

Abstract
Background:
Early life environmental exposures can have lasting effects on the function of the immune system and contribute to disease later in life. Epidemiological studies have linked early life exposure to xenobiotics that bind the aryl hydrocarbon receptor (AhR) with dysregulated immune responses later in life. Among the immune cells influenced by developmental activation of the AhR are T cells. Yet, the underlying affected cellular pathways via which activating the AhR early in life causes the responses of T cells to remain affected into adulthood remain unclear.
Objective:
Our goal was to identify cellular mechanisms that drive impaired T-cell responses later in life following maternal exposure to an exogenous AhR ligand.
Methods:
C57BL/6 mice were vertically exposed to the prototype AhR ligand, 2,3,7,8-tetrachlorodibenzo--dioxin (TCDD), throughout gestation and early postnatal life. The transcriptome and DNA methylation patterns were evaluated in T cells isolated from naïve and influenza A virus (IAV)-infected adult mice that were developmentally exposed to TCDD or vehicle control. We then assessed the influence of DNA methylation-altering drug therapies on the response of T cells from developmentally exposed mice to infection.
Results:
Gene and protein expression showed that developmental AhR activation reduced T-cell expansion and effector functions during IAV infection later in life. Furthermore, whole-genome bisulfite sequencing analyses revealed that developmental AhR activation durably programed DNA methylation patterns across the T-cell genome. Treatment of developmentally exposed offspring with DNA methylation-altering drugs alleviated some, but not all, of the impaired T-cell responses.
Discussion:
Taken together, these results indicate that skewed DNA methylation is one of the mechanisms by which early life exposures can durably change the function of T cells in mice. Furthermore, treatment with DNA methylation-altering drugs after the exposure restored some aspects of T-cell functional responsiveness. https://doi.org/10.1289/EHP7699
Introduction
An individual’s long-term health trajectory is shaped by the development of complex tissues and organ systems during early life. Development begins in the womb and, for some organ systems, such as the immune system, continues for a period of time after birth (Robin et al. 2003; Zhu and Emerson 2002). Appropriate development relies on integrated signaling networks that regulate cell fate, differentiation, and functional properties (Basson 2012; Partridge et al. 2010; Sanz-Ezquerro et al. 2017). Because this early life window is so critical, it is vulnerable to environmental influences that can durably change health and disease across the life span (Renz et al. 2017). In fact, early life exposures to environmental insults have been associated with increased incidences of obesity, cardiovascular disease, and cancers later in life (Boekelheide et al. 2012; Cao et al. 2016), and with altered function of the immune system (Boule and Lawrence 2015). This idea, which is generally referred to as the developmental origins of health and disease, has been recognized for nearly 40 y; yet we still have much to learn about the mechanisms driving these long-term changes (Boekelheide et al. 2012; de Boo and Harding 2006; Hoffman et al. 2017).
One of the barriers to understanding how early life exposures cause long-lasting consequences to health is that the cellular and molecular mechanisms underlying these durable changes remain poorly characterized. Several epidemiological studies have reported an association between early life exposures to pollutants that bind a ligand-activated transcription factor known as the aryl hydrocarbon receptor (AhR) and impaired immune responses later in life. Associations include increased incidence of respiratory tract and ear infections, increased wheezing, and decreased responses to vaccinations (Dallaire et al. 2004; Glynn et al. 2008; Heilmann et al. 2010; Hochstenbach et al. 2012; Jusko et al. 2014, 2016; Stølevik et al. 2011, 2013). An important immune cell type that is involved in all of these responses is the T cell. T cells help fight infections, bolster immunological memory, and aid in responses to vaccination (Crotty 2014; MacLeod et al. 2009; Sant and McMichael 2012; Swain et al. 2012; Zhu and Paul 2008). Given this central role of T cells, and that all immune cells, including T cells, express the AhR (Esser and Rannug 2015), we investigated how early life AhR activation modulates T-cell responses later in life.
Maternal exposure of mice to the prototype AhR ligand, 2,3,7,8-tetrachlorodibenzo--dioxin (TCDD), causes impaired T-cell responses in adult offspring (Boule et al. 2014, 2015a, 2015b; Burke et al. 2019). With specific regard to T cells, developmental AhR activation reduces conventional helper T-cell responses and increases regulatory T cell responses during influenza A virus (IAV) infection at adulthood (Boule et al. 2014). Adoptive transfer studies revealed that reduced T-cell responses were due to factors intrinsic to T cells (Boule et al. 2014). Furthermore, these changes in T-cell responses in developmentally exposed offspring were durable and observed up to 1 y after birth (Boule et al. 2015a). Given that the half-life of TCDD in mice is 7–10 d (Gasiewicz et al. 1983), differences in T-cell responses were maintained long after the exogenous AhR stimulus was cleared from the system and when AhR target genes were no longer induced (Winans et al. 2015). The fact that alterations persist well after exposure suggests that the long-term responsive capacity of T cells is programmed during development and that this programming is sensitive to environmental cues. One way that exposures can cause durable modifications is by altering the epigenetic landscape. To date, whether early life activation of the AhR by exposure to an exogenous ligand changes epigenetic modifications within T cells later in life remains unknown.
One type of epigenetic mechanism that influences cell differentiation and function is DNA methylation (Suárez-Álvarez et al. 2013; Tirado-Magallanes et al. 2017). There are several reports that early life activation of the AhR changes DNA methylation patterns in nonimmune organs such as the testes (Ding et al. 2018; Manikkam et al. 2012), ovaries (Zhang et al. 2019), placenta (Ding et al. 2018), mammary tissues (Papoutsis et al. 2015), muscle (Somm et al. 2013), and liver (Somm et al. 2013). Taken together, this information led us to hypothesize that one of the mechanisms by which early life AhR activation impairs T-cell responses later in life is via remodeling of DNA methylation patterns in T cells. To test this, mice were developmentally exposed to a vehicle control or to TCDD, which specifically activates the AhR (Boule et al. 2014; Winans et al. 2015). Unbiased whole-genome approaches were used to define how developmental AhR activation impacts DNA methylation and gene expression patterns in T cells isolated from adult animals. We also examined the causal relationship between changes in the level of DNA methylation and altered T-cell responses using pharmacological interventions that elevate or dampen DNA methylation in offspring.
Methods
Animals and Developmental Exposure
C57BL/6 () mice (5–6 wk of age) were purchased from the Jackson Laboratory. For each experiment, between 20 and 40 nulliparous females were housed with C57BL/6 males. At the initiation of experimental breeding, there were 2 or 3 females with each male. As vaginal plugs were identified, females were removed and singly housed thereafter. In the morning, females were checked daily for the presence of a vaginal plug, which was labeled as gestational day (GD) 0. Anisole was used to dissolve the TCDD ( purity; Cambridge Isotope Laboratories), which was then diluted in peanut oil. The vehicle control was peanut oil containing 0.01% anisole. Impregnated female mice were alternately assigned to treatment groups and treated with the peanut oil vehicle control (vehicle) or body weight by gavage between 1100 hours and 1300 hours on GD0, -7, and -14 and 2 d after parturition (PND2). Cages were clearly labeled to denote dam treatment and to ensure accurate dosing across pregnancy and postpartum. All members of the research team were aware of which animals were in which treatment group. Offspring were weaned at PND21, and no culling of litters was performed. Littermates were housed in same-sex groups. Offspring were assigned a number, and at the time the mice were sacrificed, only the person in charge of the experiment knew which number correlated with a specific mouse. Mice were housed at the University of Rochester in microisolator cages in a specific-pathogen free facility that is maintained in a 12-h light:dark cycle, at and 30–50% relative humidity. Mice were provided food (LabDiet 5010) and reverse osmosis purified water ad libitum. All experiments were performed using female offspring. All animal treatments had prior approval of the Institutional Animal Care and Use Committee of the University of Rochester, and followed all guidelines and regulations. The University has accreditation through the Association for Assessment and Accreditation of Laboratory Animal Care. Animals were treated humanely and with due consideration to alleviation of any distress and discomfort. All U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals guidelines for the handling of vertebrate animals were followed (https://olaw.nih.gov/policies-laws/phs-policy.htm).
S-Adenosyl Methionine and Zebularine Treatment
For some studies, at weaning, developmentally exposed offspring were randomly placed into three treatment groups. One group of mice (control) remained on drinking water provided by the vivarium. The second group (SAM) received drinking water containing S-adenosyl methionine (SAM; Sigma-Aldrich). The third group (Zeb) received drinking water containing Zebularine [Zeb; NCS309132, provided by the Developmental Therapeutics Program, National Cancer Institute, National Institutes of Health (NIH)]. These in vivo doses of SAM (Y Li et al. 2012) and Zeb (Yoo et al. 2008), administered via drinking water, have been previously shown to alter DNA methylation levels in mice. Mice were provided water ad libitum. To conserve drugs, mice within the same developmental exposure group were co-housed at up to five mice per cage. Water consumption was measured every other day, and body weight was recorded once a week (Figure S1). Mice were infected with IAV at 6–8 wk of age, and body weight was monitored daily throughout infection (Figure S1). All mice were maintained on treated water for the remainder of the experiment.
Influenza Virus Infection
IAV (strains HKx31, H3N2 or PR8, and H1N1) were initially obtained from M. Coppola (Argonex, Charlottesville, VA). Virus stocks were separately propagated in embryonated specific-pathogen free chicken eggs (Charles River), and clear allantoic fluid was aliquoted and snap frozen in liquid nitrogen, as described previously (Warren et al. 2000). Virus titers were determined by hemagglutination of erythrocytes (Barrett and Inglis 1985). Briefly, serial dilutions of allantoic fluid (1:2 to 1:8,192) were prepared in phosphate-buffered saline (PBS), and was added to U-bottom 96-well microtiter plates. To each well, of 1% (vol/vol) chicken erythrocytes (Cat. No. 31152; Colorado Serum Co.), diluted in PBS, was added, and the plate was incubated for 45 min at room temperature. PBS alone was included as a negative control, and undiluted IAV from a previously titered frozen stock was included as a positive control. To infect the mice, adult offspring (6–10 wk of age) were anesthetized with of avertin (2% 2,2,2-tribromoethanol; Sigma-Aldrich), administered by intraperitoneal injection. Infections were performed in the morning, between 0700 hours and 0900 hours. For primary infection, mice were infected intranasally with 120 hemagglutination units (HAU) of HKx31, which is a sublethal inoculum in immunocompetent mice (Warren et al. 2000; Boule et al. 2014, 2015b; Winans et al. 2015). The primary response was examined 9 d later, which is the peak of the T-cell response to acute primary IAV infection (Boule et al. 2014; Brown et al. 2006). To examine immunological memory, it is important to allow time for full resolution of the primary response and establishment of anamnestic cells. Mice were challenged with a heterosubtypic infection (0.1 HAU PR8) on Day 79 (i.e., 70 d after Day 9 post primary infection with 3 HAU HKx31). This dose of PR8 causes significant mortality during primary infection (Giannandrea et al. 2012). In all experiments, infection and tissue collection were initiated in the morning. All work with infectious agents was conducted with prior approval of the Institutional Biosafety Committee of the University of Rochester, following guidelines of the NIH and the Centers for Disease Control and Prevention (https://www.cdc.gov/labs/BMBL.html).
Tissue Collection
At the termination of each experiment, mice were sacrificed using a lethal dose of anesthetic (Avertin ) and either cervical dislocation or exsanguination. Tissue collection was initiated in the morning between 0730 hours and 0900 hours. The mediastinal lymph nodes (MLNs) were harvested, and single-cell suspensions prepared by pressing MLNs from a single mouse between the frosted ends of two microscope slides. Cells were then resuspended in cold Hanks’ balanced salt solution (HBSS) containing 2.5% fetal bovine serum (FBS) (Hyclone). Lysis solution ( ammonium chloride, sodium hydrogen carbonate, ethylenediaminetetraacetic acid) was added to the cells for 5 min at room temperature to lyse red blood cells. Lysis was quenched using cold HBSS/FBS. Debris was removed by passing cells through a filter. Cells were resuspended in cold HBSS/FBS for enumeration using a TC20 cell counter (BioRad). Cells were immediately used for analysis by flow cytometry or subjected to further treatment to enrich and purify a specific cell type and extract cellular materials. When peripheral lymph nodes were also collected, they were processed in the same manner.
Flow Cytometry
Prior to staining with fluorochrome-conjugated antibodies, single-cell suspensions containing lymph node cells were incubated for 10 min at 4°C with anti-mouse CD16/32 monclonal antibody (mAb; clone 93) to block nonspecific staining. Cells were then incubated for 20 min at 4°C with previously determined optimal concentrations of fluorochrome-conjugated antibodies against cell-surface antigens (Table 1). To identify T cells that recognize IAV, prior to extracellular antibody staining, cells were incubated for 1 h at 37°C with major histocompatibility class II tetramers containing an immunodominant peptide epitope of HKx31 [nucleoprotein (NP), ; provided by the National Institute of Allergy and Infectious Diseases, NIH Tetramer Core Facility; http://tetramer.yerkes.emory.edu]. To detect effector proteins, cells were incubated in the presence of recombinant mouse interleukin-2 (IL-2; R&D Systems) and brefeldin A (Sigma-Aldrich) for 1 h at 37°C prior to CD16/32 mAb block or extracellular antibody staining. Anti-CD107a [LAMP-1 (lysosomal-associated membrane protein 1)] was added, and the cells were incubated for an additional 4 h at 37°C prior to extracellular staining (Post et al. 2019). The cells were then fixed with a solution containing 2% formaldehyde in PBS for 20 min and resuspended in staining buffer containing 1% saponin before being incubated at room temperature with an antibody against perforin (Table 1). To detect Ki67, cyclin-dependent kinase inhibitor 2A (; p16), and T-box transcription factor (Tbet), cells were fixed/permeabilized using either the FoxP3 Staining Kit (eBioscience) or the BD Transcription Factor Buffer Set (BD Biosciences) following extracellular staining per the manufacturer’s instructions. Cells were then stained with intracellular antibodies to Ki67, p16, or Tbet for 20 min at 4°C. Prior to use, the antibody against p16 was directly conjugated to CF680 using the Mix-n-Stain Antibody Labeling Kit, following the manufacturer’s instructions (Sigma-Aldrich). Table 1 lists all antibodies used. Fluorescence minus one controls were used to determine nonspecific fluorescence staining and to define gating parameters. Data were obtained using an LSRII flow cytometer (BD Biosciences) to collect approximately 500,000 events per sample. Data were analyzed using the Flowjo software program (version 10; Tree Star).
Table 1.
Antibodies and reagents used for flow cytometry.
| Reagenta | Clone | Company | Catalog No. | Amountb |
|---|---|---|---|---|
| CD3e PE hamster anti-mouse | 145-2C11 | BD Biosciences | 553064 | |
| CD3e PE-CF594 hamster anti-mouse | 145-2C11 | BD Biosciences | 562286 | |
| CD3e PE-Cy5 hamster anti-mouse | 145-2C11 | BD Biosciences | 552774 | |
| CD4 BV650 rat anti-mouse | GK1.5 | BD Biosciences | 563232 | |
| CD4 FITC rat anti-mouse | H129.19 | BD Biosciences | 553651 | |
| CD4 PerCP-Cy5.5 rat anti-mouse | RM4-5 | Invitrogen/eBioscience | 45-0042-82 | |
| CD19 APC-R700 rat anti-mouse | 1D3 | BD Biosciences | 565473 | |
| CD19 BB515 rat anti-mouse | 1D3 | BD Biosciences | 564531 | |
| CD19 BV421 rat anti-mouse | 1D3 | BD Biosciences | 562701 | |
| CD19 BV711 rat anti-mouse | 1D3 | BD Biosciences | 563157 | |
| CD19 PE rat anti-mouse | 1D3 | BD Biosciences | 557399 | |
| CD44 BB515 rat anti-mouse | IM7 | BD Biosciences | 564587 | |
| CD44 BV421 rat anti-mouse | IM7 | BD Biosciences | 563970 | |
| CD44 BV711 rat anti-mouse | IM7 | BD Biosciences | 563971 | |
| CD45R APC rat anti-human, mouse | RA3-6B2 | Invitrogen/eBioscience | 56-0452-82 | |
| CD45R AF700 rat anti-mouse | RA3-6B2 | BD Biosciences | 557957 | |
| CD62L BV605 rat anti-mouse | MEL-14 | BD Biosciences | 563252 | |
| CD62L PE-Cy7 rat anti-mouse | MEL-14 | Invitrogen/eBioscience | 25-0621-82 | |
| CD107a (LAMP-1) PE rat anti-mouse | 1D4B | BD Biosciences | 558661 | |
| CD152 (CTLA-4) APC-R700 hamster anti-mouse | UC10-4F10-11 | BD Biosciences | 565778 | |
| CD185 (CXCR5) biotin rat anti-mouse | 2G8 | BD Biosciences | 551961 | |
| CD279 (PD-1) BV421 hamster anti-mouse | J43 | BD Biosciences | 562584 | |
| CD279 (PD-1) FITC hamster anti-mouse | J43 | Invitrogen/eBioscience | 11-9985-82 | |
| CD366 (TIM3) PE rat anti-mouse | 215008 | R&D Systems | FAB1529P | cells |
| KLRG1 PerCP-Cy5.5 hamster anti-mouse | 2F1 | BD Biosciences | 563595 | |
| MHCII tetramer [I-A(b) containing influenza A virus nucleoprotein (NP) peptide 311–325], APC | NA | NIH Tetramer Core Facility | NA | |
| Streptavidin PE | NA | BD Biosciences | 554061 | |
| Tbet PE-Cy7 mouse anti-human, mouse, rhesus monkey | 4B10 | Invitrogen/eBioscience | 25-5825-82 | |
| Perforin FITC rat anti-mouse | eBioOMAK-D | Invitrogen/eBioscience | 11-9392-82 | |
| Sigma Mix-n-Stain CF680 Antibody labeling kit | NA | Sigma-Aldrich | MX680S20 | NA |
| CDKN2A (p16INK4a) unconjugated mouse anti-human, mouse, rat | 2D9A12 | Abcam | ab54210 | |
| CD19 purified rat anti-mouse | 1D3 | BD Biosciences | 550284 | |
| Fixable Viability Dye eFluor506 | NA | Invitrogen/eBioscience | 65-0866-14 | 1:500 dilution |
| BD Horizon Brilliant Stain Buffer | NA | BD Biosciences | 563794 | NA |
| Transcription Factor Buffer Set | NA | BD Biosciences | 562574 | NA |
Note: Non-abbreviated names reflect propriety annotations used by the indicated vendor. AF, Alexa Fluor; APC, allophycocyanin; BB, brilliant blue; BV, brilliant violet; CD107a, lysosomal-associated membrane protein 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; FITC, fluorescein isothiocyanate; KLRG1, killer cell lectin-like receptor subfamily G member 1; MHCII, with major histocompatibility class II; NA, not applicable; PE, phycoerythrin; PerCP-Cy, peridinin chlorophyll protein-cyanine; p16INK4a, cyclin-dependent kinase inhibitor 2A; Tbet, T-box transcription factor; TIM3, hepatitis A virus cellular receptor 2.
Antibody information includes antigen, fluorochrome, host species, and target species.
Amount used per sample.
T Cell Purification
Pregnant mice were administered TCDD ( body weight) or peanut oil vehicle control on GD0, -7, and -14 and PND2. T cells were isolated from lymph nodes of adult female offspring (8–10 wk of age). Peripheral lymph nodes (cervical, axillary, brachial, inguinal, and sacral) were obtained from immunologically naïve mice. MLNs were harvested from mice 9 d after infection with IAV (HKx31). For naïve offspring, peripheral lymph nodes from a single mouse were combined, and there were three replicates per treatment group, with each offspring in a replicate from a unique dam. For IAV-infected offspring, MLNs were used. Mice were sacrificed 9 d after infection, and there were three replicates per treatment group, with 4–6 offspring in each unique pool of cells. All of the mice were sacrificed on the same day. Lymph nodes were processed to single-cell suspensions, and T cells were isolated using immunomagnetic negative selection to purity (Figure S2) using the MojoSort Mouse T cell kit (BioLegend), using reagents provided by the vendor and following the vendor’s instructions (https://www.biolegend.com/protocols/mojosort-isolation-kits-protocol-1/4599/), with the following change: the MagCellect Magnet (R&D Systems) was used.
RNA-Sequencing
RNA was isolated from purified T cells using the RNeasy Mini kit (Qiagen). Total RNA concentration was determined with a NanopDrop 1000 spectrophotometer (NanoDrop), and RNA quality measured using an Agilent Bioanalyzer 2100 (Agilent). RNA () was preamplified with the SMARTer Ultra Low RNA Kit for Illumina Sequencing (Clontech), following the manufacturer’s recommendations. The quantity and quality of cDNA was determined using the Qubit Fluorometer (Life Technologies), and Agilent Bioanalyzer 2100 (Agilent). complementary DNA (cDNA) () was used to generate Illumina-compatible sequencing libraries with the NexteraXT library preparation kit (Illumina), following the manufacturer’s protocols. Libraries were hybridized to the Illumina single-end flow cell and amplified using the cBot (Illumina) at a concentration of per lane. Single-end reads of 100 nucleotides (nt) were generated for each sample using Illumina’s HiSeq2500v4. Raw reads from the Illumina HiSeq2500 sequencer were demultiplexed using bcl2fastq (version 2.19.0; Illumina). Quality filtering and adapter removal were performed using Trimmomatic-0.36 (Bolger et al. 2014) with the following parameters: “TRAILING:13 LEADING:13 ILLUMINACLIP:adapters.fasta:2:30:10 SLIDINGWINDOW:4:20 MINLEN:15.” Processed/cleaned reads were then mapped to the Mus musculus reference sequence () with STAR-2.5.2b (Dobin et al. 2013) with the following parameters: “–twopassMode Basic –runMode alignReads –genomeDir –readFilesIn –outSAMtype BAM SortedByCoordinate –outSAMstrandField intronMotif –outFilterIntronMotifs RemoveNoncanonical.” The subread-1.5.0p3 (Liao et al. 2014) package (featureCounts) was used to derive gene counts given the following parameters: “-s 0 -t exon -g gene_name”. Differential expression analysis and data normalization was performed using DESeq2-1.14.1 (Love et al. 2014) with an adjusted -value threshold of 0.05 within an R (version 3.3.2; R Development Core Team) environment. Genes were considered differentially expressed (DEGs) when they had an adjusted . Pathway analysis was performed on DEGs using Ingenuity Pathways Analysis (Ingenuity Systems; http://www.ingenuity.com).
Whole-Genome Bisulfite Sequencing
DNA was isolated from purified T cells using the DNeasy blood and tissue kit (Qiagen), and processed for whole-genome bisulfite sequencing (WGBS). Genomic DNA was quantified using the Qubit fluorometer (Life Technologies). DNA integrity was determined using the TapeStation with genomics DNA tape reagents (Agilent). Methylation libraries were generated with Illumina’s TruSeq DNA Methylation Kit. Briefly, bisulfite conversion was carried out on of genomic DNA using Zymo EZ DNA Methylation Gold per the manufacturer’s instructions (Zymo Research). Conversion efficiency was determined using a Bioanalyzer 2100 (Agilent). Single-stranded cDNA was generated and tagged from bisulfite converted DNA using random hexamers containing a 5′ tag followed by terminal tagging to mark the 3′ end of the DNA. Illumina-specific adaptors were added during polymerase chain reaction amplification. DNA libraries were purified with AmpureXP beads and quantified with the Bioanalyzer 2100 and Qubit fluorometer. The amplified libraries were hybridized to the Illumina pair end flow cell and amplified using the cBot (Illumina) at a concentration of per lane. Pair end reads of 126 nt were generated on the Illumina HiSeq2500 for each sample. Raw reads generated from the Illumina HiSeq2500 sequencer were demultiplexed using bcl2fastq. Sequence reads were cleaned and adapter trimmed using trim-galore (version 0.4.3; FelixKrueger; https://github.com/FelixKrueger/TrimGalore) with the following parameters: “–paired –clip_r1 8 –clip_r2 8 –three_prime_clip_r1 8 –three_prime_clip_r2 8”. Alignment of each sample individually to the bisulfite converted mouse reference genome (GRCm38.p5) was accomplished with Bismark (version 0.18.1; Krueger and Andrews 2011) and the following parameters: “—bowtie2 –maxins 1000”. The accessory programs “filter_non_conversion” and “dedupicate_bismark” were applied to the resulting BAM files. The methylKit (version 1.4.0; Akalin et al. 2012) R package within an R (version 3.4.1) environment was used to extract methylation information in CpG context without overlap, filter loci with fewer than five reads and quality score lower than 10 out, normalize coverage across samples, and produce quality control plots. The genome was divided into 500-bp windows/tiles to assess differentially methylated regions (DMRs). Regions were united across all of the samples using the “” option (window covered in at least one sample in every group). DMRs were defined as a 500-bp window with a and a change in DNA methylation of at least 25%. The DMRs were annotated with the mm10 annotations using HOMER (version 4.9.1; Heinz et al. 2010).
Statistical Analysis
For these studies, the dam was the statistical unit. Hence, at each point in time relative to infection and in each developmental exposure group, the offspring were from a different treated dam. Except for RNASeq and WGBS data, all data were analyzed using JMP software (SAS Institute Inc.). Differences between means of vehicle or TCDD-treated groups at a single point in time were evaluated using a Student’s -test. A two-way analysis of variance, followed by a Tukey honestly significant difference post hoc test, was used to compare differences between multiple independent variables. The specific statistical test used is indicated in the legend of each figure. Differences were considered statistically significant when -values were . Error bars on all graphs and tables represent the standard error of the mean.
Data Availability
All WGBS and RNA-sequencing (RNA-seq) data have been deposited to the NIH Gene Expression Omnibus under accession number GSE143893 (WGBS) and GSE143894 (RNA-seq).
Results
Gene Expression Patterns in T Cells of Developmentally Exposed Mice
To characterize how triggering the AhR during development modifies T-cell responses later in life, the transcriptomes of T cells isolated from immunologically naïve mice and IAV-infected mice were examined using high-throughput RNA-seq (Figure 1A). In the absence of infection, gene expression profiles in T cells from mice that were developmentally exposed to vehicle control or TCDD were similar (Figure 1B). In fact, there were no DEGs in T cells from immunologically naïve adult offspring from the two treated groups of dams. However, this does not mean there was an absence of gene expression. It indicates that in the absence of infection, there were not any genes whose expression was significantly different due to maternal exposure to the AhR agonist TCDD. In contrast, following IAV infection, developmental exposure resulted in the differential expression of 163 genes (Figure S3). Thus, triggering the AhR during development, by maternal treatment with the ligand TCDD, induced differences that were not revealed until after the offspring’s immune system was challenged to respond.
Figure 1.
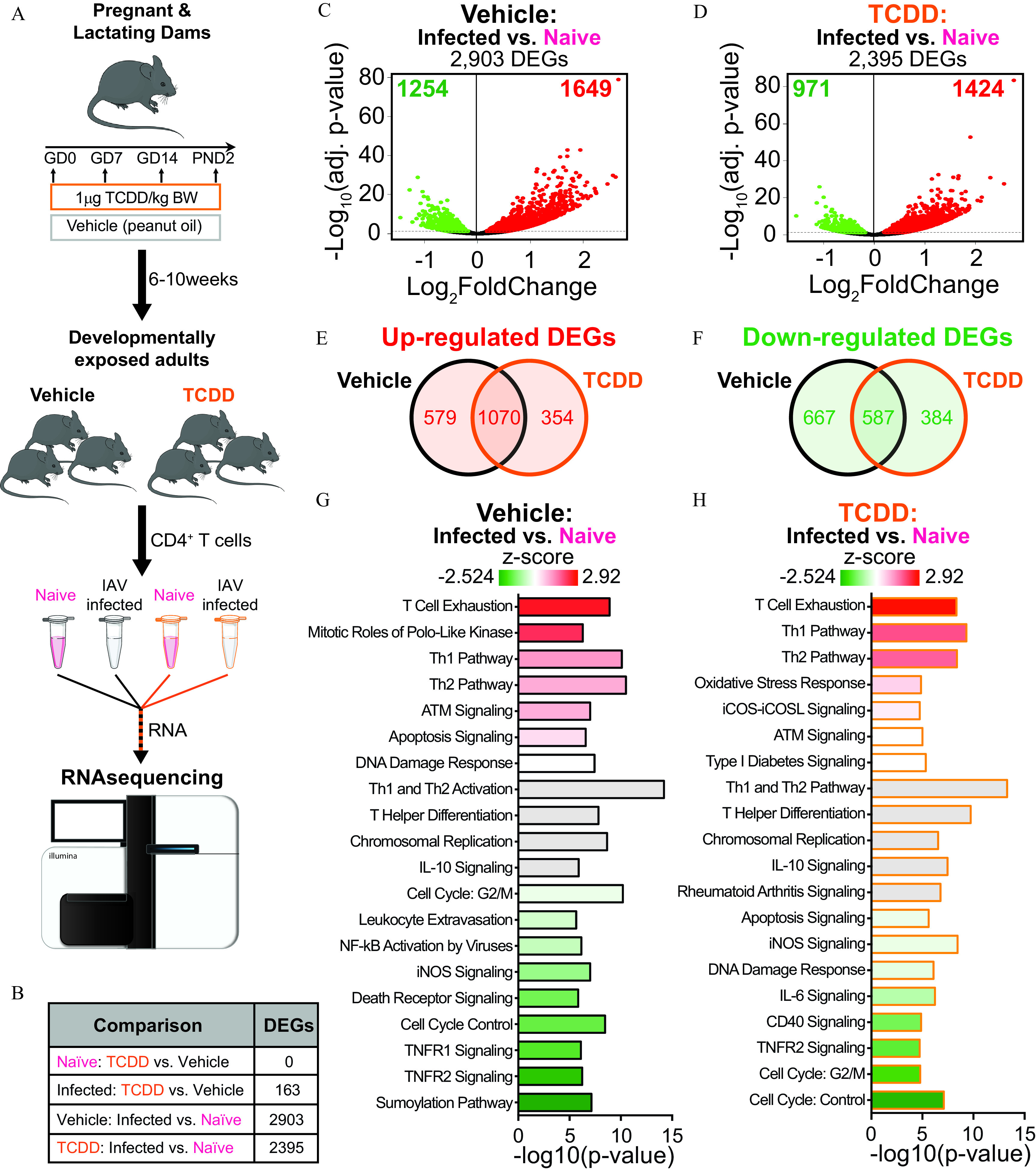
Gene expression profiles in T cells of naïve or influenza A virus (IAV)-infected mice developmentally exposed to vehicle or TCDD. (A) Pregnant mice were administered vehicle or TCDD ( body weight) on GD0, 7, and 14 and 2 d after parturition. T cells from lymph nodes of naïve or IAV-infected offspring (8–10 wk of age) were purified, RNA was isolated, and RNA-sequencing (RNA-seq) was performed. For immunologically naïve offspring, peripheral lymph nodes from a single mouse were combined, and there were a total of three replicates per treatment group, with 1 offspring from unique dams used per replicate, and 6 total mice used. For IAV-infected offspring, the mediastinal lymph nodes were used. Mice were sacrificed 9 d after infection, and there were three replicates per treatment group, with 4–6 offspring in each unique pool of cells for a total of 30 total mice used. (B) All of the mice (naïve and infected) were sacrificed on the same day with T-cell purification, RNA isolation, and RNA-seq performed in parallel. The number of differentially expressed genes (DEGs) was assessed. (C,D) Volcano plots of DEGs. On each plot, the direction of change indicates increased (right; red) or decreased (left; green) gene expression with infection compared with T cells from immunologically naïve (C) vehicle or (D) TCDD developmentally exposed mice. (E) Venn diagram of the number of DEGs that were up-regulated in both vehicle and TCDD groups, or uniquely up-regulated in only one group. (F) Venn diagram of the number of DEGs that were down-regulated by both vehicle and TCDD exposure, or uniquely down-regulated in only one group. (G,H) Ingenuity Pathway Analysis was used to identify cellular pathways affected by DEGs identified in (C) and (D). T-cell relevant pathways were ranked according to -score, in descending order of highest to lowest -score. Graphs depict pathways from (G) IAV-infected vs. naïve vehicle offspring or (H) IAV-infected vs. naïve TCDD offspring. The -axes denote (-value) for each pathway. Pathways were plotted according to -score, with highest -scores at the top (red) to lowest at the bottom (green). Gray bars were used when a -score was not predicted. Numerical details are provided in Table S1. Graphic art in (A) was manually created or adapted from Servier Medical Art templates (https://smart.servier.com) and licensed under the Creative Commons Attribution 3.0 Unported License agreement (https://creativecommons.org/licenses/by/3.0/). Note: ATM, ataxia telangiectasia mutated protein; BW, body weight; GD, gestational day; IL, interleukin; iCOS, inducible T-cell costimulator; iCOSL, inducible T-cell costimulator ligand; iNOS, inducible nitric oxide synthase; , nuclear factor kappa-light-chain-enhancer of activated B cells; PND, postnatal day; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; Th, T helper; TNFR, tumor necrosis factor receptor.
To further examine how developmental exposure shapes T-cell responses to infection, we compared DEGs in cells from naïve and IAV-infected mice within each exposure group. In T cells from IAV-infected mice that were not exposed to TCDD during development, there were 2,903 DEGs vs. immunologically naïve offspring of vehicle-treated dams (Figure 1C). In T cells from the offspring of TCDD-treated dams, there were 2,395 DEGs in infected vs. naïve mice (Figure 1D). In both developmental exposure groups, roughly 60% of the DEGs represented genes with higher expression (Figure 1C,D). Among the up-regulated genes, 1,070 DEGs were the same, regardless of maternal exposure group, whereas 354 genes with higher expression were unique to offspring of the TCDD exposed dams (Figure 1E). Similarly, more of the down-regulated DEGs were shared between T cells from vehicle and TCDD developmentally exposed offspring. Specifically, 587 infection-associated DEGs were common in vehicle and TCDD developmentally exposed offspring, whereas 384 genes were uniquely down-regulated as a result of developmental exposure (Figure 1F). Thus, when responding to antigen challenge, developmental exposure to TCDD altered the transcriptional profile of T cells, with differences that included enhanced and diminished gene expression.
Additional examination of the pathways influenced by IAV infection indicated several similarities and differences between T cells from the offspring of dams that were or were not exposed to an exogenous AhR agonist. For example, in both exposure groups, T-cell activation and T helper differentiation were among the major affected pathways (Figure 1G,H). In contrast, developmental AhR activation resulted in decreased activity of cell cycle-related pathways (e.g., cell cycle G2/M). When ranking the top 20 pathways enriched in infected vs. naïve comparisons, 14 were shared between vehicle and TCDD exposure groups (Figure S4). Therefore, IAV infection affected gene expression patterns in many similar pathways, regardless of developmental exposure. Despite this overlap, affected pathways in T cells from the offspring of control dams were at a lower rank in cells from mice developmentally exposed to TCDD (Figure S4). Thus, even among pathways shared by vehicle and TCDD groups, the manner in which they were affected was not the same.
Influence of Developmental AhR Activation on Senescence and Exhaustion
One of the most strongly enhanced pathways in both groups of developmentally exposed offspring was T-cell exhaustion (Figure 1G,H; Table S1). To determine whether developmental exposure to TCDD enhanced metrics of T-cell exhaustion, the expression of cytotoxic T-lymphocyte-associated protein 4 (CTLA4), programmed cell death protein 1 (PD1), and T cell immunoglobulin and mucin domain-3 (TIM3) was measured because increased expression of these molecules is associated with T-cell exhaustion (Akbar and Henson 2011). Flow cytometry analysis revealed that the number of T cells that expressed PD1 was significantly lower in mice that were exposed to TCDD during development (Figure 2A). Furthermore, the percentage of T cells was lower following developmental exposure to TCDD (Figure 2B). However, PD1 is also a cell-surface receptor expressed by T follicular helper (Tfh) cells. Developmental AhR activation reduced the percentage and the number of Tfh cells during IAV infection in mice (Boule et al. 2014; Burke et al. 2019). Therefore, the observation that there were fewer T cells may be due to a reduction in the number of Tfh cells, rather than a reduction in T-cell exhaustion. Similar to PD1, the number of T cells that expressed the inhibitory receptors CTLA4 (Figure 2C) and TIM3 (Figure 2E) was lower in mice developmentally exposed to TCDD. Yet, unlike PD1, the percentage of T cells that expressed CTLA4 (Figure 2D) and TIM3 (Figure 2F) were similar in mice regardless of developmental exposure group.
Figure 2.

Exhaustion markers on T cells during IAV infection following developmental AhR activation via maternal exposure to TCDD or vehicle. Vehicle (7 mice) or TCDD (8 mice) developmentally exposed mice were infected with IAV at maturity (6–8 wk of age). Nine days after infection, mice were euthanized and mediastinal lymph node cells stained for flow cytometry. (A,C,E) Bar graphs show the mean number of T cells from each exposure group that expressed (A) PD1, (C) CTLA4, or (E) TIM3. (B,D,F) Histograms depict the percentage of T cells from vehicle (gray solid line) or TCDD (orange dashed line) exposure groups that express (B) PD1, (D) CTLA4, or (F) TIM3. All values are . The numerical information, including -values, are provided in Table S2. All offspring within a treatment group were from separate dams. *, , using Student’s -test. Note: AhR, aryl hydrocarbon receptor; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; IAV, influenza A virus; PD1, programmed cell death protein 1; SEM, standard error of the mean; T, TCDD; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; TIM3, T-cell immunoglobulin and mucin domain-3; V, vehicle.
In addition to T-cell exhaustion, pathways associated with cellular senescence (Franceschi et al. 2000) were influenced by developmental exposure (Figure 1H). These include proliferation (e.g., mitotic roles for polo-like kinase, chromosomal replication, cell cycle: G2/M, and cell cycle control), and effector functions [e.g., leukocyte extravasation, nuclear factor kappa-light-chain-enhancer of activated B cells () activation by viruses, IL-6 signaling, and CD40 signaling], which were dampened or missing from the top pathways in T cells from the offspring developmentally exposed to TCDD during IAV infection. Two common markers associated with cellular senescence are killer cell lectin-like receptor subfamily G member 1 (KLRG1) and p16 (Henson and Akbar 2009; Liu et al. 2009; Suen et al. 2016). In mice that had been developmentally exposed to TCDD, there was a lower number (Figure 3A) and percentage (Figure 3B) of T cells that expressed KLRG1. Although the number was not different (Figure 3C), developmental AhR activation increased the percentage of cells that express p16 during IAV infection (Figure 3D). Effector functions of T cells during IAV infection include cytokine production and degranulation (Brown et al. 2004, 2016). Up-regulation of surface CD107a was used to evaluate degranulation (Hua et al. 2013; Lorenzo-Herrero et al. 2019). Following developmental AhR activation, the number (Figure 3E) and percentage (Figure 3F) of degranulating T cells were both lower compared with those of infected offspring of vehicle-treated dams. In addition, fewer T cells contained the cytotoxic protein perforin after early life AhR activation (Figure 3G,H).
Figure 3.

Evaluation of senescence-associated characteristics of T cells from the offspring of vehicle and TCDD-treated dams during IAV infection. Adult (6–8 wk of age) developmentally exposed mice were infected with IAV. Mediastinal lymph nodes (MLNs) were analyzed 9 d after infection. (A,C,E,G) The number of T cells from vehicle (7 mice) or TCDD (8 mice for KLRG1 and p16; 3 mice for CD107a and perforin) developmentally exposed offspring that express (A) KLRG1, (C) p16, (E) CD107a, or (F) perforin are shown as bar graphs. (B,D,F,H) Scatter plots show the percentage of (B) , (D) , (F) , or (H) T cells from infected mice that were developmentally exposed to vehicle (left) or TCDD (right). The numerical information is provided in Table S2. All offspring within a group were from separate dams. Values are . *, , using Student’s -test. Note: CD107a, lysosomal-associated membrane protein 1; IAV, influenza A virus; KLRG1, killer cell lectin-like receptor subfamily G member 1; p16, cyclin-dependent kinase inhibitor 2A; SEM, standard error of the mean; SSC-A, side scatter area; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin.
Persistence of Impaired T-cell Responses
A robust primary T-cell response to IAV infection influences anamnestic responses to subsequent infections (Sun and Braciale 2013; Wilkinson et al. 2012). Given that developmental activation of the AhR-dampened T-cell responses to primary IAV infection, we hypothesized T-cell responses would also be reduced during a subsequent IAV infection. A key metric of T-cell responsiveness is the clonal expansion of virus-specific cells (Chapman et al. 2005; Sant and McMichael 2012). Thus, the number of T cells that recognize an immunodominant epitope of IAV NP was evaluated in developmentally exposed offspring before infection, during primary IAV infection, and then prior to and after challenge with a second heterosubtypic IAV (Figure 4A). In naïve mice, the number of virus-specific T cells was similar in offspring that were developmentally exposed to vehicle or TCDD (Figure 4B). In response to primary infection (HKx31; H3N2), the number of NP-specific T cells increased in the offspring of vehicle control-treated dams. However, 9 d after infection, the number of virus-specific T cells in infected offspring of TCDD-treated dams was significantly lower than in the vehicle offspring (Figure 4C). After mice recovered from primary IAV infection, the number of virus-specific T cells was no longer significantly different between the two groups (Figure 4D). However, when they were inoculated with a heterosubtypic IAV (PR8; H1N1), expansion of virus-specific T cells was significantly lower in mice that had been developmentally exposed to TCDD.
Figure 4.

Assessment of T cells from developmentally exposed mice during primary and recall responses to IAV. (A) Scheme of experimental set up. Mice were developmentally exposed to vehicle (V) or TCDD (T) and experiment was initiated when the offspring were 6–8 wk of age. (B) The number of virus-specific T cells in the lymph nodes of naïve mice (8 vehicle; 9 TCDD) was quantified using flow cytometry. (C,D) Offspring of vehicle-treated dams and offspring of TCDD-treated dams were challenged with HKx31 (primary infection). (C) On Day 9 post primary infection, the number of virus-specific T cells in 8 mice from the vehicle exposure group and 8 mice from the TCDD group was determined using flow cytometry. (D) Mice recovered from primary IAV infection for 79 d. The number of virus-specific T cells was quantified prior to (7 vehicle; 7 TCDD) and 7 d after PR8 infection (secondary infection; 8 vehicle; 9 TCDD). The mice were 17–19 wk of age at the time of the secondary infection. The mean number of cells and -values are provided in Table S3. At each point in time, offspring within the same exposure group were from separate dams. All values are . *, , using Student’s -test. Note: IAV, influenza A virus; LN, lymph node; MLN, mediastinal lymph node; SEM, standard error of the mean; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; 1°, primary; 2°, secondary.
Influence of Developmental Exposure and Immune Challenge on DNA Methylation Patterns in T Cells
Because changes to T-cell responses and gene expression patterns were long lasting, we assessed epigenetics as an underlying mechanism. Specifically, DNA was purified from the same T cells that were used for RNA-seq in Figure 1. DNA was subjected to WGBS to evaluate DNA methylation patterns across the genome of these cells. In the absence of infection, T cells from vehicle and TCDD-exposed offspring had similar levels of global DNA methylation (Figure 5A). Compared with T cells from naïve animals, T cells from animals infected with IAV had significantly lower global DNA methylation levels, and this was more pronounced in the offspring of the dams given TCDD. Specifically, in the offspring of control dams, infection was associated with 1.8% lower overall DNA methylation (; Table S4). In T cells from mice that were exposed developmentally to TCDD, the overall DNA methylation level was lower by 2.6% (; Table S4). Thus, T cells from infected offspring that were developmentally exposed to TCDD had significantly lower levels of global DNA methylation compared with IAV-infected offspring of vehicle dams (). Thus, developmental exposure impacted DNA methylation levels in T cells, and, at a genome-wide level, differences became apparent after an immune challenge.
Figure 5.

DNA methylation patterns in T cells from naïve IAV-infected mice that were developmentally exposed to TCDD or vehicle. Mice were developmentally exposed to vehicle or TCDD, as described in Figure 1. Whole-genome bisulfite sequencing was performed using DNA from purified T cells from naïve or IAV-infected adult mice (8–10 wk of age). For naïve offspring, peripheral lymph nodes from 1 mouse were pooled, and there was a total of three replicates per treatment group, with 1 offspring from unique dams used per replicate, for a total of 6 mice. For IAV-infected offspring mediastinal lymph nodes were used. Mice were sacrificed 9 d after infection, and there were three replicates per treatment group, with 4–6 offspring in each unique pool of cells for a total of 30 total mice used. All of the mice were sacrificed at the same time. (A) Box plots show global DNA methylation levels. The horizontal line denotes the mean, and boxes depict SEM. Differences between groups were evaluated using a two-way ANOVA with Tukey’s HSD post hoc test, and -values are reported in Table S4. Groups with the same letter were not significantly different from each other. (B) The number of differentially methylated regions (DMRs) that were hypo- (blue; left side) or hyper- (red; right side) methylated are shown. The length of the bar is proportionate to the number of DMRs, and the number of DMRs is denoted on the bar segments. (C–F) Modified Manhattan plots show the distribution of hypomethylated (blue dots; left cluster) and hypermethylated (red dots; right cluster) DMRs across chromosomes in T cells. (C,D) DMRs in T cells from naïve (C) or IAV-infected TCDD (D) mice compared with vehicle controls. (E,F) DMRs in T cells from vehicle (E) or TCDD (F) mice during IAV infection compared with naïve controls. The direction of hyper- and hypomethylation in (B) top two comparisons, (C) and (D) refer to the effect of developmental exposure to TCDD, or refers to the effect of infection in (B) bottom two comparisons, (E) and (F). Global methylation levels were analyzed by ANOVA with a Tukey HSD post hoc test. (G–J) DMRs were mapped to the genome. The total numbers of DMRs are shown. The percent of DMRs in intergenic, intron, TSS, TTS, 3′UTR, exon, noncoding, and 5′UTR regions are shown. The following comparisons were made: (G) DMRs from T cells from naïve TCDD vs. naïve vehicle mice, (H) DMRs from T Cells from IAV-infected TCDD vs. IAV-infected vehicle mice, (I) DMRs from T cells from vehicle IAV-infected vs. vehicle naïve mice, and (J) DMRs from T cells from TCDD IAV-infected vs. TCDD naïve mice. Note: ANOVA, analysis of variance; HSD, honestly significant difference; IAV, influenza A virus; SEM, standard error of the mean; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; TSS, transcription start site; TTS, transcription termination site; UTR, untranslated region.
To better understand the extent to which developmental AhR activation impacted DNA methylation, DNA methylation patterns were characterized within discrete regions across the genome. DMRs were defined as having a significant methylation change of within 500 bp regions. Triggering the AhR during development caused about 22,000 DMRs in naïve T cells, and 18,752 DMRs in T cells isolated from IAV-infected mice (Figure 5B). In naïve animals, the number of DMRs that were hypo- or hypermethylated were roughly similar, which is in accordance with similar global levels of DNA methylation (Figure 5B). Moreover, DMRs spanned all chromosomes prior to and after infection, in both exposure groups (Figure 5C–F). Following infection, a greater proportion (57%) of the DMRs in the TCDD group represented hypomethylated DNA, which is consistent with the overall reduction in global DNA methylation (Figure 5B). However, infection with IAV caused 20% more DMRs (29,563) in mice developmentally exposed to TCDD compared with vehicle controls (22,911 DMRs), indicating developmental AhR activation drove dysregulation of infection-induced DNA methylation changes. Furthermore, there were 4% more hypomethylated DMRs in TCDD developmentally exposed cells (62%) compared with vehicle controls (58%) as a result of IAV infection. These results show that developmental exposure of mice to TCDD did not preferentially affect hypo- or hypermethylation of DNA in T cells but, instead, changed DNA methylation patterns overall such that DMRs throughout the entire genome of T cells included regions with greater and lower DNA methylation.
DNA methylation at different genomic features influences cellular functions, although precisely how is not fully understood (Moore et al. 2013; Schmidl et al. 2018; Tirado-Magallanes et al. 2017). To identify specific genomic features that were preferentially influenced by developmental exposure, the DMRs across the T-cell genome were annotated. This analysis showed that although the number of DMRs differed between all four comparison groups (Figure 5B), they spanned similar genomic features (Figure 5G–J). In all groups, over 60% of all DMRs were found in intergenic regions. In addition, DMRs were found in promoters, gene bodies (introns and exons), transcription termination sites (TTS), and untranslated regions (UTRs). Although promoter methylation generally has a strong influence on gene expression and function, only 2% of DMRs were found in promoter regions. These results show that both an immune challenge and developmental exposure influenced DNA methylation patterns across all genomic features in T cells.
Integrating Gene Expression and DNA Methylation
One of the best-studied ways in which DNA methylation affects cellular function is by regulating gene expression. Therefore, we integrated transcriptomic and genome-wide DNA methylation analyses. Gene expression fold changes did not correlate with DMRs across all genomic regions, or within specific regions associated with each DEG (Figure S5). Others have reported a similar lack of correlation between promoter DNA methylation and gene expression (Akemann et al. 2019). Yet, a subset of the DMRs that we observed had a strong correlation with gene expression changes (blue dots; Figure S5). This was observed regardless of maternal exposure group. For instance, compared with T cells from naïve mice, cells from IAV-infected offspring of vehicle dams had DMRs associated with 8,922 genes. Of these genes, 766 were also differentially expressed (Figure 6A). Although this accounts for only a quarter of all DEGs, these genes were involved in cellular pathways important for T-cell functions during IAV infection, including signaling, T-cell receptor signaling, and Th cell activation pathways (Figure 6B). Performing a similar comparison in T cells from developmentally exposed offspring revealed DMRs associated with 11,559 genes, of which 707 were differentially expressed (Figure 6C). Among the DEGs with DMRs, some were similar to the vehicle group, such as those involved in signaling and Th cell activation pathways (Figure 6D). Unlike vehicle offspring T cells, the TCR signaling pathway was not among the top pathways influenced by DNA methylation and gene changes in offspring that had developmental AhR activation. Furthermore, T cells from mice that were developmentally exposed to TCDD had reduced activities of co-stimulatory pathways, and Tec kinase signaling was increased.
Figure 6.

Gene expression and DNA methylation data set integration. The same cells were used for whole-genome bisulfite sequencing (WGBS) and RNA-seq. (A,C) Venn diagrams show the overlap of genes with differentially methylated regions (DMRs), assessed by WGBS, and differentially expressed genes (DEGs), assessed by RNAseq. (A) The DEG and DMR overlap shown for T cells isolated from vehicle mice prior to and during IAV infection. (C) The DEG and DMR overlap for T cells isolated from naïve and infected TCDD offspring. (B,D) Ingenuity Pathway Analysis was used to identify and rank T-cell relevant cellular pathways associated with genes with differential expression and DMRs (overlap in Venn diagrams). The pathways impacted by IAV infection in T cells from (B) vehicle or (D) TCDD developmentally exposed offspring were ranked in descending order according to -value. Pathways were colored according to -score from (diminished activity, green) to 1.606 (enhanced activity, red). Gray bars were used when a -score was not predicted. The numerical data corresponding to (B) and (D) are in Table 5S. Note: GNRH, gonadotropin-releasing hormone; IAV, influenza A virus; iCOS, inducible T-cell costimulator; iCOSL, inducible T-cell costimulator ligand; IL, interleukin; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; , nuclear factor kappa-light-chain-enhancer of activated B cells; PEDF, pigment epithelium-derived factor; , protein kinase C theta; PPARa, peroxisome proliferator-activated receptors; RNA-seq, RNA sequencing; sig, signaling; RXRa, retinoid X receptor; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; TCR, T cell receptor; Th, T helper; TNFR, tumor necrosis factor receptor.
Modifying T-cell Responses with DNA Methylation-Altering Drugs
Given that important T-cell function pathways were influenced by developmentally induced changes in DNA methylation and gene expression, we sought to determine whether influencing DNA methylation modifies the impaired T-cell responses caused by developmental exposure. To do this, we used pharmacological interventions that influence DNA methylation levels (Chen et al. 2012; TWH Li et al. 2012; Mato et al. 2013; Yoo et al. 2008). To assess the contribution of DNA hypomethylation we used SAM, which increases global DNA methylation levels by increasing the supply of methyl group donors (TWH Li et al. 2012; Mato et al. 2013). We posited that if developmental exposure affected T-cell function by reducing DNA methylation, then SAM treatment would abrogate this effect. At weaning, offspring that were developmentally exposed to vehicle or TCDD were randomly assigned to remain on untreated water (control group) or to receive water that contained SAM (Figure 7A). At maturity, mice were infected with IAV and then maintained on treated water throughout infection. SAM treatment did not influence the amount of water consumed by the mice during the treatment period (Figure S1A). Similarly, age-related weight gain was not influenced by treatment with SAM (Figure S1B). Treatment with SAM for did not impair the overall growth or health of the mice, consistent with prior reports that used DNA methylation-altering drugs in older mice for shorter periods of time (Chiang et al. 1996; TWH Li et al. 2012; Mato et al. 2013; Yoo et al. 2008). To the best of our knowledge, long-term SAM treatment has not been used in combination with IAV infection. Thus, we also determined whether drug treatment affected morbidity or mortality upon IAV infection. Upon infection, all mice lost weight (Figure S1), but there was no statistically significant difference between the groups. Furthermore, within each developmental exposure group, SAM treatment did not influence morbidity and none of the offspring died as a result of infection (Figure S1). This suggests that the overall health of the mice prior to and during IAV infection was not adversely affected by SAM treatment.
Figure 7.

Effects of treatment with S-adenosyl methionine (SAM) on the T-cell response to IAV infection in developmentally exposed mice. (A) Mice were developmentally exposed to vehicle or TCDD. At weaning (21 d of age), mice were randomly divided and given normal vivarium water to maintain DNA methylation levels or water containing SAM () to increase DNA methylation levels. At 6–8 wk of age, offspring of dams given vehicle (V; white bar) and TCDD (T; orange bar) were infected with IAV (HKx31). Mediastinal lymph nodes were harvested 9 d after infection, and cells stained for flow cytometry. (B) The total number of T cells or (C) virus-specific T cells were quantified from mice on control (black outline) or SAM (purple outline) water. The percentage of (D) activated () or (E) naïve () T cells from control or SAM mice. The percentage of (F) Th1 () or (G) Tfh () isolated from control or SAM mice. The number of mice in each group was as follows: control water vehicle (8), control water TCDD (5), SAM water vehicle (8), and SAM water TCDD (6). Due to a limited number of offspring, individual offspring were defined as the statistical unit, rather than the dam. All data shown are . *, , using a two-way ANOVA with Tukey’s HSD and Student’s -test. #, , using Student’s -test. The numerical data in the graphs, including -values are provided in Table S6. Note: ANOVA, analysis of variance, HSD, honestly significant difference; , water; IAV, influenza A virus; SEM, standard error of the mean; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; Tfh, T follicular helper; Th, T helper.
The expansion of T cells during IAV infection was evaluated in developmentally exposed mice that were and were not treated with SAM from weaning through maturity. Consistent with previous observations, developmental exposure to TCDD resulted in significantly lower total (Figure 7B) and virus-specific T cells (Figure 7C) in IAV-infected mice by half and two-thirds; respectively. In contrast, after treatment with SAM, the number of (Figure 7B) and virus-specific T cells (Figure 7C) were not significantly different in infected vehicle and TCDD offspring. Although it did not restore the number to the same level observed in infected offspring of vehicle-treated dams, treatment with SAM negated the down-modulatory effects of developmental AhR activation on these metrics of T-cell expansion upon infection.
DNA methylation also influences T-cell activation and differentiation (Durek et al. 2016; Schmidl et al. 2018). During IAV infection, developmental AhR activation resulted in a lower percentage of activated () T cells (Figure 7D) and correspondingly higher percentage of naïve () T cells (Figure 7E). A similar difference (e.g., lower numbers of activated T cells and higher numbers of naïve T cells) was observed in mice that were developmentally exposed to TCDD and treated with SAM (Figure 7D,E). Developmental exposure to TCDD resulted in a lower percentage of T cells that differentiated into T helper-1 (Th1) cells (Figure 7F) and Tfh cells (Figure 7G). Although SAM was able to restore the lower Th1 response in IAV-infected offspring of TCDD-treated dams (Figure 7F), SAM was unable to restore the Tfh response in the same mice (Figure 7G). Thus, increasing DNA methylation levels alleviated deficits in clonal expansion and some, but not all, aspects of T helper differentiation following developmental exposure of mice to TCDD.
To define the effects of developmentally associated DNA hypermethylation, similar experiments were conducted by treating offspring with Zeb (Figure 8A), which lowers global DNA methylation by blocking DNA methyltransferases (Chen et al. 2012; Yoo et al. 2008). Similar to SAM, water consumption and body weight were not affected by Zeb treatment (Figure S1A,B). Likewise, 4-wk treatment with Zeb did not adversely affect host resistance to infection (Figure S1C–E). However, unlike treatment with SAM, Zeb did not ameliorate the effects of developmental exposure on T-cell clonal expansion. That is, the numbers of (Figure 8B) and virus-specific T cells (Figure 8C) were still significantly lower in IAV-infected Zeb-treated offspring of TCDD-treated dams. In contrast, treatment with Zeb restored the skewed percentage of activated and naïve T cells (Figure 8D,E) in the offspring of TCDD-treated dams. Likewise, Zeb treatment abrogated the diminished Th1 cells (Figure 8F) and Tfh cells (Figure 8G) in TCDD developmentally exposed mice such that the frequency was similar to that of the vehicle-treated group.
Figure 8.

Effects of Zebularine (Zeb) on T cells from developmentally exposed mice after infection with IAV. (A) Mice were developmentally exposed to vehicle or TCDD. At weaning (21 d of age), mice were randomly selected be given normal vivarium water to maintain DNA methylation levels or water containing Zeb () to decrease DNA methylation levels. At 6–8 wk of age, mice developmentally exposed to vehicle (white bar) and TCDD (orange bar) were infected with IAV. Mediastinal lymph nodes (MLNs) were harvested 9 d after infection, and cells stained for flow cytometry. (B) The number of T cells or (C) virus-specific T cells were quantified from MLNs of mice on control (black outline) or Zeb (green outline) water. The percentage of (D) activated () or (E) naïve () T cells from control or Zeb mice. The percentage of (F) Th1 () or (G) Tfh () isolated from control or Zeb mice. For each group, 5–9 developmentally exposed offspring were used: control water vehicle (8), control water TCDD (5), Zeb water vehicle (9), and Zeb water TCDD (6). Due to a limited number of offspring, individual offspring were defined as the statistical unit, rather than the dam. All data shown are . *, , using a two-way ANOVA with Tukey’s HSD and Student’s -test. #, , using Student’s -test. The numerical data in the graphs and -values are provided in Table S7. Note: ANOVA, analysis of variance; HSD, honestly significant difference; , water; IAV, influenza A virus; SEM, standard error of the mean; T, TCDD; Tbet, T-box transcription factor; TCDD, 2,3,7,8-tetrachlorodibenzo--dioxin; Tfh, T follicular helper; Th, T helper; V, vehicle.
Discussion
There is growing recognition that human health is influenced not only by contemporaneous exposures, but also by the environment experienced earlier in life, including in the womb (Boekelheide et al. 2012; Cao et al. 2016; Dallaire et al. 2004; Glynn et al. 2008; Heilmann et al. 2010; Hochstenbach et al. 2012; Jusko et al. 2014, 2016; Stølevik et al. 2011; 2013). Yet, how exposures during development shape the immune system’s capacity to respond later in life are not well understood. The results of the present study show that early life exposure to the AhR agonist TCDD not only impacted the primary immune response to IAV, but also led to long-lasting consequences that also affected T-cell responses to a second immune challenge with a similar pathogen. This illustrates the durable effect that early life exposure to an AhR agonist, such as TCDD, can have on the functional capacity of T cells. Early life exposure of mice to environmentally derived AhR ligands affects T cells in an enduring manner, but the cellular mechanisms remain poorly defined (Boule et al. 2014, 2015a, 2015b; Burke et al. 2019). Our results have elucidated one of the cellular mechanisms—developmental signaling through the AhR programmed DNA methylation and gene expression profiles of T cells—and these alterations contributed to their impaired response to infection. Yet, this was not due to AhR targeting a single-cellular function or pathway. For instance, prior to infection, differences in gene expression were difficult to discern and differences in DNA methylation were modest. However, after infection, there were pronounced differences in both gene expression and DNA methylation patterns. This indicates that the AhR likely influences multiple pathways in T cells, which collectively regulate the responsive capacity of T cells in a durable manner. This also suggests that some effects of early life exposures are cryptic and are only revealed upon another challenge, such as the call to arms triggered by infection.
A key finding from this present study was that developmental AhR activation changed DNA methylation patterns in T cells prior to and during an immune challenge. This is consistent with a previous report that developmental AhR activation caused genome-wide changes in DNA methylation patterns in T cells of mice (Winans et al. 2015). Similar to T cells, T cells from TCDD developmentally exposed mice had lower levels of global DNA methylation during IAV infection compared with cells from infected vehicle control offspring (Winans et al. 2015). This suggests that a common mechanism driving durable changes to cellular function after developmental exposure to an AhR agonist may be dysregulation of DNA methylation patterns in T cells. Further support for this idea was recently described within a human population. After early life exposure to AhR ligands, changes in DNA methylation patterns in human whole blood were observed, and they persisted into adulthood (Su et al. 2019). Moreover, methylation of 11 specific genes correlated with maternal exposure levels (Su et al. 2019). Seven of these 11 genes were differentially methylated in T cells from naïve mice that were developmentally exposed to TCDD, and 5 of the 7 genes remained differentially methylated during IAV infection in mice (Table S8). Thus, in mouse and human studies, exposure to AhR-binding chemicals durably changes DNA methylation patterns in immune cells. These findings indicate that changes in DNA methylation may be a candidate biomarker for early life exposures.
Another key finding was that activation of the AhR during development modulated aspects of functional states associated with impaired responsive capacity: cellular senescence and exhaustion. These processes are characterized by poorer cellular proliferation and dysregulated effector functions, although depressed effector function is more typical of exhausted T cells (Crespo et al. 2013). Consistent with previously published reports that developmental AhR activation reduced interferon-gamma () production by T cells during IAV infection (Boule et al. 2014; Burke et al. 2019), the results presented here further demonstrate that developmental AhR activation reduced other metrics of T-cell effector function, such as CD107a and perforin levels. Cellular exhaustion and senescence are commonly observed during chronic infection and aging, respectively (Akbar and Henson 2011). However, neither should occur when nonaged animals are infected with a pathogen that does not persist or form latent reservoirs, such as IAV. Thus, observing enhancement of markers related to these processes in nonaged animals and in the absence of chronic immune challenge is indicative of dysfunctional T-cell responses (Akbar and Henson 2011; Kasakovski et al. 2018). Interestingly, there was a reduction in the number and percentage of T cells that express the exhaustion marker PD1. Using this classical marker of T-cell exhaustion proved an inconclusive sole marker to identify T-cell exhaustion because PD1 is also expressed by an important T-cell helper subset: Tfh cells. Developmental AhR activation reduced the frequency of mouse Tfh cells during an immune response to IAV (Boule et al. 2014; Burke et al. 2019). Thus, it is more likely that the lower percentage of cells observed in the present study reflects the impaired ability of T cells to differentiate into Tfh cells, rather than diminished T-cell exhaustion. A previous study showed that T cells from mice exposed to TCDD during development proliferated less in vivo after immune challenge, and changes in cellular metabolism may play a role in their reduced function (Burke et al. 2019). The work reported here extends this, and suggests that altered cellular metabolism may contribute to inappropriate T-cell exhaustion or premature senescence (Schurich and Henson 2014). In addition, analysis of DEGs in T cells from IAV-infected mice that were developmentally exposed to TCDD indicated that early life AhR activation enhanced aspects of T-cell exhaustion in T cells as well (Winans et al. 2015). Given that exhausted cells have an impaired ability to fight infections and protect the host, this could be one of the reasons that T-cell responses are reduced following developmental AhR activation.
Despite changes in gene expression in T cells from infected mice, early life exposure to TCDD was not associated with significant differences in gene expression in T cells from naïve mice. There are multiple theories that could account for this. A simplistic explanation is that this reflects the overall lack of overt changes to immune system development in mice developmentally exposed to this dose of AhR agonist (Boule et al. 2014; Vorderstrasse et al. 2004). Yet, a more likely explanation is the transcriptionally quiescent state of resting T cells in the absence of an immune challenge (Feng et al. 2010, 2011; Zhang et al. 2018). The dearth of DEGs in naïve T cells is similar to observations in T cells from uninfected offspring of TCDD and control treated dams, in which there were only a handful of DEGs (Winans et al. 2015). Thus, developmentally induced changes in the responsive capacity of T cells are cryptic or latent in naïve cells. Yet, they are revealed once the transcription machinery is stimulated, and the cells respond to an immune challenge. Pervasive differences in DNA methylation were present across all genomic features of T cells from naïve mice, which suggests that exposures during early life shape the way in which T cells are poised to respond to immune challenges encountered later in life and that DNA methylation patterns likely contribute to this poised or programmed state.
Another novel finding reported here was that developmentally induced differences in T-cell functions were modifiable later in life by DNA methylation-altering drugs. This supports the idea that changes in DNA methylation patterns are part of the causative mechanism driving altered T-cell responses that result from developmental AhR activation. Yet, altered T-cell responsive capacity was not solely due to skewing of DNA methylation in a single direction, given that increasing or decreasing overall DNA methylation affected different aspects of T-cell responses to IAV in offspring of TCDD-treated dams. Specifically, postnatal Zeb treatment restored diminished T-cell differentiation in developmentally exposed mice. This is consistent with evidence that DNA de-methylation of specific genes plays a role in naïve T-cell differentiation into helper subsets (Komori et al. 2015; Y Li et al. 2012). Zeb reduces DNA methylation by blocking DNA methyltransferases, whereas SAM increases DNA methylation by providing an additional supply of donor methyl groups (Chiang et al. 1996; Zhou et al. 2002). As a consequence, SAM’s methyl group can be transferred to both DNA and histones (Chiang et al. 1996; Detich et al. 2003; Mews and Berger 2016). Thus, perhaps it is not surprising that SAM treatment elicited different outcomes than Zeb treatment. Although Zeb treatment did not affect the metrics of clonal expansion, SAM treatment restored the reduced clonal expansion of T cells in mice that had been developmentally exposed to TCDD. This suggests that increasing DNA methylation benefits T-cell proliferation. This idea is supported by evidence from cancer studies, where aberrant DNA hypermethylation results in the silencing of tumor suppressor genes, increased proliferation, and tumor progression (Dahl et al. 2007; Gonzalez-Zulueta et al. 1995; Kane et al. 1997; Lin et al. 2017; Pilozzi et al. 2004). Although not examined in the context of immune cells, it is reasonable to hypothesize that SAM treatment restored impaired T-cell proliferation through a similar mechanism. In addition, unlike Zeb, SAM was able to restore only the depressed Th1 cell response, and not the Tfh cell response. This subset-specific effect suggests that differentiation into Th1 and Tfh cells is differentially regulated by DNA methylation. Together, these observations indicate that the mechanisms underlying changes to T-cell responses in developmentally exposed animals are complex and that DNA methylation levels influence cellular responses in discrete ways.
Given that enhancing or reducing overall DNA methylation restored some, but not all, of the impaired T-cell responses in the adult offspring of TCDD-treated dams, developmental programming is not solely controlled by DNA methylation. Transfer of naive T cells from developmentally exposed mice into recipients that have never been exposed revealed that they maintained the impaired responsive capacity (Boule et al. 2014). When considered together, these findings suggest that differences established by early life exposure to an AhR binding pollutant, such as TCDD, can be carried within the T-cell lineage and that additional regulatory mechanisms contribute to their altered functional capacity. Similar to DNA methylation marks, histone modifications can be inherited by daughter cells throughout the lifetime of an organism, contributing to long-lasting influences on gene expression patterns (Henikoff and Greally 2016; O’Kane and Hyland 2019; Zheng et al. 2016). Although not studied in the context of developmental exposure or in T cells, the AhR is capable of interacting with histone deacetylases (HDACs), including HDAC1, HDAC2, and HDAC8 (Chang et al. 2014; Gomez-Duran et al. 2008; Schnekenburger et al. 2007; Wang et al. 2017). Although these studies were performed in tumor cell lines, they suggest that AhR activation influences histone modifications. Further support that early life exposures may elicit durable changes to histone modifications stems from investigations of developmental exposure to bisphenol A, which causes long-term histone modifications in nonimmune organs (Doherty et al. 2010; Greathouse et al. 2012; Strakovsky et al. 2015). Together, these observations support the idea that in addition to DNA methylation, other epigenetic mechanisms likely contribute to the ways in which early life AhR activation influences T cells in a durable manner.
The results presented in this study have broad-spanning implications because T cells play integral roles in numerous immune responses, ranging from fighting infections and destroying tumor cells to driving allergic and autoimmune diseases (Knochelmann et al. 2018; MacLeod et al. 2009; Zhu and Paul 2008). We identified DNA methylation changes as one of the mechanisms underlying durable differences in T-cell responsive capacity in mice resulting from early life AhR activation. Given that changes to DNA methylation patterns can be passed on to future generations, our results suggest that a person’s health may be programmed not only by the environment they experience, but also by the environmental exposures experienced by their parents. The work described here expands our understanding of how early life exposures can cause long-lasting changes to the immune system. Moreover, it brings us one step closer to devising optimally targeted therapeutics by uncovering DNA methylation as a mechanism of early life exposure-induced immune dysfunction.
Supplementary Material
Acknowledgments
The authors are grateful to T. Bushnell and M. Cochran at the URMC Flow Cytometry Core and J. Ashton and M. Zanche at the UR Genomics Research Center. This work was supported by grants from the National Institutes of Health (R01-ES017250, R01-ES023260, R01-ES004862, P30-ES01247, T32-ES007026, and T32-AI007285).
References
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. 2012. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13(10):R87, PMID: 23034086, 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar AN, Henson SM. 2011. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol 11(4):289–295, PMID: 21436838, 10.1038/nri2959. [DOI] [PubMed] [Google Scholar]
- Akemann C, Meyer DN, Gurdziel K, Baker TR. 2019. Developmental dioxin exposure alters the methylome of adult male zebrafish gonads. Front Genet 9:719, PMID: 30687390, 10.3389/fgene.2018.00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Inglis SC. 1985. Growth, purification and titration of influenza viruses. In Virology: A Practical Approach. Mahy BWJ, ed. Washington, DC: IRL Press, 119–150. [Google Scholar]
- Basson MA. 2012. Signaling in cell differentiation and morphogenesis. Cold Spring Harb Perspect Biol 4(6):a008151, PMID: 22570373, 10.1101/cshperspect.a008151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekelheide K, Blumberg B, Chapin RE, Cote I, Graziano JH, Janesick A, et al. 2012. Predicting later-life outcomes of early-life exposures. Environ Health Perspect 120(10):1353–1361, PMID: 22672778, 10.1289/ehp.1204934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120, PMID: 24695404, 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule LA, Burke CG, Fenton BM, Thevenet-Morrison K, Jusko TA, Lawrence BP. 2015a. Developmental activation of the AHR increases effector CD4+ T cells and exacerbates symptoms in autoimmune disease-prone Gnaq+/– mice. Toxicol Sci 148(2):555–566, PMID: 26363170, 10.1093/toxsci/kfv203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule LA, Lawrence BP. 2015. Influence of early life environmental exposures on immune function across the life span. In: Environmental Influences on the Immune System. Esser C, ed. New York, NY: Springer Berlin Heidelberg, 21–54. [Google Scholar]
- Boule LA, Winans B, Lambert K, Vorderstrasse BA, Topham DJ, Pavelka MS Jr, et al. 2015b. Activation of the aryl hydrocarbon receptor during development enhances the pulmonary CD4+ T-cell response to viral infection. Am J Physiol Lung Cell Mol Physiol 309(3):L305–L313, PMID: 26071552, 10.1152/ajplung.00135.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule LA, Winans B, Lawrence BP. 2014. Effects of developmental activation of the AhR on CD4+ T-cell responses to influenza virus infection in adult mice. Environ Health Perspect 122(11):1201–1208, PMID: 25051576, 10.1289/ehp.1408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Dilzer AM, Meents DL, Swain SL. 2006. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol 177(5):2888–2898, PMID: 16920924, 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- Brown DM, Lampe AT, Workman AM. 2016. The differentiation and protective function of cytolytic CD4 T cells in influenza infection. Front Immunol 7:93, PMID: 27014272, 10.3389/fimmu.2016.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Román E, Swain SL. 2004. CD4 T cell responses to influenza infection. Semin Immunol 16(3):171–177, PMID: 15130501, 10.1016/j.smim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Burke CG, Myers JR, Boule LA, Post CM, Brookes PS, Lawrence BP. 2019. Early life exposures shape the CD4+ T cell transcriptome, influencing proliferation, differentiation, and mitochondrial dynamics later in life. Sci Rep 9(1):11489, PMID: 31391494, 10.1038/s41598-019-47866-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Xu X, Hylkema MN, Zeng EY, Sly PD, Suk WA, et al. 2016. Early-life exposure to widespread environmental toxicants and health risk: a focus on the immune and respiratory systems. Ann Glob Health 82(1):119–131, PMID: 27325070, 10.1016/j.aogh.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Chang CC, Sue YM, Yang NJ, Lee YH, Juan SH. 2014. 3-Methylcholanthrene, an AhR agonist, caused cell-cycle arrest by histone deacetylation through a RhoA-dependent recruitment of HDAC1 and pRb2 to E2F1 complex. PLoS One 9(3):e92793, PMID: 24658119, 10.1371/journal.pone.0092793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman TJ, Castrucci MR, Padrick RC, Bradley LM, Topham DJ. 2005. Antigen-specific and non-specific CD4+ T cell recruitment and proliferation during influenza infection. Virology 340(2):296–306, PMID: 16054188, 10.1016/j.virol.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Chen M, Shabashvili D, Nawab A, Yang SX, Dyer LM, Brown KD, et al. 2012. DNA methyltransferase inhibitor, zebularine, delays tumor growth and induces apoptosis in a genetically engineered mouse model of breast cancer. Mol Cancer Ther 11(2):370–382, PMID: 22203734, 10.1158/1535-7163.MCT-11-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, et al. 1996. S-Adenosylmethionine and methylation. FASEB J 10(4):471–480, PMID: 8647346, 10.1096/fasebj.10.4.8647346. [DOI] [PubMed] [Google Scholar]
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. 2013. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 25(2):214–221, PMID: 23298609, 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. 2014. T follicular helper cell differentiation, function, and roles in disease. Immunity 41(4):529–542, PMID: 25367570, 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl E, Wiesmann F, Woenckhaus M, Stoehr R, Wild PJ, Veeck J, et al. 2007. Frequent loss of SFRP1 expression in multiple human solid tumours: association with aberrant promoter methylation in renal cell carcinoma. Oncogene 26(38):5680–5691, PMID: 17353908, 10.1038/sj.onc.1210345. [DOI] [PubMed] [Google Scholar]
- Dallaire F, Dewailly É, Muckle G, Vézina C, Jacobson SW, Jacobson JL, et al. 2004. Acute infections and environmental exposure to organochlorines in Inuit infants from Nunavik. Environ Health Perspect 112(14):1359–1365, PMID: 15471725, 10.1289/ehp.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boo HA, Harding JE. 2006. The developmental origins of adult disease (Barker) hypothesis. Aust N Z J Obstet Gynaecol 46(1):4–14, PMID: 16441686, 10.1111/j.1479-828X.2006.00506.x. [DOI] [PubMed] [Google Scholar]
- Detich N, Hamm S, Just G, Knox JD, Szyf M. 2003. The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J Biol Chem 278(23):20812–20820, PMID: 12676953, 10.1074/jbc.M211813200. [DOI] [PubMed] [Google Scholar]
- Ding T, Mokshagundam S, Rinaudo PF, Osteen KG, Bruner-Tran KL. 2018. Paternal developmental toxicant exposure is associated with epigenetic modulation of sperm and placental Pgr and Igf2 in a mouse model. Biol Reprod 99(4):864–876, PMID: 29741588, 10.1093/biolre/ioy111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21, PMID: 23104886, 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. 2010. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: an epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer 1(3):146–155, PMID: 21761357, 10.1007/s12672-010-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durek P, Nordstrom K, Gasparoni G, Salhab A, Kressler C, de Almeida M, et al. 2016. Epigenomic profiling of human CD4+ T cells supports a linear differentiation model and highlights molecular regulators of memory development. Immunity 45(5):1148–1161, PMID: 27851915, 10.1016/j.immuni.2016.10.022. [DOI] [PubMed] [Google Scholar]
- Esser C, Rannug A. 2015. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev 67(2):259–279, PMID: 25657351, 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Feng X, Ippolito GC, Tian L, Wiehagen K, Oh S, Sambandam A, et al. 2010. Foxp1 is an essential transcriptional regulator for the generation of quiescent naive T cells during thymocyte development. Blood 115(3):510–518, PMID: 19965654, 10.1182/blood-2009-07-232694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H. 2011. Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol 12(6):544–550, PMID: 21532575, 10.1038/ni.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, et al. 2000. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci 908:244–254, PMID: 10911963, 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Gasiewicz TA, Geiger LE, Rucci G, Neal RA. 1983. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metab Dispos 11(5):397–403, PMID: 6138222. [PubMed] [Google Scholar]
- Giannandrea M, Yee M, O’Reilly MA, Lawrence BP. 2012. Memory CD8+ T cells are sufficient to alleviate impaired host resistance to influenza A virus infection caused by neonatal oxygen supplementation. Clin Vaccine Immunol 19(9):1432–1441, PMID: 22787195, 10.1128/CVI.00265-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn A, Thuvander A, Aune M, Johannisson A, Darnerud PO, Ronquist G, et al. 2008. Immune cell counts and risks of respiratory infections among infants exposed pre- and postnatally to organochlorine compounds: a prospective study. Environ Health 7:62, PMID: 19055819, 10.1186/1476-069X-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Duran A, Ballestar E, Carvajal-Gonzalez JM, Marlowe JL, Puga A, Esteller M, et al. 2008. Recruitment of CREB1 and histone deacetylase 2 (HDAC2) to the mouse Ltbp-1 promoter regulates its constitutive expression in a dioxin receptor-dependent manner. J Mol Biol 380(1):1–16, PMID: 18508077, 10.1016/j.jmb.2008.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, et al. 1995. Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 55(20):4531–4535, PMID: 7553622. [PubMed] [Google Scholar]
- Greathouse KL, Bredfeldt T, Everitt JI, Lin K, Berry T, Kannan K, et al. 2012. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol Cancer Res 10(4):546–557, PMID: 22504913, 10.1158/1541-7786.MCR-11-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann C, Budtz-Jørgensen E, Nielsen F, Heinzow B, Weihe P, Grandjean P. 2010. Serum concentrations of antibodies against vaccine toxoids in children exposed perinatally to immunotoxicants. Environ Health Perspect 118(10):1434–1438, PMID: 20562056, 10.1289/ehp.1001975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38(4):576–589, PMID: 20513432, 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Greally JM. 2016. Epigenetics, cellular memory and gene regulation. Curr Biol 26(14):R644–R648, PMID: 27458904, 10.1016/j.cub.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Henson SM, Akbar AN. 2009. KLRG1—more than a marker for T cell senescence. Age (Dordr) 31(4):285–291, PMID: 19479342, 10.1007/s11357-009-9100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstenbach K, van Leeuwen DM, Gmuender H, Gottschalk RW, Stølevik SB, Nygaard UC, et al. 2012. Toxicogenomic profiles in relation to maternal immunotoxic exposure and immune functionality in newborns. Toxicol Sci 129(2):315–324, PMID: 22738990, 10.1093/toxsci/kfs214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DJ, Reynolds RM, Hardy DB. 2017. Developmental origins of health and disease: current knowledge and potential mechanisms. Nutr Rev 75(12):951–970, PMID: 29186623, 10.1093/nutrit/nux053. [DOI] [PubMed] [Google Scholar]
- Hua L, Yao S, Pham D, Jiang L, Wright J, Sawant D, et al. 2013. Cytokine-dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J Virol 87(21):11884–11893, PMID: 23986597, 10.1128/JVI.01461-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusko TA, De Roos AJ, Lee SY, Thevenet-Morrison K, Schwartz SM, Verner MA, et al. 2016. A birth cohort study of maternal and infant serum PCB-153 and DDE concentrations and responses to infant tuberculosis vaccination. Environ Health Perspect 124(6):813–821, PMID: 26649893, 10.1289/ehp.1510101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusko TA, Sisto R, Iosif AM, Moleti A, Wimmerová S, Lancz K, et al. 2014. Prenatal and postnatal serum PCB concentrations and cochlear function in children at 45 months of age. Environ Health Perspect 122(11):1246–1252, PMID: 25051575, 10.1289/ehp.1307473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. 1997. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 57(5):808–811, PMID: 9041175. [PubMed] [Google Scholar]
- Kasakovski D, Xu L, Li Y. 2018. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol Oncol 11(1):91, PMID: 29973238, 10.1186/s13045-018-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. 2018. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol 15(5):458–469, PMID: 29563615, 10.1038/s41423-018-0004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori HK, Hart T, LaMere SA, Chew PV, Salomon DR. 2015. Defining CD4 T cell memory by the epigenetic landscape of CpG DNA methylation. J Immunol 194(4):1565–1579, PMID: 25576597, 10.4049/jimmunol.1401162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27(11):1571–1572, PMID: 21493656, 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TWH, Yang H, Peng H, Xia M, Mato JM, Lu SC. 2012. Effects of S-adenosylmethionine and methylthioadenosine on inflammation-induced colon cancer in mice. Carcinogenesis 33(2):427–435, PMID: 22159228, 10.1093/carcin/bgr295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen G, Ma L, Ohms SJ, Sun C, Shannon MF, et al. 2012. Plasticity of DNA methylation in mouse T cell activation and differentiation. BMC Mol Biol 13:16, PMID: 22642378, 10.1186/1471-2199-13-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930, PMID: 24227677, 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin RK, Hung WY, Huang YF, Chang YJ, Lin CH, Chen WY, et al. 2017. Hypermethylation of BEND5 contributes to cell proliferation and is a prognostic marker of colorectal cancer. Oncotarget 8(69):113431–113443, PMID: 29371920, 10.18632/oncotarget.22266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Ibrahim JG, et al. 2009. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8(4):439–448, PMID: 19485966, 10.1111/j.1474-9726.2009.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo-Herrero S, Sordo-Bahamonde C, Gonzalez S, López-Soto A. 2019. CD107a degranulation assay to evaluate immune cell antitumor activity. Methods Mol Biol 1884:119–130, PMID: 30465198, 10.1007/978-1-4939-8885-3_7. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550, PMID: 25516281, 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod MKL, Clambey ET, Kappler JW, Marrack P. 2009. CD4 memory T cells: what are they and what can they do? Semin Immunol 21(2):53–61, PMID: 19269850, 10.1016/j.smim.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. 2012. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One 7(9):e46249, PMID: 23049995, 10.1371/journal.pone.0046249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato JM, Martínez-Chantar ML, Lu SC. 2013. S-adenosylmethionine metabolism and liver disease. Ann Hepatol 12(2):183–189, PMID: 23396728, 10.1016/S1665-2681(19)31355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Berger SL. 2016. Exploring the dynamic relationship between cellular metabolism and chromatin structure using SILAC-mass spec and ChIP-sequencing. Methods Enzymol 574:311–329, PMID: 27423866, 10.1016/bs.mie.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G. 2013. DNA methylation and its basic function. Neuropsychopharmacology 38(1):23–38, PMID: 22781841, 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane CJ, Hyland EM. 2019. Yeast epigenetics: the inheritance of histone modification states. Biosci Rep 39(5):BSR20182006, PMID: 30877183, 10.1042/BSR20182006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF. 2015. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: preventive effects of resveratrol. Mol Carcinog 54(4):261–269, PMID: 24136580, 10.1002/mc.22095. [DOI] [PubMed] [Google Scholar]
- Partridge FA, Gravato-Nobre MJ, Hodgkin J. 2010. Signal transduction pathways that function in both development and innate immunity. Dev Dyn 239(5):1330–1336, PMID: 20131356, 10.1002/dvdy.22232. [DOI] [PubMed] [Google Scholar]
- Pilozzi E, Onelli MR, Ziparo V, Mercantini P, Ruco L. 2004. CDX1 expression is reduced in colorectal carcinoma and is associated with promoter hypermethylation. J Pathol 204(3):289–295, PMID: 15378566, 10.1002/path.1641. [DOI] [PubMed] [Google Scholar]
- Post CM, Boule LA, Burke CG, O’Dell CT, Winans B, Lawrence BP. 2019. The ancestral environment shapes antiviral CD8+ T cell responses across generations. iScience 20:168–183, PMID: 31569050, 10.1016/j.isci.2019.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renz H, Holt PG, Inouye M, Logan AC, Prescott SL, Sly PD. 2017. An exposome perspective: early-life events and immune development in a changing world. J Allergy Clin Immunol 140(1):24–40, PMID: 28673401, 10.1016/j.jaci.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Robin C, Ottersbach K, de Bruijn M, Ma X, van der Horn K, Dzierzak E. 2003. Developmental origins of hematopoietic stem cells. Oncol Res 13(6–10):315–321, PMID: 12725520, 10.3727/096504003108748519. [DOI] [PubMed] [Google Scholar]
- Sant AJ, McMichael A. 2012. Revealing the role of CD4+ T cells in viral immunity. J Exp Med 209(8):1391–1395, PMID: 22851641, 10.1084/jem.20121517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Ezquerro JJ, Münsterberg AE, Stricker S. 2017. Editorial: signaling pathways in embryonic development. Front Cell Dev Biol 5:76, PMID: 28913334, 10.3389/fcell.2017.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C, Delacher M, Huehn J, Feuerer M. 2018. Epigenetic mechanisms regulating T-cell responses. J Allergy Clin Immunol 142(3):728–743, PMID: 30195378, 10.1016/j.jaci.2018.07.014. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M, Peng L, Puga A. 2007. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim Biophys Acta 1769(9–10):569–578, PMID: 17707923, 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurich A, Henson SM. 2014. The many unknowns concerning the bioenergetics of exhaustion and senescence during chronic viral infection. Front Immunol 5:468, PMID: 25309548, 10.3389/fimmu.2014.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somm E, Stouder C, Paoloni-Giacobino A. 2013. Effect of developmental dioxin exposure on methylation and expression of specific imprinted genes in mice. Reprod Toxicol 35:150–155, PMID: 23142538, 10.1016/j.reprotox.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Stølevik SB, Nygaard UC, Namork E, Haugen M, Kvalem HE, Meltzer HM, et al. 2011. Prenatal exposure to polychlorinated biphenyls and dioxins is associated with increased risk of wheeze and infections in infants. Food Chem Toxicol 49(8):1843–1848, PMID: 21571030, 10.1016/j.fct.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Stølevik SB, Nygaard UC, Namork E, Haugen M, Meltzer HM, Alexander J, et al. 2013. Prenatal exposure to polychlorinated biphenyls and dioxins from the maternal diet may be associated with immunosuppressive effects that persist into early childhood. Food Chem Toxicol 51:165–172, PMID: 23036451, 10.1016/j.fct.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Strakovsky RS, Wang H, Engeseth NJ, Flaws JA, Helferich WG, Pan YX, et al. 2015. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol Appl Pharmacol 284(2):101–112, PMID: 25748669, 10.1016/j.taap.2015.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su KY, Li MC, Lee NW, Ho BC, Cheng CL, Chuang YC, et al. 2019. Perinatal polychlorinated biphenyls and polychlorinated dibenzofurans exposure are associated with DNA methylation changes lasting to early adulthood: findings from Yucheng second generation. Environ Res 170:481–486, PMID: 30640082, 10.1016/j.envres.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Suárez-Álvarez B, Baragaño Raneros A, Ortega F, López-Larrea C. 2013. Epigenetic modulation of the immune function: a potential target for tolerance. Epigenetics 8(7):694–702, PMID: 23803720, 10.4161/epi.25201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N, et al. 2016. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 30(8):1716–1724, PMID: 27102208, 10.1038/leu.2016.84. [DOI] [PubMed] [Google Scholar]
- Sun J, Braciale TJ. 2013. Role of T cell immunity in recovery from influenza virus infection. Curr Opin Virol 3(4):425–429, PMID: 23721865, 10.1016/j.coviro.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SL, McKinstry KK, Strutt TM. 2012. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol 12(2):136–148, PMID: 22266691, 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Magallanes R, Rebbani K, Lim R, Pradhan S, Benoukraf T. 2017. Whole genome DNA methylation: beyond genes silencing. Oncotarget 8(3):5629–5637, PMID: 27895318, 10.18632/oncotarget.13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorderstrasse BA, Cundiff JA, Lawrence BP. 2004. Developmental exposure to the potent aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs the cell-mediated immune response to infection with influenza A virus, but enhances elements of innate immunity. J Immunotoxicol 1(2):103–112, PMID: 18958643, 10.1080/15476910490509244. [DOI] [PubMed] [Google Scholar]
- Wang LT, Chiou SS, Chai CY, Hsi E, Wang SN, Huang SK, et al. 2017. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget 8(5):7489–7501, PMID: 27283490, 10.18632/oncotarget.9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren TK, Mitchell KA, Lawrence BP. 2000. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppresses the humoral and cell-mediated immune responses to influenza A virus without affecting cytolytic activity in the lung. Toxicol Sci 56(1):114–123, PMID: 10869459, 10.1093/toxsci/56.1.114. [DOI] [PubMed] [Google Scholar]
- Wilkinson TM, Li CKF, Chui CSC, Huang AKY, Perkins M, Liebner JC, et al. 2012. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 18(2):274–280, PMID: 22286307, 10.1038/nm.2612. [DOI] [PubMed] [Google Scholar]
- Winans B, Nagari A, Chae M, Post CM, Ko CI, Puga A, et al. 2015. Linking the aryl hydrocarbon receptor with altered DNA methylation patterns and developmentally induced aberrant antiviral CD8+ T cell responses. J Immunol 194(9):4446–4457, PMID: 25810390, 10.4049/jimmunol.1402044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo CB, Chuang JC, Byun HM, Egger G, Yang AS, Dubeau L, et al. 2008. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev Res (Phila) 1(4):233–240, PMID: 19138966, 10.1158/1940-6207.CAPR-07-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang X, Wang K, Xu X, Li M, Zhang J, et al. 2018. Newly generated CD4+ T cells acquire metabolic quiescence after thymic egress. J Immunol 200(3):1064–1077, PMID: 29288207, 10.4049/jimmunol.1700721. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ji M, Tan X, Yu K, Xu L, Chen G, et al. 2019. Role of epigenetic regulation of Igf2 and H19 in 2,3,7,8-tetrachlorobenzo-p-dioxin (TCDD)-induced ovarian toxicity in offspring rats. Toxicol Lett 311:98–104, PMID: 31063829, 10.1016/j.toxlet.2019.04.034. [DOI] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, et al. 2016. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63(6):1066–1079, PMID: 27635762, 10.1016/j.molcel.2016.08.032. [DOI] [PubMed] [Google Scholar]
- Zhou L, Cheng X, Connolly BA, Dickman MJ, Hurd PJ, Hornby DP. 2002. Zebularine: a novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J Mol Biol 321(4):591–599, PMID: 12206775, 10.1016/s0022-2836(02)00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Emerson SG. 2002. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 21(21):3295–3313, PMID: 12032771, 10.1038/sj.onc.1205318. [DOI] [PubMed] [Google Scholar]
- Zhu J, Paul WE. 2008. CD4 T cells: fates, functions, and faults. Blood 112(5):1557–1569, PMID: 18725574, 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All WGBS and RNA-sequencing (RNA-seq) data have been deposited to the NIH Gene Expression Omnibus under accession number GSE143893 (WGBS) and GSE143894 (RNA-seq).