

Abstract
O-Acetylated sialic acid has been found in the Neisseria meningitidis serogroup W (NmW) capsular polysaccharide (CPS) and is a required structural component of clinically used NmW CPS-based polysaccharide and polysaccharide-conjugate vaccines. The role of sialic acid O-acetylation in NmW CPS, however, is not clearly understood. This is partially due to the lack of a precise control of the percentage and the location of O-acetylation which is labile and susceptible to migration. We explore chemoenzymatic synthetic strategies of preparing N-acetylated analogs of O-acetylated NmW CPS oligosaccharides which can serve as structurally stable probe mimics. Substrate specificity studies of NmW CPS polymerase (NmSiaDW) identified 4-azido-4-deoxy-N-acetylmannosamine (ManNAc4N3) and 6-azido-6-deoxy-N-acetylmannosamine (ManNAc6N3) as suitable chemoenzymatic synthons for synthesizing N-acetyl analogs of NmW CPS oligosaccharides containing 7-O-acetyl-N-acetylneuraminic acid (Neu5,7Ac2) and/or 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2). The synthesis was achieved by NmSiaDW-dependent sequential one-pot multienzyme (OPME) strategy with in situ generation of the corresponding sugar nucleotides from simple monosaccharides or derivatives to form N3-oligosaccharides which were converted to the desired NAc-oligosaccharides by an efficient one-step chemical transformation.
Keywords: capsular polysaccharide, chemoenzymatic synthesis, glycosyltransferase, N-acetyl sialic acid, O-acetyl sialic acid
Graphical Abstract
INTRODUCTION
O-Acetylation is a common modification of carbohydrates1–2 including sialic acids which are a family of monosaccharides belonging to nine-carbon α-keto acids (so called nonulosonic acids).3–5 In addition to be an important structural component of animal glycomes, sialic acids have been found in numerous bacteria. Among more than 50 different sialic acid forms found in nature, N-acetylneuraminic acid (Neu5Ac) is the most common form. Neu5Ac in the capsular polysaccharides (CPSs) or the lipopolysaccharides (LPSs) of bacteria is often O-acetylated.2 For example, among six Neisseria meningitidis (Nm) serogroups (A, B, C, W, X, and Y) that cause life threatening invasive meningococcal diseases (IMDs) including meningitis and septicemia,6–8 four (except for serogroups A and X) have Neu5Ac in their CPSs and Neu5Ac O-acetylation has been observed for CPSs from three Nm serogroups (C, W, and Y).9 NmW strains have various levels of CPS O-acetylation.9–11 It was shown that O-acetylation of Neu5Ac in NmW CPS (a heteropolymer with a repeating unit of −6Galα1–4Neu5Acα2-) (Figure 1)12–14 may not be immunologically essential15–16 but can influence the efficacy of CPS periodate-treatment for conjugation with protein carriers in the development of conjugate vaccines.15 The World Health Organization (WHO) also specified the requirement of having minimal 0.3 mmol O-acetyl content per gram of polysaccharide in the NmW CPS polysaccharide vaccine.17
Figure 1.
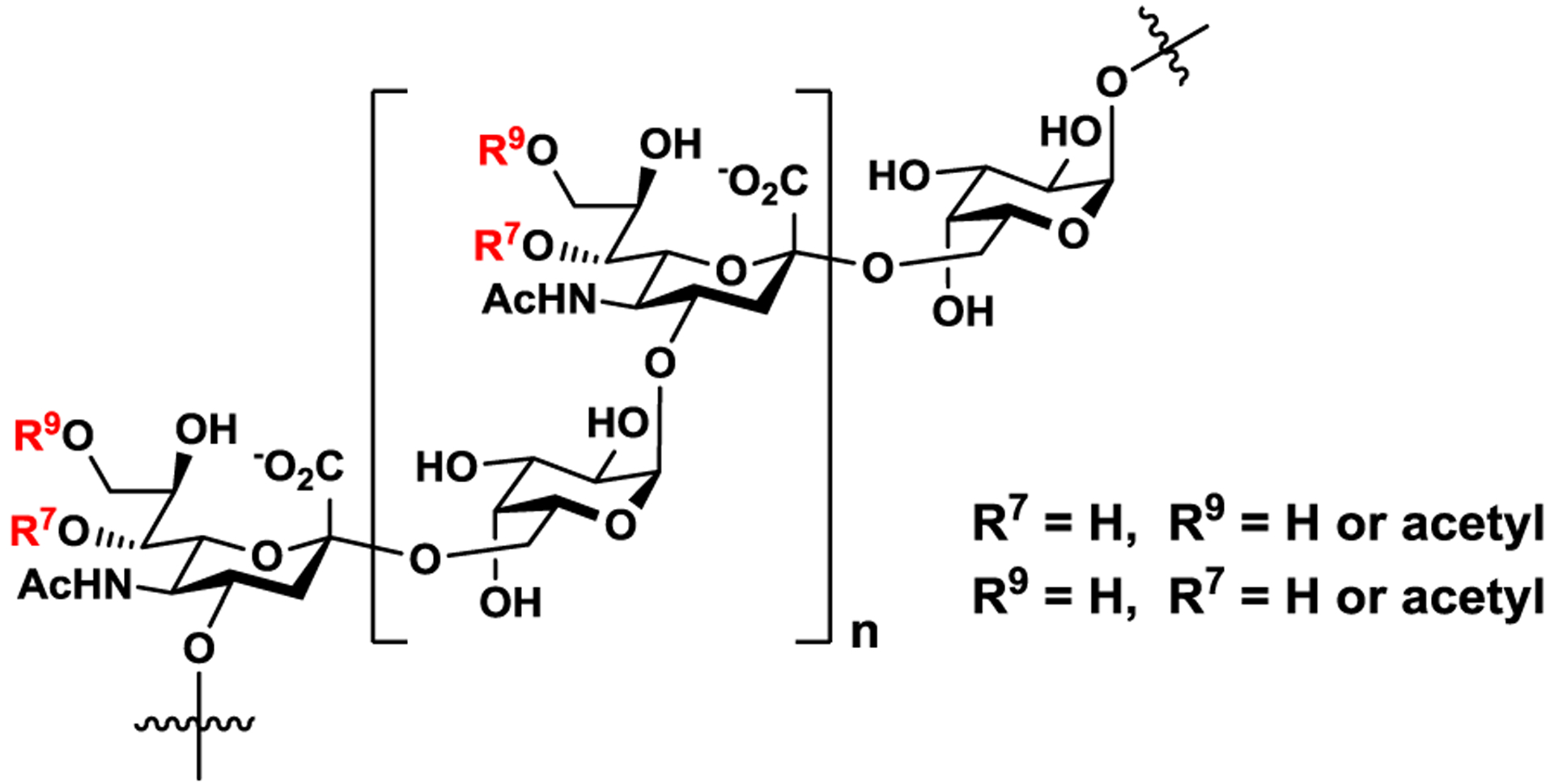
Structure of N. meningitidis serogroup W (NmW) capsular polysaccharide (CPS) containing O-acetylation at C7 or C9 of N-acetylneuraminic acid (Neu5Ac).
Nevertheless, the role of O-acetylation in NmW CPS remains unclear and the percentage of O-acetylation is suggested to be a parameter for vaccine quality control.2 The lack of the understanding is partially due to the low abundance of O-acetylation in the CPSs of some NmW strains,2 variation of O-acetylation levels due to manufacturing, instability of O-acetylation under basic conditions,10 and O-acetyl migration leading to the variation of site distribution of O-acetyl group.9
The detailed biosynthetic process for the formation of O-acetylated NmW CPS is also not clear despite the identification of the corresponding O-acetyltransferase encoded by oatWY18 and the report of its crystal structures in the presence or the absence of coenzyme A (CoA), acetyl-CoA and its non-hydrolyzable analog.19 For example, O-acetylation was observed at either C7 or C9 of Neu5Ac in NmW CPS while only one O-acetyltransferase gene oatWY was identified from NmW and NmY strains.18 The absence of a suitable acceptor in the crystal structures of NmOatWY19 and O-acetyl migration from C7 to C9 in Neu5Ac of NmW CPS observed by nuclear magnetic resonance (NMR) spectroscopy studies9 did not allow the confirmation of the specific O-acetylation site of the O-acetyltransferase NmOatWY. It was shown that NmOatWY was able to catalyze the transfer of acetyl from acetyl-CoA to NmY CPS polysaccharides and disaccharide (at a lower efficiency) but was unable to acetylate monosaccharide Neu5Ac or cytidine 5’-monophosphate-Neu5Ac (CMP-Neu5Ac).19 However, the detailed relationship of its reaction efficiency and the structures of its carbohydrate acceptor substrates is not clear. Therefore, it is impractical to obtain structurally defined NmW CPS with O-acetyl groups at designated locations by biosynthesis. Furthermore, the high-cost of acetyl-CoA, the required activated acetyl donor for acetyltransferases, prohibits large-scale enzymatic synthesis of O-acetylated NmW CPS or oligosaccharides by NmOatWY. On the other hand, chemical synthesis of O-acetylated NmW CPS oligosaccharides is possible but will be challenging. The O-acetyl migration, the sensitivity of O-acetyl group to pH variation and esterase-catalyzed cleavage20 will cause obstacles in accurately interpreting results even if pure products are used in biological studies including immunological evaluations of the vaccine candidates.
We showed previously that replacing the labile O-acetyl group in 9-O-acetylated sialosides by the more stable N-acetyl group21–22 was a promising strategy to overcome the challenges of O-acetyl cleavage and/or migration in investigating their functional roles. For example, 9-acetamido-9-deoxy-Neu5Ac (Neu5Ac9NAc) was designed as a stable mimic of 9-O-acetyl-Neu5Ac (Neu5,9Ac2).21 As determined by systematic NMR spectroscopic and molecular dynamics simulation studies, Neu5,9Ac2-containing GM3 ganglioside glycan and its Neu5Ac9NAc analog are similar on their overall secondary structures and conformations without substantial differences on the dihedral angles of their glycosidic bonds.22 Neu5Ac9NAc-containing sialosides were applied in protein-binding, cell feeding, and sialidase substrate specificity studies as good mimics of the corresponding Neu5,9Ac2-counterparts but with a higher stability.21, 23–24 Neu5Ac9NAc and 4-acetamido-4-deoxy-Neu5Ac (Neu5Ac4NAc), a stable mimic of 4-O-acetyl-Neu5Ac (Neu4,5Ac2), were also used in a protein crystallography study of viral hemagglutinin-esterases.25
Herein, we explore chemoenzymatic strategies for synthesizing structurally defined N-acetyl analogs of O-acetylated NmW CPS oligosaccharides using two one-pot multienzyme (OPME) glycosylation systems containing NmW CPS polysaccharide synthase NmSiaDW with in situ generation of uridine 5’-diphosphate-galactose (UDP-Gal) and CMP-Sia from the corresponding monosaccharides Gal and sialic acid or sialic acid precursor, respectively.26
RESULTS AND DISCUSSION
Donor substrate specificity studies of the α2–6-sialyltransferase activity of NmSiaDW.
NmSiaDW is a bifunctional polysaccharide synthase that has both α1–4-galactosyltransferase and α2–6-sialyltransferase activities for the formation of NmW CPS containing a disaccharide repeating unit of −6Galα1–4Neu5Acα2-. It was used successfully in a sequential OPME system for the synthesis of structurally defined NmW CPS oligosaccharides ranging from disaccharide to decasaccharide from a benzyloxycarbonyl (Cbz)-tagged sialylmonosaccharide 2-O-(N-benzyloxycarbonyl) aminopropyl α-N-acetylneuraminide (Neu5AcαProNHCbz).26 The hydrophobic UV-detectable Cbz-tag was shown to facilitate reaction progress monitoring, enzyme biochemical characterization, and product purification processes.26
Using previously synthesized galactosyldisaccharide Galα1–4Neu5AcαProNHCbz (G2)26 as the acceptor substrate, the donor substrate specificity study for the α2–6-sialyltransferase activity of NmSiaDW was investigated using a one-pot two-step reaction (Scheme 1) similar to that described previously.27 In step 1, CMP-sialic acid or its analog was generated in situ from a sialic acid, its analog, or its precursor in the presence of Neisseria meningitidis CMP-sialic acid synthetase (NmCSS)28–29 with or without Pasteurella multocida sialic acid aldolase (PmAldolase)30 and sodium pyruvate. The yields were determined by ultra-high performance liquid chromatography (UHPLC) and mass spectrometry (MS) analyses. In step 2, G2 and NmSiaDW were added and the reactions were carried out in a short time (10 min) with a lower concentration (10 μg/mL) of NmSiaDW (to compare the efficiency of different substrates) or a longer time (10 h) with a higher concentration (3.3 mg/mL) of NmSiaDW (to test the suitability of the compounds as potential substrates for synthesis).
Scheme 1.
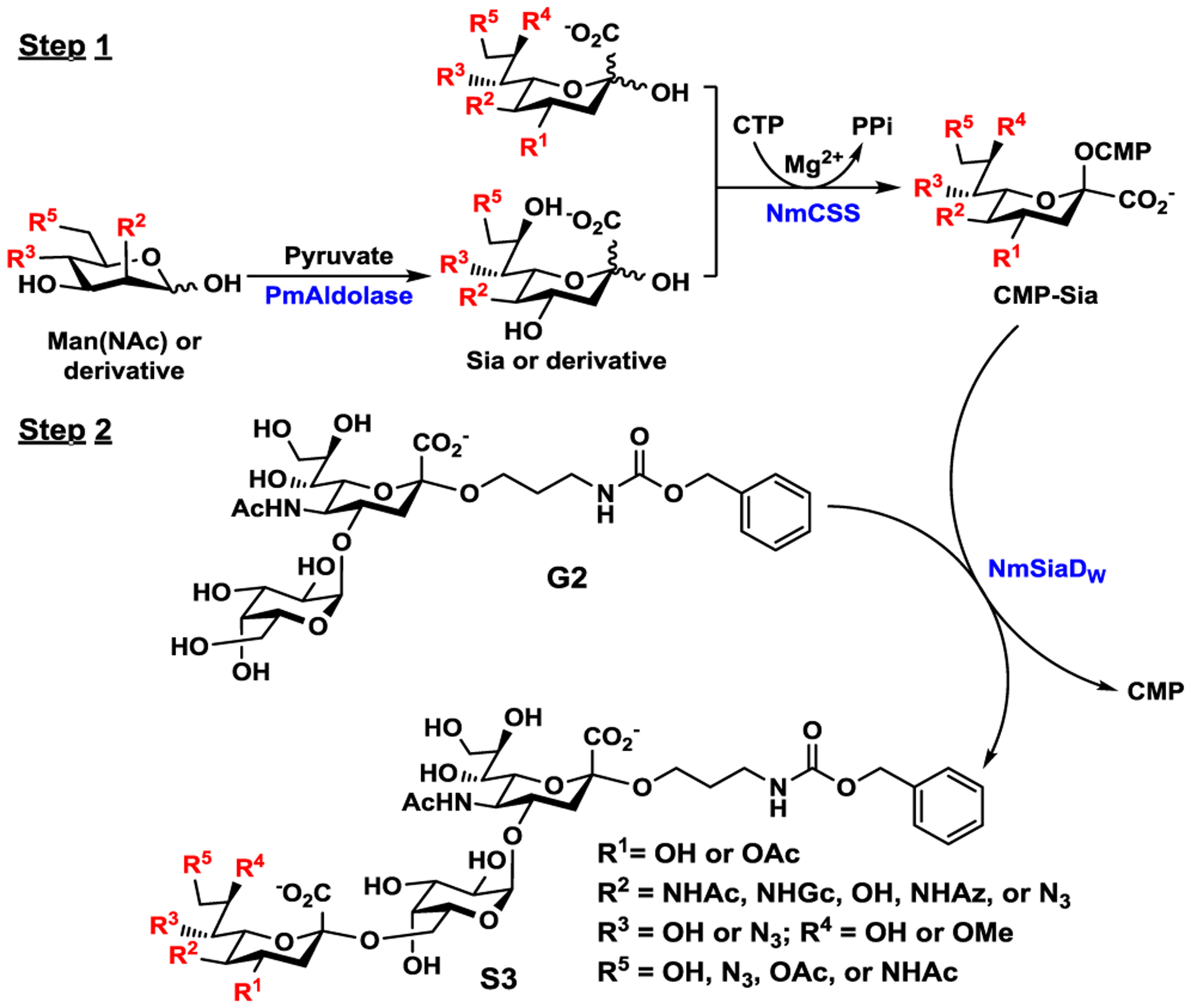
Schematic illustration of the reactions for one-pot two-step donor substrate specificity studies of the α2–6-sialyltransferase activity of NmSiaDW (in Step 2) using CMP-sialic acids and analogs generated in situ in Step 1.
As shown in Table 1, all donor precursors tested (entries 1–11, Table 1) (see Figure S1 for structures) could be catalyzed by NmCSS without (entries 1–6, Table 1) or with (entries 7–11, Table 1) PmAldolase and sodium pyruvate to produce CMP-sialic acid analogs. The α2–6-sialyltransferase activity of NmSiaDW was shown to be promiscuous towards donor substrate and derivatives. In addition to the N-acetyl group (e.g. in CMP-Neu5Ac, entry 1), N-glycolyl (e.g. in CMP-Neu5Gc, entry 2), N-azidoacetyl (e.g. in CMP-Neu5Az, entry 7), and OH (e.g. in CMP-Kdn, entry 6) at the C5 of sialic acids in CMP-sialic acids were all tolerated by NmSiaDW. A C5-N3 group substitution (e.g. in CMP-Neu5N3, entry 11) was also tolerated but to a lower extend. In addition, while an 4-O-acetyl (e.g. in CMP-Neu4,5Ac2, entry 4) or 9-O-acetyl (e.g. in CMP-Neu5,9Ac2, entry 5) was not tolerated by NmSiaDW, a 7-N3 (e.g. in CMP-Neu5Ac7N3, entry 8), 8-OMe (e.g. in CMP-Neu5Ac8OMe, entry 3), or 9-N3 (e.g. in CMP-Neu5Ac9N3, entry 9) substitution of Neu5Ac in CMP-Neu5Ac was tolerated. In comparison, 9-N-acetyl (e.g. in CMP-Neu5Ac9NAc, entry 10) was only weakly tolerated.
Table 1.
Results of donor substrate specificity studies for the α2–6-sialyltransferase activity of NmSiaDW using in situ generated CMP-sialic acids and analogs.
| Donor precursor | Percentage conversion (%) | |||
|---|---|---|---|---|
| CMP-Sialic acid | Sialyltransfer | |||
| 10 μg/mL, 10 min | 3.3 mg/mL, 10 h | |||
| 1 | Neu5Ac | aCMP-Neu5Ac (Quant.) | 30±0.3 | Quant. |
| 2 | Neu5Gc | aCMP-Neu5Gc (Quant.) | 32±2 | Quant. |
| 3 | Neu5Ac8OMe | aCMP-Neu5Ac8OMe (Quant.) | 0 | 83±7 |
| 4 | Neu4,5Ac2 | a,cCMP-Neu4,5Ac2 (Quant.) | 0 | 0 |
| 5 | Neu5,9Ac2 | a,cCMP-Neu5,9Ac2 (Quant.) | 0 | 0 |
| 6 | Kdn | aCMP-Kdn (Quant.) | 0 | 98±2 |
| 7 | ManNAz | bCMP-Neu5NAz (Quant.) | 28±2 | Quant. |
| 8 | ManNAc4N3 | bCMP-Neu5Ac7N3 (Quant.) | 0 | 78±3 |
| 9 | ManNAc6N3 | bCMP-Neu5Ac9N3 (Quant.) | 0 | 90±2 |
| 10 | ManNAc6NAc | bCMP-Neu5Ac9NAc (Quant.) | 0 | 18±1 |
| 11 | Man2N3 | bCMP-Neu5N3 (Quant.) | 0 | 64±1 |
Step 1 of the reaction was carried out with a monosaccharide (1–11, 1.2 equiv.) in the presence of NmCSS (1.5 mg/mL) and CTP (1 equiv.) for 4 h awithout (for 1–6) or bwith (for 7–11) PmAldolase (4 mg/mL) and sodium pyruvate (5 equiv.). Tris-HCl (pH 8.5) was used for all reactions except for entries 4 and 5 where cTris-HCl (pH 7.5) was used to minimize de-O-acetylation which would take place at pH 8.5.
The tolerance of CMP-Neu5Gc (entry 2), CMP-Neu5Ac8OMe (entry 3), CMP-Kdn (entry 6), CMP-Neu5Az (entry 7), CMP-Neu5Ac7N3 (entry 8), and CMP-Neu5Ac9N3 (entry 9) as donor substrates for the α2–6-sialyltransferase activity of NmSiaDW provides an opportunity for chemoenzymatic synthesis of derivatives of NmW CPS oligosaccharides. The corresponding azido-containing donor precursor analogs (ManNAz, entry 7), ManNAc4N3 (entry 8), and ManNAc6N3 (entry 9) can be further explored as potential probes for metabolic engineering of NmW CPS. The latter two (ManNAc4N331 and ManNAc6N332) can be used as chemoenzymatic synthons for synthesizing structurally defined N-acetyl analogs of 7-O- and/or 9-O-acetylated NmW CPS oligosaccharides by OPME reactions followed by conversion of the N3-groups in the oligosaccharide products to N-Ac groups. A strategy using a monosaccharide diazido derivative as a chemoenzymatic synthon has been successfully developed and applied previously by us for synthesizing glycans containing a terminal 5,7-di-N-acetyllegionaminic acid (Leg5,7diNAc, a bacterial nonulosonic acid).33 It has not, however, been tested in chemoenzymatic synthesis of bacterial polysaccharides containing internal sialic acid residues.
It is interesting to notice the lower degree of donor substrate promiscuity of the α2–6-sialyltransferase activity of NmSiaDW compared to the α2–3-sialyltransferase activity of Pasteurella multocida sialyltransferase 1 (PmST1)34 and α2–6-sialyltransferase activity of Photobacterium damselae α2–6-sialyltransferase (Pd2,6ST).35 Both PmST1 and Pd2,6ST were able to tolerate all24, 34–36 except one (CMP-Neu4,5Ac2, entry 4)37 in situ-generated CMP-Sia and analogs shown in Table 1. For example, in situ-generated CMP-Neu5,9Ac2 (entry 5)35–36 not tolerated by NmSiaDW and CMP-Neu5Ac9NAc (entry 10)24 that was only weakly accepted by NmSiaDW were both suitable donor substrates for PmST1 and Pd2,6ST.
Preparative-scale synthesis of sialyltrisaccharides containing different sialic acid forms.
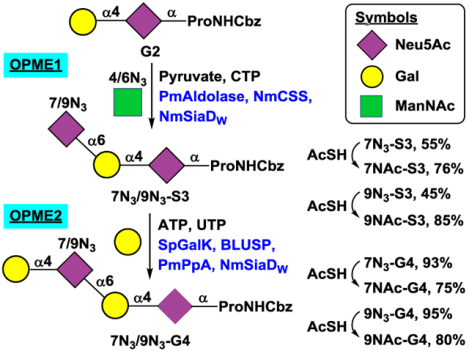
With a good understanding of the donor substrate specificity of the α2–6-sialyltransferase activity of NmSiaDW, preparative-scale chemoenzymatic synthesis of structurally defined N-acetyl analogs of O-acetyl NmW CPS oligosaccharides was carried out using previously synthesized Cbz-tagged NmW CPS galactosyldisaccharide Galα1–4Neu5AcαProNHCbz (G2)26 as the acceptor substrate and chemoenzymatic synthons ManNAc4N3 and ManNAc6N3 as donor precursors. As shown in Scheme 2, NmSiaDW was used together with PmAldolase30 and NmCSS28 in an OPME α2–6-sialylation system (OPME1) to sialylate G2 for the formation of sialyl trisaccharides containing Neu5Ac, Neu5Ac7N3, or Neu5Ac9N3. In this system, sodium pyruvate (excess amount, 5–10 equiv.) and ManNAc, ManNAc4N3, or ManNAc6N3 were used by PmAldolase to produce Neu5Ac or its monoazido-analog Neu5Ac7N3 or Neu5Ac9N3. NmCSS then used CTP to activate Neu5Ac or its derivative to form CMP-Neu5Ac or its analog which was used by the sialyltransferase activity of NmSiaDW to form α2–6-linked sialosides (Scheme 2). From 150 mg G2, trisaccharide S3 (220 mg, 97%), 7N3-S3 Neu5Ac7N3α2–6Galα1–4Neu5AcαProNHCbz (120 mg, 55%), or 9N3-S3 Neu5Ac9N3α2–6Galα1–4Neu5AcαProNHCbz (98 mg, 45%) was obtained in an excellent to a moderate yield.
Scheme 2.
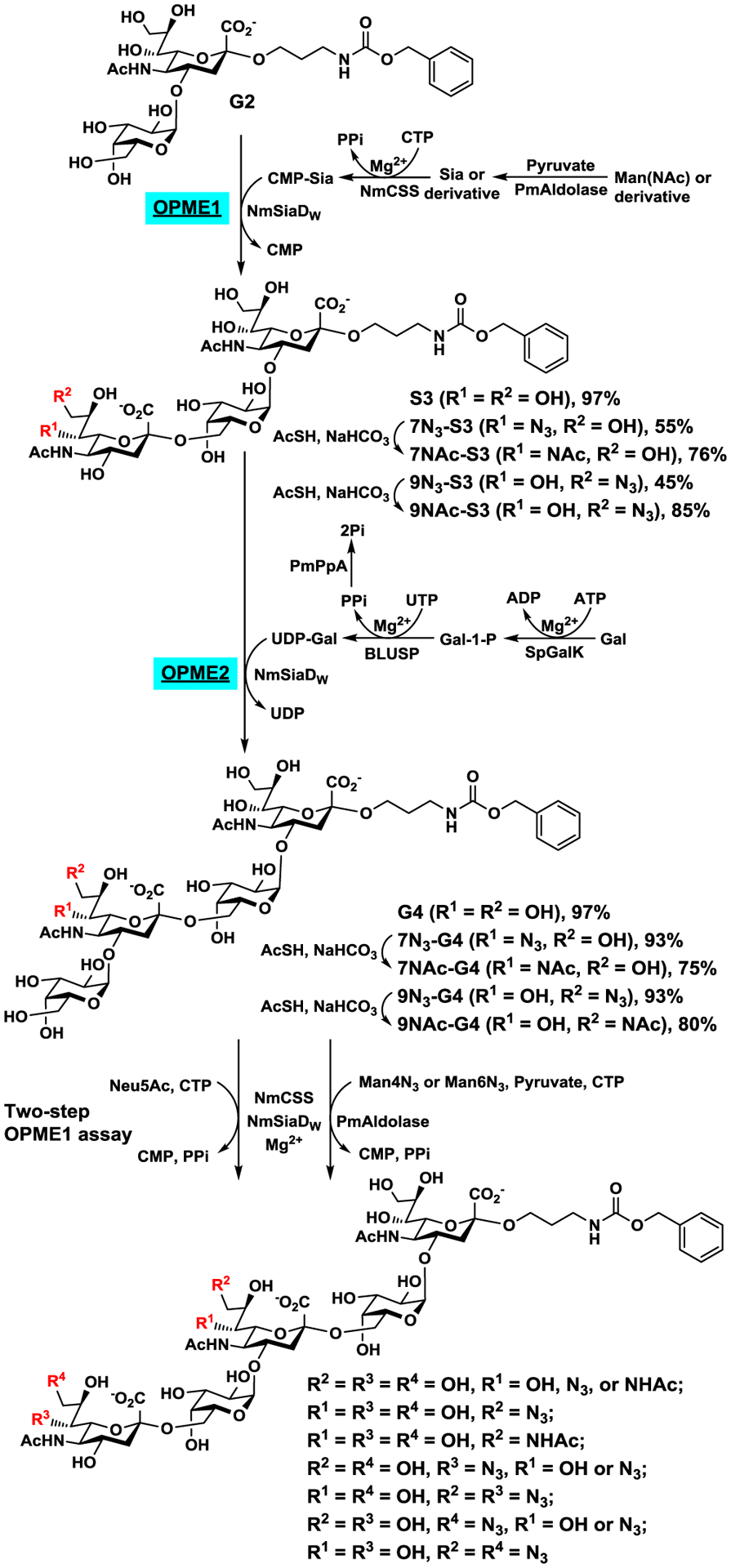
One-pot multienzyme (OPME) chemoenzymatic systems for the formation of NmW CPS trisaccharides, tetrasaccharides, pentasaccharides, and their azido or N-acetyl analogs.
The structure and the purity of the products were confirmed by nuclear magnetic resonance (NMR), high-resolution mass spectrometry (HRMS), and ultra-high performance liquid chromatography (UHPLC). For example, replacing the OH at C7 of the terminal Neu5Ac in S3 by an N3 in 7N3-S3 led to an upfield shift of the C7 13C signal from 68.24 ppm to 61.19 ppm. The influence on the C7 1H chemical shift was weaker, with 0.01 ppm difference from 3.57 ppm in S3 to 3.56 ppm in 7N3-S3 (Figure S2). For 9N3-S3, the 13C chemical shift of substituted Neu5Ac C9 moved upfield from 62.60 ppm in S3 to 52.97 ppm. Two diastereotopic protons on substituted C9 in 9N3-S3 appeared at 3.68 ppm and 3.50 ppm, respectively, both being upfield of the corresponding protons (3.86 ppm and 3.63 ppm) in S3 (Figure S3).
The azido group in trisaccharides 7N3-S3 and 9N3-S3 was readily converted to an N-acetyl group using a simple one-step reduction and simultaneous acetylation process by adding thioacetic acid in a saturated sodium bicarbonate solution33, 38 to obtain 7NAc-S3 Neu5Ac7NAcα2–6Galα1–4Neu5AcαProNHCbz (13 mg, 76%) and 9NAc-S3 Neu5Ac9NAcα2–6Galα1–4Neu5AcαProNHCbz (12 mg, 85%) in good yields.
Acceptor substrate specificity of the α1–4-galactosyltransferase activity of NmSiaDW.
Successful synthesis of longer N-acetyl analogs of O-acetylated NmW CPS oligosaccharides also depends on the acceptor substrate promiscuity of the α1–4-galactosyltransferase activity of NmSiaDW. Therefore, sialyltrisaccharides S3, 7N3-S3, 9N3-S3, 7NAc-S3, and 9NAc-S3 obtained above were tested as acceptor substrates for the α1–4-galactosyltransferase activity of NmSiaDW and uridine-5’-diphosphate galactose (UDP-Gal) was used as the donor substrate. As shown in Table 2, all sialyltrisaccharides except for 7NAc-S3 were suitable acceptors although the efficiency with 9NAc-S3 (68±3%) was lower than others (quantitative yields). These results suggest that 7N3-S3 and 9N3-S3, instead of 7NAc-S3 or 9NAc-S3, will be the preferred substrates for NmSiaDW-catalyzed enzymatic extension for the synthesis of longer N-acetyl NmW CPS oligosaccharide derivatives.
Table 2.
Results of the acceptor substrate specificity studies for the α1–4-galactosyltransferase activity of NmSiaDW using sialyltrisaccharides as potential acceptors and UDP-Gal as the donor.
| Substrates | Percentage conversion (%) | ||
|---|---|---|---|
| 20 μg/mL, 10 min | 1.2 mg/mL, 10 h | ||
| 1 | S3 | 22±1 | Quant. |
| 2 | 7N3-S3 | 11±1 | Quant. |
| 3 | 9N3-S3 | 0 | Quant. |
| 4 | 7NAc-S3 | 0 | 0 |
| 5 | 9NAc-S3 | 0 | 68±3 |
Preparative-scale synthesis of galactosyltetrasaccharides containing different sialic acid forms.
With the confirmation that both 7N3-S3 and 9N3-S3, in addition to S3, were suitable acceptor substrates for the α1–4-galactosyltransferase activity of NmSiaDW, preparative-scale synthesis of galactosyltetrasaccharides were carried out using an OPME α1–4-galactosylation system (OPME2) (Scheme 2) containing Streptococcus pneumoniae TIGR4 galactokinase (SpGalK),39 Bifidobacterium longum UDP-sugar pyrophosphorylase (BLUSP),40 Pasteurella multocida inorganic pyrophosphatase (PmPpA),41 and NmSiaDW. In this system, SpGalK was responsible for the formation of galactose-1-phosphate (Gal-1-P). It was then used by BLUSP for in situ formation of activated sugar nucleotide UDP-Gal, which was used by NmSiaDW to produce α1–4-linked galactosides. PmPpA was included to hydrolyze the inorganic pyrophosphate (PPi) formed in the BLUSP-catalyzed reaction to drive the reaction towards the formation of UDP-Gal.40 From 50 mg of S3 or its derivative 7N3-S3 or 9N3-S3, tetrasaccharide G4 (56 mg, 97%), 7N3-G4 Galα1–4Neu5Ac7N3α2–6Galα1–4Neu5AcαProNHCbz (54 mg, 93%), and 9N3-G4 Galα1–4Neu5Ac9N3α2–6Galα1–4Neu5AcαProNHCbz (55 mg, 95%) were formed in excellent yields.
Similar to that described above, the azido group in 7N3-G4 and 9N3-G4 was converted to N-acetyl group using thioacetic acid in a saturated sodium bicarbonate solution33, 38 to obtain 7NAc-G4 Galα1–4Neu5Ac7NAcα2–6Galα1–4Neu5AcαProNHCbz (20 mg, 75%) and 9NAc-G4 Galα1–4Neu5Ac9NAcα2–6Galα1–4Neu5AcαProNHCbz (21 mg, 80%), respectively, in good yields.
Donor and acceptor substrate specificity studies of the α2–6-sialyltransferase activity of NmSiaDW with galactosyltetrassacharide acceptors.
The obtained galactosyltetrasaccharides G4, 7N3-G4, 9N3-G4, 7NAc-G4, and 9NAc-G4 as well as donor precursors ManNAc4N3 and ManNAc6N3 allowed the exploration of the potential of NmSiaDW in synthesizing longer oligosaccharides with desired N-acetylation patterns. A one-pot two-step reaction process similar to that shown in Scheme 1 above for the donor substrate specificity studies of the sialyltransferase activity of NmSiaDW with G2 as the acceptor was used. As shown in Table 3, CMP-Neu5Ac, CMP-Neu5Ac7N3, and CMP-Neu5Ac9N3 were obtained readily in situ from the corresponding precursors Neu5Ac, ManNAc4N3, and ManNAc6N3, respectively, in a reaction catalyzed by NmCSS with (for ManNAc4N3 and ManNAc6N3) or without (for Neu5Ac) PmAldolase in step 1. The analysis of the NmSiaDW-catalyzed α2–6-sialyltransfer process in step 2 showed that when Neu5Ac was used as the donor precursor, all galactosyltetrasaccharides tested were well tolerated by NmSiaDW to form sialylpentasaccharide products (see Scheme 2 for structures) in high yields (83–97%, entries 1–5 in Table 3). Nevertheless, differentiation of the efficiency of the acceptors was observed when the reactions were carried out with a lower concentration of NmSiaDW in a shorter period of time. The N3-substitution at the C7 or C9 of the Neu5Ac in the acceptor was better tolerated than the corresponding N-acetyl-substitution.
Table 3.
Substrate specificity study for the α2–6-sialyltransferase activity of NmSiaDW using galactosyltetrasaccharides as well as Neu5Ac, ManNAc4N3, and ManNAc6N3.
| Acceptor | Percentage conversion (%) | |||
|---|---|---|---|---|
| CMP-Sialic acid | Transferase reaction | |||
| 10 μg/mL, 10 min | 3.3 mg/mL, 10 h | |||
| 1 | G4 | aCMP-Neu5Ac (Quant.) | 35±0.1 | 97±1 |
| 2 | 7N3-G4 | 13±1 | 83±1 | |
| 3 | 9N3-G4 | 14±0.2 | 89±3 | |
| 4 | 7NAc-G4 | 0 | 85±2 | |
| 5 | 9NAc-G4 | 0 | 97±1 | |
| 6 | G4 | bCMP-Neu5Ac7N3 (Quant.) | 0 | 90±1 |
| 7 | 7N3-G4 | 0 | 82±2 | |
| 8 | 9N3-G4 | 0 | 36±1 | |
| 9 | 7NAc-G4 | 0 | 0 | |
| 10 | 9NAc-G4 | 0 | 0 | |
| 11 | G4 | bCMP-Neu5Ac9N3 (Quant.) | 0 | 97±1 |
| 12 | 7N3-G4 | 0 | 83±2 | |
| 13 | 9N3-G4 | 0 | 31±1 | |
| 14 | 7NAc-G4 | 0 | 0 | |
| 15 | 9NAc-G4 | 0 | 0 | |
Step 1 of the reaction was carried out with aNeu5Ac, bManNAc4N3, or bManNAc6N3 (1.2 equiv.) in the presence of NmCSS (1.5 mg/mL), CTP (1 equiv.) and Tris-HCl (pH 8.5) awithout or bwith PmAldolase (4 mg/mL) and sodium pyruvate (5 equiv.).
When ManNAc4N3 (entries 6–10 in Table 3) or ManNAc6N3 (entries 11–15 in Table 3) was used as the donor precursor for the α2–6-sialyltransferase activity of NmSiaDW, G4, 7N3-G4, and 9N3-G4 were suitable acceptor substrates but 7NAc-G4 and 9NAc-G4 were not. Therefore, the data support the potential of chemoenzymatic synthesis of N-acetyl analogs of longer NmW CPS oligosaccharides with defined N-acetylation pattern although optimization is needed to further improve the yields of the reactions when 9N3-G4 was used as the acceptor substrate and ManNAc4N3 (entry 8 in Table 3) or ManNAc6N3 (entry 13 in Table 3) was used as the donor precursor.
The observation that 7NAc or 9NAc-analogs of NmW CPS oligosaccharides are poor or unsuitable substates for either the galactosyltransferase or the sialyltransferase activity of NmSiaDW indicates that NmW CPS polysaccharide is most likely formed before NmOatWY-catalyzed O-acetylation. This is consistent with the previous results from biochemical characterization of NmOatWY.19
CONCLUSIONS
In conclusion, the donor and acceptor substrate promiscuity of NmSiaDW was demonstrated. ManNAc4N3 and ManNAc6N3, the six-carbon precursors for Neu5Ac7N3 and Neu5Ac9N3, were confirmed to be suitable chemoenzymatic synthons in NmSiaDW-dependent sequential one-pot multienzyme (OPME) chemoenzymatic synthesis of structurally defined stable N-acetyl analogs of O-acetylated NmW CPS oligosaccharides. The obtained oligosaccharide products are important probes for binding studies of antibodies against NmW CPS and for investigating the functions of its O-acetylation. They can also be used to investigate host-microbe interactions. The tolerance of an azido group substitution at the C7 or C9 of Neu5Ac in the in situ-generated CMP-Neu5Ac donor derivatives by the α2–6-sialyltransferase activity of NmSiaDW and the tolerance of the resulting oligosaccharides as acceptors for the α1–4-galactosyltransferase activity of NmSiaDW showed the potential of using ManNAc4N3 and ManNAc6N3 or their per-acetylated analogs for metabolic engineering studies.42 The chemoenzymatic synthon strategy can be extended for the synthesis of the N-acetyl analogs of other oligosaccharides containing O-acetyl sialic acids.
EXPERIMENTAL SECTION
Materials and General Methods.
Chemicals were obtained from commercial suppliers and used without further purification. 1H NMR, 13C NMR, HSQC, and HSQC-TOCSY (90 ms and 10 ms) spectra were recorded on an 800 MHz Bruker Avance III spectrometer in the NMR facility of the University of California, Davis. Structural assignments were made with additional information from HSQC and HSQC-TOCSY experiments. High-resolution electrospray ionization (ESI) mass spectra were obtained using a Thermo Electron LTQ-Orbitrap Hybrid mass spectrometer at the mass spectrometry facility in the University of California, Davis or an LTQ-Orbitrap Eilte mass spectrometer at the Georgia State University. Matrix-assisted laser desorption/ionization (MALDI) mass spectra were obtained using Bruker UltraFlextreme MALDI-TOF at the mass spectrometry facility in the University of California, Davis. UHPLC assays were performed using Agilent 1290 Infinity LC with an EclipsePlus C18 (Rapid Resolution HD, 1.8 μm, 2.1×50 mm, 959757–902), an AdvanceBio Glycan Map (1.8 μm, 2.1×150 mm, 859700–913) column from Agilent Technologies or an Dionex CarboPac PA100 (8.5 μm, 4×250 mm, 043055) column from Thermo Scientific. Reverse phase chromatography purification of products was performed with a C18 column (ODS-SM, 50 mm, 120 Å, 3.0×20 cm) from Yamazen Corporation on a CombiFlash Rf 200i system. Galactose was from Fisher Scientific, Inc. N-Acetylneuraminic acid (Neu5Ac) was from Inalco (Italy). Adenosine 5’-triphosphate (ATP), cytosine 5’-triphosphate (CTP), and uridine 5’-triphosphate (UTP) were purchased from Hangzhou Meiya Pharmaceutical Co. Ltd. Recombinant enzymes Pasteurella multocida sialic acid aldolase (PmAldolase),30 Neisseria meningitidis CMP-sialic acid synthetase (NmCSS),28 Streptococcus pneumoniae TIGR4 galactokinase (SpGalK),39 Bifidobacterium longum UDP sugar pyrophosphorylase (BLUSP),40 Pasteurella multocida inorganic pyrophosphatase (PmPpA),41 Neisseria meningitidis serogroup W capsular polysaccharide polymerase (NmSiaDW)26 were expressed and purified as reported previously. ManNAc4N3,31 ManNAc6N3,32 and Galα1–4Neu5AcαProNHCbz (G2)26 were synthesized as described previously.
Two-step donor substrate specificity study for the α2–6-sialyltransferase activity of NmSiaDW using disaccharide G2 as an acceptor.
In the first step of the assay, CMP-sialic acid or its analog was synthesized from sialic acid, sialic acid analog, or its precursor. If Neu5Ac or its analog was used as a starting material, the step 1 reactions were performed in duplicate at 30 °C for 4 h in a total volume of 20 μL in a buffer (Tris-HCl, 100 mM, pH 8.5; pH 7.5 for reactions with Neu4,5Ac2 or Neu5,9Ac2) containing MgCl2 (10 mM), sialic acid or its analog (6 mM), CTP (5 mM), and NmCSS (15 μg). If a sialic acid precursor (6 mM) was used as a starting material, sodium pyruvate (25 mM) and PmAldolase (40 μg) were included in the reaction mixture. Step 2 sialylation reactions were performed in duplicate at 30 °C for 10 min (with 0.10 μg of NmSiaDW) or 10 h (with 33 μg of NmSiaDW) in a total volume of 10 μL in a buffer (Tris-HCl, 100 mM, pH 8.5; pH 7.5 for reactions with Neu4,5Ac2 or Neu5,9Ac2) containing MgCl2 (10 mM), a reaction mixture from step 1 (2.5 μL), acceptor G2 (1 mM), and NmSiaDW. Reactions were quenched by adding 10 μL of pre-chilled ethanol followed by incubation of the mixture at −20 °C for 30 min. Samples were analyzed using UHPLC with an EcilpsePlusC18 column or an AdvancBio Glycan Map column (Agilent), as well as by Bruker UltraFlextreme MALDI-TOF in a negative mode. Sialic acids and derivatives tested were Neu5Ac, Neu5Gc, Neu5Ac8OMe,43 Neu4,5Ac2,37 Neu5,9Ac2,44 and Kdn. Sialic acid precursors and derivatives tested were ManNAz,34 ManNAc4N3,31 ManNAc6N3,32 ManNAc6NAc,21 and Man2N3.35
Acceptor substrate specificity study for the α1–4-galactosyltransferase activity of NmSiaDW using sialyltrisaccharide S3 and analogs obtained.
Assays were performed in duplicate at 30 °C for 10 min (with 0.20 μg of NmSiaDW) or 10 h (with 12 μg of NmSiaDW) in a total volume of 10 μL in a buffer (MES, 100 mM, pH 6.5) containing MgCl2 (10 mM), UDP-Gal (2 mM), a sialyltrisaccharide S3 or analog (7N3-S3, 9N3-S3, 7NAc-S3, or 9NAc-S3) (1 mM), and NmSiaDW. Reactions were quenched by adding 10 μL of pre-chilled ethanol followed by incubation of the mixture at −20 °C for 30 min. Samples were analyzed using UHPLC with an EcilpsePlusC18 column (Agilent) and by Bruker UltraFlextreme MALDI-TOF in a negative mode.
Donor and acceptor substrate specificity studies of the α2–6-sialyltransferase activity of NmSiaDW using G4 or its analogs.
In the first step of the assay, CMP-sialic acid or its analog was synthesized in duplicate at 30 °C for 4 h in a total volume of 10 μL in a buffer (Tris-HCl, 100 mM, pH 8.5) containing a sialic acid or precursor (Neu5Ac, ManNAc4N3, or ManNAc6N3) (6 mM), CTP (5 mM), sodium pyruvate (25 mM), MgCl2 (10 mM), NmCSS (15 μg), and PmAldolase (40 μg). Step 2 sialylation reactions were performed in duplicate at 30 °C for 10 min (with 0.10 μg of NmSiaDW) or 10 h (with 33 μg of NmSiaDW) in a total volume of 10 μL in a buffer (Tris-HCl, 100 mM, pH 8.5) containing a reaction mixture from step 1 (2.5 μL), a galactosyltetrasaccharide G4 or analog (7N3-G4, 9N3-G4, 7NAc-G4, or 9NAc-G4) (1 mM), MgCl2 (10 mM), and NmSiaDW. Reactions were quenched by adding 10 μL of pre-chilled ethanol followed by incubation of the mixture at −20 °C for 30 min. Samples were analyzed using UHPLC with a Dionex CarboPac PA100 column (Thermo Scientific) and by Bruker UltraFlextreme MALDI-TOF in a negative mode.
OPME synthesis of Neu5Ac7N3α2–6Galα1–4Neu5AcαProNHCbz (7N3-S3).
A reaction mixture in a total volume of 10 mL containing Tris-HCl buffer (100 mM, pH 8.5), galactosyldisaccharide G2 (150 mg, 0.22 mmol), ManNAc4N3 (75 mg, 0.30 mmol), sodium pyruvate (253 mg, 2.3 mmol), CTP disodium salt (218 mg, 0.41 mmol), MgCl2 (20 mM), PmAldolase (20 mg), NmCSS (7 mg) and NmSiaDW (4 mg) was incubated in a 50 mL centrifuge tube in a shaker (100 rpm) at 30 °C for 2 days. The reaction progress was monitored by UHPLC (EclipsePlus C18, Agilent, 5–12% Acetonitrile + 0.1% TFA in water over 7 min, monitored at 215 nm). When an optimal yield was achieved, pre-chilled ethanol (10 mL) was added and the resulting mixture was incubated at 4 °C for 30 min. The precipitates were removed by centrifugation (4300 g) at 4 °C for 30 min. The supernatant was concentrated and purified by a C18 column using a CombiFlash Rf 200i system with a gradient (0–100% acetonitrile) of water with 0.1% TFA (v/v) and acetonitrile for elution. Fractions containing the product were collected, neutralized, concentrated, and further purified by a C18 column to produce 7N3-S3 as a sodium salt (120 mg, 55%).
1H NMR (800 MHz, D2O) δ 7.47–7.38 (m, 5H, Ar-H), 5.11 (s, 2H, O-CH2-Ar), 5.04 (d, J = 3.9 Hz, 1H, H”−1), 4.03 (t, J = 10.2 Hz, 1H, H’−5), 4.00–3.74 (m, 12H), 3.74–3.59 (m, 7H), 3.56 (dd, J = 9.2, 2.0 Hz, 1H, H”’−7), 3.50 (dt, J = 9.7, 6.2 Hz, 1H, O-CH2-CH2), 3.24–3.16 (m, 2H, CH2-NH), 2.87 (dd, J = 12.5, 4.7 Hz, 1H, H’−3eq), 2.72 (dd, J = 12.5, 4.7 Hz, 1H, H”’−3eq), 2.07 (s, 3H, H’-CH3-CO), 2.04 (s, 3H, H”’-CH3-CO), 1.78–1.69 (m, 3H, O-CH2-CH2-CH2-NH; H”’−3ax), 1.61 (t, J = 12.0 Hz, 1H, H’−3ax). 13C{1H} NMR (200 MHz, D2O) δ 174.53, 174.37, 173.34, 173.16, 158.37 (NH-COO), 136.56 (O-CH2-Ar), 128.75 (Ar), 128.27 (Ar), 127.56 (Ar), 100.58 (C’−2), 100.51 (C”’−2), 94.87 (C”−1), 72.89, 72.15, 71.85, 71.61, 70.97, 69.33, 69.17, 68.76, 68.37, 68.07, 67.83, 66.75 (O-CH2-Ar), 62.58, 62.54, 62.40, 62.06 (O-CH2-CH2), 61.23 (C”’−7), 52.51 (C”’−5), 49.54 (C’−5), 39.88 (C”’−3), 37.47 (CH2-NH), 36.78 (C’−3), 28.89 (O-CH2-CH2-CH2-NH), 22.43 (C’-CH3-CO), 22.11 (C”’-CH3-CO). HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C39H57N6O23 977.3475; found 977.3476.
Chemical synthesis of Neu5Ac7NAcα2–6Galα1–4Neu5AcαProNHCbz (7NAc-S3).
7N3-S3 (17 mg) was added to a round bottom flask (50 mL) containing saturated sodium bicarbonate aqueous solution (2 mL), thioacetic acid (200 μL) was then added drop-wisely. The reaction was heated in an oil bath under argon at 65 °C for 30 h. After completion of the reaction, the solvent was removed, and the compound was purified by silica gel chromatography using a mixed solvent of ethyl acetate:methanol (6:1 by volume) as an eluent and then by a C18 column in a CombiFlash Rf 200i system using CH3CN in H2O gradient as the elution solvent. 7NAc-S3 was obtained as a white solid (13 mg, 76% yield). 1H NMR (800 MHz, D2O) δ 7.46–7.39 (m, 5H), 5.11 (s, 2H), 5.09 (d, J = 3.9 Hz, 1H), 4.05 (t, J = 10.2 Hz, 1H), 3.98–3.92 (m, 3H), 3.89–3.84 (m, 5H), 3.82–3.76 (m, 3H), 3.74–3.68 (m, 3H), 3.66–3.60 (m, 3H), 3.56 (ddd, J = 9.9, 8.4, 4.5 Hz, 2H), 3.52–3.46 (m, 2H), 3.20 (h, J = 7.3 Hz, 2H), 2.91 (dd, J = 12.6, 4.7 Hz, 1H), 2.74 (dd, J = 12.4, 4.6 Hz, 1H), 2.05 (s, 3H), 1.99 (s, 3H), 1.94 (s, 3H), 1.75 (dd, J = 14.8, 9.0 Hz, 3H), 1.62 (t, J = 12.0 Hz, 1H).13C{1H} NMR (200 MHz, D2O) δ 174.4, 173.9, 173.8, 173.3, 158.4, 136.6, 128.8, 128.3, 127.6, 100.6, 100.2, 94.2, 72.4, 72.1, 71.8, 71.6, 71.6, 69.6, 69.2, 69.2, 68.6, 68.0, 67.9, 66.8, 63.8, 62.5, 62.4, 62.1, 52.0, 49.5, 49.2, 40.2, 37.5, 36.4, 28.9, 22.3, 22.1, 21.9. HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C41H61N4O24 993.3681; found 993.3696.
OPME synthesis of Neu5Ac9N3α2–6Galα1–4Neu5AcαProNHCbz (9N3-S3).
A reaction mixture in a total volume of 10 mL containing Tris-HCl buffer (100 mM, pH 8.5) galactosyldisaccharide G2 (150 mg, 0.22 mmol), ManNAc6N3 (75 mg, 0.30 mmol), sodium pyruvate (203 mg, 1.85 mmol), CTP disodium salt (194 mg, 0.37 mmol), MgCl2 (20 mM), PmAldolase (15 mg), NmCSS (5 mg), and NmSiaDW (4 mg) was incubated in a 50 mL centrifuge tube in a shaker (100 rpm) at 30 °C for 2 days. Procedures for reaction progress monitoring, centrifugation, concentration, purification, collection, and neutralization were similar to that described above for 7N3-S3. 9N3-S3 was obtained as a sodium salt (98 mg, 45%). 1H NMR (800 MHz, D2O) δ 7.46–7.37 (m, 5H, Ar-H), 5.11 (s, 2H, O-CH2-Ar), 5.05 (d, J = 3.9 Hz, 1H, H”−1), 4.06–3.99 (m, 2H), 3.95 (d, J = 3.8 Hz, 1H, H”−3), 3.89–3.74 (m, 8H), 3.73–3.60 (m, 8H), 3.58 (dd, J = 9.1, 1.8 Hz, 1H, H”’−7), 3.53–3.48 (m, 2H, O-CH2-CH2; H”’−9), 3.24–3.16 (m, 2H, CH2-NH), 2.88 (dd, J = 12.5, 4.8 Hz, 1H, H’−3eq), 2.72 (dd, J = 12.5, 4.7 Hz, 1H, H”’−3eq), 2.07 (s, 3H, H’-CH3-CO), 2.03 (s, 3H, H”’-CH3-CO), 1.75 (p, J = 6.6 Hz, 2H, O-CH2-CH2-CH2-NH), 1.70 (t, J = 12.2 Hz, 1H, H”’−3ax), 1.61 (t, J = 12.0 Hz, 1H, H’−3ax). 13C{1H} NMR (200 MHz, D2O) δ 174.93, 174.50, 173.44, 173.30, 158.37 (NH-COO), 136.56 (O-CH2-Ar), 128.75 (Ar), 128.27 (Ar), 127.56 (Ar), 100.58 (C’−2), 100.23 (C”’−2), 94.68 (C”−1), 72.80, 72.28, 72.11, 71.84, 70.23, 69.51, 69.17, 68.96, 68.87, 68.34, 68.03, 67.83, 66.75 (O-CH2-Ar), 62.87, 62.51, 62.05 (O-CH2-CH2), 53.03 (C”’−9), 51.81 (C”’−5), 49.54 (C’−5), 40.04 (C”’−3), 37.47 (CH2-NH), 36.71 (C’−3), 28.89 (O-CH2-CH2-CH2-NH), 22.39 (C’-CH3-CO), 22.01 (C”’-CH3-CO). HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C39H57N6O23 977.3475; found 977.3479.
Chemical synthesis of Neu5Ac9NAcα2–6Galα1–4Neu5AcαProNHCbz (9NAc-S3).
9N3-S3 (14 mg) was added to a round bottom flask (50 mL) containing saturated sodium bicarbonate aqueous solution (2 mL), thioacetic acid (200 μL) was then added drop-wisely. Reaction conditions and procedures for concentration and purification were similar to that described above for 7NAc-S3. 9NAc-S3 was obtained as a white solid (12 mg, 85%). 1H NMR (800 MHz, D2O) δ 7.46–7.38 (m, 5H), 5.11 (s, 2H), 5.06 (d, J = 3.9 Hz, 1H), 4.03 (t, J = 10.2 Hz, 1H), 3.95 (d, J = 3.4 Hz, 1H), 3.90 (ddd, J = 9.0, 7.9, 2.9 Hz, 1H), 3.84 (dddd, J = 26.4, 12.7, 7.7, 3.6 Hz, 6H), 3.80–3.75 (m, 2H), 3.71–3.60 (m, 7H), 3.55 (dd, J = 14.0, 2.9 Hz, 1H), 3.52–3.49 (m, 1H), 3.47 (dd, J = 9.0, 1.8 Hz, 1H), 3.31 (dd, J = 14.1, 7.9 Hz, 1H), 3.24 – 3.16 (m, 2H), 2.88 (dd, J = 12.5, 4.7 Hz, 1H), 2.72 (dd, J = 12.4, 4.7 Hz, 1H), 2.07 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.75 (p, J = 6.6 Hz, 2H), 1.70 (t, J = 12.2 Hz, 1H), 1.62 (t, J = 12.0 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.9, 174.5, 174.4, 173.5, 158.4, 136.1, 128.8, 128.3, 127.6, 100.3, 94.7, 72.8, 72.3, 72.1, 71.8, 70.0, 69.8, 69.5, 69.2, 68.9, 68.3, 68.0, 67.8, 66.4, 62.8, 62.5, 62.1, 51.8, 49.1, 42.1, 40.0, 37.5, 36.7, 28.9, 22.4, 22.0, 21.8. HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C41H61N4O24 993.3681; found 993.3691.
OPME synthesis of Galα1–4Neu5Ac7N3α2–6Galα1–4Neu5AcαProNHCbz (7N3-G4).
A reaction mixture in a total volume of 10 mL containing Tris-HCl buffer (100 mM, pH 8.5), sialyltrisacchairde 7N3-S3 (50 mg, 0.05 mmol), galactose (12 mg, 0.07 mmol), ATP disodium salt (38 mg, 0.07 mmol), UTP trisodium salt (38 mg, 0.07 mmol), MgCl2 (20 mM), SpGalK (1 mg), BLUSP (1 mg), PmPpA (1 mg), and NmSiaDW (0.5 mg) was incubated in a 50 mL centrifuge tube in a shaker (100 rpm) at 30 °C for 16 h. Procedures for reaction progress monitoring, centrifugation, concentration, purification, collection and neutralization were similar to that described above for 7N3-S3. 7N3-G4 was obtained as a sodium salt (54 mg, 93%). 1H NMR (800 MHz, D2O) δ 7.47–7.38 (m, 5H, Ar-H), 5.11 (s, 2H, O-CH2-Ar), 5.08 (d, J = 4.0 Hz, 1H, H””−1), 5.05 (d, J = 3.9 Hz, 1H, H”−1), 4.07 (t, J = 10.2 Hz, 1H, H”’−5), 4.03 (t, J = 10.3 Hz, 1H, H’−5), 4.00–3.90 (m, 5H), 3.89–3.59 (m, 20H), 3.50 (dt, J = 9.7, 6.1 Hz, 1H, O-CH2-CH2), 3.25–3.15 (m, 2H, CH2-NH), 2.90–2.85 (dd, J = 12.8, 4.8 Hz, 2H, H’−3eq; H”’−3eq), 2.08 (s, 3H, H’-CH3-CO), 2.05 (s, 3H, H”’-CH3-CO), 1.75 (p, J = 6.7 Hz, 2H, O-CH2-CH2-CH2-NH), 1.68 (t, J = 12.1 Hz, 1H, H”’−3ax), 1.61 (t, J = 12.0 Hz, 1H, H’−3ax). 13C{1H} NMR (200 MHz, D2O) δ 174.51, 174.07, 173.37, 172.81, 158.37 (NH-COO), 136.57 (O-CH2-Ar), 128.76 (Ar), 128.27 (Ar), 127.56 (Ar), 100.58 (C’−2), 100.54 (C”’−2), 95.19 (C””−1), 94.62 (C”−1), 73.06, 72.56, 72.16, 71.81, 71.43, 71.06, 70.79, 69.36, 69.22, 69.17, 69.03, 68.80, 67.99, 67.85, 67.75, 66.75 (O-CH2-Ar), 62.77, 62.48, 62.40, 62.06 (O-CH2-CH2), 61.24 (C”’−7), 60.70, 50.34 (C”’−5), 49.55 (C’−5), 37.48 (CH2-NH), 36.78 (C”’−3), 36.66 (C’−3), 28.90 (O-CH2-CH2-CH2-NH), 22.43 (C’-CH3-CO), 22.15 (C”’-CH3-CO). HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C45H67N6O28 1139.4003; found 1139.4007.
Chemical synthesis of Galα1–4Neu5Ac7NAcα2–6Galα1–4Neu5AcαProNHCbz (7NAc-G4).
To a round bottom flask containing 7N3-G4 (26 mg), saturated sodium bicarbonate solution (3 mL) was added, 300 μL of thioacetic acid was then added drop-wisely. Reaction conditions and procedures for concentration and purification were similar to that described above for 7NAc-S3. 7NAc-G4 was obtained as a white solid (20 mg, 75%). 1H NMR (800 MHz, D2O) δ 7.51–7.34 (m, 5H), 5.11 (s, 2H), 5.08 (d, J = 3.9 Hz, 1H), 5.07 (d, J = 4.0 Hz, 1H), 4.07–3.99 (m, 2H), 3.96–3.85 (m, 7H), 3.85–3.76 (m, 6H), 3.76–3.60 (m, 11H), 3.55–3.46 (m, 2H), 3.28–3.16 (m, 2H), 2.90 (ddd, J = 12.0, 6.9, 4.7 Hz, 2H), 2.06 (s, 3H), 1.99 (s, 3H), 1.95 (s, 3H), 1.75 (p, J = 6.8 Hz, 2H), 1.68 (t, J = 12.0 Hz, 1H), 1.62 (t, J = 12.0 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.4, 173.8, 173.7, 173.3, 172.9, 158.4, 136.6, 128.8, 128.3, 127.6, 100.4, 94.9, 94.3, 72.8, 72.5, 72.1, 71.8, 71.4, 71.4, 71.0, 69.6, 69.3, 69.2, 69.2, 69.0, 68.0, 67.9, 67.9, 66.8, 63.9, 62.5, 62.4, 62.1, 60.7, 49.8, 49.6, 49.3, 37.5, 37.0, 36.5, 28.9, 22.3, 22.1, 21.8. HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C47H71N4O29 1155.4209; found 1155.4214.
OPME synthesis of Galα1–4Neu5Ac9N3α2–6Galα1–4Neu5AcαProNHCbz (9N3-G4).
A reaction mixture in a total volume of 10 mL containing Tris-HCl buffer (100 mM, pH 8.5), sialyltrisacchairde 9N3-S3 (50 mg, 0.05 mmol), galactose (12 mg, 0.07 mmol), ATP disodium salt (38 mg, 0.07 mmol), UTP trisodium salt (38 mg, 0.07 mmol), MgCl2 (20 mM), SpGalK (1 mg), BLUSP (1 mg), PmPpA (1 mg), and NmSiaDW (2 mg) was incubated in a 50 mL centrifuge tube in a shaker (100 rpm) at 30 °C for 2 days. Procedures for reaction progress monitoring, centrifugation, concentration, purification, collection and neutralization were similar to that described above for 7N3-S3. 9N3-G4 was obtained as a sodium salt (55 mg, 95%). 1H NMR (800 MHz, D2O) δ 7.46–7.37 (m, 5H, Ar-H), 5.10 (d, J = 4.1 Hz, 3H, O-CH2-Ar; H””−1), 5.06 (d, J = 3.9 Hz, 1H, H”−1), 4.08 (td, J = 10.3, 5.5 Hz, 2H, H’−5; H”’−5), 4.01 (ddd, J = 8.9, 6.0, 2.7 Hz, 1H), 3.98–3.53 (m, 24H), 3.49 (dd, J = 13.2, 6.0 Hz, 1H, H”’−9), 3.22–3.18 (m, 2H, CH2-NH), 2.88–2.82 (m, 2H,, H’−3eq; H”’−3eq), 2.06 (s, 3H, H’-CH3-CO), 2.03 (s, 3H, H”’-CH3-CO), 1.78–1.69 (m, 4H, O-CH2-CH2-CH2-NH; H”’−3ax; H’−3ax). 13C{1H} NMR (200 MHz, D2O) δ 174.47, 174.33, 171.77, 171.37, 158.35 (NH-COO), 136.54 (O-CH2-Ar), 128.76 (Ar), 128.29 (Ar), 127.57 (Ar), 99.42 (C’−2), 99.23 (C”’−2), 94.72 (C””−1), 94.27 (C”−1), 72.38, 72.26, 72.15, 71.61, 71.05, 70.96, 69.77, 69.54, 69.32, 69.25, 69.06, 69.02, 68.67, 68.01, 67.88, 67.78, 66.77 (O-CH2-Ar), 63.29, 62.80, 62.06 (O-CH2-CH2), 60.72, 53.43 (C”’−9), 49.36 (C”’−5), 49.31 (C’−5),, 37.37 (CH2-NH), 36.01 (C”’−3) 35.56 (C’−3), 28.77 (O-CH2-CH2-CH2-NH), 22.30 (C’-CH3-CO), 22.11 (C”’-CH3-CO). HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C45H67N6O28 1139.4003; found 1139.4021.
Chemical synthesis of Galα1–4Neu5Ac9NAcα2–6Galα1–4Neu5AcαProNHCbz (9NAc-G4).
To a round bottom flask containing 9N3-G4 (26 mg), saturated sodium bicarbonate solution (3 mL) was added, and then 300 μL of thioacetic acid was added drop-wisely. Reaction conditions and procedures for concentration and purification were similar to that described above for 7NAc-S3. 9NAc-G4 was obtained as a white solid (21 mg, 80%). 1H NMR (800 MHz, D2O) δ 7.47–7.37 (m, 5H), 5.11 (s, 2H), 5.08 (d, J = 4.0 Hz, 1H), 5.06 (d, J = 3.9 Hz, 1H), 4.03 (td, J = 10.2, 4.5 Hz, 2H), 3.96 (ddd, J = 13.9, 3.4, 1.2 Hz, 2H), 3.92 (ddd, J = 9.0, 8.0, 2.9 Hz, 1H), 3.87–3.75 (m, 10H), 3.74–3.67 (m, 7H), 3.67–3.61 (m, 2H), 3.57 (dd, J = 14.1, 2.9 Hz, 1H), 3.50 (dt, J = 12.5, 4.4 Hz, 2H), 3.30 (dd, J = 14.1, 8.0 Hz, 1H), 3.20 (q, J = 6.8 Hz, 2H), 2.88 (ddd, J = 17.3, 12.5, 4.6 Hz, 2H), 2.07 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.75 (p, J = 6.6 Hz, 2H), 1.66 (t, J = 12.0 Hz, 1H), 1.61 (t, J = 12.0 Hz, 1H). 13C{1H} NMR (200 MHz, D2O) δ 174.5, 174.5, 174.3, 173.4, 173.1, 158.4, 136.6, 128.8, 128.3, 127.6, 100.6, 100.3, 95.0, 94.5, 73.2, 72.6, 72.1, 72.1, 71.8, 71.0, 70.1, 69.7, 69.5, 69.2, 69.0, 69.0, 68.0, 67.9, 67.8, 66.8, 63.0, 62.5, 62.1, 60.7, 49.6, 49.5, 42.1, 37.5, 36.7, 36.6, 28.9, 22.4, 22.1, 21.8. HRMS (ESI-Orbitrap) m/z: [M - H]− Calcd for C47H71N4O29 1155.4209; found 1155.4223.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the United States National Institutes of Health (NIH) Grant No. R01AI130684 and NIH Common Fund for Glycoscience Program Grant No. U01GM125288. The Bruker Avance-800 NMR spectrometer was funded by the United States National Science Foundation (Grant No. DBI-0722538). The authors would like to thank Dr. Lei Li and Dr. Ding Liu at Georgia State University for their help in obtaining HRMS data for some of the products.
Footnotes
Supporting Information
Structures of CMP-sialic acids and precursors; HSQC spectra comparison of S3 and 7N3-S3 or 9N3-S3; 1H, 13C, HSQC and HSQC-TOCSY NMR spectra of the products. The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Pauly M; Ramirez V New Insights Into Wall Polysaccharide O-Acetylation. Front. Plant Sci 2018, 9, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Berti F; De Ricco R; Rappuoli R Role of O-Acetylation in the Immunogenicity of Bacterial Polysaccharide Vaccines. Molecules 2018, 23, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Traving C; Schauer R Structure, Function and Metabolism of Sialic Acids. Cell. Mol. Life Sci 1998, 54, 1330–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Angata T; Varki A Chemical Diversity in The Sialic Acids and Related Alpha-Keto Acids: An Evolutionary Perspective. Chem. Rev 2002, 102, 439–469. [DOI] [PubMed] [Google Scholar]
- (5).Chen X; Varki A Advances in The Biology and Chemistry of Sialic Acids. ACS Chem. Biol 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Acevedo R; Bai X; Borrow R; Caugant DA; Carlos J; Ceyhan M; Christensen H; Climent Y; De Wals P; Dinleyici EC; Echaniz-Aviles G; Hakawi A; Kamiya H; Karachaliou A; Lucidarme J; Meiring S; Mironov K; Safadi MAP; Shao Z; Smith V; Steffen R; Stenmark B; Taha MK; Trotter C; Vazquez JA; Zhu B The Global Meningococcal Initiative Meeting on Prevention of Meningococcal Disease Worldwide: Epidemiology, Surveillance, Hypervirulent Strains, Antibiotic Resistance and High-risk Populations. Expert. Rev. Vaccines 2019, 18, 15–30. [DOI] [PubMed] [Google Scholar]
- (7).Batista RS; Gomes AP; Dutra Gazineo JL; Balbino Miguel PS; Santana LA; Oliveira L; Geller M Meningococcal Disease, A Clinical and Epidemiological Review. Asian Pac. J. Trop. Med 2017, 10, 1019–1029. [DOI] [PubMed] [Google Scholar]
- (8).Rouphael NG; Stephens DS Neisseria meningitidis: Biology, Microbiology, and Epidemiology. Methods Mol. Biol 2012, 799, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lemercinier X; Jones C Full 1H NMR Assignment and Detailed O-Acetylation Patterns of Capsular Polysaccharides from Neisseria meningitidis Used in Vaccine Production. Carbohydr. Res 1996, 296, 83–96. [DOI] [PubMed] [Google Scholar]
- (10).Jones C; Lemercinier X Use and Validation of NMR Assays for The Identity and O-Acetyl Content of Capsular Polysaccharides from Neisseria meningitidis Used in Vaccine Manufacture. J. Pharm. Biomed. Anal 2002, 30, 1233–1247. [DOI] [PubMed] [Google Scholar]
- (11).Longworth E; Fernsten P; Mininni TL; Vogel U; Claus H; Gray S; Kaczmarski E; Borrow R O-Acetylation Status of The Capsular Polysaccharides of Serogroup Y and W135 Meningococci Isolated in The UK. FEMS Immunol. Med. Microbiol 2002, 32, 119–123. [DOI] [PubMed] [Google Scholar]
- (12).Bhattacharjee AK; Jennings HJ; Kenny CP; Martin A; Smith IC Structural Determination of The Polysaccharide Antigens of Neisseria meningitidis Serogroups Y, W-135, and BO1. Can. J. Biochem 1976, 54, 1–8. [DOI] [PubMed] [Google Scholar]
- (13).Romanow A; Haselhorst T; Stummeyer K; Claus H; Bethe A; Muhlenhoff M; Vogel U; von Itzstein M; Gerardy-Schahn R Biochemical and Biophysical Characterization of The Sialyl-/Hexosyltransferase Synthesizing The Meningococcal Serogroup W135 Heteropolysaccharide Capsule. J. Biol. Chem 2013, 288, 11718–11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Claus H; Stummeyer K; Batzilla J; Muhlenhoff M; Vogel U Amino acid 310 Determines The Donor Substrate Specificity of Serogroup W-135 and Y Capsule Polymerases of Neisseria meningitidis. Mol. Microbiol 2009, 71, 960–971. [DOI] [PubMed] [Google Scholar]
- (15).Gudlavalleti SK; Lee CH; Norris SE; Paul-Satyaseela M; Vann WF; Frasch CE Comparison of Neisseria meningitidis Serogroup W135 Polysaccharide-Tetanus Toxoid Conjugate Vaccines Made by Periodate Activation of O-Acetylated, Non-O-Acetylated and Chemically De-O-Acetylated Polysaccharide. Vaccine 2007, 25, 7972–7980. [DOI] [PubMed] [Google Scholar]
- (16).Jin Z; Bohach GA; Shiloach J; Norris SE; Freedberg DI; Deobald C; Coxon B; Robbins JB; Schneerson R Conjugates of Group A and W135 Capsular Polysaccharides of Neisseria meningitidis Bound to Recombinant Staphylococcus aureus Enterotoxin C1: Preparation, Physicochemical Characterization, and Immunological Properties in Mice. Infect. Immun 2005, 73, 7887–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).World Health Organization, Annex 6. Requirements for Meningococcal Polysaccharide Vaccine Requiremetns for Biological Substances No. 23 1980, Addendum; 1980. [Google Scholar]
- (18).Claus H; Borrow R; Achtman M; Morelli G; Kantelberg C; Longworth E; Frosch M; Vogel U Genetics of Capsule O-acetylation in Serogroup C, W-135 and Y Meningococci. Mol. Microbiol 2004, 51, 227–239. [DOI] [PubMed] [Google Scholar]
- (19).Lee HJ; Rakic B; Gilbert M; Wakarchuk WW; Withers SG; Strynadka NC Structural and Kinetic Characterizations of The Polysialic Acid O-Acetyltransferase OatWY from Neisseria meningitidis. J. Biol. Chem 2009, 284, 24501–24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Georg Herrler; Rudolf Rott; Hans-Dieter Klenk; HansPeter Muller; Shukla Ashok K.; Schauer R. The Receptor-Destroying Enzyme of Influenza C Virus Is Neuraminate-O-Acetylesterase. EMBO J. 1985, 4, 1503–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Khedri Z; Xiao A; Yu H; Landig CS; Li W; Diaz S; Wasik BR; Parrish CR; Wang LP; Varki A; Chen X A Chemical Biology Solution to Problems with Studying Biologically Important but Unstable 9-O-Acetyl Sialic Acids. ACS Chem. Biol 2017, 12, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li W; Battistel MD; Reeves H; Oh L; Yu H; Chen X; Wang LP; Freedberg DI A combined NMR, MD and DFT Conformational Analysis of 9-O-Acetyl Sialic Acid-Containing GM3 Ganglioside Glycan and Its 9-N-Acetyl Mimic. Glycobiology 2020, 30, 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Georg Herrler; Hans-Jiirgen Gross; Angela Imhof; Reinhard Brossmer; Glenda Milks; Paulson JC A Synthetic Sialic Acid Analogue Is Recognized by Influenza C Virus as A Receptor Determinant But Is Resistant to the Receptor-Destroying Enzyme. J. Biol. Chem 1992, 267, 12501–12505. [PubMed] [Google Scholar]
- (24).Li W; Xiao A; Li Y; Yu H; Chen X Chemoenzymatic Synthesis of Neu5Ac9NAc-Containing Alpha2–3- and Alpha2–6-Linked Sialosides and Their Use for Sialidase Substrate Specificity Studies. Carbohydr. Res 2017, 451, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bakkers MJ; Zeng Q; Feitsma LJ; Hulswit RJ; Li Z; Westerbeke A; van Kuppeveld FJ; Boons GJ; Langereis MA; Huizinga EG; de Groot RJ Coronavirus Receptor Switch Explained from The Stereochemistry of Protein-Carbohydrate Interactions and A Single Mutation. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E3111–E3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li R; Yu H; Muthana SM; Freedberg DI; Chen X Size-Controlled Chemoenzymatic Synthesis of Homogeneous Oligosaccharides of Neisseria meningitidis W Capsular Polysaccharide. ACS Catal. 2020, 10, 2791–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cheng J; Yu H; Lau K; Huang S; Chokhawala HA; Li Y; Tiwari VK; Chen X Multifunctionality of Campylobacter jejuni Sialyltransferase CstII: Characterization of GD3/GT3 Oligosaccharide Synthase, GD3 Oligosaccharide Sialidase, and Trans-Sialidase Activities. Glycobiology 2008, 18, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu H; Yu H; Karpel R; Chen X Chemoenzymatic Synthesis of CMP-Sialic Acid Derivatives by a One-Pot Two-Enzyme System: Comparison of Substrate Flexibility of Three Microbial CMP-Sialic Acid Synthetases. Bioorg. Med. Chem 2004, 12, 6427–6435. [DOI] [PubMed] [Google Scholar]
- (29).Li Y; Yu H; Cao H; Muthana S; Chen X Pasteurella multocida CMP-Sialic Acid Synthetase and Mutants of Neisseria meningitidis CMP-Sialic Acid Synthetase with Improved Substrate Promiscuity. Appl. Microbiol. Biotechnol 2012, 93, 2411–2423. [DOI] [PubMed] [Google Scholar]
- (30).Li Y; Yu H; Cao H; Lau K; Muthana S; Tiwari VK; Son B; Chen X Pasteurella multocida Sialic Acid Aldolase: A Promising Biocatalyst. Appl. Microbiol. Biotechnol 2008, 79, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Khedri Z; Li Y; Muthana S; Muthana MM; Hsiao CW; Yu H; Chen X Chemoenzymatic Synthesis of Sialosides Containing C7-Modified Sialic Acids and Their Application in Sialidase Substrate Specificity Studies. Carbohydr. Res 2014, 389, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yu Hai; Chokhawala Harshal; Karpel Rebekah; Yu Hui; Wu Bingyuan; Zhang Jianbo; Zhang Yingxin; Jia Qiang; Chen X A Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for The Synthesis of Sialoside Libraries. J. Am. Chem. Soc 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- (33).Santra A; Xiao A; Yu H; Li W; Li Y; Ngo L; McArthur JB; Chen X A Diazido Mannose Analogue as A Chemoenzymatic Synthon for Synthesizing Di-N-Acetyllegionaminic Acid-Containing Glycosides. Angew. Chem. Int. Ed. Engl 2018, 57, 2929–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yu H; Chokhawala H; Karpel R; Yu H; Wu B; Zhang J; Zhang Y; Jia Q; Chen X A Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for The Synthesis of Sialoside Libraries. J. Am. Chem. Soc 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- (35).Yu H; Huang S; Chokhawala H; Sun M; Zheng H; Chen X Highly Efficient Chemoenzymatic Synthesis of Naturally Occurring and Non-Natural Alpha-2,6-Linked Sialosides: A P. damsela Alpha-2,6-Sialyltransferase with Extremely Flexible Donor-Substrate Specificity. Angew. Chem. Int. Ed. Engl 2006, 45, 3938–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chokhawala HA; Yu H; Chen X High-Throughput Substrate Specificity Studies of Sialidases by Using Chemoenzymatically Synthesized Sialoside Libraries. Chembiochem 2007, 8, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yu H; Zeng J; Li Y; Thon V; Shi B; Chen X, Effective One-Pot Multienzyme (OPME) Synthesis of Monotreme Milk Oligosaccharides And Other Sialosides Containing 4-O-Acetyl Sialic Acid. Org Biomol Chem 2016, 14, 8586–8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shangguan Ning; Katukojvala Sreenivas; Greenberg Rachel; Williams LJ The Reaction of Thio Acids with Azides: A New Mechanism and New Synthetic Applications. J. Am. Chem. Soc 2003, 125, 7754–7755. [DOI] [PubMed] [Google Scholar]
- (39).Chen M; Chen LL; Zou Y; Xue M; Liang M; Jin L; Guan WY; Shen J; Wang W; Wang L; Liu J; Wang PG Wide Sugar Substrate Specificity of Galactokinase from Streptococcus pneumoniae TIGR4. Carbohydr. Res 2011, 346, 2421–2425. [DOI] [PubMed] [Google Scholar]
- (40).Muthana MM; Qu J; Li Y; Zhang L; Yu H; Ding L; Malekan H; Chen X Efficient One-Pot Multienzyme Synthesis of UDP-sugars Using A Promiscuous UDP-sugar Pyrophosphorylase from Bifidobacterium longum (BLUSP). Chem. Commun 2012, 48, 2728–2730. [DOI] [PubMed] [Google Scholar]
- (41).Lau K; Thon V; Yu H; Ding L; Chen Y; Muthana MM; Wong D; Huang R; Chen X Highly Efficient Chemoenzymatic Synthesis of Beta1–4-Linked Galactosides with Promiscuous Bacterial Beta1–4-galactosyltransferases. Chem. Commun 2010, 46, 6066–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Mahal LK; Charter NW; Angata K; Fukuda M; Koshland DE Jr.; Bertozzi CR A Small-Molecule Modulator of Poly-alpha 2,8-Sialic Acid Expression on Cultured Neurons And Tumor Cells. Science 2001, 294, 380–381. [DOI] [PubMed] [Google Scholar]
- (43).Yu H; Cao H; Tiwari VK; Li Y; Chen X, Chemoenzymatic Synthesis of C8-Modified Sialic Acids And Related alpha2–3- And alpha2–6-linked Sialosides. Bioorg. Med. Chem. Lett 2011, 21, 5037–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Yu H; Cheng J; Ding L; Khedri Z; Chen Y; Chin S; Lau K; Tiwari VK; Chen X, Chemoenzymatic Synthesis of GD3 Oligosaccharides And Other Disialyl Glycans Containing Natural and Non-natural Sialic Acids. J. Am. Chem. Soc 2009, 131, 18467–18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.