

Abstract
Cancer immunotherapy using genetically modified immune cells such as those expressing chimeric antigen receptors has shown dramatic outcomes in patients with refractory and relapsed malignancies. NK cells as a member of the innate immune system, possessing both anti-cancer (cytotoxic) and proinflammatory (cytokine) responses to cancers rare off-target toxicities have great potential for a wide range of cancer therapeutic settings. Therefore, improving NK cell antitumor activity through genetic modification is of high interest in the field of cancer immunotherapy. However, gene manipulation in primary NK cells has been challenging because of broad resistance to many genetic modification methods that work well in T cells. Here we review recent successful approaches for genetic and epigenetic modification of NK cells including epigenetic remodeling, transposons, mRNA-mediated gene delivery, lentiviruses, and CRISPR gene targeting.
1. Viral transduction of NK cells
1.1. Retroviral vectors
Retroviral vectors are major tools to achieve stable cell transduction and, through the years, have become indispensable to study cell biology and develop cell therapy via cell engineering. However, unlike T cells or hematopoietic stem cells (HSC), NK cells are notoriously difficult to transduce.(1–3) As an example, Seillet et al. developed a transduction strategy using innate lymphoid cells (ILC) progenitors isolated from mouse bone marrow. To obtain transduced NK cells, they transduced ILCs and re-introduced them back into mice to differentiate into NK cells.(4) While this technique allowed them to study NK-cell biology in a murine model, it was clearly not applicable to a human system or for therapeutic purpose. This example illustrates the high resistance of mature NK cells to transduction and the need for more efficient transduction methods to generate engineered NK cells for cell-based therapy and/or human NK-cell biology study. Despite this difficulty, several advantages of CAR-NK have been suggested, including the duality of CAR-dependent and CAR-independent cytotoxicity of CAR-NK cells(5), and the reduced risk of graft versus host disease (GvHD) and cytokine release syndrome (CRS).
Retroviral vectors consist of simple retrovirus (Alpharetrovirus, Betaretrovirus, Gammaretrovirus) and complex retrovirus (Lentivirus). Simple retrovirus were the first vectors described for NK cell transduction, but with modest and very variable transduction rates.(6, 7) Because simple retroviruses need actively dividing cells to integrate the host cell genome, the use of an expansion system for NK cells is mandatory. While this limitation hampers the modification of freshly isolated NK, it is adaptable to the various NK amplification systems that are used in the context of cell-therapy.(8, 9) Moreover, these systems allow for multiple rounds of virus application to increase transduction rates. Indeed, Guven et al. have reported a NK-cell transduction rate averaging 50% with two rounds of gene delivery using retrovirus.(7) However, this strategy is toxic for NK cells and results in high mortality rates, thus limiting its applicability for large-scale clinical production of engineered-NK cells. This transduction rate is comparable with the best non-viral transient transfection rates achieved with electroporation in primary NK with small vector such as GFP.(10, 11) Enrichment of modified NK cells can alleviate this transfection rate constrain,(11) but the transient nature of transfection, averaging 15 days at best,(10) limiting its clinical utility. Therefore, virus-based transduction is currently favored.
Interestingly, the first published CAR-NK to advance to clinical use was based on a gammaretrovirus transduction protocol and aimed at treating leukemia,(12, 13) for which RD114-based gammaretroviral particles were used to develop cord-blood derived CAR-NK-cells.(12) Clinical responses reported for CAR-NK-cells produced by this approach are encouraging(12, 13) and advocates for the continued development NK-CAR therapies.
1.2. Genotoxicity and New Viral Vectors
Simple retroviral-based vectors were among the first to be used for cell engineering approved for clinical use,(14, 15) however, genotoxic issues have been associated with their use, especially for those based on the gammaretrovirus murine leukemia virus (MLV).(16, 17) Indeed, gammaretroviruses are known to integrate in proto-oncogene regions of the genome. Recognition of this genotoxicity led to the use and development of lentiviral and self-inactivating vectors.(18)
Self-inactivating (SIN) alpharetroviral vectors transduction protocols have been developed for NK cells. Indeed, recently a GMP-compliant NK-cell engineering protocol was recently described with the vision to be used in clinic.(19) Although this protocol resulted in acceptable NK-cell purity, viability, and recovery after thawing, the transduction rate remained in the low range (between 5% and 10% for a chimeric antigen receptor (CAR) construct).(19) Those low transduction rates increase the difficulty and the cost of production for therapeutic use, but this GMP-compliant protocol still represents a step towards a convenient use of engineered NK cells in clinic.
Even though gammaretrovirus were more common, lentiviruses are now being used more in clinic. Three generations of lentiviral vectors that were incrementally safer were gradually developed. The third-generation lentiviral vectors used today lack accessory virulence factors and are coded on different plasmid to reduce the likelihood of creating recombinant virus. Deletion in the LTRs disrupting their enhancing/promoting capacity creates a self-inactivating vector that further improve safety. Therefore, the third generation lentiviral vectors are safer than the first generation(16, 18, 20–22) in terms of risk of insertion in a proto-oncogene region(22) and systemic inflammation.(21) However, we must keep in mind that lentiviral vector still integrates in the genome and the number integrated copies is a considered a risk factor for oncogenesis. Therefore, the WHO and FDA (23) recommends an integration limit of 5 copies per cell. That being said, to date, no case of insertional oncogenesis has been reported with gene therapy using lentiviral vectors,(18) but follow-up of patients must be made diligently to assess the long-term risks. Lastly, lentiviruses also have the advantage that they do not require that the cell enters in an active division to successfully integrate the genome.(2) Third generation SIN lentiviral vectors (LV) have been successfully used in low to non-proliferative cells, such as CD34+ for gene therapy of thalassaemia (VSV-G pseudotype SNI-LV vector, PMID: 20844535), metachromatic leukodystrophy (3rd generation SNI VSV-G pseudotype vector),(24) and Wiskott-Aldrich syndrome (WAS)(3rd generation SNI VSV-G pseudotype vector (LV-w1.6W)).(25) Altogether, these lentiviral vectors become better alternatives than simple retroviruses for future clinical use.
1.3. Lentiviral vectors and the impact of pseudotypes on transduction
Tropism of lentiviral vectors can be broadened by using the envelope glycoprotein from other viruses, creating pseudotyped viruses. These pseudotypes are useful to engineer cells that are normally difficult to transduce due to the paucity of entry receptors. The most common pseudotyped lentiviral vectors (LVs) currently used in cell-engineering are the Vesicular Stomatitis Virus type-G (VSV-G)-pseudotyped, RD114-pseudotyped, and Measles Virus (MV)-pseudotyped LVs. VSV-G is the most common pseudotype used for hematopoietic stem cell (HSCT) and T cell transduction due to its ease of use and the ubiquitous expression of one of its virus entry receptor, the LDL-R.(18) However, despite the expression of the LDL-R and of other viral entry receptors such as VLDL-R, LRP1 and LRP8 on NK cells, VSV-G does not transduce NK cells efficiently.(26) The RD114 feline retrovirus envelope glycoprotein-LV was developed as an alternative to VSV-G pseudotype, as it uses a different viral entry receptor, the sodium-dependent neutral amino acid transporter 2 (ASCT-2),(27, 28) that is widely expressed in the hematopoietic cell lineage.(29) Moreover, contrary to VSV-G, it is resistant to degradation by human complement, which is an advantage when used in vivo.(29) As mentioned above, RD114 betaretrovirus has been used to produce CAR-NK cells(12) although with a low transduction efficacy. Another member of the gammaretroviruses family is the baboon endogenous retrovirus.(30) Its envelope (baboon endogenous envelope - BaEV) can also be pseudotyped and used in a lentivector setting.(30) BaEV binds both the ASCT2 and ASCT1 proteins as viral entry receptors (Figure 1b), hence broadening its tropism.(31) In the context of HSC transduction, the BaEV glycoprotein has been modified by the deletion of the fusion inhibitory R peptide (BaEVRLess glycoprotein) in order to increase viral production titers.(30) This improved-pseudotype lentiviral vector resulted in increased HSC transduction efficiency.(30) Based on these results, our team has transduced freshly isolated and activated NK cells with BaEV-LV and compared its transduction performance to other lentiviral vectors.(26) In our hands, BaEV-LVs outperformed VSV-G-, RD114- and Measles Virus (MV)-pseudotyped LVs for both freshly isolated and activated NK cells, reaching a mean of 80% transduction efficiency in activated NK cells.(26) BaEV-LV transduction allowed for a robust and efficient transduction method, permissive to CAR constructs, resulting in a sustained transgene expression.(26) This technique was compatible with NK cells activated and expanded in multiple systems, including the IL15-feeder system(8), the IL21-feeder system(9) and a feeder-cell-free system (NK-MACS Medium, Miltenyi). In light of these new results, BaEV could have a major impact on both basic research of NK-cell biology, because of its capacity to transduce non-activated NK cells, and on NK-cell-based immunotherapy, because of its high transduction rates. In addition to generating CAR-NK cells, one could also engineer NK cells to enhance their function by manipulating their signaling pathway.(1) The next critical steps will be to bring these lentiviral vectors - especially the BaEV-pseudotype LVs - to GMP and clinical standards in order to test them in clinical trials using NK-cell based immunotherapy.
Figure 1.
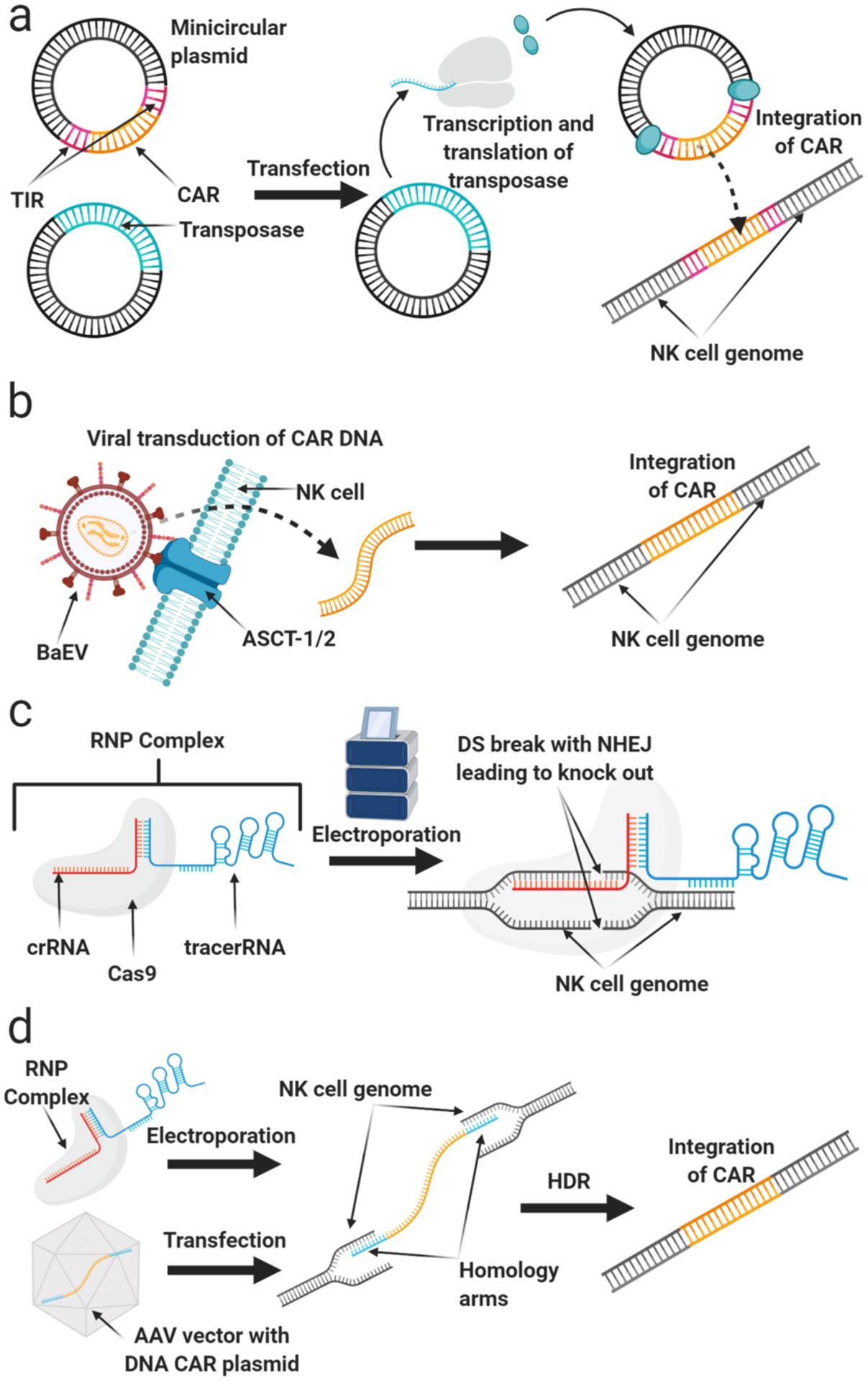
Methods for genetic modification. a) Transposase-mediated genetic modification. b) Viral transduction. c) DNA-free engineering by electroporation of Cas9/RNP. d) Combination of Cas9/RNP and AAV for targeted gene insertion.
1.4. NK cell resistance to genetic modification
The very nature of NK cells - being innate immune cells - could explain their resistance to transduction. Their role as first responders to viral infections has likely led them to evolve mechanisms of high resistance to viral infection.(32) Indeed, foreign RNA viral present during the transduction process is known to activate pathogen-associated molecular patterns (PAMPs) in NK cells, triggering a cascade of intracellular signaling that results in apoptosis.(3) Retroviruses and lentiviruses, with their double-stranded RNA, can activate TLR3, RIG-I and MDA-5 in NK cells, thus inducing toxicity in the NK cells. Interestingly, Sutlu et al. achieved a 3.8 fold boost of efficacy of NK-cell transduction by using BX795, an inhibitor of TBK1/IKKε, in their lentiviral transduction protocol.(2) The TBK1/IKKε complex is downstream of TLR3, RIG-I and MDA-5 activation cascade; hence BX795 abrogates the danger signal from the double stranded RNA present in retro- and lentivirus vectors. BX795 is minimally toxic for NK cells and could be easily implemented in existing protocols using retrovirus or lentivirus. BaEVRLess pseudotype envelope could be used together with the BX795 for a further increase in transduction efficiency which, coupled to the safety aspect of the lentiviruses, could be favorable for bringing engineered NK cells into clinic.
2. Engineering primary NK cells using transposons
DNA transposons are natural DNA transfer vehicles that can be used for DNA delivery in a manner similar to integrating viruses. In nature, they exist as well-defined elements in which the transposase gene is flanked by terminal inverted repeats (TIRs) that encode transposase binding sites. They can be used as a tool for stable genomic insertion by surrounding a gene-of-interest with TIRs and co-delivering the transposase enzyme via an expression plasmid or mRNA (Figure 1).
2.1. Transposon families
Several DNA transposons have been used in such a manner in mammalian cells. In 1997, the Sleeping Beauty (SB) transposon system was molecularly reconstructed by eliminating inactivating mutations found in members of the Tc1/mariner family of transposons isolated from fish(33). The reactivated transposon system has since been used for stable gene transfer and insertional mutagenesis in many vertebrate cell types including human cells. Subsequently, the piggyBac (PB) and Tol2 transposable elements were isolated from insects and fish, respectively, and have been optimized for enhanced activity in mammalian cells(34, 35). SB, PB, and Tol2 can be used as efficient non-viral tools for stable gene delivery, and each of these has been used for gene delivery in primary human lymphocytes(36).
2.2. Advantages of transposons
Transposons have some meaningful advantages as an alternative to viral vectors for gene therapy. Several clinical gene therapy products have been developed using genetically modified CD34+ hematopoietic stem cells or T cells. The majority of these products rely on the transduction of target cells with recombinant viruses, namely γ-retroviruses, lentiviruses, and adeno-associated viruses (AAV)(37, 38). These delivery methods carry the risk of insertional mutagenesis via activation of proto-oncogenes or inactivation of tumor suppressor genes. In addition, large-scale manufacturing of these viral vectors for clinical use can be cost-prohibitive and impede progression through clinical trials. Thus, the use of transposon systems instead of viral vectors has been pursued as an alternative due to convenient and cost-effective production and a better safety profile (17, 22, 39). Any vector that integrates into chromosomes poses the risk of insertional mutagenesis. A comparative study of the target site selection properties of SB and PB transposons as well as gammaretroviral and lentiviral systems in primary human CD4+ T cells ranked their safety profiles based multiple criteria including distance from the 5’-end of any gene and distance from any cancer-related gene. This analysis established SB as having the most favorable integration profile suggesting SB might be a safer alternative to viral vectors. (40)
2.3. Plasmid-based transposon systems
The use of transposon systems for gene delivery in human lymphocytes has been most widely studied as a method for generating human T cells engineered to express chimeric antigen receptors (CARs)(41, 42). Clinically, the SB system has been used to introduce CD19-specific CARs to patient- and donor-derived T cells(43, 44). Many preclinical studies and clinical trials thus far have introduced SB transposase and CD19 CAR by electroporation of bulk peripheral blood mononuclear cells (PBMCs)(45–48). CAR-expressing T cells were subsequently expanded over several weeks in culture using feeder cells engineered to express the CD19 antigen and co-stimulatory molecules(43). Efforts are now being made to shorten the culture time before patient infusion. One such trial is underway in which PBMCs are transferred into the patient within 2 days after electroporation with SB transposase, CD19-CAR, and membrane-bound IL15 (NCT03579888). Signaling through the CAR and mbIL15 gives genetically-modified T cells a selective advantage after transplant, ensuring their outgrowth(49).
2.4. Reduced-toxicity transposon approaches
Another approach for shortening culture time ex vivo is to electroporate T cells directly, rather than as bulk PBMCs from which T cells need to be selected. This has been challenging as transposon-based gene transfer negatively affects T cell viability due to DNA toxicity and the induction of a type I interferon (IFN) response(50, 51). Thus, mRNA and/or minicircle vectors encoding the transposase combined with minicircle transposon vectors encoding the transposon have been used to minimize the amount of DNA introduced to the T cell. Minicircle vectors are DNA delivery vehicles that do not carry a bacterial origin of replication or bacterial resistance genes, reducing the size of the vector to only that of the expression cassette (Figure 1a) (52). This approach has been used to achieve stable expression of transgenes in primary human T cells with efficiencies over 50 percent(50, 53, 54).
2.5. NK cell modification with transposons
Thus far, the use of transposons for NK cells has been mostly applied to the NK-92 cell line(55). Recently, sleeping beauty transposon has been used to transduce a CAR into cytokine-induced killer cells for targeting CD33 on chemoresistant AML in patient-derived xenografts (56). However, lessons can be learned from T cells on the use of transposons for CAR delivery to primary NK cells. The initial approach of electroporating PBMCs with the transposon-based CAR suggests that NK cells could be selectively outgrown instead of T cells. Indeed, some reports have shown outgrowth of NK cells reaching 50% of the PBMC population after co-culture with feeder cells(46). Thus, this approach could be optimized for selection of CAR-expressing NK cells, or delivery of a mixed population of CAR-T and -NK cells might be advantageous as NK cells have been shown to produce inflammatory cytokines to help shape the adaptive immune response(57).
Alternatively, the use of minicircle vectors to deliver transposons directly to purified NK cells is an option. NK cells share many properties with T cells, and delivery of DNA to NK cells has been shown to induce similar toxicity(11). Thus, reducing the amount of DNA delivered by using mRNA-encoded transposase in combination with minicircle-encoded transposon may be ideal. Our group performed proof-of-principle experiments delivering mRNA encoding SB11 or SB100x in combination with minicircle DNA encoding a GFP expressing transposon to primary human NK cells (Figure 2). We show stable expression of GFP 21 days after electroporation, with 15% efficiency using SB100x, suggesting this is a viable approach for non-viral gene delivery to NK cells.
Figure 2.
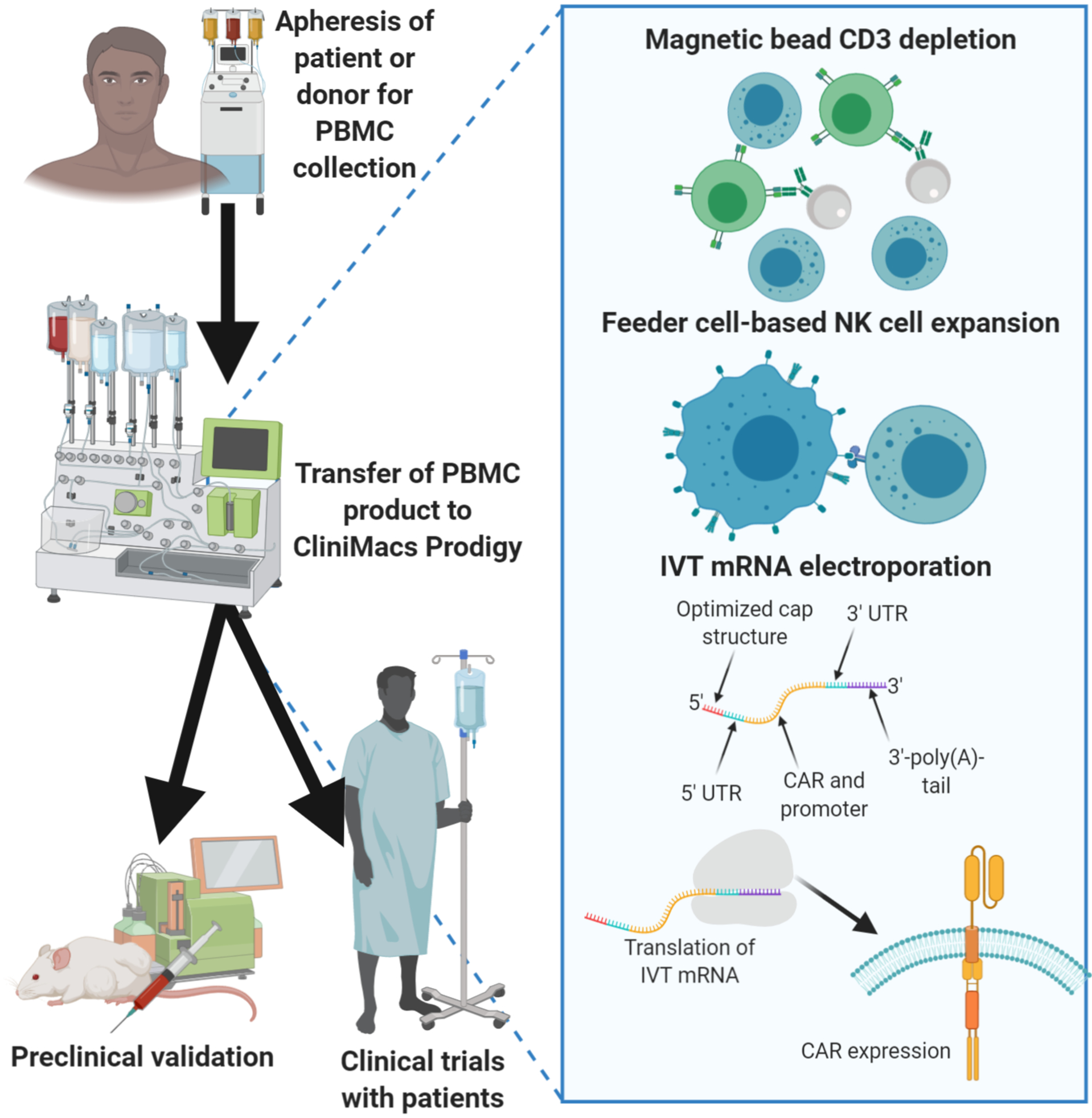
Generation of CAR-NK cells by electroporation of mRNA.
The use of transposons for engineering NK cells is not limited to the delivery of CARs. Other modifications have been proposed for enhancing aspects of NK cell activity including persistence, migration, and cytotoxicity. This includes the introduction of self-stimulating cytokine receptors(58), strong activating receptors or dominant negative versions of NK cell inhibitors(59). Such modifications could be used in combination to create an NK cell expertly-equipped to kill a broad range of tumor types. Transposons provide a non-viral strategy to introduce these transgenes that could be scaled up for clinical use.
3. Modifying NK cell function with in vitro-transcribed mRNA
Viral methods are efficient to deliver genes to proliferating cells, but their safety, immunogenicity and manufacturing challenges hamper clinical progress. The development of efficient and safe non-viral methods will greatly facilitate clinical gene therapy studies and targeted cellular therapies(60, 61). In vitro transcribed (IVT) mRNA has great therapeutic potential to transiently express targeted proteins with limited toxicity and other advantages. For example, large quantities of RNA can be easily prepared by in vitro transcription, which makes it possible to expedite manufacturing and scaling to current Good Manufacturing Practices (cGMP) products 10, enabling rapid design and testing of new therapeutic strategies, such as designing new CAR constructs. IVT mRNA has been widely used for cancer immunotherapy, vaccination against infectious diseases, allergy tolerization, protein-replacement and supplementation therapies, genome engineering, and efficiently reprogramming adult cells to form induced pluripotent stem cells (iPSC) et al (62, 63).
3.1. Optimal design of in vitro-transcribed mRNA
The template used for mRNA synthesis usually consists of: (i) the optimized cap structure, (ii) the optimized 5′ untranslated region (UTR), (iii) the codon optimized coding sequence, (iv) the optimized 3′ UTR and (v) 100–250 adenosine-containing region (3′-poly(A)-tail)(64). The 5’ 7-methylguanosine triphosphate (m7G) cap plays an important role in RNA stability(64). Design of anti-reverse cap analogs (ARCAs) with only one 3’-OH group instead of two 3’-OH groups prevented the incorporation in reverse orientation, increased RNA transcription efficiency, and improved protein expression duration and levels (65). The poly-A tail has crucial importance for the translation efficacy and mRNA stability, while the UTR’s control the translation and half-life of the mRNA(66). The order of codons and codon context also influences the efficiency of translation. Replacement of rare codons for synonymous codons is one of the ways to optimize the sequence of the coding region in mRNA(64).
IVT mRNA is produced from a linear DNA template containing a bacteriophage promotor, the optimized UTR’s and the codon optimized sequence by using a RNA polymerase (T7, T3 or SP6) and a mix of the different nucleosides (67). The ARCAs cap structure and the poly A tail are incorporated during transcription to enhance mRNA stability and translation efficacy (68).
3.2. Electroporation of mRNA
One of the main challenges associated with engineering NK cells is the low gene transfer efficiency. Electroporation is one of the nonviral strategies to transfer genes to NK cells, which is based on the generation of an electric field to induce temporary permeabilization of the cell membrane(11). Cell damage due to irreversible electroporation has been a concern. A variety of electroporators or nucleofector such as GenePulser, BTX 830 square wave electroporator, Amaxa nucleofector (Table 1), have been developed to facilitate transferring IVT mRNA to NK cells with less damage to the cells.
Table.
List of Reported Works Utilizing IVT mRNA CAR for adoptive NK cell Immunotherapy
| CAR | NK resource | CAR signaling domains | Electroporation/Nucleofection system | Diseases | Study Stage | Report Year | Ref. |
|---|---|---|---|---|---|---|---|
| Anti-CD19 CAR | Ex vivo expanded human NK or unstimulated NK | 4-1BB-CD3ζ | MaxCyte GT system | CD19+ B-lineage ALL CD19+ B-CLL | Preclinical | 2010, 2012 | (71, 72) |
| Anti-CD20 CAR | Ex vivo expanded human NK | 4-1BB-CD3ζ | Amaxa nucleofector II | CD20+ BL | Preclinical | 2015 | (73) |
| Anti-CD20 CAR | Ex vivo expanded human NK | 4-1BB-CD3ζ | MaxCyte GT system | CD20+ BL | Preclinical | 2017 | (74) |
| Anti-CD19 or anti-CD20 CAR | NK92 | CD3ζ | GenePulser II | CD19+ CLL | Preclinical | 2012 | (75) |
| Anti-CD19 CAR | IL-15 stimulated primary human NK | CD28-OX40-CD3ζ 4-1BB-CD3ζ | BTX 830 Square Wave Electroporator | CD19+ target | Preclinical | 2018 | (76) |
| Anti-ROR1 CAR | Ex vivo expanded human NK | 4-1BB-CD3ζ | Amaxa nucleofector II | Neuroblastoma Sarcoma | Preclinical | 2017 | (78) |
| Anti-ROR1 CAR | Ex vivo expanded human NK | 4-1BB-CD3ζ | MaxCyte system GT | Neuroblastoma | Preclinical | 2019 | (79) |
| NKG2D CAR | Ex vivo expanded human NK | DAP10-CD3ζ | MaxCyte system GT | Osteosarcoma | Preclinical | 2013 | (80) |
| NKG2D CAR | Autologous or allogeneic haploidentical NK cells | DAP12 or CD3ζ | NEPA21 electroporator BTX electroporator | metastatic colorectal cancer | Preclinical and Clinical Phase I (NCT03415100) | 2019 | (81) |
| CCR7 CD16F158V | Ex vivo expanded human NK | N/A | MaxCyte system GT | lymph node-associated chemokine CCL19 Lymphoma | Preclinical | 2016 | (69) |
| CD16F158V | CD38 low NK cell line | N/A | Electroporation | MM | Preclinical | 2018 | (82) |
B-CLL: B cell Chronic Lymphocytic Leukemia; BL: Burkitt Lymphoma; MM: Multiple Myeloma.
MaxCyte GT® Transfection System is a cGMP-compliant, scalable therapeutic system with unparalleled consistency, scalability, and cell loading efficiency, that is ideal for clinically-oriented cell modification (69). The MaxCyte GT® Transfection System has been used to generate CAR mRNA modified T/NK cells for clinical trials in the USA (NCT01355965, NCT01837602, NCT01897415, NCT01974479). Recently, Miltenyi has developed a closed-system CliniMACS® electroporator, which is powered and controlled by the CliniMACS Prodigy®. This closed tubing system can ensure an automated, large scale, sterile processing during the fully automated cell electroporation procedure and enables efficient delivery of DNA/mRNA to cells for clinical use (Figure 2) (70).
3.3. Introduction of CAR genes through mRNA
Anti-CD19 CAR or anti-CD20 CAR mRNA modified NK cells through electroporation or nucleofection have been investigated in preclinical settings in both B-cell Leukemia and Lymphoma (Table 1). Both Li et al and Shimasaki et al investigated the anti-CD19 CAR expression and functions in ex vivo expanded or purified unstimulated NK cells after electroporating anti-CD19-BB-z mRNA with MaxCyte GT® Transfection System (71, 72). Twenty-four hours after electroporation, the median cell viability was 90% and the anti-CD19 CAR expression was 40.3% in freshly purified and 61.3% in expanded NK cells(72). These anti-CD19 CAR NK secreted interferon (IFN)-γ in response to CD19-positive target cells and had increased cytotoxicity in vitro and in xenograft models of B-cell leukemia (71, 72). We investigated the functional activities of expanded peripheral blood NK (PBNK) cells modified by with anti-CD20 CAR mRNA using Amaxa nucleofector or MaxCyte GT® Transfection System against CD20+ B-NHL in vitro and in xenografted non-obese diabetic severe combined immunodeficiency gamma (NSG) mice (73, 74). We demonstrated that anti-CD20 CAR mRNA modified expanded NK cells (CAR+ exPBNK) had significantly enhanced in vitro cytotoxicity against rituximab-sensitive and -resistant Burkitt Lymphoma (BL) cells and extended human BL xenografted NSG survival compared to mock transfected exPBNK cells (73). Notably, CAR+ exPBNK limited BL tumour metastasis compared to mock transfected exPBNK cells (73). Consistent with previous reports that NK cells do not persist after adoptive transfer and they were detectable in the circulation for only 1–2 weeks without cytokine support (8), the administered CAR+ exPBNK cells survived around 2 weeks in NSG mice (73). Additionally, the combination therapy of anti-CD20-CAR mRNA electroporated PBNK cells and romidepsin was shown to induce synergistic anti-tumor effects both in vitro and in vivo using a semi-disseminated BL Raji xenografted NSG mouse model (74). The short lifespan/persistence of adoptively transferred CAR mRNA modified NK cells would require the repeated infusions to elicit the anti-tumor effect in the clinical.
In addition to modification of primary and expanded NK cells, the NK-92 cell line efficiently expressed anti-CD19 CAR or anti-CD20 CAR after CAR mRNA electroporation using GenePulser II electroporator to target acute lymphoblastic leukemia, lymphoma and chronic lymphocytic leukemia (CLL) cells (75). To determine if CAR engineering is influenced by the intrinsically heterogeneous functional potential in the NK-cell repertoire, Oei et al electroporated primary activated human NK with anti-CD19 CAR mRNA using a BTX 830 Square Wave Electroporator (76). They found that the redirected primary NK cells with anti-CD19 CAR mRNA were insensitive to inhibition through NKG2A/HLA-E interactions but remained sensitive to inhibition through KIR depending on the amount of HLA class I expressed on CD19+ target cells, suggesting a need to consider NK-cell diversity when optimizing efficacy of cancer immunotherapy based on CAR expressing NK cells (76).
3.4. Using mRNA transfer for rapid testing of CAR designs
The outcomes of adoptive NK cell therapies into patients with solid tumors have been dismal and extensive studies have been done to investigate different strategies to improve the NK cell function, trafficking and tumor targeting (77). We have developed anti-ROR1 CAR engineered expanded primary NK cells through CAR mRNA electroporation technology using Amaxa nucleofector II and MaxCyte GT® Transfection System to target ROR1+ solid tumors with promising in vitro anti-tumor effects (78, 79). Besides designing a CAR based on the single chain variable fragment (scFv) of a mAb again an antigen on tumor cell surface, CAR can also be formed from a NK activating receptor such as NKG2D followed by transmembrane domain and signal transduction domains (77). Chang et al designed a CAR termed NKG2D-DAP10-CD3ζ that was composed of the NK cell activating molecule NKG2D plus 2 key signaling molecules, DAP10 and CD3ζ (80). These NKG2D CAR mRNA engineered primary NK cells through electroporation showed significantly enhanced in vitro cytotoxicity against osteosarcoma cells that expressed NKG2D ligands MICA/B (80). Similarly, a group from China fused the extracellular domain of NKG2D to DAP12, to improve NK cell tumor responses (81). The expression of NKG2D-DAP12 CAR after NKG2D-DAP12 CAR mRNA electroporation significantly augmented the cytolytic activity of NK cells against several solid tumor cell lines in vitro and delayed disease progression in colorectal cancer-bearing mice (81).
3.5. Redirecting NK cell homing with mRNA
To improve NK cells homing to tumors, Carlsten et al enigineered ex vivo expanded NK cells to express high surface levels of CCR7 utilizing CCR7 mRNA electroporation with the MaxCyte system (69). CCR7 mRNA-electroporated NK cells showed marked enhanced in vitro migration capacity toward tumors cells that secreted CCL19 and CCL21 ligands, whereas non-electroporated NK cells remained incapable of migrating toward these ligands (69). The cGMP-compliant MaxCyte electroporation platform offers a method to efficiently genetically modify NK cells with chemokine receptors to enhance NK homing to tumor cells (69). To enhance antibody-dependent NK cell-mediated cytotoxicity (ADCC), NK cells obtained from CD16–158F/F donors were electroporated with mRNA coding for the high-affinity Fc receptor CD16–158V receptor (69). These CD16–158V mRNA-electroporated NK cells acquired an enhanced ADCC when cocultured with rituximab-coated EBV-transformed B-cell lymphoma cells compared to controls (69). Additionally, CD38low NK cells electroporated with CD16–158V receptor mRNA effectively killed low or high CD38 expressing multiple myeloma (MM) cells in combination therapy with daratumumab with enhanced IFNγ production, indicating CD38low CD16158V NK cells can be administered as an “off-the-shelf” cell therapy product to target both CD38low and CD38high expressing MM cells in combination with daratumumab(82).
Overall, the increasing variety and number of preclinical investigations with mRNA engineered NK cells provides a new strategy to improve the outcome of cancer immunotherapy (Table 1). With GMP-compliant electroporation equipment available, IVT mRNA becomes attractive for enabling rapid development of clinical-scale, safe, “off-the-shelf” CAR NK cells for allogeneic cell therapy.
CRISPR gene editing in primary human NK cells
The clustered regularly interspaced short palindromic repeats (CRISPR) gene editing technique has revolutionized medical science. Its ease and precision have exploded its use in basic and translational sciences. The CRISPR system contains three components: first, a crisprRNA (crRNA), which is ~20 nucleotides complementary to the gene of interest and follows by a Protospacer Adjacent Motif (PAM); Second, a tracerRNA as the backbone for gRNA and finally a Cas endonucleases protein to introduce a genomic double-stranded break (DSB). Cas9 is the most frequently used Cas endonuclease protein for genome editing. This protein uses NGG as the PAM sequence at the 3’ site of crRNA which is easy to find in target loci of the human genome. Following DSB introduced by Cas9, the cellular DNA repair machinery tries to repair the break. There are two possible DNA repair pathways that ensue-non-homologues end joining (NHEJ) which is an error-prone mechanism that typically results in nonfunctional mutations of loss of gene expression (knock-out), or homology directed repair (HDR) that restores integrity or can be manipulated for gene insertion in the presence of a DNA template encoding a gene of interest contained within homology arms for the regions flanking the DSB(83).
3.1. Hurdles in applying CRISPR to NK cells
With the precision of CRISPR and its low off-target effects on the genome, the FDA has approved several clinical trials applying CRISPR approaches to cell therapy, such as T cells with PD1 deleted to bypass checkpoint inhibition (NCT03399448). In a similar manner, CRISPR engineering of NK cells has the potential to improve their efficacy in cancer immunotherapy, but despite successful gene editing in several hard-to-modify cell types(84), gene modification in primary human NK cells was challenging. Bacterial transductions as viral transductions had shown poor efficiency and high toxicity in NK cells. This may be due to their response to viral and bacterial DNA through pathways such as RIG-I and TBK1/IKKɛ. To overcome this toxicity to cells and low efficiency, we adopted a DNA-free method of gene editing for primary and expanded NK cells. In this method, NK cells were electroporated with pre-translated Cas9 endonuclease protein and preassembled guide RNA(s) - as Cas9/RNP complexes (Figure 1c) - into human primary and expanded NK cells and showed successful gene knock-out. The Cas9/RNP as a fully-functional complex allows this approach to bypass cellular transcription/translation machinery and DNA sensing mechanisms (85). The use of pre-translated Cas9 protein is more favorable to mRNA delivery due to its rapid action and quick degradation, which also decreases the frequency of off-target effects (86). Using this method, we successfully targeted several genes which play a crucial role in the suppression of NK cell cytotoxicity, including transforming growth factor beta receptor 2 (TGFBR2) and suppressor of cytokine signaling-3 (SOCS3). We also demonstrated efficient gene editing and higher cytotoxicity of CRISPR modified NK cells against cancers (85, 87). In a similar approach, we demonstrated that CD38 can be deleted in NK cells to reduce fratricide and increase cytotoxicity for use in combination with daratumamab for multiple myeloma (88).
3.2. Alternative approaches for delivering CRISPR components
Del’Guidice et al. described an alternative approach to electroporation for introducing CRIPSR Cas9/Cpf1 RNP complexes into human primary NK cells by using membrane permeabilizing amphiphilic peptide (89). Successful gene knock-out in NK cells using the Cas9/RNP approach has also been demonstrated by electroporating pre-transcribed gRNA and mRNA-encoding Cas9 protein, which was successful in targeting PD1, CISH, and ADAM17 in NK cells (90). Electroporation of Cas9/RNP in to NK cells was also used to delete NKp46 and CIS in primary human NK cells to validate their role in antitumor activity (91).
3.3. Using CRISPR for site-directed gene knock-in
Targeted gene knock-in using a combination of Cas9/RNP and AAV vectors has been successfully tested in several cell types, including T-cells to express CAR genes(92, 93). In this approach, HDR is used for integration of DNA-encoded gene of interest at the targeted site. The DNA templates should be designed with optimal-length homology arms for the flanking region of DSB and are commonly delivered to the cells by AAV vectors to provide sufficient copy number to ensure HDR occurs (Figure 1d) (92). Using this approach, we were able to integrate reporter genes into the genome of human NK cells, and showed stable gene expression in these cells, providing proof of concept for generating CRISPR-directed CAR-NK cells ((94) and (90)). Since designing homology arms for HDR-directed gene knock-in is time-consuming approach and needs exptensive optimization, CRISPaint (CRISPR-assisted insertion tagging) is a homology-independent gene insertion approach that may also be useful for gene insertion into NK cells. This method allows for the insertion or tagging of a gene of interest into a user-defined locus with no need for designing homology arms ((95) and (94)).
It is important to recognize that the efficiency of CRISPR gene editing varies with different genomic sites and donor vectors (91). Therefore, designing the best gRNAs with highest on-target and lowest off-target effect is important to ensure adequate efficiency of CRISPR gene editing. Overall, progress in CRISPR modification of NK cells has opened a new era in cancer immunotherapy by facilitating the generation of gene-edited NK cells.
4. Epigenetic modulation of NK cells during ex vivo cultivation
NK cells are frequently low in number and function in cancer patients, resulting in suppressed antitumor immune responses. Ex vivo expansion can produce large numbers of NK cells with restored function, empowering weakened NK cells with enhanced antitumor repertoire of cytokines and receptor expression. In recent years, genetically-modified feeder cells have been developed, such as K562 expressing 41BBL and membrane-bound (mb) cytokines that support sustained NK cell proliferation, such as mbIL-15 (8) or mbIL-21 (96). In recursive stimulation of NK cells by these feeder cells, the intracellular domain of the IL-15 and IL-21R leads to the recruitment and phosphorylation of members of the signal transducer and activator of transcription (STAT) family that transduce signals for cellular events that are critical for NK cell expansion and activation. The direct impact of STAT signaling on NK cell growth, development, and function have been well-described, including cytotoxic capacity, cytokine-mediated effects and interaction with other immune effector cells (Figure 3a) (97–101). For example, NK cell cytokine production and cytotoxicity are directly affected by expression of activating and inhibitory receptors that are regulated by STAT3 (102, 103).
Figure 3.
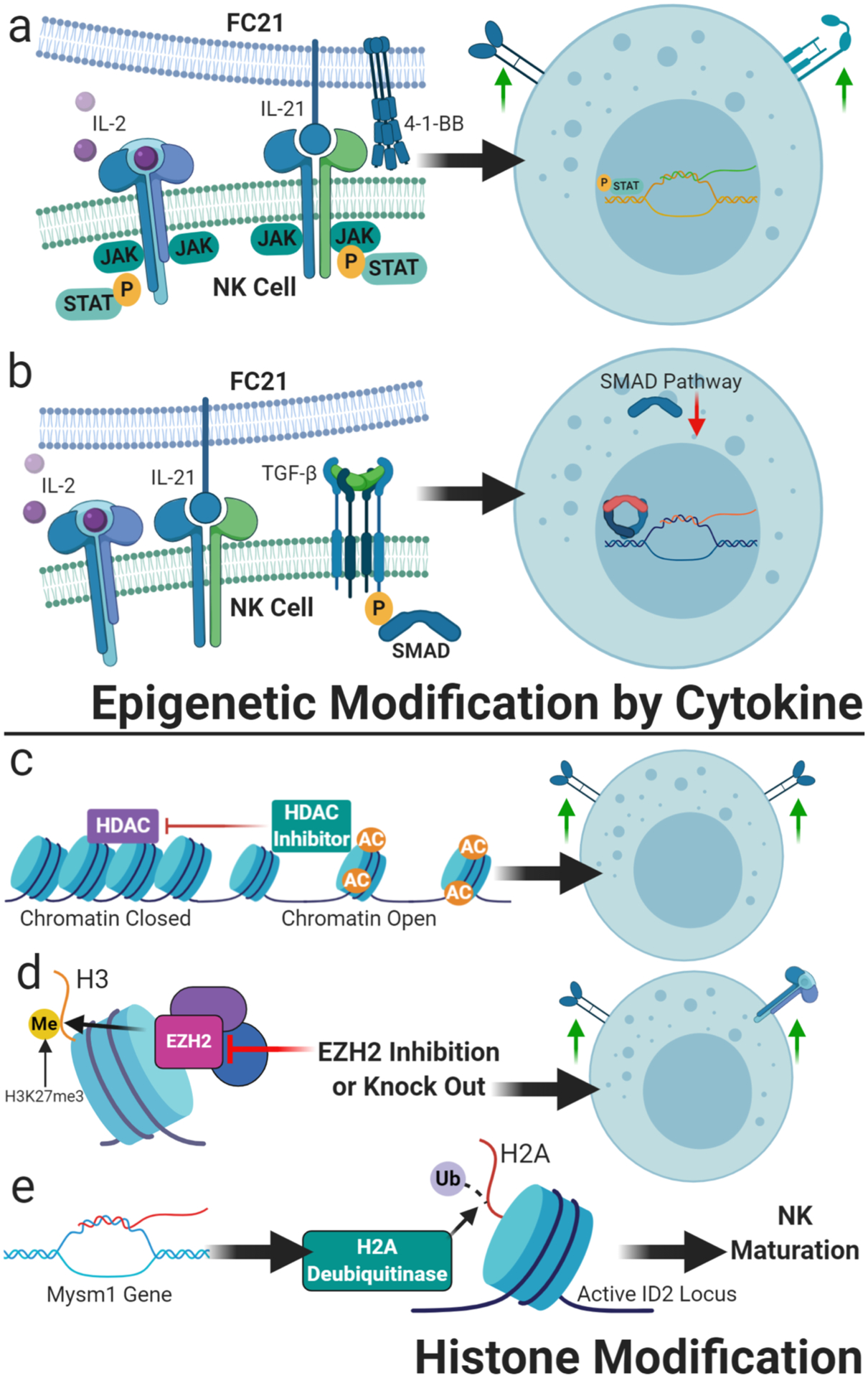
Epigenetic modification of NK cells. a) Cytokine expansion utilizing soluble and feeder cell-presented cytokines lead to epigenetic changes in the primary NK cell – such as the depicted activation of JAK and STAT by binding of the common-gamma chain cytokines IL-21 and IL-2 – to drive NK cell expansion as well as functional and phenotypic shifts making them an optimized adoptive immunotherapeutic; b) The addition of TGF beta to feeder cell expansion leads to epigenetic downregulation of the SMAD pathway, imprinting the NK cells to be resistant to further TGF-beta exposure; c) Histone deacetylase inhibitors such as valproic acid can alter chromatin conformation, maintaining accessibility of genes favorable to NK cell activation, like NKG2D; d) Modification of histone methylation as with inhibition of EZH2, can similarly enhance expression of activating receptors such as NKG2D and the high-affinity IL-2 receptor; e) Alteration of histone ubiquitination, such as through gene products of the Mysm1 gene, have been shown to have a critical role in activation of loci crucial for NK cell maturation, and could thus be a target of epigenetic NK cell modification favorable for immunotherapy.
4.1. Impact of epigenetic state on transcriptional profiles
In contrast to gene transfer or gene editing, epigenetic remodeling refers to functionally relevant modifications to the genome that impact gene expression programs to effect cellular phenotype and function, but do not involve a change in the nucleotide sequence (viz. acetylation, methylation, phosphorylation, and ubiquitylation). In addition to direct actions of transcriptional regulators such as STATs, chromatin state also has impact on regulating gene expression, thus influencing the biological processes in context of development and cellular response to environment. Chromatin is organized into large compartments, comprised of topologically-associated domains containing DNA segments that are highly transcriptionally active when open, and less transcribed when closed. These DNA domains are organized by nuclear architectural proteins (histones) that regulate transcription by tightly controlling intra- and/or inter-DNA interactions. De novo synthesis and post-translational modifications of histones during cell growth and proliferation are thought to modulate gene expression by recruiting key regulators (104). As described below, manipulating these chromatin dynamics during NK cell expansion opens a window to modulating chromatin architecture during ex vivo expansion to shape NK cell antitumor activity (105–107).
4.2. Histone acetylation modifiers
Conceptually, lysine acetylation neutralizes histone proteins enhancing the mobility of nucleosome on the DNA thus increasing the accessibility of promoter for transcription machinery, whereas deacetylation restores the positive charge of lysine and decreasing access of transcription factors to regulatory regions (108). The addition of acetyl groups to histones is regulated by histone acetyl transferases (HATs), while removal of acetyl groups is catalyzed by histone deacetylases (HDACs) (Figure 3c). By reversing the histone acetylation status, HDACs mostly inhibit gene expression. NK cells from AML patients treated with valproic acid (VPA) showed enhanced NK-mediated cell killing through upregulation of NKG2D, the immunoreceptor that binds with MICA and MICB (109). Similarly, entinostat, a narrow-spectrum HDAC inhibitor, also induced an increase in expression of NKG2D on NK cells in vitro (110). The application of histone deacetylase inhibitors during ex vivo propagation of NK cells may be used to modulate phenotype and function of therapeutic NK cells.
4.3. Histone methylation modifiers
In contrast to histone acetylation, histone methylation regulates gene expression, including tissue-specific transcriptional repression, depending on the modified residues and the number of methyl groups. Methylation status is regulated by histone methyltransferases and demethylases. The histone H3 methyltransferase enhancer of zeste homolog 2 (EZH2) is a subunit of Polycomb PRC2 multiprotein complexes that bind to chromatin and repress transcription through deposition of the H3K27me3 mark (Figure 3d) (111). EZH2 is essential for many biological processes, including the regulation of immune responses, making it an interesting target for future immunotherapies. A recent study by Yin and coworkers has highlighted the role of EZH2 in NK cell development (112). The authors have shown that in the absence of EZH2 both human and murine hematopoietic progenitors resulted in an increased commitment to the NK cell lineage. Furthermore, NK cells with EZH2−/− phenotype expressed higher levels of activating receptor NKG2D, IL2Rα, and also have increased synthesis of granzyme A and B, indicating their highly cytotoxic nature. The pharmacological inhibition of EZH2 resulted in a similar phenotype when compared to EZH2−/− NK cells. Similar findings have also been reported by other researchers (113), suggesting that EZH2 inhibitors can potentially augment NK cell growth and function by modulating the expression of various genes involved in immunosurveillance. Inhibition of methyltransferases such as EZH2 during the ex vivo NK cell expansion may lead to an increase in cell number and augment the activity of NK cells.
4.4. Histone ubiquitination modifiers
In addition to the more common histone modifications discussed above, histones can also be monoubiquitylated. Unlike polyubiquitination which promotes proteasomal degradation, attachment of a single ubiquitin moiety significantly affects the nucleosomal dynamics and serves as an epigenetic mark that regulates gene expression, DNA replication, and chromatin segregation (114), and modulates chromatin by influencing acetylation and methylation. For example, the inactivation of X chromosome is correlated with monoubiquitination of H2A that in turn affects histone methylation, thus suppressing gene transcription (115). Ubiquitination is a reversible reaction tightly controlled by the opposing actions of ubiquitin ligases and deubiquitinases. Enzymes involved in deubiquitination of histones play critical role in regulating innate immunity (116). Nandakumar and coworkers have shown that NK cell development is severely impaired in mice deficient in the histone H2A deubiquitinase Mysm1 (Myb-like, SWIRM, and MPN domains 1), (117). MYSM1 is involved in maintaining an active chromatin at the ID2 locus, a critical transcription factor for NK cell development and intrinsically controls NK cell maturation (Figure 3e).
Additionally, it has been shown that pre-activation of NK cells in vitro with IL-12/15/18 can maintain long-term antitumor activity by epigenetic imprinting(118). We also have demonstrated that TGFβ imprinting can reprogram epigenetic status of expanded NK cells through SMAD3 signaling pathway (Figure 3b). This results in generation of less sensitive NK to TGFβ as a suppressive molecule(119). Over the past decade, tremendous progress has been made in the field of immunotherapy and chromatin regulation. The systematic and integrative pursuit of epigenetic approaches to sculpt adoptive cell therapy promises a bright future for new immunotherapeutic avenues in cancer.
Summary
NK cells have broad potential for adoptive cellular immunotherapy of cancer and infectious diseases in their natural state, but recent advances in methods of gene transfer may allow for the enhanced survival, trafficking, recognition, and function that may be necessary to overcome the clever escape mechanisms of viruses and malignant transformation.
References
- 1.Freund-Brown J, Chirino L, Kambayashi T. Strategies to enhance NK cell function for the treatment of tumors and infections. Crit Rev Immunol. 2018;38(2):105–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutlu T, Nystrom S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23(10):1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Frontiers in immunology. 2019;10(1205). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seillet C, Belz GT. Assessment of Gene Function of Mouse Innate Lymphoid Cells for In Vivo Analysis Using Retroviral Transduction In: Moll J, Carotta S, editors. Target Identification and Validation in Drug Discovery: Methods and Protocols, Methods in Molecular Biology. 1953: Springer Science+Business Media, LLC, part of Springer Nature; 2019. p. 231–40. [DOI] [PubMed] [Google Scholar]
- 5.Lin C, Zhang J. Chimeric antigen receptor engineered innate immune cells in cancer immunotherapy. Sci China Life Sci. 2019;62(5):633–9. [DOI] [PubMed] [Google Scholar]
- 6.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guven H, Konstantinidis KV, Alici E, Aints A, Abedi-Valugerdi M, Christensson B, et al. Efficient gene transfer into primary human natural killer cells by retroviral transduction. Exp Hematol. 2005;33(11):1320–8. [DOI] [PubMed] [Google Scholar]
- 8.Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PLoS One. 2012;7(1):e30264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Frontiers in immunology. 2019;10:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viral Matosevic S. and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. J Immunol Res. 2018;2018:4054815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382(6):545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–3. [DOI] [PubMed] [Google Scholar]
- 15.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–72. [DOI] [PubMed] [Google Scholar]
- 16.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348(3):255–6. [DOI] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milone MC, O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberschmidt O, Morgan M, Huppert V, Kessler J, Gardlowski T, Matthies N, et al. Development of Automated Separation, Expansion, and Quality Control Protocols for Clinical-Scale Manufacturing of Primary Human NK Cells and Alpharetroviral Chimeric Antigen Receptor Engineering. Hum Gene Ther Methods. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–58. [DOI] [PubMed] [Google Scholar]
- 21.Assessment of adenoviral vector safety and toxicity: report of the National Institutes of Health Recombinant DNA Advisory Committee. Hum Gene Ther. 2002;13(1):3–13. [DOI] [PubMed] [Google Scholar]
- 22.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–9. [DOI] [PubMed] [Google Scholar]
- 23.Food and Drug Administration CfBEaR. Recommendations for Microbial Vectors Used for Gene Therapy. Maryland: Office of Communication, Outreach and Development (OCOD); 2016. [Google Scholar]
- 24.Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158. [DOI] [PubMed] [Google Scholar]
- 25.Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341(6148):1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Remon JS, et al. Efficient and robust NK-Cell transduction with Baboon Envelope pseudotyped lentivector: a major tool for immunotherapy. bioRxiv. 2019:625285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marin M, Lavillette D, Kelly SM, Kabat D. N-linked glycosylation and sequence changes in a critical negative control region of the ASCT1 and ASCT2 neutral amino acid transporters determine their retroviral receptor functions. J Virol. 2003;77(5):2936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasko JE, Battini JL, Gottschalk RJ, Mazo I, Miller AD. The RD114/simian type D retrovirus receptor is a neutral amino acid transporter. Proc Natl Acad Sci U S A. 1999;96(5):2129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandrin V, Boson B, Salmon P, Gay W, Negre D, Le Grand R, et al. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100(3):823–32. [DOI] [PubMed] [Google Scholar]
- 30.Girard-Gagnepain A, Amirache F, Costa C, Levy C, Frecha C, Fusil F, et al. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood. 2014;124(8):1221–31. [DOI] [PubMed] [Google Scholar]
- 31.Koo HM, Parthasarathi S, Ron Y, Dougherty JP. Pseudotyped REV/SRV retroviruses reveal restrictions to infection and host range within members of the same receptor interference group. Virology. 1994;205(1):345–51. [DOI] [PubMed] [Google Scholar]
- 32.Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. 2008;8(4):259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–10. [DOI] [PubMed] [Google Scholar]
- 34.Wilson MH, Coates CJ, George AL, Jr. PiggyBac transposon-mediated gene transfer in human cells. Mol Ther. 2007;15(1):139–45. [DOI] [PubMed] [Google Scholar]
- 35.Ni J, Wangensteen KJ, Nelsen D, Balciunas D, Skuster KJ, Urban MD, et al. Active recombinant Tol2 transposase for gene transfer and gene discovery applications. Mob DNA. 2016;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukahara T, Iwase N, Kawakami K, Iwasaki M, Yamamoto C, Ohmine K, et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene therapy. 2015;22(2):209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staal FJT, Aiuti A, Cavazzana M. Autologous Stem-Cell-Based Gene Therapy for Inherited Disorders: State of the Art and Perspectives. Front Pediatr. 2019;7:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staal FJ, Pike-Overzet K, Ng YY, van Dongen JJ. Sola dosis facit venenum. Leukemia in gene therapy trials: a question of vectors, inserts and dosage? Leukemia. 2008;22(10):1849–52. [DOI] [PubMed] [Google Scholar]
- 39.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467(7313):318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gogol-Doring A, Ammar I, Gupta S, Bunse M, Miskey C, Chen W, et al. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol Ther. 2016;24(3):592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68(8):2961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin Z, Maiti S, Huls H, Singh H, Olivares S, Mates L, et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene therapy. 2011;18(9):849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. 2016;126(9):3363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Srour SA, Singh H, McCarty J, de Groot E, Huls H, Rondon G, et al. Long-term outcomes of Sleeping Beauty-generated CD19-specific CAR T-cell therapy for relapsed-refractory B-cell lymphomas. Blood. 2020;135(11):862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crossland DL, Denning WL, Ang S, Olivares S, Mi T, Switzer K, et al. Antitumor activity of CD56-chimeric antigen receptor T cells in neuroblastoma and SCLC models. Oncogene. 2018;37(27):3686–97. [DOI] [PubMed] [Google Scholar]
- 46.Chicaybam L, Abdo L, Carneiro M, Peixoto B, Viegas M, de Sousa P, et al. CAR T Cells Generated Using Sleeping Beauty Transposon Vectors and Expanded with an EBV-Transformed Lymphoblastoid Cell Line Display Antitumor Activity In Vitro and In Vivo. Hum Gene Ther. 2019;30(4):511–22. [DOI] [PubMed] [Google Scholar]
- 47.Bishop DC, Xu N, Tse B, O’Brien TA, Gottlieb DJ, Dolnikov A, et al. PiggyBac-Engineered T Cells Expressing CD19-Specific CARs that Lack IgG1 Fc Spacers Have Potent Activity against B-ALL Xenografts. Mol Ther. 2018;26(8):1883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He J, Zhang Z, Lv S, Liu X, Cui L, Jiang D, et al. Engineered CAR T cells targeting mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell Immunol. 2018;329:31–40. [DOI] [PubMed] [Google Scholar]
- 49.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113(48):E7788–E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clauss J, Obenaus M, Miskey C, Ivics Z, Izsvak Z, Uckert W, et al. Efficient Non-Viral T-Cell Engineering by Sleeping Beauty Minicircles Diminishing DNA Toxicity and miRNAs Silencing the Endogenous T-Cell Receptors. Hum Gene Ther. 2018;29(5):569–84. [DOI] [PubMed] [Google Scholar]
- 51.Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, Manuri PR, et al. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J Immunother. 2009;32(8):826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darquet AM, Cameron B, Wils P, Scherman D, Crouzet J. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene therapy. 1997;4(12):1341–9. [DOI] [PubMed] [Google Scholar]
- 53.Hudecek M, Gogishvili T, Monjezi R, Wegner J, Shankar R, Kruesemann C, et al. Minicircle-Based Engineering of Chimeric Antigen Receptor (CAR) T Cells. Recent Results Cancer Res. 2016;209:37–50. [DOI] [PubMed] [Google Scholar]
- 54.Cheng C, Tang N, Li J, Cao S, Zhang T, Wei X, et al. Bacteria-free minicircle DNA system to generate integration-free CAR-T cells. J Med Genet. 2019;56(1):10–7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rotiroti MC, Buracchi C, Arcangeli S, Galimberti S, Valsecchi MG, Perriello VM, et al. Targeting CD33 in chemoresistant AML patient-derived xenografts by CAR-CIK cells engineered with an optimized Sleeping Beauty transposon version. Molecular Therapy. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331(6013):44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124(7):1081–8. [DOI] [PubMed] [Google Scholar]
- 59.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol. 2008;180(9):6392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22(12):1575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70(22):9053–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nature reviews. 2014;13(10):759–80. [DOI] [PubMed] [Google Scholar]
- 63.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7(5):618–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sergeeva OV, Koteliansky VE, Zatsepin TS. mRNA-Based Therapeutics - Advances and Perspectives. Biochemistry (Mosc). 2016;81(7):709–22. [DOI] [PubMed] [Google Scholar]
- 65.Lundstrom K Latest development on RNA-based drugs and vaccines. Future Sci OA. 2018;4(5):FSO300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11(2):121–7. [DOI] [PubMed] [Google Scholar]
- 67.Pardi N, Muramatsu H, Weissman D, Kariko K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol. 2013;969:29–42. [DOI] [PubMed] [Google Scholar]
- 68.Chu Y, Flower A, Cairo MS. Modification of Expanded NK Cells with Chimeric Antigen Receptor mRNA for Adoptive Cellular Therapy. Methods Mol Biol. 2016;1441:215–30. [DOI] [PubMed] [Google Scholar]
- 69.Carlsten M, Levy E, Karambelkar A, Li L, Reger R, Berg M, et al. Efficient mRNA-Based Genetic Engineering of Human NK Cells with High-Affinity CD16 and CCR7 Augments Rituximab-Induced ADCC against Lymphoma and Targets NK Cell Migration toward the Lymph Node-Associated Chemokine CCL19. Frontiers in immunology. 2016;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altmann T, Bergschneider E, Heckötter J, Birth K, Wild S, Andreoni CC, et al. New automated and closed electroporation system that yields cross-presenting Mo-DCs with improved functionality. Cytotherapy. 2018;20(5):S107–S8. [Google Scholar]
- 71.Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17(3):147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40. [DOI] [PubMed] [Google Scholar]
- 73.Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer immunology research. 2015;3(4):333–44. [DOI] [PubMed] [Google Scholar]
- 74.Chu Y, Yahr A, Huang B, Ayello J, Barth M, M SC. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology. 2017;6(9):e1341031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53(5):958–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer immunology research. 2018;6(4):467–80. [DOI] [PubMed] [Google Scholar]
- 77.Nayyar G, Chu Y, Cairo MS. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Frontiers in oncology. 2019;9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park H, Awasthi A, Ayello J, Chu Y, Riddell S, Rosenblum J, et al. ROR1-Specific Chimeric Antigen Receptor (CAR) NK Cell Immunotherapy for High risk Neuroblastomas and Sarcomas. Biol Blood Marrow Transplant. 2017;23(3):S136–S7. [Google Scholar]
- 79.Nayyar G, Chu Y., Negron O, Jeremy Lyngdoh J, Jeng E, Alter S, et al. Combining ROR1-Specific Chimeric Antigen Receptor (CAR) NK Cells with IL-15 Superagonist (N-803/ALT-803) to Target Chemotherapy Resistant Neuroblastoma. Biol Blood Marrow Transplant. 2019;25(3):S334. [Google Scholar]
- 80.Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–86. [DOI] [PubMed] [Google Scholar]
- 81.Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subhashis Sarkar S, Sachin Chauhan S, Arwen Stikvoort A, Alessandro Natoni A, John Daly J, Robert Henderson R, et al. CD38low Natural Killer Cells Transiently Expressing CD16F158V m-RNA Potentiates the Therapeutic Activity of Daratumumab Against Multiple Myeloma with Minimal Effector NK Cell Fratricide. Blood. 2018;132:3199. [Google Scholar]
- 83.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215(3):985–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain SA, Elmas E, Foltz JA, et al. Generation of Knock-out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J Vis Exp. 2018(136). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep. 2016;17(5):1453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naeimi Kararoudi M, Elmas E, Lamb M, Chakravarti N, Trikha P, Lee DA. Disruption of SOCS3 Promotes the Anti-Cancer Efficacy of Primary NK Cells. Blood. 2018;132(Suppl 1):5687-. [Google Scholar]
- 88.Naeimi Kararoudi M, Nagai Y, Elmas E, Pereira MSF, Ali SA, Imus PH, et al. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Del’Guidice T, Lepetit-Stoffaes JP, Bordeleau LJ, Roberge J, Theberge V, Lauvaux C, et al. Membrane permeabilizing amphiphilic peptide delivers recombinant transcription factor and CRISPR-Cas9/Cpf1 ribonucleoproteins in hard-to-modify cells. PLoS One. 2018;13(4):e0195558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pomeroy EJ, Hunzeker JT, Kluesner MT, Crosby MR, Lahr WS, Bendzick L, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. bioRxiv. 2018:430553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rautela J, Surgenor E, Huntington ND. Efficient genome editing of human natural killer cells by CRISPR RNP. bioRxiv. 2018:406934. [DOI] [PubMed] [Google Scholar]
- 92.Gaj T, Staahl BT, Rodrigues GMC, Limsirichai P, Ekman FK, Doudna JA, et al. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic acids research. 2017;45(11):e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu J, Zhou G, Zhang L, Zhao Q. Building Potent Chimeric Antigen Receptor T Cells With CRISPR Genome Editing. Frontiers in immunology. 2019;10:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kararoudi MN, Likhite S, Elmas E, Schwartz M, Meyer K, Lee DA. Highly efficient site-directed gene insertion in primary human natural killer cells using homologous recombination and CRISPaint delivered by AAV. bioRxiv. 2019:743377. [Google Scholar]
- 95.Schmid-Burgk JL, Honing K, Ebert TS, Hornung V. CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nat Commun. 2016;7:12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Somanchi SS, Lee DA. Ex Vivo Expansion of Human NK Cells Using K562 Engineered to Express Membrane Bound IL21. Methods Mol Biol. 2016;1441:175–93. [DOI] [PubMed] [Google Scholar]
- 97.Wang X, Lee DA, Wang Y, Wang L, Yao Y, Lin Z, et al. Membrane-bound interleukin-21 and CD137 ligand induce functional human natural killer cells from peripheral blood mononuclear cells through STAT3 activation. Clin Exp Immunol. 2013;172(1):104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu S, Phatarpekar PV, Denman CJ, Senyukov VV, Somanchi SS, Nguyen-Jackson HT, et al. Transcription of the activating receptor NKG2D in natural killer cells is regulated by STAT3 tyrosine phosphorylation. Blood. 2014;124(3):403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Braunschweig A, Poehlmann TG, Busch S, Schleussner E, Markert UR. Signal transducer and activator of transcription 3 (STAT3) and Suppressor of Cytokine Signaling (SOCS3) balance controls cytotoxicity and IL-10 expression in decidual-like natural killer cell line NK-92. Am J Reprod Immunol. 2011;66(4):329–35. [DOI] [PubMed] [Google Scholar]
- 100.Bedel R, Thiery-Vuillemin A, Grandclement C, Balland J, Remy-Martin JP, Kantelip B, et al. Novel role for STAT3 in transcriptional regulation of NK immune cell targeting receptor MICA on cancer cells. Cancer Res. 2011;71(5):1615–26. [DOI] [PubMed] [Google Scholar]
- 101.Cacalano NA. Regulation of Natural Killer Cell Function by STAT3. Frontiers in immunology. 2016;7:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunology and cell biology. 2011;89(2):216–24. [DOI] [PubMed] [Google Scholar]
- 103.Chester C, Fritsch K, Kohrt HE. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Frontiers in immunology. 2015;6(601). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu Z, Tee WW. Enhancers and chromatin structures: regulatory hubs in gene expression and diseases. Biosci Rep. 2017;37(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Au-Yeung N, Horvath CM. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev. 2018;44:11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gatla HR, Muniraj N, Thevkar P, Yavvari S, Sukhavasi S, Makena MR. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. Int J Mol Sci. 2019;20(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol. 2019;21(2):152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ura K, Wolffe AP. Reconstruction of transcriptionally active and silent chromatin. Methods Enzymol. 1996;274:257–71. [DOI] [PubMed] [Google Scholar]
- 109.Conte M, De Palma R, Altucci L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int J Biochem Cell Biol. 2018;98:65–74. [DOI] [PubMed] [Google Scholar]
- 110.Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, et al. The narrow-spectrum HDAC inhibitor entinostat enhances NKG2D expression without NK cell toxicity, leading to enhanced recognition of cancer cells. Pharmaceutical research. 2015;32(3):779–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chittock EC, Latwiel S, Miller TC, Muller CW. Molecular architecture of polycomb repressive complexes. Biochem Soc Trans. 2017;45(1):193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U S A. 2015;112(52):15988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li Y, Wang J, Yin J, Liu X, Yu M, Li T, et al. Chromatin state dynamics during NK cell activation. Oncotarget. 2017;8(26):41854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weake VM. Histone Ubiquitylation Control of Gene Expression In: Workman JL, Abmayr SM, editors. Fundamentals of Chromatin. New York, NY: Springer New York; 2014. p. 257–307. [Google Scholar]
- 115.Żylicz JJ, Bousard A, Žumer K, Dossin F, Mohammad E, da Rocha ST, et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell. 2019;176(1):182–97.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Panda S, Nilsson JA, Gekara NO. Deubiquitinase MYSM1 Regulates Innate Immunity through Inactivation of TRAF3 and TRAF6 Complexes. Immunity. 2015;43(4):647–59. [DOI] [PubMed] [Google Scholar]
- 117.Nandakumar V, Chou Y, Zang L, Huang XF, Chen SY. Epigenetic control of natural killer cell maturation by histone H2A deubiquitinase, MYSM1. Proc Natl Acad Sci U S A. 2013;110(41):E3927–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ni J, Holsken O, Miller M, Hammer Q, Luetke-Eversloh M, Romagnani C, et al. Adoptively transferred natural killer cells maintain long-term antitumor activity by epigenetic imprinting and CD4(+) T cell help. Oncoimmunology. 2016;5(9):e1219009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Foltz JA, Moseman JE, Thakkar A, Chakravarti N, Lee DA. TGFbeta Imprinting During Activation Promotes Natural Killer Cell Cytokine Hypersecretion. Cancers (Basel). 2018;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]