

Abstract
To understand the molecular mechanism of nitrogen use efficiency (NUE) in rice, two nitrogen (N) use efficient genotypes and two non-efficient genotypes were characterized using transcriptome analyses. The four genotypes were evaluated for 3 years under low and recommended N field conditions for 12 traits/parameters of yield, straw, nitrogen content along with NUE indices and 2 promising donors for rice NUE were identified. Using the transcriptome data generated from GS FLX 454 Roche and Illumina HiSeq 2000 of two efficient and two non-efficient genotypes grown under field conditions of low N and recommended N and their de novo assembly, differentially expressed transcripts and pathways during the panicle development were identified. Down regulation was observed in 30% of metabolic pathways in efficient genotypes and is being proposed as an acclimation strategy to low N. Ten sub metabolic pathways significantly enriched with additional transcripts either in the direction of the common expression or contra-regulated to the common expression were found to be critical for NUE in rice. Among the up-regulated transcripts in efficient genotypes, a hypothetical protein OsI_17904 with 2 alternative forms suggested the role of alternative splicing in NUE of rice and a potassium channel SKOR transcript (LOC_Os06g14030) has shown a positive correlation (0.62) with single plant yield under low N in a set of 16 rice genotypes. From the present study, we propose that the efficient genotypes appear to down regulate several not so critical metabolic pathways and divert the thus conserved energy to produce seed/yield under long-term N starvation.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-020-02631-5.
Keywords: Nitrogen use efficiency, Landraces, Metabolic pathways, Transcriptomics, Differential gene expression, SKOR transporter
Introduction
Rice is cultivated in an area of 163 million ha with ~ 15% of global nitrogen (N) fertilizer inputs resulting in the estimated production of 759.6 million tons (FAO 2018) (www.fertilizer.org). The input use efficiency of N is reported to be low (~ 30–50%) for rice (Ladha et al. 2005) and the remaining N is lost to environment. In addition to increasing the cost of cultivation, the unutilized N contributes to greenhouse gases especially, nitrous oxide affecting climate change. Nitrous oxide is 310 times more potent greenhouse gas than CO2 and 21 times more potent than methane on a 100 year time scale, though atmospheric loading of nitrous dioxide is low (IPCC 1995). Thus, improving NUE in rice has economic and environmental benefits, while maintaining the crop productivity. Genetic variation of NUE in rice germplasm has been earlier reported (Broadbent et al. 1987; Tirol-Padre et al. 1996; Vijayalakshmi et al. 2015; Rao et al. 2018). The NUE includes processes of N uptake, translocation, assimilation, and remobilization which are inherently complex and are governed by multiple genetic and environmental factors (Xu et al. 2012). Several quantitative trait loci (QTL) and genomic regions for N metabolism and NUE have been reported in rice (Vinod and Heuer 2012). Genetic approaches to improve NUE mostly included the manipulation of genes associated with N metabolism and their regulation (Li et al. 2017; Tegeder and Masclaux-Daubresse 2018).
Genome-wide expression profiling analysis using microarrays or RNA sequencing (RNA-seq) is a promising approach to understand molecular aspects of complex traits. Transcriptome analyses using microarrays (Lian et al. 2006; Cai et al. 2012; Takehisa et al. 2013; Chandran et al. 2016; Hsieh et al. 2018) and RNA-seq have identified several differentially expressed transcripts and pathways in response to differential N conditions in rice (Yang et al. 2015a, b; Sun et al. 2017; Shin et al. 2018; Sinha et al. 2018), barley (Quan et al. 2016), sorghum (Gelli et al. 2014), durum wheat (Curci et al. 2017) and maize (Chen et al. 2015). Comparative transcriptomics studies have reported a role of up-regulated high affinity nitrate transporters resulting in improved uptake efficiency and storage capacities in N efficient genotypes as compared to non-efficient genotypes in sorghum (Gelli et al. 2014), Arabidopsis (Richard-Molard et al. 2008) and wild barley (Quan et al. 2016). So far, transcriptome studies in rice under differential N conditions were reported only in seedlings under hydroponics and artificial medium (Cai et al. 2012; Yang et al. 2015a, b; Shin et al. 2018). Studies have not been conducted yet for the response of rice grown under long-term low N field conditions at the transcriptome level during reproductive stage. Comparative analysis of more number of genotypes contrasting for NUE through transcriptome studies would strengthen confidence of the identified candidate gene transcripts. Moreover, most of the rice transcriptome studies under low N only focused on a single genotype except for only one study, where two genotypes have been analysed (Sinha et al. 2018).
Rice landraces adapted to low N were reported to be promising genotype resource for identification of transcripts associated with NUE (Rao et al. 2018). It is important to study the acclimation strategy of rice to low N, especially during the reproductive stage because of the direct association of nitrogen with yield. To identify differentially expressed transcripts and pathways critical for the acclimation of rice genotypes under low N, we deployed transcriptome analyses using the data of eight sets generated from GS FLX 454 Roche and Illumina HiSeq 2000 and their de novo assembly. Thus, the objective of the present study was to identify the differentially expressed transcripts and pathways during panicle development stage using transcriptome analyses of two efficient and two non-efficient genotypes grown under long-term low N field conditions (without external N fertilization since 2011) in comparison to recommended N field conditions. To our best knowledge, this is the first study on transcriptome response of tissue at booting stage of panicle development using four contrasting genotypes under long-term low N, especially in field conditions to identify the common and exclusive up and down-regulated transcripts and pathways representing cumulative response of N uptake, assimilation and remobilization till the panicle development stage.
Methods
Plant materials and their field evaluation under differential N
Based on the earlier evaluation of 472 genotypes during two seasons under low and recommended N field conditions, 4 contrasting rice genotypes consisting of 2 efficient—Thurur Bhog (TB) (landrace) and Basmati 370 (BM) (selection of landrace) with relative higher grain yield (> 3000 kg ha−1) and 2 non-efficient—Suraksha (SK) (improved variety) and Kola Joha (KJ) (landrace) with relative lower grain yield (< 1500 kg ha−1) were selected (Rao et al. 2018). These rice genotypes were obtained from Plant Breeding, Crop improvement section, ICAR-Indian Institute of Rice Research (IIRR), Hyderabad, India. These four genotypes were grown during wet seasons of 2012, 2013 and 2015 under low and recommended N conditions for agro morphological observations. A set of 18 rice genotypes with differential yield response were also evaluated during 2015 under low and recommended N field conditions. Two field plots with different N levels viz., a nitrogen deficient plot (N low) of dimensions 19.0 m length and 24.5 m width without any artificial N inputs and a plot supplied with recommended dose of nitrogen (N rec) are being maintained since 2011 at ICAR-IIRR, (Vijayalakshmi et al. 2015). Soil properties of these two plots during wet seasons of 2012, 2013 and 2015 were analysed as per the procedures of Surekha and Satishkumar (2014) (Table S1). One-month-old seedlings of four rice genotypes raised in a nursery with regular package of practices including recommended dose of N were transplanted at a spacing of 10 × 20 cm in N low and N recommended fields. Nitrogen fertilizer @ 100 kgN ha−1 in the form of urea (46.5%) in three equal split applications was supplied only to the recommended N plots (at basal, maximum tillering and panicle initiation stages). Phosphorus as P2O5 (@40 kgha−1), potassium asK2O (@40kgha−1) and zinc as ZnO (@25 kgha−1) were applied to low and recommended N field plots and no additional nitrogen was applied to low N plot. Field management followed standard rice crop production and protection practices. For each genotype, five representative plants were harvested at maturity and were divided into vegetative and reproductive parts, dried and weighed for determining dry matter of various plant parts. Grain and straw yield were expressed in kg ha−1 and grain yield was adjusted to 14% grain moisture content. A total of 12 traits/parameters were recorded for morphological, yield and nitrogen content in low and recommended N field conditions for 3 years.
Data analysis
Three NUE indices have been calculated for low and recommended N viz., Physiological NUE (PNUE kg kg−1) (Zhao et al. 2012), Internal Efficiency (IE kg kg−1) (Dobermann and Fairhurst 2000) and Nitrogen Harvest Index (NHI %) (Singh et al. 1998). The N content of straw and grain samples after harvest was analysed with micro Kjeldahl method. The PNUE was calculated as total dry weight including straw and yield/total N uptake as N content of straw and grain samples. The IE was calculated as grain yield/total N uptake and the NHI was estimated using N content of the grain/N of the straw.
Two-way analysis of variance (ANOVA) was performed using an open source software R (R Core Team, 2012) with agricolae package.
RNA extraction, library preparation, and sequencing
The tissues of four genotypes grown during 2012 were collected from low N and recommended N plots at the booting stage by dissecting panicle out of sheath based on their flowering times. The total RNA was isolated using the Trizol method from three biological replications of developing panicle tissue and pooled. The quality and quantity of total RNA was checked on Nanodrop (Thermo fisher) as well as on 1.5% agarose gel. The quality was also assessed on Bioanalyzer (Agilent 2100) using RNA 6000 Nano chip and RNA having > 7.8 RNA Integrity Number (RIN) were further processed for GS FLX 454 Roche and Illumina HiSeq 2000 paired end sequencing.
For 454 Roche GS FLX Titanium series, rRNA removal, mRNA purification and double stranded cDNA synthesis were carried out using Roche by Genotypic Technologies Pvt. Ltd., Bangalore, India. The raw reads in fastq format were processed for removal of adaptor sequences and deposited in the NCBI's SRA database with the accession number PRJNA379890. For IlluminaHiSeq 2000, the pair-end cDNA sequencing libraries (2 × 100 bp) were prepared; clusters were generated and sequenced on the HiSeq2000 platform using sequencing by synthesis by Xcelris Genomics, Ahmedabad, India. The left and right reads for each sample were merged and raw reads were generated. The data are deposited in NCBI's SRA database with the accession numbers SAMN06718310—SAMN06718317.
Eight datasets of developing panicle transcriptome were generated comprising two efficient genotypes and two non-efficient genotypes grown under low and recommended N conditions and the differentially expressed transcripts (DETs) between low and recommended N were studied for biologically meaningful comparisons.
Pre-processing of RNA-seq data
The raw reads of Illumina were cleaned for high quality reads following the criteria of retention of bases with quality > 20 phred score and > 70% of bases. For 454, reads with > 50% of bases in a read with > 20 phred score were considered as high quality reads.
De novo assembly
The high quality 454 reads were assembled using the proprietary GS-de novo assembler (version: 2.9) and the Illumina reads were assembled using CLC Genomics Workbench 6 with default parameters. The transcripts generated from these two assemblies were clustered through cd-hit (version: 4.6.1) clustering tool to generate the final set of transcripts aptly known as the hybrid assembly at a similarity threshold of 90%. The assembly statistics were generated using N50 perl script of NGS QC toolkit.
ORF prediction and functional annotation
The transcripts were subjected to TransDecoder (version: rel16JAN2014 or TransDecoder Release v3.0.1) for identification of candidate gene coding regions within transcript sequences, i.e., Open Reading Frames (ORFs). The identified ORFs were subjected to functional annotation through Blastx (version: 2.2.28 +) against NCBI "nr" database at an e-value threshold of 1e-05. All the sequences in the nr database till 01.09.2016 were used for analysis.
Differential expression of transcripts
The Illumina reads were employed for differential transcript expression. High quality reads of Illumina were back mapped on the de novo hybrid assembly using the DESeq package. The transcripts with RC (read count) value > 0 were only considered for differential expression analysis. The "baseMean" value which is the normalized expression value was calculated from RC values and served as the basis for differential transcript expression analysis. The differential expression of transcripts was calculated as the log fold change of RC values of low/ recommended N for each genotype with criteria for differential expression as log2 fold-change ≥ 2 and false discovery rate (FDR) p-value correction of ≤ 0.05. The DETs were functionally classified and represented as heat map using Multi-experiment Viewer (MeV) v4.9.0 (Saeed et al. 2003) (http://bioinfogp.cnb.csic.es/tools/venny/) (Oliveros 2016). The number of DETs among and within conditions was plotted as Venn diagram using VennPlex https://www.nia.nih.gov/research/labs/vennplex) (Cai et al. 2013).
Alternative splicing
For understanding the alternative splicing events, high quality reads of each sample emanated from Illumina paired end sequencing were aligned to the de novo hybrid assembly as reference using MapSplice version-2.2.1 at default parameters (Wang et al. 2010). The splice variants were identified and classified as novel canonical, semi-canonical and non-canonical splice junctions, novel insertions and deletions.
Gene ontology (GO) enrichment and pathway analysis
Gene ontology (GO) enrichment analysis was performed for the functional categorization of various DETs using GOseq analysis tool, which is based on Wallenius non-central hyper-geometric distribution (Young et al. 2010). The gene ontology annotations were compared as graphs using online tool, WEGO (Web Gene Ontology Annotation Plot) (Ye et al. 2006). Pathway analysis was performed through the KEGG Automatic Annotation Server (KAAS)(http://www.genome.jp/tools/kaas/). The ORFs predicted were uploaded to KAAS and analysed using the BBH (bi-directional best hit) method. To view specific pathways related to plant metabolism, the DETs were analysed using the MapMan (version 3.5.1; http://mapman.gabipd.org/web/guest) with P value cut-off of ≤ 0.05 (Thimm et al. 2004).
Identification of gene co-expression modules
A co-expression gene network was constructed using the WGCNA software package (v1.66) in R (Langfelder and Horvath 2008) using eight datasets of developing panicle comprising two efficient and two non-efficient genotypes grown under low and recommended N (https://cran.r-project.org/web/packages/WGCNA/index.html). Transcripts with p value on and above 0.5/− 0.5 were selected for the analyses removing low expressed transcripts (reflecting noise). In each analysis, common transcripts (Table S2) were selected using merge function of plyr R package and the offending transcripts were removed by gsg (good samples genes) syntax available in WGCNA package (v1.66). A total of 4598 transcripts was used for network construction and module detection using the function block wise Modules. In brief, an adjacency matrix was created using correlation function and soft threshold of six estimated with the pick Soft Threshold function. The subsequent Topological Overlap Matrix (TOM) was used for module detection using the Dynamic Tree cut algorithm with a minimal module size of 30 and a branch merge cut height of 0.25. The module eigen transcripts with the highest correlation coefficient (> 0.9) were used to evaluate associations between the ten resulting modules and traits/parameters under low and recommended N.
Mapping DETs to known quantitative trait loci (QTL)
The genomic positions of DETs were checked for their co-localization within the QTL genome regions reported for NUE, yield and associated traits under low N condition in rice (Xu et al. 2012).
Validation of RNA-seq analysis by qPCR
The DETs on the basis of up and down regulation in efficient and non-efficient genotypes and their function in N metabolism were selected, primers were designed using QuantPrime (http://quantprime.mpimp-golm.mpg.de/) with the default parameters and were synthesized at Integrated DNA Technologies (USA) (Table S3). The stability of endogenous reference genes at booting stage was analysed using RefFinder (http://www.leonxie.com/referencegene.php) and Membrane protein (LOC_Os12g32950.1) (forward primer: GAGCGCAAAGTTCCAGAAGAA and reverse primer: CGCCACTAGTTGCCGTCCTGAT) was used for normalized relative expression of transcripts for validation (Phule et al. 2018). For qPCR, panicle tissues of 4 genotypes and 16 genotypes with single plant yield (g) ranging from 2.5 to 10.4 (Table S4) during booting stage grown during 2015 were collected as two biological replicates and immediately frozen in liquid nitrogen for RNA isolation. Panicle tissues were also similarly collected from 18 rice genotypes grown under low and recommended N during wet season 2015. Total RNA was isolated using NucleoSpin RNA Plant kit (MACHEREY–NAGEL, Germany) according to manufacturer’s protocol and the quality of the RNA was assessed using Nanodrop® ND1000 spectrophotometer (Thermo Scientific, USA). Approximately 1 µg of total RNA from each sample was used as template for the first-strand cDNA synthesis, using Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany) and diluted to 1:10 prior to use as template for qPCR. Each reaction was performed in three technical replicates containing 5 µl FastStart Universal SYBR Green Master (Roche Diagnostics, Mannheim, Germany), 2 µl template cDNA, 0.4 µl each of the primers (10 µM), and 2.2 µl RNase-free water with a total volume of 10 µl. The qPCR was performed using LightCycler 96 system (Roche Diagnostics, Mannheim, Germany) with the following program 95 °C (10 min) followed by 40 cycles of 95 °C (15 s), 58 °C (15 s) with fluorescent signal recording and 72 °C for 10 s. The melting curve was obtained using a high resolution melting profile performed after the last PCR cycle: 95 °C for 10 s followed by a constant increase in the temperature between 65 °C for 60 s and 97 °C for 1 s. The data were analysed using Light Cycler 96 system software (Roche Diagnostics, Mannheim, Germany). The relative expression levels of transcripts were calculated using the 2−∆∆Ct method according to (Schmittgen and Livak 2008) where recommended N condition was taken as control and low N was considered as treatment.
Screening of germplasm with DET of potassium channel SKOR transcript
The expression of potassium channel SKOR (LOC_Os06g14030: potassium channel stellar K + outward rectifier, putative) transcript, an up-regulated transcript in both efficient genotypes was studied in a set of 16 rice genotypes with differential yield response under low N, i.e., in efficient and non-efficient genotypes. The correlation between the fold changes of SKOR transcript and single plant yield (g) was calculated using MS Excel (Table S4).
Results
Effective assimilation and remobilization of N in efficient genotypes
Evaluation of 2 efficient genotypes (TB: ThururBhog and BM: Basmati 370) and 2 non-efficient genotypes (SK: Suraksha and KJ: Kola Joha) showed consistent performance across 3 years for 12 traits/parameters based on yield and N content. Significant differences were observed for 11 traits/parameters between N treatments, among the 4 genotypes and interaction between the treatments and varieties through ANOVA, but for Physiological Nitrogen Use Efficiency (PNUE kg kg−1) as total dry weight/total N uptake (Table 1). Variations due to years and interactions were not significant (P > 0.05). In the present study, NUE was analysed using three indices viz., PNUE to study conversion efficiency into total biomass, internal efficiency (IE kg kg−1) to study assimilation of total N taken up into grain yield and NHI (%) to study partition efficiency of N. For both efficient and non-efficient genotypes, PNUE was observed to be higher under low N than the recommended N. The IE and NHI were high in efficient genotypes in comparison to non-efficient genotypes under low N.
Table 1.
Summary of ANOVA of agro morphological traits, yield, N content and NUE indices of four genotypes grown under low and recommended N fields during 2012, 2013 and 2015
| Traits / parameters | Mean of 2012, 2013, 2015 | T | Y × T | G | Y × G | T × G | Y × T × G | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ThururBhog | Basmati370 | Suraksha | KolaJoha | |||||||||||
| Nlow | Nrec | Nlow | Nrec | Nlow | Nrec | Nlow | Nrec | |||||||
| SPAD RS | 36.0 ± 0.5 | 39.1 ± 0.3 | 34.6 ± 0.7 | 38.2 ± 0.5 | 37.9 ± 0.2 | 43.7 ± 0.9 | 33.4 ± 1.51 | 36.6 ± 0.3 | ** | ** | ** | ** | * | ** |
| PN | 345.3 ± 13.3 | 415.6 ± 5.7 | 316.5 ± 5.9 | 393.9 ± 3.6 | 271.3 ± 5.7 | 340.4 ± 2.6 | 257.1 ± 7.1 | 334.4 ± 2.5 | ** | ns | ** | ns | ** | ns |
| GY | 3616.5 ± 51.8 | 3829.6 ± 47.7 | 3398.0 ± 171.6 | 3727.3 ± 49.1 | 1630.0 ± 16.2 | 4207.2 ± 136.3 | 1378.8 ± 120.4 | 3309.0 ± 105.3 | ** | ns | ** | ns | ** | ns |
| SW | 3545.4 ± 111.8 | 4476.4 ± 8.1 | 3466.1 ± 49.4 | 4152 ± 16.3 | 3316.7 ± 134.2 | 4270.6 ± 45.1 | 2509.6 ± 84.6 | 3710.1 ± 21.9 | ** | ns | ** | ns | ** | ns |
| TDM | 7161.8 ± 150.8 | 8306 ± 52.8 | 6864.1 ± 100.8 | 7879.3 ± 64.6 | 4946.7 ± 127.1 | 8477.8 ± 93.8 | 3888.4 ± 147.0 | 7019.1 ± 31.6 | ** | ns | ** | ns | ** | ns |
| HI | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.3 ± 0.0 | 0.5 ± 0.0 | 0.4 ± 0.0 | 0.5 ± 0.0 | ** | ns | ** | ** | ** | ns |
| NG | 42.3 ± 0.1 | 53.4 ± 0.4 | 39.6 ± 0.6 | 52.2 ± 0.8 | 27.6 ± 0.4 | 52.1 ± 0.1 | 22.5 ± 0.3 | 42.4 ± 0.3 | ** | ns | ** | ns | ** | ns |
| NS | 18.7 ± 0.1 | 23.2 ± 0.4 | 17.9 ± 0.3 | 21.1 ± 0.6 | 17.5 ± 0.3 | 24.6 ± 0.2 | 14.3 ± 0.2 | 18.0 ± 0.9 | ** | ns | ** | ns | ** | ns |
| TNUP | 61.1 ± 0.1 | 76.6 ± 0.8 | 57.5 ± 0.6 | 72.3 ± 0.3 | 45.2 ± 0.7 | 76.8 ± 0.2 | 36.8 ± 0.5 | 60.4 ± 1.1 | ** | ns | ** | ns | ** | ns |
| PNUE | 12.87 ± 2.15 | 9.86 ± 1.78 | 18.18 ± 6.44 | 12.23 ± 3.29 | 78.18 ± 2.68 | 40.88 ± 6.20 | 56.34 ± 6.80 | 33.82 ± 3.73 | *** | * | ns | * | ns | ns |
| IE | 59.2 ± 0.9 | 50.0 ± 1.1 | 59.2 ± 0.3 | 51.6 ± 0.7 | 36.2 ± 0.3 | 54.8 ± 1.7 | 37.5 ± 3.0 | 54.8 ± 1.6 | ** | ns | * | ns | ** | ns |
| NHI | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.7 ± 0.0 | 0.6 ± 0.0 | 0.7 ± 0.0 | 0.6 ± 0.0 | 0.7 ± 0.0 | ** | ns | ** | ns | ** | ns |
| ANUE | 7.11 ± 0.89 | 9.38 ± 3.02 | 26.81 ± 1.66 | 19.27 ± 1.96 | ns | * | ||||||||
SPAD RS soil–plant analyses development during reproductive stage, PN panicles m−2, GY grain yield kg ha−1, SW straw weight kg ha−1, TDM total dry matter kg ha−1, HI harvest index, NG N in grain kg ha−1, NS N in straw kg ha−1, TNUP total N uptake kg ha−1, PNUE physiological N use efficiency kg kg−1, IE internal efficiency kg kg−1, NHI nitrogen harvest index, ANUE agronomic N use efficiency kg kg−1, T treatments (Nlow and Nrec: recommended), Y years (2012, 2013, 2015), G genotypes
De novo assembly and annotation of panicle transcriptome
The data were assembled de novo to identify differentially expressed transcripts and pathways during the panicle development (Figure S1). The RNA-seq by GS FLX 454 resulted in 969,535 raw reads with 829,437 (85.55%) high quality reads after filtration. IlluminaHiSeq2000 emanated 164,026,769 raw reads with 159,219,245 (97.07%) high quality reads after trimming and filtering the low quality reads (Table S5). Approximately, 20 million paired end 100 bp and 0.1 million single end high quality reads were obtained per sample with Illumina and 454 sequencing. The high quality reads of each platform were assembled through GS assembler and CLC Genomics Workbench 6 into 5770 and 107,584 transcripts, respectively (Table 2).
Table 2.
Summary of transcripts and ORF predictions of 454, Illumina and hybrid assemblies
| Particulars | 454 Data assembly GS assembler | Illumina data assembly CLC Genomics Workbench 6 |
Hybrid assembly |
|---|---|---|---|
| Number of transcripts | 5770 | 107,584 | 47,814 |
| Total length (Mb) | 3.8 | 82 | 64.0 |
| Average transcript length | 668.55 | 670.19 | 1340.38 |
| Transcript N50 | 785 | 1219 | 1701 |
| Maximum transcript length in bases | 5297 | 16,563 | 16,563 |
| Minimum transcript length in bases | 100 | 200 | 500 |
| Number of ORFs | 34,612 | ||
| Total length (Mb) | 35.33 | ||
| Average ORF length | 1020.72 | ||
| ORF N50 | 1275 | ||
| Maximum ORF length in bases | 16,200 | ||
| Minimum ORF length in bases | 297 | ||
| Number of ORFs with blast hits | 32,340 | ||
The two transcriptome data sets were de novo assembled into 47,814 transcripts totalling to 64 Mb with an average transcript length of 1340 bp and N50 value of 1,701. The total open reading frames (ORFs) in the transcripts were 34,612 (35.33 Mb) and 32,340 showed Blast similarity to the non-redundant nr database (Table 2). The functional annotation was performed for two data sets GS FLX 454 Roche and Illumina HiSeq 2000 and also for the de novo assembled transcripts for improved functional annotation (Table S6). As the four studied genotypes belong to different subgroups of rice (Basmati370—Basmati subgroup; Suraksha—indica subgroup; ThururBhog and Kola Joha—still to be sub grouped), de novo assembly improved complete assembly and increased the number of annotated transcripts.
The de novo assembled transcripts have been submitted to the NCBI, TSA with accession number SUB2593709.
Lower number of differentially expressed transcripts (DETs) in efficient genotypes under low N
The number of significant up and down-regulated transcripts in efficient genotypes was observed to be lower than 50% in non-efficient genotypes under low N as compared to recommended N (Figure S2). In efficient genotypes, the number of up-regulated transcripts was only about a half of the number of the down-regulated transcripts under low N. Contrarily, in non-efficient genotypes, the number of significant up-regulated transcripts was more than down-regulated transcripts (Table S7).
In efficient genotypes, 18 up-regulated and 191 down-regulated transcripts were observed in common with contra-regulation of 43 transcripts (Fig. 1) (Table S8). The up-regulated transcripts mostly comprised hypothetical proteins (66.6%) and a few known proteins (Table 3). The number of hypothetical proteins was also higher among the down-regulated transcripts, in addition to transcripts related metabolism of amino acids, carbon, lipids, reactive oxygen species (ROS), terpenoids and polyketides, secondary metabolites and transcription factors.
Fig. 1.
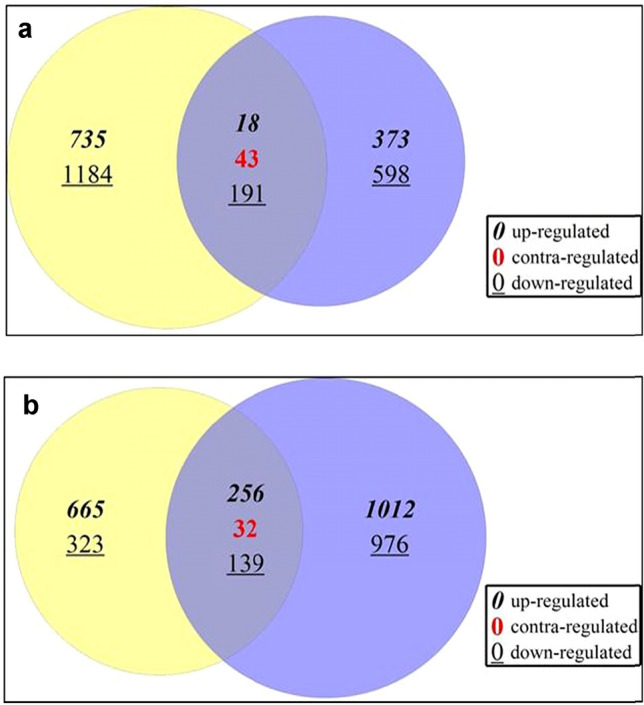
Differentially expressed transcripts (DETs) in four genotypes
Table 3.
Annotation of the common up-regulated transcripts associated with low N in two efficient genotypes
| Annotation | Fold change | ||||
|---|---|---|---|---|---|
| TB Nrec | TB Nlow |
BM Nrec |
BM Nlow | ||
| 1 | Hypothetical protein OsJ_01563 (LOC_Os01g22540) | 2.08 | 24.94 | 1.05 | 25.84 |
| 2 | Hypothetical protein OsI_17904* | 81.31 | 665.77 | 16.72 | 272.71 |
| 3 | Hypothetical protein OsJ_10779 | 91.73 | 371.26 | 52.25 | 538.72 |
| 4 | Unknown protein | 1.04 | 35.50 | 2.09 | 20.09 |
| 5 | Os06g0528700 putative flavin-containing monooxygenase | 7.30 | 49.88 | 10.45 | 87.08 |
| 6 | Os03g0184100 expressed protein | 488.88 | 3300.08 | 400.26 | 3009.38 |
| 7 | Hypothetical protein OsI_17904* | 47.95 | 313.70 | 16.72 | 123.44 |
| 8 | Putative RNA helicase/RNAseIII protein | 104.24 | 507.48 | 11.50 | 75.59 |
| 9 | Os11g0120300 unknown protein | 1.04 | 14.39 | 24.04 | 149.27 |
| 10 | Hypothetical protein OsI_17459 | 27.10 | 200.50 | 21.95 | 135.88 |
| 11 | Aspartyl proteinase (LOC_Os11g08200) | 28.14 | 146.78 | 42.85 | 254.53 |
| 12 | Hypothetical protein OsI_22386 | 130.30 | 799.12 | 335.47 | 1946.29 |
| 13 | Os07g0578300 putative subtilisin-like serine protease | 29.19 | 157.33 | 160.94 | 813.35 |
| 14 | Os01g0956200 putative glycosyltransferase | 59.42 | 330.97 | 310.38 | 1516.65 |
| 15 | Hypothetical protein OsJ_05709 (LOC_Os02g09830) | 28.14 | 123.75 | 97.19 | 466.96 |
| 16 | Hypothetical protein OsI_17459 | 93.82 | 387.57 | 70.02 | 331.08 |
| 17 | LOC_Os06g14030 SKOR protein OsJ_20828 | 51.08 | 424.02 | 140.04 | 630.58 |
| 18 | Hypothetical protein OsJ_27967 (LOC_Os08g40919) | 90.69 | 375.10 | 87.79 | 370.31 |
*Multiple predictions of ORFs on the same strand suggest alternative splicing
In non-efficient genotypes, 256 transcripts were up-regulated and 139 transcripts were down-regulated in common with 32 contra-regulated transcripts (Fig. 1) (Table S8). Most of the up-regulated transcripts belonged to the pathways of energy metabolism, environmental adaptation and cell motility and the down-regulated transcripts were mostly from pathways of amino acid metabolism.
Common biological processes across efficient and non-efficient genotypes under low N
Through GO enrichment analysis, the DETs were classified into three main categories including biological process, cellular component, and molecular function (corrected P value < 0.05). Catalytic activity, binding, metabolic and cellular processes were the most well represented sub categories in up and down-regulated DETs of efficient and non-efficient genotypes (Figure S3).
Over representation of metabolic pathways in non-efficient genotypes under low N
Using MapMan analyses, most of the transcripts involved in the metabolic pathways (69.4%) were found to be over represented in the non-efficient genotypes as compared to the efficient genotypes (Table 4) (Figure S4 A and B). Across the four genotypes under low N, (11.1%) pathways of glycolysis, N and sulphur metabolism and development were found to be up-regulated in common. The number of up-regulated transcripts in efficient genotypes in comparison to non-efficient genotypes was over represented in nucleotide, co-factor and vitamins, RNA and DNA pathways (11.1%). The number of down-regulated transcripts was higher in cell wall, lipid and metal handling metabolic pathways across four genotypes (8.3%). In efficient genotypes, higher number of down-regulated transcripts was found in fermentation, oxidative pentose phosphate pathway, amino acid, secondary, hormone, redox, biodegradation and miscellaneous metabolic pathways (22.2%) (Table 4). The analyses of sub metabolic pathways showed an interesting pattern of additional DETs in efficient and non-efficient genotypes viz., additional over representation of some sub pathways in the same direction and contra-expression of some pathways to the common up and down-regulated DETs of four genotypes (Tables S9 and S10). The additional DETs of glutathione metabolism were found to be down-regulated in efficient genotypes and up-regulated in non-efficient genotypes. Under low N, majority of DETs of the secondary metabolites pathways were found to be only down-regulated across four genotypes viz., stilbenoid, diarylheptanoid and gingerol, flavonoid, flavone and flavonol biosynthesis. Under translation, the ribosome sub metabolic pathway was found to be significantly down-regulated across four genotypes and additional DETs were also found to be down-regulated in efficient and non-efficient genotypes. Contra-regulation was also observed in the DETs of aminoacyl-tRNA biosynthesis of translation, DNA replication and nucleotide excision repair of replication and repair pathway, where a significant upregulation was found across four genotypes in common and in efficient genotypes, but significant down regulation was noted in non-efficient genotypes. Conversely, the DETs of proteasome of folding, sorting and degradation pathway were significantly down-regulated across four genotypes in common and in efficient genotypes, but significantly up-regulated in non-efficient genotypes. The DETs in efficient and non-efficient genotypes were also analysed according to a few important gene families associated with NUE (Table 5).
Table 4.
Summary of the number of the differentially expressed transcripts according to MapMan BINs in efficient and non-efficient genotypes under low N in comparison to recommended N
| Bin | Name | TB Nrec - TB Nlow |
BM Nrec - BM Nlow |
SK Nrec - SK Nlow |
KJ Nrec - KJ Nlow |
TB Nrec - TB Nlow |
BM Nrec - BM Nlow |
SK Nrec - SK Nlow |
KJ Nrec - KJ Nlow |
|---|---|---|---|---|---|---|---|---|---|
| Up-regulated | Down-regulated | ||||||||
| 1 | Photosynthesis | 85 | 150 | 64 | 149 | 122 | 57 | 143 | 58 |
| 2 | Major CHO metabolism | 70 | 88 | 92 | 70 | 71 | 51 | 50 | 70 |
| 3 | Minor CHO metabolism | 59 | 92 | 77 | 68 | 99 | 65 | 77 | 89 |
| 4 | Glycolysis | 54 | 60 | 64 | 69 | 51 | 45 | 40 | 36 |
| 5 | Fermentation | 11 | 11 | 17 | 20 | 16 | 16 | 10 | 7 |
| 6 | Gluconeogenesis / glyoxylate cycle | 10 | 6 | 10 | 12 | 8 | 12 | 18 | 6 |
| 7 | Oxidative pentose phosphate pathway | 15 | 12 | 12 | 18 | 16 | 19 | 19 | 13 |
| 8 | TCA / organic acid transformations | 32 | 53 | 55 | 39 | 49 | 26 | 25 | 40 |
| 9 | Mitochondrial electron transport / ATP synthesis | 38 | 63 | 66 | 57 | 69 | 45 | 41 | 51 |
| 10 | Cell wall | 152 | 176 | 251 | 290 | 475 | 424 | 371 | 325 |
| 11 | Lipid metabolism | 191 | 240 | 270 | 280 | 428 | 359 | 345 | 337 |
| 12 | Nitrogen metabolism | 17 | 17 | 17 | 16 | 9 | 8 | 9 | 9 |
| 13 | Amino acid metabolism | 149 | 144 | 124 | 178 | 177 | 181 | 199 | 147 |
| 14 | Sulphur assimilation | 7 | 9 | 9 | 7 | 5 | 3 | 3 | 5 |
| 15 | Metal handling | 27 | 35 | 32 | 34 | 48 | 38 | 43 | 42 |
| 16 | Secondary metabolism | 201 | 225 | 206 | 369 | 412 | 351 | 413 | 241 |
| 17 | Hormone metabolism | 255 | 267 | 314 | 382 | 399 | 365 | 339 | 269 |
| 18 | Co-factor and vitamin metabolism | 59 | 50 | 46 | 40 | 44 | 53 | 56 | 63 |
| 19 | Tetrapyrrole synthesis | 22 | 28 | 15 | 26 | 23 | 17 | 30 | 19 |
| 20 | Stress | 518 | 542 | 598 | 495 | 644 | 531 | 530 | 634 |
| 21 | Redox | 76 | 87 | 76 | 105 | 132 | 119 | 132 | 101 |
| 22 | Polyamine metabolism | 12 | 16 | 13 | 12 | 16 | 11 | 14 | 16 |
| 23 | Nucleotide metabolism | 92 | 105 | 79 | 86 | 76 | 62 | 88 | 81 |
| 24 | Biodegradation of xenobiotics | 11 | 24 | 11 | 36 | 50 | 36 | 49 | 26 |
| 25 | C1-metabolism | 20 | 18 | 14 | 22 | 20 | 22 | 26 | 18 |
| 26 | Miscellaneous | 604 | 606 | 704 | 884 | 1002 | 947 | 905 | 716 |
| 27 | RNA | 1696 | 1496 | 1487 | 1305 | 964 | 1118 | 1150 | 1337 |
| 28 | DNA | 430 | 390 | 386 | 281 | 198 | 207 | 220 | 325 |
| 29 | Protein | 2004 | 1755 | 1814 | 1654 | 1655 | 1808 | 1809 | 1967 |
| 30 | Signaling | 837 | 891 | 1120 | 911 | 1125 | 960 | 820 | 1000 |
| 31 | Cell | 401 | 446 | 463 | 346 | 468 | 413 | 401 | 518 |
| 32 | Micro RNA, Natural antisense etc | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 33 | Development | 430 | 398 | 443 | 406 | 341 | 346 | 320 | 355 |
| 34 | Transport | 638 | 774 | 829 | 772 | 819 | 645 | 611 | 670 |
| 35 | Not assigned | 7617 | 6723 | 7198 | 7217 | 7073 | 6725 | 6971 | 6986 |
| 36 | C4-photosynthesis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Table 5.
Summary of DETs and their gene families in efficient and non-efficient genotypes under low N
| Efficient | Non-efficient | |||
|---|---|---|---|---|
| Up | Down | Up | Down | |
| Nitrogen metabolism | – | EAY84173gi hypothetical protein | Os08g0468100 Nitrate reductase [NADH] 1 |
Os01g0764900 Formamidase Os03g0712800 Glutamine synthetase cytosolic |
| Vitamin and cofactor metabolism |
Os08g0245400 Bifunctionaldethiobiotinsynthetase ABA93693gi Bifunctionaldihydrofolatereductase Os02g0276500 Putative tocopherolcyclase |
– | – | – |
| Transcription factors (TF) |
Os12g0618600-CCAAT-binding TF subunit B Os03g0741400 WRKY TF ADX60281 bHLH TF Os01g0625300 Heat stress TF C-1a |
AAM27466gi Putative bHLH TF DAA05141gi WRKY TF 76 Os03g0437200 C2H2-type zinc finger protein ZFP36 CAC39058gi putative AP2-related TF Os01g0821300 WRKY TF 64-like protein |
Os10g0503100 LIM TF BAD29571gi putative TF MADS27 BAD31424gi PHD finger TF-like protein Os06g0679400 putative TF GAMyb Os08g0103900 putative OsNAC7 protein |
AAP83325gi TF Os09g0538400 putative Myb-related protein Zm38 DAA05124gi WRKY TF 59 Os02g0643200 YABBY 4 TF Os03g0764100 zinc finger TF ZF1 |
| Transporters (TPR) |
BAB17113gi ABC TPR-like Q93XI5gi potassium-sodium symporter Os01g0279700 Phosphate TPR ABA91534gi ABC TPR Os03g0195800 Sulfate TPR 1.2 |
ACN85155gi ABC TPR-like protein Os06g0228800 putative amino acid TPR BAC83382gi putative organic cation TPR AAT77331gi putative ABC TPR Os03g0150500 H/Pi co TPR |
Os05g0252000 Probable metal-nicotianamine TPR YSL4 XP_004972812gi nitrate TPR 1.1-like isoform X2 BAC83856gi putative nitrate TPR NRT1-5 Os01g0829900 Probable metal-nicotianamine TPR YSL18 Os11g0475600 hexose TPR |
Os03g0281900 ABC TPR G family member 5 AAP53801gi Sulfate TPR 3.1, putative BAB63609gi putative nitrite TPR BAB92574gi proline transport protein-like Os07g0557200 putative equilibrative nucleoside TPR ENT8 splice variant |
Nitrogen metabolism
In efficient genotypes, no common transcript related to N metabolism were found to be up-regulated and only one hypothetical protein (EAY84173) was observed to be down-regulated in efficient genotypes. Across four genotypes, there was a relatively higher number of up-regulated transcripts than down-regulated transcripts, however, only four up-regulated transcripts viz., hypothetical protein (putative function of nitrate reductase) (EEC74079); glutamine synthetase, chloroplastic (Os04g0659100); glutamate synthase 2 [NADH], chloroplastic (Os05g0555600) and a hypothetical protein (putative function of ferredoxin-dependent glutamate synthase) (EEC82609) andthree down-regulated transcripts viz., high-affinity nitrate transporter (Q94JG1), putative nitrilase (Os02g0635000) and cyanate hydratase (Os10g0471300) were in common. In non-efficient genotypes, nitrate reductase [NADH] (Os08g0468100) was up-regulated and two transcripts viz., formamidase (Os01g0764900) and glutamine synthetase, cytosolic (Os03g0712800) were down-regulated (Table S11).
Vitamin and cofactor metabolism
In efficient genotypes, among 12 up-regulated transcripts, 75% were hypothetical proteins besides bifunctional dithiobiotin synthetase (Os08g0245400), bifunctional dihydrofolate reductase-thymidylate synthase (ABA93693) and tocopherol cyclase (Os02g0276500) transcripts (Table S11).
Transcription factors
Four common up-regulated transcription factors, CCAAT-binding (Os12g0618600); WRKY DNA binding domain containing protein (Os03g0741400); bHLH (ADX60281) and heat stress (Os01g0625300) were noted in efficient genotypes. Eighteen transcription factors were found to be down-regulated in efficient genotypes comprising mostly transcription factors belonging to AP2, bHLH, WRKY and ZFP classes. In non-efficient genotypes, 17 transcription factors were up-regulated in common and, predominantly belonging to heat stress, MADS, MYB and NAC classes (Figure S5 A, B, C, and D).
Transporters
The five common up-regulated transporters in efficient genotypes included two ABC transporters (BAB17113 and ABA91534), potassium-sodium symporter (Q93XI5); phosphate transporter (Os01g0279700) and sulphate transporter (Os03g0195800). Fourteen transporters belonging to ABC, AAT and AMT families were found to be down-regulated in efficient genotypes. In non-efficient genotypes, 39 transcripts were found to be up-regulated in common comprising mostly ABC, sugar and YSL transporters (Figure S5 E, F, G, and H).
Higher number of splice junctions under low N
Approximately, 99% canonical splice junctions were identified across the genotypes and treatments (Table 6). A relatively higher number of splice junctions and canonical splice junctions were observed under low N in comparison to recommended N for three genotypes but for one non-efficient genotype. Semi-canonical splice junctions and the insertions/deletions appeared to be genotype specific. A hypothetical protein OsI_17904 with multiple predictions of ORFs on the same strand showed an upregulation in efficient genotypes suggests alternative or adjacent splicing (Table 3).
Table 6.
Total numbers of various splice variants identified under low N and their classification
| Sample name | Total splice junction | Canonical splice junction | Semi-canonical splice junction | Small deletions | Small insertions |
|---|---|---|---|---|---|
| TB Nlow | 6514 | 6491 (99.65%) | 23 (0.35%) | 57,482 | 34,944 |
| TB Nrec | 6083 | 6048 (99.42%) | 35 (0.58%) | 55,909 | 34,692 |
| BM Nlow | 6911 | 6883 (99.59%) | 28 (0.41%) | 58,048 | 35,072 |
| BM Nrec | 6825 | 6807 (99.74%) | 18 (0.26%) | 70,426 | 43,359 |
| SK Nlow | 6444 | 6419 (99.61%) | 25 (0.39%) | 55,683 | 33,674 |
| SK Nrec | 5455 | 5418 (99.32%) | 37 (0.68%) | 58,590 | 33,289 |
| KJ Nlow | 5207 | 5186 (99.60%) | 21 (0.40%) | 43,918 | 27,081 |
| KJ Nrec | 5572 | 5563 (99.84%) | 9 (0.16%) | 54,123 | 34,065 |
A negative correlation of DET clusters (modules) in efficient genotypes under low N
Gene expression network analysed using WGCNA R package resulted in 10 modules (clusters) of highly correlated DETs and their association with phenotypic traits (Fig. 2). For low N, three modules comprising both up and down-regulated transcripts were observed with only negative correlation viz., ‘pink’ with SPAD (205 DETs), ‘brown’ with panicle number and grain nitrogen (670 DETs) and ‘red’ with grain yield, internal efficiency and HI (388 DETs). The most enriched categories were amino acid metabolism in pink module, carbon metabolism, environmental adaptation and signal transduction in brown module and carbon metabolism, and signal transduction in red module (Table S12).
Fig. 2.

Module-trait relationships using WGCNA R
Co-localization of DETs with reported QTL under low N
Out of 18 common up-regulated transcripts in the 2 efficient genotypes, 10 transcripts (44.4%) were mapped to the reported QTL on chromosomes 1, 2, 3. 4, 6, 7 and 8 under low N condition. Interestingly, 75% of common down-regulated transcripts of the efficient genotypes were found to be in the region of the reported QTL for various traits under low N (Table S13).
Correlation of transcript expression between RNA-seq and qPCR
Out of 59 DETs targeted for qPCR, 52 transcripts showed a clear amplification of expected product size. The expression of transcripts fold changes through qPCR were comparable to the fold changes obtained through RNA-seq analysis (correlation coefficient r = 0.72) (Figure S6) (Table S3). The expression of SKOR (LOC_Os06g14030) has shown a positive correlation (0.62) with single plant yield (g) in a set of 16 rice genotypes selected on the basis of single plant yield ranging from 2.5 to 10.4 g under low N (Fig. 3) (Table S4).
Fig. 3.

Fold change of SKOR transcript with single plant yield (g) in a set of 16 rice genotypes under low N
Discussion
Booting stage is critical in determining rice yield, because there is degeneration of spikelets, especially under low N during this stage (Yoshida 1981). The metabolic processes should support the growth and differentiation of panicle branches and spikelets (primary and secondary) and reduce the inherent degeneration during the booting stage. Transcriptome analyses of panicle tissue of booting stage under low N have shown a predominant down regulation of transcripts in efficient genotypes with ~ 30% of the metabolic pathways over represented by down-regulated transcripts (Fig. 1, Figures S2, S3 and S4) (Tables 4, S7 and S8).
For acclimation to low N, plant needs complex and diverse physiological and biochemical changes requiring a concomitant action of numerous genes of various metabolic and regulatory pathways for its survival and propagation under limited resources. Through de novo assembly, a comprehensive transcriptome using four genotypes of two N conditions was built in the present study. The de novo assembly is useful to reduce the under estimation of variability among the genotypes and to facilitate appropriate genome wide differential gene expression analyses reference for the associated pathways (Table S6) (Kawahara et al. 2013). Our genotypes comprised landraces (proven to be adapted to low N) which represent a source of novel genes (Rao et al. 2018), we have also increased the number of genotypes (two efficient and two non-efficient) and the coverage through two kinds of analyses RNA-Seq to enhance the stringency and confidence of identified transcripts. The phenotypic observations recorded over 3 years and stringent statistical criteria employed throughout the analysis has added to the quality and accuracy of data.
The efficient genotypes could reveal relatively higher yield under low N indicating their inherent efficient uptake, remobilization and translocation abilities suggesting the rice landraces to be promising donors for NUE (Table 1) (Rao et al. 2018). In the present study, we have characterized two landrace genotypes with consistent and stable high NUE as shown by their higher internal use efficiency and remobilization of N between source (leaf) and sink (panicle). A general trend of reduction of traits was observed under low N, which was expected as N is the base material of the plant biomass (Tirol-Padre et al. 1996; Dobermann and Fairhurst 2000; Rao et al. 2018). Comparative analysis of metabolites under low N has shown an abundant decrease of major metabolites in two contrasting rice genotypes as earlier reported (Zhao et al. 2018). We suggest that the efficient genotypes appear to down regulate several not so critical metabolic pathways (Tables 4, S9 and S10) and divert the thus conserved energy to produce the seed under long-term N starvation. Co-localization of higher number of down-regulated transcripts with the reported QTL than up-regulated transcripts under low N in our study and similar observations of higher number of down-regulated transcripts in the earlier reports substantiate our observations (Lian et al. 2006; Yang et al. 2015a, b) (Table S13). Even the three modules identified through gene expression network using WGCNA R package showed a negative correlation with the six traits/parameters under low N (Fig. 2) (Table S12).
Several transcriptome data under N starvation indicated that the plants adapt different strategies for short term and long-term N deprivation. While the early (immediate) transcriptional response comprised signal transduction pathways, the late (adaptive) transcriptional response genes were related to be metabolic processes, substance transportation and physiological pathways (Hsieh et al. 2018). Under long term low N condition, the efficient genotypes through selective differential regulation of metabolic pathways were able to uphold the development of spikelets and reduce the degeneration of spikelets during the booting stage, whereas non-efficient genotypes appear to expend their energy by upregulation of genes of several pathways.
The common up and down-regulated metabolic pathways across four genotypes can be considered as a general adaptive response under long-term low N condition and it is the specific response of differentially regulated pathways in efficient genotypes, which is useful for deciphering metabolic pathways of NUE in rice. A down regulation seems to be the key strategy in efficient genotypes for NUE with upregulation of at least a few pathways for development of panicle and spikelets during the booting stage under low N. We have identified transcripts up-regulated for particular pathways viz., nucleotide, co-factor and vitamins, RNA and DNA (Tables S9 and S10). Co-expression network-based analyses reported a correlation of two of the clusters of adaptation to low N enriched with genes binding to nucleotides, purines and ATP (Coneva et al. 2014). The present data of DETs and pathways could be compared only to transcriptome studies of wheat spike, as panicle tissue transcriptome of rice under low N have not yet been reported. Even in wheat spike tissue, a predominant down regulation has been observed. Interestingly, upregulation of thiamine biosynthesis was also observed as in wheat spike (Curci et al. 2017).
Certain pathways were significantly enriched with additional transcripts either in the direction of the common expression or contra-regulated to the common expression across four genotypes (Table S9). The DETs of α-linolenic acid metabolism were found to be enriched in non-efficient genotypes, whereas there was a down regulation in common across four genotypes (Table S9). From the root transcriptomics of early response with N deficiency, α-linolenic acid genes as a part of jasmonate-mediate defense responses were reported to be induced (Huang et al. 2016; Hsieh et al. 2018). The observed up-regulated glycosyltransferases, which are a part of the major catalytic machinery for synthesis and breakage of glycosidic bonds relates N starvation to starch starvation as reported (Sinha et al. 2018). Though, there was an enhanced expression of N metabolism transcripts across four genotypes, certain differential response of transcripts related to N metabolism was observed in efficient genotypes under low N as corroborated by earlier studies (Lian et al. 2006; Zhu et al. 2006; Beatty et al. 2009). We suggest the role of transcripts underlying the metabolic sub pathways viz., nucleotide, translation, carbon, lipid, cell growth and death, replication and repair, membrane transport, secondary metabolites and transport to be critical in acclimation to the low N in rice.
A comparison of the data of panicle tissue transcriptomics in the present study to reported transcriptomics data of root and shoot tissue of the rice seedlings showed a few pathways to be similar. The phenylpropanoid biosynthesis was found to be down-regulated across four genotypes with higher a number of transcripts in efficient genotypes during the panicle development which is contrasting to the stimulation of phenylpropanoid biosynthesis in root and shoot tissues of seedlings under low N hydroponics (Richard-Molard et al. 2008; Krapp et al. 2011; Hsieh et al. 2018). Phenylpropanoid metabolism is critical for the production of several secondary metabolites (Naoumkina et al. 2010), thus a reduction of phenyl propanoid pathway appears to be an acclimation response to long term low N condition as observed in the present study.
Transcription factors (TFs) are of special interest, since they are capable of coordinating the expression of several downstream targets genes and, hence, entire metabolic and developmental pathways (Table 5). Several classes of TFs including CCAAT, WRKY, bHLH, AP2, ZFP, MADS, NAC and MYB TFs were found among DETs in this study as described earlier in rice under low N condition and in spike tissue of wheat (Yang et al. 2015a, b; Curci et al. 2017). Upregulation of WRKY and bHLH among the 63 high C: low N up-regulated transcripts was also reported in rice (Huang et al. 2016). Most of the TFs with a similar function were found to be up or down-regulated in the present study suggesting the possible compensatory mechanism of different transcripts within the same family expressing differentially to balance the gene expression and metabolism under low N (Figure S5 A, B, C, and D).
Despite the different genetic background, common transcripts were identified in the efficient genotypes unlike the earlier studies (Sinha et al. 2018). Interestingly, it was observed that most of the up-regulated transcripts in efficient genotypes are of unknown function suggesting landraces with their adaptation to low N to a promising source of the novel transcripts for NUE in rice (Tables 1 and 3). Moderate correlation of fold changes of an up-regulated transcript SKOR (LOC_Os06g14030) with single plant yield in 16 need to check genotypes with differential response under low N was observed in the present study (Table S4). Yield and NUE are complex traits involving several transcripts, thus a moderate association of SKOR transcript obtained in our study is encouraging for further identification and characterization of landraces for new transcripts/genes.
We found more alternative splicing events under low N in comparison to recommended N, while a reported study (Yang et al. 2015a) suggested no significant difference in AS of root and shoot tissues of rice seedlings under N-deficient condition (Table 6). However, AS events are reported to be organ and genotype specific (de Araujo Junior and Rosa Farias 2015). Increased levels of specifically spliced transcripts are demonstrated to have beneficial effects for development and stress acclimation (Staiger and Brown 2013). In rice, AS complexity was shown to contribute to the genetic improvement of drought resistance (Wei et al. 2017). The up-regulated hypothetical protein OsI_17904 with its two forms in efficient genotypes observed in the present study becomes an interesting candidate gene for further characterization (Table 3).
We selected the potassium transporter transcript on the basis of its upregulation in both efficient genotypes. Interestingly, the expression of this SKOR transcript was up-regulated in both efficient genotypes which prompted us to study its expression in a set of 16 genotypes differing in N efficiency and its co-relation with nitrogen uptake. The SKOR gene has been reported to function in transport of potassium, root to shoot communication and under certain stress condition responses. Moreover, the positive co-relation of nitrogen use efficiency and potassium transporter transcript reflects certain relation with respect to ion homeostasis. Its relationship with single plant yield also reflected that the expression is positively co-related with the single plant yield. It can be proposed that the expression of such transporters contributes to coping with the nitrogen deficiency.
In conclusion, two consistent NUE genotypes were identified from four rice genotypes based on their evaluation for yield, N content in straw and grain and NUE indices across 3 years. These two landrace genotypes could be deployed as donors for developing rice varieties with NUE. Using eight datasets of developing panicle transcriptome generated comprising two efficient genotypes and two non-efficient genotypes grown under low and recommended N conditions and their de novo assembly, differentially expressed transcripts and metabolic pathways were identified. Based on the differential regulation in efficient genotypes, we propose a down regulation of selective metabolic pathways to be an adaptive mechanism of rice to long-term low N acclimation. We have identified a set of 18 up-regulated transcripts and 10 differentially regulated metabolic pathways in efficient genotypes for further characterization of NUE in rice. Two up-regulated transcripts found to be promising candidate genes viz., a hypothetical protein OsI_17904 and a potassium channel SKOR transcript (LOC_Os06g14030) from the present study. The information generated about the genotypes, transcripts, pathways and mechanisms in the present study can be deployed to develop rice varieties with NUE, thus ultimately targeting the reduction of nitrous oxide emissions affecting climate change through a lower application of nitrogenous fertilizer.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by National Initiative on Climate Resilient Agriculture (NICRA), Indian Council of Agricultural Research (ICAR), Ministry of Agriculture, Govt. of India [F. No. Phy/ NICRA/2011-2012].
Abbreviations
- NUE
Nitrogen use efficiency
- IE
Internal efficiency
- NHI
Nitrogen Harvest Index
- PNUE
Physiological nitrogen use efficiency
- DET
Differentially expressed transcripts
Author contributions
CNN*—planning of the experiments, writing the manuscript, analyses, logistics. KMB—planning of some experiments, data analyses, writing the manuscript. TKK—executed the experiments. SB—executed the experiments. ISR—data collection. BS—executed the experiments. DSR—analyses. DS—analyses. PRR—support of the logistics. SRV—planning of the experiments, support of the logistics.
Compliance with ethical standards
Conflict of interest
All the authors declare that they have no conflict of interest in the publication.
References
- Beatty PH, Shrawat AK, Carroll RT, et al. Transcriptome analysis of nitrogen-efficient rice over-expressing alanine aminotransferase. Plant Biotechnol J. 2009;7:562–576. doi: 10.1111/j.1467-7652.2009.00424.x. [DOI] [PubMed] [Google Scholar]
- Broadbent FE, De DSK, Laureles EV. Measurement of nitrogen utilization efficiency in rice genotypes. Agron J. 1987;791:786–791. doi: 10.2134/agronj1987.00021962007900050006x. [DOI] [Google Scholar]
- Cai H, Lu Y, Xie W, et al. Transcriptome response to nitrogen starvation in rice. J Biosci. 2012;37:731–747. doi: 10.1007/s12038-012-9242-2. [DOI] [PubMed] [Google Scholar]
- Cai H, Chen H, Yi T, et al. VennPlex-A novel Venn diagram program for comparing and visualizing datasets with differentially regulated datapoints. PLoS ONE. 2013 doi: 10.1371/journal.pone.0053388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran AKN, Priatama RA, Kumar V, et al. Genome-wide transcriptome analysis of expression in rice seedling roots in response to supplemental nitrogen. J Plant Physiol. 2016;200:62–75. doi: 10.1016/j.jplph.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Chen Q, Liu Z, Wang B, et al. Transcriptome sequencing reveals the roles of transcription factors in modulating genotype by nitrogen interaction in maize. Plant Cell Rep. 2015;34:1761–1771. doi: 10.1007/s00299-015-1822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coneva V, Simopoulos C, Casaretto JA, et al. Metabolic and co-expression network-based analyses associated with nitrate response in rice. BMC Genomics. 2014;15:1–14. doi: 10.1186/1471-2164-15-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curci PL, Aiese Cigliano R, Zuluaga DL, et al. Transcriptomic response of durum wheat to nitrogen starvation. Sci Rep. 2017;7:1–14. doi: 10.1038/s41598-017-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo Junior AT, da Rosa Farias D. The quest for more tolerant rice: how high concentrations of iron affect alternative splicing? Transcr Open Access. 2015;03:3–7. doi: 10.4172/2329-8936.1000122. [DOI] [Google Scholar]
- Dobermann A, Fairhurst T (2000) Rice: Nutrient Disorders & Nutrient Management. Potash & Phosphate Institute (PPI), Potash & Phosphate Institute of Canada (PPIC) and International Rice Research Institute, Philippine
- FAO (2018) Food Security and Nutrition in the World.
- Gelli M, Duo Y, Konda AR, et al. Identification of differentially expressed transcripts between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genomics. 2014;15:179. doi: 10.1186/1471-2164-15-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PH, Kan CC, Wu HY, et al. Early molecular events associated with nitrogen deficiency in rice seedling roots. Sci Rep. 2018;8:1–23. doi: 10.1038/s41598-018-30632-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Sang Y, Sun W, et al. Transcriptomic analysis of responses to imbalanced carbon: nitrogen availabilities in rice seedlings. PLoS ONE. 2016;11:1–26. doi: 10.1371/journal.pone.0165732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, et al. Improvement of the oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:3–10. doi: 10.1186/1939-8433-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A, Berthome R, Mathilde O, et al. Arabidopsis roots and shoots show distinct temporal adaptation patterns toward nitrogen starvation. Plant Physiol. 2011;157:1255–1282. doi: 10.1104/pp.111.179838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladha JK, Pathak H, Krupnik TJ, et al. Efficiency of fertilizer nitrogen in cereal production: retrospects and prospects. Adv Agron. 2005;87:85–156. doi: 10.1016/S0065-2113(05)87003-8. [DOI] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. Bioinformatics. 2008 doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hu B, Chu C. Nitrogen use efficiency in crops: lessons from Arabidopsis and rice. J Exp Bot. 2017;68:2477–2488. doi: 10.1093/jxb/erx101. [DOI] [PubMed] [Google Scholar]
- Lian X, Wang S, Zhang J, et al. Expression profiles of 10,422 transcripts at early stage of low nitrogen stress in rice assayed using a cDNA microarray. Plant Mol Biol. 2006;60:617–631. doi: 10.1007/s11103-005-5441-7. [DOI] [PubMed] [Google Scholar]
- Naoumkina MA, Zhao Q, Gallego-Giraldo L, et al. Genome-wide analysis of phenylpropanoid defence pathways. Mol Plant Pathol. 2010;11:829–846. doi: 10.1111/j.1364-3703.2010.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros JC (2016) Venny 2.1.0. Venny. An Interactive Tool for Comparing Lists with Venn's Diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html
- Phule AS, Barbadikar KM, Madhav MS, et al. Transcripts encoding membrane proteins showed stable expression in rice under aerobic condition: novel set of reference transcripts for expression studies. 3 Biotech. 2018;8:383. doi: 10.1007/s13205-018-1406-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan X, Zeng J, Ye L, et al. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016;16:1–16. doi: 10.1186/s12870-016-0721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao IS, Neeraja CN, Srikanth B, et al. Identification of rice landraces with promising yield and the associated genomic regions under low nitrogen. Sci Rep. 2018;8:1–13. doi: 10.1038/s41598-018-27484-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard-Molard C, Krapp A, Francxois B, et al. Plant response to nitrate starvation is determined by N storage capacity matched by nitrate uptake capacity in two Arabidopsis genotypes. J Exp Botany. 2008;59:779–791. doi: 10.1093/jxb/erm363. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C T method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shin SY, Jeong JS, Lim JY, et al. Transcriptomic analyses of rice (Oryza sativa) transcripts and non-coding RNAs under nitrogen starvation using multiple omics technologies. BMC Genomics. 2018;19:1–20. doi: 10.1186/s12864-018-4897-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U, Ladha JK, Castillo EG, et al. Genotypic variation in nitrogen use ef ® ciency in medium- and long-duration rice. Field Crops Res. 1998;58:35–53. doi: 10.1016/S0378-4290(98)00084-7. [DOI] [Google Scholar]
- Sinha SK, Amitha Mithra SV, Chaudhary S, et al. Transcriptome analysis of two rice varieties contrasting for nitrogen use efficiency under chronic N starvation reveals differences in chloroplast and starch metabolism-related transcripts. Transcripts (Basel) 2018;9:1–22. doi: 10.3390/transcripts9040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D, Brown JWS. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell. 2013;25:3640–3656. doi: 10.1105/tpc.113.113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Di D, Li G, et al. Spatio-temporal dynamics in global rice gene expression (Oryza sativa L.) in response to high ammonium stress. J Plant Physiol. 2017;212:94–104. doi: 10.1016/j.jplph.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Surekha K, Satishkumar YS. Productivity, nutrient balance, soil quality, and sustainability of rice (Oryza sativa L.) under organic and conventional production systems. Commun Soil Sci Plant Anal. 2014;245:415–428. doi: 10.1080/00103624.2013.872250. [DOI] [Google Scholar]
- Takehisa H, Sato Y, Antonio BA, Nagamur Y. Global transcriptome profile of rice root in response to essential macronutrient deficiency. Plant Signal Behav. 2013 doi: 10.4161/psb.24409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder M, Masclaux-Daubresse C. Source and sink mechanisms of nitrogen transport and use. New Phytol. 2018;217:35–53. doi: 10.1111/nph.14876. [DOI] [PubMed] [Google Scholar]
- The AROF, Panel I, Climate ON (1995) IPCC Second Assessment Climate Change 1995.
- Thimm O, Bläsing O, Gibon Y, et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–939. doi: 10.1111/j.1365-313X.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Tirol-Padre A, Ladha JK, Singh U, et al. Grain yield performance of rice genotypes at suboptimal levels of soil N as affected by N uptake and utilization efficiency. Field Crops Res. 1996;46:127–143. doi: 10.1016/0378-4290(95)00095-x. [DOI] [Google Scholar]
- Vijayalakshmi P, Vishnukiran T, Ramana Kumari B, et al. Biochemical and physiological characterization for nitrogen use efficiency in aromatic rice genotypes. Field Crops Res. 2015;179:132–143. doi: 10.1016/j.fcr.2015.04.012. [DOI] [Google Scholar]
- Vinod KK, Heuer S. Approaches towards nitrogen- and phosphorus-efficient rice. AoB Plants. 2012 doi: 10.1093/aobpla/pls028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:1–14. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Lou Q, Xu K, et al. Alternative splicing complexity contributes to genetic improvement of drought resistance in the rice maintainer HuHan2B. Sci Rep. 2017 doi: 10.1038/s41598-017-12020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Fan X, Miller AJ. Plant nitrogen assimilation and use efficiency. Annu Rev Plant Biol. 2012;63:153–182. doi: 10.1146/annurev-arplant-042811-105532. [DOI] [PubMed] [Google Scholar]
- Yang SY, Hao DL, Song ZZ, et al. RNA-Seq analysis of differentially expressed transcripts in rice under varied nitrogen supplies. Gene. 2015;555:305–317. doi: 10.1016/j.gene.2014.11.021. [DOI] [PubMed] [Google Scholar]
- Yang W, Yoon J, Choi H, et al. Transcriptome analysis of nitrogen-starvation-responsive transcripts in rice. BMC Plant Biol. 2015;15:1–12. doi: 10.1186/s12870-015-0425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fang L, Zheng H, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S (1981) Fundamental of rice crop science. International Rice Research Institute, Los Baños, Laguna, Philippines, pp 269
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias GOseq GOseq is a method for GO analysis of RNA-seq data that takes into account the length bias inherent in RNA-seq. Genome Biol. 2010 doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SP, Zhao XQ, Shi WM. Genotype variation in grain yield response to basal N fertilizer supply among different rice cultivars. Afr J Biotechnol. 2012;11:12298–12304. [Google Scholar]
- Zhao X, Wang W, Xie Z, et al. ScienceDirect comparative analysis of metabolite changes in two contrasting rice genotypes in response to low- nitrogen stress. Crop J. 2018;6:464–474. doi: 10.1016/j.cj.2018.05.006. [DOI] [Google Scholar]
- Zhu GH, Zhuang CH, Wang YQ, et al. Differential expression of rice transcripts under different nitrogen forms and their relationship with sulfur metabolism. J Integr Plant Biol. 2006;48:1177–1184. doi: 10.1111/j.1744-7909.2006.00332.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.