

Abstract
OBJECTIVE—
Type 2 diabetes has become a global epidemic, and Asian Indians have a higher susceptibility to diabetes than Europeans. We investigated whether Indians had any metabolic differences compared with Northern European Americans that may render them more susceptible to diabetes.
RESEARCH DESIGN AND METHODS—
We studied 13 diabetic Indians, 13 nondiabetic Indians, and 13 nondiabetic Northern European Americans who were matched for age, BMI, and sex. The primary comparisons were insulin sensitivity by hyperinsulinemic-euglycemic clamp and skeletal muscle mitochondrial capacity for oxidative phosphorylation (OXPHOS) by measuring mitochondrial DNA copy number (mtDNA), OXPHOS gene transcripts, citrate synthase activity, and maximal mitochondrial ATP production rate (MAPR). Other factors that may cause insulin resistance were also measured.
RESULTS—
The glucose infusion rates required to maintain identical glucose levels during the similar insulin infusion rates were substantially lower in diabetic Indians than in the nondiabetic participants (P < 0.001), and they were lower in nondiabetic Indians than in nondiabetic Northern European Americans (P < 0.002). mtDNA (P < 0.02), OXPHOS gene transcripts (P < 0.01), citrate synthase, and MAPR (P < 0.03) were higher in Indians irrespective of their diabetic status. Intramuscular triglyceride, C-reactive protein, interleukin-6, and tumor necrosis factor-α concentrations were higher, whereas adiponectin concentrations were lower in diabetic Indians.
CONCLUSIONS—
Despite being more insulin resistant, diabetic Indians had similar muscle OXPHOS capacity as nondiabetic Indians, demonstrating that diabetes per se does not cause mitochondrial dysfunction. Indians irrespective of their diabetic status had higher OXPHOS capacity than Northern European Americans, although Indians were substantially more insulin resistant, indicating a dissociation between mitochondrial dysfunction and insulin resistance.
There is a global epidemic of type 2 diabetes (1); and while mortality from other leading causes of death, including coronary artery disease, stroke, and cancer is declining, deaths attributed to type 2 diabetes are escalating (2). It is estimated that Asian Indians have the highest world-wide prevalence of diabetes (~32 million), and it is conservatively predicted that the number of affected individuals will double in the next 30 years (3). Population growth, urbanization, aging, obesity, and physical inactivity are well recognized contributing factors for the increase in type 2 diabetes (3). In addition, Indians show several unique features, including a younger age of onset of type 2 diabetes, relatively lower BMI compared with Northern European descendants at the onset of type 2 diabetes, and lower thresholds for the other risk factors associated with type 2 diabetes (4,5). Recently, an increased prevalence of nonalcoholic fatty liver disease in association with insulin resistance has also been reported among Indians (6). Body fat distribution that causes insulin resistance is also reported to be different among South Asian Indians (7), and they show a higher body fat percentage for a given BMI in comparison with Caucasians. The underlying cause of the unusual susceptibility of Indians to type 2 diabetes remains to be determined.
We investigated whether Indians have underlying differences in energy metabolism that may render them to be more insulin resistant and contribute to their greater susceptibility to type 2 diabetes. There were several reasons to consider the involvement of energy metabolism and mitochondrial function in potentially contributing to insulin resistance and type 2 diabetes. First, Indians lived for centuries as an agrarian society with a predominantly vegetarian diet, which provided lower energy density in comparison with the predominantly meat diet of Northern Europeans. The emigration to Europe occurred ~40,000 years ago when the European continent had sparse vegetation for many months due to long winters (8,9), and the diet therefore consisted predominantly of energy-dense meat products. It has also been proposed that obesity (and type 2 diabetes) stemmed from a natural selection of our ancestors favoring a “thrifty genotype” that enabled highly efficient storage of energy during periods of food abundance (10). Similarly, a relationship between low birth weight and type 2 diabetes has been observed, epigenetically suggesting that type 2 diabetes may be attributed to a “thrifty phenotype” (11). A corollary of the above two hypotheses is that prolonged periods of low energy availability induced adaptive changes in genes and/or phenotypes that may become disadvantageous when food is plentiful and energy expenditure is minimized. Because the mitochondria is the primary organelle involved in fuel metabolism, we sought to determine whether differences in mitochondrial function may occur in the Indian population with high susceptibility to develop diabetes.
A hallmark metabolic defect of type 2 diabetes is insulin resistance, especially in skeletal muscle, which is the predominant organ involved in glucose disposal after a meal (12). Recent studies (13–17) have shown an association between insulin resistance and mitochondrial dysfunction. We therefore, sought to determine whether diabetic and nondiabetic Indians were more insulin resistant than nondiabetic Northern European Americans, who are reported to have a lesser susceptibility to type 2 diabetes than Indians (3). In addition, we determined whether the theory that insulin resistance may result from mitochondrial dysfunction is supported by studies in Indians and Northern European Americans.
RESEARCH DESIGN AND METHODS
Thirteen diabetic Indians, 13 nondiabetic Indians, and 13 nondiabetic Northern European Americans who were matched for sex (eight men and five women per group), age, and BMI (Table 1) were recruited. Type 2 diabetic participants were selected based on a known diagnosis and matched to nondiabetic control participants who had no first-degree relatives with type 2 diabetes and a fasting plasma glucose concentration <100 mg/dl. Participants were excluded after history and physical examination if there was evidence of clinically important coexisting illnesses or conditions that could have an effect on the outcome measures. Participants with serum creatinine concentrations >1.5 mg/dl, taking medications that may have an impact on energy metabolism, with liver function abnormalities, or with active coronary artery disease were excluded. All attempts were made to match participants for their activity levels. Except for one nondiabetic Indian and one nondiabetic Northern European American who were matched for their exercise programs, no other participants were involved in any regular exercise programs.
TABLE 1.
Body composition and metabolic measurements in 13 each of sex-, age-, and BMI-matched nondiabetic Northern European Americans, nondiabetic Asian Indians, and diabetic Asian Indians*
| 1. Nondiabetic European Americans | 2. Nondiabetic Asian Indians | 3. Diabetic Asian Indians | Group P | P for 1 vs. 2 | P for 1 vs. 3 | P for 2 vs. 3 | |
|---|---|---|---|---|---|---|---|
| Age (years) | 46.6 ± 2.2 | 47.2 ± 2.4 | 53.4 ± 3.2 | 0.150 | — | — | — |
| Body composition* | |||||||
| BMI (kg/m2) | 24.5 ± 0.8 | 23.8 ± 0.5 | 26.4 ± 1.0 | 0.059 | — | — | — |
| Body fat (%) | 28.3 ± 2.0 | 33.5 ± 2.1 | 37.2 ± 2.1 | 0.016 | 0.084 | 0.004 | 0.207 |
| Trunk fat (%) | 55.4 ± 1.9 | 57.3 ± 1.3 | 61.0 ± 1.0 | 0.037 | 0.381 | 0.012 | 0.087 |
| Fat free mass (kg) | 49.4 ± 0.5 | 43.2 ± 3.3 | 42.1 ± 2.4 | 0.197 | — | — | — |
| Skeletal muscle (kg) | 29.4 ± 2.2 | 26.3 ± 2.2 | 24.4 ± 1.1 | 0.243 | — | — | — |
| IMTG (μmol/g wet wt) | 3.7 ± 0.5 | 4.5 ± 0.4 | 16.4 ± 7.6 | <0.001 | 0.274 | <0.001 | 0.001 |
| Metabolic and hormone measurements† | |||||||
| Total cholesterol (mmol/l) | 4.4 ± 0.2 | 4.6 ± 0.2 | 4.2 ± 0.3 | 0.531 | — | — | — |
| HDL cholesterol (mmol/l) | 1.3 ± 0.1 | 1.0 ± 0.1 | 0.9 ± 0.1 | 0.004 | 0.009 | 0.002 | 0.213 |
| Triglycerides (mmol/l) | 1.0 ± 0.1 | 1.7 ± 0.1 | 1.3 ± 0.1 | 0.035 | 0.010 | 0.075 | 0.581 |
| Fasting glucose (mmol/l) | 4.8 ± 0.1 | 5.1 ± 0.1 | 8.9 ± 0.9 | <0.001 | 0.714 | <0.001 | <0.001 |
| Fasting glucagon (ng/l) | 65.7 ± 4.3 | 71.2 ± 4.9 | 60.4 ± 6.4 | 0.465 | — | — | — |
| 8-OH-dG (pg/μg DNA) | 0.49 ± 0.08 | 0.56 ± 0.07 | 0.39 ± 0.05 | 0.357 | — | — | — |
Data are means ± SE.
Body fat %, trunk fat % (trunk fat [kg]/fat mass [kg] × 100), fat-free mass, and skeletal muscle mass were measured by dual-energy X-ray absorptiometry. IMTGs were measured as previously described (19). ANOVA was used to test the main effect of group and body composition measures.
Metabolic measurements were log transformed to produce symmetric shaped distributions, and these measurements were analyzed via ANCOVA. The covariates included in the ANCOVA model were age, sex, and percent body fat. For all analyses, linear contrasts of the means were constructed to test our a priori hypotheses. Fisher’s restricted least significant differences criterion was used to maintain the a priori type I error rate at 0.05. 8-OH-dG, 8-hydroxy-2’-deoxyguanosine.
Participants on thiazolidinediones (2 of 13 diabetic Indians) were required to stop these medications for 3 weeks before the study. Of the other 11, 1 was on metformin alone, 2 were on sulfonylurea alone, 6 were on combination of sulfonylurea and metformin, and 2 were on diet alone. These other oral hypoglycemic agents were also stopped 5 days before the study, and during these days, their blood glucose levels were maintained between 4.4 and 7.8 mmol/l (80–140 mg/dl) by variable doses of short-acting insulin (11 of 13 diabetic Indians). Subjects were admitted to the General Clinical Research Center (current name Clinical Research Unit of Mayo Clinic CTSA) on the evening before the study. They received a standardized meal (16 kcal/kg fat-free mass [FFM] with carbohydrate:fat:protein 55:30:15) at 6:00 P.M. followed by insertion of a retrograde hand vein catheter for blood collections. A second catheter in the contralateral arm was used for infusion of insulin. Plasma glucose levels were maintained in type 2 diabetic participants between 5.0 and 6.7 mmol/l (90–120 mg/dl) using a standardized insulin infusion protocol starting at 6:00 P.M. At bedtime, a snack (5.5 kcal/kg FFM) was provided to all subjects to avoid long-term fasting. At 7:00 A.M., in both diabetic and nondiabetic participants, we collected a baseline blood sample and then started an infusion of insulin at a rate of 1.5 mU · kg−1 FFM · min−1; plasma glucose was monitored every 10 min; and a variable 40% dextrose infusion was adjusted to maintain glucose between 4.7 and 5.0 mmol/l (85–90 mg/dl) as previously described (13,18). Two vastus lateralis muscle percutaneous needle biopsies were performed in two different legs under local anesthesia as previously described (19) before and 8 h through the glucose clamp. Arterialized (20) blood samples were collected every 2 h for measurements of hormones and substrates.
Muscle mitochondrial studies.
Fresh muscle needle biopsy samples (~50 mg) (see above) were kept on ice in a saline-soaked gauze for immediate measurement of maximal mitochondrial ATP production rates (MAPR). Mitochondrial separation procedures and buffer solutions have been described previously (13). In brief, samples were homogenized in buffer A (100 mmol/l KCl, 50 mmol/l Tris, 5 mmol/l MgCl2, 1.8 mmol/l ATP, and 1 mmol/l EDTA) and spun at 1,020g in an Eppendorf 5417C centrifuge at 4°C. The supernatant was removed and spun at 10,000g. The new pellet was resuspended in buffer A and recentrifuged at 9,000g, and supernatant was removed. The resulting pellet was resuspended in buffer B (180 mmol/l sucrose, 35 mmol/l KH2PO4, 10 magnesium acetate, and 5 mmol/l EDTA) and kept on ice. This mitochondrial separation procedure yields a mitochondrial fraction that consists largely of subsarcolemmal mitochondria (19). Maximal MAPR were measured as previously described (13). The reaction mixture for MAPR measurement included a luciferin-luciferase ATP monitoring reagent (formula SL; BioThema, Dalarö, Finland), substrates for oxidation, and 35 μmol/l ADP. Substrates used were 10 mmol/l glutamate plus 1 mmol/l malate, 20 mmol/l succinate plus 0.1 mmol/l rotenone, 1 mmol/l pyruvate plus 0.05 mmol/l palmitoyl-l-carnitine plus 10 mmol/l α-ketoglutarate plus 1 mmol/l malate, 1 mmol/l pyruvate plus 1 mmol/l malate, and 0.05 mmol/l palmitoyl-l-carnitine plus 1 mmol/l malate with blank tubes used for measuring background activity. All reactions for a given sample were monitored simultaneously at 25°C for 20–25 min and calibrated with addition of an ATP standard using a BioOrbit 1251 luminometer (BioOrbit Oy, Turku, Finland). Mitochondrial integrity was monitored by measuring citrate synthase activity before and after freeze-thaw membrane disruption and the addition of Triton X-100. Accordingly, mitochondria were 94 ± 1% intact with no differences between treatments. In addition, citrate synthase activity and mitochondrial protein concentrations were determined (DC Protein Assay; Bio-Rad, Hercules, CA) as previously described (19). The MAPR values were normalized to the mitochondrial protein content.
Mitochondrial DNA copy number.
Mitochondrial DNA (mtDNA) was extracted from skeletal muscle and measured as previously described (13). Specifically, real-time PCR (Applied Biosystems 7900HT Sequence Detection System) was used to measure mtDNA copy numbers (14), using primer/probe sets to mtDNA-encoded NADH dehydrogenase 1 and cytochrome B genes. The abundance of each gene was normalized to 28S ribosomal DNA, which was co-amplified within the same reaction well.
Hormones, substrates inflammatory markers assays, and intramuscular triglycerides.
Glucose, insulin, glucagon, plasma lipids (21), and intramuscular triglyceride (IMTG) levels (22) were measured as previously described. Total adiponectin and high–molecular weight adiponectin concentrations were measured by the human adiponectin double antibody radioimmunoassay kit (Linco Research, St. Louis, MO) and the HMW Adiponectin ELISA kit (Linco Research), respectively. Highly sensitive C-reactive protein (hsCRP) was measured on the Hitachi 912 chemistry analyzer by a polystyrene particle enhanced immunoturbidimetric assay from DiaSorin (Stillwater, MN). Tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 concentrations were measured by chemiluminescent enzyme immunometric assay (Immulite, Diagnostic Products, Los Angeles, CA).
Microarray experiment.
Total RNA was extracted from skeletal muscle of individual subjects using Qiagen RNeasy Fibrous Tissue kit (Qiagen) treated with DNase and then processed for microarray experiments as previously described (15,23).
Microarray data analysis.
The arrays were normalized using invariant probe set normalization, and the expression measurement for each transcript was calculated using perfect match–only model based expression index by dChip (24). Genes with all “absent” calls by dChip across all compared samples were removed from further analysis. In addition, we did not consider the genes with average intensities ≤50 in both compared groups. Genes with a P value ≤0.05 were considered as potential candidates of differentially expressed genes between the compared groups and used as the “focus genes” for ingenuity pathway analysis (IPA). To avoid inflating pathways, only the nonredundant probe sets were used in the focus and reference gene lists.
Real-time PCR.
mRNA gene transcript levels for selected genes were examined using quantitative real-time PCR (Applied Biosystems 7900HT Sequence Detection System) as described previously (15,25). The abundance of each target gene was normalized to the signal for 28s ribosomal RNA, which was co-amplified within the same reaction well (15,25).
Immunoblotting.
The skeletal muscle protein abundance for peroxisome proliferator–activated receptor-γ coactivator (PGC) 1-α and PGC1-β were measured using standard immunoblotting techniques as previously described (15). PGC1-α and PGC1-β antibodies were purchased from Calbiochem (San Diego, CA) and Novus Biologicals (Littleton, CO), respectively.
Statistical analyses.
All statistical analyses were conducted using SAS software (version 9.1; SAS, Cary, NC). Data are presented as means ± SE. The body composition variables were analyzed on their traditional scales of measure, and these data were analyzed via ANOVA. Metabolic measurements were log transformed to produce symmetric shaped distributions, and these measurements were analyzed via ANCOVA. The covariates included in the ANCOVA model were age, sex, and percent body fat. The glucose infusion rate and insulin concentrations that were measured during the euglycemic clamp were analyzed via two-way, mixed-effects ANCOVA. The model specification for these models included parameters to estimate the group main effect, the time main effect, and group-by-time interaction on the mean response. The covariates included age, sex, and percent body fat. The ANCOVA model parameters for the glucose infusion rate and insulin concentrations during the euglycemic clamp were estimated based on the principles of restricted maximum likelihood, with the variance-covariance structure estimated in the compound symmetry form. The mRNA gene transcripts and protein abundance levels were log transformed to produce symmetric shaped distributions and were analyzed via ANOVA. For all analyses, linear contrasts of the means were constructed to test our a priori hypotheses. Fisher’s restricted least significant differences criterion was used to maintain the a priori type I error rate at 0.05.
RESULTS
Body composition.
The three groups had similar FFM and skeletal muscle mass, although the diabetic Indians had a significantly higher percentage of body fat than nondiabetic Northern European Americans (Table 1).
Hormonal, metabolic, and inflammatory measurements.
Fasting plasma glucose concentrations were higher in diabetic Indians than both nondiabetic groups, but no differences were noted between the two nondiabetic groups (Table 1). Fasting plasma insulin concentrations were higher in diabetic Indians than both nondiabetic groups and were higher in nondiabetic Indians than nondiabetic Northern European Americans (Table 1). No differences in plasma concentrations of glucagon were noted (Table 1).
Total cholesterol concentrations were not different among the three groups (Table 1), although both diabetic and nondiabetic Indians had lower HDL cholesterol than the nondiabetic Northern European Americans (Table 1). Triglyceride concentrations were higher in nondiabetic Indians than nondiabetic Northern European Americans (Table 1).
CRP, IL-6, and TNF-α concentrations were higher in diabetic Indians than in both nondiabetic groups (Fig. 1), and IL-6 concentrations were higher in nondiabetic Indians than in nondiabetic Northern European Americans (Fig. 1). Total and high molecular weight adiponectin concentrations were lower in diabetic Indians than both nondiabetic groups, and nondiabetic Indians had lower concentrations than Northern European Americans (Fig. 1).
FIG. 1.
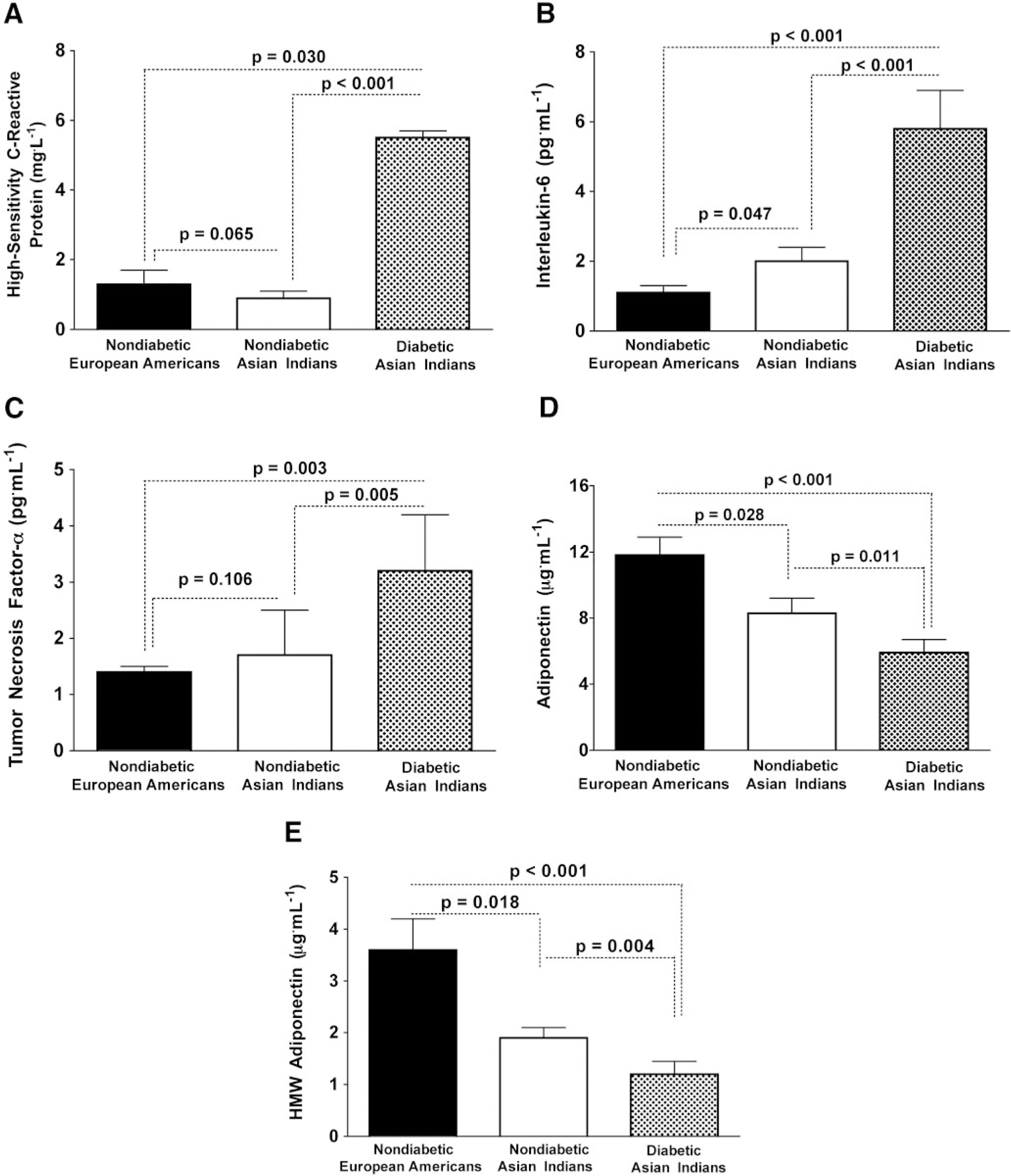
Plasma concentrations of inflammatory factors and adiponectin. Asian Indian diabetic patients have significantly higher hsCRP, IL, and TNF-α but lower total and high molecular weight adiponectin levels. Nondiabetic Indians also have lower total and high molecular weight adiponectin levels and higher IL-6 than the Northern European Americans.
Insulin sensitivity.
The glucose infusion rate needed to maintain identical plasma glucose concentrations (diabetic Indians, 5.01 ± 0.02 mmol/l; nondiabetic Indians, 5.06 ± 0.02; and nondiabetic Northern European Americans, 4.99 ± 0.03; NS) were substantially lower in diabetic Indians (4.5 ± 0.6 μmol · kg−1 FFM · min−1) than nondiabetic Indians (42.3 ± 4.0 μmol kg−1 FFM · min−1) and nondiabetic Northern European Americans (68.3 ± 4.2 μmol · kg−1 FFM · min−1) (P < 0.001) (Fig. 2). Nondiabetic Indians required a lower glucose infusion rate than the nondiabetic Northern European Americans (P < 0.002).
FIG. 2.
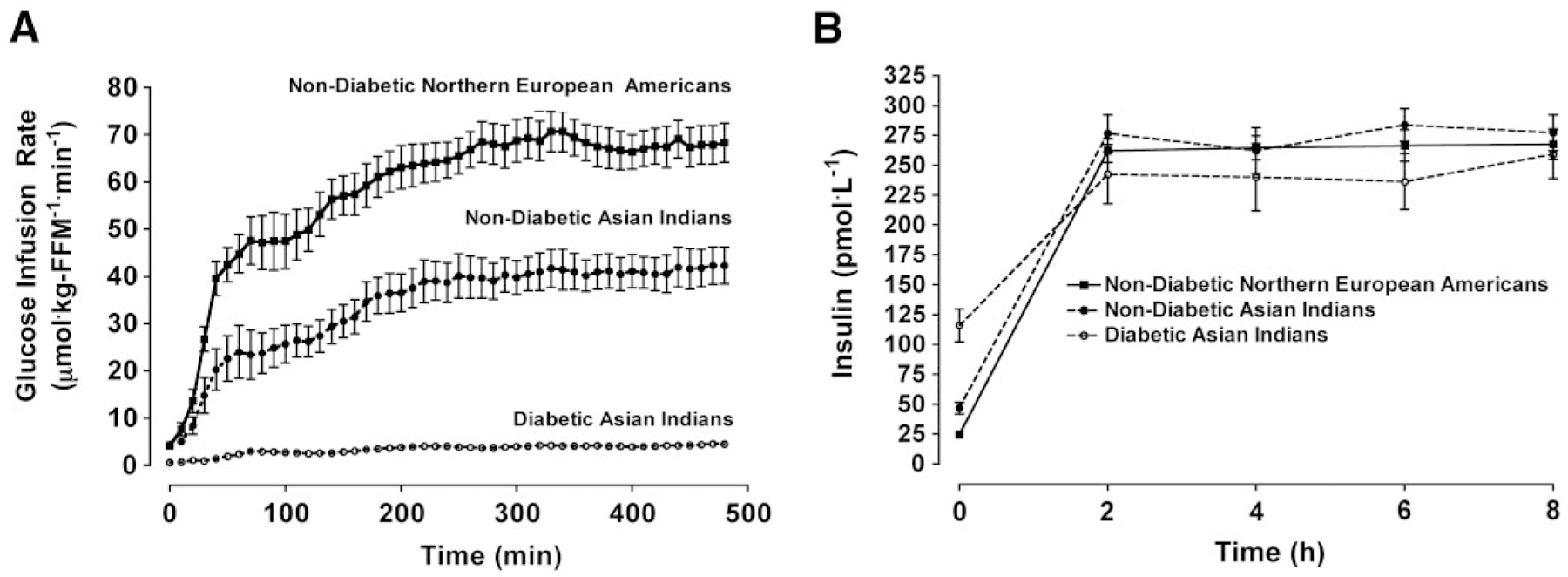
Glucose infusion rate (A) and insulin concentrations (B) during an 8-h euglycemic-hyperinsulinemic clamp in 13 each of nondiabetic European Americans, nondiabetic Asian Indians, and diabetic Asian Indians matched for sex. Data are presented as means ± SE. Mixed-effects ANCOVA models were used to test the main effect of group adjusted for age, sex, and percent fat. ANCOVA revealed a significant group × time interaction (P < 0.001) for the glucose infusion rate. Glucose area-under-the-curve values were significant different among the all three groups (P < 0.001; data not shown). ANCOVA did not reveal a significant group × time interaction (P > 0.05) for the insulin concentrations. Post hoc analyses were conducted using the Fisher’s least significant differences criterion.
Muscle mitochondrial data.
Measurements of maximal MAPR (corrected for mitochondrial protein content) using different substrate combinations in nondiabetic and diabetic Indians were significantly higher than those observed in nondiabetic Northern European Americans (P < 0.03 to <0.001 for measurements based on five substrate combinations) (Fig. 3). No differences between the Indian groups were noted. We also measured MAPR in muscle biopsy samples taken at baseline and after maintaining plasma glucose and insulin at the same concentrations for 8 h, and both showed higher ATP production rates in Indians (only baseline values shown). As expected, the 8-h infusion of insulin showed an overall trend for an insulin-induced increase in MAPR in all six substrate combinations; however, the insulin-induced elevations in MAPR did not reach the level of statistical significance among nondiabetic Northern European Americans (Fig. 3). Among nondiabetic and diabetic Indians, insulin resulted in a more variable MAPR response, and again, these insulin-induced changes in MAPR did not reach the level of statistical significance (except for the change in MAPR in the palmitate and malate condition in diabetic Indians). Moreover, there were no significant between-group differences with respect to the insulin-induced changes in MAPR. Previous studies showing increases in MAPR after insulin infusion in nondiabetic subjects occurred either when amino acids were infused (13) or when infused during somatostatin clamp (15). Muscle citrate synthase activity was also higher in Indians (P < 0.01), indicating higher mitochondrial oxidative capacity with no differences between two Indian groups (Fig. 3).
FIG. 3.
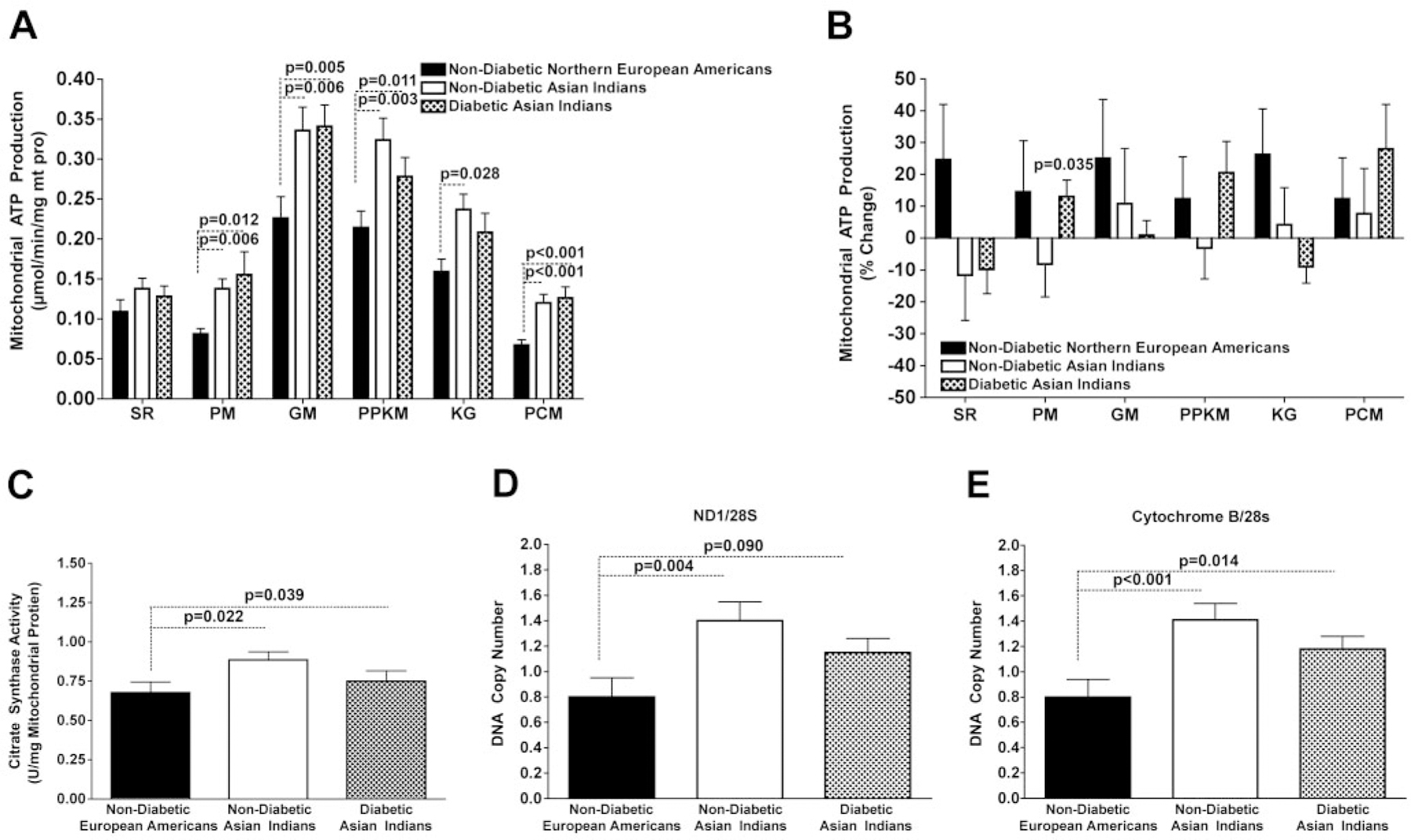
Baseline MAPRs (A) and the insulin-induced changes in MAPRs (B) obtained from 13 nondiabetic Northern European Americans, 13 nondiabetic Asian Indians, and 13 diabetic Asian Indians matched for sex. ATP production rate measurements were made in the presence of six different substrate combinations: succinate plus rotenone (SR), pyruvate plus malate (PM), glutamate plus malate (GM), palmitoyl-l-carnitine plus α-ketoglutarate plus malate (PPKM), α-ketoglutarate, and palmitoyl-l-carnitine plus malate (PCM). C: Baseline mitochondrial citrate synthase activity. D and E: mtDNA copy numbers assessed using primers and probes directed to mitochondrial-encoded genes NADH dehyrogenase 1 (D) and Cytochrome B (E) normalized to 28 s. ANCOVA was used to test the main effect of group adjusted for age, sex, and percent fat. Post hoc analyses were conducted using the Fisher’s least significant differences criterion when the main effect for group was significant at P < 0.05. The data were log normalized for analysis.
Both diabetic and nondiabetic Indians had higher mtDNA copy number (P < 0.02) than nondiabetic Northern European Americans (Fig. 3); there were no significant differences between the diabetic and nondiabetic Indians.
Gene transcript levels.
Microarray analyses were conducted on the nondiabetic groups to evaluate whether nondiabetic Indians had any pattern of gene transcript levels consistent with their enhanced mitochondrial function. Of the 19,285 present and nonredundant genes analyzed, 1,222 were differentially expressed between the nondiabetic Indians and nondiabetic Northern European Americans (Table 2). Subsequently, we used these 1,222 genes as “focus genes” for IPA, and the full set of 19,285 genes were used as reference genes for IPA. As shown in Fig. 4, the top canonical pathways associated with up- or downregulated genes in nondiabetic Indians compared with the nondiabetic Northern European Americans were listed. The complete list of altered canonical pathways by IPA is reported (Table 2). A cluster of genes (Table 3) involved in oxidative phosphorylation (OXPHOS) and the citric acid cycle were upregulated in the nondiabetic Indians compared with the nondiabetic Northern European Americans. We did not observe any significant differences in OXPHOS gene transcript levels between the nondiabetic and diabetic Indians. A list of the gene transcript levels is available in an online appendix at http://dx.doi.org/10.2337/db07-1556.
TABLE 2.
Results of IPA of differentially expressed genes between nondiabetic and diabetic Asian Indians and between nondiabetic Asian Indians and European Americans
| P | Genes | |
|---|---|---|
| Upregulated pathways in Asian Indians compared with European Americans | ||
| Inositol phosphate metabolism | 0.001 | PIK3CB, PLCD4, PIK3CD, PIP5K1C, ITPKB, OCRL |
| PI3K/AKT signaling | 0.009 | PPP2R4, PIK3CB, PIK3CD, PPP2R3B, PPM1J, RPS6KB2 |
| Oxidative phosphorylation | 0.013 | ATP5G3, COX7C, NDUFB4, NDUFAB1, COX11, COX8A, COX5A |
| Cysteine metabolism | 0.013 | LDHA, LDHC |
| Citrate cycle | 0.015 | ATP5G3, SUCLA2, IDH3G |
| Cardiac β-adrenergic signaling | 0.016 | PPP2R4, ADCY9, PLN, PPP2R3B, PPM1J |
| Pyruvate metabolism | 0.027 | LDHA, LDHC, HAGH, ACYP2 |
| IL-4 signaling | 0.030 | PIK3CB, PIK3CD, RPS6KB2 |
| ERK/MAPK signaling | 0.032 | PPP2R4, PIK3CB, PIK3CD, PPP2R3B, PTK2B, PPM1J |
| IL-2 signaling | 0.046 | PIK3CB, PIK3CD, PTK2B |
| Integrin signaling | 0.047 | PIK3CB, ARPC2, PIK3CD, ACTB, LAMB3, RALB, ITGA7 |
| Downregulated pathways in Asian Indians compared with European Americans | ||
| Methionine metabolism | 0.040 | BHMT, MAT2B, C17ORF83 |
| N-glycan degradation | 0.040 | MAN2C1, ASRGL1, FUCA2 |
| Upregulated pathways in diabetic compared with nondiabetic Asian Indians | ||
| NF-κB signaling | 0.002 | PIK3R1, NFKB2, EGF, TNFRSF1A, ZCCHC2, GSK3B, BMPR1A, CHUK, TTRAP, TRAF3 |
| PI3K/AKT signaling | 0.007 | GRB2, PIK3R1, NFKB2, YWHAQ, GSK3B, PPP2R5E, CHUK, PPP2R5A, EIF4E |
| Keratan sulfate biosynthesis | 0.011 | ST3GAL2, WDFY3, B4GALT1 |
| PTEN signaling | 0.017 | GRB2, PIK3R1, NFKB2, GSK3B, CHUK, YWHAH, PIK3R5 |
| PPAR signaling | 0.024 | PDGFA, GRB2, NFKB2, TNFRSF1A, PPARBP, CHUK |
| Selenoamino acid metabolism | 0.031 | MAT2A, AHCYL1, SEPHS2 |
| TGF-β signaling | 0.041 | TGFB2, GRB2, BMPR1A, TGFBR1, SMAD7 |
| Downregulated pathways in diabetic compared with nondiabetic Asian Indians | ||
| Serotonin receptor signaling | 0.003 | HTR6, HTR4, MAOB, HTR7, HTR2A, HTR1E, HTR3B, MAOA |
| Natural killer cell signaling | 0.014 | KIR2DL4, PRKCB1, KIR2DL5A, PIK3CD, MAPK1, SOS1, AKT3, KIR2DS1, SOS2, KIR2DL3, FCER1G, PTPN6, PRKCA, INPP5D, PAK3, ZAP70, SIGLEC7, PLCG2, PAK1 |
| Nitrogen metabolism | 0.033 | CA1 (includes EG:759), CA12, GCSH, CA6, PTPRG, CA8, CA11, GLS, TGM4, CA9 |
| Taurine and hypotaurine metabolism | 0.040 | BAAT, CDO1, CSAD, GGTL4, ACSS2 |
| Complement and coagulation cascades | 0.046 | SERPINA1, CFI, SERPINF2, F7, F2, F8, CFH, CR1, CD46, C1S, C5, FGB, FGA, VWF |
Pathways with P < 0.05 by IPA were considered significantly regulated. Genes are represented by HUGO symbols. ERK, extracellular signal–related kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; PPAR, peroxisome proliferator–activated receptor; TGF, transforming growth factor.
FIG. 4.
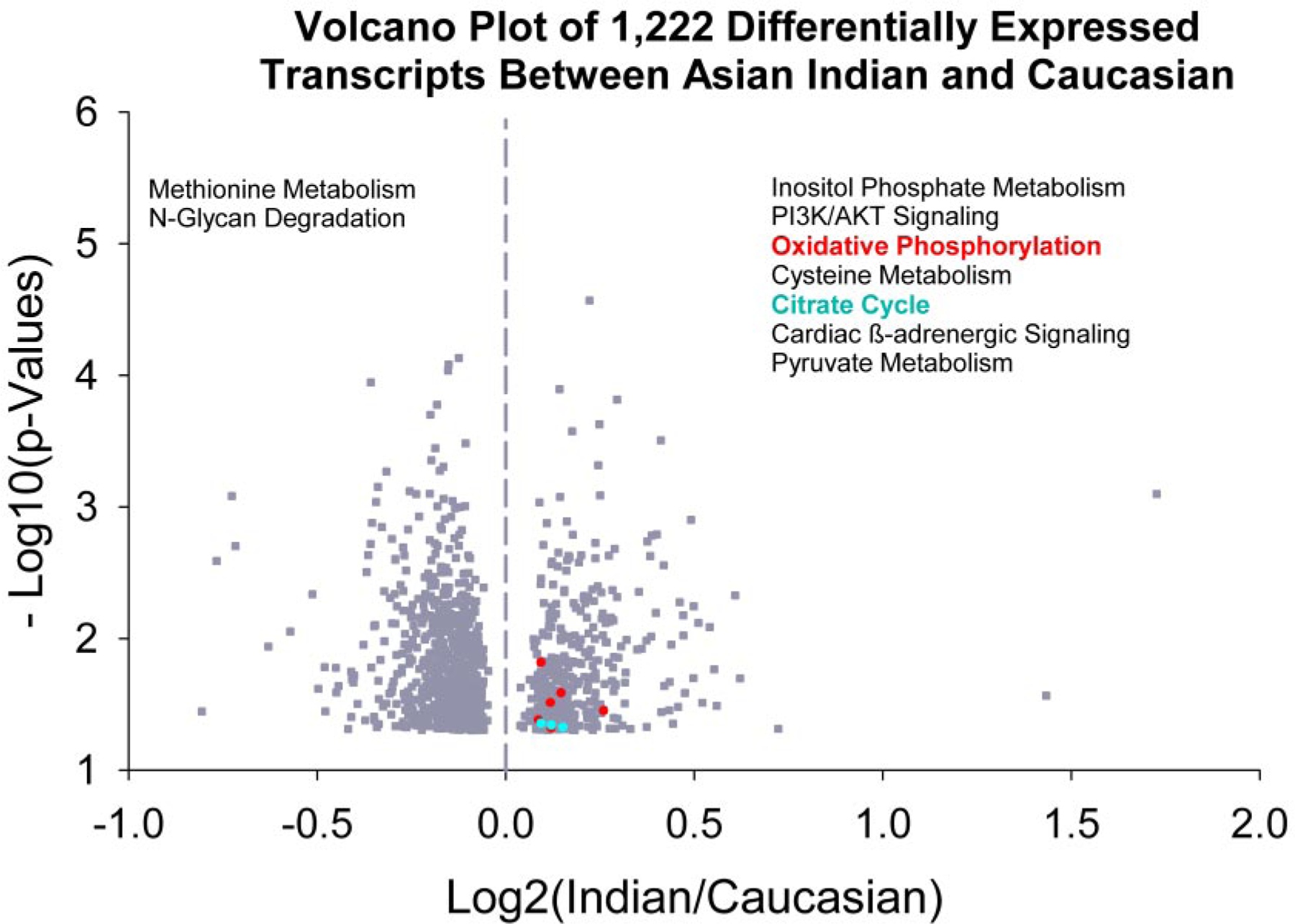
Skeletal muscle gene transcript profiles measured using Affymetrix HG-U133 plus 2.0 GeneChip arrays in nondiabetic Asian Indians and Northern European Americans. A volcano plot of 1,222 differentially expressed gene transcripts is shown. The altered canonical pathways based on IPA are shown with the pathways associated with higher expression of gene transcripts in Asian Indians on the right panel and the pathways associated with lowered expressed transcripts on the left panel. The OXPHOS (shown in red, P = 0.013) and citrate cycle (blue, P = 0.015) involving mitochondrial function gene transcripts are expressed at higher levels in Asian Indians. The list of other pathways significantly different between nondiabetic Asian Indians and Northern European Americans are given in Tables 2 and 3.
TABLE 3.
Genes involved in OXPHOS and citrate cycle, the two pathways that were upregulated in nondiabetic Asian Indians compared with nondiabetic European Americans
| Probe set | Nondiabetic European Americans | Nondiabetic Asian Indians | Nondiabetic Asian Indians/nondiabetic European Americans | P | Accession | Gene name | |
|---|---|---|---|---|---|---|---|
| Oxidative phosphorylation, P = 0.013 | |||||||
| NDUFB4 | 218226_s_at | 7659.66 | 8172.12 | 1.067 | 0.0151 | NM_004547 | NADH dehydrogenase (ubiquinone) 1 β subcomplex 4, |
| COX8A | 201119_s_at | 4522.81 | 5004.64 | 1.107 | 0.0259 | NM_004074 | Cytochrome c oxidase subunit 8A (ubiquitous) |
| COX11 | 239760_at | 95.54 | 103.71 | 1.085 | 0.0307 | AI198212 | COX11 homolog |
| COX5A | 229426_at | 127.38 | 152.44 | 1.197 | 0.0354 | BF196691 | Cytochrome c oxidase subunit Va |
| COX7C | 213846_at | 2179.92 | 2314.60 | 1.062 | 0.0416 | AA382702 | Cytochrome c oxidase subunit VIIc |
| ATP5G3 | 228168_at | 321.71 | 357.21 | 1.110 | 0.0472 | AU153583 | ATP synthase, H + transporting, mitochondrial F0 complex, subunit C3 (subunit 9) |
| NDUFAB1 | 202077_at | 3916.67 | 4256.75 | 1.087 | 0.0478 | NM_005003 | NADH dehydrogenase (ubiquinone) 1, α/β subcomplex, 1, |
| Citrate cycle, P = 0.015 | |||||||
| SUCLA2 | 202930_s_at | 1874.18 | 1999.18 | 1.067 | 0.0442 | NM_003850 | Succinate-CoA ligase, ADP-forming, β-subunit |
| IDH3G | 202471_s_at | 610.71 | 663.92 | 1.087 | 0.0454 | NM_004135 | Isocitrate dehydrogenase 3 (NAD+)-γ |
| ATP5G3 | 228168_at | 321.71 | 357.21 | 1.110 | 0.0472 | AU153583 | ATP synthase, H + transporting, mitochondrial F0 complex, subunit C3 (subunit 9) |
P values associated with each pathway were calculated by IPA. P values associated with each gene were calculated using paired t test.
RT-PCR and immunoblotting.
Table 4 presents the mRNA and transcript levels of selected target genes and the protein abundance for PGC1-α and PGC-β by study group. There were no significant differences in mRNA transcripts for PGC1-α, mitochondrial transcription factor-A (TFAM), nuclear respiratory factor-1 (NRF-1), estrogen-related receptor-α (ERR-α), myosin heavy chain (MHC)-I, MHC-IIa, or MHC-IIx among the three groups. In contrast, nondiabetic Northern European Americans had significantly higher levels of GLUT4 gene transcripts than both Indian groups (Table 4). There were no significant differences in protein abundance for PGC1-α and PGC1-β among the three groups.
TABLE 4.
Real-time PCR (mRNA) and Western blot (protein abundance) measurements in 13 each of sex-, age-, and BMI-matched nondiabetic Northern European Americans, nondiabetic Asian Indians, and diabetic Asian Indians
| 1. Nondiabetic European Americans | 2. Nondiabetic Asian Indians | 3. Diabetic Asian Indians | Group P | P for 1 vs. 2 | P for 1 vs. 3 | P for 2 vs. 3 | |
|---|---|---|---|---|---|---|---|
| mRNA* | |||||||
| PGC1-α | 0.53 ± 0.05 | 0.50 ± 0.04 | 0.48 ± 0.04 | 0.862 | — | — | — |
| TFAM | 1.19 ± 0.16 | 1.18 ± 0.13 | 1.30 ± 0.13 | 0.594 | — | — | — |
| NRF1 | 0.81 ± 0.06 | 0.86 ± 0.11 | 0.07 ± 0.11 | 0.249 | — | — | — |
| ERR-α | 0.97 ± 0.06 | 0.94 ± 0.06 | 1.06 ± 0.09 | 0.591 | — | — | — |
| MHC-I | 1.04 ± 0.09 | 1.12 ± 0.09 | 0.92 ± 0.10 | 0.244 | — | — | — |
| MHC-IIa | 1.08 ± 0.08 | 1.03 ± 0.11 | 1.30 ± 0.10 | 0.515 | — | — | — |
| MHC-IIx | 1.36 ± 0.29 | 0.90 ± 0.16 | 1.45 ± 0.20 | 0.113 | — | — | — |
| GLUT-4 | 1.34 ± 0.08 | 1.13 ± 0.05 | 1.06 ± 0.05 | 0.014 | 0.039 | 0.005 | 0.390 |
| Protein abundance† | |||||||
| PGC1-α | 1.60 ± 0.50 | 1.20 ± 0.20 | 2.00 ± 0.50 | 0.304 | — | — | — |
| PGC1-β | 1.30 ± 0.20 | 1.50 ± 0.20 | 1.90 ± 0.30 | 0.308 | — | — | — |
Data are means ± SEM.
mRNA measured by real-time PCR normalized to 28 s.
Protein abundance measured by Western blot normalized to a loading control. Measurements were log transformed to produce symmetric shaped distributions, and these measurements were analyzed via ANOVA. For all analyses, linear contrasts of the means were constructed to test our a priori hypotheses. Fisher’s restricted least significant differences criterion was used to maintain the a priori type I error rate at 0.05.
DISCUSSION
The current study demonstrated that nondiabetic Northern European Americans were substantially more insulin sensitive than both diabetic and nondiabetic Indians. However, irrespective of their diabetic status, the Indians had higher skeletal muscle mitochondrial OXPHOS capacity as demonstrated by the higher abundances of mtDNA, mRNA of OXPHOS genes, oxidative enzyme activity (citrate synthase), and maximal ATP production rate. Despite being more insulin-resistant than nondiabetic Indians, the diabetic Indians had similar skeletal muscle mitochondrial OXPHOS capacity.
We measured mtDNA copy number using two different primer/probe sets and found that Indians, irrespective of their diabetic status had significantly higher mtDNA copy number than nondiabetic Northern European Americans. No differences were noted in mtDNA copy numbers between diabetic and nondiabetic Indians, which is consistent with what has been observed in diabetic and nondiabetic Northern European American populations (15). A close correlation has been previously observed between mtDNA abundance, its transcript levels, and ATP production capacity in skeletal muscle of people of wide range of age groups (14). The observation of higher maximal MAPR corrected for mitochondrial protein content in Indians using different substrates and citrate synthase activity support greater capacity to produce ATP, and this greater capacity was related to increased mtDNA abundance. It is well known that mtDNA only encodes 13 proteins involved in mitochondrial functions, whereas the rest of proteins are encoded by nuclear genes. The results from gene array analysis, which measures nuclear encoded gene transcripts, demonstrated that clusters of genes involved in OXPHOS and the citric acid cycle were upregulated in the nondiabetic Indians compared with the nondiabetic Northern European Americans. Among the differentially expressed genes between nondiabetic Indians and Northern European Americans are upregulation of genes involved in pyruvate metabolism and citric acid cycle, which are consistent with observation of increase in OXPHOS pathway. Upregulation of integrin pathway (26) is consistent with the associated upregulation of ERK/MAPK signaling and may be involved in cell proliferation. Similarly, the cytokine pathways (IL-2 and IL-6) are inflammatory pathways and are consistent with the overall increase in circulating inflammatory factors. Although cytokines and integrin pathways may be involved in cell cycling, it is not clear whether they may have contributed to increased mtDNA copy number. It is well established that both nuclear and mitochondrial genes involved in energy metabolism are well regulated and coordinated (27), and the results from the current study clearly support the hypothesis that higher mtDNA copy number and nuclear-encoded gene transcript levels cause the increased mitochondrial oxidative capacity.
In the current study, we measured mRNA and protein expression of PGC-1α in skeletal muscle and could not detect any significant differences among diabetic Indians, nondiabetic Indians, or nondiabetic Northern European Americans. Moreover, the finding that there was not a significant difference in mRNA transcript levels between diabetic and nondiabetic Indians is consistent with our findings in diabetic and nondiabetic Northern European Americans (15). We also did not find any differences of those nuclear regulators of mitochondrial biogenesis between Northern European Americans and Indians. Based on previous studies (15,28), it appears that mRNA levels of these regulatory genes change based on insulin levels. Both diabetic and nondiabetic Indians had lower GLUT4 mRNA transcript levels than nondiabetic Northern European Americans, which is consistent with their level of insulin resistance. However, we did not observe a significant difference between diabetic and nondiabetic Indians.
The potential implication of higher mitochondrial OXPHOS capacity on energy needs of Indians remains to be determined. Previous studies across various species, including yeast (29), rodents (30), and humans, (31), demonstrated enhanced mitochondrial biogenesis in response to caloric restriction. However, it remains unclear from the current study whether observed higher muscle mitochondrial oxidative capacity in Indians represents an adaptive process or whether other genetic factors are involved in this metabolic pattern among Indians.
The current results clearly demonstrated that irrespective of their diabetic status, Indians were substantially less insulin sensitive than their nondiabetic Northern European American counterparts, despite having enhanced mitochondrial function and higher mtDNA copy numbers. However, the current study did not specifically address the site (i.e., liver versus skeletal muscle) of insulin resistance in Indians. It is possible a component of insulin resistance among Indians could be partly localized to liver because it has been shown that Indians have high prevalence of nonalcoholic fatty liver (6). Insulin resistance and reduced muscle MAPR have been reported to occur with aging (14,16). This age-related muscle mitochondrial dysfunction, however, occurs in association with a concomitant reduction in mtDNA abundance (14); and in a selected cohort of offspring of type 2 diabetic patients, reduced mitochondrial density was associated with reduced MAPR and insulin resistance (32). In contrast, we previously reported that nondiabetic and type 2 diabetic Northern European Americans, have similar muscle MAPR and mtDNA copy numbers at postabsorptive insulin levels, however, the diabetic Northern European American subjects failed to increase their MAPR in response to low levels of insulin and only increased when insulin levels reached high physiological levels (13,15). This failure to increase muscle MAPR occurred in association with reduced insulin-induced glucose disposal, indicating insulin resistance (15). A reduction in skeletal muscle mRNA abundance of OXPHOS genes has been reported (28,33,34) by insulin treatment (33).
Consistent with the previous reports of association between insulin resistance and IMTG (35), we found that diabetic Indians have substantially higher IMTG levels than their nondiabetic counterparts (Table 1). Despite observing substantially lower insulin sensitivity in nondiabetic Indians, we did not find any higher IMTG in this group in comparison with nondiabetic Northern European Americans. We also observed that the nondiabetic Northern European Americans had higher plasma concentrations of total and high molecular weight adiponectin than the nondiabetic and diabetic Indians. Moreover, the nondiabetic Indians had higher total and high molecular weight adiponectin concentrations than the diabetic Indians. Adiponectin, a hormone secreted from fat cells, has been shown to correlate with insulin sensitivity (36). The inflammatory markers such as CRP, TNF-α, and IL-6 were higher in the diabetic Indians (Table 1), and the nondiabetic Indians also tended to have higher levels of these inflammatory markers (37) than the nondiabetic Northern European Americans, consistent with the reported link between insulin resistance and inflammation (38). Finally, the observation of lower concentrations of HDL cholesterol in the Indians is also consistent with the reported higher susceptibility to coronary artery disease among Indians (39).
In conclusion, the present data indicate that despite being more insulin resistant, diabetic Indians had similar muscle OXPHOS capacity as nondiabetic Indians, demonstrating that diabetes per se does not cause mitochondrial dysfunction. Moreover, irrespective of their diabetic status, Indians had higher OXPHOS capacity than Northern European Americans, despite being substantially more insulin resistant, indicating that mitochondrial dysfunction cannot account for insulin resistance in Asian Indians. The present data also suggest that Asian Indians might have a greater propensity for intramyocellular triglyceride accumulation than Northern European Americans, although the molecular mechanisms remain to be elucidated.
Supplementary Material
ACKNOWLEDGMENTS
B.A.I. has received Public Service Grant T32-DK-07352-28. K.S.N. has received a David Murdock Dole Professorship. This work has been supported by Public Service Grants UL1 RR24150 (Mayo Clinic Center for Clinical and Translational Research-CTSA), R01-DK-41973, and Centers for Disease Control and Prevention Grant 10 awarded through the American Association of Physicians of Indian Origin (AAPI).
We thank the Mayo Clinic Clinical Research Unit nursing and nutrition staff, Immunochemistry and Advanced Genomic Technology Center, Jane Kahl and Dawn Morse for skilled technical assistance, and Joseph Melton, MD, PhD, and James Patrie, MS, for useful discussions.
Glossary
- CRP
C-reactive protein
- FFM
fat-free mass
- hsCRP
highly sensitive CRP
- IL
interleukin
- IMTG
intramuscular triglyceride
- IPA
ingenuity pathway analysis
- MAPR
mitochondrial ATP production rate
- mtDNA
mitochondrial DNA
- OXPHOS
oxidative phosphorylation
- PGC
peroxisome proliferator–activated receptor-γ coactivator
- TNF-α
tumor necrosis factor-α
REFERENCES
- 1.Zimmet P, Alberti KGMM, Shaw J: Global and societal implications of the diabetes epidemic. Nature 414:782–787, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Novak K: Group calls for big increase in diabetes research. Nat Med 5:364, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Wild S, Roglic G, Green A, Sicree R, King H: Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Ramachandran A, Snehalatha C, Mary S, Mukesh B, Bhaskar AD, Vijay V, Indian Diabetes Prevention Program: The Indian Diabetes Prevention Programme shows that lifestyle modification and metformin prevent type 2 diabetes in Asian Indian subjects with impaired glucose tolerance (IDPP-1). Diabetologia 49:289–297, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran A, Snehalatha C, Vijay V: Low risk threshold for acquired diabetogenic factors in Asian Indians. Diabetes Res Clin Pract 65:189–195, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, Cobelli C, Shulman G: Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci U S A 103:18273–18277, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandalia M, Lin P, Seenivasan T, Livingston EH, Snell PG, Grundy SM, Abate N: Insulin resistance and body fat distribution in South Asian men compared to Caucasian men. PLoS ONE 2:e812, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedges SB: A start for population genomics. Nature 408:652–653, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Ingman M, Kaessmann H, Pääbo S, Gyllensten U: Mitochondrial genome variation and the origin of modern humans. Nature 408:708–712, 2000 [DOI] [PubMed] [Google Scholar]
- 10.Neel JV: Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress. ” Am J Hum Genet 4:353–362, 1962 [PMC free article] [PubMed] [Google Scholar]
- 11.Hales CN, Barker DJ: Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35:595–601, 1992 [DOI] [PubMed] [Google Scholar]
- 12.DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP: The effect of insulin on the disposal of intravenous glucose: results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30:1000–1007, 1981 [DOI] [PubMed] [Google Scholar]
- 13.Stump CS, Short KR, Bigelow ML, Schimke JC, Nair KS: Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc Natl Acad Sci U S A 100:7996–8001, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Short KR, Bigelow ML, Kahl JC, Singh R, Coenen-Schimke JM, Raghavakaimal S, Nair KS: Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 102:5618–5623, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow M, Nair KS: Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 55:3309–3319, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Petersen KF, Befroy D, Sufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman G: Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300:1140–1142, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petersen KF, Shulman GI: Etiology of insulin resistance. Am J Med 119:S10–S16, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeFronzo RA, Tobin JD, Andres R: Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab 237:E214–E223, 1979 [DOI] [PubMed] [Google Scholar]
- 19.Rooyackers OE, Adey DB, Ades PA, Nair KS: Effect of age in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A 93:15364–15369, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Copeland KC, Kenney FA, Nair KS: Heated dorsal hand vein sampling for metabolic studies: a reappraisal. Am J Physiol Endocrinol Metab 263: E1010–E1014, 1992 [DOI] [PubMed] [Google Scholar]
- 21.Dhatariya KK, Bigelow ML, Nair KS: Effect of dehydroepiandrosterone replacement on insulin sensitivity and lipids in hypoadrenal women. Diabetes 54:765–769, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Guo Z, Mishra P, Macura S: Sampling the intramyocellular triglycerides from skeletal muscle. J Lipid Res 42:1041–1048, 2001 [PubMed] [Google Scholar]
- 23.Sreekumar R, Halvatsiotis P, Schimke JC, Nair KS: Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 51:1913–1920, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Li C, Wong WH: Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A 98:31–36, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balagopal P, Schimke JC, Ades PA, Adey D, Nair KS: Age effect on transcript levels and synthesis rate of muscle MHC and response to resistance exercise. Am J Physiol Endocrinol Metab 280:E203–E208, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Giancotti FG, Ruoslahti E: Integrin signaling. Science 285:1028–1033, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Puigserver P, Spiegelman BM: Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ: Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100:8466–8471, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L: Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418:344–348, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Sreekumar R, Unnikrishnan J, Fu A, Nygren J, Short KR, Schimke JC, Barazzoni R, Nair KS: Effects of caloric restriction on mitochondrial function and gene transcripts in rat muscle. Am J Physiol Endocrinol Metab 283:38–43, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, Smith SR, Ravussin E, for the CALERIE Pennington Team: Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Medicine 4:e76, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI: Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 115:3587–3593, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sreekumar R, Halvatsiotis P, Nair KS: Gene expression profile in skeletal muscle of type 2 diabetic patients: a study using genechip array (Abstract). Diabetes 49 (Suppl. 1):A84, 2000 [Google Scholar]
- 34.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC: PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34:267–273, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Petersen KF, Shulman GI: Etiology of insulin resistance. Am J Med 119:S10–S16, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY: APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Shoelson SE, Lee J, Goldfine AB: Inflammation and insulin resistance. J Clin Invest 116:1793–1801, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotamisligil GS: Inflammation and metabolic disorders. Nature 444:860–867, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Mohan V, Shanthirani CS, Deepa M, Deepa R, Unnikrishnan RI, Datta M: Mortality rates due to diabetes in a selected urban south Indian population: the Chennai Urban Population Study (CUPS-16). J Assoc Physicians India 54:113–117, 2006 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.