C) and photoperiod conditions (12/12 h light/dark cycle) with food and water ad libitum. All of the mice had a C57BL/6 background. ERα+/- mice (Jackson Laboratory, Bar Harbor, ME, USA) were mated to produce ERα−/− mice. ERαadipoKO mice were generated by crossing adiponectin-Cre mice (Jackson Laboratory, Bar Harbor, ME, USA) with mice containing ERα-floxed alleles [25]. Mice with ERα-floxed alleles without Cre were used as controls for the ERαadipoKO mice. Genotype was determined at weaning by PCR of extracted DNA. The WT and ERα−/− mice were fed HFD (60 kcal% fat [high fat]) or NCD at 4 months or 6 months for 4 weeks with or without EPO (recombinant human EPO, Epogen, Amgen, Thousand Oaks, CA, USA) treatment (3000 units/kg three times/week). The ERαadipoKO and their littermate control mice were fed HFD at 4 months for 3 weeks with saline or EPO treatment (3000 units/kg three times/week). To generate obese mice, the ERαadipoKO and their littermate control mice were fed the HFD for 12 weeks and the last 2 weeks were treated with saline or EPO (3000 units/kg three times/week). EPO was administrated by intraperitoneal injection. Food intake and activity levels monitored by indirect colorimetry were determined using the comprehensive laboratory animal monitoring system described [19]. Body composition was measured using a 3-in-1 EchoMRI (Echo Medical Systems). Hematocrit was measured manually before and every week after EPO administration.
2.2. Glucose and insulin tolerance tests
Glucose (GTT) and insulin tolerance tests (ITT) were conducted as described [19].
2.3. Quantitative real-time RT-PCR
Quantitative real-time RT-PCR analyses were carried out using gene-specific primers (Supplementary Table S1) and fluorescently labeled SYBR Green dye (Roche) in a 7900 Sequence Detector (PE Applied Biosystems, Foster City, CA, USA). For relative mRNA quantification, Ct values were normalized with RPL13a as an internal control using the delta–delta CT method.
2.4. Histology and microscopy
Gonadal fat pads were fixed in 10% formalin. Hematoxylin and eosin (H&E)-stained sections were imaged to measure fat cell sizes and crown-like structures. H&E-sectioned slides were scanned by a NonoZoomer digital scanner and analyzed with NDP viewing software and ImageJ. At least 2000 cells were counted for each group.
2.5. Serum parameter analysis
Leptin concentrations were determined by a multiplex assay (MMHMAG-44k, EMD Millipore, Billerica, MA, USA). The adiponectin concentration was determined by ELISA (BioVision, Inc.).
2.6. Western blotting
Cells were lysed in denaturing radioimmunoprecipitation (RIPA) assay buffer supplemented with protease and phosphatase inhibitors (Roche). Lysates were resolved by 4–20% Tris-glycine SDS/PAGE, transferred to nitrocellulose membranes, and blotted using an XCell SureLock Mini-Cell system (Invitrogen) and visualized using protein-specific antibodies (Supplementary Table S2). Quantitative analysis was performed by measuring the integrated density with an NIH ImageJ system and normalized with β-actin.
2.7. Preadipocyte differentiation
3T3L1 cells (American Type Culture Collection, Manassas, VA, USA) were grown in 5% CO2, DMEM, 10% FBS, and 1% pen/strep (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). For preadipocyte differentiation, cells at 100% confluency were stimulated in DMEM medium supplemented with 0.5 mM of 3-isobutyl-1-methylxanthine, 5 μg/ml of insulin, and 1 μM of dexamethasone (Sigma, Burlington, MA, USA) in the absence or presence of EPO (5 unit/ml) or 10 nM of 17-β estradiol. After 2 days, maintenance medium (DMEM, 10% FBS, 1% pen/strep, and 5 μg/ml insulin) was used for 2 days. After 2 days, regular medium (DMEM, 10% FBS, and 1% pen/strep) was used until more than 90% of the 3T3L1 adipocytes expressed the mature adipocyte phenotype. Fresh EPO and 17-β estradiol were added at medium changes. Between 7 or 8 days, differentiated 3T3L-1 cells were serum-deprived for 12 h prior to the experiments. After serum deprivation, 5 unit/ml of EPO, 10 nM of 17-β estradiol, and 5 μg/ml of insulin were added for 24 h and mature 3T3L1 adipocytes were harvested.
2.8. Statistical analysis
The data are expressed as mean ± s.e.m. Comparisons between two groups were made using Student's two-tailed non-paired t-test. Statistical differences between three or more groups were evaluated by two-way ANOVA. P values of <0.05 were regarded as statistically significant.
3. Results
3.1. EPO protected against body weight gain and improved glucose tolerance in the ERα−/− mice
The female ERα−/− mice had similar body weights to the WT mice after weaning (5 weeks), followed by a disproportionate increase in body weight. By 16 weeks, they were 33% larger than the female WT mice (Figure 1A). In contrast, the male ERα−/− mice after weaning had 17% less body weight than the male WT mice but by 16 weeks, their body weights were similar to the male ERα−/− and WT mice. Fat mass was greater in the male and female ERα−/− mice than the WT mice at 10 and 16 weeks (Figure 1B). However, the female ERα−/− mice had more lean mass than the female WT mice while the male ERα−/− mice had less lean mass than the WT mice (Figure 1C). To determine the EPO's treatment ability to regulate body weight gain, the adult WT and ERα−/− mice at 4 months on a normal chow diet (NCD) or high-fat diet (HFD) were treated with high doses of EPO (3000 U/kg 3 times per week) for 4 weeks. On the NCD, EPO treatment did not change body weight or fat mass in the female WT or ERα−/− mice (Figure 1D,G). On the HFD, only the female ERα−/− mice showed significantly decreased body weight and fat mass accumulation with EPO treatment (Figure 1E,H). Similarly, at 6 months, the female ERα−/− mice on the HFD exhibited significantly decreased body weight and fat mass accumulation with EPO treatment, but not the female WT mice (Supplementary Figure S1A–B). These data indicated that EPO treatment had a greater effect on regulating the increase in body weight and fat mass in the female ERα−/− mice that had more accumulated weight and fat mass than the WT mice. In contrast, the male WT and ERα−/− mice with body weights comparable to the female ERα−/− mice at 4 months of age showed significant reductions in body weight and fat mass with EPO treatment (Figure 1F,I). These results demonstrate that EPO had a significant effect in reducing fat mass gain in the female ERα−/−, male ERα−/−, and male WT but not in the female WT, suggesting that ERα in the female mice protected against HFD-induced obesity and reduced the ability of EPO to regulate body weight gain. EPO treatment had little or no effect on lean mass (Supplementary Figs. S2A–D) and the increase in hematocrits was comparable in the WT and ERα−/− mice (Supplementary Figure S2E–H). Decreased food intake and increased activity promoted a lean phenotype. We assessed the food intake and locomotor activity in the 6-month-old female WT and ERα−/− mice. EPO treatment in the female ERα−/− but not the WT mice showed a tendency toward decreased food intake (Supplementary Figure S2I). A trend of increased total activity with EPO treatment was also observed in the female ERα−/− but not the WT mice (Supplementary Figure S2J). The trends of EPO-stimulated reduced food intake and increased total activity in the female ERα−/− mice followed the EPO response of the WT male mice previously reported [19], providing further support for EPO regulation of energy homeostasis. These data suggested that EPO regulation of body weight/fat mass and energy homeostasis was more effective with the loss of ERα, particularly in the female mice that lost estrogen-associated protective activity against HFD-induced obesity. Obese ERα−/− mice are reported to exhibit reduced energy expenditure without changes in energy intake and lower metabolic activity [9]. We found that EPO treatment in the female WT and ERα−/− mice on the HFD did not affect oxygen consumption or respiratory quotient determined by indirect calorimetry at 22 °C (Supplementary Figure S3).
Figure 1.

EPO's effects on body weight and fat mass accumulation in the ERα−/− mice. (A) Body weight of the WT mice and ERα−/− mice are indicated for female and male mice. Symbols indicate the female WT (filled circle, solid line) and ERα−/− (open square, dotted line) mice and male WT (filled triangle, dashed line) and ERα−/− (open inverted triangle, dash-dotted line) mice. (B–C) Fat mass (B) and lean mass (C) in the ERα−/− mice and WT littermate control mice at 10 weeks and 16 weeks of age for the female and male mice (n = 12/group, ∗p < 0.05, ∗∗p < 0.01, and ∗ means WT vs ERα−/− mice). (D–I) Measurement of body weight (D–F) and fat mass (G–I) in the WT and ERα−/− mice treated with EPO or PBS for 4 weeks for females at 4 months on the normal chow diet (NCD; D and G), females at 4 months on the high-fat diet (HFD; E and H), and males at 4 months on the HFD (F and I). Symbols indicate WT PBS (filled circle, solid line) and EPO (open square, thin line)-treated mice and ERα−/− PBS (filled triangle, solid line) and EPO (open inverted triangle, dotted line)-treated mice. (n = 6–9/group, ∗p < 0.05, ∗∗p < 0.01, ∗ means PBS vs EPO in the ERα−/− mice, and † means PBS vs EPO in the WT mice).
Animal studies suggest that EPO improves glucose tolerance and insulin resistance and is protective in diet-induced obesity, particularly in male mice [[22], [23], [24]]. EPO's effects on glucose metabolism in the ERα−/− mice were determined by GTT and ITT. The ERα−/− mice exhibited reduced glucose tolerance and insulin tolerance in both the females and males (Figure 2A–E) compared to the WT mice. Furthermore, with EPO treatment, the ERα−/− mice showed a greater improvement in glucose and insulin tolerance than the WT mice on the HFD and NCD at 4 months (Figure 2A–E) and the female ERα−/− mice at 6 months compared with the WT mice (Supplementary Figure S1C–D). In the female mice on the HFD, fasting blood glucose levels of the ERα−/− mice were significantly higher than those of the WT mice, but with EPO treatment, the blood glucose levels of the WT and ERα−/− mice decreased and the blood glucose levels of the EPO-treated WT and ERα−/− mice became similar (Figure 2E). These results illustrated that the improvement in glucose tolerance associated with EPO administration [19,22] overcame the decrease in glucose tolerance resulting from the loss of ERα, particularly in the females. The proportional improvement in glucose tolerance with EPO treatment was greater with the loss of ERα, suggesting that ERα activity may blunt or interfere with EPO's response with respect to regulation of both fat mass and glucose tolerance, especially in the female mice at 4 months on the HFD.
Figure 2.

Glucose metabolism changes in the ERα−/− mice after EPO treatment for 4 weeks on the HFD or NCD. (A–C) Glucose tolerance tests (GTT) were performed in the WT and ERα−/− mice treated with EPO or PBS for the females at 4 months on the HFD (A), females at 4 months on the NCD (B), and males at 4 months on the HFD (C). (D) Insulin tolerance tests (ITT) in the female WT and ERα−/− mice at 6 months on the HFD. Symbols indicate the WT PBS (filled circle, solid line) and EPO (open square, thin line)-treated mice and ERα−/− PBS (filled triangle, solid line) and EPO (open inverted triangle, dotted line) treated mice. (n = 6–9/group, ∗, †, ‡, §p < 0.05, ∗∗, ††, ‡‡, §§p < 0.01, ∗ means PBS vs EPO in the ERα−/− mice, † means PBS vs EPO in the WT mice, ‡ means EPO in the ERα−/− vs EPO in WT, § means PBS in the ERα−/− vs PBS in WT). (E) Fasting glucose levels in the females at 4 months on the HFD, females at 4 months on the NCD, and males at 4 months on the HFD with EPO or PBS. (F) Expression of insulin receptor, FGF21, FOXO1, Glut1, and Glut4 genes in inguinal fat pads from the females at 4 months on the HFD (n = 5–6/group).
EPO can increase metabolic activity and promote the expression of brown fat-associated genes in white adipocytes of male WT mice [19,23]. The expression levels of genes related to glucose metabolism were assessed in inguinal fat pads from the female mice at 4 months on the HFD to determine the potential mechanism associated with EPO glucose regulation (Figure 2F). Little or no change was observed in Glut1 expression. While insulin receptor, FGF21, FOXO1, and Glut4 showed a significant increase in gene expression in the EPO-treated ERα−/− mice compared with the untreated WT mice, only the expression of insulin receptor and Glut4 increased significantly with EPO treatment in the ERα−/− mice, suggesting that EPO activity modulated insulin receptor and Glut4 in white adipose tissue (WAT) to improve glucose tolerance, especially with the loss of ERα.
3.2. Adipocyte size and white adipose tissue-associated gene expression with EPO treatment
Adipocyte size generally correlates with body weight in obese mice [26]. The female ERα−/− mice with increased fat mass and body weight compared with the WT mice on the NCD and HFD exhibited a shift in adipocytes toward larger sizes in their gonadal fat pads (Figure 3A–F). The male ERα−/− mice with increased fat mass on the HFD also exhibited a shift toward increased adipocyte size (Supplementary Figure S4A–C). In comparison, the mice with EPO receptor restricted to the erythroid tissue exhibited decreased adipocyte sizes in their gonadal fat pads despite the obese phenotype and increased fat mass resulting in a 30%–50% increase in body weight compared with the age-matched non-obese wild-type mice [19]. These data suggested that regulation of adipocyte size mediated by the expression of EPO receptor in non-hematopoietic tissue was distinct from size regulation in WT adipocytes and/or by expression of ERα. With EPO treatment during HFD feeding, both the male and female WT mice showed a significant decrease in adipocyte sizes accompanying the reduced trend in body fat accumulation (Figure 3A and C-D; Supplementary Figs. S4A–C). In contrast, while EPO treatment during HFD feeding showed a comparable or greater decrease in body weight and fat mass in the ERα−/− mice compared with the WT mice (Figure 1E–F and H–I), adipocyte sizes in the ERα−/− mice showed minimal decreases compared with the female and male WT mice (Figure 3A and C-D; Supplementary Figure S4A–C). Minimal changes in adipocyte sizes were observed with EPO treatment in the female WT or ERα−/− mice on the NCD, respectively (Figure 3B and 3E-F). These data suggested that adipocyte size was differentially regulated with the loss of ERα compared with the WT mice and distinct from regulation with the loss of EPO receptor.
Figure 3.

Adipocyte sizes and white fat associated gene expression in the female ERα−/− mice after EPO treatment for 4 weeks on the HFD or NCD. Representative hematoxylin and eosin staining of gonadal fat pad sections (scale bar: 50 μm) from the WT and ERα−/− mice at 4 months on the HFD (A) and NCD (B). (C–F) Analysis of adipocyte cell sizes from the WT and ERα−/− gonadal fad pads at 4 months on the HFD (C–D) and NCD (E–F) by ImageJ. Symbols indicate the WT PBS (filled circle, solid line) and EPO (open square, dotted line)-treated mice and ERα−/− PBS (red filled triangle, solid line) and EPO (red open inverted triangle, dotted line)-treated mice. (G–J) Serum leptin (G) and adiponectin (I) levels in the 4-month-old WT and ERα−/− mice on the HFD and NCD (G, I) and leptin receptor gene (H) and adiponectin gene (J) expression levels by quantitative real-time PCR in inguinal fat pads of the 4-month-old WT and ERα−/− mice on the HFD. (n = 5–6/group, ∗p < 0.05, and ∗∗p < 0.01). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Adipokines secreted by WAT, leptin, and adiponectin regulate endogenous glucose production, fatty acid oxidation, and food intake by binding to their specific receptors in target organs such as the brain, liver, and muscle. Serum leptin levels appeared to be proportional to WAT mass and were higher in the ERα−/− mice than the female and male WT mice, respectively (Figure 1, Figure 3G; Supplementary Figure S4D). EPO treatment significantly reduced serum leptin levels in the female ERα−/− mice on the HFD and NCD (Figure 3G). Leptin receptor mRNA levels showed an increasing trend with EPO treatment in the female WT and ERα−/− mice on the HFD, respectively (Figure 3H). In the males on the HFD treated with EPO, a significant reduction in serum leptin levels was observed in the WT and ERα−/− mice (Supplementary Figure S4D). Leptin receptor mRNA levels also showed an increasing trend with EPO treatment in the male WT and ERα−/− mice on the HFD (Supplementary Figure S4E). Circulating levels of adiponectin in the serum were higher in the female WT than the ERα−/− mice on the HFD and NCD, and with EPO treatment, only the ERα−/− mice showed elevated serum adiponectin levels with an increasing trend in adiponectin mRNA expression in WAT (Figure 3I–J). These results suggested that EPO regulation of WAT-derived leptin and adiponectin, particularly in the female ERα−/− mice, contributed to the EPO's response in glucose metabolism, food intake, and fat mass accumulation. Examination of other adipokines in WAT of the female and male WT and ERα−/− mice on the HFD showed increased proinflammatory cytokine MCP-1 expression in the ERα−/− mice, primarily in the females, and EPO treatment decreased the expression of proinflammatory cytokines, tumor-necrosis factor-α, and MCP-1 in the ERα−/− mice (Supplementary Figure S5A–B), raising the possibility that the loss of ERα increased WAT inflammation, affecting glucose tolerance, which was reduced with increasing fat mass and that EPO treatment promoted a shift toward an anti-inflammatory state in the ERα−/− mice as previously observed in male WT mice [24].
3.3. EPO reversed the increase in WAT-associated gene expression and promoted brown fat-associated gene expression and UCP1 protein in the ERα−/− mice
WAT plays a critical role in regulating energy and glucose homeostasis by modulating lipid turnover and adipocyte morphology as well as through inflammation [27]. The global transcriptome in human WAT can also be influenced by obesity through body weight changes [28]. Obesity is associated with an increase in WAT specialized to store excess energy and associated gene expression in contrast to mitochondria-rich brown adipose tissue with gene expression specialized for its thermogenic capacity [29,30]. The increased fat mass in the female ERα−/− mice on the HFD was accompanied by increased expression of WAT-associated genes, annexinA1, Ednra, Psat1, Wdnm1-like, and resistin in inguinal fat pads compared with the female WT mice (Figure 4A). EPO treatment significantly decreased the expression of WAT-associated gene Psat1, which plays a role in the regulation of insulin sensitivity, Wdnm-1like, and Serpina3k in the ERα−/− mice with decreasing trends suggested in the WT mice (Figure 4A). The expression of mitochondrial biogenesis genes showed a significant increase in the EPO-treated ERα−/− mice compared with the untreated WT mice (Supplementary Figure S6A). Significant decreases in the expression of some WAT-associated genes were also observed with EPO treatment in the male WT mice but not in the female WT mice (Figure 4A and Supplementary Figure S6B).
Figure 4.

White fat, brown fat, and estrogen-associated gene expression in inguinal fat pads from the ERα−/− mice after EPO treatment for 4 weeks on the HFD. (A–C) Expression of white adipose tissue-related genes (A), brown fat-associated genes (B), and estrogen-related genes (C) in inguinal fat pads from the female WT and ERα−/− mice at 4 months (n = 5–6/group). (D) Protein expression of UCP1, Cidea, PRDM16, pERK/ERK, pAKT/AKT, and pJNK/JNK in inguinal fat pads from the female WT and ERα−/− mice at 4 months (n = 3–4/group, ∗p < 0.05, and ∗∗p < 0.01).
Increasing brown adipose tissue (BAT)-associated gene expression in WAT has emerged as a therapeutic target in obesity [31] and EPO can promote brown fat-associated gene expression in white adipose tissue of male mice [23]. EPO treatment in the female ERα−/− mice on the HFD but not in the female WT mice significantly increased the expression of brown fat-like genes, PGC-1α, Cidea, PPAR-α, and PRDM16 (Figure 4B). Interestingly, the protein level of the brown fat marker uncoupling protein 1 (UCP1) increased markedly with EPO treatment in the WAT of the female ERα−/− mice compared with the untreated ERα−/− mice, although increases in UCP1 gene expression were not evident with EPO treatment in the WT and ERα−/− mice (Figure 4B,D). The increased PRDM16 protein levels in the EPO-treated ERα−/− mice were consistent with the PRDM16 gene expression patterns (Figure 4B,D). These data indicated a shift from a WAT-associated program of gene expression toward brown fat-like characteristics by EPO, especially in the female mice with the loss of ERα.
EPO stimulation of STAT5 activation has recently been reported in mouse primary isolated adipocytes and adipose tissue in mice on high-fat diets [32]. To further examine the mechanism of EPO activity, we considered potential EPO receptor downstream signaling molecules ERK and JNK, which are also associated with ERα activation [33]. We previously observed ERK activation in white adipose tissue from WT mice but not from mice with EPO receptor restricted to erythroid tissue [19]. EPO also stimulated ERK activation in preadipocyte differentiation cultures of mouse embryonic fibroblasts from WT mice but not from mice with EPO receptor restricted to erythroid tissue [19]. We found that EPO treatment induced significant increases in pERK and pJNK in inguinal fat pads of the female ERα−/− mice but not the WT mice (Figure 4D), indicating that EPO may regulate the ERK and JNK signaling pathways in female WAT only with the loss of ERα. This suggested an overlap between the EPO signaling and ERα signaling pathways. Increased pAKT by EPO in the WT and ERα−/− mice (Figure 4D) also suggested that EPO activity modulates AKT activation, which may affect insulin signaling.
We confirmed the loss of ERα expression in inguinal fat pads in the ERα−/− mice by analyzing the expression of Esr1, the gene that encodes ERα, and glutathione peroxidase GPX3, a target gene of ERα. The expression of Esr1 and GPX3 was downregulated by an order of magnitude in the female ERα−/− mice compared with the WT mice (Figure 4C). While EPO treatment did not appear to affect Esr1 expression in WAT, GPX3 expression increased by EPO in the ERα−/− mice, indicating GPX3 regulation by other processes in addition to ERα signaling. Bone morphogenetic protein 4 (BMP4), which promotes the accumulation of more metabolically active inguinal fat and reduction in visceral fat, also increased only in the female ERα−/− mice with EPO treatment (Figure 4C).
3.4. EPO improved glucose tolerance in the mice with ERα deleted in WAT
In non-hematopoietic tissues, a high level of EPO receptor is expressed in white adipose tissue compared with muscle and liver and contributes to EPO's metabolic response [19,23]. ERα is the main estrogen receptor expressed in white adipose tissue and plays a direct role for estrogen regulation of fat mass as suggested by changes in white adipose tissue gene expression, including genes involved in lipogenesis with estrogen treatment [[34], [35], [36]]. Therefore, mice with targeted deletion of ERα in their adipocytes were generated to assess the interference of estrogen signaling with EPO activity in adipocytes in the regulation of fat mass in female mice. Mice containing ERα-floxed alleles [25] were mated with mice containing a Cre-recombinase gene controlled by adiponectin promotor [37] to generate mice with a fat-specific deletion of ERα (ERαadipoKO mice), and littermates with ERα-floxed alleles without Cre were used as controls (designated WT). From 6 to 16 weeks, the female ERαadipoKO mice exhibited similar body weights with a trend toward lower fat mass compared with the female WT mice on the NCD (Figure 5A–B). On the HFD from 6 to 16 weeks, a trend toward increased body weight and fat mass was observed in the female ERαadipoKO mice relative to the WT mice (Figure 5C–D), suggesting that the female ERαadipoKO mice may have been more susceptible to diet-induced obesity. In the female mice at 4 months fed the HFD for 3 weeks with EPO treatment (3000 U/kg 3 times per week), only the ERαadipoKO mice but not the WT mice showed a significant decrease in body weight and fat mass with EPO treatment for 15 and 21 days, respectively (Figure 5E–F).
Figure 5.

Glycemic control in the female WT and ERαadipoKO mice treated with EPO. (A–B) Body weight (A) and fat mass (B) of the WT and ERαadipoKO females on the NCD for 10 weeks. Symbols indicate the WT (filled circle, solid line) and ERα−/− (open square, dotted line) mice (n = 12/group). (C–D) Measurement of body weight (C) and fat mass (D) in the WT and ERαadipoKO females at 4 months with EPO or PBS treatment for 3 weeks on the HFD. Symbols indicate the WT PBS (filled circle, solid line) and EPO (open square, thin line)-treated mice and ERα−/− PBS (filled triangle, solid line) and EPO (open inverted triangle, dotted line)-treated mice. (n = 6–9/group, ∗ means PBS vs EPO in the ERα−/− mice, ∗p < 0.05, and ∗∗p < 0.01). (E–F) Body weight (E) and fat mass (F) of the WT and ERαadipoKO females on the HFD for 10 weeks. Symbols indicate the WT (filled circle, solid line) and ERα−/− (open square, dotted line) mice (n = 12/group). (G–H) Measurement of body weight (G) and fat mass (H) in the WT and ERαadipoKO females at 4 months on the HFD for 12 weeks. All mice were treated with EPO or PBS for the last 2 weeks (n = 6). Symbols indicate the WT PBS (filled circle, solid line) and EPO (open square, thin line)-treated mice and ERα−/− PBS (filled triangle, solid line) and EPO (open inverted triangle, dotted line)-treated mice. (I–L) The WT and ERαadipoKO females at 4 months on the HFD for 12 weeks with EPO or PBS treatment for the last 2 weeks were used. GTT (I) and ITT (J) were performed (n = 6–9/group, ∗, †, ‡, §p < 0.05, ∗∗, ††, ‡‡, §§p < 0.01, ∗ means PBS vs EPO in the ERα−/− mice, † means PBS vs EPO in the WT mice, ‡ means EPO in ERα−/− vs EPO in WT, § means PBS in the ERα−/− vs PBS in WT). Fasting glucose levels (K) and expression of insulin receptor gene (L) were measured.
We previously showed that obese male WT mice on an HFD for 12 weeks and treated with EPO for the last two weeks had improved glucose tolerance and insulin resistance and a decrease in white adipose tissue inflammation compared with saline treatment [24]. Similarly, obesity was induced in female mice with 10 weeks of HFD feeding beginning at 6 weeks of age and then treated with EPO (3000 U/kg 3 times per week) or saline for the last two weeks on HFD feeding. As observed previously in male mice, female mice did not exhibit a change in body weight or fat mass with EPO treatment compared with saline treatment. Body weights and fat mass were similar in the ERαadipoKO mice compared with littermate control (WT) mice (Figure 5G–H), indicating that as with the obese male mice [24], two weeks of EPO treatment was not sufficient to affect changes in body weight/fat mass in the obese female ERαadipoKO or WT mice. However, two weeks of EPO treatment in the obese females demonstrated a greater improvement in glucose and insulin tolerance in the ERαadipoKO mice compared with the WT mice (Figure 5I–J). EPO treatment also decreased fasting glucose levels in the obese ERαadipoKO mice but not in the WT mice (Figure 5K). A trend toward increased insulin receptor gene expression in the EPO-treated ERαadipoKO mice compared with the untreated ERαadipoKO mice was also observed and a significant increase between the EPO-treated ERαadipoKO mice and untreated WT mice (Figure 5L). Reduction in ERα in WAT of the ERαadipoKO mice due to the loss of adipocyte ERα was verified by quantification of the ERα (Esr1) gene expression in inguinal fat pads of the ERαadipoKO mice and their littermate control mice with and without EPO treatment (Supplementary Figure S7A), which indicated the contribution of other non-adipocyte cells including macrophages to ERα expression in WAT.
3.5. EPO stimulated reduction in WAT adipocyte cell sizes and inflammation required ERα
To assess the effect of EPO treatment on adipocyte sizes in the obese mice, the female WT and ERαadipoKO mice were fed a HFD for 12 weeks, resulting in comparable increases in adipocyte sizes in the WT and ERαadipoKO mice (Figure 6A–C). The resultant adipocyte sizes after 12 weeks of HFD were markedly larger than the WT and ERα−/− mice on the NCD or four weeks of HFD feeding (Figure 3A–F). Although EPO treatment during the last 2 weeks of HFD feeding did not significantly alter body weight or fat mass (Figure 5G–H), the profile of adipocyte sizes shifted to smaller cells in the obese WT mice and less so in the ERαadipoKO mice (Figure 6C). This was analogous to the changes in adipocyte size distribution observed for 4 weeks of EPO treatment concomitant with HFD feeding in the WT and ERα−/− mice (Figure 3D). Although EPO regulation of fat mass accumulation in the female mice was greater with reduced estrogen signaling as demonstrated by the ERα−/− mice (Figure 1H) and ovariectomized mice [22], these results suggested that the reduction in WAT adipocyte size with EPO treatment during HFD feeding was greatest in the male and female WT mice, especially with the adipocyte expression of ERα.
Figure 6.

Adipocyte sizes and gonadal fat pad gene expression in the mice with targeted deletion of ERα in adipocyte tissues. (A–C) Adipocyte size distribution from female gonadal fat pad sections of the WT and ERαadipoKO mice at 4 months on the HFD for 12 weeks with 2 weeks of EPO treatment. Representative hematoxylin and eosin staining (A; scale bar: 50 μm), average cell size (B), and cell size distribution (C). Symbols indicate the WT PBS (filled circle, solid line) and EPO (open square, dotted line)-treated mice and ERα−/− PBS (red filled triangle, solid line) and EPO (red open inverted triangle, dotted line)-treated mice. (D–E) Expression of proinflammatory genes (D; n = 6–9/group) and iNOS protein levels (E; n = 3–4/group) in inguinal fat pads from the WT and ERαadipoKO females at 4 months on the HFD for 12 weeks with EPO or PBS treatment for the last 2 weeks. ∗p < 0.05 and ∗∗p < 0.01. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
EPO treatment promotes an anti-inflammatory response in WAT in WT mice, particularly during HFD-induced obesity [24], and changes with EPO administration in pro-inflammatory associated expression in WAT in the female ERαadipoKO mice were assessed. More crown-like structures were observed in WAT of the ERαadipoKO mice than the WT mice (Supplementary Figure S7B), with a corresponding increase in MCP1 expression (Figure 6D). EPO treatment reduced TNFα and iNOS gene expression with a trend toward decreased iNOS protein in the ERαadipoKO but not WT mice (Figure 6D–E). These EPO-stimulated changes in WAT of the ERαadipoKO mice were consistent from WAT of the female ERα−/− mice on the HFD showing reduced TNF-α expression with EPO treatment (Supplementary Figure S5B). In 3T3L1 cells, EPO decreased MCP1 expression, and with insulin, both EPO and estradiol decreased MCP1 expression (Supplementary Figure S5C). These results demonstrated that the loss of adipocyte-specific ERα contributed to EPO-regulated glucose metabolism and anti-inflammatory responses independent of EPO activity to regulate fat mass accumulation.
3.6. Increased WAT-associated gene expression in ERαadipoKO WAT was suppressed by EPO
In the absence of ERα in WAT adipocytes, the expression of WAT-associated genes in WAT fat pads showed increasing trends, reaching significance for Serpina3k and Wdnm1-like in the female obese ERαadipoKO mice compared with the WT mice (Figure 7A). This provided evidence of estrogen's protective effects against diet-induced obesity. EPO treatment for 2 weeks in the obese ERαadipoKO mice but not the WT mice decreased the expression of these WAT-associated genes, Ednra, Psat1, resistin, and Wdnm1-like, with a decreasing trend in Serpina3k expression (Figure 7A). These changes in WAT-associated gene expression with EPO treatment and analogous results in WAT-associated gene expression with EPO treatment and HFD feeding in the ERα−/− mice (Figure 4A) illustrated the protective effect of EPO against diet-induced obesity that was also observed in the ovariectomized mice [22]. These observations suggested that EPO-stimulated decreases in WAT-associated gene expression were blocked by endogenous estrogen signaling in the female mice, especially mediated by ERα in WAT adipocytes.
Figure 7.

Fat pad gene expression and signaling with EPO treatment in the mice with targeted deletion of ERα in adipocyte tissues. (A–B) Expression of white fat-associated genes in inguinal fat pads from the female WT and ERαadipoKO mice (n = 5–6/group) at 4 months on the HFD for 12 weeks with 2 weeks EPO treatment (A) and in 3T3L1 cells (B) treated with EPO, E2, or insulin (n = 4/group). In (B), ∗ means PBS vs treatment group without insulin treatment and † means PBS vs treatment group with insulin treatment. (C–D) Expression of brown fat-associated genes (C; n = 5–6/group) and protein levels of UCP1 and pERK/ERK (D; n = 3–4/group) in inguinal fat pads from the female WT and ERαadipoKO mice at 4 months on the HFD for 12 weeks with 2 weeks EPO treatment. ∗, †p < 0.05, ∗∗, ††p < 0.01.
Culture studies of 3T3L1 cells were used to examine the direct effect of EPO or estrogen or the combination of EPO and estrogen on adipocyte expression of WAT-associated gene expression (Figure 7B). EPO treatment downregulated the expression of resistin and Wdnm1-like genes. Estrogen also decreased the expression of resistin and Wdnm1-like genes, and the combination of EPO and estrogen further decreased the expression of these genes. In addition, the decrease in resistin and Wdnm1-like gene expression by insulin was further decreased by the combination of EPO and insulin but was not affected by estrogen alone. Estrogen also decreased the expression of other WAT-associated genes including Ednra, Psat1, and Serpina3k; however, no further decrease in expression was seen with the combination of estrogen and EPO or estrogen and insulin.
The ERαadipoKO mice treated with EPO for two weeks showed a marked increase in UCP1 compared to saline treatment, and an increasing trend in Cidea expression that was significant compared with the untreated WT mice (Figure 7C). Western blotting confirmed the corresponding EPO-stimulated increase in UCP1 protein in the obese female ERαadipoKO mice (Figure 7D) that also indicated an increasing trend of EPO activation of pERK (Figure 7D). Increased expression of UCP1 mRNA and protein by EPO also was observed in inguinal fat pads of the ERαadipoKO mice but not the WT mice. These results provided evidence that EPO promoted brown fat-like characteristics through active UCP1, especially in white adipocyte with targeted deletion of ERα, which might have been related to the EPO-mediated metabolic improvement observed.
4. Discussion
While previous animal studies demonstrated that EPO administration contributes to weight loss and reduced body fat, several of these were carried out predominantly in male rodents and limited understanding of the sex dimorphic metabolic response to EPO treatment [19,20,23,38]. In female mice, estrogen induction of EPO in the uterus contributes to estrus cycle remodeling [39]. Estrogen also contributes to increasing environmental hypoxia and ventilatory response in females mediated via EPO activity in the brain and carotid body that is sensitive to estrogen and sex hormones [[40], [41], [42]]. Estrogen-mediated gender-specific EPO metabolic response was demonstrated for regulating body fat accumulation and weight gain, including the contribution of cerebral regulation of body mass and hypothalamic inflammation observed in male mice [22,43]. In this study, we used mouse models with ERα global knockout, ERα−/−, fat tissue-specific ERα knockout, and ERαadipoKO, demonstrating that the gender-differential metabolic response to EPO is associated with estrogen activity, especially in WAT. Endogenous estrogen and its receptors contribute to maintaining normal fat mass, and we found ERα expression in WAT consistent with previous reports [9,44]. Based on our observation, EPO treatment on an HFD decreased body weight gain and fat accumulation in the female mice only in the absence of ERα. Therefore, we propose that in addition to endogenous estrogen activity in WAT conferring protection against obesity and associated metabolic syndrome, estrogen in WAT contributes to gender-differential EPO metabolic responses to regulate fat accumulation.
Weight gain and increased fat accumulation that cause metabolic disturbances result when energy intake exceeds energy expenditure and excess calories increase fat storage. In the female mice, only the HFD-fed ERα−/− mice and not the HFD-fed WT mice showed reduced food intake and a trend of increased total activity by EPO treatment. Leptin and adiponectin are implicated in the regulation of gluconeogenesis, fatty acid oxidation, food intake, and energy expenditure [45]. Leptin levels increase in obesity and fat accumulation while the expression of adiponectin decreases with increases in adiposity.
The leptin hormone is produced primarily in white adipose tissue that critically regulates body weight and metabolism and is produced in proportion to the amount of body fat [46]. Leptin binds to its receptors on neurons, particularly in the hypothalamus, to suppress appetite and food intake and increase energy expenditure [47,48]. Leptin stimulation activates the hypothalamic JAK2/STAT3 pathway and requires SH2B1 for regulation of body weight and core body temperature [48,49]. With EPO treatment in the mice, serum leptin levels remained proportional to fat mass (Figure 3G), and fat mass and serum leptin decreased while leptin mRNA per microgram of tissue in white adipose tissue remained unchanged [19]. In the mice with EPO restricted to erythroid tissue and those with fat-specific knockout of EPO receptor, serum leptin levels increased proportionate to the increase in fat mass [19,23]. In cultures of wild-type mouse embryonic fibroblast cells, EPO treatment did not change leptin gene expression under undifferentiated (d0) and preadipocyte differentiated (d7) conditions [19]. These results suggest that EPO has no effect on leptin gene expression. Furthermore, EPO treatment of ob/ob mice significantly decreased fat mass, providing evidence that EPO regulation of fat mass is independent of leptin activity [19,20]. Overall, decreased serum leptin with EPO treatment is a consequence of decreased fat mass.
In our study, serum leptin levels appeared proportional to fat mass in both male and female mice on the HFD and NCD (Figure 3G). In the female mice, EPO treatment reduced leptin levels only in the absence of ERα concomitant with decreased fat mass (Figure 3G). The loss of ERα increased adiposity and suppressed serum adiponectin, and EPO treatment elevated adiponectin in the female mice only with the loss of ERα. Collectively, these observations illustrated that impaired ERα signaling in the female mice allowed for EPO protection of fat accumulation to help stabilize energy homeostasis. The comparable increase in hematocrits with EPO treatment among the various groups of mice independent of genotype and diet demonstrated that the regulatory effect of EPO and estrogen on fat mass accumulation was independent of erythropoiesis.
Obesity is linked to insulin resistance caused by dysfunction in critical target tissues, adipose tissue, skeletal muscle, liver, and the brain. In obesity and type 2 diabetes, adipocytes and skeletal muscle have decreased insulin-stimulated glucose transport and metabolism, while liver tissue has impaired regulation of hepatic glucose production [50]. Adipocytes are one of the most highly insulin-responsive cell types, and importantly adipose tissue can produce estrogen [51]. In our study, ERα−/− mice showed impaired glucose tolerance and insulin resistance as previously reported [9]. During HFD feeding, EPO treatment improved insulin sensitivity to a greater degree in the ERα−/− mice compared with the WT mice that was also accompanied by a reduction in fat mass accumulation, especially in the ERα−/− mice. Interestingly, in the female ERα−/− mice on the NCD treated with EPO or in the obese ERαadipoKO on the HFD (12 weeks) treated with EPO for the last 2 weeks, no changes in body weight or fat mass were evident. However, improvements in glucose tolerance were observed that were greater in the female ERα−/− and ERαadipoKO mice than in the female WT mice, suggesting that EPO regulation of glucose metabolism in adipose tissue did not require EPO-stimulated reduction in fat mass accumulation observed with EPO treatment in the female ERα−/− mice on the HFD. EPO did not affect Esr1 expression in adipose tissue, although EPO treatment in the female ERα−/− mice on the HFD induced GPX3 that may have contributed to EPO-stimulated improvement in insulin resistance. GPx3 is a direct target gene of ERα and is downregulated in adipose tissue of female ERα−/− mice [36]. GPx3 is a selenocysteine-containing protein and in adipose tissue, insulin receptor expression correlates with expression of GPx3 and other selenoproteins [52]. GPX3 expression in adipose tissue was reduced in animal models of obesity and db/db mice, and treatment with antioxidant, antidiabetic drug rosiglitazone, or selenium-enriched HFD increased GPx3 expression in adipose tissue, improved insulin resistance, and reduced inflammatory gene expression [52,53].
Impaired glucose homeostasis may result from impaired insulin signaling in part in adipocytes and skeletal muscle by downregulating the major insulin-responsive glucose transporter GLUT4 and reduced insulin binding to its receptor [50]. Obese humans with type 2 diabetes show decreased IRS-1 expression and INSR protein, which results in reduced IRS-2-associated PI3k activity, especially in adipocytes [54,55]. In this study using adipose tissue from the ERα−/− mouse model, we determined that EPO enhances insulin sensitivity with increased GLUT4 and insulin receptor in the absence of ERα. This provided evidence that estrogen might interfere with EPO's response to elevated insulin signals through activation of GLUT4 and insulin binding to its receptor. However, in the fat-specific ERα knockout mice (ERαadipoKO), we did not see increased GLUT4 expression with EPO treatment, although a trend of increased insulin receptor was observed. ERα in skeletal muscle may contribute to EPO's protective response of improved insulin signaling through GLUT4 regardless of the loss of ERα in WAT, while the loss of ERα in WAT enhances EPO activity with respect to improved insulin sensitivity through increased insulin binding in WAT.
Adipocytes play a role as endocrine cells for insulin action, which helps maintain metabolic homeostasis. Adipocytes express and secrete numerous peptide hormones and cytokines, including TNF-α. TNF-α signaling impairs insulin signaling in part through serine phosphorylation of IRS-1 and can reduce GLUT4 gene expression [56,57]. In several animal models of obesity-associated insulin resistance, the knockout of TNF-α or TNF-α receptor genes improved insulin sensitivity [58]. We demonstrated that in the female mice, EPO reduced TNF-α gene expression in WAT from the ERα−/− and ERαadipoKO mice but not from the WT mice, and EPO improved GTT and ITT considerably in the ERα−/− and ERαadipoKO mice compared to the WT mice. These data provided evidence that the absence of ERα mediated EPO activity to reduce TNF-α signaling, especially in WAT, which might have resulted in enhanced insulin sensitivity.
We previously showed that male and female WT mice treated with EPO showed improvement in GTT as demonstrated by measurements of serum glucose levels and blood glucose levels [22]. Glucose can readily diffuse across red blood cell membranes, and the difference between serum glucose levels and blood glucose levels is due largely to the volume taken up by hemoglobin molecules in red blood cells. With EPO treatment, the change in GTT determined by blood glucose is greater than GTT determined from serum glucose due to the increase in hematocrits. For example, in female mice on an HFD, EPO treatment increased hematocrits from 46% to 71% and reduced peak serum glucose levels in GTT measurement by 30% and peak blood glucose levels by 41% compared with PBS treatment [22]. In this study, glucometer measurements were used to determine blood glucose levels for GTT.
Obesity is associated with adipose tissue dysfunction characterized by adipocyte hypertrophy and proinflammatory responses closely associated with the development of insulin resistance in adipose tissue [[59], [60], [61]]. The ERα−/− and ERαadipoKO mice showed enlarged adipocyte sizes with impaired insulin resistance compared to the WT mice, suggesting that adipocyte hypertrophy due to the loss of ERα might be associated with impaired insulin resistance. While the female ERα−/− mice and mice with EPO receptor restricted to erythroid tissue exhibited an obese phenotype on the NCD, regulation of adipocyte sizes were distinct between these two mouse models. Adipocyte size distribution in the obese ERα−/− mice and WT HFD obese mice shifted toward larger cell sizes in contrast to smaller adipocyte sizes in the mice with EPO receptor restricted to erythroid tissue [19].
ERα−/− mice have been known to possess greater adipocyte numbers and visceral fat pad areas compared to WT mice [9] and decreased fat accumulation in ERα−/− by EPO may result from reduced adipocyte numbers and minimal change in cell sizes. This is supported by a previous observation that EPO treatment during preadipocyte differentiation reduces preadipocyte differentiation [19]. It has been reported that improved insulin sensitivity via increasing serum adiponectin may result from changes in the large adipocyte subfraction [62].With EPO treatment, the shift of adipocyte size distribution toward smaller sizes, especially adipocytes with diameters less than 60 μm, was greater in the WT mice than the ERα−/− mice, although improved insulin sensitivity and reduction of fat accumulation by EPO was greater in the ERα−/− mice. The shift to smaller adipocyte sizes and increased subfraction with diameters less than 60 μm were evident in the obese WT mice by two weeks of EPO treatment that improved glucose tolerance and underscored that EPO can reduce adipocyte sizes and alter glucose metabolism without an overall decrease in fat mass. While EPO treatment in the ERα−/− and ERαadipoKO mice on the HFD improved glucose tolerance and insulin sensitivity, changes in adipocyte sizes were less remarkable and lacked an increased subfraction of small adipocytes (diameter <60 μm) seen in the WT mice, indicating that regulation of adipocyte size by EPO treatment was not able to overcome the increase in adipocyte sizes due to the loss of adipocyte ERα in the mice on the HFD.
Activation of BAT is associated with various physiological improvements such as improved glucose homeostasis, increased resting energy expenditure, and reduced body weight [63]. Not only the activation of BAT but also browning of white adipose tissue producing beige/brite cells has gained interest because of the potential for stimulating the development of beige adipocytes in WAT to reduce adverse effects of WAT and possibly improve metabolic health [64]. In our study, in the female mice, EPO suppressed WAT-associated genes such as resistin, which is known to promote insulin resistance [65], Ednra, Psat1, Wdnm1-like protein, and Serpina3k [66,67], and EPO activated brown fat-specific mitochondrial protein and UCP1 protein levels only in the female ERα−/− and ERαadipoKO mice but not the WT mice. EPO also increased the expression of brown adipocyte-enriched factors, such as PGC-1α, PRDM16, PPAR-α, and Cidea in the female ERα−/− mice. EPO receptor expression was reported in 3T3L1 cells to increase during adipocyte differentiation, reaching levels comparable with K562 cells in mature 3T3L1 adipocytes, and EPO stimulation increased AKT activation [68,69]. Shown here in 3T3L1 cultures, EPO and estrogen treatment downregulated the expression of resistin and Wdnm1-like genes. In particular, WAT-selective genes can be suppressed by PRDM16 [67]. The Ednra pathway through elevating Gq signaling can reduce UCP1 expression in BAT, whole-body energy expenditure, and the number of brown-like/beige cells in WAT [67]. Therefore, this study suggests a mechanism by which EPO response in adipocytes that lack ERα and estrogen signaling suppresses WAT-enriched factors, Ednra, Psat1, Wdnm1-like protein, and Serpina3k, and contributes to browning of WAT, possibly mediating the regulation of UCP1. Sirt1 knockdown has been shown to attenuate EPO response in adipose tissue [23]. The participation of SIRT1 to mediate the transcriptional network with EPO stimulation in adipocytes that lack ERα to regulate WAT-enriched factors and brown adipocyte-enriched factors remains to be determined.
We previously reported that endogenous EPO contributes to fat metabolism in mice and humans [19,21]. Normal plasma EPO levels range from 0.01 to 0.03 U/ml and increase to 1000-fold or more during hypoxia or anemia. In treatments for anemia in adult chronic renal failure patients, a retrospective study of hemodialysis patients from the Centers for Medicare and Medicaid Services End-Stage Renal Disease Clinical Performance Measure Project from 1997 to 2005 determined the mean EPO dose per week was 223 U/kg [70]. We found that in mice, EPO treatment as low as 150 U/kg three times weekly, approximately double the mean EPO for dialysis patients, significantly reduced body weight and fat mass [38]. The EPO dose administered to mice in the current study was 20 times higher at 3000 U/kg three times weekly. Studies of EPO treatment, primarily in young healthy males, provide evidence of EPO's response when administered closer to therapeutic doses. Eight weeks of EPO treatment beginning at 5000 U resulted in increased hematocrits, increased VO2max, and increased skeletal muscle mitochondrial respiratory capacity [71,72]. Whole body maximal fat oxidation did not change after treatment [72]. Resting energy expenditure significantly increased after 10 weeks of EPO treatment in men with a significant increase in VO2max and a concomitant increase in serum free fatty acid levels, although changes in BMI were not detected [73]. A single EPO dose of 400U/kg in healthy young men was also found to significantly increase resting energy expenditure [74].
5. Conclusion
Our findings demonstrate the complexity of hormone responses and the interplay between EPO and estrogen in gender-specific regulation of metabolism and molecular mechanisms of obesity and associated metabolic disorders. The potential gender-specific relationship between endogenous EPO levels and body weight changes is suggested by analysis of southwestern Native Americans with increased susceptibility to type 2 diabetes [21]. Males exhibit a negative association between endogenous circulating EPO levels and annual weight changes, which is not observed in females. In contrast, the positive association between EPO levels and weight changes in females may be related to nutritional needs associated with pregnancy and estrogen regulation of EPO production in female reproductive organs [21,39,75]. Together with the animal studies presented herein, these observations are consistent with estrogen as a major regulator of sex-specific modulators of EPO protection against fat mass accumulation and diet-induced obesity.
Acknowledgments
The authors thank Oksana Gavrilova and the NIDDK Mouse Metabolism Core Laboratory for their advice and assistance. This study was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and Division of Intramural Research of the NIEHS 1ZIAES070065 to KSK.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.101142.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Xu Y., Lopez M. Central regulation of energy metabolism by estrogens. Mol Metab. 2018;15:104–115. doi: 10.1016/j.molmet.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi H., Clegg D.J. Sex differences in the regulation of body weight. Physiology & Behavior. 2009;97(2):199–204. doi: 10.1016/j.physbeh.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vistisen D., Witte D.R., Tabak A.G., Brunner E.J., Kivimaki M., Faerch K. Sex differences in glucose and insulin trajectories prior to diabetes diagnosis: the Whitehall II study. Acta Diabetologica. 2014;51(2):315–319. doi: 10.1007/s00592-012-0429-7. [DOI] [PubMed] [Google Scholar]
- 4.Paschou S.A., Marina L.V., Spartalis E., Anagnostis P., Alexandrou A., Goulis D.G. Therapeutic strategies for type 2 diabetes mellitus in women after menopause. Maturitas. 2019;126:69–72. doi: 10.1016/j.maturitas.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Yang W., Lu J., Weng J., Jia W., Ji L., Xiao J. Prevalence of diabetes among men and women in China. New England Journal of Medicine. 2010;362(12):1090–1101. doi: 10.1056/NEJMoa0908292. [DOI] [PubMed] [Google Scholar]
- 6.Mauvais-Jarvis F., Manson J.E., Stevenson J.C., Fonseca V.A. Menopausal hormone therapy and type 2 diabetes prevention: evidence, mechanisms, and clinical implications. Endocrine Reviews. 2017;38(3):173–188. doi: 10.1210/er.2016-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bitoska I., Krstevska B., Milenkovic T., Subeska-Stratrova S., Petrovski G., Mishevska S.J. Effects of hormone replacement therapy on insulin resistance in postmenopausal diabetic women. Open Access Maced J Med Sci. 2016;4(1):83–88. doi: 10.3889/oamjms.2016.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riant E., Waget A., Cogo H., Arnal J.F., Burcelin R., Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150(5):2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- 9.Heine P.A., Taylor J.A., Iwamoto G.A., Lubahn D.B., Cooke P.S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proceedings of the National Academy of Sciences of the U S A. 2000;97(23):12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Z., Moore T.M., Drew B.G., Ribas V., Wanagat J., Civelek M. Estrogen receptor alpha controls metabolism in white and brown adipocytes by regulating Polg1 and mitochondrial remodeling. Science Translational Medicine. 2020;12(555) doi: 10.1126/scitranslmed.aax8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yasrebi A., Rivera J.A., Krumm E.A., Yang J.A., Roepke T.A. Activation of estrogen response element-independent ERalpha signaling protects female mice from diet-induced obesity. Endocrinology. 2017;158(2):319–334. doi: 10.1210/en.2016-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barros R.P., Gabbi C., Morani A., Warner M., Gustafsson J.A. Participation of ERalpha and ERbeta in glucose homeostasis in skeletal muscle and white adipose tissue. American Journal of Physiology. Endocrinology and Metabolism. 2009;297(1):E124–E133. doi: 10.1152/ajpendo.00189.2009. [DOI] [PubMed] [Google Scholar]
- 13.Hart-Unger S., Arao Y., Hamilton K.J., Lierz S.L., Malarkey D.E., Hewitt S.C. Hormone signaling and fatty liver in females: analysis of estrogen receptor alpha mutant mice. International Journal of Obesity. 2017;41(6):945–954. doi: 10.1038/ijo.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arao Y., Hamilton K.J., Lierz S.L., Korach K.S. N-terminal transactivation function, AF-1, of estrogen receptor alpha controls obesity through enhancement of energy expenditure. Mol Metab. 2018;18:68–78. doi: 10.1016/j.molmet.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teng R., Calvert J.W., Sibmooh N., Piknova B., Suzuki N., Sun J. Acute erythropoietin cardioprotection is mediated by endothelial response. Basic Research in Cardiology. 2011;106(3):343–354. doi: 10.1007/s00395-011-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burger D., Lei M., Geoghegan-Morphet N., Lu X., Xenocostas A., Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovascular Research. 2006;72(1):51–59. doi: 10.1016/j.cardiores.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 17.Sakanaka M., Wen T.C., Matsuda S., Masuda S., Morishita E., Nagao M. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proceedings of the National Academy of Sciences of the U S A. 1998;95(8):4635–4640. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernaudin M., Nedelec A.S., Divoux D., MacKenzie E.T., Petit E., Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. Journal of Cerebral Blood Flow and Metabolism. 2002;22(4):393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Teng R., Gavrilova O., Suzuki N., Chanturiya T., Schimel D., Hugendubler L. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nature Communications. 2011;2:520. doi: 10.1038/ncomms1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katz O., Stuible M., Golishevski N., Lifshitz L., Tremblay M.L., Gassmann M. Erythropoietin treatment leads to reduced blood glucose levels and body mass: insights from murine models. Journal of Endocrinology. 2010;205(1):87–95. doi: 10.1677/JOE-09-0425. [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt M., Dey S., Tom Noguchi C., Zhang Y., Krakoff J., Thearle M.S. Non-hematopoietic effects of endogenous erythropoietin on lean mass and body weight regulation. Obesity. 2016;24(7):1530–1536. doi: 10.1002/oby.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Rogers H.M., Zhang X., Noguchi C.T. Sex difference in mouse metabolic response to erythropoietin. The FASEB Journal. 2017;31(6):2661–2673. doi: 10.1096/fj.201601223RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L., Teng R., Di L., Rogers H., Wu H., Kopp J.B. PPARalpha and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes. 2013;62(12):4122–4131. doi: 10.2337/db13-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alnaeeli M., Raaka B.M., Gavrilova O., Teng R., Chanturiya T., Noguchi C.T. Erythropoietin signaling: a novel regulator of white adipose tissue inflammation during diet-induced obesity. Diabetes. 2014;63(7):2415–2431. doi: 10.2337/db13-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hewitt S.C., Kissling G.E., Fieselman K.E., Jayes F.L., Gerrish K.E., Korach K.S. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. The FASEB Journal. 2010;24(12):4660–4667. doi: 10.1096/fj.10-163428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Beek L., van Klinken J.B., Pronk A.C., van Dam A.D., Dirven E., Rensen P.C. The limited storage capacity of gonadal adipose tissue directs the development of metabolic disorders in male C57Bl/6J mice. Diabetologia. 2015;58(7):1601–1609. doi: 10.1007/s00125-015-3594-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Longo M., Zatterale F., Naderi J., Parrillo L., Formisano P., Raciti G.A. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. International Journal of Molecular Sciences. 2019;20(9) doi: 10.3390/ijms20092358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwok K.H.M., Ryden M., Andersson D.P., Beauchef G., Guere C., Vie K. Prospective analyses of white adipose tissue gene expression in relation to long-term body weight changes. International Journal of Obesity. 2020;44(2):377–387. doi: 10.1038/s41366-019-0385-1. [DOI] [PubMed] [Google Scholar]
- 29.Inagaki T., Sakai J., Kajimura S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nature Reviews Molecular Cell Biology. 2016;17(8):480–495. doi: 10.1038/nrm.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shapira S.N., Seale P. Transcriptional control of Brown and beige fat development and function. Obesity. 2019;27(1):13–21. doi: 10.1002/oby.22334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh A.M., Zhang L., Avery J., Yin A., Du Y., Wang H. Human beige adipocytes for drug discovery and cell therapy in metabolic diseases. Nature Communications. 2020;11(1):2758. doi: 10.1038/s41467-020-16340-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J., Yang M., Yu Z., Tian J., Du S., Ding H. Kidney-secreted erythropoietin lowers lipidemia via activating JAK2-STAT5 signaling in adipose tissue. EBioMedicine. 2019;50:317–328. doi: 10.1016/j.ebiom.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khariv V., Acioglu C., Ni L., Ratnayake A., Li L., Tao Y.X. A link between plasma membrane calcium ATPase 2 (PMCA2), estrogen and estrogen receptor alpha signaling in mechanical pain. Scientific Reports. 2018;8(1):17260. doi: 10.1038/s41598-018-35263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundholm L., Moverare S., Steffensen K.R., Nilsson M., Otsuki M., Ohlsson C. Gene expression profiling identifies liver X receptor alpha as an estrogen-regulated gene in mouse adipose tissue. Journal of Molecular Endocrinology. 2004;32(3):879–892. doi: 10.1677/jme.0.0320879. [DOI] [PubMed] [Google Scholar]
- 35.D'Eon T.M., Souza S.C., Aronovitz M., Obin M.S., Fried S.K., Greenberg A.S. Estrogen regulation of adiposity and fuel partitioning. Evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. Journal of Biological Chemistry. 2005;280(43):35983–35991. doi: 10.1074/jbc.M507339200. [DOI] [PubMed] [Google Scholar]
- 36.Lundholm L., Putnik M., Otsuki M., Andersson S., Ohlsson C., Gustafsson J.A. Effects of estrogen on gene expression profiles in mouse hypothalamus and white adipose tissue: target genes include glutathione peroxidase 3 and cell death-inducing DNA fragmentation factor, alpha-subunit-like effector A. Journal of Endocrinology. 2008;196(3):547–557. doi: 10.1677/JOE-07-0277. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z.V., Deng Y., Wang Q.A., Sun K., Scherer P.E. Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology. 2010;151(6):2933–2939. doi: 10.1210/en.2010-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foskett A., Alnaeeli M., Wang L., Teng R., Noguchi C.T. The effects of erythropoietin dose titration during high-fat diet-induced obesity. Journal of Biomedicine and Biotechnology. 2011:373781. doi: 10.1155/2011/373781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasuda Y., Masuda S., Chikuma M., Inoue K., Nagao M., Sasaki R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. Journal of Biological Chemistry. 1998;273(39):25381–25387. doi: 10.1074/jbc.273.39.25381. [DOI] [PubMed] [Google Scholar]
- 40.Soliz J., Khemiri H., Caravagna C., Seaborn T. Erythropoietin and the sex-dimorphic chemoreflex pathway. Advances in Experimental Medicine & Biology. 2012;758:55–62. doi: 10.1007/978-94-007-4584-1_8. [DOI] [PubMed] [Google Scholar]
- 41.Gassmann M., Pfistner C., Doan V.D., Vogel J., Soliz J. Impaired ventilatory acclimatization to hypoxia in female mice overexpressing erythropoietin: unexpected deleterious effect of estradiol in carotid bodies. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2010;299(6):R1511–R1520. doi: 10.1152/ajpregu.00205.2010. [DOI] [PubMed] [Google Scholar]
- 42.Gassmann M., Tissot van Patot M., Soliz J. The neuronal control of hypoxic ventilation: erythropoietin and sexual dimorphism. Annals of the New York Academy of Sciences. 2009;1177:151–161. doi: 10.1111/j.1749-6632.2009.05028.x. [DOI] [PubMed] [Google Scholar]
- 43.Dey S., Cui Z., Gavrilova O., Zhang X., Gassmann M., Noguchi C.T. Sex-specific brain erythropoietin regulation of mouse metabolism and hypothalamic inflammation. JCI Insight. 2020;5(5) doi: 10.1172/jci.insight.134061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clegg D., Hevener A.L., Moreau K.L., Morselli E., Criollo A., Van Pelt R.E. Sex hormones and cardiometabolic health: role of estrogen and estrogen receptors. Endocrinology. 2017;158(5):1095–1105. doi: 10.1210/en.2016-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stern J.H., Rutkowski J.M., Scherer P.E. Adiponectin, leptin, and fatty acids in the maintenance of metabolic homeostasis through adipose tissue crosstalk. Cell Metabolism. 2016;23(5):770–784. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y., Rui L. Leptin signaling and leptin resistance. Frontiers of Medicine. 2013;7(2):207–222. doi: 10.1007/s11684-013-0263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peelman F., Zabeau L., Moharana K., Savvides S.N., Tavernier J. 20 years of leptin: insights into signaling assemblies of the leptin receptor. Journal of Endocrinology. 2014;223(1):T9–T23. doi: 10.1530/JOE-14-0264. [DOI] [PubMed] [Google Scholar]
- 48.Wauman J., Zabeau L., Tavernier J. The leptin receptor complex: heavier than expected? Frontiers in Endocrinology. 2017;8:30. doi: 10.3389/fendo.2017.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang L., Su H., Wu X., Shen H., Kim M.H., Li Y. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nature Communications. 2020;11(1):1517. doi: 10.1038/s41467-020-15328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kahn B.B., Flier J.S. Obesity and insulin resistance. Journal of Clinical Investigation. 2000;106(4):473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson L.R., Bulun S.E. Estrogen production and action. Journal of the American Academy of Dermatology. 2001;45(3 Suppl):S116–S124. doi: 10.1067/mjd.2001.117432. [DOI] [PubMed] [Google Scholar]
- 52.Hauffe R., Stein V., Chudoba C., Flore T., Rath M., Ritter K. GPx3 dysregulation impacts adipose tissue insulin receptor expression and sensitivity. JCI Insight. 2020;5(11) doi: 10.1172/jci.insight.136283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee Y.S., Kim A.Y., Choi J.W., Kim M., Yasue S., Son H.J. Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Molecular Endocrinology. 2008;22(9):2176–2189. doi: 10.1210/me.2008-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cignarelli A., Genchi V.A., Perrini S., Natalicchio A., Laviola L., Giorgino F. Insulin and insulin receptors in adipose tissue development. International Journal of Molecular Sciences. 2019;20(3) doi: 10.3390/ijms20030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rondinone C.M., Wang L.M., Lonnroth P., Wesslau C., Pierce J.H., Smith U. Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proceedings of the National Academy of Sciences of the U S A. 1997;94(8):4171–4175. doi: 10.1073/pnas.94.8.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cawthorn W.P., Sethi J.K. TNF-alpha and adipocyte biology. FEBS Letters. 2008;582(1):117–131. doi: 10.1016/j.febslet.2007.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plomgaard P., Bouzakri K., Krogh-Madsen R., Mittendorfer B., Zierath J.R., Pedersen B.K. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54(10):2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- 58.Petersen M.C., Shulman G.I. Mechanisms of insulin action and insulin resistance. Physiological Reviews. 2018;98(4):2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J.I., Huh J.Y., Sohn J.H., Choe S.S., Lee Y.S., Lim C.Y. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Molecular and Cellular Biology. 2015;35(10):1686–1699. doi: 10.1128/MCB.01321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verboven K., Wouters K., Gaens K., Hansen D., Bijnen M., Wetzels S. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Scientific Reports. 2018;8(1):4677. doi: 10.1038/s41598-018-22962-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tandon P., Wafer R., Minchin J.E.N. Adipose morphology and metabolic disease. Journal of Experimental Biology. 2018;221(Pt Suppl 1) doi: 10.1242/jeb.164970. [DOI] [PubMed] [Google Scholar]
- 62.Pasarica M., Tchoukalova Y.D., Heilbronn L.K., Fang X., Albu J.B., Kelley D.E. Differential effect of weight loss on adipocyte size subfractions in patients with type 2 diabetes. Obesity. 2009;17(10):1976–1978. doi: 10.1038/oby.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava S., Veech R.L. Brown and brite: the fat soldiers in the anti-obesity fight. Frontiers in Physiology. 2019;10:38. doi: 10.3389/fphys.2019.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartelt A., Heeren J. Adipose tissue browning and metabolic health. Nature Reviews Endocrinology. 2014;10(1):24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- 65.Banerjee R.R., Rangwala S.M., Shapiro J.S., Rich A.S., Rhoades B., Qi Y. Regulation of fasted blood glucose by resistin. Science. 2004;303(5661):1195–1198. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- 66.Altshuler-Keylin S., Shinoda K., Hasegawa Y., Ikeda K., Hong H., Kang Q. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metabolism. 2016;24(3):402–419. doi: 10.1016/j.cmet.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kajimura S., Seale P., Tomaru T., Erdjument-Bromage H., Cooper M.P., Ruas J.L. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes & Development. 2008;22(10):1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pan Y., Shu J.L., Gu H.F., Zhou D.C., Liu X.L., Qiao Q.Y. Erythropoietin improves insulin resistance via the regulation of its receptor-mediated signaling pathways in 3T3L1 adipocytes. Molecular and Cellular Endocrinology. 2013;367(1–2):116–123. doi: 10.1016/j.mce.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 69.Broudy V.C., Lin N., Egrie J., de Haen C., Weiss T., Papayannopoulou T. Identification of the receptor for erythropoietin on human and murine erythroleukemia cells and modulation by phorbol ester and dimethyl sulfoxide. Proceedings of the National Academy of Sciences of the U S A. 1988;85(17):6513–6517. doi: 10.1073/pnas.85.17.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright D.G., Wright E.C., Narva A.S., Noguchi C.T., Eggers P.W. Association of erythropoietin dose and route of administration with clinical outcomes for patients on hemodialysis in the United States. Clinical Journal of the American Society of Nephrology. 2015;10(10):1822–1830. doi: 10.2215/CJN.01590215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Plenge U., Belhage B., Guadalupe-Grau A., Andersen P.R., Lundby C., Dela F. Erythropoietin treatment enhances muscle mitochondrial capacity in humans. Frontiers in Physiology. 2012;3:50. doi: 10.3389/fphys.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guadalupe-Grau A., Plenge U., Helbo S., Kristensen M., Andersen P.R., Fago A. Effects of an 8-weeks erythropoietin treatment on mitochondrial and whole body fat oxidation capacity during exercise in healthy males. Journal of Sports Science. 2015;33(6):570–578. doi: 10.1080/02640414.2014.951872. [DOI] [PubMed] [Google Scholar]
- 73.Christensen B., Nellemann B., Larsen M.S., Thams L., Sieljacks P., Vestergaard P.F. Whole body metabolic effects of prolonged endurance training in combination with erythropoietin treatment in humans: a randomized placebo controlled trial. American Journal of Physiology. Endocrinology and Metabolism. 2013;305(7):E879–E889. doi: 10.1152/ajpendo.00269.2013. [DOI] [PubMed] [Google Scholar]
- 74.Christensen B., Vendelbo M.H., Krusenstjerna-Hafstrom T., Madsen M., Pedersen S.B., Jessen N. Erythropoietin administration acutely stimulates resting energy expenditure in healthy young men. Journal of Applied Physiology. 2012;112(7):1114–1121. doi: 10.1152/japplphysiol.01391.2011. (1985) [DOI] [PubMed] [Google Scholar]
- 75.Chikuma M., Masuda S., Kobayashi T., Nagao M., Sasaki R. Tissue-specific regulation of erythropoietin production in the murine kidney, brain, and uterus. American Journal of Physiology. Endocrinology and Metabolism. 2000;279(6):E1242–E1248. doi: 10.1152/ajpendo.2000.279.6.E1242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.