

Abstract
The DNA damage response (DDR) is a complex signaling network that relies on cascades of protein phosphorylation, which are initiated by three protein kinases of the family of PI3‐kinase‐related protein kinases (PIKKs): ATM, ATR, and DNA‐PK. ATM is missing or inactivated in the genome instability syndrome, ataxia‐telangiectasia (A‐T). The relative shares of these PIKKs in the response to genotoxic stress and the functional relationships among them are central questions in the genome stability field. We conducted a comprehensive phosphoproteomic analysis in human wild‐type and A‐T cells treated with the double‐strand break‐inducing chemical, neocarzinostatin, and validated the results with the targeted proteomic technique, selected reaction monitoring. We also matched our results with 34 published screens for DDR factors, creating a valuable resource for identifying strong candidates for novel DDR players. We uncovered fine‐tuned dynamics between the PIKKs following genotoxic stress, such as DNA‐PK‐dependent attenuation of ATM. In A‐T cells, partial compensation for ATM absence was provided by ATR and DNA‐PK, with distinct roles and kinetics. The results highlight intricate relationships between these PIKKs in the DDR.
Keywords: ataxia‐telangiectasia, ATM, DNA damage response, phosphoproteomics, PIKKs
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics;
Targeted proteomics via selected reaction monitoring defines the relative contributions of the PI3K‐related master kinases ATM, ATR and DNA‐PK to DNA damage response signaling in wild‐type and ataxia‐telangiectasia patient cells.

Introduction
The integrity of organismal DNA is constantly challenged by endogenous and exogenous DNA damaging agents. Cells respond to DNA damage by activating an elaborate signal transduction network termed the DNA damage response (DDR), which includes DNA repair pathways and many special circuits that temporarily modulate cellular metabolism until damage repair is completed (Chatterjee & Walker, 2017; Lanz et al, 2019). The DDR is vigorously activated by the highly cytotoxic DNA double‐strand breaks (DSBs; Goodarzi & Jeggo, 2013; Goldstein & Kastan, 2015). The apical transducers that orchestrate the DSB response are three serine‐threonine protein kinases, ataxia‐telangiectasia, mutated (ATM), ataxia‐telangiectasia and Rad3‐related (ATR) and DNA‐dependent protein kinase (DNA‐PK), which belong to a family of PI3‐kinase‐related protein kinases (PIKKs; Lovejoy & Cortez, 2009; Blackford & Jackson, 2017; Menolfi & Zha, 2019).
Double‐strand breaks incite activation of all three PIKKs, but ATM is considered the chief transducer of the DSB response multi‐branched network (Shiloh & Ziv, 2013; Paull, 2015). DNA‐PK’s most documented role is its coordination of DSB repair via nonhomologous end‐joining (NHEJ)—the major DSB repair pathway in mammalian cells (Davis et al, 2014; George et al, 2019). ATR has a prominent role in coordinating the cellular response to replication stress (Flynn & Zou, 2011; Saldivar et al, 2017). The three PIKKs maintain complex functional relationships in these responses to genotoxic stress (Blackford & Jackson, 2017). In view of the cardinal roles of these PIKKs in the DDR, their inhibitors are regarded as potential cancer drugs (O'Connor, 2015; Brown et al, 2017).
ATM, mutated null alleles cause the autosomal recessive genome instability syndrome, ataxia‐telangiectasia (A‐T), whose hallmarks are cerebellar degeneration, immunodeficiency, chronic lung disease, premature aging, chromosomal instability, cancer predisposition, and acute sensitivity to DSB‐inducing agents (Savitsky et al, 1995; Rothblum‐Oviatt et al, 2016). Null alleles of ATR cause embryonic lethality in homozygotes, and hypomorphic ATR mutations lead to the genome instability syndrome, ATR‐Seckel (O'Driscoll et al, 2003). Hypomorphic mutations in the PRKDC gene encoding the catalytic subunit of DNA‐PK lead to severe combined immunodeficiency (SCID; van der Burg et al, 2009; Woodbine et al, 2013). The crosstalk between the three PIKKs may affect the phenotypes associated with their corresponding mutations. For example, it has been suggested that ATR and/or DNA‐PK might partially compensate for ATM absence in A‐T patients (Tomimatsu et al, 2009; Shiloh & Ziv, 2013).
Previous phosphoproteomic surveys of the DDR have provided a broad overview of its numerous branches by identifying damage‐induced phosphorylations, many of which were subsequently studied in detail (Matsuoka et al, 2007; Bennetzen et al, 2010; Bensimon et al, 2010; Beli et al, 2012). In such studies, it is important to discriminate between PIKK‐mediated phosphorylations, which typically occur on the S/TQ motifs (Matsuoka et al, 2007; Blackford & Jackson, 2017) and PIKK‐dependent events, which can also occur on other motifs and be mediated by PIKK‐activated, downstream kinases. PIKK‐dependent events can be identified using PIKK inhibitors, which are constantly being improved. For example, our previous phosphoproteomic analysis of the DSB response (Bensimon et al, 2010), demonstrated that only 60% of DSB‐induced phosphorylations in a human melanoma cell line were ATM‐dependent, leaving the dependence of the rest in question. Information is still lacking about the division of labor between the three major PIKKs in the cellular response to genotoxic stress. The emergence of increasingly specific PIKK inhibitors (Leahy et al, 2004; Golding et al, 2009; Foote et al, 2013), coupled with technological advances in discovery phosphoproteomics (Junger & Aebersold, 2014), and targeted phosphoproteomics (Aebersold et al, 2016) provide now advanced experimental approach to this question.
We conducted a label‐free quantitative phosphoproteomic analysis after induction of DSBs or replication stress in the presence of inhibitors against each of the above three PIKKs. The experiments were carried out in WT and A‐T lymphoblasts. The results of this high‐throughput screen were substantiated using selected reaction monitoring (SRM)—a quantitative targeted proteomic technique with accuracy and reproducibility that are superior to those of non‐targeted proteomic approaches (Picotti & Aebersold, 2012; Picotti et al, 2013). This experimental setup enabled us to address several cardinal questions concerning PIKK regulation and crosstalk in response to DNA damage, one of which is PIKK redundancy and possible compensation for the absence of one by the other two. We show that, while some ATM‐dependent phosphorylations are indeed missing in A‐T cells, others are taken over by ATR or DNA‐PK. ATR‐ and DNA‐PK‐dependent compensation for ATM absence occurs in distinct kinetics and involves different subsets of ATM substrates. Interestingly, ATR/DNA‐PK compensation for chemical inhibition of ATM is more modest than such compensation when ATM is absent altogether. We also obtained evidence for DNA‐PK‐dependent attenuation of ATM’s response to DSBs, in agreement with a previous suggestion of such mechanism. Our results demonstrate fine‐tuned relationships among the three PIKKs and present them as a coordinated functional module at the core of the cellular response to genotoxic stress. Comparison of our results with an extensive meta‐analysis of 34 published screens highlights proteins with repeated appearance as strong candidates for novel DDR factors.
Results
Dissection of phosphoproteome dynamics in response to DSBs
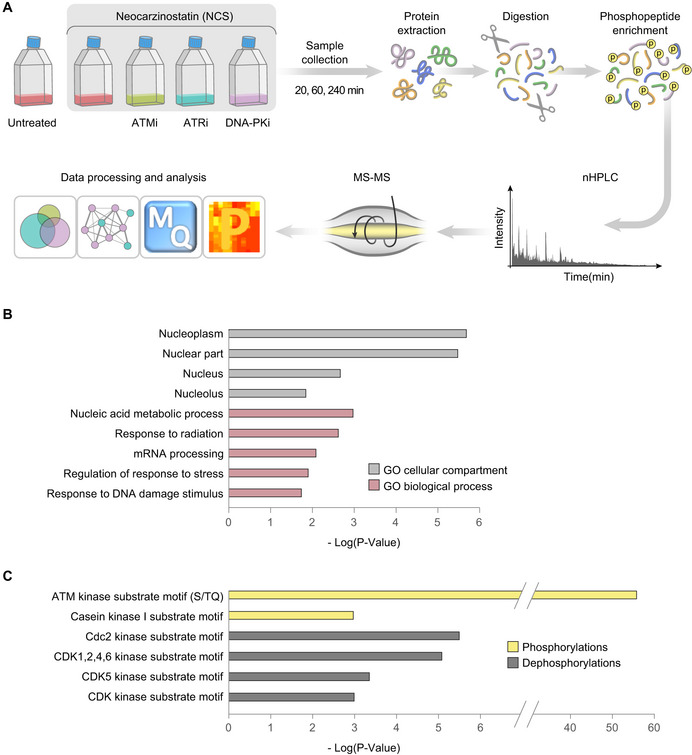
Global quantitative phosphoproteomic analysis (Fig 1A) was carried out in human lymphoblast lines, NL‐550 (derived from a healthy individual) and AT59RM (derived from an A‐T patient). Cells were treated with the radiomimetic drug, neocarzinostatin (NCS), in the presence of selective inhibitors against ATM (KU60019, denoted hereafter ATMi; Golding et al, 2009), ATR (AZ20, ATRi; Foote et al, 2013), and DNA‐PK (NU7441, DNA‐PKi) (Leahy et al, 2004; Fig EV1). Samples were collected 20, 60, and 240 min after NCS addition and subjected to the phosphoproteomic analysis pipeline shown in Fig 1A.
Figure 1. Modulation of the cellular phosphoproteome following NCS treatment.
-
ASchematic workflow of the phosphoproteomic experiment. Cells were treated with 20 ng/ml NCS in the presence of selective inhibitors of ATM (KU60019, 5 µM), ATR (AZ20, 0.5 µM), DNA‐PK (NU7441, 5 µM), or DMSO (inhibitor solvent). Inhibitors were added 30 min prior to NCS treatment, and samples were collected 20, 60, and 240 min following NCS addition. Protein extracts were digested into peptides and subsequently enriched for phosphopeptides, which were measured using LC‐MS/MS and subjected to label‐free quantification and subsequent data processing and analysis using the MaxQuant (Cox & Mann, 2008; Tyanova et al, 2016a) and the Perseus (Tyanova et al, 2016b) software. An FDR of 0.05 together with a minimal fold change of twofold was applied to determine regulation in response to NCS and PIKK inhibitors.
-
BEnriched GO cellular compartments (gray) among NCS‐induced phosphorylations and dephosphorylations, and GO biological processes (purple‐red) among NCS‐induced phosphorylations, in WT cells. Enrichment was tested using the Fisher exact test implemented in Perseus (Tyanova et al, 2016b).
-
CEnriched motifs among NCS‐induced phosphorylations (yellow) and dephosphorylations (gray) in WT cells. Enrichment was tested using the Fisher exact test implemented in Perseus (Tyanova et al, 2016b).
Figure EV1. Dose‐dependent action of PIKK inhibitors.
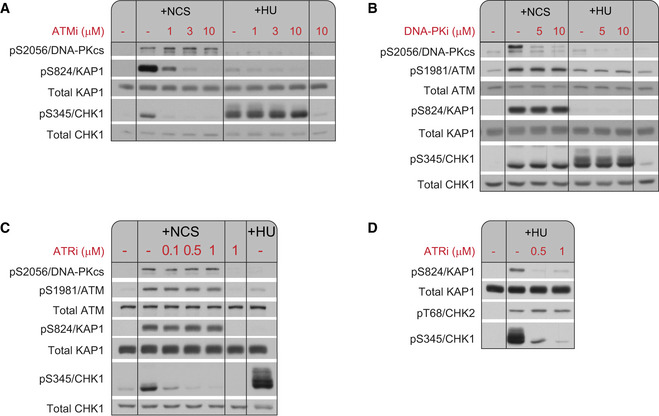
Western blots of WT (NL‐550) cells treated for 1 h with neocarzinostatin (NCS; 20 ng/ml) or hydroxyurea (HU; 1 mM) in the presence of the indicated inhibitors. The inhibitors were added to the cultures 30 min prior to the addition of NCS or HU.
-
AATMi inhibited ATM in a dose‐dependent manner, as demonstrated by the phosphorylation of S824/KAP‐1.
-
BDNA‐PKi inhibited DNA‐PK in a dose‐dependent manner, as demonstrated by the autophosphorylation of S2056/DNA‐PKcs.
-
C, DATRi inhibited ATR in a dose‐dependent manner, as demonstrated by HU‐induced phosphorylation of S345/CHK1.
We identified a total of 9,690 phosphopeptides corresponding to 2,818 proteins. 1,322 phosphopeptides were modulated in response to NCS in WT cells, out of which 598 represented phosphorylations and 724 represented dephosphorylations in at least one time point (Table EV1, Appendix Fig S1). Of note, some of the phosphorylations identified in this study were well‐documented DNA damage‐responsive sites, such as pS343/NBS1 (Gatei et al, 2000), pS824/KAP‐1 (Ziv et al, 2006), pS3205/DNA‐PK (Douglas et al, 2002), pS831/TP53BP1 (Jowsey et al, 2007), pS139/H2AX (Rogakou et al, 1998), pS183/PPM1G (Beli et al, 2012), pS272/RAD9 (Chen et al, 2001), pS635/RAD50 (Linding et al, 2007), pS317/CHK1 (Zhao & Piwnica‐Worms, 2001), pS114/PNKP (Segal‐Raz et al, 2011), and pS1083/SMC3 (Luo et al, 2008).
Initial gene‐ontology (GO) analysis showed that NCS‐responsive phosphorylation and dephosphorylation targets were highly enriched for nuclear compartment proteins. NCS‐induced phosphorylations were also enriched for DNA damage‐related biological processes and mRNA processing players (Fig 1B). In order to identify possible involvement of protein kinases other than the PIKKs in the DSB response, we performed linear motif analysis using the Perseus software (Tyanova et al, 2016b; Fig 1C). The most significantly enriched phosphorylation motif was S/TQ—the common PIKK target motif (FDR q‐value: 4*10−56). Nevertheless, this motif was included in only 21% of NCS‐induced phosphorylation sites, in agreement with previous studies (Bensimon et al, 2010), implicating other kinases in this phosphorylation cascade. Notable enriched motifs among phosphorylated sites were those of casein kinases 1 (FDR q‐value: 1.1*10−3). Dephosphorylated sites were enriched for motifs recognized by cell cycle kinases, among them CDC2 and CDKs (FDR q‐value: 3.1*10−6 and 8.2*10−6, respectively). This result probably further unfolds the robust regulation of the damaged‐induced cell cycle checkpoints via protein phosphorylation (Shaltiel et al, 2015).
Identification of strong, novel DDR factor candidates
Many high‐throughput functional and proteomic screens were carried out over the past two decades aimed at discovering key players in the DDR. The wealth of information obtained through these screens can be further amplified if the data of these screens are collated, allowing prioritization of candidate DDR factors for in‐depth functional studies in view of their repeated appearance in screens. We constructed a database that contains manually curated information provided by the data of 34 such screens (Kolas et al, 2007; Matsuoka et al, 2007; Stokes et al, 2007; Lovejoy et al, 2009; Paulsen et al, 2009; Bennetzen et al, 2010; Bensimon et al, 2010; Chou et al, 2010; Hurov et al, 2010; O'Connell et al, 2010; O'Donnell et al, 2010; Piwko et al, 2010; Smogorzewska et al, 2010; Cotta‐Ramusino et al, 2011; Kondo & Perrimon, 2011; Menzel et al, 2011; Stirling et al, 2011; Adamson et al, 2012; Beli et al, 2012; Floyd et al, 2013; Jungmichel et al, 2013; Sirbu et al, 2013; Benzina et al, 2015; Boucas et al, 2015; Elia et al, 2015; Herr et al, 2015; Izhar et al, 2015; Kavanaugh et al, 2015; Raschle et al, 2015; Boeing et al, 2016; Kozlov et al, 2016; Lopez‐Saavedra et al, 2016; Baranes‐Bachar et al, 2018; Olivieri et al, 2020). Queries to this database provide a “profile” for a candidate protein according to the screens in which it was identified. Repeated appearance in various types of screens strongly suggests the involvement of the candidate protein in central DDR pathways. Using this approach, we recently identified the DDR roles of the RNA processing factor, PABPN1 (Gavish‐Izakson et al, 2018) and the proteasome chaperone, ubiquilin 4 (Jachimowicz et al, 2019).
We matched the list of phosphorylation/dephosphorylation targets obtained in our screen with this database and obtained such profiles for our hits (Table EV2). We expect this analysis to make our data an interesting and useful resource for investigators interested in identifying new players in the DDR arena using the candidate approach.
PIKK control of phosphoproteome dynamics in response to DSBs
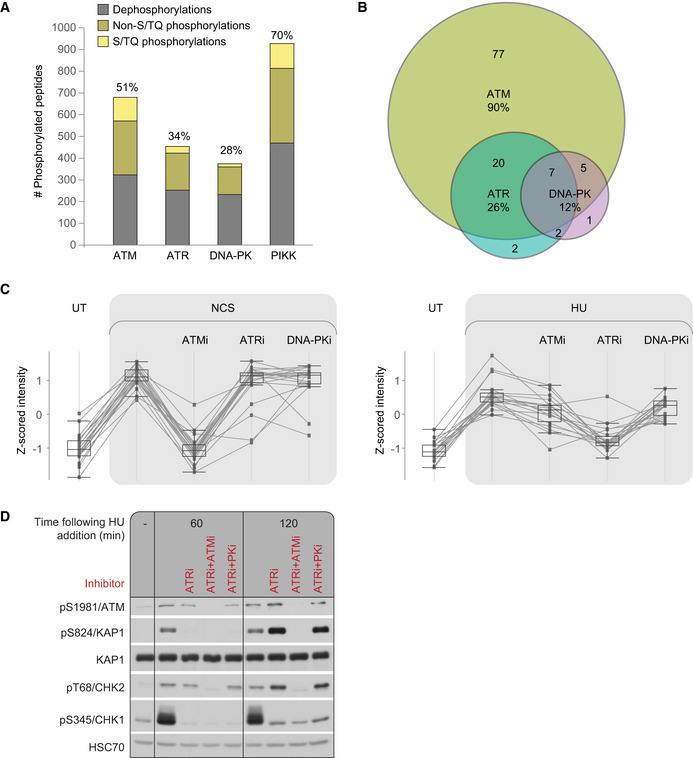
Using PIKK‐directed chemical inhibitors, we determined the relative share of ATM, ATR and DNA‐PK in NCS‐induced phosphoproteome dynamics (Table EV1). Jointly, the three PIKKs governed 70% of the NCS‐response (Fig 2A). Fifty‐one percent of the NCS‐induced changes in the phosphoproteome were ATM‐dependent, roughly similar to a previous study from our lab (Bensimon et al, 2010). ATRi and DNA‐PKi influenced 34% and 28% of NCS‐induced phosphoproteome alterations, respectively. Only 25% of the PIKK‐dependent phosphorylations following NCS treatment occurred on the S/TQ motif, indicating that a large portion of the PIKK‐dependent effect on the phosphoproteome was likely indirect. Notably, there are documented PIKK‐mediated phosphorylations which are not on the PIKK‐canonical substrate motif, predominantly by DNA‐PK, suggesting that some of the non‐S/TQ sites might still be direct PIKK targets (Lees‐Miller & Meek, 2003; Jette & Lees‐Miller, 2015). Ninety‐four percent of NCS‐induced phosphorylations on the S/TQ motif were PIKK‐dependent, and some of these phosphorylations were modulated by more than one PIKK. Remarkably, 90% of them were ATM‐dependent and only 26% were ATR‐dependent and 12%—DNA‐PK‐dependent (Fig 2B), pointing to the central role of ATM in direct damage‐induced phosphorylations.
Figure 2. PIKK dependencies of DNA damage‐induced dynamics of the phosphoproteome in WT cells.
-
ARelative share of the three PIKKs in NCS‐responsive phosphorylations.
-
BVenn diagram depicting the relative share of the three PIKKs in NCS‐induced phosphorylations occurring on S/TQ sites.
-
CThe profile of a group of substrates targeted by ATM 20 min after neocarzinostatin (NCS) addition and by ATR 1 h after hydroxyurea (HU) addition. Box plots depict 20 phosphopeptides measured in two independent biological replicates. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend to the maximum and minimum values which are not farther than 1.5 times the interquartile range (IQR).
-
DWestern blotting analysis confirming that the ATM substrate, pS824/KAP1 is targeted by ATR in response to HU. pS345/CHK1—an established ATR substrate—served as a positive control for ATR activation.
Source data are available online for this figure.
PIKK involvement in the cellular response to replication stress
ATM is regarded as the major transducer of the DSB response while ATR is considered a major coordinator of the response to replication stress (Flynn & Zou, 2011). We compared the share of the load by the three PIKKs in the response to DSBs and the response to replication stress, which we induced using hydroxyurea (HU) treatment. NL‐550 human lymphoblastoid cells were treated with HU for 60 min in the presence or absence of PIKK inhibitors and subjected to a similar processing and analysis as in the NCS‐experiment (Fig 1A). Interestingly, ATM‐dependent phosphorylation sites identified in the previous experiment, 20 min after NCS treatment, were enriched for ATR‐dependent sites that we subsequently found to be phosphorylated in response to HU (enrichment factor 3.1, FDR q‐value: 4.9*10−8; Fig 2C, Table EV3). This set of substrates was enriched for the S/TQ motif (enrichment factor 4.7, FDR q‐value: 2.7*10−5), presumably representing direct ATM/ATR‐mediated phosphorylations. For example, Western blotting analysis confirmed that 1 h after HU treatment, pS824/KAP‐1, which is ATM‐dependent following NCS treatment, was ATR‐dependent (Fig 2D). ATM‐dependent pT68/CHK2, which is typical for the DSB response (Zhou et al, 2000), was still ATM‐ rather than ATR‐dependent in response to HU, possibly representing a response to DSBs that follow replication fork collapse. Interestingly, when ATR was inhibited, pS824/KAP‐1 and pT68/CHK2 showed ATM‐dependent hyper phosphorylation 2 h following HU treatment, which might stem from ATM activation due to collapsed replication forks.
Elevated response of ATM to DSBs upon DNA‐PK inhibition
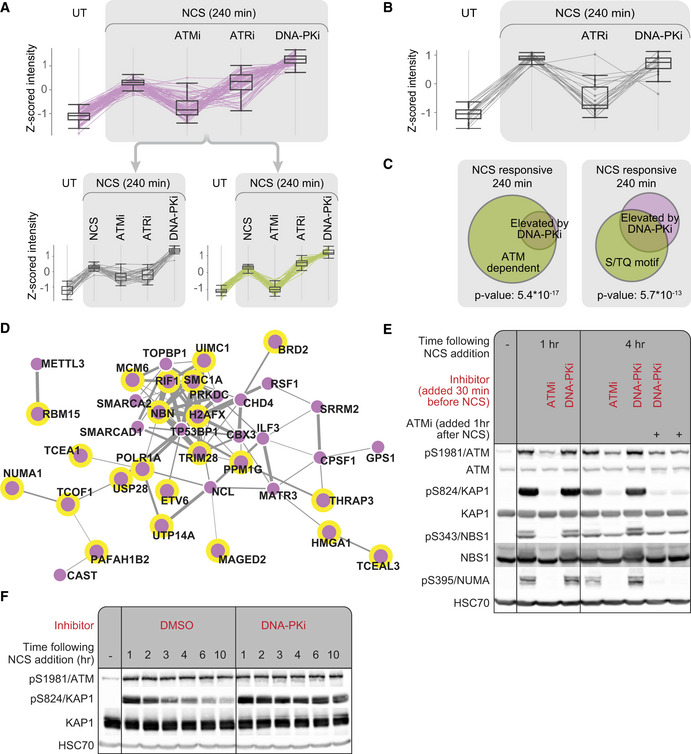
Our attention was caught by a subset of NCS‐induced phosphorylations that displayed a unique pattern: 4 h after NCS treatment, their level was further elevated in WT cells treated with DNA‐PKi compared with cells that were not treated with this inhibitor (Fig 3A, Table EV4). Importantly, this phenomenon was not observed in A‐T cells, suggesting it represented ATM‐dependent phosphorylations (Fig 3B). We used K‐means clustering to divide this cluster into two sub‐clusters, with strong or moderate ATM dependence (Fig 3A, bottom two clusters).
Figure 3. Chemical inhibition of DNA‐PK enhances ATM‐mediated phosphorylations in WT cells.
-
AA cluster of 71 neocarzinostatin (NCS)‐induced phosphorylations that were enhanced upon continuous inhibition of DNA‐PK. Untreated cells are marked UT. This cluster breaks down into two distinct groups according to their ATM dependence 240 min after NCS addition. Clusters were obtained using K‐means algorithm implemented in Perseus on Z‐scored intensities. Box plots depict 71 phosphopeptides measured in two independent biological replicates. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend from the box to the maximum and minimum values which are not farther than 1.5 times the interquartile range (IQR).
-
BDepicted are phosphopeptides from (A) that responded to NCS treatment in A‐T cells. No significant elevation was observed in these phosphorylations in A‐T cells following continuous inhibition of DNA‐PK. Box plots depict 19 phosphopeptides measured in two independent biological replicates. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend from the box to the maximum and minimum values which are not farther than 1.5 time the interquartile range (IQR).
-
CThe cluster depicted in (A) was enriched for ATM‐dependent phosphorylations and the S/TQ phosphorylation motifs. Enrichment was tested using the Fisher exact test implemented in Perseus (Tyanova et al, 2016b).
-
DSTRING‐Network representation of the proteins in the same cluster. Proteins phosphorylated on the S/TQ motif are marked with a yellow border, and the thickness of connecting lines represents the combined score for interaction confidence according to STRING.
-
EWestern blotting analysis of selected phosphorylations in the above cluster showing DNA‐PKi‐induced elevation.
-
FTemporal dynamics of DNA‐PK‐dependent attenuation of pS824/KAP‐1.
Source data are available online for this figure.
To further substantiate the ATM‐dependent nature of this cluster’s elevated phosphorylations, we examined its enrichment for sites that we previously found to be ATM‐dependent. Indeed, 94% of the phosphorylations in this cluster were ATM‐dependent in at least one of the time points after NCS treatment (enrichment FDR q‐value: 5.4*10−17; Fig 3C). Using linear motif analysis, we found that the S/TQ motif was highly enriched in this subset (48% of the phosphorylations, enrichment FDR q‐value: 5.7*10−13; Fig 3C). Interestingly, 46% of the proteins included in this subset were annotated as involved in DNA or chromatin binding (GO terms: DNA binding, chromatin binding, histone binding; enrichment FDR q‐value: 1.8*10−5; Table EV4). Notably, some of the sites whose phosphorylation was enhanced by DNA‐PKi were found on well‐documented DDR players; e.g., H2AX, NBS1, TOPBP1, KAP‐1, SMC1, and RIF1 (Fig 3D). Further validation of this pattern was carried out in NL‐550 and U2‐OS cells focusing on three ATM targets in this group, pS824/KAP‐1, pS343/NBS1, and pS395/NUMA (Vidi et al, 2014; Salvador Moreno et al, 2019). For this purpose, we used Western blotting analysis based on phospho‐specific antibodies directed against these sites (Figs 3E and EV2). Collectively, these results suggest that DNA‐PK might negatively regulate ATM or positively regulate a factor that counteracts ATM's activity. Alternatively, continuous inhibition of DNA‐PK, which has a central role in the NHEJ repair pathway (Davis & Chen, 2013), might lead to accumulation of unrepaired DSBs and thus enhance ATM‐mediated signaling.
Figure EV2. DNA‐PKi elevates the ATM response in U2‐OS cells.
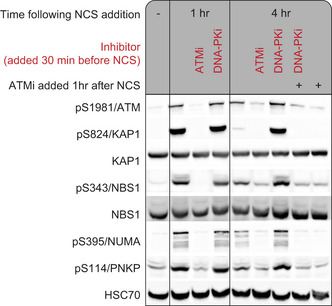
Western blotting analysis of selected phosphorylations showing their DNA‐PKi‐dependent elevation 4 h after neocarzinostatin (NCS) addition.
We examined the temporal dynamics of this phenomenon by monitoring the S824/KAP‐1 phosphorylation site. This phosphorylation in DNA‐PKi‐treated cells reached its usual peak level but did not exhibit its subsequent normal decline (Fig 3F). Furthermore, when in the same experimental setup we added ATMi after these phosphorylations had reached their peak level, one hr following NCS addition, the phosphorylations rapidly declined, suggesting that ATM continues to phosphorylate these sites at the time they normally decay (Fig 3E). Moreover, a combination of pretreatment with DNA‐PKi and treatment with ATMi one hr after NCS addition, showed that DNA‐PKi‐dependent continuous phosphorylation of the tested substrates depended on sustained ATM activity (Fig 3E).
ATR and DNA‐PK partially compensate for ATM's absence in A‐T cells
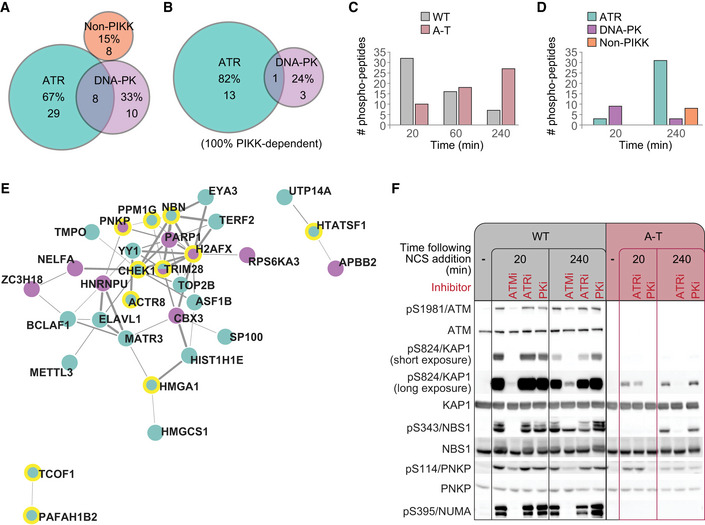
ATM, ATR, and DNA‐PK share redundant roles in the DDR while keeping their own non‐redundant functions. It has been suggested that complete loss of one of them, such as that of ATM in A‐T patients, might be partly compensated for by one or both the other two (Shiloh & Ziv, 2013). We examined this possibility by carrying out our experiments concomitantly in WT and A‐T cells. While 259 of the 314 strictly ATM‐dependent sites in WT cells did not respond to NCS treatment in A‐T cells, 55 did respond (Table EV5). Since these phosphorylations were strictly ATM‐dependent in WT cells, their occurrence in A‐T cells presumably reflects the action of other kinases. Indeed, 85% of the “compensated sites” in A‐T cells were ATR‐ or DNA‐PK‐dependent, of which 67% were ATR‐dependent and 33% DNA‐PK‐dependent (Fig 4A). Importantly, the exclusively ATM‐dependent S/TQ sites represented a more prominent role for ATR in their phosphorylation in A‐T cells, where 82% of them depended on ATR (Fig 4B). Interestingly, however, the kinetics of the “compensated site” phosphorylation was slower in A‐T cells than in WT cells (Fig 4C).
Figure 4. PIKK‐dependent compensation for ATM absence in A‐T cells.
-
AA subset of exclusive ATM‐dependent sites in WT cells, which responded to neocarzinostatin (NCS) also in A‐T cells. Their dependencies in A‐T cells are shown.
-
BDependencies of S/TQ phosphorylations within the same subset.
-
CThe first time points at which “compensated sites” from (A) responded to NCS in WT and A‐T cells are presented.
-
DTemporal kinase dependencies of the “compensated sites” in A‐T cells. Shown are the numbers of "compensated sites" in A‐T cells, which depended on each kinase at the 20 and 240 min time points. The early ones depend mainly on DNA‐PK, while the later ones—on ATR.
-
ESTRING‐Network representation of the proteins that were included in the early, DNA‐PK‐dependent compensation (purple) or the late, ATR‐dependent compensation (green). Proteins phosphorylated on the S/TQ motif are highlighted by yellow margins. The thickness of connecting lines represents the combined score for interaction confidence according to STRING.
-
FWestern blotting analysis confirming high‐throughput results for selected ATM substrates.
We examined whether the extent of compensation by ATR and DNA‐PK varied over a time course by following the PIKK dependence of the “compensated sites” at 20 and 240 min after NCS addition. The early and late phases of the compensation for ATM absence showed distinct profiles (Fig 4D). The early phase was largely DNA‐PK‐dependent and included some well‐documented ATM substrates, such as pS139/H2AX, pS824/KAP‐1, and pS114/PNKP (Fig 4D and E). The late phase was largely ATR‐dependent, encompassed a larger number of targets, and also included known ATM substrates such as pS343/NBS1, pS824/KAP‐1, pS183/PPM1G, and pS317/CHK1 (Fig 4D and E). Western blotting analysis confirmed the pattern: pS824/KAP‐1 and pS114/PNKP showed early compensation, which was DNA‐PK‐dependent, while pS824/KAP‐1 and pS343/NBS1 showed late compensation, which was ATR‐dependent (Fig 4F). This analysis also confirmed that some substrates, such as pS395/NUMA, were not compensated for ATM absence. Similar compensation patterns by ATR/DNA‐PK were observed in an additional A‐T cell line (L‐119; Appendix Fig S2). Taken together, our results demonstrate that some phosphorylation targets that depend exclusively on ATM in WT cells can be targeted in A‐T cells by DNA‐PK or ATR. Of these, very few can be compensated in A‐T cells as early as 20 min following NCS treatment, and these phosphorylations are largely DNA‐PK‐dependent. Late‐phase compensation is more substantial and is largely ATR‐dependent.
Compensation for chemical inhibition of ATM in WT cells
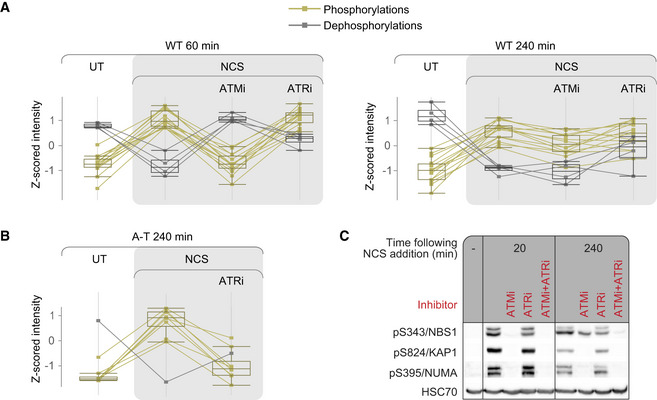
Since a certain degree of compensation for ATM’s absence was observed in A‐T cells, we asked whether this phenomenon could also be observed in WT cells treated with an ATM inhibitor. We first asked whether certain early ATM‐dependent substrates had lost their responsiveness to ATM inhibitor at a late phase of the DSB response. Such phenomenon could represent ATR/DNA‐PK‐dependent compensation. We identified a group of 18 exclusively ATM‐dependent sites, which were ATM‐dependent 1 h after NCS addition, and were still NCS‐responsive at 4 h, but lost their responsiveness to ATMi at that time point (Fig 5A, Table EV6). Since they were not affected by ATRi nor DNA‐PKi at any time point, we asked whether some of them might be compensated by one or two of these kinases only upon inhibition of ATM. We speculated that overlapping this group of sites with the one compensated by ATR/DNA‐PK in A‐T cells might direct us to the compensating kinase. This group was enriched for substrates that were compensated by ATR in A‐T cells (enrichment factor: 15.8, FDR q‐value: 5.3*10−10, against a background of the WT NCS‐responsive phosphoproteome; Fig 5B). Interestingly, this group was also enriched for substrates that were ATR‐dependent following HU treatment (enrichment factor: 9.4, FDR q‐value: 1.8*10−5, against a background of the WT NCS‐responsive phosphoproteome). Since this group was enriched for substrates that could be targeted by ATR in different situations (lack of ATM in A‐T cells or replication stress in WT cells), we surmised that these sites represented ATR‐dependent compensation under ATM inhibition.
Figure 5. Compensation for chemical inhibition of ATM in WT cells.
-
AA group of exclusively ATM‐dependent sites that were ATM‐dependent 1 hr after NCS addition and were still phosphorylated or dephosphorylated 4 h after treatment, but were not affected by ATMi at that time point. Box plots depict 18 phosphopeptides measured in two independent biological replicates. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend from the box to the maximum and minimum values which are not farther than 1.5 times the interquartile range (IQR).
-
BThis group was enriched for sites that were compensated in an ATR‐dependent manner in A‐T cells. Box plots depict nine phosphopeptides measured in two independent biological replicates. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend from the box to the maximum and minimum values which are not farther than 1.5 times the interquartile range (IQR).
-
CWestern blotting analysis of pS343/NBS1, which is compensated upon ATM inhibition, and pS824/ KAP‐1 and pS395/NUMA, which are not compensated under these conditions.
Using Western blotting analysis and double inhibition of ATM and ATR, we found that pS343/NBS1, which was included in the group of sites that were compensated upon ATM inhibition, was indeed ATM‐dependent at the beginning of the response, but ATMi’s effect on its phosphorylation was largely lost at the later phase of the DSB response (Fig 5C). Nevertheless, it did not demonstrate strong ATR dependency at this time point, and only concomitant inhibition of ATM and ATR led to complete loss of its phosphorylation. We therefore suggest that this phosphorylation is ATR‐dependent only upon ATM inhibition. Notably, this ATR‐mediated compensation did not depend on NCS dose (Appendix Fig S3). It should also be noted that this pattern was unique to a subset of ATM substrates that was smaller than the one compensated for in A‐T cells, and was not demonstrated by ATM targets such as pS824/KAP‐1 or pS395/NUMA (Fig 5C), although pS824/KAP‐1 did show compensation in A‐T cells (Fig 4F).
SRM confirms the patterns identified by shotgun proteomics
Our shotgun phosphoproteomic analysis revealed novel PIKK targets and various patterns of PIKK‐dependent phosphorylation in response to genotoxic stresses. Results of high‐throughput proteomic analysis are usually validated by testing the dynamics of individual proteins or PTMs using Western blotting analysis, as we did for some phosphorylation targets. However, this validation strategy is highly restricted by the availability of high quality phospho‐specific antibodies against the corresponding sites. SRM is a quantitative mass‐spectrometric technology that enables the validation of putative phosphorylations identified using high‐throughput approaches, without the need for substrate‐specific antibodies. This targeted proteomic technique provides mass‐spectrometric measurement of predefined peptides with accuracy and reproducibility superior to shotgun proteomics (Picotti & Aebersold, 2012; Picotti et al, 2013). In order to validate specific phosphorylations and phosphorylation patterns using SRM, we carried out a second, independent large‐scale experiment in which we treated WT (NL‐550) and A‐T (AT59RM) cells with NCS in the presence of PIKK inhibitors, including combinations of two inhibitors, and collected samples at 20 and 240 min following NCS addition. Protein extracts were digested into peptides and enriched for phosphopeptides, and samples were spiked‐in with specific heavy peptides to enable accurate quantification. We applied this approach to 99 phosphopeptides—including known DDR targets and control irrelevant sites that should not be modulated in response to DNA damage (Table EV7). Importantly, the SRM results were in agreement with the shotgun data, concerning both the phosphorylation of individual proteins and the dynamic patterns reflected in the shotgun experiments.
Thirty‐eight sites were modulated in response to NCS in WT cells (Table EV8). Seven of them were hyper‐phosphorylated following long inhibition of DNA‐PK, among them pS9/HMGA1, pS183/PPM1G, pS824/KAP1, pS243/ACINUS, and pS3205/DNA‐PKcs (Fig 6A, C, D, E and F). Out of 28 sites that were exclusively ATM‐dependent in WT cells, 17 were NCS‐responsive in A‐T cells as well. Of these, two showed early‐phase compensation for ATM absence in A‐T cells, which was DNA‐PK‐dependent, with pS824/KAP1 being a prominent example (Fig 6D). Thirteen sites demonstrated late‐phase compensation in A‐T cells, which was ATR‐dependent, among them p140/RAP80, pS183/PPM1G, and pS824/KAP1 (Fig 6B–D). The compensated sites typically reached lower intensity in A‐T cells compared with WT cells. Of note, 11 sites that responded to NCS in an exclusively ATM‐dependent manner in WT cells did not respond at all in A‐T cells, as exemplified by pS243/ACINUS and pS3205/DNA‐PK (Fig 6E and F). Some of the substrates that were compensated by ATR in A‐T cells, such as p140/RAP80, also exhibited ATR‐dependent compensation in WT cells upon ATM inhibition, which was evident in cells treated with both ATMi and ATRi (Fig 6B). On the other hand, some substrates, such as pS183/PPM1G, were compensated in A‐T cells, but not in WT cells under ATM inhibition, demonstrating the lesser scope of compensation upon chemical ATM inhibition compared with the compensation upon ATM absence (Fig 6C).
Figure 6. Representative SRM measurements of specific phosphorylation sites.
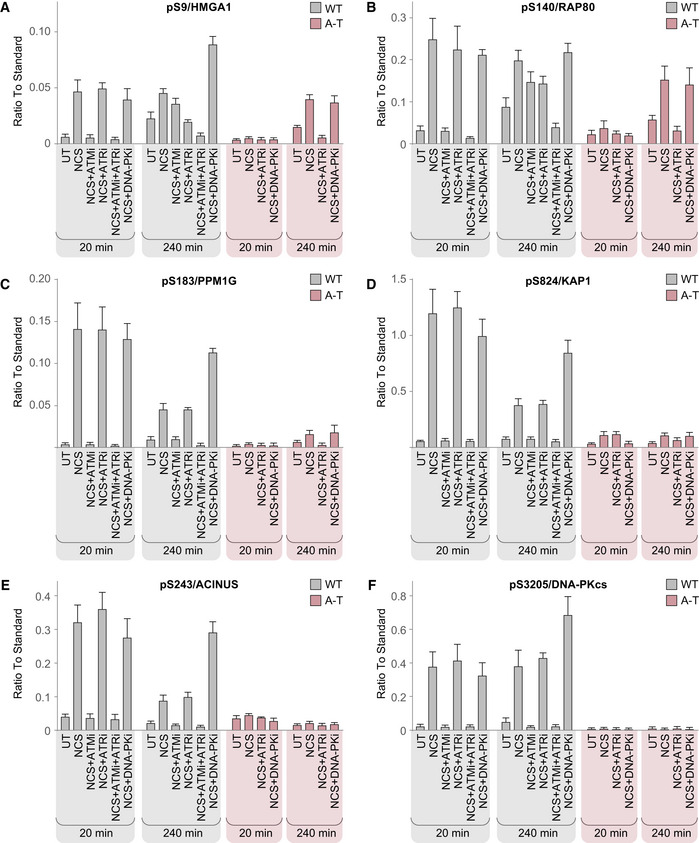
Cells were either untreated (UT) or treated with neocarzinostatin (NCS) in the presence or absence of PIKK inhibitors. Intensities from three independent biological replicates were normalized against reference synthetic standards and are depicted as mean ± SEM.
To account for possible off‐target effects of the ATM inhibitor that we used, we tested the dynamics of 21 phosphopeptides in the presence of another ATM inhibitor, KU55933 (Hickson et al, 2004), applied to HeLa cells. Eighteen out of them were elevated in response to NCS in both datasets, 18 and 17 of them showed ATM dependency when tested with KU60019 or KU55933, respectively (Fig EV3, Table EV9). ATM substrates that responded to NCS in A‐T cells are particularly interesting with regard to possible inhibitor off‐targets. Out of the 21 phosphopeptides, there were 11 KU60019‐inhibited phosphopeptides that responded to NCS in A‐T cells, 10 of these were inhibited also by KU55933. These results demonstrate high correlation between substrates suppressed by both inhibitors and largely reduce the possibility that our results were confounded by inhibitor off‐target effects.
Figure EV3. SRM measurements with KU55933.
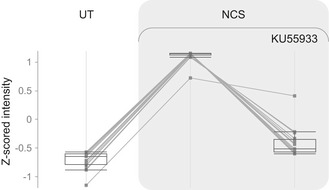
HeLa cells were either untreated (UT) or treated with 10 µM KU55933 and 50 ng/ml neocarzinostatin (NCS) and harvested 1 h following NCS treatment. Depicted are the z‐scored intensities of 18 phosphopeptides that were elevated at least twofold following NCS. Box plots depict 18 phosphopeptides measured in one replicate. The box indicates the range from first to third quartiles, and the central band represents the median. Upper and lower whiskers extend from the box to the maximum and minimum values which are not farther than 1.5 times the interquartile range (IQR).
Discussion
We explored the dynamics of the cellular phosphoproteome following DSB induction and its dependence on the PIKKs, ATM, ATR, and DNA‐PK. The successive application of shotgun proteomics and targeted proteomics enabled us to discover and extensively validate new patterns of PIKK regulation and crosstalk (Fig 7). The pivotal role of ATM, ATR, and DNA‐PK in the cellular response to genotoxic stress was strongly reflected in our results, and ATM remained the dominant PIKK in the DSB response network. It was interesting to identify among ATM targets in this network some ATR targets in the response to replication stress.
Figure 7. A model of PIKK collaboration in the cellular response to genotoxic stress, based on the results of this study.
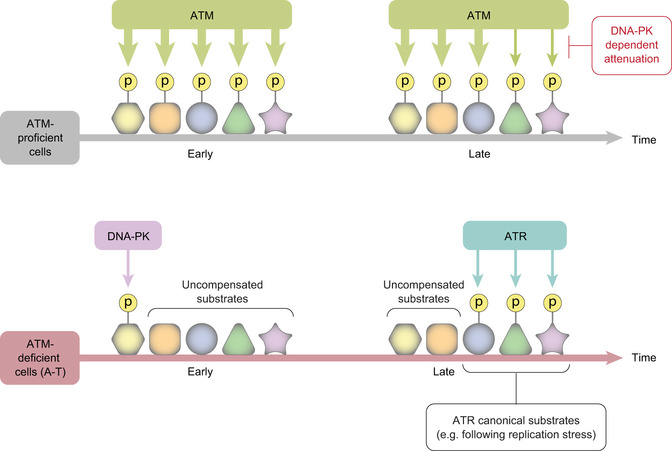
ATM‐proficient cells respond to DSBs with initial, robust ATM‐mediated phosphorylation of numerous substrates, after which ATM’s activity is attenuated in a DNA‐PK‐dependent manner. In ATM‐deficient cells, some ATM substrates are targeted by DNA‐PK at the early phase of the response, or by ATR in the later phase, but to a lesser extent than in WT cells. ATR‐dependent compensation for ATM absence in A‐T cells includes significantly more substrates than that of DNA‐PK and involves sites that ATR usually targets in response to genotoxic stresses other than DSBs. A few of these can be targeted by ATR also when ATM is chemically inhibited in WT cells.
We were intrigued by the phenomenon of enhanced and sustained phosphorylation of certain ATM substrates upon chemical inhibition of DNA‐PK. A similar observation was made also upon prolonged inhibition of ATR in HU‐treated cells. A simple explanation could be that DNA‐PK inhibition leaves many DSBs unrepaired, causing persistent ATM‐mediated phosphorylations. Alternatively, this phenomenon might reflect negative regulation of ATM by DNA‐PK, which is relieved upon DNA‐PK inhibition. Indeed, such mechanism was recently reported (Finzel et al, 2016; Zhou et al, 2017). Zhou et al (2017) showed that DNA‐PK phosphorylates ATM on multiple sites, leading to attenuation of its activity. They suggested that the excitatory effect of DNA‐PKi on ATM activity does not result from impaired activity of the NHEJ pathway of DSB repair, since down‐regulation of the genes encoding central NHEJ players such as XRCC4 or LIG4 did not yield a similar effect. A third possibility is DNA‐PK‐dependent activation of one or more phosphatases. However, our finding that this process involves sustained ATM activity argues against this mechanism and favors the one based on DNA‐PK‐mediated attenuation of ATM activity (Finzel et al, 2016; Zhou et al, 2017), further demonstrating the delicate crosstalk between the PIKKs in maintenance of genome stability.
An important question concerning the PIKKs’ role in the cellular response to genotoxic stress is the extent of redundancy among them. An extreme situation for testing such redundancy is provided by A‐T cells: ATM is absent from the outset in these cells and DNA‐PK and ATR function in an ATM‐deficient environment. Do they function differently than in ATM‐proficient cells? The severe clinical and cellular phenotype of A‐T indicate that even if ATR/DNA‐PK‐dependent compensation does occur, it is far from substantial replacement of ATM. Our data do show that in A‐T cells, DNA‐PK and ATR phosphorylate limited subsets of bona fide ATM substrates, albeit not at the same kinetics and extent as ATM‐mediated phosphorylation. This phenomenon attests to a potential of DNA‐PK and ATR to act on ATM substrates, which is not implemented when ATM is present. It has been demonstrated that certain substrates can be phosphorylated by more than one PIKK in vitro, despite being targeted by a single PIKK in cells. For example, pS15/p53 is phosphorylated in vitro by both ATM and DNA‐PK (Lees‐Miller et al, 1992), but after the induction of DSBs in cells this site is not targeted by DNA‐PK (Jimenez et al, 1999). pS251/XLF is readily phosphorylated by DNA‐PK in vitro, but is an ATM target in cells (Yu et al, 2008). The balance between the action of different PIKKs on the same substrates in WT as in A‐T cells is apparently affected by the degree of PIKK activation in response to different stresses and their relative affinity to specific substrates in response different stimuli. Presumably, ATM absence in A‐T cells alters this balance and allows the other two PIKKs to approach targets they can phosphorylate but do not do so in the presence of ATM. In this regard, it is interesting that ATM target sites phosphorylated by ATR in A‐T cells were enriched for sites that were ATR‐dependent following HU in WT cells, but were not targeted by ATR in NCS‐treated WT cells. Thus, inherent capability of ATR that is normally activated in response to other genotoxic stresses is exploited in A‐T cells. ATR‐dependent compensation in ATM‐null cells might contribute to the extreme chemo‐ and radiosensitivity observed in ATM‐deficient cancer cells treated with ATRi (Weber & Ryan, 2015).
Notably, chemical inhibition of ATM led to some ATR‐dependent compensation involving significantly fewer substrates taken care of by ATR than in A‐T cells. For example, of the two major ATM substrates, pS343/NBS1 and pS824/KAP‐1, which were phosphorylated by ATR in A‐T cells, only the first one was phosphorylated in an ATR‐dependent manner in ATMi‐treated WT cells. This might reflect different rewiring of cellular signaling circuits following short‐term absence of ATM activity vs. permanent total lack of ATM. It should also be noted that the physiological results of lack of ATM and presence of inactive ATM in cells are extremely different, with the latter situation leading to a more severe cellular and organismal phenotype. In mice, the Atm‐knockout genotype is viable with variable recapitulation of the A‐T phenotype, while expression of catalytically inactive Atm is embryonic lethal (Daniel et al, 2012; Shiloh & Ziv, 2012; Yamamoto et al, 2012). This striking difference might stem from a more severe disruption of the DDR by the presence of “kinase‐dead” Atm at the damage sites compared with lack of ATM altogether (Daniel et al, 2012; Shiloh & Ziv, 2012; Yamamoto et al, 2012). Our findings suggest that this could be due to the more limited compensation for an inactive ATM, compared with the wider compensation for total absence of ATM.
The limited compensation for ATM absence in A‐T patients by DNA‐PK and ATR raises the possibility that without such compensation the A‐T phenotype would have been more severe. It also suggests that drugs that further enhance the action of DNA‐PK and ATR on ATM targets could potentially be used to alleviate some of the symptoms of this disease.
Materials and Methods
Cell culture
NL‐550 (WT), AT59RM (A‐T), and L‐119 (A‐T) lymphoblastoid cell lines were grown in RPMI medium supplemented with 15% fetal calf serum (FCS), penicillin, and streptomycin, at 37°C in 5% CO2 atmosphere. U2‐OS and HeLa cells were grown in DMEM medium supplemented with 10% FCS, penicillin, and streptomycin at 37°C in 5% CO2 atmosphere.
Chemical treatments
NCS and HU (Sigma‐Aldrich, St. Louis, MO, USA) were diluted in phosphate‐buffered saline (PBS) prior to addition to the culture medium. The inhibitors against ATM, ATR, and DNA‐PK (KU60019, AZ20, and NU7441, respectively) were obtained from Tocris Bioscience (Bristol, UK) and dissolved in DMSO prior to addition to the culture medium. Unless otherwise specified, the chemicals were added to the following final concentrations in the culture medium: 20 ng/ml NCS, 1 mM HU, 5 µM KU60019, 0.5 µM AZ20, and 5 µM NU7441. All chemical treatments remained in the culture throughout the experiments and were not washed.
Western blotting analysis
Cells were washed in PBS and subsequently lysed for 30 min at 4°C in RIPA lysis buffer. Cell lysates were clarified by centrifugation, and the protein concentration was determined using the Bradford assay. Lysates were separated using SDS–PAGE and transferred onto nitrocellulose or PVDF membranes. Membranes were then incubated overnight at 4°C with primary antibodies diluted in Tween–tris‐buffered saline (TTBS) containing 1% bovine serum albumin (BSA) and 0.02% azide.
Anti‐ATM (cat #2873), anti‐pS345/CHK1 (cat #2348), anti‐pT68/CHK2 (cat #2197), and anti‐pS395/NUMA (cat #3429) antibodies were purchased from Cell Signaling Technology (Cell Signaling Technology, Beverly, MA). Anti‐CHK1 (cat #ab80615), anti‐pS1981/ATM (cat #2152‐1), and anti‐pS2056/DNA‐PK (cat #ab18192) antibodies were purchased from Abcam (Cambridge, UK). Anti‐PNKP (cat #A300‐258A), anti‐pS824/KAP1 (cat #A300‐767A), and anti‐pS114/PNKP (cat #BL3846) antibodies were purchased from Bethyl Laboratories (Montgomery, TX, USA). Anti‐HSC70 (cat #sc‐7298) was purchased from Santa‐Cruz Biotechnology (Dallas, TX, USA). Anti‐KAP1 (cat #610680) was purchased from BD Biosciences (San Jose, CA, USA), and Anti‐NBS1 (cat # NB100‐143) was purchased from Novus Biologicals (Littleton, CO, USA).
The membranes were then washed with TTBS buffer. This was followed by incubation for 1 h with peroxidase‐conjugated secondary antibodies in TTBS containing 1% skim‐milk, and subsequent washes in TTBS. Chemiluminescence was performed using Luminata HRP substrate and detected either by films or by the Fusion FX imaging system.
Discovery phosphoproteomics
Sample preparation
Samples were prepared in biological duplicates. Cells were collected at the indicated time points following NCS or HU treatment, washed twice in ice‐cold phosphate‐buffered saline, and spun down for 5 min at 1,000 g. Cell pellets were re‐suspended in 8 M urea solution containing 0.1 M ABC, 0.1% RapiGest, and cocktail 2 and 3 phosphatase inhibitors. Disulfide bonds were reduced with tris(2‐carboxyethyl)phosphine (TCEP) at a final concentration of 10 mM at 37°C for 30 min. Free thiols were alkylated with 20 mM iodoacetamide at room temperature for 30 min in the dark. The bicinchoninic acid protein assay was used to measure protein concentration. A volume corresponding to 1.1 mg total protein was subsequently diluted to a final concentration of 1 M urea and digested overnight at 37°C with sequencing grade modified trypsin at a protein‐to‐enzyme ratio of 75:1. Peptides were desalted on a C18 Sep‐Pak cartridge and dried under vacuum.
Phosphopeptides were isolated from digested lysates with titanium‐di‐oxide (TiO2) resin (GL Sciences, Tokyo, Japan) by a protocol modified from (Bodenmiller et al, 2007; Zhou et al, 2013). The dried peptides were rigorously dissolved in a solution of 80% acetonitrile (ACN) and 6% trifluoroacetic acid (TFA) solution, followed by incubation with the TiO2 resin in rotation for 1 h at room temperature. The resin was washed twice with 80% ACN/6% TFA solution, twice with 80% ACN/0.1% TFA, and finally twice with 0.1% TFA. Phosphopeptides were eluted with a 0.3 M NH4OH solution and desalted using C18 ultramicrospin columns. Prior to sample injection, samples were re‐suspended in a 2% ACN/0.1% FA buffer that contained the iRT retention time kit (Biognosys AG, Schlieren, Switzerland; Escher et al, 2012).
LC‐MS/MS analysis
LC‐MS/MS analysis was performed using an Easy‐nLC 1000 HPLC system coupled to the Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Peptides were separated by reversed‐phase chromatography on an Acclaim PepMap 100 RSLC C18 column (150 × 0.075 mm, 2 Å particle size, Thermo Fisher Scientific) at a flow rate of 300 nl/min. Gradient elution was performed using mobile phases A (water/acetonitrile/formic acid, 98:2:0.15) and B (acetonitrile/water/formic acid, 98:2:0.15), with a gradient from 5% to 30% B in 180 min. The mass spectrometer was operated in data‐dependent acquisition mode, with one full MS scan in the orbitrap analyzer (scan range 350–1,600 m/z, resolution 120,000) followed by 15 dependent MS/MS scans in the linear ion trap. Collision‐induced dissociation was performed at 35% normalized collision energy, and singly charged precursors or precursors of unknown charge state were excluded for fragmentation. Dynamic exclusion was enabled for 30 s.
Database search and label‐free quantification
Raw data were analyzed with MaxQuant (Cox & Mann, 2008; Tyanova et al, 2016a) software (version 1.5.3.5) and the Andromeda search engine (Cox et al, 2011), with an FDR threshold of 0.01 (PSM and phosphosite level), against the complete human UniProt database. Minimal score for modified peptides was 40. For in silico digestion, trypsin was used as the protease with a maximum of two missed cleavages. The minimal peptide length was set to eight amino acids. The peptide search included carbamidomethyl‐cysteine as a fixed modification and phosphorylation on serine/ threonine/ tyrosine, N‐terminal acetylation, and methionine oxidation as variable modifications. Peptide mass tolerance was set to 20 and 4.5 ppm for the first search and main search, respectively. A maximum of two gapped scans was allowed. The “match between runs” feature was enabled.
Data processing and peptide filtering
MS2 spectra of phosphopeptides do not always provide enough information to correctly localize the phosphate group. This poses an even greater problem in experiments with a large number of samples, which reduces the overlap of localized sites between samples. We therefore considered phosphopeptides with the same sequence and an identical number of phosphate groups and acetylations as a single entity, and in cases in which several different phosphate localizations were identified for this entity, we assigned to it the one with the highest probability. In cases in which the same sequence with the same amount of phosphate groups and acetylations appeared with different oxidation status, we considered the form with the lowest number of missing values.
The resulting data was analyzed using Perseus (Tyanova et al, 2016b) software. The data were filtered out for peptides identified in the decoy database and potential contaminants. The log2 transformed intensities were normalized to the median of each sample. To examine which phosphopeptides responded to stimuli, we first determined which responded to NCS or HU at each time point separately. For this purpose, we filtered for phosphopeptides that had at least two true values in either NCS/HU‐treated or NCS/HU‐untreated samples of the specific time point. Missing values were then imputed by replacing them with random, low intensity values that formed a normal distribution with a width of 30%, and downshift of 1.9 standard deviations of the general data distribution. To account for batch effect between the replicates, we next performed principle component analysis (PCA) and subtracted the first component. Student's t‐test was applied with a Benjamini–Hochberg FDR of 5%, and only phosphopeptides that changed at least twofold were considered. Phosphopeptides that were inconsistently phosphorylated and dephosphorylated in different time points were filtered out of subsequent analysis. To determine PIKK dependence, NCS/HU‐treated samples were compared with samples treated with NCS/HU in the presence of a specific kinase inhibitor, with a Benjamini–Hochberg FDR of 5% and a minimal fold change of twofold.
Annotation enrichment analysis
Annotations enrichment was examined using Fisher exact test implemented in Perseus (Tyanova et al, 2016b), with an FDR threshold of 0.02. GO annotation enrichments were tested against the entire set of phosphorylated proteins identified in this study as background, and the enrichment was calculated relative to proteins, meaning that phosphopeptides belonging to the same protein were counted as one entity.
Motif analysis
Linear motif analysis was carried out using Fisher exact test implemented in Perseus (Tyanova et al, 2016b), with an FDR threshold of 0.02, against the entire set of phosphopeptides identified in this study as background. Only phosphorylation sites with localization probability greater than 0.75 were considered for motif analysis.
Network analysis
The interaction network was generated by the STRING database (Szklarczyk et al, 2019) and processed with Cytoscape (Shannon et al, 2003). The network was filtered for interactions with a confidence score higher than 0.4, with the intensity of the edges indicative of the confidence score.
Targeted phosphoproteomics
Sample preparation
Samples were prepared in biological triplicates by a procedure similar to the one used to prepare the samples for discovery phosphoproteomics, with the following modifications: From a starting amount of 0.7 mg total protein mass, phosphopeptides were enriched using TiO2 by a protocol modified from Bodenmiller et al (2007) and Zhou et al (2013). Briefly, the dried peptides were dissolved in an 80% acetonitrile (ACN), 2.5 % TFA, 1 M glycolic acid solution, and then transferred to a 200 µl tip pre‐loaded with TiO2 and processed as described in (Zhou et al, 2013). The enriched phosphopeptides were eluted, desalted, and re‐suspended in a 2% ACN/0.1% FA buffer that contained diluted synthetic reference peptide mix (see below) and iRT retention time kit (Biognosys AG, Schlieren, Switzerland; Escher et al, 2012).
The samples were analyzed on a 5500 QTRAP hybrid triple quadrupole/ion trap mass spectrometer (SCIEX, Framingham, MA, USA) equipped with a nanoelectrospray ion source. Chromatographic separation was performed by a nanoLC AS2 (SCIEX) coupled to a 15‐cm (75 μm ID) fused silica emitter (MSwil, Zurich, Switzerland), self‐packed with prontoSIL C18 AQ 3 μm resin (WICOM, Heppenheim, Germany). Peptides were separated at a flow rate of 300 nL/min, in a gradient of solvent A (98% water/2% ACN/0.1% FA) and solvent B (98% ACN/2% water/0.1% FA) from 2% to 35% in 65 min (65 min gradient, 2–35%). The instrument was operated in scheduled positive SRM mode at a unit resolution (0.7 m/z half‐maximum peak width) for both Q1 and Q3 analyzers. Unless further optimized, collision energies (CEs) were calculated according to the formulas: CE = 0.044 × m/z precursor + 5.5 and CE = 0.051 × m/z precursor + 0.55, for doubly and triply charged precursor ions, respectively.
The SRM experiment reported in Fig EV3 was conducted with the following modifications to the procedure described above: Samples were prepared as a single replicate and enriched for phosphopeptides with Ti‐IMAC beads (a generous gift from Dr. Mingliang Ye, Dalian Institute of Chemical Physics, China). Synthetic reference phosphopeptides were utilized for identification and relative quantification unless otherwise specified in Table EV9.
Targeted assay generation
Targeted assays were generated using synthetic phosphopeptides (Thermo Fisher) labeled with heavy isotopes at the C‐terminal Lys (+8 Da) or Arg (+10 Da) (unless otherwise mentioned in Table EV7). The successful synthesis of each peptide was first confirmed by identification of the peptide in shotgun proteomic analysis. Peptides were mixed in pools and analyzed on a 5500 QTRAP to generate full MS2 fragment ion chromatograms of the y‐ion and b‐ion series for the 2+ and 3+ precursors. For selected phosphopeptides, neutral loss (−98 Da) ions were also included in the transition list. Skyline (MacLean et al, 2010) and Panorama (Sharma et al, 2014) were used to generate a library from the acquired full MS2 fragment ion chromatograms. For each synthetic phosphopeptide, the 4‐8 most intense fragment ions were selected for the targeted assays, taking into account phosphorylation site localization.
Targeted data analysis
Skyline (MacLean et al, 2010) was used for targeted data analysis (version 4.0.9.11707). SRM peak integration was manually confirmed. The reference synthetic standards were used to validate peptide identity by analogy of chromatographic and fragmentation properties (rdotp > 0.9), and interfered transitions were removed for quantification. Relative quantification and statistical analysis were performed using MSstats (Choi et al, 2014) (version 3.12.3) with the following modifications to default parameters: Normalization was performed by equalizing the medians of the reference synthetic standards; Tukey’s median polish (TMP) was used as summary method. We selected an adjusted FDR q‐value of 0.05 as a cutoff for significance.
The experiment described in Fig EV3 was analyzed with Skyline (version 20.1.0.31). A twofold change was selected as a cutoff that deems a phosphopeptide as regulated, and no further statistical analysis was performed on those samples.
Author contributions
Conceptualization: SS‐B, YS, YZ, AB, and RA; Methodology: SS‐B, YZ, AB, RA, TG, MH, and YS; Formal analysis of discovery proteomics: SS‐B; Formal analysis of targeted proteomics: AB, SS‐B; Investigation: SS‐B, AB; Writing—original draft: SS‐B; Writing—review and editing: SS‐B, AB, YZ, RA, TG, and YS; Visualization: SS‐B; Supervision: YS, YZ, RA, and TG; Funding acquisition: YS, RA, and SS‐B.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Alexander Leitner for technical assistance, Ludovic Gillet for technical and computational advice, Ran Elkon for discussions concerning the statistical analysis, Megi Cemel‐David for experimental advice, Ayelet Klartag for logistic assistance, and Mingliang Ye for the Ti‐IMAC beads. This work was funded by research grants from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and The Israel Cancer Research Fund (to Y.S.). S.S.‐B. was supported by a Tel Aviv University Global Research and Training Fellowship (GRTF) provided by the Naomi Prawer Kadar Foundation. Work in the group of RA was supported by the Swiss National Science Foundation grant 31003A_166435 and by SystemsX.ch, the Swiss Initiative for Systems Biology. Y.S. is a Research Professor of the Israel Cancer Research Fund.
The EMBO Journal (2021) 40: e104400.
Data availability
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository (https://www.ebi.ac.uk/pride/) with the dataset identifiers PXD015455, PXD020884.
References
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ (2012) A genome‐wide homologous recombination screen identifies the RNA‐binding protein RBMX as a component of the DNA‐damage response. Nat Cell Biol 14: 318–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, Bensimon A, Collins BC, Ludwig C, Sabido E (2016) Applications and developments in targeted proteomics: from SRM to DIA/SWATH. Proteomics 16: 2065–2067 [DOI] [PubMed] [Google Scholar]
- Baranes‐Bachar K, Levy‐Barda A, Oehler J, Reid DA, Soria‐Bretones I, Voss TC, Chung D, Park Y, Liu C, Yoon JB et al (2018) The ubiquitin E3/E4 ligase UBE4A adjusts protein ubiquitylation and accumulation at sites of DNA damage. Facilitating double‐strand break repair. Mol Cell 69: 866–878.e867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 46: 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS (2010) Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics 9: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, Aebersold R, Shiloh Y (2010) ATM‐dependent and ‐independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal 3: rs3 [DOI] [PubMed] [Google Scholar]
- Benzina S, Pitaval A, Lemercier C, Lustremant C, Frouin V, Wu N, Papine A, Soussaline F, Romeo PH, Gidrol X (2015) A kinome‐targeted RNAi‐based screen links FGF signaling to H2AX phosphorylation in response to radiation. Cell Mol Life Sci 72: 3559–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R (2007) Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods 4: 231–237 [DOI] [PubMed] [Google Scholar]
- Boeing S, Williamson L, Encheva V, Gori I, Saunders RE, Instrell R, Aygun O, Rodriguez‐Martinez M, Weems JC, Kelly GP et al (2016) Multiomic analysis of the UV‐induced DNA damage response. Cell Rep 15: 1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucas J, Fritz C, Schmitt A, Riabinska A, Thelen L, Peifer M, Leeser U, Nuernberg P, Altmueller J, Gaestel M et al (2015) Label‐free protein‐RNA interactome analysis identifies Khsrp signaling downstream of the p38/Mk2 kinase complex as a critical modulator of cell cycle progression. PLoS One 10: e0125745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, O'Carrigan B, Jackson SP, Yap TA (2017) Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 7: 20–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Walker GC (2017) Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58: 235–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY (2001) ATM‐dependent phosphorylation of human Rad9 is required for ionizing radiation‐induced checkpoint activation. J Biol Chem 276: 16580–16586 [DOI] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry‐based proteomic experiments. Bioinformatics 30: 2524–2526 [DOI] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ (2010) A chromatin localization screen reveals poly (ADP ribose)‐regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA 107: 18475–18480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotta‐Ramusino C, McDonald ER 3rd, Hurov K, Sowa ME, Harper JW, Elledge SJ (2011) A DNA damage response screen identifies RHINO, a 9‐1‐1 and TopBP1 interacting protein required for ATR signaling. Science 332: 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Daniel JA, Pellegrini M, Lee BS, Guo Z, Filsuf D, Belkina NV, You Z, Paull TT, Sleckman BP, Feigenbaum L et al (2012) Loss of ATM kinase activity leads to embryonic lethality in mice. J Cell Biol 198: 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Chen DJ (2013) DNA double strand break repair via non‐homologous end‐joining. Transl Cancer Res 2: 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Chen BP, Chen DJ (2014) DNA‐PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst) 17: 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees‐Miller SP (2002) Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA‐dependent protein kinase. Biochem J 368: 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia AE, Boardman AP, Wang DC, Huttlin EL, Everley RA, Dephoure N, Zhou C, Koren I, Gygi SP, Elledge SJ (2015) Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol Cell 59: 867–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12: 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzel A, Grybowski A, Strasen J, Cristiano E, Loewer A (2016) Hyperactivation of ATM upon DNA‐PKcs inhibition modulates p53 dynamics and cell fate in response to DNA damage. Mol Biol Cell 27: 2360–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ et al (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Zou L (2011) ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 36: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote KM, Blades K, Cronin A, Fillery S, Guichard SS, Hassall L, Hickson I, Jacq X, Jewsbury PJ, McGuire TM et al (2013) Discovery of 4‐{4‐((3R)‐3‐Methylmorpholin‐4‐yl)‐6‐(1‐(methylsulfonyl)cyclopropyl)pyrimidin‐2‐y l}‐1H‐indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J Med Chem 56: 2125–2138 [DOI] [PubMed] [Google Scholar]
- Gatei M, Young D, Cerosaletti KM, Desai‐Mehta A, Spring K, Kozlov S, Lavin MF, Gatti RA, Concannon P, Khanna K (2000) ATM‐dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet 25: 115–119 [DOI] [PubMed] [Google Scholar]
- Gavish‐Izakson M, Velpula BB, Elkon R, Prados‐Carvajal R, Barnabas GD, Ugalde AP, Agami R, Geiger T, Huertas P, Ziv Y et al (2018) Nuclear poly(A)‐binding protein 1 is an ATM target and essential for DNA double‐strand break repair. Nucleic Acids Res 46: 730–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George VC, Ansari SA, Chelakkot VS, Chelakkot AL, Chelakkot C, Menon V, Ramadan W, Ethiraj KR, El‐Awady R, Mantso T et al (2019) DNA‐dependent protein kinase: Epigenetic alterations and the role in genomic stability of cancer. Mutat Res 780: 92–105 [DOI] [PubMed] [Google Scholar]
- Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M, Rigoreau L, Menear KA et al (2009) Improved ATM kinase inhibitor KU‐60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 8: 2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Kastan MB (2015) The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med 66: 129–143 [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Jeggo PA (2013) The repair and signaling responses to DNA double‐strand breaks. Adv Genet 82: 1–45 [DOI] [PubMed] [Google Scholar]
- Herr P, Lundin C, Evers B, Ebner D, Bauerschmidt C, Kingham G, Palmai‐Pallag T, Mortusewicz O, Frings O, Sonnhammer E et al (2015) A genome‐wide IR‐induced RAD51 foci RNAi screen identifies CDC73 involved in chromatin remodeling for DNA repair. Cell Discov 1: 15034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC (2004) Identification and characterization of a novel and specific inhibitor of the ataxia‐telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159 [DOI] [PubMed] [Google Scholar]
- Hurov KE, Cotta‐Ramusino C, Elledge SJ (2010) A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 24: 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano‐Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, Elledge SJ (2015) A systematic analysis of factors localized to damaged chromatin reveals PARP‐dependent recruitment of transcription factors. Cell Rep 11: 1486–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jachimowicz RD, Beleggia F, Isensee J, Velpula BB, Goergens J, Bustos MA, Doll MA, Shenoy A, Checa‐Rodriguez C, Wiederstein JL et al (2019) UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell 176: 505–519.e522 [DOI] [PubMed] [Google Scholar]
- Jette N, Lees‐Miller SP (2015) The DNA‐dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol 117: 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez GS, Bryntesson F, Torres‐Arzayus MI, Priestley A, Beeche M, Saito S, Sakaguchi K, Appella E, Jeggo PA, Taccioli GE et al (1999) DNA‐dependent protein kinase is not required for the p53‐dependent response to DNA damage. Nature 400: 81–83 [DOI] [PubMed] [Google Scholar]
- Jowsey P, Morrice NA, Hastie CJ, McLauchlan H, Toth R, Rouse J (2007) Characterisation of the sites of DNA damage‐induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair (Amst) 6: 1536–1544 [DOI] [PubMed] [Google Scholar]
- Junger MA, Aebersold R (2014) Mass spectrometry‐driven phosphoproteomics: patterning the systems biology mosaic. Wiley Interdiscip Rev Dev Biol 3: 83–112 [DOI] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML (2013) Proteome‐wide identification of poly(ADP‐Ribosyl)ation targets in different genotoxic stress responses. Mol Cell 52: 272–285 [DOI] [PubMed] [Google Scholar]
- Kavanaugh G, Ye F, Mohni KN, Luzwick JW, Glick G, Cortez D (2015) A whole genome RNAi screen identifies replication stress response genes. DNA Repair (Amst) 35: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM et al (2007) Orchestration of the DNA‐damage response by the RNF8 ubiquitin ligase. Science 318: 1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Perrimon N (2011) A genome‐wide RNAi screen identifies core components of the G(2)‐M DNA damage checkpoint. Sci Signal 4: rs1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov SV, Waardenberg AJ, Engholm‐Keller K, Arthur JW, Graham ME, Lavin M (2016) Reactive oxygen species (ROS)‐activated ATM‐dependent phosphorylation of cytoplasmic substrates identified by large‐scale phosphoproteomics screen. Mol Cell Proteomics 15: 1032–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, Smolka MB (2019) DNA damage kinase signaling: checkpoint and repair at 30 years. The EMBO J 38: e101801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC (2004) Identification of a highly potent and selective DNA‐dependent protein kinase (DNA‐PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett 14: 6083–6087 [DOI] [PubMed] [Google Scholar]
- Lees‐Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW (1992) Human DNA‐activated protein kinase phosphorylates serines 15 and 37 in the amino‐terminal transactivation domain of human p53. Mol Cell Biol 12: 5041–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees‐Miller SP, Meek K (2003) Repair of DNA double strand breaks by non‐homologous end joining. Biochimie 85: 1161–1173 [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jorgensen C, Miron IM, Diella F, Colwill K, Taylor L, Elder K et al (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129: 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Saavedra A, Gomez‐Cabello D, Dominguez‐Sanchez MS, Mejias‐Navarro F, Fernandez‐Avila MJ, Dinant C, Martinez‐Macias MI, Bartek J, Huertas P (2016) A genome‐wide screening uncovers the role of CCAR2 as an antagonist of DNA end resection. Nat Commun 7: 12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Cortez D (2009) Common mechanisms of PIKK regulation. DNA Repair (Amst) 8: 1004–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, Sirbu BM, Titus LC, Shyr Y, Cortez D (2009) Functional genomic screens identify CINP as a genome maintenance protein. Proc Natl Acad Sci USA 106: 19304–19309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Li Y, Mu JJ, Zhang J, Tonaka T, Hamamori Y, Jung SY, Wang Y, Qin J (2008) Regulation of intra‐S phase checkpoint by ionizing radiation (IR)‐dependent and IR‐independent phosphorylation of SMC3. J Biol Chem 283: 19176–19183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- Menolfi D, Zha S (2019) ATM, DNA‐PKcs and ATR: shaping development through the regulation of the DNA damage responses. Genom Instabil Dis 1: 47–68 [Google Scholar]
- Menzel T, Nahse‐Kumpf V, Kousholt AN, Klein DK, Lund‐Andersen C, Lees M, Johansen JV, Syljuasen RG, Sorensen CS (2011) A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep 12: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ, Harper JW (2010) A genome‐wide camptothecin sensitivity screen identifies a mammalian MMS22L‐NFKBIL2 complex required for genomic stability. Mol Cell 40: 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor MJ (2015) Targeting the DNA damage response in cancer. Mol Cell 60: 547–560 [DOI] [PubMed] [Google Scholar]
- O'Donnell L, Panier S, Wildenhain J, Tkach JM, Al‐Hakim A, Landry MC, Escribano‐Diaz C, Szilard RK, Young JT, Munro M et al (2010) The MMS22L‐TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40: 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz‐Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia‐telangiectasia and Rad3‐related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Olivieri M, Cho T, Alvarez‐Quilon A, Li K, Schellenberg MJ, Zimmermann M, Hustedt N, Rossi SE, Adam S, Melo H et al (2020) A genetic map of the response to DNA damage in human cells. Cell 182: 481–496.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT (2015) Mechanisms of ATM activation. Annu Rev Biochem 84: 711–738 [DOI] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow‐Cordero DE et al (2009) A genome‐wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R (2012) Selected reaction monitoring‐based proteomics: workflows, potential, pitfalls and future directions. Nat Methods 9: 555–566 [DOI] [PubMed] [Google Scholar]
- Picotti P, Bodenmiller B, Aebersold R (2013) Proteomics meets the scientific method. Nat Methods 10: 24–27 [DOI] [PubMed] [Google Scholar]
- Piwko W, Olma MH, Held M, Bianco JN, Pedrioli PG, Hofmann K, Pasero P, Gerlich DW, Peter M (2010) RNAi‐based screening identifies the Mms22L‐Nfkbil2 complex as a novel regulator of DNA replication in human cells. EMBO J 29: 4210–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K et al (2015) DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross‐links. Science 348: 1253671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Rothblum‐Oviatt C, Wright J, Lefton‐Greif MA, McGrath‐Morrow SA, Crawford TO, Lederman HM (2016) Ataxia telangiectasia: a review. Orphanet J Rare Dis 11: 159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador Moreno N, Liu J, Haas KM, Parker LL, Chakraborty C, Kron SJ, Hodges K, Miller LD, Langefeld C, Robinson PJ et al (2019) The nuclear structural protein NuMA is a negative regulator of 53BP1 in DNA double‐strand break repair. Nucleic Acids Res 47: 2703–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar‐Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S et al (1995) A single ataxia telangiectasia gene with a product similar to PI‐3 kinase. Science 268: 1749–1753 [DOI] [PubMed] [Google Scholar]
- Segal‐Raz H, Mass G, Baranes‐Bachar K, Lerenthal Y, Wang SY, Chung YM, Ziv‐Lehrman S, Strom CE, Helleday T, Hu MC et al (2011) ATM‐mediated phosphorylation of polynucleotide kinase/phosphatase is required for effective DNA double‐strand break repair. EMBO Rep 12: 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltiel IA, Krenning L, Bruinsma W, Medema RH (2015) The same, only different ‐ DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci 128: 607–620 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B et al (2014) Panorama: a targeted proteomics knowledge base. J Proteome Res 13: 4205–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y (2012) The ATM protein: the importance of being active. J Cell Biol 198: 273–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210 [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu‐Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D (2013) Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 288: 31458–31467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP et al (2010) A genetic screen identifies FAN1, a Fanconi anemia‐associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 39: 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Bloom MS, Solanki‐Patil T, Smith S, Sipahimalani P, Li Z, Kofoed M, Ben‐Aroya S, Myung K, Hieter P (2011) The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet 7: e1002057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, Yang V, Beausoleil SA, Gygi SP, Livingstone M et al (2007) Profiling of UV‐induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA 104: 19855–19860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P et al (2019) STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 47: D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Burma S (2009) Distinct roles of ATR and DNA‐PKcs in triggering DNA damage responses in ATM‐deficient cells. EMBO Rep 10: 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi‐Yano K, Mari PO, Tezcan I, Chen DJ, Zdzienicka MZ et al (2009) A DNA‐PKcs mutation in a radiosensitive T‐B‐ SCID patient inhibits Artemis activation and nonhomologous end‐joining. J Clin Invest 119: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidi PA, Liu J, Salles D, Jayaraman S, Dorfman G, Gray M, Abad P, Moghe PV, Irudayaraj JM, Wiesmuller L et al (2014) NuMA promotes homologous recombination repair by regulating the accumulation of the ISWI ATPase SNF2h at DNA breaks. Nucleic Acids Res 42: 6365–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AM, Ryan AJ (2015) ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 149: 124–138 [DOI] [PubMed] [Google Scholar]
- Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, Dobyns WB, Ogi T, Meek K, Davies EG et al (2013) PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest 123: 2969–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Wang Y, Jiang W, Liu X, Dubois RL, Lin CS, Ludwig T, Bakkenist CJ, Zha S (2012) Kinase‐dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol 198: 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Mahaney BL, Yano K, Ye R, Fang S, Douglas P, Chen DJ, Lees‐Miller SP (2008) DNA‐PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 7: 1680–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Piwnica‐Worms H (2001) ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK (2000) Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia‐telangiectasia‐mutated kinase activity. J Biol Chem 275: 10342–10348 [DOI] [PubMed] [Google Scholar]
- Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck AJ, Zou H, Mohammed S (2013) Robust phosphoproteome enrichment using monodisperse microsphere‐based immobilized titanium (IV) ion affinity chromatography. Nat Protoc 8: 461–480 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lee JH, Jiang W, Crowe JL, Zha S, Paull TT (2017) Regulation of the DNA damage response by DNA‐PKcs inhibitory phosphorylation of ATM. Mol Cell 65: 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker‐Jensen S, Bartek J, Shiloh Y (2006) Chromatin relaxation in response to DNA double‐strand breaks is modulated by a novel ATM‐ and KAP‐1 dependent pathway. Nat Cell Biol 8: 870–876 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository (https://www.ebi.ac.uk/pride/) with the dataset identifiers PXD015455, PXD020884.