

Summary
Mutations in the p53 tumor suppressor are frequent causes of cancer. Because p53 aggregates appear in some tumor cells, it has been suggested that p53 could also cause cancer by forming self-replicating protein aggregates (prions). Here, using yeast, we show that transient p53 overexpression induced the formation of p53 prion aggregates that were transmitted for >100 generations, found in lysate pellets, stained with Thioflavin T, and transmitted by cytoplasmic transfer, or transfection with lysates of cells carrying the prion or with p53 amyloid peptide. As predicted for a prion, transient interruption of p53 expression caused permanent p53 prion loss. Importantly, p53 transcription factor activity was reduced by prion formation suggesting that prion aggregation could cause cancer. p53 has also been found in liquid-like nuclear droplets in animal cell culture. In yeast, we found that liquid-like p53 foci appear in response to stress and disappear with stress removal.
Subject areas: Biochemistry, Cell Biology, Cancer
Graphical abstract
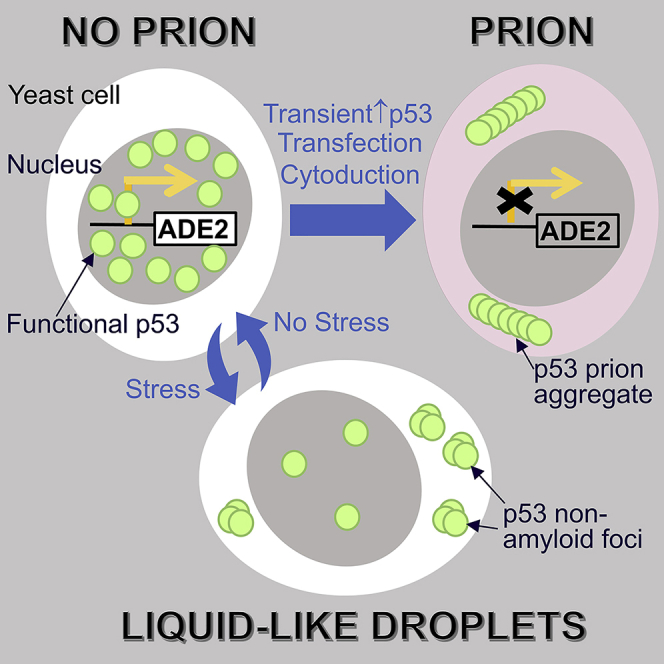
Highlights
-
•
A published yeast model of functional nuclear human p53 tumor suppressor was used
-
•
Upon transient overexpression p53 loses its transcription function and aggregates
-
•
These p53 aggregates are cytoplasmic and behave like stable heritable prions
-
•
Stress induces p53 to form liquid-like droplets that are unstable and not prion-like
Biochemistry; Cell Biology; Cancer
Introduction
Mutations in the human p53 transcription factor are a frequent cause of cancer. Cancers occur when a normal cell undergoes a series of mutations that transform it into a cell that grows without control. The most important tumor suppressor gene is likely the p53 transcription factor dubbed the “guardian of the genome” and “policemen of the oncogenes” (Lane, 1992; Levine and Oren, 2009; Levine et al., 2011; Ozaki and Nakagawara, 2011). Indeed, in more than 50% of human cancer cells, at least one of the two p53 alleles carries a mutation. Furthermore, people born without p53 always get cancer (Iwakuma et al., 2005).
It has been hypothesized that inactivation of p53 leading to tumor formation may be caused not only by mutations in p53 but also when p53 forms punctate inclusions that have been seen in tumor cell lines and cancer biopsies with certain p53 mutations (reviewed in de Oliveira et al., 2020; Navalkar et al., 2020). Interestingly, these puncta have some of the biochemical characteristics of amyloid, which is found in many prion aggregates. Also, certain mutant p53 can form fibers in vitro that stain with dyes suggestive of amyloid (Congo red and Thioflavin T [ThT]) and have an X-ray diffraction characteristic of amyloid. Although the kinetic properties of p53 aggregation in vitro are not equivalent to classic nucleated growth expected of amyloid (Wilcken et al., 2012), wild-type p53 aggregation can be stimulated by the addition of mutant p53 fibers (Ano Bom et al., 2012).
Furthermore, a p53 peptide of eight residues (amino acid [aa] 250–257, PILTIITL called P8) forms amyloid fibers in vitro and seeds the formation of a larger “core” domain of p53 (aa. 94–312) to form amyloid. Fibers of this peptide, when internalized by SH-SY5Y neuroblastoma cells, caused the appearance of intracellular p53 aggregates, and lysates of these cells internalized by virgin cells caused de novo p53 aggregation (Ghosh et al., 2014, 2017). Thus p53 aggregates can enter cells and seed aggregation of endogenous p53. However, this could occur if intra-cellular p53 decorates the infecting aggregates as has been seen for prion-like proteins such as Huntingtin that do not form heritable prions (Osherovich et al., 2004).
Real prion formation also requires that the newly aggregated p53 fragments in the cell create an exponential increase in propagon number that can then infect other cells. The goal of this work was to determine if p53 can form such a prion and, importantly, if this is associated with loss of p53 function, which would likely cause cancer.
Prions are altered conformations of a protein that seed other molecules of that protein to rapidly adapt the prion conformation. At a given concentration in the cell these proteins remain soluble in the absence of seed but aggregate in the presence of seed. What causes the occasional de novo switch from a soluble protein to a prion seed in the absence of an infecting propagon is largely unknown. Once formed, however, prion aggregates increase in number when they fragment creating new seeds with ends that attract and template soluble conformers of that protein to rapidly switch to the prion conformation. Importantly, only a limited number of proteins, which generally have unstructured domains, are able to form amyloid in cells, even though all proteins can misfold into an amyloid conformation under non-physiological conditions (Chiti and Dobson, 2006). Furthermore, not all proteins that form amyloid in cells fragment to form heritable prions (Caudron and Barral, 2013; Marchante et al., 2017).
Prions have been found in yeast where they control endogenous traits (Liebman and Chernoff, 2012). These yeast proteins can lose, or change, their function when they switch to an aggregated, often amyloid conformation. Studies of prion proteins in yeast have contributed much to our knowledge about this phenomenon in human disease (Armakola et al., 2011; Bagriantsev and Liebman, 2006; Bonini and Gitler, 2011; Chernoff et al., 1995; Liebman and Chernoff, 2012; Park et al., 2011; Treusch et al., 2011). Indeed, the prion hypothesis was first proved when transfection of prions, in the form of pure amyloid fibers, into yeast cells caused prion infection transmitted for many generations (King and Diaz-Avalos, 2004; Tanaka et al., 2004). Interestingly, the majority of yeast prions and human disease proteins with prion-like domains are, like p53, nucleic acid-binding proteins (March et al., 2016).
Although yeast do not have endogenous p53, a robust assay for transcription factor activity of human p53 expressed in yeast has been used for decades (Fronza et al., 2000; Inga et al., 1997; Monti et al., 2019). High-level expression of wild-type p53 has been shown to cause reduced growth of some yeast strains under certain growth conditions. As loss-of-function p53 mutants did not have this effect, it was suggested that the growth inhibition is caused by the functional p53's aberrant transcription of endogenous yeast genes (Bischoff et al., 1992; Inga and Resnick, 2001; Kumar et al., 2017; Nigro et al., 1992). Thus, we reasoned that inactivation of p53 by prion formation might reduce its ability to inhibit growth.
Transient overexpression of yeast prion proteins induces the proteins to form aggregates that are self-seeding and efficiently fragment, so they propagate even after overexpression is removed (Alberti et al., 2009; Byers and Jarosz, 2017; Chakrabortee et al., 2016; Chakravarty et al., 2020; Derkatch et al., 1996; Du et al., 2008; Harvey et al., 2020; Itakura et al., 2020; Liebman and Chernoff, 2012; Patel et al., 2009; Wickner, 1994). Thus, transient overexpression of mutant or wild-type p53 could increase the likelihood of de novo p53 prion formation and inactivation, leading to a chain reaction of p53 aggregation and aggregate replication. This could explain why inactive wild-type p53 is found in the cytoplasm of some tumors (Elledge et al., 1994; Moll et al., 1995, 1997) and why melanoma cells expressing high levels of wild-type p53 are often associated with inactive p53 (Houben et al., 2011).
In addition to forming aggregates that are amyloid-like, p53 has also been shown to be present in non-membrane-bound liquid-like nuclear droplets, including Cajal and promyelocytic leukemia protein bodies (Banani et al., 2017; Cioce and Lamond, 2005; Fogal et al., 2000; Guo et al., 2000). p53 can also form liquid-like droplets in vitro (Kamagata et al., 2020). Other proteins with disordered sequences capable of forming amyloid aggregates can also form cellular and in vitro liquid-like droplets under certain conditions. These droplets appear to form by liquid-liquid phase separation (LLPS). Unlike amyloid-like prion aggregates, liquid droplets are not stable. They often appear and disappear in the cell in response to protein-specific stress triggers (Boeynaems et al., 2018; Boncella et al., 2020; Buchan et al., 2011; Chakravarty and Jarosz, 2018; Grousl et al., 2013; Itakura et al., 2018; Jin et al., 2017; Lee et al., 2018; Narayanaswamy et al., 2009). In particular, P-bodies, highly conserved RNA-protein granules that increase in number to help cells divert resources to survival in response to stress, have been shown to be liquid-like droplets in yeast (Kroschwald et al., 2015). Although the relationship between liquid droplets and amyloid aggregates is unknown, it has been suggested that the temporary concentration of a protein with an unstructured domain into a liquid droplet may enhance its chance of forming a more stable prion-like aggregate (Boeynaems et al., 2018; Taylor et al., 2016).
Here, we show that wild-type human p53 expressed in yeast and normally found to be diffuse in the nucleus can be induced to form cytoplasmic foci of two types. Transient overexpression of p53 resulted in the appearance of stable transmissible prions with amyloid-like structure, whereas the stresses of ethanol, heat, or glucose starvation caused the appearance of unstable non-amyloid foci.
Results
Establishment of a yeast strain with stable p53 cytoplasmic foci
We obtained stable heritable aggregates of p53 by screening for isolates with impaired p53 activity and increased p53 aggregation, following transient overexpression of p53. We used strain yIG397, containing an ADE2 reporter controlled by a promoter that is dependent upon p53 binding, and is therefore diagnostic for functional p53 activity (Inga et al., 1997). To obtain strain L3671, in which p53 expression could be controlled, yIG397 was co-transformed with a CEN plasmid constitutively expressing untagged wild-type p53 driven by the constitutive ADH promoter (pADH1-p53), as well as with a CEN plasmid expressing p53-EYFP from an inducible GAL promoter (pGAL-p53-EYFP). L3671 showed the expected ADE2 reporter phenotypes for functional non-prion wild-type p53: cells were white, indicative of full ADE2 expression resulting from p53 activity. We expected cells with a prion form of p53 to have reduced p53 function due to aggregation, resulting in reduction of ADE2 expression leading to reduced growth and a red or pink color on adenine-limiting media (Figure 1A).
Figure 1.

Wild-type p53 forms stable cytoplasmic aggregates that reduce p53 activity
(A) Cartoon of strain L3671 in which prion candidates were obtained showing cells with and without prion grown on glucose versus galactose. Shown are plasmids pADH1-p53 and pGAL1-p53-EYFP and ADE2 reporter gene under p53 control, contained in L3671. The nucleus is depicted as a blue oval. Even though the plasmids are nuclear, they are shown in the cytoplasm to make it easier to see them. In the absence of prion, p53 is in the nucleus where it is diffuse and turns on the ADE2 gene, making cells white and able to grow in the absence of adenine. In the presence of prion, p53 forms cytoplasmic puncta, reducing the amount of p53 in the nucleus so that ADE2 is not transcribed efficiently, and the cells are reddish in color on most media and grow poorly in the absence of adenine. L3671 grown on glucose only expresses untagged p53, from the constitutive ADH1 promoter. L3671 grown on galactose also expresses p53 tagged with EYFP from the GAL1 promoter. The nuclei in cells without the prion grown on galactose (but not glucose) show diffuse green fluorescence. Likewise, the cytoplasmic p53 foci in prion cells are only fluorescently tagged when grown on galactose.
(B) p53 prion candidates have reduced p53 activity. Cells shown are on plasmid-selective, adenine-limiting, glucose medium. The L3671 control (C) and prion candidates (1–28) are shown. Reduced expression of ADE2, and therefore reduced p53 activity, is seen as a pinkish color and reduced growth on media with limited adenine. Expression of p53 cannot be estimated in candidates 1, 2, or 12, because they are [rho−] and this loss of mitochondria caused them to become dark red.
(C) Reduced ADE2 expression is stably maintained in a p53 prion candidate. Following extensive culturing, normalized suspensions of subclones of prion candidate #7 (L3672), non-prion control (L3671) and empty vector (e.v.) control strain (yIG397), were one-tenth serially diluted in water and spotted on YPD (complex glucose) with and without additional adenine, on plasmid-selective synthetic glucose (glucose) with and without adenine, and on plasmid-selective 2% galactose medium with adenine plates. These plates were photographed after 3–5 days of incubation at 30°C. Red papillations on the non-plasmid-selective YPD plate without excess adenine result from loss of the p53 plasmid.
(D) p53 sediments more in lysates of prion candidates than in non-prion controls. Non-prion control (L3671) and prion (L3672) cells grown in plasmid-selective glucose or galactose media were lysed as described in Transparent Methods. Total lysates (T) were separated into Supernatant (S) and Pellet (P) by centrifugation at 80K RPM for 30 min. Samples were run on SDS-PAGE, and gels were immunoblotted with anti-p53, stripped, and reprobed with anti-PGK for a loading control. Detection of p53-EYFP monomer (~70 kD) or dimer and PGK (44.7 kD) on western blot was based on migration of Precision Plus Protein Dual Color (Bio Red, Hercules, CA).
(E) Prion candidate has p53-EYFP cytoplasmic foci. Both non-prion control (L3671) and prion candidate (L3672) repeatedly subcloned cells, grown on galactose to induce expression of GAL1-p53-EYFP, were examined under a fluorescent microscope and photographed (top). The fraction of cells with nuclear fluorescence (Nuclear), cytoplasmic aggregates (Cytoplasmic Foci), or vacuolar fluorescence (Vacuolar) was determined, counted, and converted into % fraction (bottom). Trypan blue was used to stain dead cells. Standard error of the mean bars shown were based on nine replicates and 200–700 cells were scored for each replica. The prion and non-prion cultures were statistically different (p < 0.05%) for fractions of cells with nuclear fluorescence, cytoplasmic puncta, and vacuolar fluorescence.
(F) p53 prion-like aggregates stain with Thioflavin T. Fluorescence of non-prion control (L3671) and prion cells (L3672) both containing pADH1-p53 and pGAL1-p53-EYFP grown on plasmid-selective galactose (left) or glucose (right) plates, fixed and stained (+ThT) or not stained (- ThT) with Thioflavin T versus EYFP fluorescence were, respectively, detected under CFP versus YFP filters. Cells expressing EYFP that were stained with Thioflavin T exhibited reduced, but detectable, EYFP fluorescence. Scale bar, 5 μM. Note: dead cells often look bright due to increased autofluorescence.
See Figures S1 and S2 for additional characterization of p53 non-prion and prion strains and Tables S1 and S2 for a description of strains and plasmids.
L3671 cells grown on galactose, to overexpress p53-EYFP, or glucose, as control, were transferred to glucose medium with limiting adenine to end the p53-EYFP expression (see Transparent Methods for details). As untagged p53 was still expressed from the ADH1 promoter, these cells were expected to remain white unless the p53 activity was reduced, e.g., by forming a prion. A few small dark red colonies were seen whether the cells were previously grown on glucose or galactose. These colonies were all [rho−], which frequently arise in yeast cultures due to the loss of functional mitochondria. However, on the plates that were replica-plated from galactose, but not from glucose, there were also larger pink colonies. Cells from 25 such colonies were patched and replica-plated to glucose media along with non-prion control L3671 that had never been grown on galactose (Figure 1B).
To visualize aggregation of p53, the L3671 control and five of the pink derivatives were grown on galactose to allow expression of the p53-EYFP. In control cells p53-EYFP was generally diffuse in nuclei (Figure S1), with less than 1% of the cells containing cytoplasmic fluorescent dots. In contrast, fewer of the pink cells had p53 in nuclei and many had cytoplasmic fluorescent dots. Nuclear fluorescent foci were seen in some preparations but were hard to reliably distinguish from control nuclei. To determine if the elevated presence of cytoplasmic dots was stable, we sequentially subcloned and repeatedly patched one of these isolates (#7, named L3672 prion subclone) on plasmid-selective glucose media for over 100 generations and they remained pink, indicative of prion retention (Figure 1C). Also, even in the presence of adenine, the prion subclones grew slower than the non-prion control (Figure 1C), an effect frequently seen for other prions and prion-like aggregates (Armakola et al., 2011; Liebman and Chernoff, 2012; Park et al., 2017; Pezza et al., 2014).
We then grew the white L3671 (non-prion control) and pink L3672 (prion subclones) on galactose medium to induce p53-EYFP and found that <1% of the non-prion control cells had cytoplasmic dots and >99% had nuclear, generally diffuse, p53-EYFP, excluding dead cells, cells that had no fluorescence, and cells that only had fluorescence in the vacuole. In contrast, about 30%–50% of cells in the prion subclones had cytoplasmic dots and only about 70%–50% had nuclear p53-EYFP (Figure 1E). Finally, many of the cells in the prion cultures had fluorescence only visible in the vacuole.
Yeast prion aggregates, e.g., [PSI+] and [PIN+], are stained by ThT, which causes them to fluoresce with a CFP filter (Arslan et al., 2015; Summers and Cyr, 2011). Likewise, ThT stains aggregates of the amyotrophic lateral sclerosis-associated human proteins FUS and CREST, but not TDP-43, when expressed in yeast (Park et al., 2019). Figure 1F shows that the p53-EYFP cytoplasmic foci present in L3672 are stained by ThT. This is not caused by bleed through from the p53-EYFP, because in the absence of ThT no such CFP fluorescence is visible. This staining suggests that the cytoplasmic p53-EYFP aggregates are amyloid-like, a characteristic of many prions.
Furthermore, when the prion subclones, which showed p53-EYFP fluorescent aggregates on galactose, were transferred to glucose where only the untagged p53 is expressed, ThT staining still revealed the presence of amyloid-like cytoplasmic foci (Figure 1F). This suggests that the p53-EYFP aggregates were prions that seeded the untagged p53 to also form amyloid-like prion aggregates. Also as predicted, when control cells that showed mostly nuclear diffuse p53-EYFP on galactose, were transferred to glucose, ThT staining did not reveal cytoplasmic foci. Nuclei, even if shown to contain p53-EYFP, never stained with ThT in either control or prion-containing cells. This suggests that unlike the cytoplasmic p53 aggregates, the p53 in nuclei is not amyloid-like.
One of the hallmarks of prion aggregation in yeast is the conversion of a soluble protein to one that is found in the pellet when lysates are centrifuged (Liebman and Chernoff, 2012). Likewise, we found that p53 is soluble when expressed in yeast in the non-prion form but is largely found in the pellet in cells with the p53 prion (Figure 1D). Another hallmark of prions in yeast is dependence on chaperones. Most but not all yeast prions are dependent on the presence of the HSP104 chaperone that is not found in mammals (Liebman and Chernoff, 2012). Other yeast prions have been shown to be dependent on HSP90 (Chakrabortee et al., 2016). We found that neither 5 mM guanidine hydrochloride, which inactivates the HSP104 chaperone, nor deletion of HSP104 cured cells of the p53 prion (Figures S2A and S2B). Likewise addition of radicicol, which inactivates HSP90 (Sharma et al., 1998), did not cause loss of the p53 prion (Figure S2C).
Wild-type p53 cytoplasmic aggregates can be transferred by cytoduction to both wild-type and R175H mutant p53
Cytoduction was used to transfer cytoplasm from the MATα [RHO+] donor control (L3671) and isogenic p53 prion subclones (L3672) to MATa kar1 [rho−] recipient cells constitutively expressing p53-EYFP or p53-R175H-EYFP, which contains a cancer hotspot structural mutation. Before cytoduction, fluorescence in these recipient cells was largely diffuse in nuclei, but occasional cytoplasmic dots were more frequently seen in p53-R175H-EYFP versus p53-EYFP cells. The kar1 mutation prevents nuclear fusion so buds from the zygote with the recipient nucleus and mixed cytoplasm “cytoductants” can be selected (Conde and Fink, 1976) (Figure 2A). Cytoductants were identified because they were [RHO+], and therefore have donor cytoplasm, but have the unique auxotrophic markers and mating type diagnostic of the recipient. We then examined EYFP fluorescence in cytoductants (Figure 2B). In 38 of 44 p53 wild-type cytoductants obtained with the prion donor, ∼30% of the cells had cytoplasmic foci. In 23 of 28 p53 analogous mutant cytoductants cytoplasmic foci were observed in ∼40% cells. For the wild-type p53 prion cytoductants there were frequently more than two cytoplasmic dots per cell and some were large and irregular in shape. For the mutant p53 prion cytoductants most cells with foci contained only one or two dots and these were small, round, and of high intensity. Furthermore, the frequency and appearance of foci in the wild-type or mutant cytoductants were unchanged after >100 generations. Cytoductants obtained with control non-prion donor to either wild-type or mutant p53 recipients contained only about 5% cells with fluorescent cytoplasmic dots. The ability to cytoduce traits indicates that they are not dependent upon nuclear genes and is therefore diagnostic for cytoplasmic prions (Liebman and Chernoff, 2012). Thus, the finding of successful cytoduction of stable p53-EYFP aggregates strongly supports the idea that they are prions. Furthermore, the data show that wild-type p53 prions can induce mutant p53-R175H proteins to form a prion as efficiently as it induces wild-type p53 to form a prion.
Figure 2.

p53 prion-like aggregates can be transferred by cytoduction, from prion donor cells, to non-prion recipient cells expressing either wild-type p53 or mutant p53-R175H
(A) Cartoon illustrating cytoduction. Donor with green nucleus and pink dotted cytoplasm is mated with recipient with black nucleus and blue diagonals cytoplasm. Upon cell fusion the cytoplasms mix, but often nuclei fail to fuse because of the presence of the kar1 mutation that inhibits karyogamy. Selection is for cytoductants that bud off of the fused heterokaryon, which contain just the recipient's nucleus but a fusion of the donor and recipient cytoplasms.
(B) Data showing transmission of p53 prion via cytoduction. Subclones of L3672 containing p53 prions as well as isogenic control L3671 lacking prions were used as cytoplasmic donors by crossing them to a recipient non-prion kar1 strain, L3663, transformed with pGPD-p53-EYFP or pGPD-p53-R175H-EYFP. Cytoductants, with mixed cytoplasm but recipient nuclei, were assayed for cytoplasmic fluorescent foci. All those obtained from the non-prion donor showed >95% cells with only nuclear fluorescence, whereas 38/44 and 23/28 of the cytoductants obtained from the prion donor, respectively, into wild-type or R175H p53 recipients showed ~30%–40% cells with cytoplasmic fluorescent foci. The dramatic difference in the fraction of cells with foci (~30%–40% versus approximately <5%) seen in the cytoductants made it easy to score them as containing or lacking the prion, as there were no cytoductants with an intermediate level of foci. Scale bar, 5 μM.
See also Tables S1 and S2 for a description of strains and plasmids.
We next asked if all cells in the cytoductant cultures had the prion, even though foci were only detected in ∼30% or ∼40% of the prion-containing cells. To do this we obtained 20 subclones from both a p53 wild-type and an R175H mutant cytoductant with aggregates. We found that 19 of 20 wild-type and all 20 mutant subclones contained the prion. Subclones scored as containing a prion showed the same ∼30% or ∼40% of the progeny with foci seen in the original cytoductants. The presence of nuclear fluorescence and absence of cytoplasmic aggregates in 95% of progeny from one of the 20 wild-type subclones indicates that the prion is occasionally lost, as has been seen for other prions in yeast (Liebman and Chernoff, 2012). In contrast, in control cytoductants without foci all the 20 wild-type and 18 of 20 mutant subclones had 95% of progeny with nuclear p53 fluorescence and no cytoplasmic foci. The remaining two mutant subclones contained only about 10% cells with foci, in agreement with our general observations that non-prion cells with the p53-R175H mutant contain more cytoplasmic foci than cells with wild-type p53. Taken together, the results show that most cells in the wild-type or mutant cytoductant cultures with foci contain the prion, even though we only see aggregates, respectively, in ∼30% and ∼40% of the wild-type and mutant cells.
Non-aggregated p53-EYFP in recipient cells becomes aggregated when transfected with lysates of strains with heritable p53 aggregates or with p53 amyloid-like protein peptide
Lysates of the isogenic strains with (L3672) and without (L3671) the p53 prion were co-transfected, along with plasmid pGPD-p53-EYFP, into recipients (L3628 or L3719) that that lacked p53 (Figure 3). Of 50 pGPD-p53-EYFP L3628 transformants with the control lysate, none contained more than 1% cells with cytoplasmic aggregates (Figure 3B). In contrast, 97 of 248 transfectants with prion lysate had more than 10% cells with cytoplasmic foci and without nuclear p53 fluorescence (Figures 3A and 3B).
Figure 3.

p53 prion-like aggregates can be transferred by transfection
(A) Cell lysate from p53 prion yeast, or in vitro-made P8 polymer, promote the appearance of p53 cytoplasmic foci in transfectants. Cleared cell lysates of p53 prion (L3672) or control non-prion cells (L3671) or P8 polymer of the p53 peptide or empty control were co-transformed with pGPD-p53-EYFP into ADE2 p53 reporter strains L3628 or L3719 lacking p53. Successful transfection was scored by the appearance of cytoplasmic fluorescent foci. Retention of foci was scored after >100 generations. Scale bar, 5 μM.
(B) The frequency of prion transfection is ~ 25%–50%. Shown are data for plasmid transformants into strain L3628, which were considered to be transfectants of prion lysates or P8 that seeded p53 aggregation if they had greater than or approximately 10% cells with cytoplasmic p53-EYFP foci. Plasmid transformants scored as not being transfected looked like controls with minimal foci.
(C) Frequency of cells with foci in transfectants and transformation controls. Each bar represents counts of 150–300 cells for each of the three L3719 transfectants or controls. Standard error is shown. Blue bars are data from initial transfectants or controls. Orange bars are data from their subclones after >100 generations. ∗p=<0.05.
Also, when pGPD-p53-EYFP and in vitro-made amyloid fibers of the p53 fragment P8 (aa 250–257) were co-transfected into the L3628 recipient, fluorescent aggregates were frequently present in the transformants: 24 of 50 transformants showed about >10% cells with p53-EYFP cytoplasmic foci (Figure 3B). Controls transformed with pGPD-p53-EYFP without P8 showed essentially only nuclear fluorescence without cytoplasmic foci. Thus, amyloid seed made of a p53 peptide is sufficient to induce in vivo p53-EYFP aggregation. Induction of heritable p53-EYFP aggregation by transformation with a p53 peptide aggregate is strong evidence for protein-based prion inheritance.
To ask if the transfectants could maintain the p53 prion we propagated three transfectants of L3719 obtained from prion or non-prion lysates, and three obtained from P8 polymer or plasmid only, each for >100 generations. Both before and after the 100-generation propagation there were statistically significant increases in the percent of cells with foci in the transformants relative to control cultures p= <0.05 (Figure 3C). There was no statistical difference (p=>0.05) between the frequency of cells with foci in transformant cultures obtained from prion lysates whether they were examined soon after transformation or following 100 generations, but there was a reduction (p=<0.05) in the frequency of cells with foci in P8 transfectants after 100 generations. This is not surprising because unlike transfection with the lysates containing aggregates of a single heritable prion variant, transfection with the P8 polymer would be inducing the de novo formation of different types (variants) of prion-like aggregates, and these are often unstable (Liebman and Chernoff, 2012).
Transient interruption of p53 expression causes loss of the p53 prion
Transmission of a prion requires the continued presence of the prion seed. Thus, one test for a prion is to show that inhibiting expression of the prion protein, which causes loss of all prion seeds, prevents re-seeding of the prion protein when it is re-expressed (Wickner, 1994). To perform this test for the p53 prion we cultured the prion containing isolate L3672 and its isogenic non-prion control L3671, both carrying pADH-p53 (LEU2) and pGAL-p53-EYFP (TRP1), on non-plasmid-selective glucose medium for many generations. We then plated for individual cells and obtained five colonies from L3672 that lost pADH-p53 (were Leu−) but retained pGAL-p53-EYFP (were Trp+). These cells did not express p53 when grown on glucose. When returned to galactose, they all failed to form fluorescent foci, indicating p53-EYFP was not seeded to form a prion (Figure 4A). Furthermore, as seen in Figure 4B, their (rows 7 and 8) growth on galactose looked identical to isolates from the non-prion control strain that lost pADH-p53 (rows 3 and 4) and better than subclones that retain the prion (rows 5 and 6). Thus, as expected for a prion, transient interruption of p53 expression by losing the pADH-p53 plasmid in a prion strain and growing it on glucose where pGAL-p53-EYFP could not express p53-YFP, caused permanent loss of the prion when the cells were returned to galactose where p53-EYFP was expressed.
Figure 4.

Transient loss of p53 cures cells of p53 prion
(A) Cartoon showing how transient loss of p53 cures cells of the prion. A prion containing cell, grown on galactose, is shown on the left. After loss of the ADH1-p53 plasmid this strain is grown on glucose, where no p53 is expressed (middle). Then upon return to galactose (right) there is no remaining prion seed, so the newly made tagged p53 remains diffuse in the nucleus and is able to turn on expression of the ADE2 reporter.
(B) Data showing that transient absence of p53 causes permanent loss of the p53 prion. Prion strain L3672, and its isogenic non-prion control L3671, both carrying pADH-p53 (LEU2) and pGAL-p53-EYFP (TRP1), were grown on non-plasmid-selective glucose medium for many generations to allow for loss of the pADH-p53 plasmid. Cells that were Leu− indicating loss of pADH-p53 (-pADH-p53) but Trp+ indicating retention of pGAL-p53-EYFP were grown and spotted along with independent controls that retained pADH-p53 (pADH-p53) from cells with and without the prion (upper). Only pADH-p53, but not -pADH-p53, cells express any p53 when grown on glucose. When returned to galactose the -pADH-p53 isolates from the prion culture looked identical to non-prion control -pADH-p53, growing better on galactose medium with limiting adenine than prion pADH-53 controls that retained the prion. Two representative isolates of each type are shown. Furthermore, all -pADH-p53 isolates from the prion cells failed to form cytoplasmic fluorescent foci when grown on galactose, indicating that no p53 prion was present (bottom). Scale bar, 5 μM.
See also Tables S1 and S2 for a description of strains and plasmids.
P53 forms reversible cytoplasmic foci that do not stain with ThT in response to various stresses
When yIG397 transformed with pGPD-p53-EYFP was grown on plasmid-selective glucose media, cells showed largely nuclear diffuse fluorescence (Figures 5A left and S1). However, 30 min after transfer of the cells to medium with 2% ethanol instead of glucose, cytoplasmic fluorescent foci appeared. Nuclear foci were also sometimes seen following stress (Figure 5A) but were hard to score and distinguish from unstressed nuclei. We counted foci in the cytoplasm of about 25% of cells (Figure 5A middle), and the formation of these foci was independent of the HSP104 chaperone (Figure S2D). These foci remained as long as the cells were retained on ethanol medium. However, after 30 min of return to glucose medium both the nuclear and cytoplasmic foci disappeared (Figure 5A right). Similar results were obtained when cells were stressed with heat or glucose starvation (Figure 5A). These stress-induced p53 foci are reminiscent of unstable liquid-like droplets formed by liquid-liquid phase separation of a variety of proteins with intrinsically disordered regions often in response to stress.
Figure 5.

p53 can also form unstable foci
(A) Ethanol, glucose starvation, and heat stress lead to the formation of p53 foci that disappear when the stress is removed. Shown (three upper rows) is yIG397 transformed with pGPD-p53-EYFP grown on plasmid-selective glucose media (left), following stress: 30 min in medium with 2% ethanol, 30 min of glucose starvation or 10 min at 46°C (middle), and then after removal of stress (right). Graph shows the mean of counts from 3 independent transformants ± SEM. Three lower rows show yIG397 transformed with pGPD-p53-175H-EYFP under the same stresses. Arrowhead shows nucleus with foci on top of diffuse fluorescence. Scale bar, 5 μM.
(B) Foci caused by stress do not stain with Thioflavin T. Shown are cells with stress-induced p53-EYFP foci (obtained as in A above) or cells with prion p53-EYFP foci (obtained as in Figure 1E). Cells were fixed, stained with ThT, and examined for EYFP (YFP) fluorescence or ThT staining (CFP).
(C) Hexanediol causes the disappearance of stress-induced, but not prion foci. Shown are cells with stress-induced p53-EYFP foci (obtained as in A above) or cells with prion p53-EYFP foci (obtained as in Figure 1E) before (-Hex) and after (+Hex) 5-min treatments with 10% 1,6-hexanediol. Graph shows counts from three independent transformants ± SEM.
(D) Circularity of stress versus prion foci. Circularity of cytoplasmic aggregates in cells with stress or prion foci obtained as described in (C) (except 15 min of heat was given) was measured. ImageJ and the formula (4 ∗π ∗ area)/perimeterˆ2 was used (n = 40 for each). A value of 1.0 indicates a perfect circular shape, and the lower the value, the less circular the aggregate. Shown are mean ± SEM. ∗∗p calculated with one-tailed t test as <0.005 is shown.
See also Figure S2 and Tables S1 and S2 for a description of strains and plasmids.
As short exposure to the aliphatic alcohol 1,6-hexanediol has been shown to dissolve liquid-like droplets but not solid protein assemblies (Kroschwald et al., 2017) we examined the effect of 1,6-hexanediol on the cytoplasmic stress-induced p53-EYFP foci when compared with the stable prion p53-EYFP foci. A 5-min treatment with 1,6-hexanediol caused the disappearance of >90% of the stress-induced foci but had no effect on prion foci (Figure 5C). A short exposure was required because, similar to a previous report (Kroschwald et al., 2017), longer treatment with 1,6-hexanediol actually caused the appearance of cytoplasmic fluorescent foci in all cells with wild-type p53-EYFP (Figure S3). Another difference we found between the prion foci and the unstable stress-induced foci is that only the former stain with ThT (Figure 5B). This suggests that the prion foci, but not the stress foci, are amyloid-like. Also, we found that the stress foci had the high circularity level expected of a liquid droplet (Kroschwald et al., 2015) unlike the prion foci that were less circular (Figure 5D). Curiously, we found that the cancer hotspot structural mutation p53-R175H prevents formation of the stress foci (Figure 5A).
P53 stress-induced cytoplasmic foci co-localize with P-bodies
Strain yIG397 was co-transformed with pGPD-p53-EYFP to allow for detection of p53-EYFP stress foci and either pEdc3-mCH or pDcp2-RFP to allow for detection of P-bodies. The cells were subjected to ethanol, heat, or glucose starvation to induce the appearance of both EYFP-marked p53 foci and mCh- or RFP-marked P-bodies. These two types of foci were seen to partially co-localize (Figure 6).
Figure 6.

p53 stress foci co-localize with cellular P-bodies
Strain yIG397 was co-transformed with pGPD-p53-EYFP to allow for detection of p53-EYFP stress foci and either pEdc3-mCH or pDcp2-RFP to allow for detection of P-bodies. The cells were subjected to ethanol, heat, or glucose starvation to induce the appearance of both EYFP-marked p53 foci and mCh- or RFP-marked P-bodies and were photographed, respectively, with YFP and mCh filters. Scale bar, 5 μM.
See also Tables S1 and S2 for a description of strains and plasmids.
Discussion
In this work we show that wild-type p53 expressed in living yeast not only has the ability to propagate as a true prion accompanied by the appearance of stable cytoplasmic foci and reduction in p53 activity but also can also form unstable foci in response to stress. Inactivation of p53 via prion formation has a direct implication for the onset of cancer. Whether the unstable stress-induced foci affect the appearance of p53 prions remains to be determined.
Previous evidence suggested that p53 aggregates in some cancer cells (Ano Bom et al., 2012; De Smet et al., 2017; Gitler and Lehmann, 2012; Levy et al., 2011; Xu et al., 2011). Often p53 amyloid is found in nuclei of tumor cells as oligomers, leading to the suggestion that amyloid p53 oligomers in the nucleus could result in an oncogenic gain of function (Pedrote et al., 2020). Possibly fibrillar p53 would be present in the cytoplasm. One clear example of cytoplasmic p53 is the accumulation of amyloid Δ40p53 in endometrial carcinoma cells (Melo Dos Santos et al., 2019). Our data show that in yeast p53 can form stable, ThT stainable, cytoplasmic aggregates. Although foci are sometimes seen in nuclei, we have never observed nuclear staining with ThT. Why we observe largely cytoplasmic amyloid p53 in yeast rather than the nuclear p53 amyloid seen in tumor cells is unknown. As RNA is known to inhibit p53 foci formation (Kovachev et al., 2017) possibly the excess RNA in yeast nuclei inhibits p53 from forming amyloid.
The p53 cytoplasmic aggregates that we observe in yeast have the characteristics of prions: they not only self-seed but also continually replicate the seeds, causing the prion to be stable for over 100 generations. Also, we show that the prion causes loss of p53 function. Our finding that overexpression of p53 triggers it to form a heritable prion in yeast with reduced transcriptional activity suggests that increased levels of p53 may likewise trigger prion formation and concomitant p53 inactivation in animal cells, leading to cancer. Indeed high levels of p53 are often found in tumor cells (Houben et al., 2011), and stresses such as DNA damage that induce cancer often lead to an increase in the cellular level of p53 (Lavin and Gueven, 2006).
In addition, the ability of p53 to form a prion upends the classic idea that overexpression of wild-type p53 via gene therapy could cure cancers caused by p53-inactivating mutations. Such a treatment would not be expected to help if the inactivating mutations result in prion seeds that attract and therefore inactivate wild-type p53. Also, overexpression of wild-type p53 could generate prion seeds de novo that, instead of providing p53 activity, cause aggregation of the p53 mutant protein causing increased loss of activity. p53 prion protein could also trap and inactivate its related transcription factors, p63 and p73 (Billant et al., 2017), which could increase cancer pathology. In contrast, prion-like aggregates that do not multiply would have a more limited capacity to inactivate WT p53, p63, or p73.
Overexpression of p53 allowed us to isolate a strain of yeast that had cytoplasmic p53 aggregates and reduced p53 activity. These aggregates appear to be prions because they stained with ThT, suggestive of amyloid, and were stable for over 100 generations. Following centrifugation of cell lysates, the p53 aggregates were found in pellet fractions as seen for other prions. Also, like other prions, the cytoplasm of the p53 prion strain infected recipient cells with the prion via cytoduction. Also, the p53 prion was transferred to recipients by transfecting them with lysates from prion cells or with amyloid fibers of a p53-derived peptide (P8) made in vitro.
The question of how p53, when aggregated or non-aggregated, affects yeast growth is complicated and remains unresolved. As reported by others (Bischoff et al., 1992; Inga and Resnick, 2001; Kumar et al., 2017; Nigro et al., 1992), we have seen growth inhibition associated with overexpression of wild-type non-aggregated but not mutant p53 in some strain backgrounds. Assuming the inhibition is caused by excess p53 transcription factor activity, one would expect that the reduced activity associated with prion formation would rescue cells from the growth inhibition. Indeed, this hypothesis is supported by our finding that following transient overexpression of p53, pink colonies with inactive p53 and p53-EYFP aggregates pop out on a background of cells with active p53 following weeks of room-temperature incubation. However, even though there is no increase in dead cells among prion subclones (Figure 1E), in spot tests the prion causes a reduced growth rate even in the presence of adenine (Figure 1C). It is likely that p53 is largely non-functional and lacks toxic p53 amyloid in the subset of cells in the prion culture that have essentially only vacuolar fluorescence. These cells may grow better than cells in the prion culture with cytoplasmic dots because they lack significant toxic amyloid aggregates. They also may grow better than prion or non-prion cells with nuclear fluorescence because of their reduced level of nuclear p53 and therefore reduced toxicity associated with excess nuclear p53 (Bischoff et al., 1992; Inga and Resnick, 2001; Kumar et al., 2017; Nigro et al., 1992). These cells could arise if p53 prion intermediate structures were directed to the vacuole for degradation before a stable prion was formed.
Whether wild-type p53 aggregates seed aggregation of heterologous proteins can now be determined. We show that one cancer-causing p53 mutant, p53-R175H, is seeded to form a prion by the wild-type p53 prion. Whether other p53 mutants are able to form prions can now be established. It should also be possible to use yeast to select for p53 mutants that cannot join prion aggregates but retain transcription factor activity. If such mutants were used in gene therapy, they would avoid the prion associated problems described above and could provide a useful therapy for cancer. Also, our results have the potential to be used for high-throughput screening of compounds with potential antitumoral activity for drugs that target aggregation of p53. This would complement recent targeted approaches for such drugs (Rangel et al., 2019; Silva et al., 2018; Soragni et al., 2016).
Interestingly, non-membrane-bound foci of a variety of proteins, including P-bodies, have been described that appear to arise from LLPS. p53 has also been shown to be present in such non membrane-bound liquid-like nuclear droplets, including Cajal and promyelocytic leukemia protein bodies (Banani et al., 2017; Cioce and Lamond, 2005; Fogal et al., 2000; Guo et al., 2000). p53 can also form liquid-like droplets in vitro (Kamagata et al., 2020). Now we show that p53 can form unstable foci in live yeast in response to ethanol, heat, and glucose starvation. These stresses also induce the appearance of P-bodies previously shown to be liquid droplets in yeast (Kroschwald et al., 2015), and we show that some p53 stress foci co-localize with the P-bodies. Like P-bodies, the p53 stress foci are distinct from amyloid-like stable foci because they disappear rapidly when the stresses are removed, do not stain with ThT, and are dissolved by 1,6-hexanediol. The foci located in the cytoplasm are unlikely to promote transcription, but the effect of the nuclear stress foci on transcription is unknown.
Increasingly LLPS, or related structures (McSwiggen et al., 2019), have been found to affect transcription. Enhanced or reduced activity of transcription factors has been seen when they join LLPS foci (Cho et al., 2018; Chong et al., 2018; Sabari et al., 2018; Tarczewska and Greb-Markiewicz, 2019). Our demonstration that the transcription factor p53 forms unstable foci in yeast in response to stress opens the possibility that p53 activity is likewise regulated by phase transition. Possibly stresses that promote the formation of the foci enhance or reduce the activity of p53, leading to important cellular ramifications. Indeed, we have shown that mutations in p53 can prevent it from forming stress foci: the p53-175H mutation associated with cancer that inactivates p53 fails to form stress foci.
Limitations of the study
This work used an yeast model to show that p53 can form a stable prion as well as liquid-like droplets. Although yeast is a eukaryote and has proved to be a useful model for mammalian diseases, there are many differences between yeast and mammalian cells, differences in chaperones.
Resource availability
Lead contact
Susan W. Liebman sliebman@unr.edu can be contacted for all strains and plasmids used.
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
This study did not generate the datasets.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Joshua Killinger and Kaitlin Robertson for help with these experiments and Irina Derkatch for helpful comments on the manuscript. We also thank Alberto Inga, University of Trento, Italy; Richard Iggo, Bergonie Cancer Institute, France; Samir K Maji, Indian Institute of Technology, Bombay, India; Reed Wickner, National Institutes of Health; and Ross Buchan, University of Arizona, for sharing strains and plasmids and Michael Resnick, NIEHS, for stimulating conversations. This work was supported by a grant from the US Army, W911NF-18-1-0151.
Author contributions
S.-K.P., S.P., and C.P. conducted experiments. S.-K.P. and S.W.L. designed experiments. S.-K.P., S.P., and S.W.L. prepared the figures and edited the paper. S.W.L. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.102000.
Supplemental information
References
- Alberti S., Halfmann R., King O., Kapila A., Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano Bom A.P., Rangel L.P., Costa D.C., de Oliveira G.A., Sanches D., Braga C.A., Gava L.M., Ramos C.H., Cepeda A.O., Stumbo A.C. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J. Biol. Chem. 2012;287:28152–28162. doi: 10.1074/jbc.M112.340638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armakola M., Hart M.P., Gitler A.D. TDP-43 toxicity in yeast. Methods. 2011;53:238–245. doi: 10.1016/j.ymeth.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arslan F., Hong J.Y., Kanneganti V., Park S.K., Liebman S.W. Heterologous aggregates promote de novo prion appearance via more than one mechanism. PLoS Genet. 2015;11:e1004814. doi: 10.1371/journal.pgen.1004814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev S., Liebman S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006;4:32. doi: 10.1186/1741-7007-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billant O., Blondel M., Voisset C. p53, p63 and p73 in the wonderland of S. cerevisiae. Oncotarget. 2017;8:57855–57869. doi: 10.18632/oncotarget.18506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff J.R., Casso D., Beach D. Human p53 inhibits growth in Schizosaccharomyces pombe. Mol. Cell. Biol. 1992;12:1405–1411. doi: 10.1128/mcb.12.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncella A.E., Shattuck J.E., Cascarina S.M., Paul K.R., Baer M.H., Fomicheva A., Lamb A.K., Ross E.D. Composition-based prediction and rational manipulation of prion-like domain recruitment to stress granules. Proc. Natl. Acad. Sci. U S A. 2020;117:5826–5835. doi: 10.1073/pnas.1912723117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini N.M., Gitler A.D. Model organisms reveal insight into human neurodegenerative disease: ataxin-2 intermediate-length polyglutamine expansions are a risk factor for ALS. J. Mol. Neurosci. 2011;45:676–683. doi: 10.1007/s12031-011-9548-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan J.R., Yoon J.H., Parker R. Stress-specific composition, assembly and kinetics of stress granules in Saccharomyces cerevisiae. J. Cell Sci. 2011;124:228–239. doi: 10.1242/jcs.078444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers J.S., Jarosz D.F. High-throughput screening for protein-based inheritance in S. cerevisiae. J. Vis. Exp. 2017;8:56069. doi: 10.3791/56069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudron F., Barral Y. A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell. 2013;155:1244–1257. doi: 10.1016/j.cell.2013.10.046. [DOI] [PubMed] [Google Scholar]
- Chakrabortee S., Byers J.S., Jones S., Garcia D.M., Bhullar B., Chang A., She R., Lee L., Fremin B., Lindquist S. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell. 2016;167:369–381 e312. doi: 10.1016/j.cell.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty A.K., Jarosz D.F. More than just a phase: prions at the crossroads of epigenetic inheritance and evolutionary change. J. Mol. Biol. 2018;430:4607–4618. doi: 10.1016/j.jmb.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty A.K., Smejkal T., Itakura A.K., Garcia D.M., Jarosz D.F. A non-amyloid prion particle that activates a heritable gene expression program. Mol. Cell. 2020;77:251–265 e259. doi: 10.1016/j.molcel.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff Y.O., Lindquist S.L., Ono B., Inge-Vechtomov S.G., Liebman S.W. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Cho W.K., Spille J.H., Hecht M., Lee C., Li C., Grube V., Cisse Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361:412–415. doi: 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S., Dugast-Darzacq C., Liu Z., Dong P., Dailey G.M., Cattoglio C., Heckert A., Banala S., Lavis L., Darzacq X. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361:eaar2555. doi: 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioce M., Lamond A.I. Cajal bodies: a long history of discovery. Annu. Rev. Cell Dev. Biol. 2005;21:105–131. doi: 10.1146/annurev.cellbio.20.010403.103738. [DOI] [PubMed] [Google Scholar]
- Conde J., Fink G.R. A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proc. Natl. Acad. Sci. U S A. 1976;73:3651–3655. doi: 10.1073/pnas.73.10.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira G.A.P., Petronilho E.C., Pedrote M.M., Marques M.A., Vieira T., Cino E.A., Silva J.L. The status of p53 oligomeric and aggregation states in cancer. Biomolecules. 2020;10:548. doi: 10.3390/biom10040548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet F., Saiz Rubio M., Hompes D., Naus E., De Baets G., Langenberg T., Hipp M.S., Houben B., Claes F., Charbonneau S. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: a hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2017;242:24–38. doi: 10.1002/path.4872. [DOI] [PubMed] [Google Scholar]
- Derkatch I.L., Chernoff Y.O., Kushnirov V.V., Inge-Vechtomov S.G., Liebman S.W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z., Park K.W., Yu H., Fan Q., Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008;40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge R.M., Clark G.M., Fuqua S.A., Yu Y.Y., Allred D.C. p53 protein accumulation detected by five different antibodies: relationship to prognosis and heat shock protein 70 in breast cancer. Cancer Res. 1994;54:3752–3757. [PubMed] [Google Scholar]
- Fogal V., Gostissa M., Sandy P., Zacchi P., Sternsdorf T., Jensen K., Pandolfi P.P., Will H., Schneider C., Del Sal G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fronza G., Inga A., Monti P., Scott G., Campomenosi P., Menichini P., Ottaggio L., Viaggi S., Burns P.A., Gold B. The yeast p53 functional assay: a new tool for molecular epidemiology. Hopes and facts. Mutat. Res. 2000;462:293–301. doi: 10.1016/s1383-5742(00)00011-9. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Ghosh D., Ranganathan S., Anoop A., P S.K., Jha N.N., Padinhateeri R., Maji S.K. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry. 2014;53:5995–6010. doi: 10.1021/bi500825d. [DOI] [PubMed] [Google Scholar]
- Ghosh S., Salot S., Sengupta S., Navalkar A., Ghosh D., Jacob R., Das S., Kumar R., Jha N.N., Sahay S. p53 amyloid formation leading to its loss of function: implications in cancer pathogenesis. Cell Death Differ. 2017;24:1784–1798. doi: 10.1038/cdd.2017.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler A.D., Lehmann R. Modeling human disease. Science. 2012;337:269. doi: 10.1126/science.1227179. [DOI] [PubMed] [Google Scholar]
- Grousl T., Ivanov P., Malcova I., Pompach P., Frydlova I., Slaba R., Senohrabkova L., Novakova L., Hasek J. Heat shock-induced accumulation of translation elongation and termination factors precedes assembly of stress granules in S. cerevisiae. PloS one. 2013;8:e57083. doi: 10.1371/journal.pone.0057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A., Salomoni P., Luo J., Shih A., Zhong S., Gu W., Pandolfi P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- Harvey Z.H., Chakravarty A.K., Futia R.A., Jarosz D.F. A prion epigenetic switch establishes an active chromatin state. Cell. 2020;180:928–940 e914. doi: 10.1016/j.cell.2020.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houben R., Hesbacher S., Schmid C.P., Kauczok C.S., Flohr U., Haferkamp S., Muller C.S., Schrama D., Wischhusen J., Becker J.C. High-level expression of wild-type p53 in melanoma cells is frequently associated with inactivity in p53 reporter gene assays. PloS one. 2011;6:e22096. doi: 10.1371/journal.pone.0022096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inga A., Cresta S., Monti P., Aprile A., Scott G., Abbondandolo A., Iggo R., Fronza G. Simple identification of dominant p53 mutants by a yeast functional assay. Carcinogenesis. 1997;18:2019–2021. doi: 10.1093/carcin/18.10.2019. [DOI] [PubMed] [Google Scholar]
- Inga A., Resnick M.A. Novel human p53 mutations that are toxic to yeast can enhance transactivation of specific promoters and reactivate tumor p53 mutants. Oncogene. 2001;20:3409–3419. doi: 10.1038/sj.onc.1204457. [DOI] [PubMed] [Google Scholar]
- Itakura A.K., Chakravarty A.K., Jakobson C.M., Jarosz D.F. Widespread prion-based control of growth and differentiation strategies in Saccharomyces cerevisiae. Mol. Cell. 2020;77:266–278 e266. doi: 10.1016/j.molcel.2019.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura A.K., Futia R.A., Jarosz D.F. It pays to Be in phase. Biochemistry. 2018;57:2520–2529. doi: 10.1021/acs.biochem.8b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakuma T., Lozano G., Flores E.R. Li-Fraumeni syndrome: a p53 family affair. Cell Cycle. 2005;4:865–867. doi: 10.4161/cc.4.7.1800. [DOI] [PubMed] [Google Scholar]
- Jin M., Fuller G.G., Han T., Yao Y., Alessi A.F., Freeberg M.A., Roach N.P., Moresco J.J., Karnovsky A., Baba M. Glycolytic enzymes coalesce in G bodies under hypoxic stress. Cell Rep. 2017;20:895–908. doi: 10.1016/j.celrep.2017.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamagata K., Kanbayashi S., Honda M., Itoh Y., Takahashi H., Kameda T., Nagatsugi F., Takahashi S. Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains. Sci. Rep. 2020;10:580. doi: 10.1038/s41598-020-57521-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C.Y., Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319–323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- Kovachev P.S., Banerjee D., Rangel L.P., Eriksson J., Pedrote M.M., Martins-Dinis M., Edwards K., Cordeiro Y., Silva J.L., Sanyal S. Distinct modulatory role of RNA in the aggregation of the tumor suppressor protein p53 core domain. J. Biol. Chem. 2017;292:9345–9357. doi: 10.1074/jbc.M116.762096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S., Maharana S., Mateju D., Malinovska L., Nuske E., Poser I., Richter D., Alberti S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife. 2015;4:e06807. doi: 10.7554/eLife.06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S., Shovamayee M., Alberti S. Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. 2017;3 [Google Scholar]
- Kumar A., Dandekar J.U., Bhat P.J. Fermentative metabolism impedes p53-dependent apoptosis in a Crabtree-positive but not in Crabtree-negative yeast. J. Biosci. 2017;42:585–601. doi: 10.1007/s12038-017-9717-2. [DOI] [PubMed] [Google Scholar]
- Lane D.P. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- Lavin M.F., Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- Lee H.Y., Chao J.C., Cheng K.Y., Leu J.Y. Misfolding-prone proteins are reversibly sequestered to an Hsp42-associated granule upon chronological aging. J. Cell Sci. 2018;131:jcs220202. doi: 10.1242/jcs.220202. [DOI] [PubMed] [Google Scholar]
- Levine A.J., Oren M. The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A.J., Tomasini R., McKeon F.D., Mak T.W., Melino G. The p53 family: guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011;12:259–265. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- Levy C.B., Stumbo A.C., Ano Bom A.P., Portari E.A., Cordeiro Y., Silva J.L., De Moura-Gallo C.V. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011;43:60–64. doi: 10.1016/j.biocel.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Liebman S.W., Chernoff Y.O. Prions in yeast. Genetics. 2012;191:1041–1072. doi: 10.1534/genetics.111.137760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March Z.M., King O.D., Shorter J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016;1647:9–18. doi: 10.1016/j.brainres.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchante R., Beal D.M., Koloteva-Levine N., Purton T.J., Tuite M.F., Xue W.F. The physical dimensions of amyloid aggregates control their infective potential as prion particles. Elife. 2017;6:e27109. doi: 10.7554/eLife.27109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen D.T., Mir M., Darzacq X., Tjian R. Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 2019;33:1619–1634. doi: 10.1101/gad.331520.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo Dos Santos N., de Oliveira G.A.P., Ramos Rocha M., Pedrote M.M., Diniz da Silva Ferretti G., Pereira Rangel L., Morgado-Diaz J.A., Silva J.L., Rodrigues Pereira Gimba E. Loss of the p53 transactivation domain results in high amyloid aggregation of the Delta40p53 isoform in endometrial carcinoma cells. J. Biol. Chem. 2019;294:9430–9439. doi: 10.1074/jbc.RA119.007566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll U.M., LaQuaglia M., Benard J., Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc. Natl. Acad. Sci. U S A. 1995;92:4407–4411. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll U.M., Valea F., Chumas J. Role of p53 alteration in primary peritoneal carcinoma. Int. J. Gynecol. Pathol. 1997;16:156–162. doi: 10.1097/00004347-199704000-00012. [DOI] [PubMed] [Google Scholar]
- Monti P., Bosco B., Gomes S., Saraiva L., Fronza G., Inga A. Yeast as a chassis for developing functional assays to study human P53. J. Vis. Exp. 2019 doi: 10.3791/59071. [DOI] [PubMed] [Google Scholar]
- Narayanaswamy R., Levy M., Tsechansky M., Stovall G.M., O'Connell J.D., Mirrielees J., Ellington A.D., Marcotte E.M. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. U S A. 2009;106:10147–10152. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navalkar A., Ghosh S., Pandey S., Paul A., Datta D., Maji S.K. Prion-like p53 amyloids in cancer. Biochemistry. 2020;59:146–155. doi: 10.1021/acs.biochem.9b00796. [DOI] [PubMed] [Google Scholar]
- Nigro J.M., Sikorski R., Reed S.I., Vogelstein B. Human p53 and CDC2Hs genes combine to inhibit the proliferation of Saccharomyces cerevisiae. Mol. Cell. Biol. 1992;12:1357–1365. doi: 10.1128/mcb.12.3.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich L.Z., Cox B.S., Tuite M.F., Weissman J.S. Dissection and design of yeast prions. PLoS Biol. 2004;2:E86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T., Nakagawara A. p53: the attractive tumor suppressor in the cancer research field. J. Biomed. Biotechnol. 2011;2011:603925. doi: 10.1155/2011/603925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Park S.K., Watanabe N., Hashimoto T., Iwatsubo T., Shelkovnikova T.A., Liebman S.W. Calcium-responsive transactivator (CREST) toxicity is rescued by loss of PBP1/ATXN2 function in a novel yeast proteinopathy model and in transgenic flies. PLoS Genet. 2019;15:e1008308. doi: 10.1371/journal.pgen.1008308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.K., Hong J.Y., Arslan F., Kanneganti V., Patel B., Tietsort A., Tank E.M.H., Li X., Barmada S.J., Liebman S.W. Overexpression of the essential Sis1 chaperone reduces TDP-43 effects on toxicity and proteolysis. PLoS Genet. 2017;13:e1006805. doi: 10.1371/journal.pgen.1006805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.K., Pegan S.D., Mesecar A.D., Jungbauer L.M., LaDu M.J., Liebman S.W. Development and validation of a yeast high-throughput screen for inhibitors of Abeta(4)(2) oligomerization. Dis. Models Mech. 2011;4:822–831. doi: 10.1242/dmm.007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel B.K., Gavin-Smyth J., Liebman S.W. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 2009;11:344–349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrote M.M., Motta M.F., Ferretti G.D.S., Norberto D.R., Spohr T., Lima F.R.S., Gratton E., Silva J.L., de Oliveira G.A.P. Oncogenic gain of function in glioblastoma is linked to mutant p53 amyloid oligomers. iScience. 2020;23:100820. doi: 10.1016/j.isci.2020.100820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezza J.A., Villali J., Sindi S.S., Serio T.R. Amyloid-associated activity contributes to the severity and toxicity of a prion phenotype. Nat. Commun. 2014;5:4384. doi: 10.1038/ncomms5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel L.P., Ferretti G.D.S., Costa C.L., Andrade S., Carvalho R.S., Costa D.C.F., Silva J.L. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019;294:3670–3682. doi: 10.1074/jbc.RA118.004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari B.R., Dall'Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.V., Agatsuma T., Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene. 1998;16:2639–2645. doi: 10.1038/sj.onc.1201790. [DOI] [PubMed] [Google Scholar]
- Silva J.L., Cino E.A., Soares I.N., Ferreira V.F., G A.P.d.O. Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc. Chem. Res. 2018;51:181–190. doi: 10.1021/acs.accounts.7b00473. [DOI] [PubMed] [Google Scholar]
- Soragni A., Janzen D.M., Johnson L.M., Lindgren A.G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A.B., Lu J., Jiang L., Faull K.F. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers D.W., Cyr D.M. Use of yeast as a system to study amyloid toxicity. Methods. 2011;53:226–231. doi: 10.1016/j.ymeth.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M., Chien P., Naber N., Cooke R., Weissman J.S. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- Tarczewska A., Greb-Markiewicz B. The significance of the intrinsically disordered regions for the functions of the bHLH transcription factors. Int. J. Mol. Sci. 2019;20:5306. doi: 10.3390/ijms20215306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J.P., Brown R.H., Jr., Cleveland D.W. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S., Hamamichi S., Goodman J.L., Matlack K.E., Chung C.Y., Baru V., Shulman J.M., Parrado A., Bevis B.J., Valastyan J.S. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner R.B. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- Wilcken R., Wang G., Boeckler F.M., Fersht A.R. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. U S A. 2012;109:13584–13589. doi: 10.1073/pnas.1211550109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Reumers J., Couceiro J.R., De Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.C. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011;7:285–295. doi: 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate the datasets.