

Discovery of a new HDAC6 inhibitor reveals role for HDAC6 in the regulation of glycolytic metabolism.
Abstract
Triple-negative breast cancer (TNBC) is a subtype of breast cancer without a targeted form of therapy. Unfortunately, up to 70% of patients with TNBC develop resistance to treatment. A known contributor to chemoresistance is dysfunctional mitochondrial apoptosis signaling. We set up a phenotypic small-molecule screen to reveal vulnerabilities in TNBC cells that were independent of mitochondrial apoptosis. Using a functional genetic approach, we identified that a “hit” compound, BAS-2, had a potentially similar mechanism of action to histone deacetylase inhibitors (HDAC). An in vitro HDAC inhibitor assay confirmed that the compound selectively inhibited HDAC6. Using state-of-the-art acetylome mass spectrometry, we identified glycolytic substrates of HDAC6 in TNBC cells. We confirmed that inhibition or knockout of HDAC6 reduced glycolytic metabolism both in vitro and in vivo. Through a series of unbiased screening approaches, we have identified a previously unidentified role for HDAC6 in regulating glycolytic metabolism.
INTRODUCTION
Triple-negative breast cancer (TNBC) is a highly aggressive subtype of breast cancer that lacks expression of estrogen receptor, progesterone receptor, and human epidermal growth factor 2 (HER2) (1). Numerous “omic” studies have tried to molecularly characterize the heterogeneous disease and identify “driver” mutations to target therapeutically. The standard of care for both recently diagnosed patients and patients with advanced disease is cytotoxic chemotherapy (2), and targeted therapies are not routinely used in the treatment of TNBC. While chemotherapy is effective in a subset of patients (3), there is a large proportion of patients (60 to 70%) that are refractory to chemotherapy and have poorer survival (4). Novel therapeutic strategies are urgently required for patients with TNBC with chemoresistant disease.
The development of chemoresistance is multifactorial; potential contributors include drug pharmacokinetics, protective microenvironments, drug target alterations, and dysfunctional cell death signaling (5). There is evidence that metabolic rewiring in cancer is one of the mechanisms underlying therapeutic resistance (6), in which glycolytic metabolism may play a key role. Aerobic glycolysis, otherwise known as the “Warburg effect,” is the utilization of glycolysis even in the presence of abundant oxygen by converting glucose to pyruvate and then to lactate (7). The reliance of cancer cells on aerobic glycolysis has been directly linked to oncogene activation including c-MYC (8) and RAS (9), along with mutant tumor-suppressor genes (e.g., TP53) (10). TNBC cell lines and patient samples are characterized by elevated glycolysis and have evidence of c-MYC amplification and TP53 mutation, especially in the relapsed setting (11, 12). It has been suggested that the glycolytic potential of TNBC is associated with chemotherapeutic resistance, as exposing TNBC cells to increasing levels of glucose decreases the efficacy of metformin-induced apoptosis (13).
Furthermore, dysfunctional cell death signaling is also a major contributor to chemoresistance. The mitochondrion is a central apoptotic signaling node for responses to diverse cytotoxic chemotherapy (14). DNA damage or microtubule stabilization ultimately results in changes in the BCL-2 family of proteins, and if these changes are of sufficient magnitude, the proapoptotic proteins BAX and BAK oligomerize at the mitochondrion leading to mitochondrial outer membrane permeabilization (MOMP) and programmed cell death (15). Tumor resistance to cytotoxic chemotherapy is frequently acquired through deregulation of expression of the antiapoptotic BCL-2 family members (BCL-2, MCL-1, and BCL-XL), either by genetic or epigenetic mechanisms (15). The antiapoptotic protein MCL-1 is one of the most frequently amplified genes, along with BCL-xL, across all cancers (16). There is evidence of MCL-1 amplification in TNBC patient samples following neoadjuvant chemotherapy, highlighting that altered mitochondrial apoptosis is a mechanism of chemoresistance in vivo (12).
To identify alternative therapeutic targets for the treatment of mitochondrial apoptotic-resistant TNBC, we performed a phenotypic screen of small molecules. The aim was to identify therapeutic targets in TNBC that were independent of the mitochondrial cell death pathway using an unbiased screening approach. To do this, we knocked down two essential mitochondrial-apoptotic proteins (BAX and BAK) in the TNBC cell line MDA-MB-231 and screened for agents that selectively killed the apoptotic-resistant TNBC cells but did not kill nontransformed MCF10a cells. We identified a novel, highly selective HDAC6 inhibitor. Using state-of-the-art acetylome and interactome mass spectrometry (MS) approaches, we show that HDAC6 binds and regulates four key glycolytic enzymes. We confirmed this role for HDAC6 in regulating glycolysis both in vitro and in vivo in two distinct syngeneic mouse models.
RESULTS
Identification of a compound that selectively kills apoptotic-resistant TNBC cells
Given the reliance of cytotoxic chemotherapy responses on the mitochondrial cell death pathway (14), we set up a phenotypical screen to identify potential therapeutic targets in TNBC that were independent of the mitochondrial apoptotic pathway (Fig. 1A). We knocked down BAX/BAK, two essential proteins for mitochondrial apoptosis (fig. S1A). The MDA231 BAX/BAK knockdown (KD) cells were, as expected, broadly resistant to apoptosis-inducing agents, including etoposide (fig. S1B) and ABT-737 (an antagonist of BCL-2/BCL-XL and BCL-w) (fig. S1C). BH3 profiling confirmed that the BAX/BAK KD cells were unprimed (14) and no longer sensitive to the addition of either activator or sensitizer BH3 peptides (fig. S1D).
Fig. 1. Identification of a compound that kills cancer cells independent of BAX/BAK.
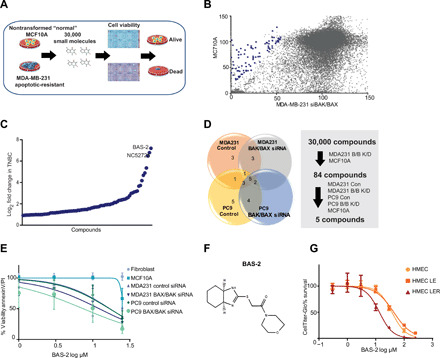
(A) Schematic of the phenotypical screen. MDA231 cells were transfected with two siRNAs against BAX and BAK proteins. The MDA231 and MCF10a were treated with 30,000 small molecules in duplicate from diverse libraries. (B) Cell viability of the MCF10a and MDA231 (BAX/BAK siRNA) cells for each of the 30,000 compounds test. Blue dots indicate the compounds that were cherry picked for a secondary screen. (C) The fold change in response of the MDA231 cells to cherry-picked compounds compared with MCF10a cells. (D) Pie chart represents the number of compounds that showed a response in the indicated cells. (E) Cell survival of the indicated cell lines following a 48-hour treatment with BAS-2 (n = 3, mean ± SEM). (F) Chemical structure of the lead compound, BAS-2. (G) Cell survival of the indicated cell lines following a 48-hour treatment with BAS-2 (n = 3, mean ± SEM).
We counterscreened the nontransformed MCF10a cells to identify compounds that were selectively toxic to the cancer cells. We screened over 30,000 small molecules, comprising U.S. Food and Drug Administration–approved drugs, natural compounds, and synthetically derived libraries, for their ability to preferentially kill the MDA231 BAX/BAK KD but not the MCF10a cell line (Fig. 1B). A secondary screen was conducted on “hit” compounds selected on the basis of the following criteria: a difference of 20% in viability and a fold change loss in viability greater than 1.9 between the MDA231 cells compared with the MCF10a cells (Fig. 1C and data file S1). In the secondary screen, 84 compounds were screened for cancer selectivity across both the MDA231 and PC9 cells (lung cancer cell line) (Fig. 1D and fig. S1, E and F). From this secondary screen, five compounds were analyzed in low-throughput assays (Fig. 1E and fig. S1G), and the lead compound, BAS 0485063 from the Assenix 1 library, referred to as BAS-2 (Fig. 1F), selectively killed the breast and lung cancer cells irrespective of BAX/BAK status more readily than normal cells (Fig. 1E). To confirm the selectivity of BAS-2 for cancer cells, we used the in vitro model for studying the tumorigenic conversion of human mammary epithelial cells (HMECs) (17). The transformed HMEC LER cells, with oncogenic RAS, were more sensitive to BAS-2 than the nontransformed HMEC and HMEC LE cells (Fig. 1G), indicating a selectivity of BAS-2 for cancer cells. Through a phenotypic screen, we identified a compound, BAS-2, that selectively killed TNBC cancer cells independent of mitochondrial apoptosis.
A functional genetic approach revealed BAS-2 as a HDAC6 inhibitor that impedes TNBC growth in vitro and in vivo
One challenge when performing a phenotypic screen is trying to identify the molecular target that the small molecule is inhibiting. We used a published functional genetic approach to try and cluster the hit small molecules from the screen, with the mechanism of action of known drugs (18). Briefly, Eμ-Myc lymphoma cells are infected with eight different green fluorescent protein (GFP)–labeled short hairpin RNAs (shRNAs) and treated with drugs of known mechanism of action or novel small molecules. The pattern of resistance and sensitivity is monitored by depletion/enrichment of GFP. By comparing patterns, we can create hypotheses about biochemical modes of action (Fig. 2A). Following treatment of the infected cells with BAS-2, it emerged that BAS-2 had a novel mechanism of action, which most closely resembled that of suberoylanilide hydroxamic acid (SAHA), a pan–histone deacetylase (HDAC) inhibitor (Fig. 2B and fig. S2A).
Fig. 2. Functional genetic approach to establish the target of BAS-2.
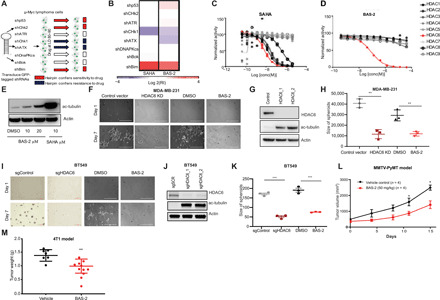
(A) Schematic of the GFP-based competition assays. LD, lethal dose. (B) Heatmap showing the response of cells expressing the indicated shRNAs to SAHA and BAS-2. (C and D) Inhibition of trifluoroacetyllysine substrate processing by suberoylanilide hydroxamic acid (SAHA) (C) and BAS-2 (D) (mean of triplicate measurements). (E) Western blot for acetylated tubulin following indicated treatments with BAS-2 and SAHA. (F) Images of MDA231 cells grown in Matrigel for 7 days with HDAC6 KD or following treatment with 30 μM BAS-2. (G) Western blot of HDAC6 and acetylated tubulin levels in control and HDAC6 KD cells. (H) Dot plots show the size of colonies in all treatment groups (n = 3, mean ± SEM). (I) Images of BT-549 cells grown as described in (F). (J) Western blot of HDAC6 and acetylated tubulin levels in control or HDAC6 KO cells. (K) Dot plots show the size of colonies in all treatment groups (n = 3, mean ± SEM). (L) MMTv-PyMT tumors were measured and plotted as average total tumor burden following randomization to vehicle [dimethyl sulfoxide (DMSO)] or BAS-2 (50 mg/kg; n = 4, mean ± SEM). (M) BALB/cJ mice were subcutaneously inoculated with 4T1 cells and treated with BAS-2 (50 mg/kg) for 14 days, and tumor weight was plotted (n = 10, mean ± SEM). **P < 0.01 and ***P < 0.001.
HDACs are a family of enzymes that modulate their substrates by removing the acetyl group from lysine residues. Using an in vitro HDAC inhibitor screen (19), we demonstrated that SAHA inhibits the activity of HDACs 1, 2, 6, 7, and 8 (Fig. 2C). BAS-2 inhibited only the isozyme HDAC6 with a half-maximal inhibitory concentration (IC50) of 76 nM (Fig. 2D). While there are other highly selective HDAC6 inhibitors, such as tubacin and CAY10603, BAS-2 only inhibited HDAC6 in the in vitro HDAC assay (>250-fold selectivity) (20, 21). We confirmed an increase in the acetylated tubulin in cells, a known substrate of HDAC6, following treatment with increasing doses of BAS-2 (Fig. 2E). BAS-2 treatment did not cause an increase in acetylation of histone 4, indicating selective inhibition of HDAC6 in cells (fig. S2B). To ensure that the compound could induce cell death, we measured the IC50 values by annexin V/propidium iodide staining across a panel of TNBC cell lines and estrogen receptor–positive breast cancer cells lines and found equivalent IC50 values (fig. S2C). Phenotypic changes induced by HDAC6 inhibition include prevention of migration and invasion of cancer cells (22). We detected a significant reduction in both cell migration and invasion following HDAC6 KD in MDA231 cells (fig. S2, D and F), and when treated with BAS-2 both in the MDA231 and BT-549 cells (fig. S2, E and G). There was a reduction in three-dimensional (3D) spheroid formation following HDAC6 KD in MDA231 and following treatment with BAS-2 (Fig. 2, F to H). Similar results were found in the BT-549 cells following HDAC6 knockout (KO) and BAS-2 treatment (Fig. 2, I to K) and in the mouse breast cancer cell line 4T1 (fig. S2, H to J). No change was found in the proliferation rate of MDA231 following HDAC6 KD (fig. S2K).
Next, we assessed the in vivo efficacy of BAS-2, as a single agent, using both a syngeneic breast mouse model and a genetic breast mouse model (MMTV-pyMT). Following 2 weeks of treatment at BAS-2 (50 mg/kg), we found a reduction in tumor volume across the 10 tumors per mouse in the aggressive MMTV-pyMT mouse model (Fig. 2L) and a reduction in tumor weight and tumor volume in the 4T1 syngeneic mouse model (Fig. 2M and fig. S3A). We observed similar effects in the syngeneic KrasLSL-G12D p53−/− (KP) non–small cell lung cancer (NSCLC) mouse mode (fig. S3B) treated over 2 weeks with BAS-2 (50 mg/kg). We did not detect any overt evidence of toxicity, as assessed by weight of the mice while on treatment (fig. S3, C and D).
Combined, using a functional genetic approach, we identified BAS-2 as a HDAC6 inhibitor and confirmed similar phenotypic responses between HDAC6 KD or HDAC6 KO and BAS-2 treatment in TNBC cells. In addition, these data suggest that this small molecule, which has not yet been chemically developed, has moderate inhibitory effects as a single agent both in vitro and in vivo.
Measuring the acetylome of HDAC6 using MS
Since we had identified a highly selective HDAC6 inhibitor, we decided to use the compound as a chemical probe to determine HDAC6 substrates in TNBC cells. We used advanced MS techniques to identify the acetylome of the cells following HDAC6 inhibition with BAS-2 and compared it with the HDAC6 KD acetylome (23). Since HDAC6 removes acetyl groups from lysine residues, inhibition of HDAC6 should result in an increase in the acetylation levels of its substrates. Following treatment of MDA231 cells with BAS-2 or following HDAC6 KD, we extracted and trypsin digested proteins. The digested peptides were then incubated with anti-acetyllysine to enrich for acetylated peptides. Liquid chromatography–tandem MS (LC-MS/MS) was performed on acetylated peptides to identify them and determine relative abundance (Fig. 3A). LCMS-MaxQuant analysis revealed increased acetylation of 85 proteins in the presence of BAS-2, when compared with the dimethyl sulfoxide (DMSO) control (Fig. 3B and data file S2). Known substrates of HDAC6, including heat shock protein 90 and tubulin (24), showed increased acetylation by MS in the presence of BAS-2, validating our approach (Fig. 3B). To assess the functional implications of the altered acetylation, we conducted pathway analysis on the acetyl-proteome using the PANTHER analysis and found the greatest alteration to be in the metabolic process pathway (Fig. 3C). This was confirmed using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, which demonstrated the acetyl-proteome significantly affected the pathogenic Escherichia coli infection and glycolysis/gluconeogenesis pathways (Fig. 3D). In the HDAC6 KD cells, we observed a significant increase in the acetylation of a total of 101 proteins compared with the control vector, and again, we identified some known substrates of HDAC6 (Fig. 3E and data File S3). The functional implications of the altered acetyl-proteome also pointed toward the metabolic process (Fig. 3F), and the KEGG pathway enrichment analysis showed significant effect on the glycolysis/gluconeogenesis pathway (Fig. 3G). Notably, we found an increase in the acetylation of 33 of the exact same peptides, from 31 distinct proteins, at the same lysine residue, overlapping between the BAS-2 and HDAC6 KD experiments (examples shown in Fig. 3, H and M, and fig. S4A).
Fig. 3. Quantitative proteomics of the BAS-2 acetylome and HDAC6 KD converges on the glycolytic pathway.
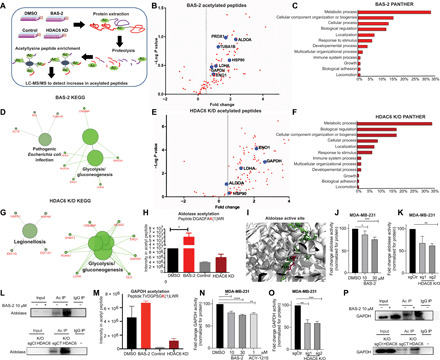
(A) Proteomics workflow. (B) Volcano plot showing enhanced acetylated peptides following BAS-2 treatment (30 μM) for 48 hours. (C) Bar graph of biological pathways with increased acetylated peptides, as assessed by Panther (C) and as assessed by KEGG pathway (D). (E to G) The same as (B) to (D) but for HDAC6 KD compared with control. (H) Bar graph representing intensity of acetyl peptide of aldolase following BAS-2 treatment or HDAC6 KD. (I) Image of aldolase crystal structure extracted from the Protein Data Bank. The peptide from (H) shown in green (inserted by Pymol), and the lysine residue that is acetylated is in pink. (J and K) Measurement of aldolase enzymatic activity in MDA231 cells following 24-hour BAS-2 treatment (J) and following HDAC6 KO (K). (L) Western blots for immunoprecipitated (IP) acetylated proteins in MDA231 cells following BAS-2 treatment (10 μM) for 24 hours or with HDAC6 KO. IgG, immunoglobulin G. (M) Bar graph representing intensity of the acetyl peptide of GAPDH following BAS-2 treatment or HDAC6 KD. (N and O) Measurement of GAPDH enzymatic activity in MDA231 cells following 24-hour BAS-2 treatment (N) and HDAC6 KO (O). (P) Western blots for immunoprecipitated acetylated proteins in MDA231 cells following BAS-2 treatment (10 μM) for 24 hours or with HDAC6 KO (n = 3, mean ± SEM). *P < 0.05, **P < 0.01, and ***P < 0.001.
Similarly, the exact same pathways were altered between the BAS-2 and the HDAC6 KD following PANTHER analysis (Fig. 3, C and F). This suggests that the novel inhibitor, BAS-2, has a high selectivity for HDAC6 in cells. Excitingly, when mapping the aldolase peptide (DGADFAKWR) (Fig. 3I, shown in green), the lysine residue (lysine-201) (shown in pink) that had increased acetylation following BAS-2 treatment or HDAC6 KD (Fig. 3H) was located in the active site of the aldolase protein (Fig. 3I). Therefore, we measured the activity of aldolase following BAS-2 treatment and found a dose-dependent significant reduction in aldolase enzymatic activity in the MDA231 cells (Fig. 3J) and in the BT-549 cells (fig. S4E). We also confirmed a significant reduction in aldolase activity in the HDAC6 KO using CRISPR-Cas9 (fig. S4B) in the MDA231 (Fig. 3K) and in the BT-549 cells (Fig. S4F). We confirmed using immunoprecipitation with acetyl beads that HDAC6i with BAS-2 (10 μM, 24-hour treatment), or HDAC6 KO increased the acetylation of aldolase (Fig. 3L). Gyceraldehyde-3-phosphate dehydrogenase (GAPDH) was also acetylated at K192 in both the HDAC6i BAS-2–treated cells and the HDAC6 KD cells (Fig. 3M). Similarly, we detected a dose-dependent reduction in GAPDH activity following HDAC6i with BAS-2 (Fig. 3N) and following HDAC6 KO in MDA231 (Fig. 3O) and BT-549 (fig. S4, G and H), along with increased acetylation of GAPDH following BAS-2 treatment and following HDAC6 KO (Fig. 3P). Protein expression of aldolase or GAPDH was not altered and did not account for the reduced enzymatic activity (fig. S4I). Combined, these data show validation of altered activity and acetylation in two of the glycolytic enzymes identified by MS following HDAC6i with BAS-2 and HDAC6 KD.
The glycolytic pathway is identified in the interactome of HDAC6
To investigate whether substrates that showed increased acetylation following inhibition of HDAC6 or HDAC6 KD actually interacted with HDAC6, we also measured the interactome of HDAC6 using immunoprecipitation and MS (Fig. 4A). HDAC6 was immunoprecipitated from the MDA231 cells, and interacting proteins were detected by LC-MS/MS. A total of 332 proteins were identified to significantly interact with HDAC6 compared with immunoglobulin G control (Fig. 4, B and C, and data file S4). Known targets such as heat shock protein 90, tubulin, and peroxiredoxin-2 were identified. KEGG pathway analysis was used to cluster HDAC6-interacting proteins into biological pathways. HDAC6 significantly interacted with proteins involved in protein production including protein processing, ribosomes, and the proteasome pathway (Fig. 4D). In agreement with acetylome experiments, glycolysis was also identified as a key novel pathway that HDAC6 significantly interacted with (Fig. 4D and fig. S4C). Since this suggested a functional overlap between proteins that had increased acetylation following HDAC6 inhibition and proteins that physically interacted with HDAC6, we compared both sets of proteins. In total, 15 proteins were statistically significantly altered in common between the three MS datasets (fig. S4D). When we applied the Cytoscape pathway enrichment analysis to these overlapping proteins, we found glycolysis/gluconeogenesis to be the only common significantly altered pathway (Fig. 4E). HDAC6 significantly interacted with GAPDH, enolase, and lactate dehydrogenase A (LDHA), along with a large fold change in interaction with aldolase. In the acetyl-proteome experiments, HDAC6 inhibition either by BAS-2 or HDAC6 KD caused an increase in the acetylation of aldolase, GAPDH, enolase, and LDHA (Figs. 3, B and E, and 4E). Using published RNA-sequencing data from TCGA (The Cancer Genome Atlas) we found that three of the four glycolytic enzymes are highly expressed in TNBC breast cancer subtypes (Fig. 4F) (25). In conclusion, using complementary proteomic approaches, we have shown that HDAC6 interacts with key glycolytic enzymes, and inhibition of HDAC6 causes an increase in the acetylation of these glycolytic enzymes.
Fig. 4. HDAC6 interactome shows binding with glycolytic enzymes.
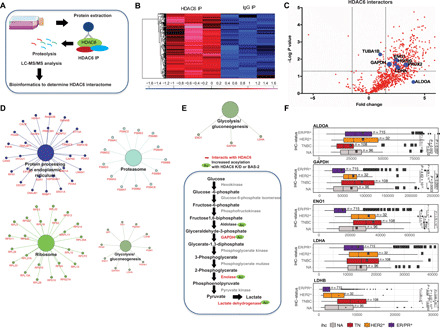
(A) Proteomic workflow. (B) Heatmap of z-score log-transformed ion current intensities of significantly enriched proteins of HDAC6 immunoprecipitation. (C) Volcano plot illustrating the proteins interacting with HDAC6. (D) Significantly enriched KEGG pathways from HDAC6-interacting proteins. (E) Significantly enriched KEGG pathways from the 15 overlapping proteins from HDAC6 IP and acetylation experiments. Glycolytic pathway showing the proteins that HDAC6 interacts with and the proteins that have increased acetylation following BAS-2 treatment or HDAC6 KD. (F) Analysis of the expression of ALDOA, GAPDH, ENO1, LDHA, and LDHB in patients with TNBC in published RNA-sequencing data from TCGA. IHC, immunohistochemistry; ALDOA, aldolase A; ENO1, enolase 1; LDHA, lactate dehydrogenase A.
Inhibition and knockout of HDAC6 alter glycolytic metabolism in TNBC cells
Next, we aimed to dissect whether the altered acetylation of the glycolytic enzymes leads to a global alteration to glycolytic metabolism following HDAC6 KO and inhibition of HDAC6 with BAS-2.
First, we measured the extracellular acidification rate of MDA231 and MCF10a cells following treatment with 10 μM BAS-2 for 24 hours. As is evident, BAS-2 reduced the glycolytic capacity of the MDA231 cells, but it did not affect the glycolysis or glycolytic capacity of the nontransformed MCF10a cell line (Fig. 5, A and B). These data suggest a cancer selectivity for the reduction in glycolysis, but this would need further investigation. A reduction in extracellular acidification rate (ECAR) was also observed in the MDA231 treated with the HDAC6 selective inhibitor, tubacin (fig. S5A). Next, we assessed whether HDAC6 KO led to a change in the glycolytic capacity of MDA231 cells. Following HDAC6 KO, there was a significant reduction in ECAR, which is clearly evident with decreased glycolysis and glycolytic capacity in MDA231 cells (Fig. 5, C and D). We did not detect any reduction in glycolysis or glycolytic capacity in the HDAC6 KO cells following BAS-2 treatment (Fig. 5, E and F), highlighting that HDAC6 is required for the BAS-2–induced reduction in glycolysis.
Fig. 5. Chemical inhibition and knockout of HDAC6 reduces glycolysis.
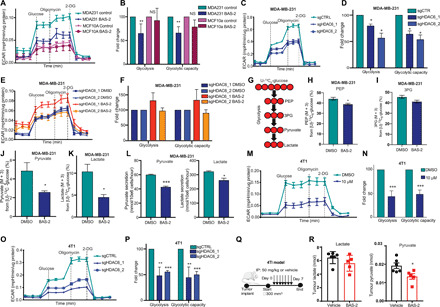
(A) Representative ECAR traces (mpH min−1 μg−1 of protein) are shown (mean ± SEM) for MCF10a (n = 2) and MDA231 (n = 4) cells treated with DMSO or BAS-2 (10 μM) for 24 hours. (B) Fold change in glycolysis and glycolytic capacity (n = 2/4, mean ± SD). NS, not significant. (C and E) Representative ECAR traces for MDA231 cells following HDAC6 KO (C) and treated with BAS-2 (10 μM) for 24 hours (E). (D and F) Fold change in glycolysis and glycolytic capacity (n = 3, mean ± SEM). (G) MDA231 cells were traced with 10 mM U13C6 glucose following 10 μM BAS-2 treatment for 24 hours. (H to K) Percentages of phosphoenolpyruvate (PEP; M + 3) (H), 3-phosphoglyceric acid (3PG) (M + 3) (I), pyruvate (M + 3) (J), and lactate (K) from glucose are shown (n = 3, mean ± SEM). (L) Lactate and pyruvate secreted from MDA231 cells treated with 10 μM BAS-2 for 24 hours (n = 3, mean ± SEM). (M) Representative ECAR traces for 4T1 cells treated with DMSO or BAS-2 for 24 hours. (N) Fold change in glycolysis and glycolytic capacity (n = 3, mean ± SEM). (O) ECAR values (mpH min−1 μg−1 of protein) shown for 4T1 following HDAC6 KO. (P) Fold change in glycolysis and glycolytic capacity (n = 3, mean ± SEM). (Q) Schematic of in vivo experiment. (R) Metabolites extracted from tumors were analyzed (n = 5, mean ± SD). *P < 0.05, **P < 0.01, and ***P < 0.001.
To further confirm the effect of HDAC6i on the glycolytic enzymes, we performed stable isotope tracing and analyzed cell extracts by gas chromatography MS (GC-MS). Using U-13C6-glucose, we observed a significant reduction in carbon incorporation from glucose in downstream glycolytic metabolites phosphoenolpyruvate (PEP), 3-phosphoglyceric acid (3PG), lactate, and pyruvate following HDAC6i in the MDA231 cells (Fig. 5, G to K). We also detected a significant reduction in the secretion of lactate and pyruvate when the MDA231 cells were treated with BAS-2 (Fig. 5L), further suggesting a decrease in glycolysis.
To confirm that the reduction in glycolysis observed with HDAC6 inhibition and KO was not unique to the MDA231 cell lines, we also examined the effects on the 4T1 cell line. We found that both HDAC6 inhibition with BAS-2 and CRISPR-Cas9 KO of HDAC6 resulted in a decrease in glycolysis and glycolytic flux in the 4T1 cell line (Fig. 5, M to P). A similar reduction in ECAR was observed in the 4T1 cells treated with tubacin (fig. S5C). We also demonstrated that in the 4T1 HDAC6 KO cells, we did not detect any reduction in glycolysis or glycolytic capacity following BAS-2 treatment (fig. S5, E and F), again demonstrating that HDAC6 is required for the BAS-2–induced reduction in glycolysis. We confirmed all the above results in the BT-549 cell line, with the further addition of ACY-241, another HDAC6-selective inhibitor (fig. S5, G to O). Altering glycolysis in the BT-549 cells either through removal of glucose for 24 hours or the addition of galactose, which maintains glycolysis through the LeLoir pathway, increased the cell death induced by BAS-2 (fig. S5P). These data suggest that BAS-2–induced cell death can be enhanced by altering glycolysis. In summary, in three different cell lines, we have confirmed that loss of HDAC6 expression and HDAC6i reduces glycolysis and glycolytic capacity in TNBC cells, by two distinct assays.
To ensure that the changes we observed in glycolytic flux were not merely a by-product of cell death, we assessed cell viability and mitochondrial membrane potential following BAS-2 treatment. We found no loss of mitochondrial membrane potential (fig. S6, A and B) or cell viability (fig. S6, C and D) at 10 μM BAS-2 treatment, despite initial changes in glycolytic metabolism, suggesting that the reduced glycolytic flux occurs before cell death.
Next, we aimed to investigate the effect of HDAC6 inhibition on other metabolic pathways, including the pentose phosphate pathway and mitochondrial oxidative respiration. To measure the pentose phosphate pathway, we used a 1,2-13C glucose tracer and analyzed differential label incorporation in downstream glycolytic metabolites lactate and pyruvate (fig. S6E). Analysis of label incorporation into these metabolites would suggest a shift toward slightly increased pentose phosphate pathway activity (fig. S6F). We also measured the oxygen consumption rate (OCR), an indicator of mitochondrial oxidative respiration, following BAS-2 treatment in MDA231 and BT-549 cells, and we did not observe any significant changes in OCR or in basal respiration and ATP (adenosine 5′-triphosphate) production at the lower 10 μM dose (fig. S6, G to J).
Last, to determine whether HDAC6 inhibition with BAS-2 could lead to reduced glycolytic metabolism in vivo, we used the 4T1 syngeneic breast cancer model and quantified tumor metabolites following BAS-2 treatment using GC-MS (Fig. 5Q). Our results showed a significant reduction in intratumoral pyruvate levels following BAS-2 treatment (Fig. 5R) and a trend toward decreased intratumoral lactate, although this can be difficult to show due to abundance in lactate found in the tumor (Fig. 5R). We observed similar results in tumors excised from the syngeneic KP NSCLC mouse model treated over 2 weeks with BAS-2 (fig. S6, K and L).
Collectively, these findings confirm a role for HDAC6i in regulating glycolysis both in vitro and in vivo.
DISCUSSION
The discovery of novel biology often stems from unbiased screening approaches like small-molecule, genetic, or proteomic screens, where probing the biological system illuminates underlying mechanism of action of previously unknown processes (26). The majority of both cytotoxic and targeted therapeutics kill cells via the mitochondrial pathway of apoptosis (14). We took an unbiased approach and used a small-molecule screen to identify new therapeutic targets in apoptosis-resistant TNBC cells. We feel that this represents a paradigm shift from the conventional approach to drug discovery, which, intentionally or not, has been mainly effective at identifying therapeutics that kill via mitochondrial apoptotic mechanisms. This type of small-molecule phenotypic screen was previously conducted in oncogenic RAS-transformed fibroblasts and led to the discovery of errastin that induced an iron-dependent cell death, called ferroptosis (26). While phenotypic screens have the advantage of being cell based, one issue is in identifying the mechanism of action of the small molecule. Using a functional-genetic approach, along with cell-free and cell-based assays, we confirmed that BAS-2 was a highly selective HDAC6 inhibitor. While we show that BAS-2 inhibits HDAC6, as it is a small molecule, we cannot rule out alternative target proteins within the cell. We showed that both HDAC6 KD and KO, similar to HDAC6 inhibition with BAS-2, prevented 3D spheroid formation and reduced glycolytic metabolism. This is in contrast to a recent publication that stated that HDAC6 KO lacked a phenotype in tumor cells, including TNBC (27). BAS-2 did not alter glycolysis in cells that lacked HDAC6. The selectivity of the novel inhibitor, along with advanced MS approaches, allowed us to decipher a new phenotype and an additional function for HDAC6 in regulating glycolytic metabolism.
Much effort in the drug discovery field has concentrated on developing selective HDAC inhibitors to reduce the clinical side effects of pan-HDAC inhibitors, in particular the cardiovascular effects (28). Currently, a series of HDAC6-selective inhibitors have been developed including ACY-241, ACY-1215, and KA2507, and there are ongoing clinical trials assessing the efficacy of the inhibitors in combination treatments in both solid and hematological cancers (29–31). We assessed the selectivity of our inhibitor both in vitro (>250-fold selectivity for HDAC6) and in cells by measuring increased acetylation of peptides using cutting-edge MS approaches (32). We found that over 33 of the exact same peptides, from 31 distinct proteins, had increased acetylation, at the exact same lysine residue, in both the BAS-2–treated and HDAC6 KD cells. A similar approach was taken by Schölz et al. (23) to assess the acetylome of a series of HDAC inhibitors in mouse embryonic fibroblasts. Similar pathways were found to be altered, as in our study, following treatment with tubacin, a selective HDAC6i, validating our approach (23). When we immunoprecipitated HDAC6 and looked at the interactome, we found over 300 proteins to interact with HDAC6, which was much greater than that of the acetylome of HDAC6. This could be due to pulling down protein complexes with HDAC6, limitations of the technologies used, or the additional function of HDAC6 as a ubiquitin binding protein (33). Pathway analysis identified glycolytic metabolism as a key pathway in which HDAC6 interacts with. Furthermore, from the 33 peptides that showed increased acetylation in the measured acetylome following HDAC6i and HDAC6 KD, 4 of these were glycolytic enzymes (aldolase, enolase, GAPDH, and LDHA).
Our study is the first to report, to the best of our knowledge, an additional role for HDAC6 in regulating glycolysis. Collectively, our experiments suggest that following HDAC6, inhibition or HDAC6 KO glycolysis is reduced; however, we have not conclusively distinguished whether this is a driver or a passenger effect in the reduction in cell growth. While HDAC6 KO reduces glycolysis, in 2D, the cells appear to grow at a similar rate, and yet in 3D, there is an extensive reduction in the number and size of spheroids. This difference between 2D and 3D cell growth is something we are interested in following up on. Our study shines light on a new function for HDAC6 in modulating glycolytic metabolism; however, the consequence of these alterations does require further elucidation.
It is known that sirtuins, the class III NAD (nicotinamide adenine dinucleotide)–dependent HDACs, are involved in regulating metabolism in response to nutrient availability (34). In particular, sirt1 has been shown to deacetylate the glycolytic enzyme phosphoglycerate mutase, reducing its activity (35). In addition, it was previously shown that valproate, a pan-HDAC inhibitor, caused a reduction in the glucose transporter 1 and a reduction in hexokinase II activity, but this was not due to increased acetylation (36). The acetyllysine alteration that we identified on the aldolase peptide is located in the active site, an alteration that is known to negatively regulate aldolase activity and reduce glycolysis (37). We confirmed that both aldolase and GAPDH had increased acetylation following HDAC6i and HDAC6 KO. Using an aldolase activity assay, we showed that HDAC6 inhibition with BAS-2/ACY-1215, or following HDAC6 KO, reduced aldolase activity in two separate cell lines. We found a similar reduction in GAPDH activity following HDAC6i and HDAC6 KO. Enolase was acetylated at K5, while LDHA was acetylated at K81, and follow-up studies are required to determine the effect of HDAC6i on their specific activity, along with rescue experiments using mutants insensitive to acetylation. However, upon global glycolytic flux analysis, we showed that HDAC6 inhibition caused an overall reduction in flux in vitro in two separate cell lines. It is reported that metabolic dependencies vary in vitro and in vivo (38). However, we determined a reduction in glycolytic metabolites, pyruvate, and lactate in vivo in two different animal models following treatment with the HDAC6i BAS-2.
Overall, our analysis suggests a new additional function for HDAC6 in the regulation of glycolytic metabolism in TNBC. There are numerous reports describing the importance of aerobic glycolysis in TNBC cell lines and patient samples (11, 13). TNBC cell lines are reported to harbor high glycolytic flux, when compared with luminal A and HER2-positive breast cancer cells (11, 39). We present evidence of increased expression of glycolytic enzymes in TNBC, compared with other subtypes of breast cancer, using mRNA data from the TCGA (25). Understanding the underlying metabolic dependencies in tumor cells and identifying important regulators of the metabolic pathways, such as our discovery of HDAC6 regulating glycolysis, provide important developments for clinically targeting metabolism in the clinic.
MATERIALS AND METHODS
Cell culture
MDA231, BT-549, HCC1143, HCC1937, HS574T, PC9, 4T1, MCF10a, MDA134, MCF-7, ZR751, TD47, and fibroblast cells were purchased from the American Type Culture Collection and maintained in RPMI (Thermo Fisher Scientific, 118757093) or Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, D5546) supplemented with 10% (v/v) fetal calf serum (Sigma-Aldrich, F2442), 10 mM l-Glu (Sigma-Aldrich G6392), and penicillin/streptomycin (5 mg/ml; Sigma-Aldrich P4458). For the glucose-free experiments, glucose-free RPMI (Sigma-Aldrich R1383) was used, and glucose (Sigma-Aldrich G7528) or galactose 10 mM (Sigma-Aldrich G5388) was added 12 hours before drug treatment. HMECs were maintained in a mammary epithelial growth media (MEGM) bullet kit (Lonza CC-3150). The cell lines were recently authenticated by short-term tandem repeat profiling in January 2017.
Western blotting
Protein samples for Western blot analysis were separated by 12% SDS–polyacrylamide gel electrophoresis (PAGE) gels. Following separation on the gel, proteins were transferred using electrophoresis onto a nitrocellulose membrane and blocked at room temperature shaking in 5% milk (w/v) in tris-buffered saline (TBS) containing 0.5% Tween 20 (TBS-T). Membranes were incubated overnight at 4°C with primary antibody. Horseradish peroxidase (HRP)–tagged secondary antibodies were diluted in TBS-T/5% milk for 1 hour. Antibody-reactive bands were detected with LAS-3000, Fujifilm. Fluorescent secondary antibodies were diluted 1:10,000 in TBS-T/5% milk for 1 hour. Antibody-reactive bands were detected with the Odyssey infrared imaging system (LI-COR Biosciences). IRDye 680LT and 800CW Infrared Dye coupled anti-rabbit or anti-mouse (LI-COR Biosciences).
Antibodies
The following were the antibodies used: anti-mouse actin (Sigma-Aldrich, A1978), anti-rabbit BAX (Cell Signaling, 3792S), anti-rabbit BAK (Cell Signaling, 2774S), anti-rabbit acetyl alpha tubulin (Cell Signaling, 5335S), anti-rabbit HDAC6 (Cell Signaling, 7888S), anti-rabbit aldolase A (Cell Signaling, 3188S), anti-rabbit GAPDH (Cell Signaling, 5174S), anti-acetyllysine agarose (ImmuneChem, ICP0388), anti-mouse HRP secondary antibody (LI-COR Biosciences, 926-80010), and anti-rabbit HRP secondary antibody (LI-COR Biosciences, 926-80011); IRDye 680LT and 800CW Infrared Dye coupled anti-rabbit or anti-mouse (LI-COR).
Generation of HDAC6 KO cell line
Oligonucleotide sequences for specific short guide RNA (sgRNA) pLentiCRISPRv2 KO of HDAC6 were designed using the MIT (Massachusetts Institute of Technology) CRISPR oligo algorithm. Two different sgRNA plasmids were developed for two different target sequences within the open reading frame (sgScramble, sgHDAC6-1, and sgHDAC6-2). Sequences were located in exon 1 of the open reading frame and directly followed by a NGG protospacer adjacent motif sequence on the 3′ end. Each 20-mer oligonucleotide was ligated into the digested lentiviral pLentiCRISPRv2 vector (40). Lentiviral transduction was used to transduce recipient MDA231, BT-549, and 4T1 cells with the respective pLentiCRISPR sgHDAC6 and sgSCR KO plasmids, and selected for with puromycin (Thermo Fisher Scientific, A1113802) to generate stable HDAC6 KO cell lines.
Establishment of stable HDAC6 KD MDA231 cells
Scrambled and HDAC6 pLKO shRNA vectors were provided by T. Hideshima from Dana-Farber Cancer Institute (DFCI). Human embryonic kidney (HEK) 293T cells were transiently transfected with recombinant lentivirus following a standard protocol. The MDA231 were seeded and incubated for 16 hours with culture supernatants from HEK293T cells. The next day, the cells were washed with media and selected for antibiotic resistance for 1 week.
Small interfering RNA transfection
Cells were transiently transfected with small interfering RNA (siRNA) oligonucleotide against BAK (Dharmacon, L-003308-01-0005), BAX (Dharmacon, L-003305-00-0005), or nontarget region (Dharmacon, D-001810-03-05) using Lipofectamine PLUS (Biosciences, 31985047). Cells were incubated in Opti-MEM (Biosciences, 13778150) supplemented with 5% fetal bovine serum (FBS) for 24 hours following transfection.
Cell viability assays
Cells were treated with BAS-2 for 24 or 48 hours where indicated. Apoptosis was assessed by annexin V/propidium iodide (BioLegend, 640906, Sigma-Aldrich P4170) staining using Biosciences LSR II and FACSCanto. Cell viability was detected by CellTiter-Glo (My Bio, G3582) according to the manufacturer’s protocol. Mitochondrial membrane potential was measured by adding tetramethylrhodamine ethyl ester (TMRE) (Biosciences, T669) at 20 nM for 30 min at 37°C and assessed by flow cytometry with FACSCanto II.
Drug screen
Day 1: MDA-MB-231 cells were seeded at 1.75 × 104 per well in the presence of 0.05 μl of RNAi MAX and 20 nM BAX and BAK siRNA, and the MCF10a cells were seeded at 2.25 × 104 per well in 40 μl of appropriate media (MDA-MB-231 Opti-MEM no penicillin/streptomycin and MCF10a MEGM). Day 2: 20 μl of media was added to each well (Opti-MEM and penicillin/streptomycin). Day 3: Pin transfer of small molecules from drug plate in duplicate and addition of positive controls (10 μM ABT-737 and 20 μM gossypol) and negative control (DMSO) by electronic pipette. Day 5: Viability was measured by addition of 10 μl of CellTiter-Glo (My Bio, G3582) for 20 min, and luminescence was read on Envision 3. The screen was done in the ICCB-Longwood screening facility. BAS 04850653 was the lead hit identified from the Assenix 1 library and referred throughout the paper as BAS-2, and for low-throughput assays, it was purchased from MCULE (7388843487).
Genetic approach for drug target
Eμ-Myc p19Arf−/−cells were infected with retroviruses encoding eight shRNAs (P53, CHK2, CHK1, ATX, ATR, DNAPK, BOK, and BIM) in pMLS and were infected at 10 to 20% GFP+ proportion as previously described (18). Briefly, individual infected cell populations were seeded at 1 million cells per milliliter in 48-well plates and treated with drugs. Cells are dosed with equivalent lethal doses rather than equivalent molarities of compound. Cell death was measured by propidium iodide (PI) exclusion at 48 hours. Eighty percent to 90% cell death is used to measure GFP enrichment or depletion relative to a vector control at 72 hours. To avoid outgrowth of untreated control cells, the cells were typically seeded at 0.25 million/ml, and 75% of medium was replaced at 24 and 48 hours. Relative resistance index was calculated as described in (18).
HDAC inhibitory assay
The inhibition of HDAC proteins (HDAC1–9) was determined using a kinetic assay to measure trifluoroacetyllysine substrate processing, as reported previously (19).
Migration and invasion assays
Cell migration and invasion experiments were performed using the Boyden Chamber Assay (Corning, CLS3464). A total of 500,000 cells were seeded in serum-free RPMI media (Thermo Fisher Scientific, 118757093) in the upper chamber, and RPMI medium containing 10% FBS (Sigma-Aldrich, F2442) was placed in the lower chamber. For invasion experiments, the upper chamber was coated with Matrigel (Corning). After 4 hours, cells were treated with BAS-2 (30 μM). Following 24 hours (migration experiments) or 48 hours (invasion experiments) of treatment, cells were stained with 0.5% crystal violet.
3D cell cultures
Individual wells of a six-well plate was coated with Matrigel (Corning) and placed in an incubator at 37°C for 30 min. A total of 50,000 cells/ml were resuspended in RPMI supplemented with 2% Matrigel. Cells were placed in Matrigel-coated wells for 30 min at 37°C, after which RMPI supplemented with 2% Matrigel was added to the cultures. Cells were treated with BAS-2 (30 μM) after 48 hours. Cells were maintained in culture for 7 days in an incubator at 37°C, 5% CO2, and cultures were imaged every 24 hours with a Nikon Eclipse TE300.
Mouse experiments
All experiments involving the mammary tumor virus (MMTV)–polyoma virus middle T-antigen (PyMT) animals were reviewed and approved by the DFCI Institutional Animal Care and Use committee. All experiments involving 4T1 breast mouse model and KP NSCLC mouse model were reviewed and approved by the Institutional Animal Care and Use Committee at the New York University Langone Health (NYULH). For the breast cancer transgenic mouse model experiments, virgin female mice carrying the polyoma virus middle T-antigen (PyMT) under the control of the mammary tumor virus (MMTV) promoter were used. The mice had a least one tumor with size of 300 to 500 mm3 before randomization of treatment of either vehicle (DMSO) or BAS-2 (50 mg/kg) intraperitoneal injection with 5 days on and 2 days off. For the 4T1 mouse model, 1 × 106 4T1 cells were implanted into the right and left mammary lower fat pad of BALB/C mice. The mice had a least one tumor with size of 100 to 150 mm3 before randomization of treatment of either vehicle (DMSO) or BAS-2 (50 mg/kg) intraperitoneal injection every day. KP cell lines were established as described previously (41). KP cells (5 × 105) were subcutaneously injected into both flanks of C57BL/6J mice. The mice had a least one tumor with size of 100 to 150 mm3 before randomization of treatment of either vehicle (DMSO) or BAS-2 (50 mg/kg) intraperitoneal injection every day.
For all mouse studies, caliper measurements were used to measure tumor volume [(length × width2)/2] and were done in a blinded fashion. The total sum of the tumor burden was calculated by adding the volume of each of the tumors. At the end of treatment, the mice were euthanized humanely in a CO2 chamber.
MS-based detection of protein acetylation
Preparation of cell lysates
MDA231 cells were cultured in the presence of BAS-2 (30 μM) or DMSO for 48 hours before lysis. MDA231 HDAC6 KD and control vector cells were seeded 24 hours before the experiment. Cell lysates were prepared in urea lysis buffer [20 mM Hepes (pH 8.0), 9 M urea (Sigma-Aldrich, U5378), 1 mM sodium orthovanadate (Sigma-Aldrich, S6508), 2.5 mM sodium pyrophosphate (Sigma-Aldrich, 221368), and 1 mM β-glycerophosphate (Sigma-Aldrich, G9422)] and sonicated.
In-solution protein digestion and reverse-phase solid-phase extraction of digests
Sonicated cell lysates were cleared by centrifugation at 20,000g, and proteins were reduced with dithiothreitol (DTT) (Sigma-Aldrich, 10197777001) at a final concentration of 1.25 M and alkylated with iodacetamide (Sigma-Aldrich, 16125). For digestion with trypsin, protein extracts were diluted in 20 mM Hepes (pH 8.0) to a final concentration of 2 M urea, and trypsin was added to digest overnight. Trifluoroacetic acid (TFA) (Bioscience, 28901) was added to protein digest to a final concentration of 1%, precipitate was removed by centrifugation at 2000g for 5 min, and digests were loaded onto Sep-Pak C-18 columns (Waters, WAT051910) that were equilibrated with 0.1% TFA. Acidified and cleared digests were added before washing columns with TFA and acetonitrile (ACN; Sigma-Aldrich, 271004). Peptides were eluted with 0.1% TFA in 40% ACN. All peptide fractions were lyophilized.
Immunoaffinity purification of acetylated peptides and LC-MS/MS
Lyophilized peptides were resuspended in IAP (immunoaffinity purification) buffer [50 mM MOPS (pH 7.2), 10 mM sodium phosphate, and 50 mM NaCl] and cleared by centrifugation at 10,000g. Acetylated peptides were enriched using pan-specific anti-acetylated lysine beads (ImmuneChem, ICP0388). Supernatant was mixed with anti-acetylated lysine beads for 3 hours at 4°C and centrifuged at 2000g. Beads were washed, and peptides were eluted with 0.15% TFA (Bioscience, 28901). Eluted peptides were concentrated and purified using Stage Tips (Pierce, 87782) and resuspended in 0.1% TFA. LC-MS/MS was performed on the resuspended immunoaffinity-purified acetylated peptides.
The samples were analyzed by Systems Biology Ireland and Mass Spectrometry Resource in the University College Dublin on a Thermo Scientific Q Exactive mass spectrometer connected to a Dionex Ultimate 3000 (RSLCnano) chromatography system. Peptides were separated on a C18 homemade column (C18RP Reposil-Pur, 100 mm × 0.075 mm × 1.9 μm) over 150 min at a flow rate of 250 nl/min with a linear gradient of increasing ACN from 1 to 97%. The mass spectrometer was operated in a data-dependent mode; a high-resolution (70,000) MS scan [mass/charge ratio (m/z), 300 to 1600] was performed to select the 12 most intense ions and fragmented using high-energy C-trap dissociation for MS/MS analysis.
MS identification of HDAC6 interactions
MDA231 cells were lysed [50 mM tris-HCL (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and protease inhibitors]. Cell lysates were precleared and incubated with anti-HDAC6 antibody (Cell Signaling, 7888S) and A-agarose beads (Pierce, 20333) and incubated at 4°C overnight. The agarose resin was washed with lysis buffer, and immunoprecipitated proteins were frozen and shipped to TDI Mass Spectrometry Laboratory at the University of Oxford for MS analysis. Peptides were analyzed on a Thermo Scientific Q Exactive mass spectrometer connected to a Dionex Ultimate 3000 (RSLCnano) chromatography system. Peptides were loaded in 0.1% TFA in 2% ACN onto a trap column (PepMAP C18, 300 μm × 5 mm, 5-μm particle, Thermo Fisher Scientific) and separated on an Easy Spray column (PepMAP C18, 75 μm × 500 mm, 2-μm particle, Thermo Fisher Scientific) with a gradient 2% ACN to 95% ACN in 0.1% formic acid in 5% DMSO. The mass spectrometer was operated in data-dependent mode; a high-resolution (70,000) MS scan (m/z, 380 to 1800) was performed to select the 15 most intense ions and fragmented using high-energy C-trap dissociation for MS/MS analysis.
Data processing and bioinformatics
Raw data from the Q-Exactive were processed using the MaxQuant (version 1.6.2.10) incorporating the Andromeda search engine (42). To identify peptides and proteins, MS/MS spectra were matched against Uniprot Homo sapiens database (2018_04) containing 73,045 entries. All searches were performed using the default setting of MaxQuant, with trypsin as specified enzyme allowing two missed cleavages and a false discovery rate of 1% on the peptide and protein levels. The database searches were performed with carbamidomethyl (C) as fixed modification, and acetylation (protein N terminus), oxidation (M), and acetylation (K) as variable modifications. For the generation of label-free quantitative ion intensities for protein profiles, signals of corresponding peptides in different nano–HPLC MS/MS runs were matched by MaxQuant in a maximum time window of 1 min. The Perseus computational platform (version 1.6.2.3) was used to process MaxQuant results (43). Data were log transformed. For visualization of data using volcano plots, data were log transformed, and missing values were inputted with values from a normal distribution, and a cutoff of t test difference of 1.5 and a −log(P value) of 1.3 were applied. For visualization of acetylated peptides, a t test difference cutoff of 0.5 was applied. To evaluate pathway annotation networks of enriched peptides (t test difference, >0.5) from BAS-2– and HDAC6-treated acetylome and significantly enriched proteins (t test difference, >1.5; P value of 0.05 or –log P value of 1.3) from HDAC6 immunoprecipitation, pathway enrichment analysis was performed using the ClueGo (v2.5.2) (44) and Cluepedia (v1.5.2) (45) plugins in Cytoscape (v3.6.1) (46) with the H. sapiens (9606) marker set. The KEGG functional pathway databases, consisting of 7425 genes, were used (47). Gene ontology (GO) tree levels (min = 3; max = 8) and GO term restriction (min#genes = 3, min% = 1%) were set, and terms were grouped using a Kappa score threshold of 0.4. The classification was performed by the two-side hypergeometric statistic test, and its probability value was corrected by the Bonferroni method (adjusted % term P value <0.05).
Immunoprecipitation
MDA231 cells were treated with BAS-2 (10 μM) for 24 hours. Cell lysates were prepared in lysis buffer [50 mM tris-HCL (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and protease inhibitors]. Protein was quantified, and 500 μg of protein was added to 20 μl of A-agarose beads (Pierce, 20333) and allowed to rotate for 1 hour at 4°C and 3000 rpm. Proteins were centrifuged for 3 min at 3000 rpm at 4°C. Supernatant was added to 500 μl of lysis buffer, 30 μl of pan-specific anti-acetylated lysine beads (ImmuneChem, ICP0388), or control agarose beads, and made up to 1 ml with dH2O. Samples were rotated overnight at 4°C at 3000 rpm. Samples were washed with lysis buffer and were resuspended in 30 μl of 2× IP dye with beta-mercaptoethanol and boiled and ran out on 12% SDS-PAGE gel.
TCGA glycolytic enzyme analysis
The RNA-sequencing and clinical data were downloaded from TCGA (25). Male patients, patients with an overall survival time of ≤0 days, and history of other malignancy were removed. For every gene (plot), a Kruskal-Wallis rank sum test was performed and a pairwise Wilcoxon rank sum tests between groups if the former is statistically significant. P values of the pairwise Wilcoxon rank sum test were adjusted for multiple comparison using the Benjamini and Hochberg (48) method.
Aldolase and GAPDH activity assay
For aldolase and GAPDH activity assay, 1 × 106 MDA231 cells were treated with BAS-2 for 24 hours before experiment. The activity assay was performed as per the manufacturer’s instructions (aldolase: Biovision, K665; GAPDH: Sigma-Aldrich, MAK277). Values were normalized to protein concentrations, and fold change was calculated relative to DMSO.
Extracellular flux assay
The bioenergetic function of cells in response to drug treatments was determined using a Seahorse Bioscience XF96 Extracellular Flux Analyzer (Seahorse Bioscience). Cells were seeded in specialized V7 Seahorse tissue culture plates (Agilent, 102601-100) for 24 hours. Cells were then treated with the indicated concentrations of BAS-2 for further 24 hours. One hour before the experiment, cells were washed and changed to XF Base medium (Agilent, 10353-100) adjusted to pH 7.4. For oxidative phosphorylation experiments, medium was supplemented with pyruvate (1 mM), l-glutamine (2 mM), and glucose (10 mM). Three baseline OCR measurements were taken, followed by three measurements after each injection of oligomycin (1 μM), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP; 0.5 μM), and rotenone and antimycin A (0.5 mM) (Agilent, 103015-100), respectively. For glycolysis experiments, medium was supplemented with l-glutamine (1 mM). Three baseline measurements were taken for ECAR, followed by three measurements after each injection of glucose (10 mM), oligomycin (1 μM), and 2-DG (50 mM) (Agilent, 103020-100), respectively. For all analysis, values were normalized to protein concentration before baseline measurements were subtracted. The fold change was calculated relative to DMSO.
GC-MS analysis
Isotope labeling
Cells were seeded in 12-well tissue culture plates and left to adhere overnight. Cells were then treated with 10 μM BAS-2 for further 24 hours. Following treatment, medium was removed, and cells were washed with 1× phosphate-buffered saline before adding basic RPMI (Thermo Fisher Scientific, 11879020) supplemented with dialyzed FBS (Gibco, 26400044) and either 10 mM [U]-13C6–labeled glucose (Cambridge Isotopes, CLM-1396) for 30 min or tracing media containing 10 mM 1,2-13C–labeled glucose (Cambridge Isotopes, CLM-504) for 12 hours. Cellular metabolites were extracted after a brief wash with 0.9% ice-cold saline solution using a methanol/water/chloroform extraction method, as previously described (49).
Metabolite secretion
Medium was collected from cells seeded in six-well plates and cultured in basic RPMI (Thermo Fisher Scientific, 11879020) supplemented with 10% dialyzed FBS (Gibco, 26400044) after a 24-hour treatment with 10 μM BAS-2. Medium was centrifuged at 1000g to pellet cell debris, and 5 μl of supernatant was extracted in 80% ice-cold methanol mixture containing isotope-labeled internal standards for lactate (Cayman Chemicals, CLM-1579) and pyruvate (Cayman Chemicals, CLM-2440) and evaporated to dryness.
Tumor metabolites
Following 7- or 14-day treatment, mice were fasted for 6 to 8 hours before euthanasia and tumor harvest. Tumor metabolites were extracted in methanol/water/chloroform using the Qiagen TissueLyser LT, evaporated to dryness, and resuspended in 80% methanol. The equivalent volume for 5 mg of tissue was further extracted in 80% ice-cold methanol mixture containing isotope-labeled standards for lactate (Cayman Chemicals, CLM-1579) and pyruvate (Cayman Chemicals, CLM-2440) and evaporated to dryness.
Metabolite derivatization
Metabolite derivatization using MOX-tBDMS was conducted as previously described (50). Derivatized samples were analyzed by GC-MS using a DB-35MS column (30 m × 0.25 mm inner diameter × 0.25 μm) installed in an Agilent 7890B GC interfaced with an Agilent 5977B mass spectrometer as previously described (51) and corrected for natural abundance using in-house algorithms adapted from (52).
BH3 profiling
The sequence of the BH3-only peptides used and method of synthesis are as previously described (14). BH3 profiling was performed using plate-based fluorimetry. Briefly, BH3 peptides (BIM, 0.3 μM BIM, BCL-2 associated agonist of cell death, p53 upregulated modulator of apoptosis, BCL-2 modifying factor, and positive-control FCCP) at 70 μM, unless stated otherwise, were plated in triplicate on a black 384-well plate. Cells were gently permeabilized with 0.005% digitonin and loaded with the mitochondrial dye 0.5 μM JC-1. The cells were plated on top of the peptides. The loss of mitochondrial potential was measured on the Tecan Safire2 Percentage mitochondrial depolarization, and that for the peptides was calculated by normalization to the solvent-only control DMSO (0%) and the positive control FCCP (100%) using area under the curve.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA) unless stated otherwise. The IC50 for the dose-response curves following drug treatments was calculated using linear regression curve fit (log inhibitor versus normalized response). Significance between two groups was determined using an unpaired two-tailed t test. Significance between more than two groups was determined using a two-way analysis of variance (ANOVA) with the Tukey’s post hoc test. For all statistical analysis, differences were considered to be statistically significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank everyone at the ICCB at Harvard Medical School for the help with the small-molecule screening. Last, we thank T. Hideshima from the Dana-Farber Cancer Institute for the gift of the pLKO HDAC6 vectors. Funding: This work was funded by grants from SFI-HRB-Wellcome Trust 202079/Z/16/Z, Breast Cancer Now 2016NovPR849, L’Oréal-UNESCO UK & I FWIS Fellowship, and Science Foundation Ireland 19/FFP/6461. C.M.D. is funded by the Irish Research Council Postdoctoral Fellowship GOIPD/2017/1271 and the H2020 MSCA Global Fellowship (799724). A.G.L. is funded by NIH R35CA242427. Author contributions: T.N.C. conceived the project, designed the experiments, and wrote the paper. C.M.D. performed the experiments, analyzed the data, and wrote the paper. K.E.R.H., A.D.G., J.P., H.Z., E.T.D., K.H., E.P., A.K.M., R.B., A.U.L., B.M., H.D., K.W., and J.L.G. performed the experiments and analyzed the data. D.O., J.H.M.P., M.H., K.-K.W., J.E.B., A.G.L., A.C.K., and G.C. analyzed the data and edited the manuscript. G.C. analyzed the data and helped with the writing of the manuscript. Competing interests: T.N.C. receives research support from AbbVie but not in relation to this project. J.E.B. is president of the Novartis Institute of Biomedical Research. A.G.L. discloses that he has worked as a consultant for and his laboratory has received research support from AbbVie, Novartis, and AstraZeneca. A.G.L. is an equity holder and cofounder of Flash Therapeutics and Zentalis Pharmaceuticals. A.G.L. is an equity holder and advisor for Dialectic Therapeutics. J.L.G. is a consultant for Glaxo-Smith Kline (GSK) and Array BioPharma Codagenix, Verseau, and Kymera and receives sponsored research support from GSK, Eli Lilly, and Array BioPharma. K.-K.W. is a founder and equity holder of G1 Therapeutics and has consulting and sponsored research with AstraZeneca, Janssen, Pfizer/Array Biopharma, Novartis, Merck, and Zentalis, as well as sponsored research (only) with Takeda, BMS, Mirati, Alkermes, Merus, Amgen, Ansun Biopharma, Eliven Therapeutics, Tvardi Therapeutics, Delfi Diagnostics, and Dracen Pharmaceuticals. A.C.K. has financial interests in Vescor Therapeutics LLC and Rafeasel/Cornerstone Pharmaceuticals. A.C.K. is an inventor on patents pertaining to KRAS-regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the SAB of Rafael/Cornerstone Pharmaceuticals. A.C.K. is on the SAB of Vescor Therapeutics LLC and Rafael/Cornerstone Pharmaceuticals. A.C.K. has been a consultant for Deciphera Pharmaceuticals. The other authors declare that they have no competing interests. US provisional patent application filed 15 December 2020. Reference number 129147.00021. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabc4897/DC1
REFERENCES AND NOTES
- 1.Perou C. M., Sørlie T., Eisen M. B., van de Rijn M., Jeffrey S. S., Rees C. A., Pollack J. R., Ross D. T., Johnsen H., Akslen L. A., Fluge Ø., Pergamenschikov A., Williams C., Zhu S. X., Lønning P. E., Børresen-Dale A.-L., Brown P. O., Botstein D., Molecular portraits of human breast tumours. Nature 406, 747–752 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Bianchini G., Balko J. M., Mayer I. A., Sanders M. E., Gianni L., Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carey L., Winer E., Viale G., Cameron D., Gianni L., Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 7, 683–692 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Liedtke C., Mazouni C., Hess K. R., André F., Tordai A., Mejia J. A., Symmans W. F., Gonzalez-Angulo A. M., Hennessy B., Green M., Cristofanilli M., Hortobagyi G. N., Pusztai L., Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–1281 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Holohan C., Van Schaeybroeck S., Longley D. B., Johnston P. G., Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Vander Heiden M. G., Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 10, 671–684 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Warburg O., On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 8.Shim H., Chun Y. S., Lewis B. C., Dang C. V., A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. U.S.A. 95, 1511–1516 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J. K. V., Markowitz S., Zhou S., Diaz L. A. Jr., Velculescu V. E., Lengauer C., Kinzler K. W., Vogelstein B., Papadopoulos N., Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vousden K. H., Ryan K. M., p53 and metabolism. Nat. Rev. Cancer 9, 691–700 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Lim S.-O., Li C.-W., Xia W., Lee H.-H., Chang S.-S., Shen J., Hsu J. L., Raftery D., Djukovic D., Gu H., Chang W.-C., Wang H.-L., Chen M.-L., Huo L., Chen C.-H., Wu Y., Sahin A., Hanash S. M., Hortobagyi G. N., Hung M.-C., EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 76, 1284–1296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balko J. M., Giltnane J. M., Wang K., Schwarz L. J., Young C. D., Cook R. S., Owens P., Sanders M. E., Kuba M. G., Sánchez V., Kurupi R., Moore P. D., Pinto J. A., Doimi F. D., Gómez H., Horiuchi D., Goga A., Lehmann B. D., Bauer J. A., Pietenpol J. A., Ross J. S., Palmer G. A., Yelensky R., Cronin M., Miller V. A., Stephens P. J., Arteaga C. L., Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 4, 232–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zordoky B. N. M., Bark D., Soltys C. L., Sung M. M., Dyck J. R. B., The anti-proliferative effect of metformin in triple-negative MDA-MB-231 breast cancer cells is highly dependent on glucose concentration: Implications for cancer therapy and prevention. Biochim. Biophys. Acta 1840, 1943–1957 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Chonghaile T. N., Sarosiek K. A., Vo T.-T., Ryan J. A., Tammareddi A., Moore V. D. G., Deng J., Anderson K. C., Richardson P., Tai Y.-T., Mitsiades C. S., Matulonis U. A., Drapkin R., Stone R., De Angelo D. J., Mc Conkey D. J., Sallan S. E., Silverman L., Hirsch M. S., Carrasco D. R., Letai A., Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334, 1129–1133 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letai A. G., Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 8, 121–132 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y.-J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M.-S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elenbaas B., Spirio L., Koerner F., Fleming M. D., Zimonjic D. B., Donaher J. L., Popescu N. C., Hahn W. C., Weinberg R. A., Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang H., Pritchard J. R., Williams R. T., Lauffenburger D. A., Hemann M. T., A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat. Chem. Biol. 7, 92–100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradner J. E., West N., Grachan M. L., Greenberg E. F., Haggarty S. J., Warnow T., Mazitschek R., Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X.-X., Wan R.-Z., Liu Z.-P., Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 143, 1406–1418 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Hideshima T., Qi J., Paranal R. M., Tang W., Greenberg E., West N., Colling M. E., Estiu G., Mazitschek R., Perry J. A., Ohguchi H., Cottini F., Mimura N., Görgün G., Tai Y.-T., Richardson P. G., Carrasco R. D., Wiest O., Schreiber S. L., Anderson K. C., Bradner J. E., Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 113, 13162–13167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wedel S., Hudak L., Seibel J.-M., Makarević J., Juengel E., Tsaur I., Wiesner C., Haferkamp A., Blaheta R. A., Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin. Exp. Metastasis 28, 479–491 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Schölz C., Weinert B. T., Wagner S. A., Beli P., Miyake Y., Qi J., Jensen L. J., Streicher W., McCarthy A. R., Westwood N. J., Lain S., Cox J., Matthias P., Mann M., Bradner J. E., Choudhary C., Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 33, 415–423 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Boyault C., Sadoul K., Pabion M., Khochbin S., HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26, 5468–5476 (2007). [DOI] [PubMed] [Google Scholar]
- 25.The Cancer Genome Atlas Network , Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W. S., Stockwell B. R., Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin A., Giuliano C. J., Palladino A., John K. M., Abramowicz C., Yuan M. L., Sausville E. L., Lukow D. A., Liu L., Chait A. R., Galluzzo Z. C., Tucker C., Sheltzer J. M., Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 11, eaaw8412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones P. A., Issa J.-P. J., Baylin S., Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Ferrarelli L. K., HDAC inhibitors in solid tumors and blood cancers. Sci. Signal. 9, ec216 (2016). [Google Scholar]
- 30.Yau H. L., Ettayebi I., De Carvalho D. D., The cancer epigenome: Exploiting its vulnerabilities for immunotherapy. Trends Cell Biol. 29, 31–43 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Laino A. S., Betts B. C., Veerapathran A., Dolgalev I., Sarnaik A., Quayle S. N., Jones S. S., Weber J. S., Woods D. M., HDAC6 selective inhibition of melanoma patient T-cells augments anti-tumor characteristics. J. Immunother. Cancer 7, 33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M., Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Hook S. S., Orian A., Cowley S. M., Eisenman R. N., Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. U.S.A. 99, 13425–13430 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang H.-C., Guarente L., SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 25, 138–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hallows W. C., Yu W., Denu J. M., Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J. Biol. Chem. 287, 3850–3858 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wardell S. E., Ilkayeva O. R., Wieman H. L., Frigo D. E., Rathmell J. C., Newgard C. B., McDonnell D. P., Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 23, 388–401 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris A. J., Tolan D. R., Lysine-146 of rabbit muscle aldolase is essential for cleavage and condensation of the C3-C4 bond of fructose 1, 6-bis (phosphate). Biochemistry 33, 12291–12297 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Davidson S. M., Papagiannakopoulos T., Olenchock B. A., Heyman J. E., Keibler M. A., Luengo A., Bauer M. R., Jha A. K., O’Brien J. P., Pierce K. A., Gui D. Y., Sullivan L. B., Wasylenko T. M., Subbaraj L., Chin C. R., Stephanopolous G., Mott B. T., Jacks T., Clish C. B., Heiden M. G. V., Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelicano H., Zhang W., Liu J., Hammoudi N., Dai J., Xu R.-H., Pusztai L., Huang P., Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 16, 434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelson T., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F., Huang Q., Luster T. A., Hu H., Zhang H., Ng W.-L., Khodadadi-Jamayran A., Wang W., Chen T., Deng J., Ranieri M., Fang Z., Pyon V., Dowling C. M., Bagdatlioglu E., Almonte C., Labbe K., Silver H., Rabin A. R., Jani K., Tsirigos A., Papagiannakopoulos T., Hammerman P. S., Velcheti V., Freeman G. J., Qi J., Miller G., Wong K.-K., In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov. 10, 270–287 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M., Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., Mann M., Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., Galon J., ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bindea G., Galon J., Mlecnik B., CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 29, 661–663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 49.Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., Stephanopoulos G., Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis C. A., Parker S. J., Fiske B. P., Closkey D. M., Gui D. Y., Green C. R., Vokes N. I., Feist A. M., Heiden M. G. V., Metallo C. M., Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grassian A. R., Parker S. J., Davidson S. M., Divakaruni A. S., Green C. R., Zhang X., Slocum K. L., Pu M., Lin F., Vickers C., Joud-Caldwell C., Chung F., Yin H., Handly E. D., Straub C., Growney J. D., Heiden M. G. V., Murphy A. N., Pagliarini R., Metallo C. M., IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 74, 3317–3331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernandez C. A., Des Rosiers C., Previs S. F., David F., Brunengraber H., Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 31, 255–262 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/3/eabc4897/DC1