

Abstract
Elevated body temperature (Tcore) is associated with poor outcome after subarachnoid hemorrhage (SAH). Brain temperature (Tbrain) is usually higher than Tcore. However, the implication of this difference (Tdelta) remains unclear. We aimed to study factors associated with higher Tdelta and its association with outcome. We included 46 SAH patients undergoing multimodal neuromonitoring, for a total of 7879 h of averaged data of Tcore, Tbrain, cerebral blood flow, cerebral perfusion pressure, intracranial pressure and cerebral metabolism (CMD). Three-months good functional outcome was defined as modified Rankin Scale ≤2. Tbrain was tightly correlated with Tcore (r = 0.948, p < 0.01), and was higher in 73.7% of neuromonitoring time (Tdelta +0.18°C, IQR −0.01 – 0.37°C). A higher Tdelta was associated with better metabolic state, indicated by lower CMD-glutamate (p = 0.003) and CMD-lactate (p < 0.001), and lower risk of mitochondrial dysfunction (MD) (OR = 0.2, p < 0.001). During MD, Tdelta was significantly lower (0°C, IQR −0.2 – 0.1; p < 0.001). A higher Tdelta was associated with improved outcome (OR = 7.7, p = 0.002). Our study suggests that Tbrain is associated with brain metabolic activity and exceeds Tcore when mitochondrial function is preserved. Further studies are needed to understand how Tdelta may serve as a surrogate marker for brain function and predict clinical course and outcome after SAH.
Keywords: Subarachnoid haemorrhage, outcome studies, microdialysis, neurocritical care, clinical practice
Introduction
Fever is a common complication after acute brain injury affecting up to 40% of patients suffering from spontaneous subarachnoid haemorrhage (SAH) already within the first 48 h. Early development of fever is commonly referred to as non-infectious fever, whereas the majority of SAH patients develop fever during hospitalization (up to 72%) which represents both neurogenic and infectious fever.1,2 Recent data suggest that fever is associated with more complications during hospitalization, including a higher rate of delayed cerebral ischemia (DCI), leading to poor long-term functional outcome after SAH.3–5
Although guideline recommendations are not very precise on targeting normothermia in neurocritical care patients, most intensivists have adapted a protocol for aggressive fever management in their intensive care units (ICUs) which is supported by expert recommendations.1,6,7 The targeted temperature is thoroughly referenced on the body core temperature (Tcore), although it is well known that brain temperature (Tbrain) may differ by up to 2.5°C after brain injury.8–15 Expert clinical scientists recommended that direct measurement of Tbrain should be performed in neurocritical care patients, as none of the body temperature sites can adequately serve as a surrogate for Tbrain.16 Little is known on how Tbrain is regulated after acute brain injury in humans. Previous animal and human studies suggested that Tbrain is usually higher but strongly correlated with Tcore and is linked to metabolic activity and changes in cerebral blood flow.8,14,17–19 Furthermore, a temperature gradient may exist between different brain regions, which may even be higher in patients with focal brain injury.20
Measurement of brain temperature is difficult as non-invasive techniques using, e.g. neuro-imaging are not readily available for standard clinical use in severely brain injured patients.21--24 Measurement of Tbrain by implanted catheters is limited by its local measurement and invasiveness, which restricts the use to poor grade comatose patients. However, multimodal neuromonitoring with invasive techniques can be used to further elucidate on mechanisms of Tbrain regulation by studying metabolic activity (using cerebral microdialysis, CMD) and cerebral blood flow (CBF) in the vicinity to the brain tissue where Tbrain is measured. Recently, we could show that Tbrain increases in timely association with occurrence of cortical spreading depolarizations (SD), which resemble highly metabolic active processes and contribute to secondary brain injury and poor outcome.25
In the current study, we aimed to examine mechanisms of Tbrain regulation in poor-grade SAH patients using systemic (Tcore) and local information (CBF, intracranial pressure (ICP), metabolism) derived from advanced neuromonitoring techniques. Our hypothesis was that a higher Tdelta is associated with preserved brain metabolic activity which relates to a higher chance of better outcome after SAH.
Materials and methods
Patients and ethical approval
This is a prospective cohort study with retrospective data analysis including 46 consecutive aneurysmal SAH patients in whom multimodal neuromonitoring was implanted. In all patients, an aneurysm could be identified. All patients were admitted to the neurological intensive care unit (NICU) at the Medical University of Innsbruck in Austria, between 2010 and 2016. The conduct of this study was approved by the ethics committee of the Medical University of Innsbruck (AN3898 285/4.8, AM4091-292/4.6, UN3898 285/4.8) and informed consent was obtained from all patients according to Austrian law and in accordance with the Declaration of Helsinki. Inclusion criteria were (1) admission with aneurysmal SAH, (2) ≥18 years of age, and (3) invasive neuromonitoring comprising at least continuous contemporaneous measurements of brain and core temperature.
Data collection and neuromonitoring
Patient characteristics, interventions, complications and outcome were prospectively recorded in our institutional SAH database. Invasive multimodal neuromonitoring was instituted, according to the local institutional protocol, to patients who presented with: (1) Initial poor grade (H&H 4-5) SAH or neuro-worsening to poor clinical grade within 24 h; (2) estimated prolonged need for mechanical ventilation (>48 h) (based on clinical and neuroimaging findings); (3) and/or clinical or radiological signs suggestive of increased intracranial pressure; (4) no anticoagulant treatment; (5) likely to survive >48 h. The protocol is in compliance with the Helsinki Declaration and has been approved by the local ethics committee (UN3898 285/4.8). All continuous parameters were saved on a 5-min average interval in our patient data management system (CentricityTM Critical Care 7.0 SP2; GE Healthcare Information Technologies, Dornstadt, Germany) and the values up to the first 14 days of hospitalization were aggregated to 1-h intervals to match the microdialysis sampling time. Day 1 was defined as the first 24 h after admission to the intensive care unit. A parenchymal probe (NEUROVENT-P-TEMP; Raumedic®, Helmbrechts, Germany) was inserted for the measurement of intracranial pressure (ICP) and Tbrain, either through a frontal approach using a triple-lumen bolt, or, together with the CMD-probe (71 High Cut-Off Brain Microdialysis Catheter, membrane length 1 cm, pore size 100 kDa; M Dialysis AB, Stockholm, Sweden), tunneled and placed in the white matter of the frontal watershed ipsilateral to the aneurysm or the vascular territory exhibiting the maximal pathology. In the same manner, a Hemedex probe (Hemedex® Cambridge, Massachusetts, USA) was inserted for the continuous measurement of CBF in a subgroup of 15 patients.
Isotonic perfusion fluid (Perfusion Fluid CNS; M Dialysis AB, Stockholm, Sweden) was pumped through the microdialysis system at a flow rate of 0.3 μl/min. Microdialysis samples were obtained hourly and immediately analyzed with CMA 600 and Iscusflex (M Dialysis AB, Stockholm, Sweden) for CMD-glucose, CMD-pyruvate, CMD-lactate and CMD-glutamate concentrations. The first sample was drawn at least 1 h after probe insertion and discarded, to avoid artifacts caused by the intervention. After the bedside analysis, samples were stored at −80°C.
Serial CMD samples were available for 34 patients. The thresholds for normal values were CMD-LPR <30, CMD-lactate <4 mmol/l, CMD-pyruvate >120 µmol/l, CMD-glucose >0.7 mmol/l, CMD-glutamate <10 µmol/l and CMD-glycerol <50 µmol/l, as previously defined in the literature.26 The presence of mitochondrial dysfunction was defined as CMD-LPR >30 together with CMD-pyruvate >70 µmol/l.26
Grading, radiologic definitions, neuromonitoring and patient care
Admission disease severity was graded using the Hunt & Hess scale.27 Computed tomography (CT) of the brain was performed on admission, after aneurysm repair and when clinically needed. CT scans were rated by an independent clinician, blinded to clinical data, using the modified Fisher score, the SAH sum score and the intraventricular hemorrhage (IVH) sum score, and were assessed for the presence of global cerebral edema (GCE).28–30 Microdialysis probe location was defined as “perilesional” if the gold tip of the probe was within 1 cm to a focal hypodensity (edema/infarction) or hyperdensity (hematoma) on CT, or otherwise as “normal-appearing brain tissue”.
Fever definitions and patient management
Body temperature was measured by the temperature sensor of the bladder catheter. Fever was defined as Tcore > 38.3°C based on previously published guidelines.31 Temperatures between 36.5°C and 37.5°C were defined as normothermia, and the interval between 37.5°C and 38.3°C as sub-febrile temperature range. Brain hyperthermia was accordingly defined as Tbrain > 38.3°C. Tdelta was defined as the difference between each time associated measurements of brain temperature (Tbrain) and core temperature (Tcore) given as 1-h mean values over the time of neuromonitoring.
Core normothermia was targeted based on an institutional protocol including first- and second-line pharmacological therapies, and non-pharmacological invasive (CoolGard 3000 or ThermoGard XP®, ZOLL) and non-invasive cooling devices (ARCTIC SUN®, Bard Medical), as described.1
Clinical care of SAH patients conformed to current guidelines,32,33 although prophylactic nimodipine was generally provided as a continuous intravenous infusion at the rate of 1–2 mg/h. Ruptured aneurysms were treated by surgical clipping or endovascular coiling. Intravenous fluids (crystalloids and colloids), vasopressors (noradrenaline, phenylephrine) and dobutamine were used for hemodynamic stabilization and to achieve a cerebral perfusion pressure (CPP) of more than 70 mmHg.34 Intracranial hypertension was defined as sustained ICP ≥ 20 mmHg. Rebleeding was defined as a new hemorrhage on CT scan associated with clinical deterioration. Anemia was diagnosed when hemoglobin levels were less than 8 g/dL. Pneumonia was defined as the occurrence of a new infectious state associated with consistent chest clinical, microbiological or imaging findings. Sepsis was defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.35 All patients were comatose during the period of invasive neuromonitoring and routinely received continuous intravenous midazolam and sufentanil to facilitate mechanical ventilation. Ketamine was used when clinically needed. Patients were followed with transcranial color-coded duplex sonography (TCD, LOGIQ S8; GE Healthcare, Chicago, IL) for detection of vasospasm. Vasospasm was defined as elevation of mean velocities greater than 120 cm/s in the middle or anterior cerebral artery or daily change in mean TCD velocities greater than 50 cm/s. Severe vasospasm (>200 cm/s) was further confirmed by catheter cerebral angiogram. Delayed cerebral ischemia (DCI) was defined as new infarct on CT or MRI, not attributable to other causes. Functional outcome was assessed three months after SAH using the modified Rankin Scale (mRS). Good functional outcome was defined conventionally as mRS ≤2.
Statistical analysis
Continuous variables are reported as median and interquartile range (IQR) unless indicated otherwise. Categorical variables are reported as count and proportions in each group. Time-series data were analyzed by generalized estimating equations to account for repeated measurements within one patient. Different working correlation matrices were chosen accordingly to the quasi-likelihood ratio that identified the best correlation matrix to describe the dependency structure between the observations and to account for the different lengths of monitoring.36
Spearman’s rho was used to analyze the correlations between data of brain and core temperature, cerebral perfusion (cerebral blood flow (CBF), CPP, ICP and cerebral metabolism (CMD-glutamate, CMD-glucose, CMD-lactate, CMD-pyruvate, CMD-lactate-to-pyruvate ratio and CMD-glycerol).
Our primary analysis aimed to associate fluctuations of Tdelta with data of brain metabolism, hemodynamic parameters or the presence of mitochondrial dysfunction. As secondary outcome parameter, we studied the association with functional outcome at three months.
The association between percentage of time in mitochondrial dysfunction and outcome was assessed using a generalized linear model.
For outcome analysis Tbrain, Tcore and Tdelta, the presence of mitochondrial dysfunction and common risk factors for poor outcome were included, according to previous literature. For the multivariable analysis, the following variables were added: age, intubation days, modified Fisher scale, Hunt and Hess score, occurrence of DCI or mitochondrial dysfunction. All analysis was performed with IBM-SPSS V24 (SPSS Inc., Chicago, IL, USA). The threshold for statistical significance was set at a p-value of <0.05.
Data availability statement
Anonymized data used for this study are available from the corresponding author on reasonable request.
Results
Baseline characteristics, disease severity, hospital complications and functional outcome of 46 consecutive patients are summarized in Table 1. Neuromonitoring was initiated on day 1 (IQR 1–2) after SAH with a median recording time of 178 h (IQR 107–269 h) per patient. Microdialysis probes were identified in normal-appearing brain tissue or perilesional in each half of the patients (N = 23, 50%).
Table 1.
Baseline characteristics, complications, and outcome.
| Clinical characteristics | N = 46 | |
|---|---|---|
| Age (years) | 53 (46–67) | |
| Gender (female) | 32 (69.6%) | |
| Admission H&H grade | 2 | 3 (6.5%) |
| 3 | 16 (34.8%) | |
| 4 | 5 (10.9%) | |
| 5 | 22 (47.8%) | |
| Loss of consciousness | 31 (67.4%) | |
| Admission radiological characteristics | ||
| mFisher scale | 1 | 1 (2.2%) |
| 2 | 4 (8.7%) | |
| 3 | 8 (17.4%) | |
| 4 | 33 (71.7%) | |
| SAH sum score | 23.5 (16.75–27) | |
| IVH sum score | 4 (2–7) | |
| Aneurysm size above 10 mm | 11 (23.9%) | |
| Generalized cerebral edema | 17 (37%) | |
| Surgical procedures | ||
| Hydrocephalus requiring EVD | 32 (69.6%) | |
| Clipping | 26 (56.5%) | |
| Hemicraniectomy | 8 (17.4%) | |
| Complications | ||
| Pneumonia | 36 (78.3%) | |
| Sepsis | 18 (39.1%) | |
| Vasospasm | 36 (78.3%) | |
| Delayed cerebral ischemia | 13 (28.3%) | |
| Anemia requiring transfusion | 15 (32.6%) | |
| Aneurysm rebleeding | 5 (10.9%) | |
| Hyperosmolar therapy | 27 (58.7%) | |
| Outcome characteristics | ||
| Length of hospital stay (days) | 36 (10–107) | |
| 3-month mRS | 0 | 2 (4.3%) |
| 1 | 7 (15.2%) | |
| 2 | 5 (10.9%) | |
| 3 | 5 (10.9%) | |
| 4 | 6 (13%) | |
| 5 | 13 (28.3%) | |
| 6 | 8 (17.4%) | |
H&H: Hunt&Hess; mFisher: modified Fisher; ICH: intracerebral hemorrhage; SAH: subarachnoid hemorrhage; mRS: modified Rankin Scale; EVD: extraventricular drainage.
Brain and body temperature
Simultaneous recording of Tbrain and Tcore was available for 7879 h (328 patient days) and was highly correlated (r = 0.948; p < 0.01). Median Tcore was 36.9°C (IQR 36.4–37.6 °C), and episodes of fever were observed in 28 patients (61%) and 6.5% of monitoring time (512 h). Median Tbrain was 37.2°C (IQR 36.6–37.8°C). Brain hyperthermia (>38.3°C) was recorded more often (766 h, 9.7% of neuromonitoring time) and occurred in 30/46 patients (65%).
Overall, Tbrain was higher by a Tdelta of 0.18°C (IQR −0.01 to 0.37°C) compared to Tcore. Interestingly, this difference was more evident when the brain metabolic profile indicated preserved mitochondrial function (median 0.23°C, IQR 0.06–0.42), whereas during episodes of brain mitochondrial dysfunction, the Tdelta was close to zero (median −0.02°C, IQR −0.15 −0.09) (p < 0.001, Figure 1). In 26% of neuromonitoring time, Tbrain was lower than Tcore.
Figure 1.
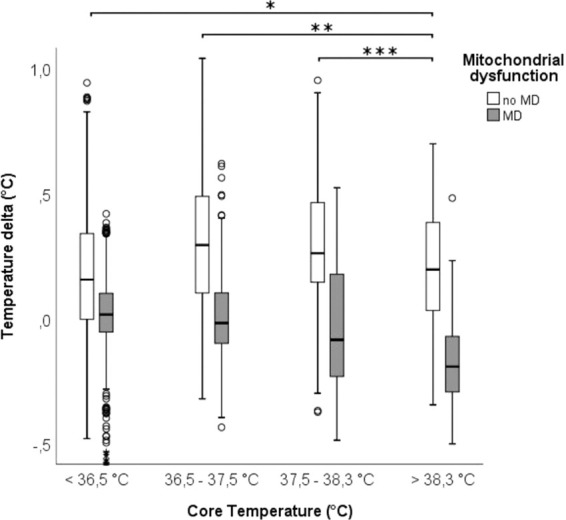
Core temperature (X-axis; hypothermia (<36.5°C), normothermia (36.5–37.5°C), sub-febrile (37.5–38.3°C) and fever (>38.3°C)) and the difference between core and brain temperature (Y-axis, a positive value indicates higher brain temperature compared to core temperature) during 7879 neuromonitoring hours in 46 poor grade SAH patients. Groups represent hours with (grey square) and without (white square) mitochondrial dysfunction defined as CMD-LPR >30 and CMD-pyruvate >70 µmol/l. CMD: cerebral microdialysis; LPR: lactate-to-pyruvate ratio (between episodes with and without MD for each group, p < 0,001; between core temperature groups as shown in the graph, during periods without MD *p < 0.05 **p = 0.001 ***p < 0.001; during MD all comparisons are significant at p < 0.001).
When analysing differences in Tdelta based on probe location, we found a Tdelta of 0.2°C (IQR 0.1–0.4) in the healthy appearing brain tissue, a Tdelta of 0°C (IQR −0.1–0.1) in the perilesional brain tissue and the highest Tdelta of 0.4°C (IQR −0.2−0.5) in patients with GCE. Tdelta was slightly higher during normothermia (median MD 0.3°C, IQR 0.1 to 0.5°C) when compared to episodes of fever (median 0.2°C, IQR 0 to 0.4°C).
In the presence of mitochondrial dysfunction, Tdelta was lower, with a median of 0°C IQR 0.1 to 0.1°C during normothermia, compared to a median of −0.2°C, IQR −0.3 to −0.1°C during fever (p < 0.001, Figure 1).
Temperature delta and brain metabolism
Overall brain metabolic monitoring revealed moderate abnormal values with a median CMD-LPR of 29 (IQR 22–38), CMD-lactate of 3.8 mmol/L (IQR 2.2–6.3 mmol/L), CMD-pyruvate of 132 µmol/L (IQR 94–184 µmol/L), CMD-glucose of 1.5 mmol/L (IQR 0.9–2.3 mmol/L), CMD-glutamate of 6 µmol/L (IQR 2–42 µmol/L), and CMD-glycerol of 41 µmol/L (IQR 24–78 µmol/L). A larger Tdelta, but not absolute temperature, was associated with lower CMD-glutamate (p = 0.003) and lower CMD-lactate (p < 0.001) (Figures 2 and 3). Furthermore, we found a correlation between lower CMD-glycerol and higher Tdelta (r = −0.4 p = 0.031), Tbrain (r = −0.4 p = 0.016) and Tcore (r = −0.35 p = 0.006) as well as lower CMD-LPR and higher Tdelta (r = −0.56 p = 0.007). Despite the significant correlation, we did not find group differences when accounting for repeated measures within patients (p > 0.05). There were no relevant associations between temperature variables and CMD-glucose.
Figure 2.

Absolute values of CMD-derived metabolites (y-axis) at high (>0.18°C) and low (≤0.18°C) temperature delta (difference between brain temperature and core temperature (Tdelta, x-axis). The horizontal lines indicate thresholds for pathological absolute CMD levels, as indicated in literature: (a) glucose <0,7 mmol/L, (b) glutamate >10 µmol/L, (c) glycerol >50 µmol/L, (d) lactate >4 mmol/L, (e) LPR >30 reflecting mitochondrial dysfunction, (f) pyruvate <120 µmol/L. *Indicates statistical significance, p values are specified in the graph. CMD: cerebral microdialysis; LPR: lactate-to-pyruvate ratio.
Figure 3.

Daily mean (±95% confidence intervals) levels of CMD-glutamate (Panel A) in µmol/L and CMD-lactate (Panel B) in mmol/L (y-axis) in patients with high (triangle) and low (grey circle) difference between brain temperature and core temperature (Tdelta), divided at the median Tdelta = 0.18°C. CMD: cerebral microdialysis.
Mitochondrial dysfunction was detected in 16% of neuromonitoring time in 76% (26/34) of patients. In 13/34 patients (38%), MD was evident in more than half of the neuromonitoring time. During episodes of MD, median Tdelta was significantly lower (0°C, IQR −0.2 to 0.1) when compared to episodes without MD (0.2°C, IQR 0.1 to 0.4) (p < 0.001) (Figure 4).
Figure 4.
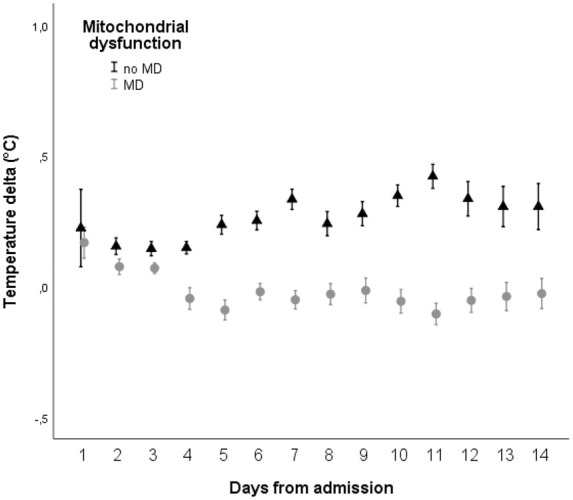
Daily mean (± 95% confidence intervals) difference between brain and core temperature (Tdelta, y-axis, positive values indicating higher brain temperature compared to core temperature) during episodes with (grey circles) and without (triangles) mitochondrial dysfunction, defined as CMD-LPR >30 and CMD-pyruvate >70 µmol/l. CMD: cerebral microdialysis; LPR = lactate-to-pyruvate ratio (p < 0.001).
Temperature delta, CPP, ICP and CBF
Overall, patients had a median CPP of 74 mmHg (IQR 68–81mmHg), ICP of 11 mmHg (IQR 7–15mmHg), and CBF of 21 ml/100 g/min (IQR 11–31). Tbrain, Tcore or Tdelta were not associated with changes in CPP, ICP or CBF (p > 0.05). However, during mitochondrial dysfunction, CBF was negatively associated with Tdelta (r = −0.53 p < 0.01) but not with absolute temperature levels (p > 0.05).
Temperature, mitochondrial dysfunction and outcome
One-third of patients had a favourable three-months outcome (14/46, 30%). There was no association between absolute Tbrain or Tcore and neurological outcome. However, a larger Tdelta was associated with a higher probability of good functional outcome at three months (per 1°C increase OR = 7.7, 95% CI = 1.4–43.6 p = 0.02). This association remained significant after adjusting for intubation days, age, gender, DCI, Hunt & Hess grade and modified Fisher grade (per 1°C increase adjOR = 1.6, 95% CI = 1–2.5 p = 0.047).
Tdelta > 0.2°C was associated with a lower risk of mitochondrial dysfunction (OR = 0.2, 95% CI = 0.13–0.33 p < 0.001).
Furthermore, the percentage of time in mitochondrial dysfunction was associated with poor three-months outcome (medians 5% (IQR 0%–25%) for good outcome and 50% (IQR 7%–90%) for poor outcome (p = 0.022). The association of outcome with the overall percentage of time in mitochondrial dysfunction remained significant even after adjusting for age, gender, intubation days, DCI, Hunt & Hess grade and modified Fisher scale (p = 0.017).
Discussion
The main finding of this study is that brain temperature is highly associated with brain metabolic activity and exceeds body core temperature only when mitochondrial function is preserved. Moreover, a larger difference between body and brain temperature was associated with favourable brain metabolic state as indicated by lower CMD-lactate levels and decreased excitotoxicity. Preserved mitochondrial function and a higher delta brain temperature were independently associated with better functional outcome.
Tbrain is tightly regulated by Tcore, however may exceed Tcore by up to 2.5°C in patients with acute brain injury.8–15 The temperature gradient is even higher in cortical areas when compared to deep brain structures including the white matter,11,12,19,37,38 which seems to be further aggravated after acute brain injury. This concept has been previously described as “brain thermopooling”.39
The measured Tbrain reflects the balance between mechanisms of heat production and dissipation with the ultimate goal to prevent excessive overheating. The main contributors to this balance are the arterial inflow, the brain metabolic activity and the venous outflow; however, the dynamic interactions between these mechanisms are complex.8,10,13–15,18,19,37,40–44 In the current study, we found a minor overall difference between Tbrain and Tcore by only about 0.2°C, which is consistent with previously reported data in patients with TBI, SAH or comatose patients after cardiac arrest.8,14,45 One explanation may be that we follow the concept of normothermia in our SAH patients by aggressive fever control, as a higher Tdelta is usually observed at higher temperature levels.
Interestingly, we found that Tbrain was significantly lower during episodes of mitochondrial dysfunction resulting in Tbrain closely following Tcore. Our data suggest that brain mitochondrial dysfunction is associated with a decrease of local heat production. The brain itself is a highly metabolic active organ and mitochondria are the main source of energy production through respiratory chain reactions.46 The energy is required not only for electrical activity of neurons, endothelial cells and glia, but also for intracellular processes such as synthesis of macromolecules and proton transport across mitochondrial membrane, all of which may be disturbed after acute brain injury.9–11 Around 60% of the energy is used for the production of ATP, the rest is dissipated as heat.42 In this line we found evidence for energy and heat production when the metabolic profile suggests preserved mitochondrial function.
On average, around 0.7 J/g brain tissue are generated per minute, which would result in an increase by 0.2°C in Tbrain per minute if compensatory heat removing mechanisms would fail.10,11,17,47 Heat transfer normally occurs via radiation, conduction, convection and evaporation, but in the CNS the main regulator is through washout of CBF.8–11 In fact, the temperature of the venous outflow has been previously reported to be as much as 0.3°C higher than the arterial inflow.8,10 CBF is also known to increase with brain activation, hence serving the dual-purpose of sustaining metabolism with nutrients delivery and buffering the resulting heat production.8,37 This complex relationship between CBF and brain temperature explains why the increase in CBF linked to metabolic activation does not necessarily lead to a relevant change in Tdelta or Tbrain.38,44 However, during flow-metabolism mismatch and in pathological conditions when variations in CBF are unrelated to heat production, a rise in CBF may be associated with a significant reduction in Tbrain.8,15,19,38 This is consistent with our results, considering that the correlation between CBF and Tdelta became negative during episodes of mitochondrial dysfunction, when energy production fails.
The association between fever and higher long-term morbidity and mortality is well established in neurocritical care patients.3–5 The role of elevated Tbrain and its association with outcome remains less clear. Although monitoring of Tbrain is recommended, it is rarely performed in clinical routine and the therapeutic relevance is still debatable. It is well known that extremely elevated Tbrain >40°C can be detrimental by direct neurotoxic effects, but also through indirect mechanisms including potentiation of glutamate-induced and reactive oxygen species toxicities.9,37,42 Higher Tbrain has also been associated with increased blood–brain barrier permeability, contributing to brain edema.9,10 In contrast to that, we found that a higher Tdelta was associated with improved brain metabolic profile. This is of interest, however temperature differences in our study were minor compared to the detrimental mechanism described above occurring at higher Tbrain levels
At a cellular level, general pathogenetic mechanisms can be identified regardless of the underlying neurological injury, including mitochondrial dysfunction, reactive oxygen species (ROS) generation and inadequate ROS scavenging.8 Both, the downregulation of antioxidant cellular capabilities and the increased production of ROS due to ischemia-mediated metabolic disruption of mitochondrial structures underlie the increased oxidative stress observed in SAH patients.48 It has been suggested that the oxyhemoglobin-induced calcium influx through L-type voltage-dependent calcium channel might be responsible for depolarizing the mitochondrial membrane, with consequent increased ROS production and cellular damage, and this may also cause disruption of the mitochondrial dynamics by activating Drp1, favouring mitochondrial fragmentation and hence dysfunction.49
In addition, other mechanisms like neuroinflammation and glutamate toxicity seem to play an important role to promote secondary injury.42,50 Based on the ‘sandwich model’, all of these mechanisms are cumulative and may reach a level of cellular toxicity leading to cell death.51 In this context, the activation of mitochondrial uncoupling proteins (UCP) following brain injury would seem to counteract these processes by dissipating the increased ROS produced and the accumulated mitochondrial membrane potential through heat, thereby increasing the temperature locally in the damaged brain.42,51
When comparing differences in Tdelta by probe location, we found the highest Tdelta in patients with GCE, suggesting a hypermetabolic response with local heat production which was previously shown in a microdialysis study of SAH patients.52 In contrast, there was no difference between body and brain temperature when Tbrain was measured in the perilesional area suggesting a more severely injured brain and energy failure.
We did not find an association between Tbrain and ICP or CPP. Although the association with fever and high ICP is well described, our patients were mostly treated within the range of normothermia. This may also explain why neither Tbrain nor Tcore were associated with functional outcome in our cohort. Interestingly, we found an association between higher Tdelta and better long-term functional outcome. One explanation for this may reflect improved brain metabolic activity through preserved mitochondrial function. Previous reports support the hypothesis that in case of spontaneous equalizing of Tbrain and Tcore, the patients’ outcome is worse.13,15,37,42,44,53,54 This spontaneous lowering of Tbrain towards Tcore has been linked to an increased risk of hospital death as it may indicate failure of metabolic heat production and decreased CBF towards irreversibility.38,43 Of note, during targeted normothermia, the same reversal was recorded due to lower cerebral metabolic rates but was associated with favourable prognosis.37,38,53,55 Consequently, given the strict institutional fever protocol (preventive or reactive), we might have underestimated the true effect of a higher brain temperature on metabolic changes. The role of Tdelta as a biomarker is so far not well defined, but ultimately might serve as a marker of retained electro-metabolic brain activity in response to the acute injury. More research is needed to dissect the implication of a higher Tdelta and whether this can be translated to a different clinical management strategies (e.g. optimizing brain perfusion, improving mitochondrial activity, etc.).
This study has several limitations. It was single-centered, even though we included data from a relatively large cohort including 46 SAH patients. We included only poor-grade SAH patients with invasive, therefore localized, brain temperature measurement which limits the generalizability of our results. In this context, it is important to mention that all other multimodal neuromonitoring parameters were obtained in the vicinity of the localized Tbrain measurement site, which allowed us to analyse associated metabolic and perfusion parameters. Another limitation is that we did not include depth of sedation or the occurrence of seizures or cortical spreading depolarizations in this analysis. This could have influenced our findings as episodes of cortical spreading depolarizations (CSD) were associated with timely increases in brain temperature. We previously reported dynamic short-term changes in brain temperature associated with CSD. These are most likely related to brain activation during this highly energy-consuming event.25 However, this was only observed for approximately 1 h and cannot be claimed for the overall higher brain temperature observed in our patients. In addition, we used a strict fever protocol and targeted normothermia in our patients, therefore pharmacological and non-pharmacological interventions may have influenced our findings. Still, our data represent physiologic and pathophysiologic changes in brain and body temperature reflecting clinical practice in poor-grade SAH patients.
In conclusion, our data suggest that brain temperature is closely regulated by body temperature and may exceed Tcore by 0.2°C when mitochondrial dysfunction is preserved. During episodes of mitochondrial dysfunction, Tbrain seems to passively follow Tcore which was associated with worse outcome after SAH. Further studies are needed to better understand the physiologic and pathophysiologic processes resulting in differences in Tdelta, and to further elucidate the impact of Tdelta on brain metabolism and the chance of recovery after SAH.
Acknowledgements
We wish to thank all nurses in the NICU who substantially contribute to our patients’ outcome. Moreover, we would like to thank all patients and family members for their support in our ongoing research work despite being affected by the severity of the acute brain injury.
Footnotes
Authors’ contributions: AA was involved in the acquisition of data, statistical analysis, interpretation of data, study design, writing and manuscript drafting. RH was involved in the study design, interpretation of data, statistical analysis, manuscript writing and drafting, and final revision of the manuscript. MG, AS, MK, BI, VR contributed to acquisition of data, statistical analysis, interpretation of data and manuscript drafting. ES, BP, RB, AL and GB participated in the acquisition and interpretation of data. CT was involved in the study design and data acquisition. All authors discussed the results, commented on the manuscript and approved the final version.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Scaravilli V, Tinchero G, Citerio G, et al. Fever management in SAH. Neurocrit Care 2011; 15: 287–294. [DOI] [PubMed] [Google Scholar]
- 2.Suehiro E, Sadahiro H, Goto H, et al. Importance of early postoperative body temperature management for treatment of subarachnoid hemorrhage. J Stroke Cerebrovasc Dis 2016; 25: 1482–1488. [DOI] [PubMed] [Google Scholar]
- 3.Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage*. Crit Care Med 2006; 34: 617–623. [DOI] [PubMed] [Google Scholar]
- 4.Oliveira-Filho J, Ezzeddine MA, Segal AZ, et al. Fever in subarachnoid hemorrhage: relationship to vasospasm and outcome. Neurology 2001; 56: 1299–1304. [DOI] [PubMed] [Google Scholar]
- 5.Park YK, Yi HJ, Choi KS, et al. Predictive factors of fever after aneurysmal subarachnoid hemorrhage and its impact on delayed cerebral ischemia and clinical outcomes. World Neurosurg 2018; 114: e524–e531. [DOI] [PubMed] [Google Scholar]
- 6.Madden LK, Hill M, May TL, et al. The implementation of targeted temperature management: an evidence-based guideline from the neurocritical care society. Neurocrit Care 2017; 27: 468–487. [DOI] [PubMed] [Google Scholar]
- 7.Lopez GA.Temperature management in the neurointensive care unit. Curr Treat Options Neurol 2016; 18: 12. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Wang B, Normoyle KP, et al. Brain temperature and its fundamental properties: a review for clinical neuroscientists. Front Neurosci 2014; 8: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiyatkin EA.Brain temperature fluctuations during physiological and pathological conditions. Eur J Appl Physiol 2007; 101: 3–17. [DOI] [PubMed] [Google Scholar]
- 10.Kiyatkin EA.Brain temperature homeostasis physiological fluctuations and pathological shifts. Front Biosci 2010; 15: 73–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertolizio G, Mason L, Bissonnette B.Brain temperature: heat production, elimination and clinical relevance. Paediatr Anaesth 2011; 21: 347–358. [DOI] [PubMed] [Google Scholar]
- 12.McIlvoy L.Comparison of brain temperature to core temperature: a review of the literature. J Neurosci Nurs 2004; 36: 23–31. [DOI] [PubMed] [Google Scholar]
- 13.Huschak G, Hoell T, Wiegel M, et al. Does brain temperature correlate with intracranial pressure? J Neurosurg Anesthesiol 2008; 20: 105–109. [DOI] [PubMed] [Google Scholar]
- 14.Rossi S, Zanier ER, Mauri I, et al. Brain temperature, body core temperature, and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry 2001; 71: 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Childs C.Human brain temperature: regulation, measurement and relationship with cerebral trauma: part 1. Br J Neurosurg 2008; 22: 486–496. [DOI] [PubMed] [Google Scholar]
- 16.Childs C, Wieloch T, Lecky F, et al. Report of a consensus meeting on human brain temperature after severe traumatic brain injury: its measurement and management during pyrexia. Front Neurol 2010; 1: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu M, Ackerman JJ, Yablonskiy DA.Body and brain temperature coupling: the critical role of cerebral blood flow. J Comp Physiol B 2009; 179: 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serota HM, Gerard RW.Localized thermal changes in the cat’s brain. J Neurophysiol 1938; 1: 115–124. [Google Scholar]
- 19.Hayward JN, Baker MA.A comparative study of the role of the cerebral arterial blood in the regulation of brain temperature in five mammals. Brain Res 1969; 16: 417–440. [DOI] [PubMed] [Google Scholar]
- 20.Karaszewski B, Carpenter TK, Thomas RG, et al. Relationships between brain and body temperature, clinical and imaging outcomes after ischemic stroke. J Cereb Blood Flow Metab 2013; 33: 1083–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rieke V, Butts Pauly K.MR thermometry. J Magn Reson Imaging 2008; 27: 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozak LR, Bango M, Szabo M, et al. Using diffusion MRI for measuring the temperature of cerebrospinal fluid within the lateral ventricles. Acta Paediatr 2010; 99: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karaszewski B, Wardlaw JM, Marshall I, et al. Measurement of brain temperature with magnetic resonance spectroscopy in acute ischemic stroke. Ann Neurol 2006; 60: 438–446. [DOI] [PubMed] [Google Scholar]
- 24.Verius M, Frank F, Gizewski E, et al. Magnetic resonance spectroscopy thermometry at 3 Tesla: importance of calibration measurements. Ther Hypothermia Temp Manag 2019; 9: 146–155. [DOI] [PubMed] [Google Scholar]
- 25.Schiefecker AJ, Kofler M, Gaasch M, et al. Brain temperature but not core temperature increases during spreading depolarizations in patients with spontaneous intracerebral hemorrhage. J Cereb Blood Flow Metab 2018; 38: 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helbok R, Kofler M, Schiefecker AJ, et al. Clinical use of cerebral microdialysis in patients with aneurysmal subarachnoid hemorrhage-state of the art. Front Neurol 2017; 8: 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunt WE, Hess RM.Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28: 14–20. [DOI] [PubMed] [Google Scholar]
- 28.Claassen J, Bernardini GL, Kreiter K, et al. Effect of cisternal and ventricular blood on risk of delayed cerebral ischemia after subarachnoid hemorrhage: the Fisher scale revisited. Stroke 2001; 32: 2012–2020. [DOI] [PubMed] [Google Scholar]
- 29.Claassen J, Carhuapoma JR, Kreiter KT, et al. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke 2002; 33: 1225–1232. [DOI] [PubMed] [Google Scholar]
- 30.Hijdra A, Brouwers PJ, Vermeulen M, et al. Grading the amount of blood on computed tomograms after subarachnoid hemorrhage. Stroke 1990; 21: 1156–1161. [DOI] [PubMed] [Google Scholar]
- 31.O’Grady NP, Barie PS, Bartlett JG, et al. Guidelines for evaluation of new fever in critically ill adult patients: 2008 update from the American College of Critical Care Medicine and the Infectious Diseases Society of America. Crit Care Med 2008; 36: 1330–1349. [DOI] [PubMed] [Google Scholar]
- 32.Bederson JB, Connolly ES, Jr., Batjer HH, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke 2009; 40: 994–1025. [DOI] [PubMed] [Google Scholar]
- 33.Connolly ES, Jr., Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43: 1711–1737. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt JM, Ko SB, Helbok R, et al. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke 2011; 42: 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016; 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziegler A, Vens M.Generalized estimating equations. Notes on the choice of the working correlation matrix. Methods Inf Med 2010; 49: 421–425; discussion 426–432. [DOI] [PubMed] [Google Scholar]
- 37.Mrozek S, Vardon F, Geeraerts T.Brain temperature: physiology and pathophysiology after brain injury. Anesthesiol Res Pract 2012; 2012: 989487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soukup J, Rieger A, Holz C, et al. Temperature gradient between brain tissue and arterial blood mirrors the flow-metabolism relationship in uninjured brain: an experimental study. Acta Anaesthesiol Scand 2007; 51: 872–879. [DOI] [PubMed] [Google Scholar]
- 39.Arrica M, Bissonnette B.Therapeutic hypothermia. Semin Cardiothorac Vasc Anesth 2007; 11: 6–15. [DOI] [PubMed] [Google Scholar]
- 40.Hayward JN, Baker MA.Role of cerebral arterial blood in the regulation of brain temperature in the monkey. Am J Physiol 1968; 215: 389–403. [DOI] [PubMed] [Google Scholar]
- 41.Verlooy J, Heytens L, Veeckmans G, et al. Intracerebral temperature monitoring in severely head injured patients. Acta Neurochir 1995; 134: 76–78. [DOI] [PubMed] [Google Scholar]
- 42.Rango M, Arighi A, Bresolin N.Brain temperature: what do we know? Neuroreport 2012; 23: 483–487. [DOI] [PubMed] [Google Scholar]
- 43.Otawara Y, Ogasawara K, Kubo Y, et al. Brain and systemic temperature in patients with severe subarachnoid hemorrhage. Surg Neurol 2003; 60: 159–164. [DOI] [PubMed] [Google Scholar]
- 44.Soukup J, Zauner A, Doppenberg EM, et al. The importance of brain temperature in patients after severe head injury: relationship to intracranial pressure, cerebral perfusion pressure, cerebral blood flow, and outcome. J Neurotrauma 2002; 19: 559–571. [DOI] [PubMed] [Google Scholar]
- 45.Coppler PJ, Marill KA, Okonkwo DO, et al. Concordance of brain and core temperature in comatose patients after cardiac arrest. Ther Hypothermia Temp Manag 2016; 6: 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jezek P, Zackova M, Ruzicka M, et al. Mitochondrial uncoupling proteins–facts and fantasies. Physiol Res 2004; 53(Suppl 1): S199–211. [PubMed] [Google Scholar]
- 47.Yablonskiy DA, Ackerman JJH, Raichle ME.Coupling between changes in human brain temperature and oxidative metabolism during prolonged visual stimulation. Proc Natl Acad Sci U S A 2000; 97: 7603–7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ayer RE, Zhang JH.Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm. Acta Neurochir Suppl 2008; 104: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Chen J, Sun X, et al. Evaluation of the neuroprotective effect of EGCG: a potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct 2018; 9: 6349–6359. [DOI] [PubMed] [Google Scholar]
- 50.Helbok R, Schiefecker AJ, Beer R, et al. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Crit Care 2015; 19: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mattiasson G, Sullivan PG.The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid Redox Signal 2006; 8: 1–38. [DOI] [PubMed] [Google Scholar]
- 52.Zetterling M, Hallberg L, Hillered L, et al. Brain energy metabolism in patients with spontaneous subarachnoid hemorrhage and global cerebral edema. Neurosurgery 2010; 66: 1102–1110. [DOI] [PubMed] [Google Scholar]
- 53.Suehiro E, Fujisawa H, Koizumi H, et al. Significance of differences between brain temperature and core temperature (delta T) during mild hypothermia in patients with diffuse axonal injury. Neurol Med Chir 2011; 51: 551–555. [DOI] [PubMed] [Google Scholar]
- 54.Fountas KN, Kapsalaki EZ, Feltes CH, et al. Disassociation between intracranial and systemic temperatures as an early sign of brain death. J Neurosurg Anesthesiol 2003; 15: 87–89. [DOI] [PubMed] [Google Scholar]
- 55.Fischer M, Schiefecker A, Lackner P, et al. Targeted temperature management in spontaneous intracerebral hemorrhage: a systematic review. Curr Drug Targets 2017; 18: 1430–1440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data used for this study are available from the corresponding author on reasonable request.