

Abstract
A prevalent model of Alzheimer’s disease (AD) pathogenesis postulates the generation of neurotoxic fragments derived from the amyloid precursor protein (APP) after its internalization to endocytic compartments. The molecular pathways that regulate APP internalization and intracellular trafficking in neurons are incompletely understood. Here, we report that 5xFAD mice, an animal model of AD, expressing signaling‐deficient variants of the p75 neurotrophin receptor (p75NTR) show greater neuroprotection from AD neuropathology than animals lacking this receptor. p75NTR knock‐in mice lacking the death domain or transmembrane Cys259 showed lower levels of Aβ species, amyloid plaque burden, gliosis, mitochondrial stress, and neurite dystrophy than global knock‐outs. Strikingly, long‐term synaptic plasticity and memory, which are completely disrupted in 5xFAD mice, were fully recovered in the knock‐in mice. Mechanistically, we found that p75NTR interacts with APP at the plasma membrane and regulates its internalization and intracellular trafficking in hippocampal neurons. Inactive p75NTR variants internalized considerably slower than wild‐type p75NTR and showed increased association with the recycling pathway, thereby reducing APP internalization and co‐localization with BACE1, the critical protease for generation of neurotoxic APP fragments, favoring non‐amyloidogenic APP cleavage. These results reveal a novel pathway that directly and specifically regulates APP internalization, amyloidogenic processing, and disease progression, and suggest that inhibitors targeting the p75NTR transmembrane domain may be an effective therapeutic strategy in AD.
Keywords: A‐beta, amyloid, BACE, endocytosis, neurodegeneration
Subject Categories: Neuroscience, Signal Transduction
APP tethering to the cell surface by signaling‐deficient variants of neurotrophin receptor p75 prevents its internalization and processing, restoring memory and reducing amyloid plaque burden and disease pathology in AD mouse models.

Introduction
A central tenet of the amyloid hypothesis of AD pathogenesis is the generation of neurotoxic fragments of APP through a series of proteolytic cleavages (Karran et al, 2011; Selkoe & Hardy, 2016). APP cleavage by BACE (beta‐site APP cleaving enzyme) at an extracellular site close to the plasma membrane leaves a transmembrane C‐terminal stub (beta‐carboxyterminal fragment or CTFβ) that serves as a substrate for further intramembrane cleavage by gamma‐secretase, a multisubunit complex that includes the aspartyl protease presenilin‐1 (PS‐1) as its catalytic subunit. Cleavage by the gamma‐secretase complex liberates a soluble CTFβ (referred to as either sCTFβ or APP intracellular domain, AICD) and a small N‐terminal fragment of 40 or 42 amino acids in length known as the amyloid beta peptide or Aβ, the main component of the amyloid plaques that accumulate in the AD brain (Karran et al, 2011; Selkoe & Hardy, 2016). The majority of familial AD cases are caused by mutations in the genes encoding APP or PS‐1 (Karran et al, 2011; Selkoe & Hardy, 2016), supporting the amyloid hypothesis of AD pathogenesis. APP can also be cleaved by cell surface alpha‐secretases, most notably ADAM10, in an extracellular site very close to the plasma membrane, but C‐terminal to the site of BACE cleavage. Thus, alpha‐secretase cleavage precludes the generation of all BACE‐derived products, including Aβ, and constitutes the non‐amyloidogenic pathway in APP processing. Cleavage by alpha‐secretase generates a soluble N‐terminal fragment (sAPPα) and a C‐terminal stub (sCTFα) that can be further processed by gamma‐secretase.
Recent studies have indicated that, while cleavage by alpha‐secretases occurs at the plasma membrane, proteolytic processing by BACE requires APP internalization from the cell surface and thus mainly takes places in intracellular, endocytic compartments (Schneider et al, 2008; Sannerud et al, 2011; Haass et al, 2012). Several studies have linked APP internalization to Aβ production (Koo & Squazzo, 1994; Selkoe et al, 1996), and complementary lines of evidence support this notion, including the requirement of low pH for BACE optimal catalytic activity (Vassar et al, 2009), the fact that genetic or pharmacological inhibition of APP internalization reduces Aβ generation (Perez et al, 1999; Carey et al, 2005), and evidence that neuronal activity enhances Aβ production by inducing APP internalization and trafficking to BACE‐containing endosomes (Das et al, 2013, 2016). Thus, the localization and intracellular trafficking of APP appear to be critical for the balance between competing amyloidogenic and non‐amyloidogenic pathways of APP processing. Aside from main components of the endocytic machinery, such as dynamin, our knowledge of the molecular pathways that can regulate APP internalization and intracellular trafficking in neurons is incomplete.
p75NTR is a member of the death receptor superfamily, characterized by the presence of a death domain (DD) in their intracellular region (Liepinsh et al, 1997), which also includes the tumor necrosis factor receptor 1 (TNFR1), CD40, Fas, and others (Ibáñez & Simi, 2012). Many of these receptors can induce cell death pathways as a mechanism for clearing damage produced after a lesion or insult. However, after severe injury or disease, they can also amplify tissue damage as a result of overactivation and/or overexpression. Upon neural injury or cellular stress, p75NTR signaling can contribute to neuronal death, axonal degeneration, and synaptic dysfunction (Ibáñez & Simi, 2012). p75NTR can function as a receptor of neurotrophins, a family of neurotrophic growth factors, that includes nerve growth factor (NGF), brain‐derived neurotrophic factor (BDNF), and others, as well as other ligands unrelated to the neurotrophins, including Aβ (for review, see (Underwood & Coulson, 2008)). Notably, expression of p75NTR is increased in the brain of AD patients (Ernfors et al, 1990; Mufson & Kordower, 1992; Hu et al, 2002; Chakravarthy et al, 2012) as well as animal models of AD (Chakravarthy et al, 2010; Wang et al, 2011). Aβ can induce rapid cell death in cultured neurons through direct interaction with p75NTR and downstream activation of cell death pathways (Rabizadeh et al, 1994; Yaar et al, 1997; Perini et al, 2002; Sotthibundhu et al, 2008; Knowles et al, 2009), but the relevance of in vitro overnight effects is unclear, as neuronal degeneration occurs during long periods of time in AD patients, and it is in fact seldom observed in animal models of AD. In line with this, elimination of p75NTR affords a rather limited improvement in those models (Knowles et al, 2009).
In this study, we used the 5xFAD model of AD (Oakley et al, 2006) to investigate neuropathological effects in different strains of knock‐in and knock‐out p75NTR mutant mice. Unexpectedly, we found that knock‐in mutations that inactivate p75NTR signaling, but leave normal levels of receptor expression, conferred much higher protection from AD‐associated neuropathology than a global knock‐out. In our efforts to understand how an inactive receptor can afford greater neuroprotection than the absence of the receptor, we discovered a novel mechanism by which p75NTR regulates APP internalization, intracellular trafficking, and its localization to endocytic compartments containing BACE.
Results
Reduced Aβ content and histopathology in the hippocampus of 5xFAD mice carrying inactive p75NTR variants
The transmembrane domain of p75NTR contains a highly conserved cysteine residue (Cys259 in mouse) which is critical for p75NTR activation and signaling in response to neurotrophin ligands (Vilar et al, 2009). Neurons from knock‐in mice carrying a Cys to Ala mutation at this position (C259A), or lacking the receptor death domain (ΔDD), are resistant to cell death induced by pro‐neurotrophins and neurodegeneration induced by epileptic seizures, to a similar extent as knock‐out (KO) neurons lacking p75NTR entirely (Tanaka et al, 2016). Cortical neurons derived from these 3 lines also show comparable resistance to Aβ‐mediated toxicity in vitro (Fig 1A–C). In order to study the contribution of p75NTR activity to AD neuropathology in vivo, we crossed each of the 3 lines of p75NTR mutant mice to the 5xFAD mouse model of AD. These mice express a human APP transgene carrying three mutations found in AD patients and a human PS‐1 transgene with two AD mutations, both under regulatory sequences of a Thy1 transgene, thus primarily directing expression to neurons (Oakley et al, 2006). 5xFAD mice develop cerebral amyloid plaques starting at 2 months of age, achieve substantial Aβ burden with gliosis and neurite degeneration by 4 months, and show significant memory impairment by 6 months. The severity and accelerated progression of AD pathology displayed by this model served as a stringent test for assessing potential protective effects of the different p75NTR mutations. At 5 months of age, 5xFAD mice showed increased levels of p75NTR in the hippocampus compared to wild‐type mice, with prominent expression in dendrites of pyramidal neurons (Fig 2A). At 6 months, Western blots of hippocampal extracts indicated approximately 3‐fold increase in p75NTR protein levels in 5xFAD mice compared to wild‐type controls (Fig 2B and C).
Figure 1. Neurons lacking p75NTR transmembrane Cys259, the death domain, or the whole receptor are equally resistant to Aβ neurotoxicity in vitro .
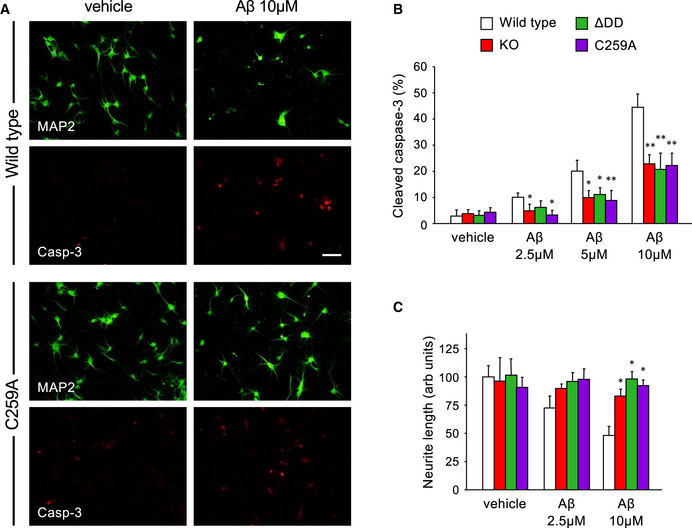
- Representative photomicrographs of MAP2 (green) and cleaved caspase‐3 (red) immunocytochemistry of embryonic cortical neurons from wild‐type (upper panels) or C259A mice treated with vehicle or 10 µM Aβ oligomers for 14 h. Scale bar 15 µm.
- Quantification of active (cleaved) caspase‐3 in cultured cortical neurons of the indicated genotypes treated for 14 h with vehicle or Aβ oligomers (2.5, 5, or 10 µM). Data are expressed as percentage cleaved caspase‐3 and MAP2 double positive cells over the total number of MAP2 positive cells. Error bars indicate SD. N = 3 independent experiments each performed in duplicate. *P < 0.05; **P < 0.01 versus vehicle (Student’s t‐test).
- Quantification of neurite length in cultured cortical neurons of the indicated genotypes treated for 24 h with vehicle or Aβ oligomers (2.5, or 10 µM). Data are expressed as mean neurite length in individual neurons, relative to wild‐type vehicle (set to 100 units). Error bars indicate SD. N = 3 independent experiments, n ≥ 35 neurons per experiment. *P < 0.05 versus vehicle (Student’s t‐test).
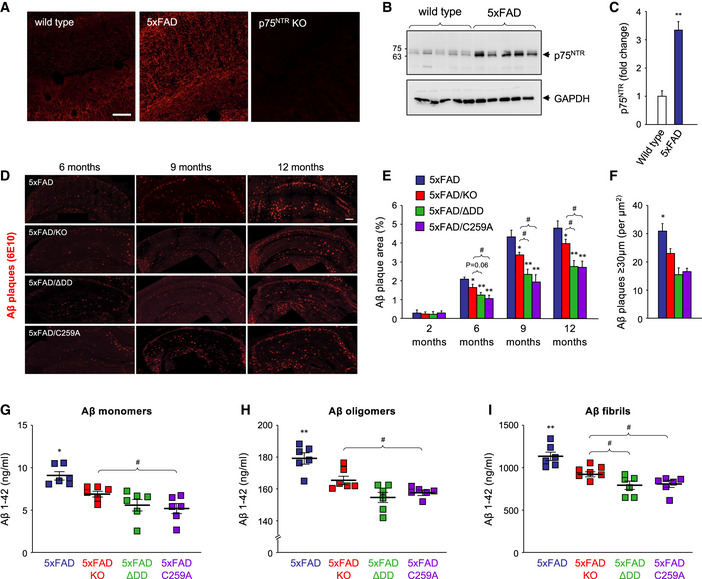
Figure 2. Reduced Aβ content in the hippocampus of 5xFAD mice carrying inactive p75NTR variants.
-
AImmunohistochemistry for p75NTR in hippocampal CA1 of 5‐month‐old wild type, 5xFAD, and p75NTR knock‐out (KO) mice. Scale bar, 20 μm.
-
BWestern blot analysis of p75NTR expression in total lysates of hippocampus from 6‐month‐old wild‐type (WT) and 5xFAD mice assessed using antibody GT15057 against p75NTR extracellular domain (Table S1). The lower panel shows reprobing for GAPDH to control for equal gel loading.
-
CQuantification of p75NTR expression in mouse hippocampus relative to GAPDH. Mean ± SEM; **P < 0.01; N = 5 mice per group (one‐way ANOVA followed by post hoc test).
-
DImmunostaining for Aβ plaques with 6E10 antibody in coronal sections through the hippocampus of 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A mice of the indicated ages. Scale bar, 400 μm.
-
EQuantification of Aβ plaque burden in the hippocampus of 5xFAD mouse strains carrying different p75NTR variants as indicated. Histogram shows the percentage of hippocampal area occupied by Aβ plaques (mean ± SEM, N = 5 mice per group). *P < 0.05 and **P < 0.01 versus 5XFAD. #P < 0.05 versus 5XFAD/KO (one‐way ANOVA followed by post hoc test).
-
FQuantification of the number of Aβ plaques larger than 30 µm in diameter per µm2 in coronal sections through the hippocampus of 9‐month‐old 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A mice. Color codes are as in panel (E). Histogram shows mean ± SEM, N = 4 mice per group; *P < 0.05 KO versus knock‐in genotypes.
-
G–IELISA determinations of Aβ1‐42 content in hippocampus of 5xFAD mouse strains carrying different p75NTR variants as indicated. Aβ monomers (G) refer to the soluble fraction after Tris‐buffered saline extraction, Aβ oligomers (H) to the soluble fraction after RIPA buffer extraction of the Tris‐buffered saline pellet, and Aβ fibrils (I) to the soluble fraction after formic acid treatment of the RIPA pellet. See Materials and Methods for details. Shown is mean ± SEM; squares denote individual animals. *P < 0.05 and **P < 0.01 versus 5XFAD. #P < 0.05 versus 5XFAD/KO (one‐way ANOVA followed by post hoc test).
Source data are available online for this figure.
5xFAD mice developed progressively increased Aβ plaque burden as detected histologically in sections through the hippocampus; at 12 months of age, 5% of the area of the hippocampus was occupied by Aβ plaques (Fig 2D and E). No Aβ immunoreactivity could be detected at any age in wild‐type mice (not shown). In the 5xFAD background, all three p75NTR mutants (5xFAD/KO, 5xFAD/C259A, and 5xFAD/ΔDD, respectively) showed reduced levels of Aβ plaque burden at all the ages examined (6, 9, and 12 months) compared to 5xFAD mice expressing wild‐type p75NTR (Fig 2E). However, the two knock‐in mutants afforded greater protection than the knock‐out, with differences against 5xFAD/KO mice reaching statistical significance, specially at 9 and 12 months of age (Fig 2E). In addition, all p75NTR mutants showed significantly reduced number of large Aβ plaques (> 30 μm in diameter) compared to 5xFAD mice that were wild type for p75NTR (Fig 2F). Next, we quantified the levels of Aβ1–42 monomers, oligomers, and fibrils by ELISA, after differential detergent and acid extraction from hippocampus of 9‐month‐old mice. All three p75NTR mutations significantly lowered the levels of Aβ1–42 species in 5xFAD mice, with, again, the knock‐in variants showing a stronger effect than the knock‐out (Fig 2G–I).
Astrogliosis and microgliosis were significantly increased between 6 and 12 months of age in the hippocampus of 5xFAD mice, reaching 15–20% and 6–10% of the hippocampus area, respectively, compared to 5–10% and 2–4% in wild‐type controls (Fig 3A–D). Astrocytes and microglial cells with morphology indicative of an activated state were found concentrated around Aβ plaques in the hippocampus of 5xFAD animals (Fig 3A and C). All three p75NTR mutations significantly reduced both forms of gliosis in the 5xFAD hippocampus. However, the knock‐in strains showed the lowest levels of gliosis, reaching statistically significant differences compared to 5xFAD/KO at 9 and 12 months (Fig 3B and D). Next, we assessed the extent of neurite dystrophy in dendritic arbors of pyramidal hippocampal neurons, as assessed by accumulation of reticulon 3 (RTN3). Previous studies have shown RTN3 aggregates to be markedly accumulated in dystrophic neurites in the brains of AD patients and APP transgenic mice (Hu et al, 2007). At 9 months, RTN3‐positive neurites appeared as bright spots concentrated in and around Aβ plaques in the hippocampus of 5xFAD mice (Fig 3E). No RTN3 immunoreactivity could be detected in wild‐type mice (not shown). RTN3 immunoreactivity in 5xFAD hippocampus was significantly reduced by all three p75NTR mutations, with the strongest effects observed in the knock‐in strains (Fig 3F). We note that, while soluble RTN3 has been reported to negatively regulate BACE1 activity (He et al, 2004), its aggregation appears to correlate with enhanced amyloidogenic cleavage of APP and production of β‐amyloid (Shi et al, 2009). Finally, we looked at mitochondrial dysfunction, a well‐known feature of AD neuropathology (Swerdlow, 2018), as assessed by MitoSOX staining, a mitochondrial superoxide indicator widely used to assess mitochondrial stress (Dikalov & Harrison, 2014). MitoSOX staining was significantly increased at 2 and 6 months in neurons of the pyramidal layer of the hippocampus of 5xFAD mice compared to wild‐type controls (Fig 3G and H). At 2 months of age, all three p75NTR mutations reduced MitoSOX levels significantly in the 5xFAD background, almost to the low levels found in wild‐type mice (Fig 3H). However, at 6 months, there was a significant advantage of the p75NTR knock‐in strains over 5xFAD/KO mice (Fig 3H).
Figure 3. Reduced histopathology in the hippocampus of 5xFAD mice carrying inactive p75NTR variants.
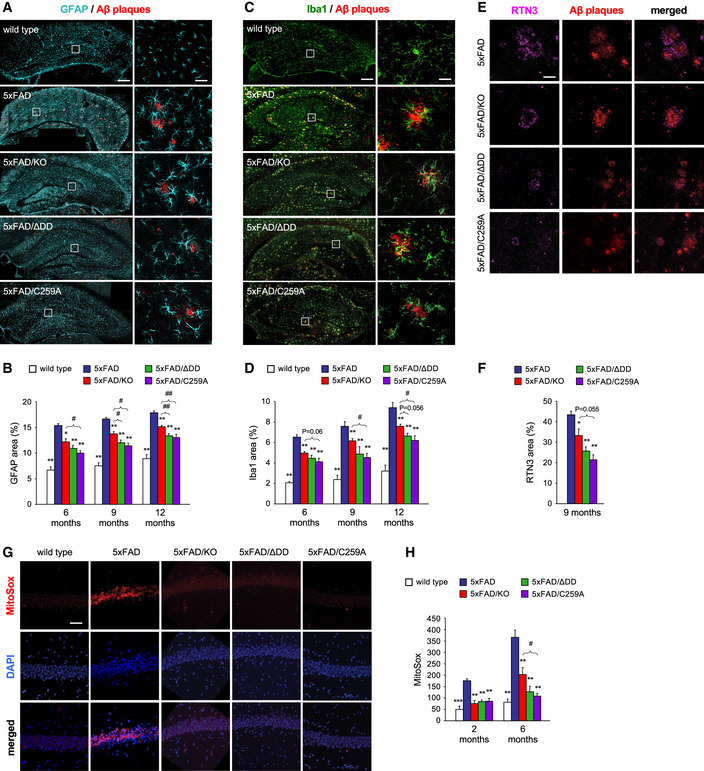
- Immunostaining for glial fibrillary acidic protein (GFAP), a marker of astrocytes, and Aβ plaques in coronal sections through the hippocampus of 6‐month‐old wild type, 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A mice. Scale bar, 300 μm. Right‐hand panels show high magnification of the indicated areas. Scale bar, 50 μm.
- Quantification of GFAP area in the hippocampus of wild type and 5xFAD mouse strains carrying different p75NTR variants as indicated. Histogram shows the percentage of hippocampal area occupied by GFAP immunostaining (mean ± SEM, N = 5 mice per group). *P < 0.05 and **P < 0.01 versus 5XFAD. #P < 0.05 and ##P < 0.01 versus 5XFAD/KO (one‐way ANOVA followed by post hoc test).
- Immunostaining for Ionized calcium binding adaptor molecule 1 (Iba1), a marker of microglial cells, and Aβ plaques in coronal sections through the hippocampus of 6‐month‐old wild type, 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A. Scale bar, 300 μm. Right‐hand panels show high magnification of the indicated areas. Scale bar, 10 μm.
- Quantification of Iba1 area in the hippocampus of wild type and 5xFAD mouse strains carrying different p75NTR variants as indicated. Histogram shows the percentage of hippocampal area occupied by Iba1 immunostaining (mean ± SEM, N = 5 mice per group). *P < 0.05 and **P < 0.01 versus 5XFAD. #P < 0.05 versus 5XFAD/KO. Other P values are indicated (one‐way ANOVA followed by post hoc test).
- Immunostaining of reticulon 3 (RTN3), a marker of dystrophic neurites, and Aβ plaques in coronal sections through the hippocampus of 6‐month‐old 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A mice. Scale bar, 40 μm.
- Quantification of RTN3‐positive dystrophic neurite area in the hippocampus of 5xFAD mouse strains carrying different p75NTR variants as indicated. Histogram shows the percentage of Aβ plaque area that overlapped with RTN3 immunostaining (mean ± SEM, N = 5 mice per group). *P < 0.05 and **P < 0.01 versus 5XFAD. Other P values are indicated (one‐way ANOVA followed by post hoc test).
- MitoSOX staining, a mitochondrial superoxide indicator, and DAPI in coronal sections through the hippocampus of 6‐month‐old wild type, 5xFAD, 5xFAD/KO, 5xFAD/ΔDD, and 5xFAD/C259A mice. Scale bar, 60 μm.
- Quantification of MitoSOX signal in the pyramidal cell layer of hippocampus of wild type and 5xFAD mouse strains carrying different p75NTR variants as indicated. Histogram shows MitoSOX mean fluorescence intensity in arbitrary units (mean ± SEM, N = 5 mice per group). **P < 0.01 and ***P < 0.001 versus 5XFAD. #P < 0.05 versus 5XFAD/KO. (one‐way ANOVA followed by post hoc test).
Together, these studies indicated a significantly higher level of neuroprotection in mouse strains carrying signaling‐deficient p75NTR variants compared to knock‐out mice lacking the receptor. This was in contrast to the results of the in vitro assay of Aβ neurotoxicity (Fig 1), in which neurons from all three strains were equally resistant, suggesting that additional mechanisms must operate to account for the differences observed in vivo.
Inactive p75NTR variants, but not the knock‐out, fully rescue synaptic deficits and memory impairment in 5xFAD mice
We asked whether the beneficial effects on AD histopathology afforded by the different p75NTR mutants had an impact on synaptic function and cognitive behavior of 5xFAD mice. Previous studies have shown that AD is associated with deficits in various forms of synaptic plasticity, including hippocampal long‐term potentiation (LTP), in human patients and animal models (Selkoe, 2002; Palop & Mucke, 2010; Koch et al, 2012; Mango et al, 2019). In our studies, we used a form of late LTP induced by theta‐burst stimulation (TBS‐LTP) in hippocampal area CA1. Hippocampal slices from 6‐month‐old wild‐type mice showed pronounced potentiation induced by TBS that was sustained for at least 4 h (Fig 4A). In contrast, synaptic potentiation was not maintained and declined rapidly in slices derived from 5xFAD mice, indicating a profound deficit in LTP induction (Fig 4A). Slices from 5xFAD/KO mice showed intermediate levels of potentiation that declined slowly over the course of the 4h recording, indicating a partial recovery (Fig 4A). Remarkably, TBS‐LTP in slices from 5xFAD/C259A and 5xFAD/ΔDD mice was strong in terms of induction, persistent, and maintained throughout the whole recording period of 4h, and was essentially indistinguishable from that recorded in wild‐type slices (Fig 4A). Quantification of the change in field excitatory post‐synaptic potential (fEPSP) revealed a stronger rescue of synaptic function in the p75NTR knock‐in strains, reaching statistically significant differences from 5xFAD mice throughout the recording period, unlike the knock‐out (Fig 4B). We note that TBS‐LTP in the absence of the 5xFAD transgene was normal in all three mutant strains and identical to that of wild‐type animals (Fig EV1A and B).
Figure 4. Inactive p75NTR variants, but not the knock‐out, fully rescue synaptic deficits and memory impairment of 5xFAD mice.
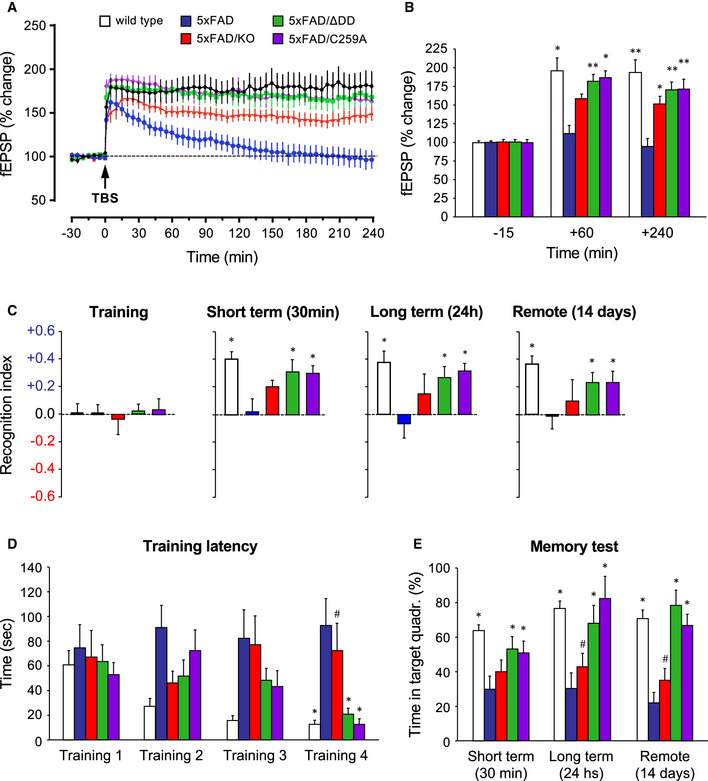
- Percentage of change in field excitatory post‐synaptic potential (fEPSP) recorded after theta‐burst stimulation (TBS) in Schaffer collaterals of hippocampal slices from 6‐month‐old wild type, 5xFAD, and 5xFAD/p75NTR mutant mice, as indicated. Results are presented as mean % change normalized to t = 0 ± SEM. N = 6 (wild type), 5 (5xFAD), 9 (5xFAD/KO), 7 (5xFAD/ΔDD), and 8 (5xFAD/C259A) slices from 3 mice per genotype, respectively.
- Quantification of fEPSP (mean % change ± SEM) in the indicated genotypes at 3 time points. *P < 0.05; **P < 0.01 versus 5xFAD (two‐way ANOVA followed by post hoc test). N numbers as in (A).
- Behavior in the novel object recognition (NOR) test of 6‐month‐old wild type, 5XFAD, and 5XFAD/p75NTR mutant mice, as indicated. Histograms show mean recognition index ± SEM during training, and 30 min, 24 h, and 14 days after training, corresponding to measures of short‐term, long‐term, and remote memory, respectively. Bar color codes are as in panel (A). *P < 0.05 versus 5xFAD (two‐way ANOVA followed by post hoc test). N = 12 (wild type, 5xFAD/ΔDD, and 5xFAD/C259A), 10 (5xFAD), and 8 (5xFAD/KO) mice per genotype, respectively.
- Training latency in the Barnes maze test of 6‐month‐old wild type, 5XFAD, and 5XFAD/p75NTR mutant mice, as indicated. Histograms show mean latency in seconds to find the platform hole ± SEM in 4 consecutive training sessions. Bar color codes are as in panel (A). *P < 0.05 versus 5xFAD; #P < 0.05 versus wild type, 5xFAD/ΔDD or 5xFAD/C259A (two‐way ANOVA followed by post hoc test). N = 14 (wild type and 5xFAD/ΔDD), 10 (5xFAD, 5xFAD/KO and 5xFAD/C259A) mice per genotype, respectively.
- Percentage of time spent (mean ± SEM) in the target quadrant of the Barnes maze test 30 min, 24 h, and 14 days after training. *P < 0.05 versus 5xFAD; #P < 0.05 versus wild type, 5xFAD/ΔDD, or 5xFAD/C259A (two‐way ANOVA followed by post hoc test). N numbers as in (D).
Figure EV1. Normal TBS‐LTP in p75NTR mutant mice.
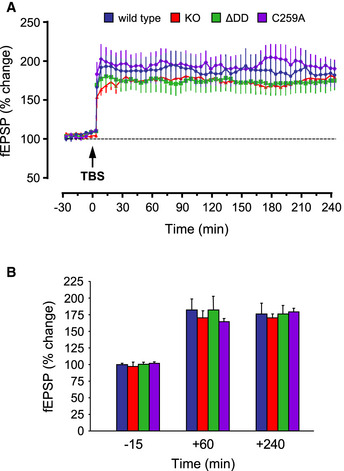
- Percentage of change in field excitatory post‐synaptic potential (fEPSP) recorded after theta‐burst stimulation (TBS) in Schaffer collaterals of hippocampal slices from WT and p75NTR mutant mice.
- Quantification of fEPSP (% change) in different genotypes at 3 time points. There was no significant difference between them (n = 7 slices from N = 3 mice per condition). Data are shown as average ± SEM.
Next, we assessed learning and memory in 6‐month‐old animals using two different paradigms based on novelty and spatial memory, respectively, namely the novel object recognition test (NOR) and the Barnes maze. There was no difference between strains during training in the NOR test, when the animals are confronted with two identical objects, with recognition index (RI) close to zero, indicating no preference between the objects (Fig 4C). Probe tests were conducted 30 min, 24 h, and 14 days after training, to assess short‐term, long‐term, and remote memory, respectively, by confronting animals to a new object placed besides a familiar object. Wild‐type mice spent approximately 40% more time (RI≈0.4) on the new object at all three probe time points, which is the expected response of normal mice in the NOR test (Fig 4C). In contrast, 5xFAD mice showed no indication that they recognized the new object (RI close to zero) at any time point (Fig 4C), indicating severe memory disruption. Knock‐out of p75NTR afforded some degree of rescue, with average RI values between 0 and 0.15, but without reaching statistically significant differences compared to 5xFAD mice expressing p75NTR at any time point, despite the large number of animals used (Fig 4C). On the other hand, the deficits of 5xFAD mice were nearly completely rescued by either of the two p75NTR knock‐in alleles, showing average RI values that were not statistically different from those of wild‐type mice (Fig 4C). The differences in cognitive performance between p75NTR knock‐in and knock‐out strains were more striking in the Barnes maze, a test of spatial memory strongly dependent on hippocampal function. During the training phase, wild‐type animals showed the expected learning performance, with progressively decreasing latencies in finding the escape hole, reaching 10 s or less by the 4th training session (Fig 4D). In contrast, 5xFAD mice showed no reduction of training latency, spending well over a minute to find the escape hole even after 4 training sessions (Fig 4D), indicating a very strong learning deficit. As before, probe tests were conducted 30 min, 24 h, and 14 days after training to assess memory of the previous location of the escape hole. While wild‐type animals spent most (60–70%) of the probe time in the target quadrant, the performance of 5xFAD mice was not different from chance (≈25% in the target quadrant, Fig 4E) at all time points, indicating no memory of the previous location of the escape hole. Remarkably, knock‐out of p75NTR did not confer any benefit to 5xFAD mice in this test. Latencies remained high and unchanged during training, and probe memory tests did not show statistically significant differences from 5xFAD mice expressing p75NTR at any time point (Fig 4E). In contrast, the p75NTR knock‐in alleles almost completely reverted the deficits of 5xFAD mice (Fig 4E). After a small delay, these animals reached similar latencies as wild‐type mice during training, and spent most of the probe time (50–80%) in the target quadrant at all time points tested (Fig 4E), indicating normal memory performance. We conclude from these studies that, although loss of p75NTR afforded some level of protection from AD‐associated histopathology and synaptic deficits—though not in cognitive performance, greater neuroprotection and, in fact, nearly full synaptic and behavioral recovery were only observed in 5xFAD mice that expressed the C259A and ΔDD variants.
Signaling‐deficient p75NTR variants favor non‐amyloidogenic processing of APP
The fact that signaling‐deficient p75NTR variants afforded greater neuroprotective effects than the knock‐out in vivo but not in vitro prompted us to investigate additional mechanisms. We speculated that the lower Aβ plaque burden and reduced levels of Aβ species in the brain of 5xFAD mice carrying p75NTR variants could be due to reduced amyloidogenic APP processing in the mutants. In order to investigate this, we examined the levels of CTFβ, a product of APP cleavage by BACE1, in 9‐month‐old hippocampus of the different strains (Fig 5A). This analysis revealed reduced levels of CTFβ in 5xFAD mice carrying mutant alleles of p75NTR (Fig 5B). Interestingly, 5xFAD/ΔDD and 5xFAD/C259A mice showed significantly lower levels of CTFβ than 5xFAD/KO mice, indicating reduced amyloidogenic APP cleavage in the knock‐in strains. We note that expression of full‐length APP was indistinguishable between all 5xFAD strains (Fig EV2A), demonstrating comparable transgene expression levels. We assessed mRNA and protein levels of the beta‐secretase BACE1 in hippocampal extracts of the four 5xFAD strains, but did not detect any significant differences (Fig EV2B and C). We reasoned that reduced amyloidogenic cleavage could have been due to increased non‐amyloidogenic processing by alpha‐secretase. We therefore assessed the levels of sAPPα, a product of the competing, non‐amyloidogenic pathway (Fig 5C). We found significantly increased levels of sAPPα in hippocampal extracts of 5xFAD mice carrying mutant p75NTR alleles compared to 5xFAD animals expressing wild‐type p75NTR (Fig 5D). Mirroring the effects observed on CTFβ, 5xFAD/ΔDD, and 5xFAD/C259A mice showed significantly higher levels of sAPPα than 5xFAD/KO mice, indicating that non‐amyloidogenic APP cleavage is more prevalent in the knock‐in strains. mRNA and protein levels of ADAM10, the main alpha‐secretase, in hippocampal extracts of the four 5xFAD strains were comparable (Fig EV2D and E). Together, these results indicated a bias favoring non‐amyloidogenic alpha‐processing of APP in 5xFAD strains carrying signaling‐deficient alleles of p75NTR, without altered levels of the main secretases or the substrate.
Figure 5. Reduced amyloidogenic processing in hippocampal neurons carrying signaling‐deficient p75NTR variants.
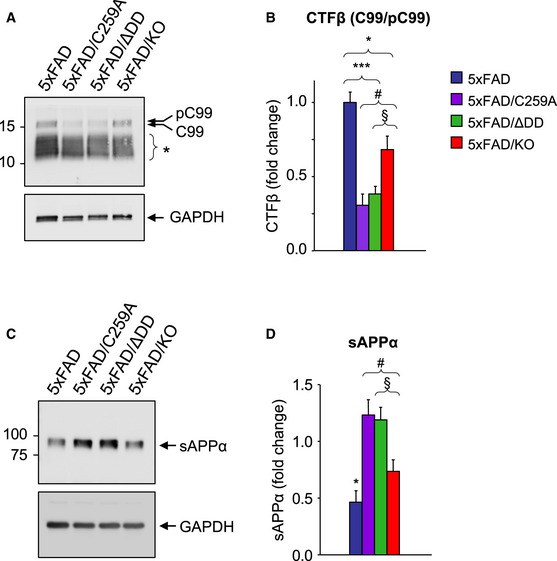
- Western blot analysis of CTF beta (CTFβ) in hippocampal lysates of 9‐month‐old 5xFAD mice carrying different p75NTR alleles detected using anti‐APP‐CTF antibody (A8717, Table S1). Arrows point to phospho‐C99 and C99 CTFβ species as previously assigned by Buxbaum et al, (1998) and Kwart et al, (2019). Asterisk denotes different species of native and phosphorylated alpha and beta CTFs of lower molecular weights (based on Figs 3F and 5A in Kwart et al, (2019)). Lower panel shows reprobing for GAPDH.
- Quantification (mean ± SEM) of phospho‐C99/C99 CTFβ species, normalized to GAPDH and expressed relative to levels in 5xFAD mice. *P < 0.05 5xFAD versus 5xFAD/KO; ***P < 0.001 5xFAD versus 5xFAD/ΔDD and 5xFAD/C259A; #P < 0.05 5xFAD/C259A versus 5xFAD/KO; §P = 0.056 5xFAD/ΔDD versus 5xFAD/KO. N = 5 mice per group.
- Western blot analysis of soluble APP alpha (sAPPα) in the soluble Tris‐buffered saline fraction (see Materials and Methods) of hippocampal homogenate made from 9‐month‐old 5xFAD mice carrying different p75NTR alleles detected using 6E10 antibody (Table S1). Lower panel shows reprobing for GAPDH.
- Quantification (mean ± SEM) normalized to GAPDH and expressed relative to levels in 5xFAD mice. *P < 0.05 5xFAD versus 5xFAD/KO, 5xFAD/ΔDD and 5xFAD/C259A; #P < 0.05 5xFAD/C259A versus 5xFAD/KO; §P < 0.05 5xFAD/ΔDD versus 5xFAD/KO. N = 9 mice per group.
Source data are available online for this figure.
Figure EV2. Unaltered expression of full‐length APP, ADAM10, and BACE1 in hippocampus of 9‐month‐old 5xFAD mouse strains.
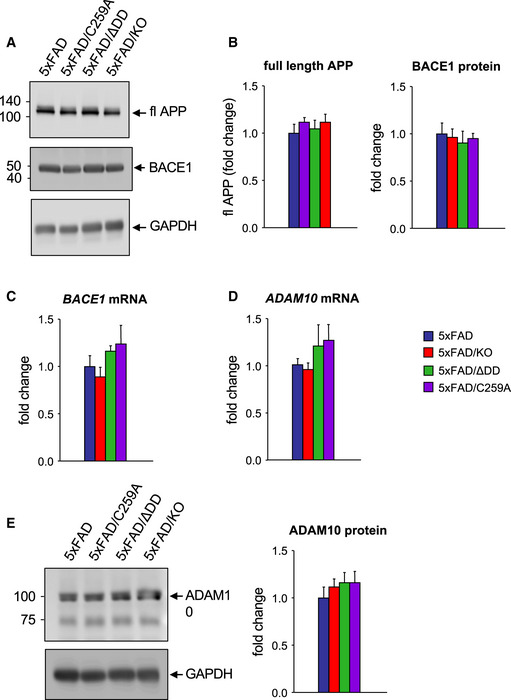
- Western blot analysis of full‐length APP (fl APP) and BACE1 expression (after reprobing of the same membrane) in hippocampal lysates of 5xFAD mice carrying different p75NTR alleles. Lower panel shows reprobing for GAPDH as control.
- Quantification showing mean ± SEM normalized to GAPDH expressed relative to 5xFAD levels. N = 6 mice per group.
- Expression of BACE mRNA in hippocampus of 5xFAD mice carrying different p75NTR alleles. N = 6.
- Expression of ADAM10 mRNA in hippocampus of 5xFAD mice carrying different p75NTR alleles assessed by qPCR, normalized to Gapdh mRNA, and expressed as fold change over 5xFAD. N = 6.
- Western blot analysis of ADAM10 expression in hippocampal lysates. ADAM10 precursor runs at ≈100kDa, processed ADAM10 at ≈75kDa. The lower panel shows reprobing for GAPDH. Quantification shown to the right normalized to GAPDH. N = 6 mice per group.
Reduced APP internalization in hippocampal neurons carrying inactive p75NTR variants
Realizing that the competing alpha‐ and beta‐cleavage pathways of APP processing are known to occur in different subcellular compartments, namely plasma membrane versus intracellular endosomes, respectively, we hypothesized that perhaps substrate availability, rather than overall substrate or secretase levels, may underlie the bias favoring non‐amyloidogenic APP processing in p75NTR mutant mice. Following this line of thought, we reasoned that changes in APP internalization could differentially affect the access of alpha‐ and beta‐secretases to APP. In order to test this idea, we established cultures of hippocampal neurons infected with a lentivirus expressing human APP (hAPP) carrying the 3 mutations found in 5xFAD mice. This afforded us a higher throughput of culture preparations, as well as allowing control of APP specificity in PLA experiments (see below). Live neuron cultures were fed with anti‐human APP antibodies on ice to label cell surface hAPP molecules. The steady‐state levels of surface hAPP were comparable among hippocampal neurons from all four p75NTR genotypes (Fig EV3A). Following antibody feeding, neuron cultures were rapidly washed and placed at 37°C for different periods of time to allow internalization (Fig EV3B). After internalization, cultures were quickly rinsed with low pH solution (acid wash) to eliminate any residual antibody from the cell surface (see Materials and Methods for details). We found striking differences in hAPP internalization among the different p75NTR genotypes (Fig 6A and B). In wild‐type neurons, maximal internalization of cell surface hAPP was close to 100% (Imax≈90%) and relatively fast (T1/2≈2 min) (Fig 6C). In p75NTR knock‐out neurons, overall APP internalization was lower (Imax≈75%) but of comparable speed (T1/2≈2.3 min) (Fig 6C). These result suggested that p75NTR is a positive regulator of APP internalization. Interestingly, maximal APP internalization was further reduced in hippocampal neurons derived from ΔDD and C259A knock‐in mice (Imax≈65% and 63%, respectively) and considerably slower than in wild‐type neurons (T1/2≈3.2 and 2.6 min, respectively) (Fig 6C). The fact that APP internalization was even lower in neurons expressing signaling‐deficient variants of p75NTR suggested that receptor activity contributes to the effects of p75NTR on APP internalization. In order to test this further, we assessed the consequences of enhancing p75NTR signaling through exogenous addition of ligand to cultures of wild‐type hippocampal neurons, reasoning that such treatment should have the opposite effect, namely, accelerate APP internalization. Indeed, we found that NGF significantly accelerated APP internalization in wild‐type hippocampal neurons (Fig 6D and E). In contrast, NGF treatment had no effect on the kinetics of APP internalization in either knock‐out, ΔDD or C259A p75NTR mutant neurons (Fig EV4A–F), further confirming that p75NTR signaling regulates APP internalization in neurons.
Figure EV3. Internalization of 5xFAD hAPP and p75NTR in wild‐type mouse hippocampal neurons.
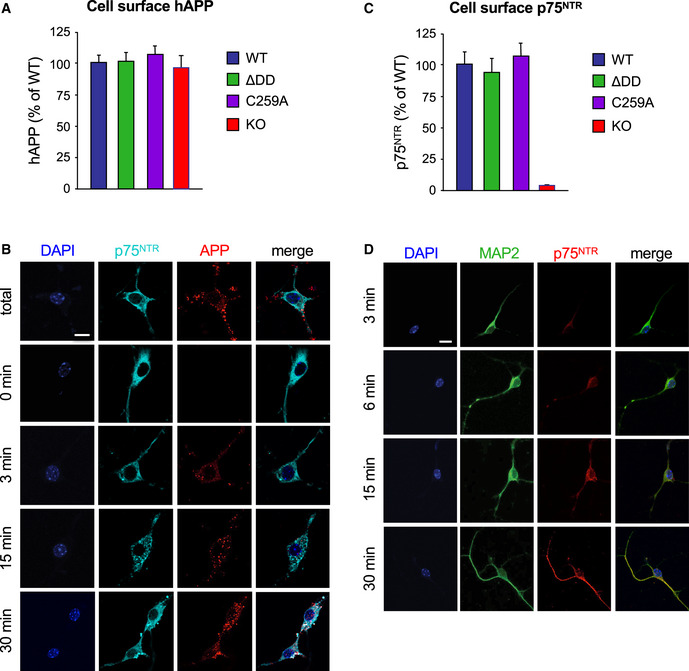
- Quantification of cell surface hAPP in wild type and p75NTR mutant neurons after 6E10 antibody feeding on ice followed by fixation. Values were normalized to levels in wild‐type neurons and are expressed as percentage ± SEM. N = 3 independent experiments.
- Internalization of 5xFAD hAPP in wild‐type mouse hippocampal neurons. Live neuron cultures were fed with anti‐human APP antibodies (6E10) on ice, washed, and then placed at 37°C for different periods of time to allow internalization. The reaction was stopped by a quick acid wash followed by fixation. Total staining (100%) was determined by direct fixation after antibody feeding. Baseline (t = 0 min) was obtained by acid wash directly after antibody feeding. Counterstaining for p75NTR (antibody GT15057, see Table S1) and DAPI is also shown. Scale bar, 10 μm.
- Quantification of cell surface p75NTR in wild type and mutant neurons after antibody feeding on ice followed by fixation. Values were normalized to levels in wild‐type neurons and are expressed as percentage ± SEM. N = 3 independent experiments.
- Internalization of p75NTR in wild‐type mouse hippocampal neurons. Live neuron cultures were fed with anti‐mouse p75NTR antibodies on ice, washed, and then placed at 37°C for different periods of time to allow internalization. Counterstaining for MAP2 and DAPI is also shown. Scale bar, 10 μm.
Figure 6. Reduced APP internalization in hippocampal neurons carrying signaling‐deficient p75NTR variants.
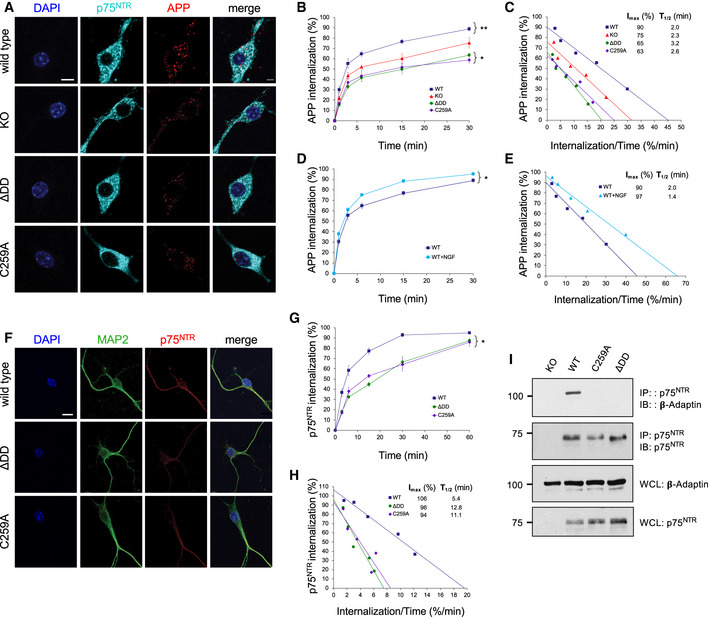
- Internalization of triple mutant hAPP in wild‐type, p75NTR knock‐out (KO), ΔDD, and C259A hippocampal neurons (15‐min time point). Counterstaining for p75NTR and DAPI is also shown. Scale bar, 10 μm.
- Kinetics of internalization of triple mutant hAPP in hippocampal neurons from wild‐type, p75NTR knock‐out (KO), ΔDD, and C259A mice. Shown is mean ± SEM of percentage internalization of total surface APP (set to 100%). N = 3 independent experiments each performed in duplicate; *P < 0.05 knock‐in versus KO mutants; **P < 0.01, WT versus all other genotypes (2‐way ANOVA).
- Linear transformation of hAPP internalization kinetics shown in (B). IMAX denotes maximal internalization in %. T1/2 denotes time for half maximal internalization in minutes.
- Kinetics of internalization of p75NTR in wild‐type, hippocampal neurons in the presence or absence of NGF. Shown is mean ± SEM of percentage internalization of total surface p75NTR (set to 100%). N = 3 independent experiments each performed in duplicate; *P < 0.05 ± NGF (2‐way ANOVA).
- Linear transformation of p75NTR internalization kinetics shown in (G). IMAX denotes maximal internalization in %. T1/2 denotes time for half maximal internalization in minutes.
- Internalization of endogenous p75NTR in wild‐type hippocampal neurons (30 min time point). Counterstaining for MAP2 and DAPI is also shown. Scale bar, 10 μm.
- Kinetics of internalization of p75NTR in hippocampal neurons from wild type, ΔDD, and C259A mice. Shown is mean ± SEM of percentage internalization of total surface p75NTR (set to 100%). N = 3 independent experiments each performed in duplicate; *P < 0.05 knock‐in mutants versus WT (2‐way ANOVA).
- Linear transformation of p75NTR internalization kinetics shown in (G). IMAX denotes maximal internalization in %. T1/2 denotes time for half maximal internalization in minutes.
- Co‐immunoprecipitation between p75NTR and β‐adaptin in hippocampal extracts from 6‐month‐old wild type and p75NTR mutant mice as indicated. Results shown are representative from 2 independent experiments. Molecular weights are in kDa. IP: immunoprecipitation; IB: immunoblotting; WCL, whole cell lysate.
Source data are available online for this figure.
Figure EV4. Internalization of 5xFAD hAPP in mouse hippocampal neurons from p75NTR mutant mice in the presence and absence of NGF.
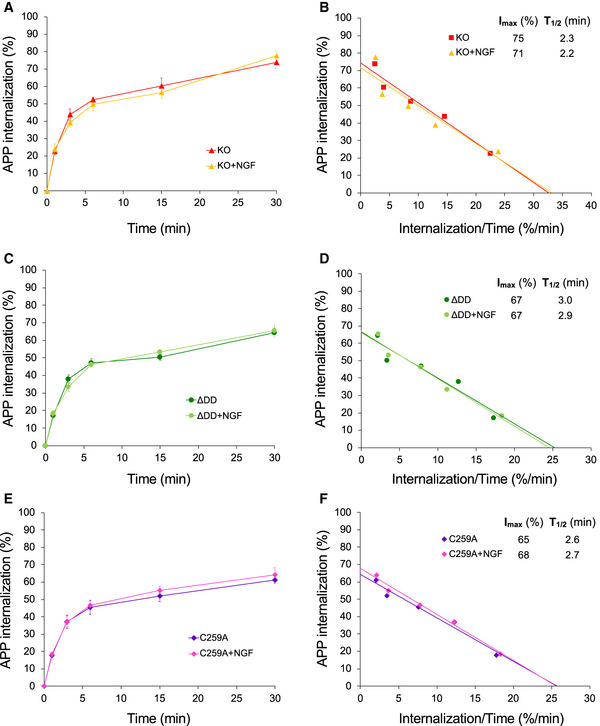
-
A, C, EInternalization of hAPP in hippocampal neurons from knock‐out (A) ΔDD (C) and C259A (E) mice. Shown are averages ± SEM of percentage internalization of total surface hAPP (set to 100%). N = 3 independent experiments each performed in duplicate.
-
B, D, FLinear transformation of hAPP internalization kinetics shown in panels (A, C, and E) respectively. IMAX denotes maximal internalization in %. T1/2 denotes time for half maximal internalization (in minutes).
To begin addressing how signaling‐deficient variants of p75NTR differentially affected APP internalization, we first evaluated the internalization of p75NTR itself in hippocampal neurons derived from wild type, ΔDD, and C259A mice, respectively, applying similar methodology as above, but using anti‐mouse p75NTR antibodies instead (Fig 6F). The steady‐state levels of p75NTR at the cell surface were comparable among hippocampal neurons from all three p75NTR genotypes (Fig EV3C). After antibody feeding, internalization was allowed to proceed at 37°C as before for up 60min (Fig EV3D). Maximal internalization of cell surface p75NTR was close to saturation for wild type as well as ΔDD and C259A variants (Imax≈106, 96 and 94%, respectively) (Fig 6G and H). However, ΔDD and C259A were internalized at much slower speeds, approximately half, compared to wild‐type p75NTR (T1/2≈12.8 and 11.1 min, respectively, compared to 5.4 min in wild‐type neurons) (Fig 6G and H). The slower internalization of signaling‐deficient p75NTR variants is in agreement with earlier studies in PC12 cells and sympathetic neurons showing that p75NTR internalization is enhanced by receptor activity (Bronfman et al, 2003; Saxena et al, 2004, 2005; Escudero et al, 2014). As neurotrophin‐mediated internalization of p75NTR has been shown to involve clathrin‐coated vesicles and recruitment of the AP‐2 adaptor (Deinhardt et al, 2007; Escudero et al, 2014), we investigated whether the reduced internalization of ΔDD and C259A p75NTR variants may have been due to inefficient recruitment of AP‐2 adaptor components, such as β‐adaptin. Indeed, co‐immunoprecipitation studies in hippocampal extracts showed no detectable recruitment of β‐adaptin by either ΔDD or C259A, in contrast to wild‐type p75NTR (Fig 6I), suggesting that these p75NTR variants internalize in a clathrin‐independent manner, perhaps from lipid rafts (Hibbert et al, 2006; Deinhardt et al, 2007).
APP and p75NTR interact and co‐internalize in hippocampal neurons
The slower internalization speeds of ΔDD and C259A p75NTR correlated with the overall lower levels of APP internalization observed in neurons expressing these variants, prompting us to consider whether ΔDD and C259A may affect APP internalization by holding up APP molecules through direct interaction. An earlier study had reported that APP and p75NTR can be co‐immunoprecipitated from transfected cells and total brain extracts (Fombonne et al, 2009), but did not use bait controls or knock‐out tissue, and left open the question of whether the two proteins can interact directly, in situ and under physiological conditions on the plasma membrane of living neurons. We first confirmed by co‐immunoprecipitation from hippocampal extracts that both ΔDD and C259A p75NTR could be recovered in APP immunoprecipitates from 5xFAD mice to a similar extent as wild‐type p75NTR (Fig 7A), suggesting that wild type and mutant p75NTR molecules interact with similar affinity with APP. No co‐immunoprecipitation was observed in the absence of either protein (Fig 7A). To assess whether the interaction between APP and p75NTR can be visualized directly on neurons, we used the proximity ligation assay (PLA) in fixed and permeabilized mouse hippocampal neurons infected with lentivirus expressing triple mutant hAPP (Fig 7B). Quantification of PLA signals over the entire cell revealed comparable levels of APP/p75NTR complexes in neurons expressing either wild type, ΔDD or C259A p75NTR (Fig 7C), further indicating that all three receptor variants are able to interact with APP to the same extent. We did not detect any PLA signals in wild‐type neurons that were not infected with hAPP‐expressing lentivirus, nor p75NTR knock‐out neurons expressing hAPP (Fig 7B and C). Having demonstrated that hAPP and p75NTR can interact directly in hippocampal neurons, we sought to determine whether the two molecules can also internalize together from the plasma membrane. To this end, we adapted the internalization assay to assess the trafficking from the cell surface of complexes labeled with both anti‐hAPP and anti‐mouse p75NTR antibodies (Fig 7C). Antibodies against p75NTR and APP were applied simultaneously to live neuron cultures on ice and, following a washing step, internalization was initiated by placing cultures at 37°C. A set of cultures was fixed directly without internalization to establish total PLA labeling at the plasma membrane. Quantification of cell surface PLA signals showed comparable steady‐state levels of APP/p75NTR complexes for all three p75NTR receptor variants (Fig 7D), in agreement with the results for the individual molecules (Fig EV3A and C). We note that normal steady‐state levels at the cell surface despite slower internalization would suggest a slower turnover of APP and p75NTR molecules in mutant neurons. After different times at 37°C, any residual antibodies on the cell surface were removed by a quick acid wash, and PLA reaction was performed on fixed cultures to visualize internalized APP/p75NTR complexes (Fig 7E). APP/p75NTR complexes internalized to a lower extent in neurons form p75NTR mutant mice compared to wild type (Fig 7F). The internalization kinetics of complexes between APP and wild‐type p75NTR was very similar to that of total APP, with Imax≈86% and T1/2≈2.2 min (Fig 7G). In contrast, internalization of APP/p75NTR complexes was markedly reduced, as well as slower, in neurons derived from ΔDD and C259A mice, with Imax≈68 and 66% and T1/2≈4.3 and 3.9 min, respectively (Fig 7G).
Figure 7. 5xFAD hAPP and p75NTR interact and co‐internalize in hippocampal neurons.
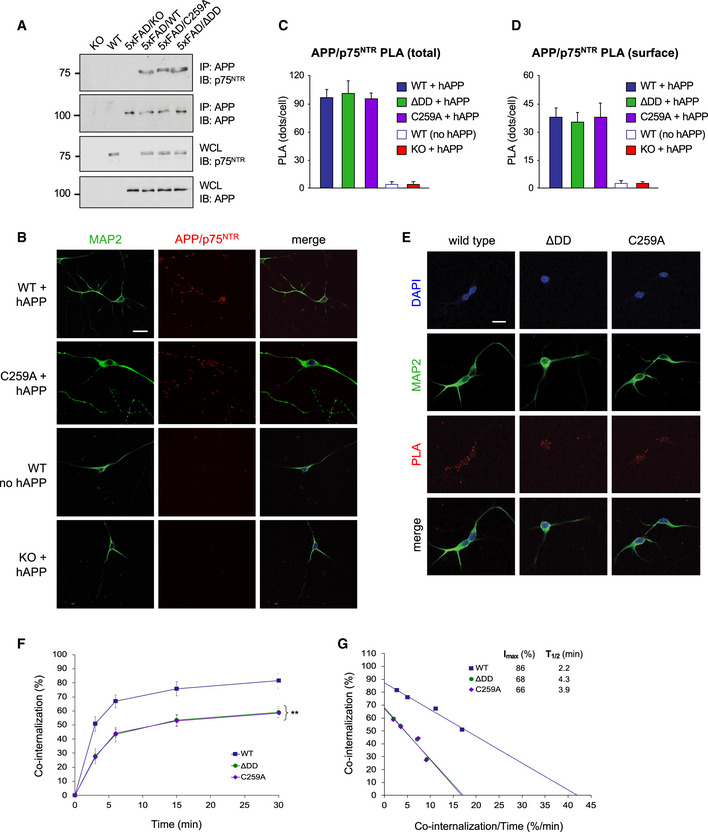
- Co‐immunoprecipitation between hAPP and p75NTR in hippocampal extracts from 6‐month‐old 5xFAD and p75NTR mutant mice as indicated. Results shown are representative from 3 independent experiments. Molecular weights are indicated in kDa. IP: immunoprecipitation; IB: immunoblotting; WCL, whole cell lysate.
- Proximity ligation assay (PLA) between triple mutant hAPP and p75NTR in hippocampal neurons from wild type and C259A mice (first and second rows, respectively). The third row (no APP) shows control PLA reaction in neurons that did not receive hAPP lentivirus. The fourth row (KO) shows control PLA reaction in neurons from p75NTR knock‐out mice infected with hAPP lentivirus. Counterstaining for MAP2 is also shown. Scale bar, 10 μm.
- Quantification of total PLA signals in wild type and p75NTR mutant neurons in the presence or absence of hAPP lentivirus, as indicated. Values were normalized to levels in wild‐type neurons and are expressed as mean PLA puncta per cell ± SEM from at least 25 neurons in 3 independent experiments.
- Quantification of cell surface PLA signals in wild type and p75NTR mutant neurons in the presence or absence of triple mutant hAPP lentivirus, as indicated. Live neuron cultures were fed with anti‐mouse p75NTR and anti‐hAPP antibodies on ice, washed after staining, fixed, and developed with PLA reaction. Values were normalized to levels in wild‐type neurons and are expressed as mean PLA puncta per cell ± SEM from at least 25 neurons in 3 independent experiments.
- Internalization of hAPP/p75NTR PLA signals in hippocampal neurons of wild type and p75NTR mutant mice. Live neuron cultures were fed with anti‐mouse p75NTR and anti‐hAPP antibodies on ice, washed, and plates placed at 37°C for different periods of time to allow internalization. Internalization was stopped by acid wash, followed by fixation and PLA reaction. Counterstaining for MAP2 and DAPI is also shown. Scale bar, 10 μm.
- Kinetics of co‐internalization of hAPP and p75NTR in hippocampal neurons from wild type, ΔDD, and C259A mice. Shown is mean ± SEM of percentage internalization of total surface PLA signal (set to 100%). N = 3 independent experiments each performed in duplicate; **P < 0.01 knock‐in mutants versus WT (2‐way ANOVA).
- Linear transformation of p75NTR internalization kinetics shown in (D).
Source data are available online for this figure.
Reduced APP trafficking to intracellular compartments containing BACE1 in neurons expressing ΔDD and C259A p75NTR alleles
We investigated whether decreased APP internalization in neurons expressing mutant ΔDD and C259A p75NTR alleles translated into reduced encounters between APP and BACE1 in intracellular compartments, the first step in the amyloidogenic pathway. To this end, we used structured illumination microscopy and deconvolution to assess the extent to which internalized APP was localized to endosomes containing BACE1 in hippocampal neurons expressing different p75NTR variants. Internalized hAPP was labeled as before by feeding live neuron cultures infected with triple mutant hAPP lentivirus with anti‐hAPP antibodies, then allowing this label to traffic intracellularly at 37°C. This was followed by acid wash and immunostaining for BACE1 (Fig 8A). As expected, we found significantly lower levels of BACE1 co‐localized with internalized hAPP in ΔDD and C259A mutant neurons compared to wild‐type neurons (Fig 8B), in agreement with the lower levels of internalized APP in the knock‐in mutants. Interestingly, the proportion of internalized APP that co‐localized with BACE1 was also significantly lower in mutant neurons (Fig 8C), suggesting altered intracellular trafficking of APP in neurons expressing ΔDD and C259A p75NTR variants.
Figure 8. Reduced 5xFAD hAPP trafficking to intracellular compartments containing BACE in neurons expressing ΔDD and C259A alleles.
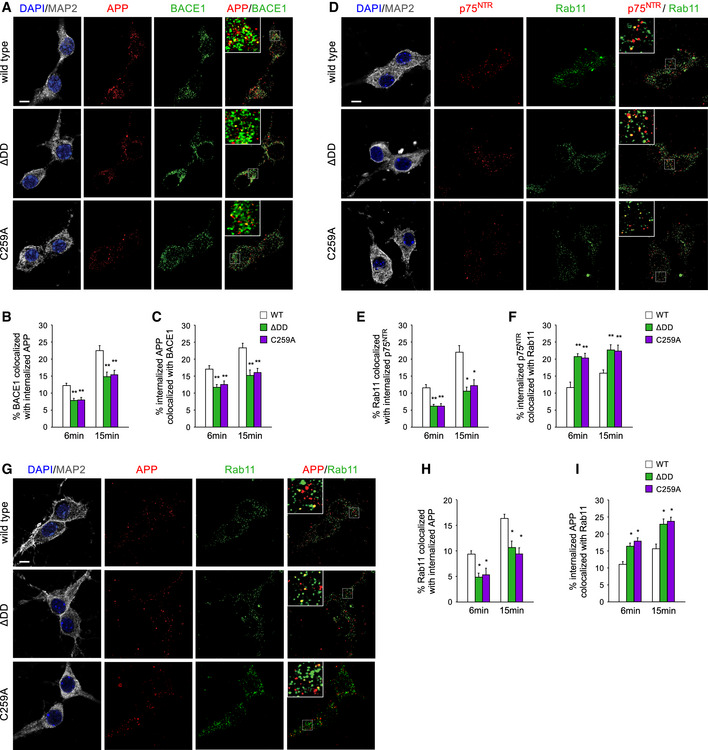
- Micrographs of triple mutant hAPP after 15‐min internalization (red), BACE1 immunocytochemistry (green), and their superimposition in wild‐type hippocampal neurons infected with triple mutant hAPP lentivirus. DAPI and MAP2 staining is shown in the left column. Insets show higher (3.5x) magnification of the areas indicated in the main images. Scale bar, 5 μm.
- Quantification of the proportion of BACE1 that co‐localized with hAPP after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % BACE co‐localized with hAPP. N = 3 independent experiments, each performed in duplicate; **P < 0.01 versus WT (one‐way ANOVA followed by post hoc test).
- Quantification of the proportion of internalized hAPP that co‐localized with BACE1 after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % internalized hAPP co‐localized with BACE1. N = 3 independent experiments, each performed in duplicate; *P < 0.05; **P < 0.01 versus WT (one‐way ANOVA followed by post hoc test).
- Micrographs of p75NTR after 15‐min internalization (red), Rab11 immunocytochemistry (green), and their superimposition in wild‐type hippocampal neurons. DAPI and MAP2 staining is shown in the left column. Insets show higher (3.5x) magnification of the areas indicated in the main merged images. Scale bar, 5 μm.
- Quantification of the proportion of Rab11 that co‐localized with p75NTR after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % Rab11 co‐localized with p75NTR. n = 4; *P < 0.05; **P < 0.01 versus WT (one‐way ANOVA followed by post hoc test).
- Quantification of the proportion of internalized p75NTR that co‐localized with Rab11 after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % internalized p75NTR co‐localized with Rab11. n = 4; **P < 0.01 versus WT (one‐way ANOVA followed by post hoc test).
- Micrographs of hAPP after 15‐min internalization (red), Rab11 immunocytochemistry (green), and their superimposition in wild‐type hippocampal neurons infected with triple mutant hAPP lentivirus. DAPI and MAP2 staining is shown in the left column. Insets show higher (3.5x) magnification of the areas indicated in the main merged images. Scale bar, 5 μm.
- Quantification of the proportion of Rab11 that co‐localized with hAPP after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % Rab11 co‐localized with hAPP. n = 4; *P < 0.05 versus WT (one‐way ANOVA followed by post hoc test).
- Quantification of the proportion of internalized hAPP that co‐localized with Rab11 after 6 and 15 min of internalization at 37°C. Results are expressed as mean ± SEM of % internalized hAPP co‐localized with Rab11. n = 4; *P < 0.05 versus WT (one‐way ANOVA followed by post hoc test).
In order to further elucidate how signaling‐deficient p75NTR variants affect APP intracellular trafficking, we considered the possibility that, in addition to internalize at slower rates, the mutant molecules may also show enhanced trafficking toward recycling endosomes, due to their inability to engage signaling pathways. It has been previously shown in cultured motor neurons that, while p75NTR constitutively internalizes and recycles back to the plasma membrane, ligand‐induced activation redirects a fraction of plasma membrane receptors away from the recycling pathway and toward early (Rab5+) endosomes for subsequent trafficking by axonal retrograde transport (Deinhardt et al, 2007). We tested this notion by assessing the co‐localization of internalized p75NTR molecules with Rab11, a marker of recycling endosomes, in hippocampal neurons from knock‐in mutants and wild‐type mice (Fig 8D). As expected, the proportion of Rab11 carriers that overlapped with internalized p75NTR was lower in ΔDD and C259A mutant neurons, in line with the overall lower levels of internalization of these variants (Fig 8E). However, a significantly larger fraction of internalized p75NTR molecules co‐localized with Rab11 in mutant ΔDD and C259A neurons compared to wild‐type neurons (Fig 8F), indicating a differential engagement with the recycling pathway. Importantly, a similar pattern was found when we investigated the co‐localization between internalized APP and Rab11 (Fig 8G–I). Hippocampal neurons carrying ΔDD and C259A p75NTR variants displayed a larger fraction of their internalized APP in Rab11 compartments compared to wild‐type neurons (Fig 8I), indicating increased APP trafficking to the recycling pathway in the mutant neurons. Intriguingly, the increased fraction of hAPP that co‐localized with Rab11 in ΔDD and C259A neurons was quantitatively similar to the reduction observed in hAPP/BACE1 co‐localization (compare Fig 8C and I), further supporting the notion of altered APP intracellular trafficking in hippocampal neurons carrying signaling‐deficient p75NTR variants.
Discussion
In this study, we describe a new pathway that directly and specifically regulates APP internalization, its amyloidogenic processing, and its impact on disease progression. We find that the death receptor p75NTR contributes to AD neuropathology by promoting internalization of APP to intracellular compartments containing BACE1. p75NTR interacts directly with APP in hippocampal neurons and enables higher levels of APP internalization compared to neurons lacking the receptor. A striking finding of this study, however, is the ability of signaling‐deficient variants of p75NTR to confer greater neuroprotection than the elimination of the receptor. This unexpected result can be explained by the significantly reduced internalization and bias toward the recycling pathway of these variants, which, through their interaction with APP, leads to reduced APP internalization, enhanced APP cleavage by alpha‐secretases, decreased APP trafficking to BACE1‐containing endosomes, and decreased cleavage by beta‐secretase. We note that ΔDD and C259A could also reduce Aβ plaque burden by other mechanisms, including clearance of Aβ peptides (Wang et al, 2011), but such mechanism would also be present in neurons expressing wild‐type p75NTR, and hence less likely to account for the differences observed here. Moreover, the reduced levels of CTFβ and increased levels of sAPPα observed in 5xFAD/ΔDD and 5xFAD/C259A mice suggest reduced amyloidogenic processing, rather than enhanced clearance, as the predominant mechanism of Aβ plaque reduction in these mutants.
Several reports have linked the intracellular accumulation of CTFβ to early neurodegenerative processes in AD (Lauritzen et al, 2012, 2016; Xu et al, 2016; Pera et al, 2017; Kwart et al, 2019). We found that the ΔDD and C259A alleles of p75NTR significantly reduced the accumulation of this fragment in the hippocampus of 5xFAD mice, which, together with decreased Aβ plaque formation, could also contribute to their neuroprotective effects. Interestingly, the levels of CTFβ and Aβ in the hippocampus of 5xFAD mice carrying different p75NTR alleles also correlated with the extent of APP internalization. This novel mechanism is independent of the ability of p75NTR to mediate some of the neurotoxic effects of Aβ oligomers, including cell death and neurite degeneration. On the other hand, these in vitro effects fail to explain both the time course and several aspects of the disease observed in AD patients and animal models.
Neurotrophin binding enhances p75NTR endocytosis in PC12 cells and sympathetic neurons (Bronfman et al, 2003; Saxena et al, 2004, 2005; Escudero et al, 2014), underlying the important role of receptor signaling in intracellular trafficking. It has also been shown that only ligand‐mediated internalization of p75NTR is clathrin‐dependent, while spontaneous receptor internalization and recycling takes place in a clathrin‐independent manner from lipid rafts (Deinhardt et al, 2007). In agreement with these observations, we found that p75NTR variants that cannot be activated by neurotrophins did not associate with components of the endocytic machinery that mediate clathrin‐dependent endocytosis, such as β‐adaptin, providing a possible rationale for their inefficient internalization. Moreover, ΔDD and C259A variants displayed greater association with the recycling pathway, a notion that is also in agreement with their signaling impairment. Strikingly, this resulted in a greater fraction of internalized APP localized to recycling compartments labeled by the small GTPase Rab11. We propose that the ΔDD and C259A variants of p75NTR affect APP intracellular trafficking by at least two mechanisms: On the one hand, they reduce APP internalization, and on the other hand, they redirect a fraction of the internalized APP to Rab11+ recycling endosomes. Intriguingly, it has been shown that BACE1 can traffic from early endosomes to Rab11+ endosomes for recycling to the plasma membrane, from where it can be internalized to replenish the early endosome pool (Chia et al, 2013; Buggia‐Prévot et al, 2014; Toh et al, 2018), currently believed to be the main site of BACE1/APP interaction and beta‐cleavage (Rajendran & Annaert, 2012; Toh & Gleeson, 2016). On the other hand, and unlike early endosomes, Rab11+ endosomes are not acidic, as they lack functional vacuolar ATPase (Gagescu et al, 2000; Schmidt & Haucke, 2007), and although both BACE1 and APP can traffic through the recycling pathway, beta‐cleavage is not believed to occur in Rab11+ endosomes. Our results showing redirection of a fraction of internalized APP from BACE1+ to Rab11+ endosomes in p75NTR mutant neurons would suggest that BACE1 traffics more rapidly through recycling endosomes, or else APP and BACE1 transit through different pools of Rab11+ endosomes in those cells. At any rate, these data support the idea that inactive p75NTR variants can alter the intracellular trafficking of APP in detriment to the amyloidogenic cleavage pathway. We suggest that the ability of p75NTR and APP to interact with each other can be seen as a kind of double‐edged sword: Active p75NTR enhances APP internalization and amyloidogenic processing, but, if inactive, it will reduce APP endocytosis and trafficking to BACE1+ endosomes. Interestingly, a recent paper presented evidence suggesting an interaction between p75NTR and BACE1 which resulted in increased co‐localization of APP and BACE1 in early endosomes (Saadipour et al, 2017), although it was not clear whether such interaction was direct or through APP. As hippocampal neurons produce neurotrophins endogenously, particularly BDNF, the reduced internalization and increased association with recycling endosomes of ΔDD and C259A variants are likely linked to their inability to signal in response to endogenous ligands. Taken to its logical conclusion, such notion has an interesting corollary: Through their effects on p75NTR internalization and trafficking, neurotrophins may enhance APP intracellular trafficking to BACE endosomes, and hence Aβ production. In agreement with this, we found that NGF treatment enhanced APP internalization in a p75NTR‐dependent manner in wild‐type hippocampal neurons.
The 5xFAD mouse model of AD displays enhanced and accelerated AD‐like neuropathology and is perhaps one of the most severe AD models in mice. Importantly, however, 5xFAD mice do not display any pathology that is not found in the AD patient population. Remarkably, we found a considerable reduction in the histopathology and nearly complete recovery of synaptic function and cognitive behavior in this rather strong model of AD after deletion of the p75NTR death domain or mutation of its transmembrane Cys259. We find quite striking that a single amino acid change in the mouse genome can have such dramatic effects on the course of this disease. The majority of previous efforts to identify small molecules targeting p75NTR have focused on the extracellular domain of the receptor, with the intent of either mimicking or inhibiting neurotrophin binding. This has proven difficult, due to the large interfaces involved. The results of the present study suggest that targeting the transmembrane domain of p75NTR may be a more promising strategy. We have recently provided proof‐of‐principle of this general concept in a recent report (Goh et al, 2018), paving the way for larger scale screenings of compound collections that may enable the discovery of substances mimicking the effects of the C259A mutation.
In summary, the results of the present study reveal an unexpected mechanism by which p75NTR affects the generation of neurotoxic APP fragments through its ability to interact with APP and regulate APP internalization and trafficking to intracellular compartments containing the amyloidogenic protease BACE. The fact that a single point mutation in a gene other than APP can have such strong neuroprotective effects in such severe model of AD highlights the importance of the transmembrane domain in the activation and function of p75NTR, and should encourage efforts to target this mechanism as a means to limit neurodegeneration in AD.
Materials and Methods
Animals
Mice were housed in a 12‐h light–dark cycle and fed a standard chow diet. The mouse lines utilized in this study have been described previously and are as follows: 5xFAD (Oakley et al, 2006); p75NTR exon 3 knock‐out (Lee et al, 1992); and ΔDD and C259A p75NTR knock‐in mice with deletion of the death domain or a Cys259Ala substitution, respectively (Tanaka et al, 2016). All strains were back‐crossed for at least 10 generations to a C57BL/6J background (considered as wild type). All animal procedures were approved by the National University of Singapore Institutional Animal Care and Use Committee.
Primary culture of cortical and hippocampal neurons
Pregnant female mice were euthanized on the 17th day of gestation by injection of sodium pentobarbital followed by cervical dislocation. Cerebral cortical or hippocampal structures were aseptically removed from the embryos and digested in Papain (Sigma‐Aldrich) for 30 min at 37°C and rinsed in neuronal maintenance media. Neurons were triturated into a single cell suspension, counted with a hemocytometer, and then transferred to coverslips coated with 0.01% poly‐D‐lysine (Sigma‐Aldrich) and 1 μg/ml mouse Cultrex® Laminin (R&D Systems). Cultured hippocampal neurons were maintained in serum‐free defined Neurobasal media supplemented with B27 (Invitrogen), GlutaMAX (Invitrogen), and 50 μg/ml gentamicin (Invitrogen) at 37°C in 5% CO2.
Lentivirus generation and transduction
Expression of human APP carrying the Swe, Flo, and Lon AD mutations in cultured hippocampal neurons was made through a lentiviral vector containing promoter sequences from the human Synapsin 1 gene and an EGFP marker gene separated by an IRES sequence (pESL‐hSyn1‐hAPP(Swe/Flo/Lon)‐IRES2‐EGFP). HEK293FT cells (Invitrogen) were maintained in OptiMEM with 5% fetal bovine serum and antibiotics (Invitrogen). Cells were co‐transfected with lentiviral constructs for overexpression with the packaging vectors Δ8.9 and VSV‐G (Addgene) using FuGENE®‐6 (Promega). Supernatants containing viral particles were aseptically collected 3 days post‐transfection, filtered using 0.4‐μm PES filters (Sartorius), concentrated 50–100 times, and dialyzed into sterile Dulbecco’s phosphate buffer saline (DPBS) by centrifugation in 100 kDA cut‐off Amicon Ultra‐15 centrifugal filters (Millipore). Viruses were aliquoted and snap‐frozen in liquid nitrogen. Viruses were titrated by serial dilution on primary dissociated cortical neurons and quantified for EGFP expression with a Ti‐E inverted fluorescence microscope. Lentiviral transduction of hippocampal neurons was performed after 1 day in vitro (DIV1) at a multiplicity of infection (MOI) = 5 and left for 24 h before the media was changed. The infected cultures were used at 5 days post‐infection.
Protein fractionation from mouse hippocampus and Aβ ELISA
For extraction of Aβ monomers, oligomers, and fibrils, we followed a fractionation protocol previously described (Sherman & Lesné, 2011) with modifications as follows. Dissected frozen mouse hippocampal tissue (9 months old) was weighed, thawed on ice, and homogenized using a manual Dounce homogenizer, in Tris‐buffered saline (25 mM Tris, 140 mM NaCl, pH 7.2–7.6) containing protease and phosphatase inhibitor (Nacalai Tesque) at a ratio of 1:9 (tissue:buffer, w/v). Homogenates were then ultracentrifuged (Himac CS150GXL, Hitachi) at 100,000 g for 1h at 4°C. The supernatant, which constitutes the soluble materials containing Aβ monomers, was collected as the Tris‐buffered saline fraction. The pellet of the Tris‐buffered saline fraction was resuspended in RIPA buffer (50 mM Tris, pH 8, 150 mM NaCl, 1% NP40, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors by trituration 10–15 times with a 1ml pipette followed by a 27G syringe needle. Samples were incubated at 4°C for 1h with agitation and ultracentrifuged at 100,000 g for another hour at 4°C. The supernatant constituted the RIPA fraction containing Aβ oligomers. The pellet of the RIPA fraction was dissolved in 70% formic acid (FA), and the homogenate was sonicated on ice for 30 s (VCX 130, Sonics & Materials Inc; amplitude 40%, 3 × 10 s of sonication). Sonicated samples were subjected to ultracentrifugation at 100,000 g for 30 min at 4°C. The supernatant was collected and brought to neutral pH using neutralization buffer (1 M Tris, 0.5 M Na2HPO4, 0.05% NaN3) at 1:20 dilution factor. These samples constituted the FA fraction containing Aβ fibrils and were stored at room temperature to avoid precipitation at lower temperature. The Tris‐buffered saline and RIPA fractions were stored at −20°C until further analysis. The content of Aβ1‐42 in the Tris‐buffered saline, RIPA, and FA fractions was assessed using the Human Aβ1‐42 enzyme‐linked immunosorbent assay (ELISA) Kit (Merck) according to the manufacturer’s instructions. The concentration of Aβ1‐42 was determined by the absorbance value detected using a microplate reader (BioTek Cytation 5, US) at 450 and 590 nm.
Western blotting
Tissue lysates and protein fractions were mixed with 5X sample buffer (250 mM Tris–HCl pH6.8, 10% SDS, 30% glycerol, 5% β‐mercaptoethanol, 0.02% bromophenol blue) and boiled at 95°C for 5 min before electrophoresis on polyacrylamide gels. 16.5% Tris‐Tricine gels (Bio‐Rad) were used to resolve APP CTF fragments. Proteins were blotted on polyvinylidene fluoride (PVDF) membranes (0.2 µm; Amersham). Membranes were blocked using 5% non‐fat milk (Bio‐Rad) in TBST (0.1% Tween‐20) and incubated overnight at 4°C with primary antibodies as listed in Table 1. Immunoreactivity was visualized using horseradish peroxidase (HRP)‐conjugated secondary antibodies at 1:10,000 dilution. Immunoblots were developed using the SignalFire ECL Reagent (Cell Signaling) or SuperSignal West Femto (Pierce) and exposed to CL‐Xposure Film (Thermo Fisher). Densitometric analysis of x‐ray films was done using ImageJ software (NIH).
Cell death and neurite length assays in response to Aβ
Aβ‐induced neurotoxicity has been shown to correlate with the extent of beta sheet structure in Aβ oligomers (Simmons et al, 1994). These were formed by 24‐h incubation at 37°C of a 1 mg/ml solution of Aβ 1–42 peptide (Sigma) in PBS. Aβ oligomers were added to cortical neuron cultures at different concentrations, and the cultures were maintained at 37°C in a CO2 atmosphere. Cell death was assessed after overnight incubation by the appearance of cleaved caspase‐3 positive neurons as detected by immunocytochemistry. Neurite length was assessed after 24‐h incubation by MAP2 immunostaining followed by image analysis using ImageJ software.
Internalization assay
Primary antibody incubation was performed using mouse 6E10 monoclonal antibody (BioLegend) against human Aβ diluted (1:200) in artificial cerebrospinal fluid (ACSF: 124 mM NaCl, 3.7 mM KCl, 1.0 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 24.6 mM NaHCO3, and 10mM D‐glucose) for 1 h at 4°C to label surface hAPP. For p75NTR internalization, GT15057 antibody (Neuromics) against the receptor extracellular domain was used instead. The cultures were then washed in ACSF and incubated at 37°C to allow internalization. At different time points, the internalization was stopped by quick wash in 70% formic acid (acid wash). Total staining (100%) was determined by direct fixation after antibody feeding, without acid wash. Baseline (t = 0 min) was obtained by acid wash directly after antibody feeding without 37°C incubation. The cultures were then fixed by addition of 4% PFA in PBS for 15 min at room temperature, washed with PBS, and incubated in blocking buffer for 30 min at room temperature. Labeled hAPP and p75NTR were detected by incubation with appropriate secondary antibodies at 1:2,000 dilution in blocking buffer for 1h at room temperature. Cells were washed three times with PBS and mounted in Flouromount. In all experiments, cells were visualized on a Leica SP8 confocal microscope.
Proximity ligation assay (PLA)
Hippocampal neurons were fixed for 15 min in 4% PFA, permeabilized, and blocked in 10% normal donkey serum and 0.3% Triton X‐100 in PBS. Cells were then incubated overnight at 4°C with rabbit anti‐p75NTR (1:200, AB1554), mouse anti‐human Aβ (6E10; 1:1,000), and chicken anti‐MAP2 (1:2,000) antibodies in PBS supplemented with 3% BSA. The Duolink In Situ Proximity Ligation kit (Sigma) was used as per the manufacturer’s instructions with fluorophore‐conjugated secondary antibody to recognize MAP2 (1:2,000) included during the amplification step. The cultures were imaged with a Leica SP8 confocal microscope to detect PLA signals. PLA puncta were quantified using ImageJ software. Internalization of PLA signals followed the protocol described in the previous section, except that live neuron cultures were simultaneously fed with antibodies against APP (6E10) and p75NTR (AB1554). MAP2 counterstaining was performed after internalization was completed and following fixation of the cultures.
Immunocytochemistry and immunohistochemistry
Hippocampal neurons cultured on coverslips were briefly washed in PBS, fixed for 15min in 4% paraformaldehyde solution (Sigma‐Aldrich), and blocked in PBS containing 0.2% gelatin and 0.25% Triton X‐100. Fixed cells were then incubated overnight at 4°C with the appropriate antibodies as listed in Table 1, followed by incubation in fluorophore‐conjugated secondary antibodies. Coverslips were mounted onto microscopy slides using fluoromount‐G (SouthernBiotech). Confocal laser scanning microscopy was performed on a Leica SP8 microscope. For immunohistochemistry, cryostat sections (30 μm) of mouse brain prepared as previously described were fixed 30 min at room temperature with 4% paraformaldehyde (PFA), washed in PBS, treated 10 min with 70% formic acid (when mouse 6E10 was used), and pre‐incubated in PBS containing 0.2% gelatin and 1% Triton X‐100 for 30 min. Sections were then processed for immunostaining by overnight incubation at 4°C with primary antibodies as indicated in Table 1, washed in PBS, and incubated in secondary donkey anti‐rabbit and anti‐mouse IgG antibodies conjugated with different Alexa FluoR (Invitrogen) diluted 1:2,000 in PBS. Sections were washed, mounted in Fluoromount, and examined with a Leica SP8 confocal microscope. Detection of mitochondrial superoxide was performed in acute brain slices (400 μm) from 6‐month‐old mice by incubation with 5 μM MitoSOX Red (Life Technologies) in ACSF for 10 min. Slices were then fixed with 4% paraformaldehyde at 4°C for 1 h. After washing and permeabilization in PBS, slices were further incubated with TO‐PRO‐3 Iodide (diluted in PBS, 1:2,000, Life Technologies) at RT for 10 min, rinsed, and mounted onto slides with Fluoromount. The slices were examined using a Leica SP8 confocal microscope with a 40× objective.
Image analysis
For each mouse, five brain coronal sections spaced 120 μm were quantified. In each brain section, mosaic images in hippocampus were captured. ImageJ software was used to quantify positive signal area for the different markers. For Aβ, GFAP, and Iba1, percentage of positive signal area was normalized to total area of hippocampus. For MitoSOX staining, mean fluorescence intensity in a fixed area within the hippocampal pyramidal cell layer was quantified. For RTN3, positive signal area within Aβ plaques was for quantification and expressed as percentage of total Aβ plaque area. For quantification of MitoSOX signal, stacks of 22 consecutive confocal images taken at 0.5‐μm intervals were acquired sequentially with two lasers (405 nm for TO‐PRO and 561 nm for MitoSOX). All parameters were held constant for all the sections. Four images covering the pyramidal layer visualized with TO‐PRO nuclear staining in CA1 hippocampal formation were captured for each mouse (three age‐matched mice per genotype). ImageJ software was used to quantify the mean fluorescence intensity in the pyramidal cell layer. Imaging for analyses of co‐localization between internalized APP or p75NTR with either BACE1 or Rab11 was performed on a Leica TCS SP8 X microscope equipped with structured illumination. Deconvolution was done using the Leica HyVolution module, which can resolve down to 130nm, under the supervision of a Leica engineer. Images were captured using a 63x/1.4NA oil immersion objective and 4X zoom. Ten super‐resolution images were acquired for each group. Co‐localization was analyzed using Fiji software (NIH Image).
Electrophysiology
Our electrophysiological procedures are described in greater detail in Shetty et al (Shetty et al, 2015). Briefly, mice were decapitated after anesthesia with CO2 and the brains were quickly removed into cold (4°C) artificial cerebrospinal fluid (ACSF: 124 mM NaCl, 3.7 mM KCl, 1.0 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 24.6 mM NaHCO3, and 10 mM d‐glucose) equilibrated with 95% O2/5% CO2 (carbogen; total consumption 16L/h). From each mouse, 6–8 transverse hippocampal slices (400 μm thick) were prepared from the right hippocampus by using a manual tissue chopper. Slices were incubated at 32°C in an interface chamber (Scientific System Design) at an ACSF flow rate of 1 ml/min. One monopolar, lacquer coated, stainless steel electrode (5 MΩ; AM Systems) was positioned at an adequate distance within the stratum radiatum of the CA1 region for stimulating synaptic inputs of one neuronal population, thus evoking field excitatory post‐synaptic potential (fEPSP) from Schaffer collateral‐commissural‐CA1 synapses. For recording, another electrode was placed in the CA1 apical dendritic layer. The signals were amplified by a differential amplifier (Model 1700, AM Systems) and were digitized using a CED 1401 analog‐to‐digital converter (Cambridge Electronic Design). After the pre‐incubation period of 2 h, an input–output curve (afferent stimulation versus fEPSP slope) was plotted prior to experiments. To set the test stimulus intensity, a fEPSP of 40% of its maximal amplitude was determined. Biphasic constant‐current pulses were used for stimulation. Late long‐term potentiation (L‐LTP) was induced using a theta‐burst stimulation (TBS) protocol which consists of 50 bursts (consisting of 4 stimuli) at an inter‐stimulus interval of 10 ms. The 50 bursts were applied over a period of 30s at 5 Hz (or at an inter‐burst interval of 200 ms). The slopes of fEPSPs were monitored online. The baseline was recorded for 30 min. For baseline recording and testing at each time point, four 0.2 Hz biphasic constant‐current pulses (0.1 ms/polarity) were used. fEPSPs were recorded every 5 min from 30 min before stimulation up to 240 min after stimulation across the CA1‐CA3 Schaffer collaterals and normalized against t = 0.
Behavior tests
The novel object recognition (NOR) test for mice consists of 3 days of exposure training, followed by a short‐term memory (STM) test 20min after training, a long‐term memory (LTM) test 24h later, and a remote memory test 2 weeks later. The objects are chosen based on similarities in dimensions and complexity. Tests are carried out in an acrylic box (20.32 × 40.5 × 16 cm LxWxH) that is sanitized, together with the objects, with 70% ethanol between each experiment. The time spent with an object includes direct visual orientation toward an object within half a body length of the object, sniffing, touching, or climbing on the object. The tests are video‐recorded, and preference scores are calculated as time spent with novel object minus time spent with familiar object divided by the total time spent with both objects. Positive scores indicate preferences for the novel object; negative scores show preferences for the familiar object.
For the Barnes maze spatial memory test, spatial cues were placed around the maze and these were kept constant throughout the study. On the first day of training, the mouse was placed in the escape box for 1 min. The animal was then placed in the center of the maze inside a black chamber. As in all subsequent sessions, the chamber was removed after 10 s, whereupon a buzzer (80 dB) and a light (400 lux) were turned on, and the mouse was free to explore the maze for 3 min or until the mouse entered the escape tunnel. The tunnel was always located underneath the same hole, which was randomly determined for each mouse. The platform was moved every day by 90° to avoid any odorant cue but the spatial cues and the tunnel position remained the same. Mice were trained using this protocol once daily for 4 days. For the test sessions, the escape tunnel was removed, and the mouse was allowed to freely explore the maze for a maximum of 3 min to assess spatial memory. The short‐term memory test was conducted 20–30 min after the first training. Long‐term and remote memory tests were conducted 24 h and 14 days after the last training, respectively. To quantify the preference for the trained target quadrant, time spent in the target quadrant and time spent in the other quadrants was measured.
Statistical analysis
Statistics analyses were performed using Prism 7 software (GraphPad, SPSS IBM corporation) and Microsoft Excel (Microsoft). Results are presented as mean ± standard error of the mean (SEM). Student’s t‐test, one‐way ANOVA, or two‐way ANOVA was performed to test statistical significance according to the requirements of the experiment. One‐way ANOVA followed by post hoc test was used for statistical analysis of immunohistopathology data. Behavioral data were analyzed using two‐way ANOVA with post hoc analysis. Statistical significance: *P < 0.05; **P < 0.01 and ***P < 0.001.
Author contributions
CY and KYG performed all experiments, except cell death assay, performed by KT, and electrophysiology, performed by LWW. CY and CFI designed the experiments. CFI and SS directed research. CFI wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2B
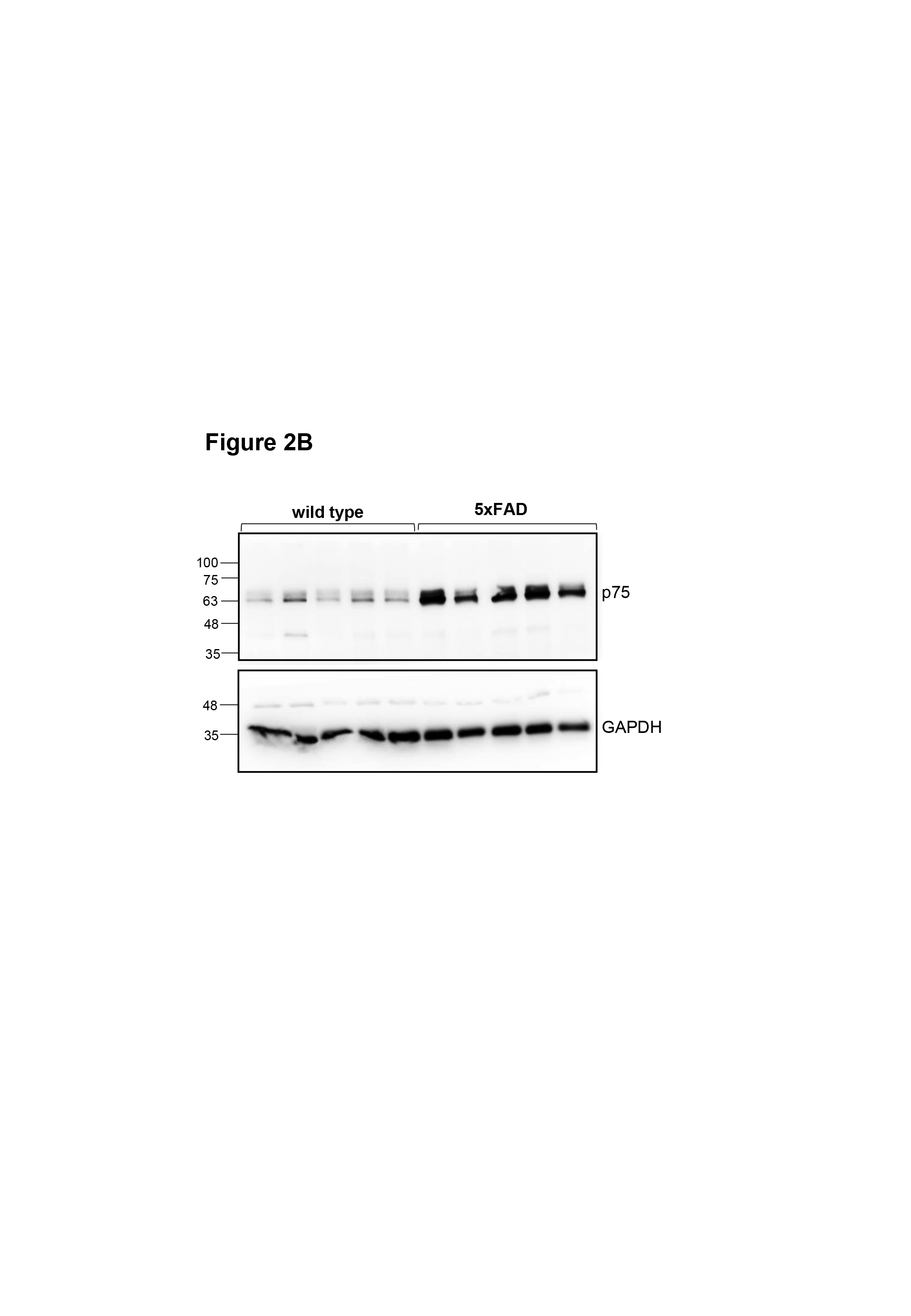{kind=link}
Source Data Figure 5A, C
{kind=link}
Source Data for Figure 6I
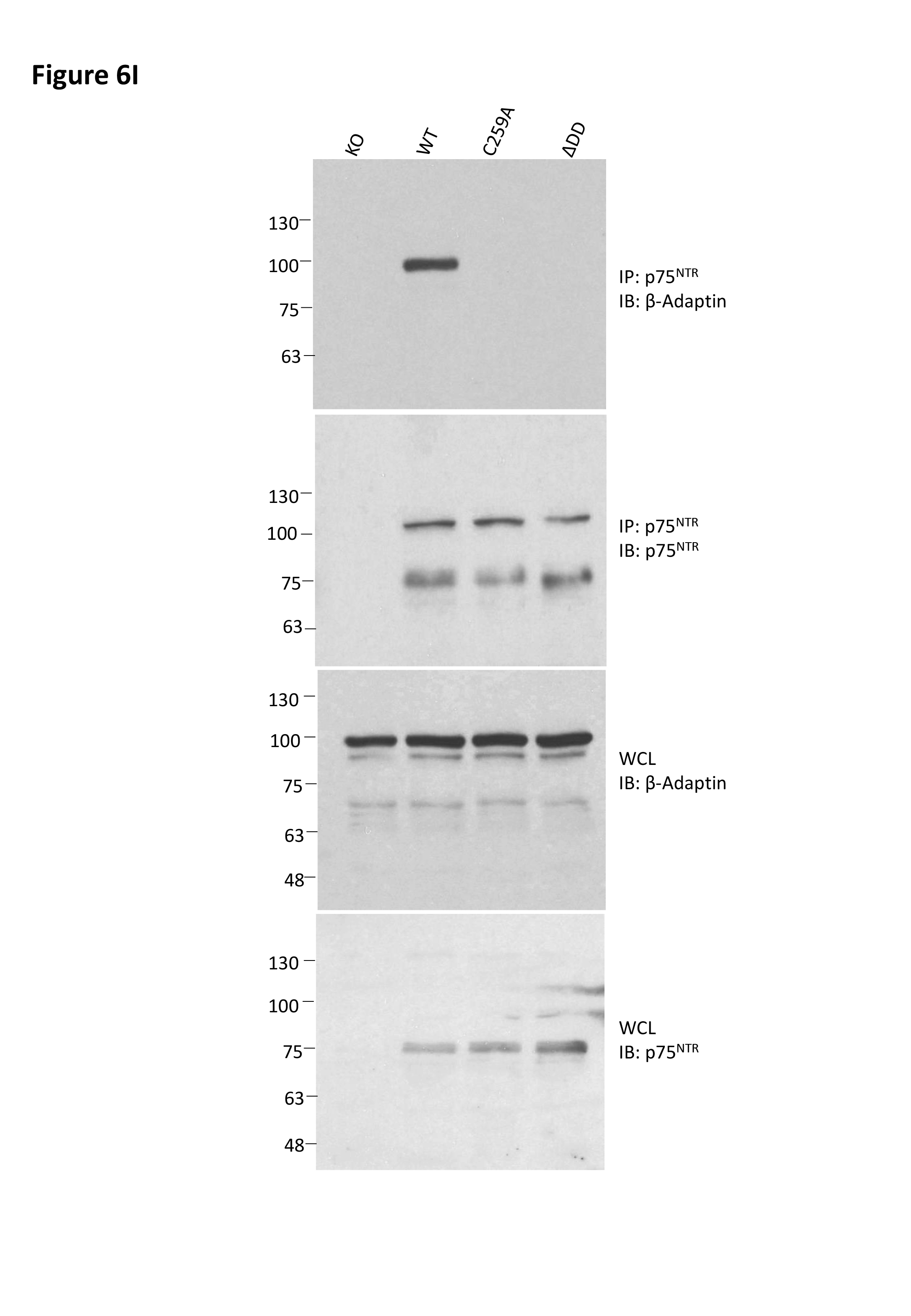{kind=link}
Source Data for Figure 7A
{kind=link}
Acknowledgements
We thank Peiyan Wong for assistance with behavioral studies, the Harbin Institute of Technology, Shenzhen and The First Affiliation Hospital of Guangzhou Medical University, Guangzhou, China, for access to microscopy facilities, Yi Rao for help with access to microscope facility, Jason Tann for help with lentivirus generation, and Eddie Koo, Gopal Thinakaran and Kika Bronfman for comments and critical reading of the manuscript. This research was funded by grants NMRC/CBRG/0107/2016 (to C.F.I.) and NMRC/OFIRG/0037/2017 (to S.S.) from the Singapore National Medical Research Council, MOE2018‐T2‐1‐129 (to C.F.I.) and MOE2017‐T3‐1‐002 (to S.S.) from Singapore Ministry of Education, ODPRT Strategic Programme Award (to C.F.I. and S.S.) and Aspiration Fund World Class (to C.F.I.) from the National University of Singapore, and VR‐2016‐01538 (to C.F.I.) from the Swedish Research Council.
The EMBO Journal (2021) 40: e104450.
Data availability
This study includes no data deposited in external repositories.
References
- Bronfman FC, Tcherpakov M, Jovin TM, Fainzilber M (2003) Ligand‐induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J Neurosci 23: 3209–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggia‐Prévot V, Fernandez CG, Riordan S, Vetrivel KS, Roseman J, Waters J, Bindokas VP, Vassar R, Thinakaran G (2014) Axonal BACE1 dynamics and targeting in hippocampal neurons: a role for Rab11 GTPase. Mol Neurodegener 9: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, Sisodia SS (1998) Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci 18: 9629–9637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RM, Balcz BA, Lopez‐Coviella I, Slack BE (2005) Inhibition of dynamin‐dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 6: 30–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy B, Gaudet C, Ménard M, Atkinson T, Brown L, Laferla FM, Armato U, Whitfield J (2010) Amyloid‐beta peptides stimulate the expression of the p75(NTR) neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J Alzheimers Dis 19: 915–925 [DOI] [PubMed] [Google Scholar]
- Chakravarthy B, Ménard M, Ito S, Gaudet C, Dal‐Pra I, Armato U, Whitfield J (2012) Hippocampal membrane‐associated p75NTR levels are increased in Alzheimer's disease. J Alzheimers Dis 30: 675–684 [DOI] [PubMed] [Google Scholar]
- Chia PZC, Toh WH, Sharples R, Gasnereau I, Hill AF, Gleeson PA (2013) Intracellular itinerary of internalised β‐secretase, BACE1, and its potential impact on β‐amyloid peptide biogenesis. Traffic 14: 997–1013 [DOI] [PubMed] [Google Scholar]
- Das U, Scott DA, Ganguly A, Koo EH, Tang Y, Roy S (2013) Activity‐induced convergence of APP and BACE‐1 in acidic microdomains via an endocytosis‐dependent pathway. Neuron 79: 447–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang L, Ganguly A, Saikia JM, Wagner SL, Koo EH, Roy S (2016) Visualizing APP and BACE‐1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19: 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, Reversi A, Berninghausen O, Hopkins CR, Schiavo G (2007) Neurotrophins Redirect p75NTR from a clathrin‐independent to a clathrin‐dependent endocytic pathway coupled to axonal transport. Traffic 8: 1736–1749 [DOI] [PubMed] [Google Scholar]
- Dikalov SI, Harrison DG (2014) Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal 20: 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Lindefors N, Chan‐Palay V, Persson H (1990) Cholinergic neurons of the nucleus basalis express elevated levels of nerve growth factor receptor mRNA in senile dementia of the Alzheimer type. Dementia 1: 138–145 [Google Scholar]
- Escudero CA, Lazo OM, Galleguillos C, Parraguez JI, Lopez‐Verrilli MA, Cabeza C, Leon L, Saeed U, Retamal C, Gonzalez A et al (2014) The p75 neurotrophin receptor evades the endolysosomal route in neuronal cells, favouring multivesicular bodies specialised for exosomal release. J Cell Sci 127: 1966–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE (2009) Selective vulnerability in Alzheimer's disease: amyloid precursor protein and p75(NTR) interaction. Annu Neurol 65: 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagescu R, Demaurex N, Parton RG, Hunziker W, Huber LA, Gruenberg J (2000) The recycling endosome of Madin‐Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol Biol Cell 11: 2775–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh ETH, Lin Z, Ahn BY, Lopes‐Rodrigues V, Dang N‐H, Salim S, Berger B, Dymock B, Senger DL, Ibáñez CF (2018) A small molecule targeting the transmembrane domain of death receptor p75NTRInduces melanoma cell death and reduces tumor growth. Cell Chem Biol 25: 1485–1494.e1485 [DOI] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2: a006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Lu Y, Qahwash I, Hu X‐Y, Chang A, Yan R (2004) Reticulon family members modulate BACE1 activity and amyloid‐beta peptide generation. Nat Med 10: 959–965 [DOI] [PubMed] [Google Scholar]
- Hibbert AP, Kramer BMR, Miller FD, Kaplan DR (2006) The localization, trafficking and retrograde transport of BDNF bound to p75NTR in sympathetic neurons. Mol Cell Neurosci 32: 387–402 [DOI] [PubMed] [Google Scholar]
- Hu X‐Y, Zhang H‐Y, Qin S, Xu H, Swaab DF, Zhou J‐N (2002) Increased p75(NTR) expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Exp Neurol 178: 104–111 [DOI] [PubMed] [Google Scholar]
- Hu X, Shi Q, Zhou X, He W, Yi H, Yin X, Gearing M, Levey A, Yan R (2007) Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J 26: 2755–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez CF, Simi A (2012) p75 neurotrophin receptor signaling in nervous system injury and degeneration: paradox and opportunity. Trends Neurosci 35: 431–440 [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10: 698–712 [DOI] [PubMed] [Google Scholar]
- Knowles JK, Rajadas J, Nguyen T‐VV, Yang T, LeMieux MC, Griend LV, Ishikawa C, Massa SM, Wyss‐Coray T, Longo FM (2009) The p75 neurotrophin receptor promotes amyloid‐beta(1–42)‐induced neuritic dystrophy in vitro and in vivo. J Neurosci 29: 10627–10637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch G, Di Lorenzo F, Bonnì S, Ponzo V, Caltagirone C, Martorana A (2012) Impaired LTP‐ but not LTD‐Like cortical plasticity in Alzheimer's disease patients. J Alzheimers Dis 31: 593–599 [DOI] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL (1994) Evidence that production and release of amyloid beta‐protein involves the endocytic pathway. J Biol Chem 269: 17386–17389 [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy E, Paquet D, Duffield M, Fak J, Olsen O, Darnell R, Tessier‐Lavigne M (2019) A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β‐CTFs, Not Aβ. Neuron 104: 256–270.e5 [DOI] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi‐Piquard R, Bauer C, Brigham E, Abraham J‐D, Ranaldi S, Fraser P, St‐George‐Hyslop P, Le Thuc O, Espin V et al (2012) The β‐secretase‐derived C‐terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple‐transgenic mouse hippocampus. J Neurosci 32: 16243–21655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I, Pardossi‐Piquard R, Bourgeois A, Pagnotta S, Biferi M‐G, Barkats M, Lacor P, Klein W, Bauer C, Checler F (2016) Intraneuronal aggregation of the β‐CTF fragment of APP (C99) induces Aβ‐independent lysosomal‐autophagic pathology. Acta Neuropathol 132: 257–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe A, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity Ngf receptor P75 leads to deficits in the peripheral sensory nervous‐system. Cell 69: 737–749 [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Ilag LL, Otting G, Ibáñez CF (1997) NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 16: 4999–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mango D, Saidi A, Cisale GY, Feligioni M, Corbo M, Nisticò R (2019) Targeting synaptic plasticity in experimental models of Alzheimer's disease. Front Pharmacol 10: 778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Kordower JH (1992) Cortical neurons express nerve growth factor receptors in advanced age and Alzheimer disease. Proc Natl Acad Sci USA 89: 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet‐Bongaarts A, Ohno M, Disterhoft J, Van Eldik L et al (2006) Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010) Amyloid‐beta‐induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13: 812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera M, Larrea D, Guardia‐Laguarta C, Montesinos J, Velasco KR, Agrawal RR, Xu Y, Chan RB, Di Paolo G, Mehler MF et al (2017) Increased localization of APP‐C99 in mitochondria‐associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J 36: 3356–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH (1999) Mutagenesis identifies new signals for beta‐amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem 274: 18851–18856 [DOI] [PubMed] [Google Scholar]
- Perini G, Della‐Bianca V, Politi V, Valle DG, Dal‐Pra I, Rossi F, Armato U(2002) Role of p75 neurotrophin receptor in the neurotoxicity by beta‐amyloid peptides and synergistic effect of inflammatory cytokines. J Exp Med 195: 907–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabizadeh S, Bitler CM, Butcher LL, Bredesen DE (1994) Expression of the low‐affinity nerve growth factor receptor enhances beta‐amyloid peptide toxicity. Proc Natl Acad Sci USA 91: 10703–10706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Annaert W (2012) Membrane trafficking pathways in Alzheimer's disease. Traffic 13: 759–770 [DOI] [PubMed] [Google Scholar]
- Saadipour K, Mañucat‐Tan NB, Lim Y, Keating DJ, Smith KS, Zhong J‐H, Liao H, Bobrovskaya L, Wang Y‐J, Chao MV et al (2017) p75 neurotrophin receptor interacts with and promotes BACE1 localization in endosomes aggravating amyloidogenesis. J Neurochem 144: 302–317 [DOI] [PubMed] [Google Scholar]
- Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B et al (2011) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci USA 108: E559–E568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Howe CL, Cosgaya JM, Hu M, Weis J, Kruttgen A (2004) Differences in the surface binding and endocytosis of neurotrophins by p75NTR. Mol Cell Neurosci 26: 292–307 [DOI] [PubMed] [Google Scholar]
- Saxena S, Howe CL, Cosgaya JM, Steiner P, Hirling H, Chan JR, Weis J, Kruttgen A (2005) Differential endocytic sorting of p75NTR and TrkA in response to NGF: a role for late endosomes in TrkA trafficking. Mol Cell Neurosci 28: 571–587 [DOI] [PubMed] [Google Scholar]
- Schmidt MR, Haucke V (2007) Recycling endosomes in neuronal membrane traffic. Biol Cell 99: 333–342 [DOI] [PubMed] [Google Scholar]
- Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M (2008) Flotillin‐dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci 28: 2874–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C(1996) The role of APP processing and trafficking pathways in the formation of amyloid beta‐protein. Ann N Y Acad Sci 777: 57–64 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789–791 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MA, Lesné SE (2011) Detecting aβ*56 oligomers in brain tissues. Methods Mol Biol 670: 45–56 [DOI] [PubMed] [Google Scholar]
- Shetty MS, Sharma M, Hui NS, Dasgupta A, Gopinadhan S, Sajikumar S (2015) Investigation of synaptic tagging/capture and cross‐capture using acute hippocampal slices from rodents. J Vis Exp 103: e53008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Hu X, Prior M, Yan R (2009) The occurrence of aging‐dependent reticulon 3 immunoreactive dystrophic neurites decreases cognitive function. J Neurosci 29: 5108–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN (1994) Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol 45: 373–379 [PubMed] [Google Scholar]
- Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ (2008) beta‐amyloid(1–42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 28: 3941–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH (2018) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62: 1403–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Kelly CE, Goh KY, Lim KB, Ibáñez CF (2016) Death domain signaling by disulfide‐linked dimers of the P75 neurotrophin receptor mediates neuronal death in the CNS. J Neurosci 36: 5587–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh WH, Gleeson PA (2016) Dysregulation of intracellular trafficking and endosomal sorting in Alzheimer's disease: controversies and unanswered questions. Biochem J 473: 1977–1993 [DOI] [PubMed] [Google Scholar]
- Toh WH, Chia PZC, Hossain MI, Gleeson PA (2018) GGA1 regulates signal‐dependent sorting of BACE1 to recycling endosomes, which moderates Aβ production. Mol Biol Cell 29: 191–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood CK, Coulson EJ (2008) The p75 neurotrophin receptor. Int J Biochem Cell Biol 40: 1664–1668 [DOI] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC (2009) The beta‐secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 29: 12787–12794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar M, Charalampopoulos I, Kenchappa RS, Simi A, Karaca E, Reversi A, Choi S, Bothwell M, Mingarro I, Friedman WJ et al (2009) Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide‐linked receptor dimers. Neuron 62: 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y‐J, Wang X, Lu J‐J, Li Q‐X, Gao C‐Y, Liu X‐H, Sun Y, Yang M, Lim Y, Evin G et al (2011) p75NTR regulates Abeta deposition by increasing Abeta production but inhibiting Abeta aggregation with its extracellular domain. J Neurosci 31: 2292–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Weissmiller AM, White JA, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S et al (2016) Amyloid precursor protein‐mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest 126: 1815–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA (1997) Binding of beta‐amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest 100: 2333–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2B
Source Data Figure 5A, C
Source Data for Figure 6I
Source Data for Figure 7A
Data Availability Statement
This study includes no data deposited in external repositories.