

Summary
Astrocytes tile the central nervous system and are widely implicated in brain diseases, but the molecular mechanisms by which astrocytes contribute to brain disorders remain incompletely explored. By performing astrocyte gene expression analyses following 14 experimental perturbations of relevance to the striatum, we discovered that striatal astrocytes mount context-specific molecular responses at the level of genes, pathways, and upstream regulators. Through data mining, we also identified astrocyte pathways in Huntington’s disease (HD) that were reciprocally altered with respect to the activation of striatal astrocyte G-protein coupled receptor (GPCR) signaling. Furthermore, selective striatal astrocyte stimulation of the Gi-GPCR pathway in vivo corrected several HD-associated astrocytic, synaptic, and behavioural phenotypes with accompanying improvement of HD-associated astrocyte signaling pathways, including those related to synaptogenesis and neuroimmune functions. Overall, our data show that astrocytes are malleable, utilising context-specific responses that can be dissected molecularly and used for phenotypic benefit in brain disorders.
Keywords: astrocyte, striatum, RNA-sequencing, HD, GPCR, synaptogenic, behaviour, neuroimmune
Graphical Abstract
eTOC Blurb
Using astrocyte RNA-sequencing, Yu and Nagai et al. demonstrate context-specific molecular responses of striatal astrocytes to multiple experimental perturbations. They identify reciprocal changes between Huntington’s disease (HD) and activation of the astrocyte Gi-GPCR pathway. Selective stimulation of the striatal astrocyte Gi-GPCR pathway corrects several HD-associated cellular and behavioral phenotypes.
Introduction
Astrocytes tile the entire central nervous system and form close interactions with neurons, blood vessels and other glial cells. Astrocytic contributions to physiology and disease have been discussed for decades ever since these cells were discovered (Barres, 2008). Such roles are critical to understand, because pathophysiological and genetic studies show that astrocytes are broadly involved in brain disorders, including Huntington’s disease (HD) (Grubman et al., 2019; Itoh et al., 2018; Kelley et al., 2018; Mathys et al., 2019; Song et al., 2019; Velmeshev et al., 2019; Wilton and Stevens, 2020).
Despite progress, major open questions remain to be addressed and relate to how astrocytes respond to different challenges, how they contribute to specific diseases, and if their contributory molecular pathways can be identified and utilised for disease modification. In other words, how does a defined population of astrocytes respond to different challenges, can such responses be quantified molecularly, and can they be exploited for correction of disease phenotypes with contributory astrocytic roles? We addressed these questions for striatal astrocytes and their roles in HD with a focus on mouse models of this disorder.
HD is a progressive neurodegenerative disease characterized by motor, cognitive, and psychiatric symptoms. HD is caused by a polyglutamine expansion in Huntingtin (HTT) that results in the expression of a mutant protein (mHTT) (Bates et al., 2015). mHTT is expressed in cells throughout the body, but the striatum especially undergoes marked dysfunction and atrophy. Striatal astrocytes express mHTT in HD mouse models and in postmortem human tissue (Bradford et al., 2009; Bradford et al., 2010), and several astrocytic mechanisms that contribute to HD are known (Khakh et al., 2017). Recently, systems biology approaches have begun to shed light on broader astrocyte alterations in HD (Al-Dalahmah et al., 2020; Diaz-Castro et al., 2019; Hodges et al., 2006; Lee et al., 2020). These insights raise the possibility that it might be possible to molecularly dissect, understand and exploit astrocyte mechanisms for beneficial effect and for exploring HD pathogenesis. Furthermore, since HD is caused by a known molecular defect and affects well-characterized neural circuitry, exploring astrocytes in HD may provide insights of relevance to other brain disorders.
Since astrocytes differ markedly between brain nuclei (Khakh and Deneen, 2019), we challenged astrocytes in vivo and studied their responses in a single brain area – the adult mouse striatum (Graybiel and Grafton, 2015; Khakh, 2019). We used fourteen carefully selected and controlled experimental perturbations (EPs) associated with different physiological pathways and disease contexts. The EPs were chosen to be relevant to striatal astrocytes and their contributions to disease, with an emphasis on HD (STAR Methods). By performing experiments to evaluate astrocyte gene expression changes following each of the fourteen EPs, we found that the molecular responses of striatal astrocytes were context-specific at the level of genes, pathways, and upstream transcriptional signaling regulators. Detailed evaluations of the EPs revealed pathways in HD that were reciprocally transformed with respect to striatal astrocyte G-protein coupled receptor (GPCR) signaling in mice. This allowed us to design an experimental approach in HD model mice using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) (Roth, 2016), which led to the finding that selective stimulation of the Gi-GPCR pathway in striatal astrocytes corrected several HD-associated phenotypes partly through the activation of a synaptogenic cue and rescue of excitatory synaptic transmission, and partly through additional responses such as those related to neuroimmune function. By mining striatal astrocyte gene expression data, we identified new endogenous astrocyte GPCRs that could be explored in future studies as potential targets for therapeutics and for modifying disease phenotypes in HD and possibly other disorders.
Results
This study has three parts. In the first part (Figures 1–2), we explored how striatal astrocytes responded at a molecular level to multiple EPs (STAR Methods). In the second part, we used those data to identify a GPCR-based strategy to explore if the changes observed for striatal astrocytes could be manipulated to produce beneficial effects for a HD mouse model (Figures 3, 4, and 5). In the third part, we investigated molecular mechanisms underlying improvements in HD model mice following astrocyte GPCR activation (Figures 6–7) and identified endogenous GPCRs as targets for future study (Figure 8). All the gene expression data are in Excel files S1, S2 and S5 and are deposited at the Gene Expression Omnibus (Table S1). We included analysis of three recently published datasets (Diaz-Castro et al., 2019; Nagai et al., 2019; Yu et al., 2018) and the generation of ten new gene expression datasets (Key Resources Table). Raw data values and statistical analyses are in the figures and in Excel files S3 & S4.
Figure 1: Context-specific striatal astrocyte molecular responses following physiological and pathological experimental perturbations (EPs).
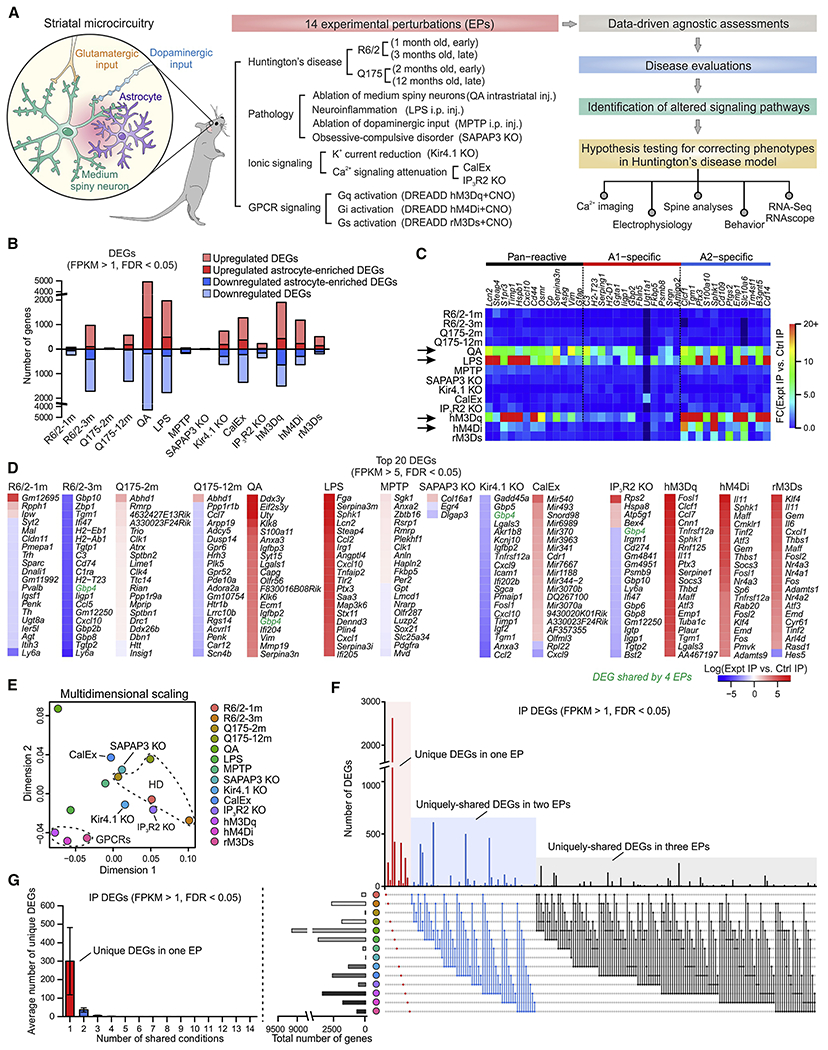
(A) Schematic of the experimental strategy with 14 EPs from four groups, assessing HD, cellular pathology, altered ionic signaling and GPCR signaling. We assessed EPs in relation to HD to identify altered common pathways and used such evaluations to test hypotheses using physiology and behaviour. n = 4 mice were used for each EP for RNA-seq along with 4 controls. (B) Numbers of differentially expression genes (DEGs) (FDR < 0.05, FPKM > 1) that were upregulated and downregulated across 14 EPs. DEGs that were more than 2-fold higher in the IP than the input were designated as astrocyte-enriched. (C) Heat map of relative expression levels of 38 known reactive astrocyte markers in striatal astrocytes compared with control across 14 EPs. (D) Top 20 DEGs (FPKM > 5 with FDR < 0.05) in striatal astrocytes ranked by differential expression log ratio compared with control levels, revealing most profound changes triggered by different EPs. (E) Multidimensional scaling plot based on the top 1000 variable genes showed clear differences between 14 EPs. (F) The UpSet plot shows numbers of DEGs under different combinations of EPs up to three conditions. The most numbers of DEGs were found to be uniquely expressed under a single EP (Figure S3). (G) Average numbers of unique DEGs (FPKM > 1 with FDR < 0.05) in striatal astrocytes shared between various numbers of EPs. Data are shown as mean ± SEM. See also Figure S4. Lists of DEGs and WGCNA modules are reported in Excel files S1 and S2.
Figure 2: Similarity between mouse and human striatal astrocytes, relationship to disease, and reciprocal pathway changes between Gi-GPCR activation and HD.
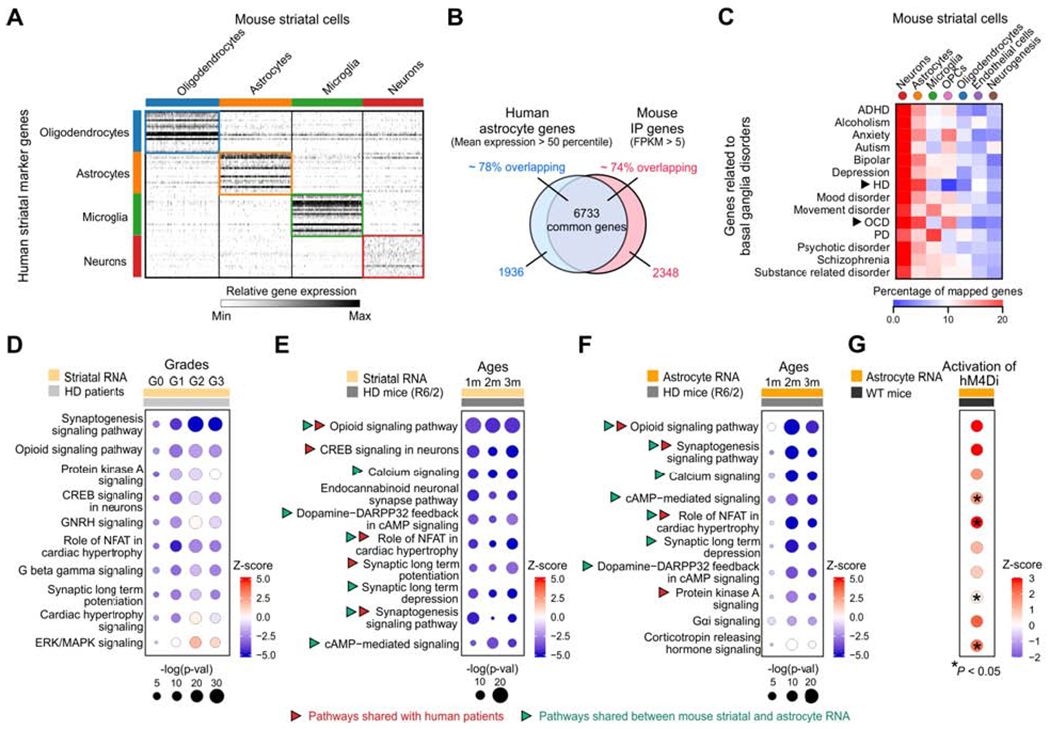
(A) Heat map of relative expression of human striatal marker genes for oligodendrocytes, astrocytes, microglia and neurons in mouse striatal scRNA-seq data (Figure S5). (B) The Venn diagram of overlap between highly expressed astrocyte genes in the human striatum (mean expression > 50 percentile) and mouse striatal IP sample (FPKM > 5). (C) The percentage genes related to basal ganglia disorders mapped onto mouse striatal scRNA-seq data based on the top 1000 cell type marker genes for the 7 major cell types. Arrows indicated the two disorders (HD and OCD) with the highest of astrocyte genes mapped. (D) Dot plots of the top 10 common canonical pathways in caudate nucleus RNA from HD patients that were altered at different grades (Diaz-Castro et al., 2019; Hodges et al., 2006), identified by IPA. Sizes of dots represent log-transformed p-value and colors indicate either pathway activation (red) or inhibition (blue) based on z-scores. (E,F) Top 10 common canonical pathways from striatal tissue bulk RNA-seq (E) and astrocyte-specific RNA-seq data (F) that were altered in HD mouse model (R6/2) at different ages (1 m, 2 m and 3 m of age). Red arrows indicate pathways shared in human and mouse RNA-seq data, while green arrows indicate pathways shared between mouse striatal RNA and astrocyte RNA. (G) Effects of hM4Di DREADD activation on the top 10 altered common pathways identified from astrocyte RNA-seq of R6/2 mice.
Figure 3: Astrocyte Gi-GPCR pathway activation corrected astrocyte alterations in HD mice.
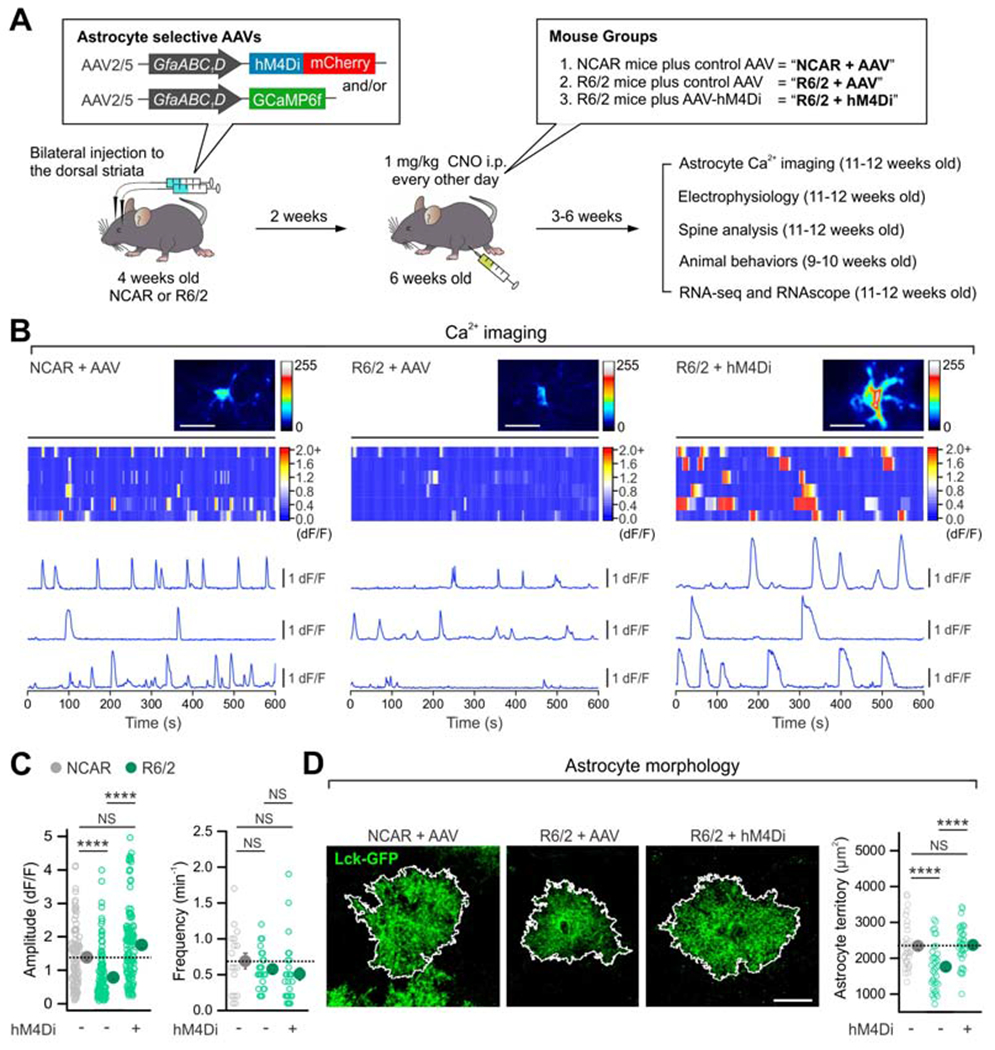
(A) Schematic of experimental design. (B) Maximum intensity projection images, kymographs and ΔF/F traces of astrocyte intracellular Ca2+ signals from indicated groups. (C) The reduced peak amplitude of astrocyte Ca2+ signals in R6/2 mice was rescued with hM4Di activation (left). The frequency of Ca2+ signals was not different between the groups (right). n = 111-138 Ca2+ events from 18-27 astrocytes from 4 mice. (D) hM4Di activation in striatal astrocytes increased the territory size of Lck-GFP-expressing striatal astrocytes in R6/2 mice (n = 33-37 astrocytes from 4 mice per group). Scale bars: 20 μm. Data are shown as mean ± SEM. In some cases, the SEM symbol is smaller than the symbol for the mean. Full details of n numbers, precise P values and statistical tests and raw values are reported in Excel files S3 and S4. **** indicates P < 0.0001, NS indicates not significantly different.
Figure 4: Astrocyte Gi-GPCR pathway activation corrected MSN synaptic dysfunctions in HD mice.
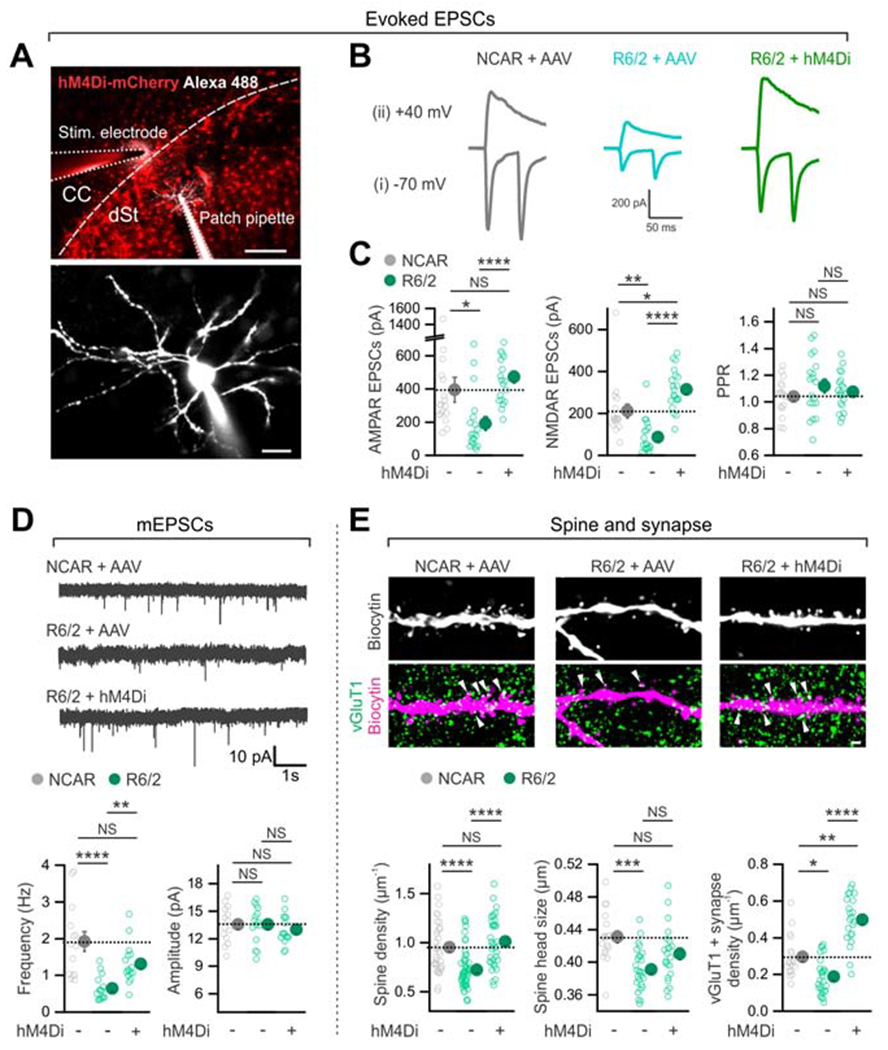
(A) Top, representative image of a parasagittal slice of mouse brain showing the expression of hM4Di in dorsal striatal astrocytes, the stimulating electrode and the location of a MSN that was recorded with whole-cell patch clamp. The MSN was filled through the patch-pipette with Alexa Fluor 488. CC, the corpus callosum. dSt, the dorsal striatum. Bottom, a high magnification image of the MSN shown in the top panel. (B) Traces of evoked AMPA receptor-mediated EPSCs due to paired stimuli at −70 mV (i) and NMDA receptor-mediated EPSCs due to single stimuli at +40 mV (ii) from the indicated three experimental groups. (C) Astrocyte hM4Di activation significantly increased amplitudes of AMPAR and the NMDAR EPSCs in R6/2 mice. There was no significant change in PPR. (D) Decreased mEPSC frequency in the R6/2 + AAV group was rescued by hM4Di activation in striatal astrocytes in R6/2 mice (n = 14-16 MSNs from 4 mice per group). There was no difference in mEPSC amplitudes between groups. (E) The decreased density and size of MSN spines and vGluT1-positive cortico-striatal excitatory synapses onto MSNs in R6/2 mice was rescued by astrocyte hM4Di activation in R6/2 mice (n = 34-54 dendritic segments from 4 mice per group for the left graph and 18-27 dendritic segments from 4 mice per group for the middle and right graphs). Scale bars: 200 μm in upper panel of A, 20 μm in lower panel of A, and 2 μm in E. Data are shown as mean ± SEM. In some cases, the SEM ymbol is smaller than the symbol for the mean. Full details of n numbers, precise P values and statistical tests and raw values are reported in Excel files S3 and S4. * indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.001, **** indicates P < 0.0001, NS indicates not significantly different.
Figure 5: Astrocyte Gi-GPCR pathway activation corrected behavioral phenotypes in HD mice.
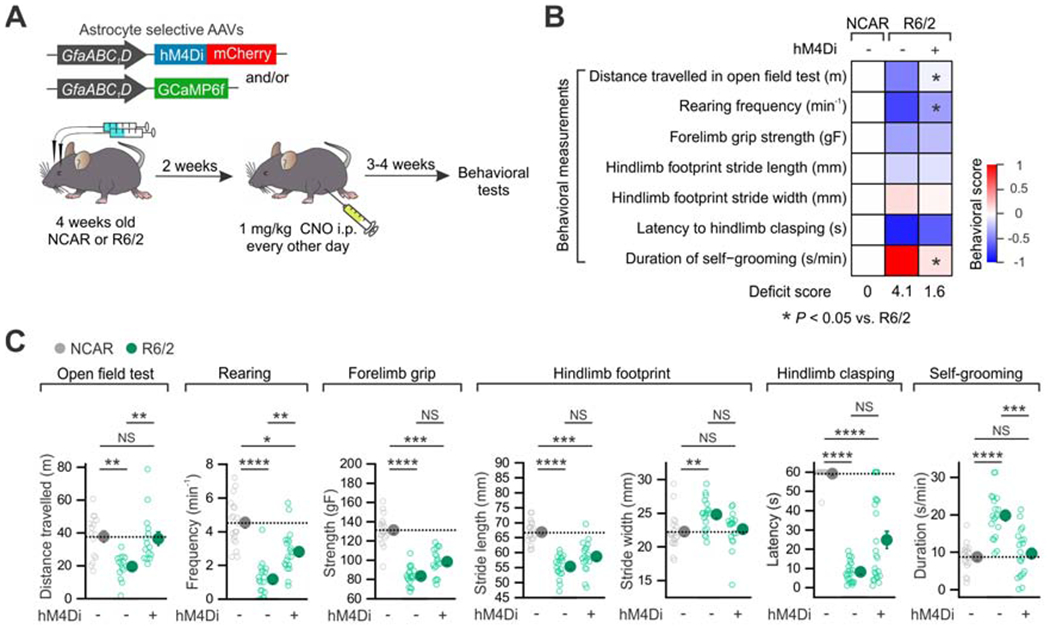
(A) Experimental procedure of AAV delivery, drug treatment and behavioral evaluations. (B) Heat map summarizes the 7 behavioral parameters assessed in the indicated groups: (i) the distance travelled over 30 min, (ii) the frequency of rearing in an open field chamber, (iii) the grip strength of forelimbs, (iv) the stride length, (v) the stride width of hind limbs, (vi) the latency to clasp hind limbs upon tail suspension, and (vii) the duration of self-grooming (n = 14-24 mice each). Activation of astrocyte hM4Di in R6/2 produced significant improvement in three behavioral deficits, which is summarized by the aggregate deficit score and by the behavioral score heat map (see STAR Methods). (C) Individual data points of seven behavioral parameters assessed. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Full details of n numbers, precise P values, statistical tests and the raw values are reported in Excel files S3 and S4.
Figure 6: Astrocyte Gi-GPCR pathway activation reversed disease-associated astrocyte transcriptomes in R6/2 mice.
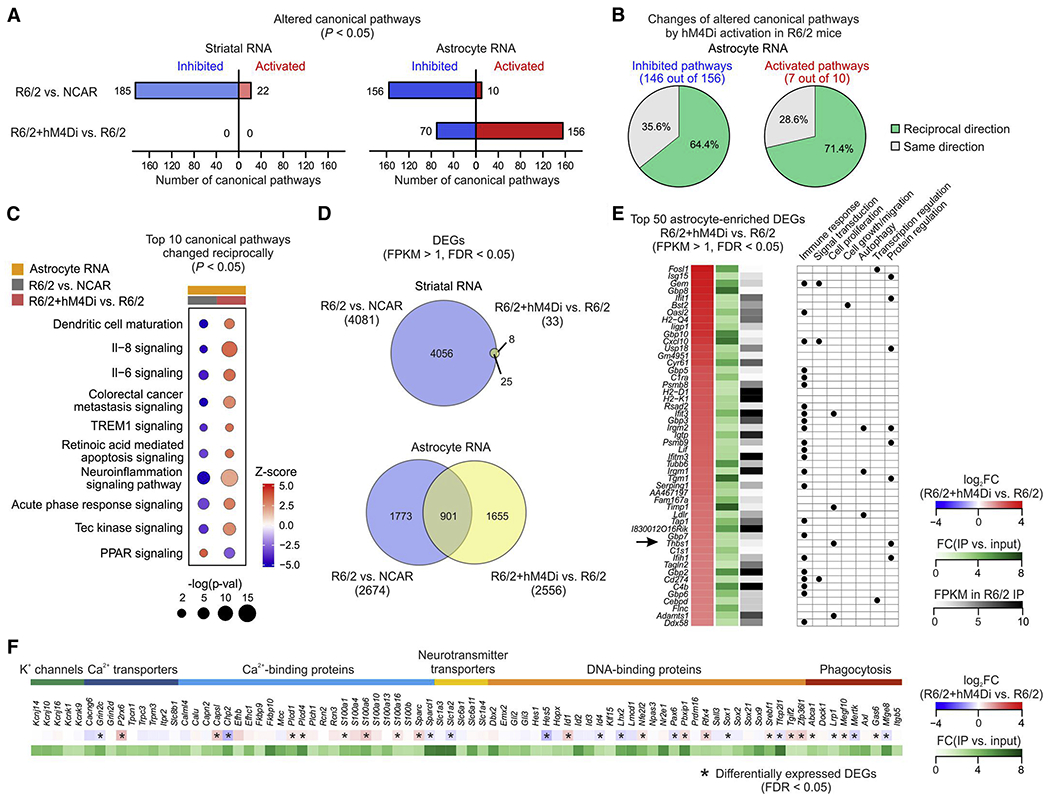
(A) Number of altered canonical pathways (P < 0.05) in striatal bulk RNA and astrocyte RNA. Colors indicate either pathway activation (red) or inhibition (blue) based on z-scores. (B) Pie charts showing percentages of inhibited (left) and activated (right) canonical pathways identified in R6/2 mice that were changed in the reciprocal direction or the same direction by astrocyte hM4Di. (C) Top 10 altered canonical pathways in astrocyte RNA-seq from R6/2 mice that were changed reciprocally by hM4Di activation. Sizes of dots represent log-transformed p-value and colors indicate either pathway activation (red) or inhibition (blue) based on z-scores. (D) Venn diagrams showing hM4Di activation induced changes of DEGs (FPKM > 1, FDR < 0.05) identified in striatal RNA-seq (top) and astrocyte RNA-seq (bottom). (E) Left, top 50 astrocyte-enriched DEGs (FPKM > 1, FDR < 0.05) by astrocyte hM4Di in R6/2 mice, ranked by fold change when comparing R6/2 + hM4Di IP with R6/2 IP. All listed DEGs displayed at least 2-fold astrocyte enrichment when comparing IP vs. input. Right, summary table of selected gene ontology terms (biological processes) that are associated with each DEGs. (F) List of genes that encode K+ permeable channels, proteins regulating Ca2+ signaling, neurotransmitter transporters, DNA-binding proteins and proteins involved in phagocytic pathways. All the genes have FPKM > 1 in both NCAR and R6/2 and at least 2-fold astrocyte enrichment.
Figure 7: Several phenotypic improvements in HD model mice were gabapentin sensitive.
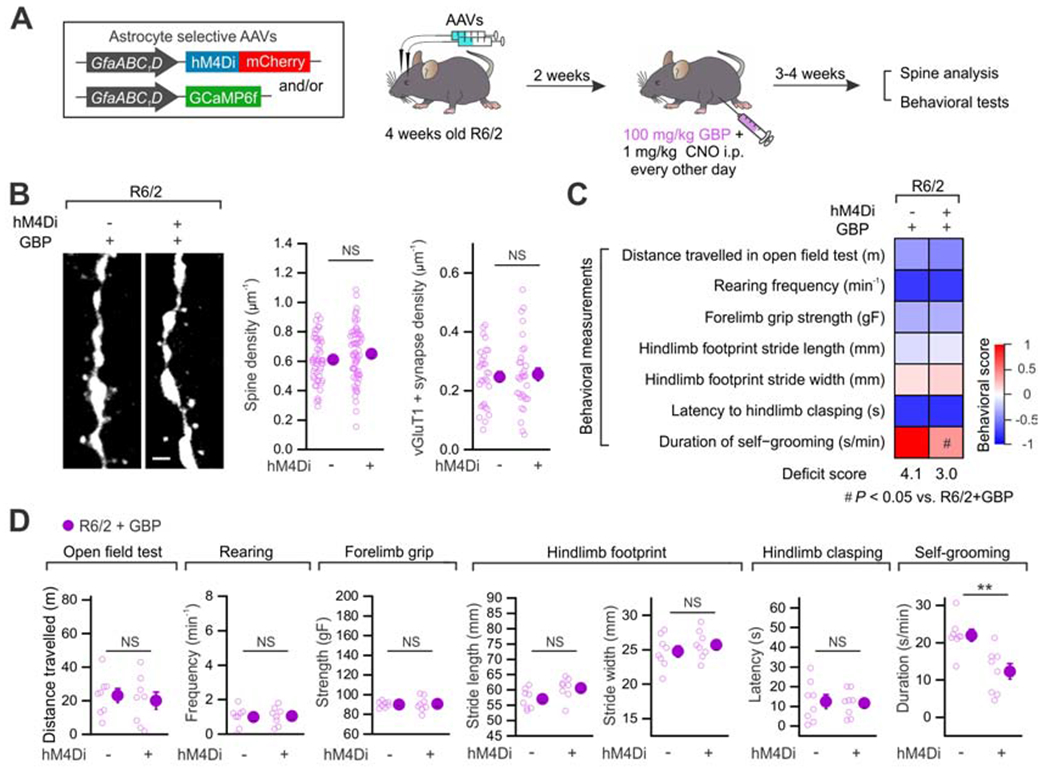
(A) Experimental procedure of AAV delivery, drug treatment and subsequent experimental evaluations in R6/2 mice. (B) Left, representative images of spines in R6/2 and R6/2 + hM4Di groups in the presence of GBP. Right, overall spine density and vGluT1+ synapse density with GBP administration. (C) Heat map summarizes the seven behavioral parameters assessed in R6/2 groups with and without GBP treatment (n = 8 mice in each group). The beneficial effects of astrocyte hM4Di in R6/2 mice were significantly attenuated by GBP administration, which is summarized by the aggregate deficit score and by the behavioral score heat map (see STAR Methods). (D) Individual data points of seven behavioral parameters assessed. Data are shown as mean ± SEM. **P < 0.01. Full details of n numbers, precise P values, statistical tests and the raw values are reported in Excel files S3 and S4.
Figure 8: Identification of endogenous GPCRs in HD mice as potential therapeutic targets.
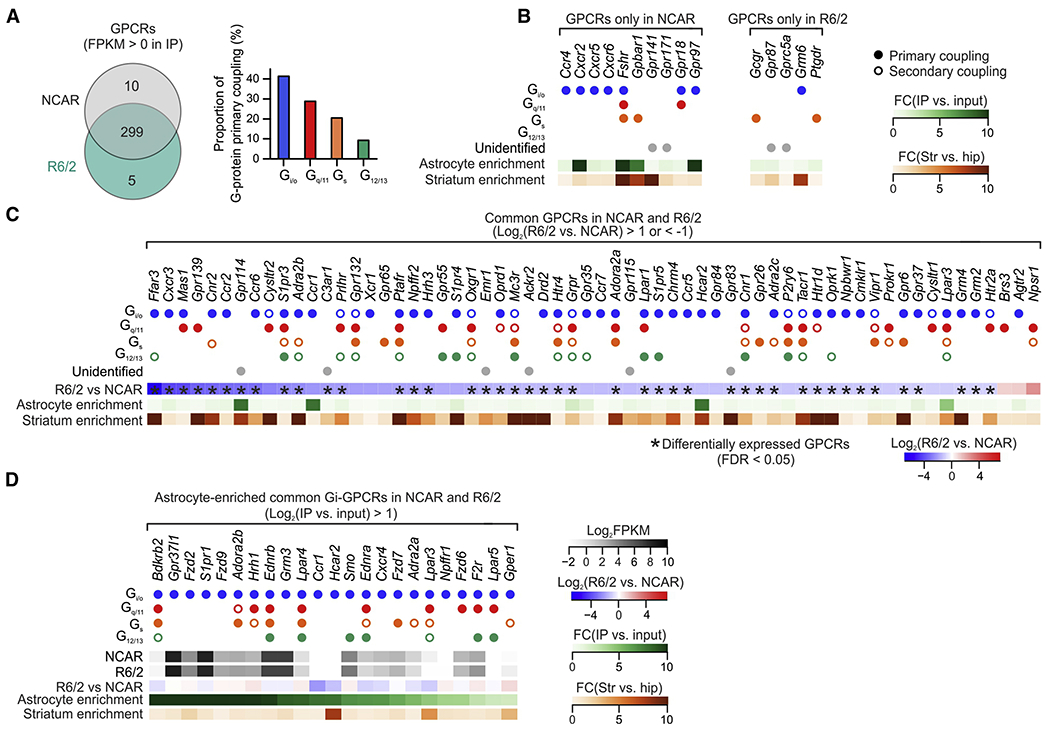
(A) GPCRs identified from striatal astrocyte RNA-seq data in NCAR and R6/2 at 3 months of age. The bar graph showing the proportion of primary coupling to G-proteins (Gi/o, Gq/11, Gs and G12/13). (B) Unique GPCRs that were present in NCAR and R6/2. Filled dots indicate primary G-protein coupling, while open dots indicate secondary G-protein coupling. Astrocyte enrichment was calculated as fold-change between striatal IP and striatal input RNA-seq data. Striatum enrichment was calculated as fold-change between striatal IP and hippocampal IP RNA-seq data. (C) Common GPCRs in the striatal astrocytes with > 2-fold increase or decrease between R6/2 and NCAR. Differentially expressed GPCRs (FDR < 0.05) are highlighted by asterisks. (D) GPCRs primarily coupled to Gi that were enriched in astrocytes and present in both R6/2 and NCAR. All listed GPCRs displayed >2-fold astrocyte enrichment and were not significantly altered in R6/2 compared with NCAR (FDR > 0.05). FPKM values presented as log2FPKM. All GPCRs reported in Excel file S5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-S100β | Sigma-Aldrich | Cat# S2532; RRID:AB_477499 |
| mouse anti-NeuN (clone A60) | Millipore | Cat# MAB377; RRID: AB_2298772 |
| chicken anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| rabbit anti-Kir4.1 | Alomone | Cat# APC-035; RRID: AB_2040120 |
| rabbit anti-DARPP-32 | Abcam | Cat# ab40801; RRID: AB_731843 |
| mouse anti-tyrosine hydroxylase | Immunostar | Cat# 22941; RRID:AB_572268 |
| rabbit anti-SAPAP3 | Welch et al., 2007 | N/A |
| rabbit anti-c-Fos | Millipore | Cat# ABE457; RRID: AB_2631318 |
| guinea pig anti-vGluT1 | Synaptic Systems | Cat# 135302; RRID: AB_887875 |
| Alexa Fluor 488 goat anti-chicken | Molecular Probes | Cat# A11039; RRID: AB_2534096 |
| Alexa Fluor 488 goat anti-rabbit | Molecular Probes | Cat# A11008; RRID: AB_143165 |
| Alexa Fluor 546 goat anti-mouse | Molecular Probes | Cat# A11003; RRID: AB_2534071 |
| Alexa Fluor 546 goat anti-chicken | Molecular Probes | Cat# A11040; RRID: AB_2534097 |
| Alexa Fluor 594 goat anti-rabbit | Molecular Probes | Cat# R37117; RRID: AB_2556545 |
| Alexa Fluor 647 goat anti-rabbit | Molecular Probes | Cat# A21245; RRID: AB_ 2535812 |
| streptavidin conjugated Alexa 647 | Molecular Probes | Cat# S21374; RRID: AB_2336066 |
| Bacterial and Virus Strains | ||
| AAV2/5 GfaABC1D cyto-GCaMP6f | Haustein et al., 2014 | Addgene Vectors #52925-AAV5 RRID:Addgene_52925 |
| AAV5 GfaABC1D tdTomato | Shigetomi et al., 2013 | Addgene Vectors #44332-AAV5 RRID:Addgene_44332 |
| AAV2/5 GfaABC1D Rpl22HA | Yu et al., 2018 | Addgene Vectors #111811 RRID:Addgene_111811 |
| AAV5 GfaABC1D-mCherry-hPMCA2w/b | Yu et al., 2018 | Addgene Vectors #111568 RRID:Addgene_111568 |
| AAV2/5 GfaABC1D hM3Dq-mCherry | Chai et al., 2017 | Addgene Vectors #92284 RRID:Addgene_92284 |
| AAV2/5 GfaABC1D hM4Di-mCherry | Chai et al., 2017 | Addgene Vectors #92286 RRID:Addgene_92286 |
| AAV2/5 GfaABC1D rM3Ds-mCherry | Chai et al., 2017 | Addgene Vectors #92285 RRID:Addgene_92285 |
| AAV2/5 GfaABC1D PI-Cre | Nagai et al., 2019 | Addgene Vectors #105603 RRID:Addgene_105603 |
| AAV2/5 GfaABC1D Lck-GFP | Shigetomi et al., 2013 | Addgene Vectors #105598-AAV5 RRID:Addgene_105598 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formalin, Buffered, 10% | Fisher Chemical | Cat# SF100-20 |
| Pronase | Sigma-Aldrich | Cat# P6911 |
| Fetal bovine serum | Thermo Fisher Scientific | Cat# 10437028 |
| BSA | Sigma-Aldrich | Cat# A8806 |
| Actinomycin D | Sigma-Aldrich | Cat# A1410 |
| TTX | Cayman Chemical Company | Cat# 14964 |
| Alexa fluor 568 hydrazide | Thermo Fisher Scientific | Cat# A10441 |
| Clozapine N-oxide (CNO) | Tocris | Cat# 4936 |
| γ-aminobutyric acid | Sigma-Aldrich | Cat# A2129 |
| Biocytin | Tocris | Cat# 3349 |
| Deposited Data | ||
| RNA-seq data: HD | Diaz-Castro et al., 2019 | GEO: GSE124846 |
| RNA-seq data: CalEx | Yu et al., 2018 | GEO: GSE114757 |
| RNA-seq data: hM4Di | Nagai et al., 2019 | GEO: GSE119058 |
| RNA-seq data: QA and saline control | This paper | GEO: GSE143475 |
| RNA-seq data: LPS and saline control | This paper | GEO: GSE143475 |
| RNA-seq data: Kir4.1 KO and wild-type control | This paper | GEO: GSE143475 |
| RNA-seq data: IP3R2 KO and wild-type control | This paper | GEO: GSE143475 |
| RNA-seq data: hM3Dq and parallel control | This paper | GEO: GSE143475 |
| RNA-seq data: rM3Ds and parallel controls | This paper | GEO: GSE143475 |
| RNA-seq data: MPTP and saline control | This paper | GEO: GSE153791 |
| RNA-seq data: SAPAP3 KO and wild-type control | This paper | GEO: GSE153791 |
| RNA-seq data: R6/2 mice with hM4Di and parallel controls | This paper | GEO: GSE153791 |
| Striatal single cell RNA-seq data | This paper | GEO: GSE156628 |
| Raw data values used to generate figures | This paper | Excel Files S3 and S4 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6CBA-Tg(HDexon1)62Gpb/3J | Jackson Laboratory | Stock# 006494; RRID:IMSR_JAX:006494 |
| Mouse: B6J.129S1-Htttm1.1Mfc/190ChdiJ | Jackson Laboratory | Stock# 370832 |
| Mouse: B6.129-Dlgap3tm1Gfng/J | Jackson Laboratory | Stock# 008733; RRID:IMSR_JAX:008733 |
| Mouse: B6.129- Kcnj10tm1Kdmc/J | Jackson Laboratory | Stock# 026826; RRID:IMSR_JAX: 026826 |
| Mouse: Itpr2tm1.1Chen | Srinivasan et al., 2015 | RRID:MGI:3641042 |
| Mouse: C57Bl/6NJ | Jackson Laboratory | Stock# 005304; RRID: IMSR_JAX:005304 |
| Mouse: C57Bl/6NTac inbred mice | Taconic | Stock# B6; RRID:IMSR_TAC:b6 |
| Oligonucleotides | ||
| Mm-Thbs1-C3 | ACDBio | Cat# 45891-C3 |
| Software and Algorithms | ||
| OriginPro 2016 | Origin Lab Corporation | RRID:SCR_015636 |
| pCLAMP10.4 | Molecular Devices | RRID:SCR_011323 |
| ClampFit10.4 | Molecular Devices | N/A |
| Fluoview FV10-ASW | Olympus | N/A |
| ImageJ v1.51h | NIH | RRID:SCR_003070 |
| Ethovision XT | Noldus Information Technology | RRID:SCR_000441 |
| CorelDraw X7 | Corel Corporation | RRID:SCR_014235 |
| Labview 2011 | National Instruments | RRID:SCR_014325 |
| Bioconductor | Law et al., 2014 | http://www.bionconductor.org |
| R v3.5.2 | RCoreTeam, 2016 | https://cran.r-project.org/bin/windows/base/old/3.5.2/ |
| Python 3.6.7 | The Python Software Foundation | https://www.python.org/downloads/ |
| Scanpy | Wolf et al., 2018 | https://github.com/theislab/scanpy |
Well-defined experimental perturbations of relevance to the striatum
Astrocytes change substantially during development and aging, and vary between brain regions (Haim and Rowitch, 2017; Khakh and Deneen, 2019). In order to explore astrocyte molecular mechanisms, we performed a series of well-defined EPs related to the core functions of striatal astrocytes in adult mice (Figure 1A, Table S1, Figure S2). Given the brain region-dependent nature of astrocytes, our strategy aimed to avoid errors that can arise from pooling gene expression data from studies that are usually not astrocyte or brain-region specific, or which rely on data of variable coverage from unmatched tissue samples frequently from different ages, and reliant on different methods and sequencing platforms.
The EPs comprised four groups encompassing striatum-relevant disease and physiology with parallel age-matched controls (STAR Methods). The first group, included assessments for HD (R6/2 and Q175 mouse models at presymptomatic and symptomatic disease stages). For subsequent hypothesis testing, we focussed on R6/2 mice. The second group assessed astrocytes under several pathological conditions: MSN ablation, neuroinflammation, dopamine depletion, and in a mouse model of obsessive-compulsive disorder. The third group assessed the impact of altered astrocyte K+ and Ca2+ ionic signalling. The fourth group evaluated astrocytes after activating specific astrocyte GPCR pathways (Gq, Gi and Gs) in striatal astrocytes.
To assess astrocyte molecular signatures agnostically, we performed astrocyte-specific RNA-sequencing (RNA-seq). The RNA-seq data from the IP samples were enriched with astrocyte markers and depleted of markers for other cells (Figure S1). Differentially expressed genes (DEGs) in striatal astrocytes were identified by comparing the astrocyte RNA-seq data between each EP and its control. Gene expression data were used for assessments of signalling pathways and upstream regulators to reveal astrocyte transcriptomic responses to the various EPs. This information was then used to evaluate astrocytes in the context of basal ganglia related diseases by using striatal single-cell RNA-seq data (scRNA-seq). We then focused on HD and identified altered astrocyte signaling, which allowed us to form and test a hypothesis related to HD phenotypes in mice (Figure 1A).
Context-specific astrocyte molecular responses in vivo
The number of astrocyte-enriched, and the total number of up- and down-regulated DEGs, varied markedly by ~100-fold between the EPs (FPKM > 1, FDR < 0.05; Figure 1B), each of which were validated (Table S1; Figure S2). Furthermore, we observed little effect on putative 38 pan-reactive, neurotoxic-A1 and neuroprotective-A2 reactivity genes (Liddelow et al., 2017) from ten of the EPs (Figure 1C). This recalls work for HD, where HD astrocytes were not associated with proposed A1 and A2 reactivity phenotypes (Diaz-Castro et al., 2019; Lee et al., 2020). In four EPs, the effects were EP-specific (arrows in Figure 1C) and there was no evidence for uniquely A1 or A2 reactive astrocytes for any EP. The simplest explanation for the differences between our LPS evaluations and past work (Zamanian et al., 2012) is that we only assessed astrocytes from the dorsolateral striatum.
We next evaluated the top 20 DEGs from each EP that reflect the most pronounced changes in gene expression (FPKM > 5, FDR < 0.05; Figure 1D; Excel file S1). From the identities of the DEGs and from the magnitude of the changes observed (fold-change Log2 ratio relative to control; Figure 1D), there was negligible overlap between the EPs. There was only one DEG (Gbp4) shared by four EPs, and no significantly altered genes that were shared in more than four EPs (Figure 1D). Weighted Gene Co-expression Network Analysis (WGCNA) was next used to identify gene clusters and associated pathways, revealing 47 modules of highly correlated genes associated with the EPs. Similar to the DEGs, the WGCNA modules also displayed a high degree of EP-specificity (Excel file S2).
We performed multidimensional scaling analyses for the 1000 most variable genes for each EP. In Figure 1E, closely positioned dots indicate similar gene expression profiles between EPs, for instance the dots representing activation of Gq, Gi and Gs DREADDs were clustered. This shows that they evoke similar, but non-identical molecular changes: when the three EPs of GPCR activation were analyzed independently of the other EPs, they were separated in a principal component analysis (Figure S3A) and had few DEGs shared by all three GPCR activations (Figure S3B). The dots representing the other eleven EPs were dispersed indicating marked dissimilarity between them (Figure 1E). To explore this further, we reviewed the numbers of shared DEGs (FPKM > 1, FDR < 0.05) under all possible combinations of EPs, from pairwise comparisons to all fourteen EPs (Figure 1F; Figure S3C). The greatest numbers of unique DEGs were for single EPs (red) and few were shared by greater than three EPs (Figure 1F; Figure S3C): the average number of unique DEGs from 1 to 14 shared EP combinations fell precipitously after two EPs (Figure 1G). There were no common DEGs shared by more than 10 EPs. Further support for EP-specific molecular responses derived from the top 20 unique DEGs, from the top 3 canonical pathways identified by the DEGs, and from the top 10 putative unique upstream regulators that were significantly associated with each EP (Figure S4A–C, P < 0.05). These upstream regulators included several highly EP-specific transcriptional regulators, growth factors and GPCRs. Similar to the DEGs, there were no canonical pathways shared by all EPs. There was only one common upstream regulator shared across all EPs, which was the transcriptional regulator HTT – the Huntingtin protein (Figure S4D). Polyglutamine expansions in HTT cause HD (Bates et al., 2015).
Together, comprehensive analyses show that in striatal astrocytes, (i) the number of DEGs, (ii) the magnitude of their dynamics, (iii) their identities, (iv) their overlap, (v) gene networks and their associated pathways, and (vi) upstream regulators of astrocyte gene expression are all poorly shared between EPs (Figures 1, S3, S4 and Excel files S1 and S2). These data show that striatal astrocytes display remarkable flexibility in the responses they mount in response to different perturbations.
HD-related evaluations and assessment of altered astrocyte signaling pathways
To assess the potential contribution of striatal astrocytes to neurological and psychiatric disorders with known genetic associations, we performed single cell RNA-sequencing (scRNA-seq) of striata from adult mice (8-9 weeks) (Wu et al., 2017). scRNA-seq yielded profiles of 20,912 striatal cells (n = 3 mice) and identified 11 transcriptomic clusters corresponding to the major striatal cell types (Figure S5), which were essentially the same as a published mouse scRNA-seq dataset (Saunders et al., 2018). We related our mouse striatal scRNA-seq data for oligodendrocytes, astrocytes, microglia, and neurons to the top 20 marker genes for these cells from human striatal RNA-seq data (Kelley et al., 2018). The normalized relative expression heat map (Figure 2A) showed strong concordance across these cell types between mouse and human. Furthermore, when we compared 8669 human striatal astrocyte genes with expression in the top 50 percentile (Kelley et al., 2018), with 9081 mouse striatal astrocyte genes with FPKM > 5, we found 6733 were shared, i.e. 78% overlap (Figure 2B). We next used the top 1000 genes for the seven most populous striatal cell types (neurons, astrocytes, microglia, OPCs, oligodendrocytes, endothelial cells, and neurogenic cells; Figure S5) and mapped known basal ganglia disease-associated genes to explore disease associations to these cells (Figure 2C). For a range of neurological and psychiatric disorders, disease-related genes mapped onto striatal cells with the following order: neurons > astrocytes > microglia > OPCs > oligodendrocytes > endothelial cells > neurogenic cells. While neurons were dominant, some currently known disease-related genes mapped to astrocytes, including for HD and OCD (arrowheads in Figure 2C).
Since HD was a hot spot for disease-related gene associations with potential astrocyte contributions, we further probed HD mouse and human gene expression data (Diaz-Castro et al., 2019). Our aim was to determine (i) if astrocyte pathway alterations could be identified in HD and (ii) how they related to the changes evoked by the 14 EPs. The dot plots in Figure 2D report the top 10 common pathways from gene expression analyses of human HD samples that were altered with increasing severity from neuropathological grade G0 to G3. In such plots, the larger dots indicate greater significance and the colors indicate pathway activation (red) or inhibition (blue) based on the z-scores values. Marked disease-progression dependent changes in human HD data were detected (Figure 2D). We repeated these analyses for R6/2 HD model mice at 1, 2 and 3 months that also track with disease severity for both bulk tissue and for striatal astrocyte-specific RNA-seq (Figure 2E,F). Significant disease-phenotype dependent changes were observed and several pathways were related to synaptic plasticity, as well as GPCR and Ca2+ signaling, which recalls expected and known HD pathophysiology (Cepeda et al., 2010; Khakh et al., 2017). Moreover, most genes were downregulated in HD, consistent with loss of essential functions (Diaz-Castro et al., 2019).
HD-related evaluations suggest contributions of astrocyte Gi-GPCR signaling pathways
Of the top pathways that were downregulated in HD astrocytes (Figure 2F), several related to GPCR signaling, including opioid, dopamine, Ca2+, cAMP, protein kinase A. More specifically, Gαi signaling was consistently inhibited in HD astrocytes (Figure 2F). This prompted us to evaluate these 10 pathways in the EP where astrocyte Gi signaling was stimulated in vivo using hM4Di DREADD (Figure 1). Remarkably, all ten pathways that were downregulated in HD were upregulated following Gi-GPCR activation in vivo using hM4Di DREADD (Figure 2F,G). Moreover, four out of ten pathways were significantly upregulated for Gi-GPCR activation (Figure 2F,G). Together, these data show altered signaling pathways in striatal astrocytes in HD and also show opposing effects on these pathways that were triggered by Gi-DREADD activation in striatal astrocytes in vivo.
Gi-GPCR pathway activation corrects HD-related cellular phenotypes of astrocytes
We hypothesized that activation of astrocyte Gi-GPCRs in HD model mice may produce beneficial effects, because astrocyte pathway alterations in HD model mice and following Gi-GPCR pathway activation were reciprocal (Figure 2F,G). Endogenous astrocyte Gi-GPCRs exist in multiple cell types, therefore drugs for such receptors cannot be used selectively in vivo. To specifically explore the consequences of striatal astrocyte Gi-GPCR pathway activation in R6/2 HD mice, we expressed hM4Di DREADD (Roth, 2016) (with GCaMP6f) in astrocytes. Two weeks after AAV microinjections into the striatum, we administered CNO to activate striatal astrocyte hM4Di (1 mg/kg i.p.; Figure 3A). The experiments were performed in parallel for four groups of mice. For simplicity, the three most relevant groups are reported in Figures 3–5, but all data and statistics are provided (Excel files S3 and S4). The first group comprised control non-carrier (NCAR) mice that received control AAVs (“NCAR + AAV”). The second group comprised R6/2 mice that also received control AAVs (“R6/2 + AAV”). The third group comprised the test group, whereby R6/2 mice received hM4Di AAVs (“R6/2 + hM4Di”). All groups were identically treated with CNO to control for potential off-target effects. We evaluated HD-related phenotypes between NCAR + AAV and R6/2 + AAV, and compared R6/2 + hM4Di to both NCAR + AAV and R6/2 + AAV to evaluate the consequences of hM4Di activation in HD model mice.
We analyzed astrocyte spontaneous Ca2+ signals in striatal slices, because they are attenuated in HD model mice (Jiang et al., 2016). We detected reduced astrocyte Ca2+ event amplitudes in the R6/2 + AAV group relative to the NCAR + AAV group (Figure 3B,C, P < 0.0001, 24 & 18 astrocytes, 4 mice) and found such reductions were significantly restored in the R6/2 + hM4Di group (Figure 3B,C; P < 0.0001; 27 astrocytes, 4 mice) to levels in the NCAR + AAV group (P = 0.051, NCAR + AAV vs. R6/2 + hM4Di). There were no significant differences in the Ca2+ event frequency (Figure 3B,C; P = 0.50, R6/2 + AAV vs. R6/2 + hM4Di; P = 0.12, NCAR + AAV vs. R6/2 + hM4Di). Next, we assessed astrocyte territory sizes with membrane targeted Lck-GFP. Consistent with past studies (Octeau et al., 2018), astrocyte territory areas (2338 ± 110 μm2; 33 astrocytes, 4 mice in the NCAR + AAV mice) were smaller in the R6/2 + AAV mice (1761 ± 105 μm2; 37 astrocytes, 4 mice), and this difference was also rescued in the R6/2 + hM4Di mice (2366 ± 90 μm2; 36 astrocytes, 4 mice; Figure 3D; P = 0.0002, R6/2 + AAV vs. R6/2 + hM4Di; P > 0.99, NCAR + AAV vs. R6/2 + hM4Di).
Gi-GPCR pathway activation corrects some HD-related MSN cellular phenotypes
Astrocytes and neurons both contribute to neurodegenerative disorders and astrocytes regulate neurons through a variety of mechanisms, prompting us to explore if astrocyte Gi-pathway activation in HD model mice affected MSN dysfunctions (Cepeda et al., 2010; Khakh et al., 2017). We stimulated cortico-striatal axons to assess glutamatergic fast excitatory postsynaptic currents (EPSCs) onto MSNs (Figure 4A). We recorded AMPA receptor-mediated evoked EPSCs, paired-pulse responses (at −70 mV) and NMDA receptor-mediated evoked EPSCs (at +40 mV; Figure 4B,C). The significantly reduced AMPA and NMDA EPSCs in the R6/2 + AAV group relative to the NCAR + AAV group were restored in the R6/2 + hM4Di group (Figure 4B,C, P < 0.0001; from −191 ± 41 pA to −471 ± 36 pA for AMPA EPSCs and from +87 ± 19 pA to +313 ± 21 pA for NMDA EPSCs; 19 & 20 MSNs, 4 mice). The restoration of AMPA EPSCs was to the control level (P = 0.24, NCAR + AAV vs. R6/2 + hM4Di), but NMDA EPSCs were higher than control (P = 0.037, NCAR + AAV vs. R6/2 + hM4Di) (Figure 4B,C). There was no change in the evoked AMPA EPSC paired-pulse ratios (P > 0.99). We also detected significantly decreased miniature EPSC (mEPSC) frequency in R6/2 + AAV compared to NCAR + AAV mice, and this was restored with astrocyte Gi-GPCR pathway activation in the R6/2 + hM4Di mice (Figure 4D, P = 0.0032, R6/2 + AAV vs. R6/2 + hM4Di; from 0.6 ± 0.1 Hz to 1.3 ± 0.1 Hz; 15 & 16 MSNs, 4 mice; P = 0.80, NCAR + AAV vs. R6/2 + hM4Di). There were no significant changes in mEPSC amplitudes (Figure 4D, P > 0.99). Next, when we evaluated MSN dendritic spines, we detected significantly reduced cortico-striatal MSN spine density, spine head size and density of vGluTl-positive MSN synapses in R6/2 + AAV as compared to NCAR + AAV groups. The reduced density of total spines and vGluT1-positive synapses were restored in the R6/2 + hM4Di group (Figure 4E, P < 0.0001, R6/2 + AAV vs R6/2 + hM4Di). Such rescue was complete for spine density (P = 0.94, NCAR + AAV vs R6/2 + hM4Di), but the numbers of vGluT1-positive synapses were higher than control levels in R6/2 + hM4Di mice (P = 0.0041).
We mention neuronal aspects that we assessed, but that were not altered in the R6/2 + hM4Di group. The complexity of MSN dendritic morphology, HD-related changes in intrinsic MSN intrinsic excitability and action potential properties were not affected by astrocyte Gi-GPCR pathway activation (Figure S6A,B; Excel files S3 and S4). Furthermore, striatal tissue shrinkage and atrophy was unaffected by Gi-GPCR pathway activation in astrocytes (Figure S6C; 4 mice). Neuronal loss has proven hard to detect in HD mouse models, but a consequence of striatal tissue shrinkage is that the apparent density of neurons per unit area increases in HD model mice, but this too was not affected by astrocyte Gi-GPCR pathway activation (Figure S6D; 4 mice). We also measured a decrease in the size of NeuN positive neuronal somata in the R6/2 mice relative to controls, but this was not affected by astrocyte Gi-GPCR pathway activation (Figure S6D; 4 mice). Thus, activation of astrocyte Gi-GPCRs in vivo rescued several, but not all, HD-related neuronal dysfunctions. This is expected since mHTT is found in all cells of the body and some neuronal dysfunctions are caused by intrinsic HD-related pathophysiology within neurons themselves.
Astrocyte Gi-GPCR activation corrects behavioral deficits in HD model mice
Since stimulation of the astrocyte Gi-GPCR pathway restored some cellular dysfunctions in HD model mice (Figures 3 and 4), we explored if it also altered HD-related mouse behaviors (Figure 5A). The R6/2 + hM4Di mice showed several significantly (P < 0.0042, R6/2 + AAV vs R6/2 + hM4Di) improved behaviors relative to the R6/2 + AAV group (Figure 5B,C). These restorations were to the level of controls for the distance travelled in the open field, rearing frequency, and duration of self-grooming (Figure 5B,C, n = 14-20 mice; Excel file S3). In addition, improvement in gait was also observed as the hind limb stride width in the R6/2 + hM4Di mice was indistinguishable from that in the NCAR + AAV group (Figure 5C, P = 0.84, NCAR + AAV vs. R6/2 + hM4Di, n = 18-19 mice; Excel file S3). However, body weight loss observed in HD mice did not improve (Figure S6E; P = 0.14, two-way ANOVA repeated measures, n = 19-20 mice). Thus, activation of astrocyte Gi-GPCRs produced significant improvements in some, but not all, whole animal HD-related phenotypes.
In regards to the effects of Gi-GPCR pathway activation in HD-model mice, in total we made 18 assessments at the cellular, electrophysiological and behavioral level. Of these, two responses recovered to levels higher than in the controls following Gi-GPCR activation in HD-model mice. These were NMDA EPSCs and the numbers of vGluT1-positive synapses (Figure 4C, E). We do not have a satisfying explanation for this, but if our approach were to be pursued as a potential therapeutic strategy this overshoot response in 2/18 assessments should be carefully considered.
Assessing molecular modifications following astrocyte Gi-GPCR activation in HD model mice
To investigate the underlying molecular modifications induced by astrocyte Gi-GPCR activation in HD model mice, we performed RNA-seq. In these experiments we evaluated both the striatal bulk tissue RNA that represents the soup of all cells as well as astrocyte-specific RNA in age-matched NCAR, R6/2, and R6/2 + hM4Di mice. Alterations in signaling pathways as well as gene expression were compared between R6/2 vs. NCAR and R6/2 + hM4Di vs. R6/2 to uncover the transcriptional impact of astrocyte Gi-GPCR activation in HD mice on astrocytes and whole striatal tissue.
As expected, based on gene expression differences, R6/2 mice displayed significantly altered signaling pathways in both striatal RNA and astrocyte RNA data compared with NCAR mice (Figure 6A). The majority of the altered pathways were inhibited in R6/2 mice, i.e. 185 out of 207 pathways and 156 out of 166 pathways were inhibited in bulk striatal and astrocyte samples, respectively – recalling previous HD analyses (Figure 2D–F). In contrast, astrocyte Gi-GPCR activation in R6/2 mice produced no effect on bulk striatal samples, but induced marked alterations in signaling pathways from astrocyte RNA with a shift towards significantly more pathways being activated (Figure 6A). Specifically, 64.4% of inhibited astrocyte pathways of R6/2 mice were found to be activated by hM4Di (Figure 6B). A similar reciprocal change was also observed in 71.4% of activated pathways in R6/2, which were significantly inhibited by astrocyte hM4Di (Figure 6B). Surprisingly, several of the top 10 signaling pathways that were changed reciprocally by hM4Di activation were associated with neuroinflammatory responses, such as IL-8/6 and TREM1 signaling (Figure 6C).
Consistent with the pathway analysis, DEG analysis also revealed substantial changes in transcriptional profiles by hM4Di activation in astrocytes, but not in the bulk tissue of R6/2 mice (Figure 6D; 33 DEGs in striatal RNA vs. 2556 DEGs in astrocyte RNA). Since the greatest effects of hM4Di activation were found within the striatal astrocytes themselves, we further focused on astrocyte-enriched DEGs that were altered by hM4Di activation in R6/2 mice (FPKM > 1, FDR < 0.05, fold enrichment IP vs. input > 2). When ranked by the degree of expression level change induced by hM4Di, the top 50 astrocyte-enriched DEGs were all found to be significantly upregulated (Figure 6E). The biological functions that are associated with these pronounced astrocyte changes included immune responses, regulation of transcription and translation, cell proliferation and growth (Figure 6E). To summarize these findings, RNA-seq of bulk tissue and astrocytes following Gi-GPCR activation in HD model mice showed significant reversal of HD-related molecular signatures in astrocytes, but only modest changes in bulk tissue. This confirms cellular specificity of our approach, and also suggests that the improved cellular phenotypes as a result of Gi-GPCR activation in HD model mice are downstream of astrocytes.
We considered if the beneficial effects of Gi-GPCR activation in HD model mice might be at the expense of core astrocyte functions. We surveyed a panel of genes that are related to astrocyte core functions, including ion homeostasis, neurotransmitter uptake, DNA regulation, and phagocytosis. Genes that were enriched in striatal astrocytes and shared in both NCAR and R6/2 mice were listed for each category (Figure 6F; FPKM > 1). The expression levels of most of these genes were not significantly altered by hM4Di activation. In addition, the changes that were observed for the DEGs were moderate (0.52 to 1.84 fold when compared with R6/2 mice without hM4Di).
Gabapentin sensitive improvements following astrocyte Gi-GPCR activation in HD mice
Among the top 50 astrocyte-enriched DEGs that were significantly increased by hM4Di in R6/2 mice, was enhanced expression of Thbs1 (arrow in Figure 6E). Thbs1 encodes Thrombospondin-1 (TSP1) that was previously found to promote synaptogenesis and is astrocyte enriched (Christopherson et al., 2005; Eroglu et al., 2009; Kim et al., 2016; Nagai et al., 2019; Zhang et al., 2014). Consistent with the RNA-seq data, we detected significantly increased Thbs1 expression in striatal astrocytes from R6/2 + hM4Di groups using RNAscope and immunohistochemistry relative to NCAR + AAV and R6/2 + AAV groups (Figure S6F; P < 0.0001; 20-24 astrocytes, 4 mice). However, we could not perform immunohistochemistry for TSP1 as the antibodies were unreliable (Christopherson et al., 2005; Eroglu et al., 2009; Nagai et al., 2019; Risher et al., 2018).
Since an increase in TSP1 following astrocyte Gi-GPCR activation in the adult striatum was found to modify synaptic transmission (Nagai et al., 2019), we next explored cellular and behavioral improvements in HD model mice by using gabapentin (GBP) administered along with CNO (Figure 7A). Our use of GBP in these regards comes with several considerations. First, it is often used as an antagonist of the putative TSP1 receptor α2δ–1 on neurons and is proposed to block TSP1 actions in vivo in adult mice (Crawford et al., 2012; Eroglu et al., 2009; Nagai et al., 2019). In this context, although α2δ–1 is a calcium channel auxiliary subunit, it is suggested that TSP1 and α2δ-1 effects on synapse formation are independent of voltage-dependent calcium channels (Eroglu et al., 2009). The assumption is that gabapentin may have little effect on calcium channel currents directly: there is evidence for and against this view because small effects on calcium channels have been reported in some settings, but not others (Dolphin, 2016). Second, gabapentin is used clinically for certain types of chronic pain and for seizures and therefore may have other attendant pharmacological actions (Calandre et al., 2016), which should be thoughtfully considered in the interpretation of our data for this specific experiment. In the presence of GBP, hM4Di activation in the striatal astrocytes failed to increase the MSN spine density and the number of vGluT1+ synapses onto MSNs (Figure 7B; P = 0.24 and 0.95, 4 mice in each group). Furthermore, improved behavioral outcomes in locomotive activity and rearing as a result of astrocyte Gi-GPCR activation (Figure 5B,C) were not observed when GBP was administered (Figure 7C,D; P = 0.57 and 0.86, 8 mice in each group). However, self-grooming was not affected by GBP in the R6/2 + hM4Di group (Figure 7C,D; P = 0.003, 8 mice in each group), suggesting GBP-insensitive downstream mechanisms of Gi-GPCR activation that mediate different behavioral improvements in HD mice.
Identifying GPCRs as potential targets by mining of astrocyte gene expression data
Our data show that identification of molecular changes within astrocytes (Figures 1 & 2) and their selective targeting corrects several disease-related phenotypes in HD model mice (Figures 3–5), indicating that Gi-DREADDs could potentially be used therapeutically (Keenan et al., 2017; Lieb et al., 2019; Urban and Roth, 2015; Weston et al., 2019). We next mined striatal astrocyte RNA-seq data in order to identify potential endogenous GPCR targets that may mimic the effects seen with Gi-GPCR activation (Flock et al., 2017; Harding et al., 2018; Inoue et al., 2019; Pándy-Szekeres et al., 2018b) (see STAR Methods). We compared GPCRs identified using striatal astrocyte RNA-seq from NCAR and R6/2 mice at 3 months of age: 299 GPCRs were shared, whereas 10 and 5 were unique to NCAR and R6/2 mice, respectively (Figure 8A,B). When grouped by coupling to Gi/o, Gq/11, Gs and G12/13 G-proteins, ~40% of the putative GPCRs in striatal astrocytes were identified as Gi/o coupled (Figure 8A). For GPCRs that were common between NCAR and R6/2, we first analyzed the most differentially expressed GPCRs (FDR < 0.05) and found they were downregulated in R6/2 compared with NCAR (Figure 8C). Consistent with our data, 29/43 of these were primarily Gi/o-protein coupled, which offers an explanation for why Gi-GPCR activation was beneficial (Figures 3–5). Second, we identified 23 Gi-coupled GPCRs that were astrocyte enriched and were not downregulated in HD model mice (Figure 8D; Excel file S5). Based on our findings, we propose that these astrocyte-enriched GPCRs might represent drug targets for endogenous Gi-pathway activation to correct astrocyte-dependent cellular and behavioral dysfunctions in HD and potentially other disorders (e.g. Gpr37l1, Slp1r, Lpar3, Ednrb).
Discussion
Our data provide direct evidence that multiple, experimental perturbations produce astrocyte molecular responses that are context specific for a defined population of astrocytes in vivo. Such responses can be teased apart to identify a Gi-GPCR mechanism that produced beneficial effects at molecular, cellular and behavioral levels for HD model mice. A cure for HD would entail removing the mutant protein from all relevant cells of the body. However, our data indicate that harnessing astrocyte mechanisms in vivo can alter cellular properties sufficiently to result in improvements in HD-associated phenotypes, which could be explored as a therapeutic strategy in HD and other disorders.
In our work, astrocyte responses were not stereotyped and did not conform to known forms of pan-reactive, A1 or A2 astrocyte reactivity (Liddelow et al., 2017). However, we emphasize that our aim was not to study A1 and A2 reactivity, but rather to assess astrocyte responses broadly. Our data show that striatal astrocytes display molecular changes that depend strongly on how they are challenged. This finding of heterogeneous astrocyte molecular changes dependent on perturbation context has broad relevance for exploring fundamental glial biology in health and disease. As far as we know, this has not hitherto been reported for a defined population of astrocytes in the adult CNS in vivo, prompting us to explore the relevance of such responses in HD model mice, which display astrocytic and striatal dysfunctions (Introduction).
The heterogeneity of astrocytes between brain areas (Haim and Rowitch, 2017; Khakh and Deneen, 2019) raised the possibility that astrocytes in brain regions associated with specific diseases could potentially be manipulated to evoke beneficial effects in a disease-specific manner. Based on evaluations of RNA-seq datasets from 14 EPs, we were able to design experiments to test this hypothesis using manipulation of astrocytes in the striatum. It was previously found that the nature of major astrocyte pathway downregulation in human HD was shared with mouse models (Diaz-Castro et al., 2019; Lee et al., 2020). In the present study we found that downregulated signaling pathways in HD could be reciprocally upregulated following astrocyte Gi-GPCR activation. In consequence, striatal astrocyte Gi-GPCR pathway activation in HD model mice corrected several disease-related phenotypes, and led to the identification of potential new astrocyte GPCR therapeutic targets, including the possible clinical use of DREADDs themselves (Keenan et al., 2017; Lieb et al., 2019; Urban and Roth, 2015; Weston et al., 2019).
We place our findings in context. First, it is important to remember that our analyses of the 14 EPs was based on gene expression. In the future, it would be valuable to develop methods that allow for unbiased evaluation of protein expression selectively from astrocytes. Such methods do not yet exist for astrocytes. Second, since astrocytes change in separable ways at a molecular level in the striatum in response to different EPs, it is possible that their brain region specific gene expression patterns also reflect local cues that drive transcriptomic signatures. Since all the data in the current study were gathered for the striatum, we cannot address this possibility directly. However, as additional datasets for astrocytes in other brain areas emerge, it should be possible to assess this topic in due course. It is possible that brain-region specific astrocyte properties reflect a combination of local and developmental cues.
To begin to explore the molecular processes following astrocyte Gi-GPCR activation in HD model mice, we analyzed the transcriptional profiles of striatal astrocytes and made several observations: (i) reciprocal regulation of gene expression and signaling pathways; (ii) an enhancement of neuroimmune/inflammatory responses; and (iii) increased gene expression of TSP1 that may have synaptogenic roles in some of the responses that we measured. We started to explore the potential contribution of TSP1 receptor α2δ1 by using GBP and found significant attenuation of some improvements evoked by astrocyte Gi-GPCR activation. Our use of GBP in these regards carries the caveat that this drug may have other attendant actions, including possible regulation of calcium channels, that may have influenced our experiments (Calandre et al., 2016; Dolphin, 2016). It is problematic to rule out such potential confounds: interpretation of this one experiment must therefore be considered thoughtfully with this caveat in mind. Irrespectively, however, the key point is that outcomes following astrocyte Gi–GPCR activation in HD model mice were beneficial rather than deleterious across several measures at the cellular, circuit and behavioral level, i.e. the HD-related pathophysiology was improved. The data also illustrate the potential for using astrocytes to delay or remedy synaptic dysfunction by activating relevant mechanisms such as their synaptogenic potential, e.g. via Gi-GPCR stimulation. However, our results should not be over interpreted to indicate that TSP1 is the main mechanism and further studies are necessary. Our data provide insights and databases for such additional mechanisms in the context of HD following Gi-GPCR activation, which we discuss presently.
Neuroinflammation has been well-documented in HD patients, including non-symptomatic individuals (Agus et al., 2019; Hodges et al., 2006; Labadorf et al., 2015; Lee et al., 2020; Rodrigues et al., 2016). The occurrence of this phenomenon has spurred clinical trials of anti-inflammatory drugs with the assumption that neuroinflammatory responses exacerbate HD pathogenesis (Colpo et al., 2017; Roch et al., 2016). However, to date, these drugs have not markedly improved clinical outcomes. Thus, the issue of whether neuroinflammatory responses serve damaging or protective functions during HD pathogenesis is still open (Wilton and Stevens, 2020). In these regards, there is evidence to suggest that some forms of neuroinflammation may be protective in HD. IL-6, for example, was found to have neuroprotective effects in two rodent models of HD (Bensadoun et al., 2001; Wertz et al., 2020) as well as other neurological diseases (Yang et al., 2012; Zigmond, 2012). In accord, our experiments and analyses consistently revealed significant enhancement of the IL-6 signaling pathways by astrocyte Gi-GPCR activation, which otherwise were attenuated in HD model mice. Moreover, recent studies have demonstrated that the neuroimmune system functions to maintain homeostasis and neuromodulation (Alves de Lima et al., 2020; Da Mesquita et al., 2018; Norris and Kipnis, 2019). Brain neuroimmune responses in HD are also expected to be accompanied by blood brain barrier breakdown and infiltration of peripheral immune signals and cells (Sweeney et al., 2018). Since mHTT is found in every cell, combined with the fact that multiple cellular changes are known to occur in HD, we suggest that it would be misleading to promote, or expect, a single beneficial molecular mechanism in this complex disorder. From these perspectives, our data identify several astrocytic mechanisms that can now be explored in HD with the view to producing beneficial effects by exploring tissue homeostasis, neuroimmune, and cell-cell interactions. New types of tools are needed to plan such experiments, but our data provide evidence from agnostic evaluations of large datasets to suggest potential protective roles for synaptogenic cues and neuroimmune signaling in HD that can be triggered by appropriately stimulating astrocytes. We also list potential astrocyte GPCRs to target in order to explore such effects in follow up studies.
As far as we know, this study represents the first direct experimental evaluation of how a defined population of astrocytes responds to multiple stimuli in vivo. Taken together, the data reveal that striatal astrocytes are remarkably malleable and that they utilise context-specific responses that can be dissected molecularly to identify signalling mechanisms. Our experiments show that astrocyte molecular mechanisms are also exploitable for HD phenotype correction, portending the exploration of therapeutic strategies in HD and potentially for additional brain diseases. The extensive RNA-seq data also represent a valuable resource for exploring specific hypotheses concerning fundamental astrocyte biology in the striatum and in other brain areas.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Baljit S. Khakh (bkhakh@mednet.ucla.edu).
Material availability statement
All unique/stable reagents generated in this study are available upon request from the Lead Contact without restriction.
Data and code availability
All data and codes are available upon request from the Lead Contact. Excel file S4 reports raw replicates used for the figures. RNA-seq data are deposited at the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) with accession numbers provided in Table S1 and Key Resources Table. Lists of DEGs for each EPs are provided in Excel file S1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All animal experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. Male and female mice were used in this study. Most experiments were for mice aged 3 months, but for a specific set of experiments mice aged between 1- and 12-months old were used (ages specified in Table S1). Mice were housed in the vivarium managed by the Division of Laboratory Animal Medicine at University of California, Los Angeles (UCLA) with a 12 h light/dark cycle and ad libitum access to food and water. Wild-type C57BL/6NTac mice were generated from in house breeding colonies or purchased from Taconic Biosciences. R6/2 and non-carrier control mice at 2- and 3-months old were purchased from the Jackson Laboratory (Strain B6CBA-Tg(HDexon1)62Gpb/3J; JAX #006494). R6/2 and non-carrier control mice at 1 month old were bred and provided by Dr. Michael Levine’s laboratory at UCLA. Wild type (WT) and heterozygous zQ175 mice were acquired from Jackson Laboratory (Strain B6J.129S1-Htttm1.1Mfc/190ChdiJ; JAX #370832). Sapap3 knockout mice were purchased from the Jackson Laboratory (Strain B6.129-Dlgap3tm1Gfng/J; JAX #008733). Kcnj10flox/flox mice were purchased from the Jackson Laboratory (Strain B6.129- Kcnj10tm1Kdmc/J; JAX #026826). Ip3r2 knockout mice (Itpr2tm1.1Chen) were originally obtained from Dr. Ju Chen at University of California-San Diego and maintained as a heterozygous line (Srinivasan et al., 2015).
METHODS DETAILS
Strategy using experimental perturbations (EPs)
We provide full details and rationale for the EPs. The EPs employed comprised four groups encompassing striatum-relevant disease and physiology with parallel age-matched controls, including assessments for HD, cellular pathology, altered ionic signaling, and GPCR signalling.
The first group assessed astrocytes in two widely used HD mouse models (Diaz-Castro et al., 2019) including a transgenic R6/2 model that likely represents juvenile-onset HD and a knock-in heterozygous Q175 model that more closely represents adult-onset HD in human patients (Diaz-Castro et al., 2019). We examined presymptomatic and symptomatic disease stages to assess gene expression changes during disease progression. For subsequent hypothesis testing, we focussed on R6/2 mice.
The second group assessed astrocytes under several pathological conditions of relevance to the striatum and to HD. Striatal medium spiny neurons (MSNs) were ablated with quinolinic acid, because this is relevant to striatal dysfunction in HD where tissue volume loss occurs (Beal et al., 1986; Vonsattel, 2007). Neuroinflammation was induced by the bacterial cell wall endotoxin lipopolysaccharide (LPS) since molecular changes associated with neuroinflammatory processes accompany several striatal disorders including HD (Diaz-Castro et al., 2019; Lee et al., 2020; Rodrigues et al., 2018). Neuroinflammation is also pertinent to severe streptococcal infections that are proposed to change striatal function (Esposito et al., 2014). Since dysfunction of the nigrostriatal circuitry is associated with HD and Parkinson’s disease (PD), we used a mouse model of dopaminergic input loss generated with MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (Jackson-Lewis and Przedborski, 2007). Striatal deficits are implicated in the pathogenesis of obsessive-compulsive disorder (OCD), which is also a clinical comorbidity of prodromal HD. To investigate how astrocytes are altered in a model of OCD-like behavior, we used a well-characterized genetic mouse model called the Sapap3 knockout mice (SAPAP3 KO)(Welch et al., 2007).
The third group assessed the impact of altered astrocyte ionic signalling. Astrocyte potassium channel Kir4.1 (Kcnj10) was conditionally deleted in striatal astrocytes to reduce astrocyte K+ currents (Nwaobi et al., 2016). This is relevant because K+ channel loss is associated with pathophysiology in HD and several other diseases (Tong et al., 2014). To attenuate astrocyte intracellular Ca2+ signalling, we used two strategies: a CalEx AAV that reduces striatal astrocyte Ca2+ signaling (Yu et al., 2018) and transgenic Ip3r2 knockout mice that display strongly reduced intracellular Ca2+ signals (Jiang et al., 2016). These are relevant, because loss of Ca2+ signals is associated with HD and several other neurodegenerative diseases (Jiang et al., 2016).
The fourth group evaluated astrocytes after activating specific astrocyte G-protein coupled receptor (GPCR) pathways (Gi, Gq and Gs) by using DREADDs expressed in striatal astrocytes (Chai et al., 2017; Yu et al., 2020). These evaluations were relevant as GPCRs represent major therapeutic targets in brain diseases and astrocytes use GPCR signaling.
Differentially expressed genes (DEGs) in striatal astrocytes were identified by comparing the IP RNA-seq data between each EP and its cognate control. Gene expression data were used for assessments of signalling pathways and upstream regulators to reveal astrocyte transcriptomic responses to the various EPs. This information was then used to evaluate astrocytes in the context of basal ganglia related diseases by using striatal single-cell RNA-seq data (scRNA-seq). We then focused on HD and identified altered astrocyte signaling, which allowed us to form and test a hypothesis related to amelioration of HD phenotypes in mice. Figure 1A schematizes the workflow and overall approach we used. In order to generate a testable hypothesis from gene expression analyses, we exploited three recently published RNA-seq datasets (Table S1) (Diaz-Castro et al., 2019; Nagai et al., 2019; Yu et al., 2018). In addition, our study generated ten new RNA-seq datasets including nine striatal astrocyte-specific RNA-seq datasets with parallel striatal bulk tissue RNA-seq and one scRNA-seq dataset for the cells that comprise the mouse striatum. This new information is summarized in Table S1 and in the Key Resources Table and is available in the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo).
Surgical procedure of in vivo microinjection
Surgical procedures for viral microinjections have been described previously (Chai et al., 2017; Nagai et al., 2019; Yu et al., 2018). In brief, mice were anesthetized and placed onto a stereotaxic frame (David Kopf Instruments, Tujunga CA). Continuous anesthesia using isoflurane was carefully monitored and adjusted throughout the surgery. Mice were injected with buprenorphine (Buprenex; 0.1 mg/kg) subcutaneously before surgery. Scalp incisions were made and craniotomies (~1 mm in diameter) above the left parietal cortex were created using a high-speed drill (K.1070; Foredom) for unilateral viral injections while two craniotomies were made above both parietal cortices for bilateral viral injections. Beveled glass pipettes (1B100–4; World Precision Instruments) filled with viruses were placed into the striatum (0.8 mm anterior to the bregma, 2.0 mm lateral to the midline, and 2.4 mm from the pial surface). AAVs were injected at 200 nl/min using a syringe pump (Pump11 PicoPlus Elite; Harvard Apparatus). Glass pipettes were withdrawn after 10 min and scalps were cleaned and sutured with sterile surgical sutures. Mice were allowed to recover in clean cages with food containing Trimethoprim/Sulfamethoxazole and water for 7 days. Subsequent experiments were performed at least three weeks after surgeries.
Viruses used in this study included: AAV2/5 GfaABC1D-Rp122-HA (RiboTag AAV), AAV2/5 GfaABC1D-PI-Cre, AAV2/5 GfaABC1D-mCherry-hPMCA2w/b (CalEx AAV), AAV2/5 GfaABC1D-hM3Dq-mCherry, AAV2/5 GfaABC1D-hMADi-mCherry, AAV2/5 GfaABC1D-rM3Ds-mCherry, AAV2/5 GfaABC1D-GCaMP6f, AAV2/5 GfaABC1D-Lck-GFP and AAV2/5 GfaABC1D-tdTomato. All of these have been previously characterized for the striatum at the ages used in this study for wild type and the HD-model mice (Chai et al., 2017; Diaz-Castro et al., 2019; Haustein et al., 2014; Jiang et al., 2016; Nagai et al., 2019; Shigetomi et al., 2013; Yu et al., 2018). Viruses were diluted with saline when necessary and injected with a total volume of 0.5-1 μl per site to deliver ~0.5~2.5 x 1010 genome copies into the dorsal striatum. To sparsely label astrocytes for morphological analysis, AAV2/5 GfaABC1D-Lck-GYP was diluted to deliver 1 x 109 genome copies.
Details of the fourteen experimental perturbations (EPs)
To explore astrocyte molecular mechanisms in the striatum, we designed four groups of fourteen EPs including striatum-relevant disease and physiology. Each EP had its own age-matched cognate controls. The first group assessed astrocytes in two Huntington’s disease (HD) mouse models. A transgenic R6/2 model models juvenile-onset HD and a knock-in heterozygous Q175 model models adult-onset HD in human patients. Presymptomatic (1 m R6/2 and 2 m Q175) and symptomatic disease stages (2 m R6/2 and 12 m Q175) were examined to assess gene expression changes over the disease progression. Age-matched non-carrier (NCAR) mice were used as controls for R6/2 mice while wild-type (WT) mice were used as controls for Q175 mice. The second group assessed astrocytes under pathological conditions. Striatal medium spiny neurons (MSNs) were ablated with quinolinic acid (QA, 30 nM) through microinjection in the dorsal striatum one week before tissue harvest. In controls, vehicle (PBS) was microinjected into the dorsal striatum one week before tissue harvest. To induce neuroinflammation, lipopolysaccharide (LPS, 5 mg/kg body weight) was administered by intraperitoneal (i.p.) injection 24 hours before tissue harvest. Control mice received i.p. injection of vehicle (PBS) 24 hours before tissue harvest. To ablate dopaminergic neurons, mice received one i.p. injection of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) at 20 mg/kg body weight every 2 hours for a total of four doses one week before tissue harvest. This approach has been used to induce Parkinsonism in mice (Jackson-Lewis and Przedborski, 2007). In the control group, mice received injections of saline using the same regimen. Sapap3 knockout mice (SAPAP3 KO) were used as a genetic mouse model for obsessive-compulsive disorder (OCD) (Welch et al., 2007). Because OCD-like behavioral phenotypes developed after 4 months old of age, RiboTag AAV was injected into the dorsal striatum at 4.5 months old of age of both KO and WT littermates. The third group assessed the impact of altered astrocyte ionic signalling. To reduce astrocyte K+ current, 0.5 μl of AAV2/5 GfaABC1D-Cre was co-injected with RiboTag AAV into the dorsal striatum of Kcnj10flox/flox mice to conditionally delete astrocyte potassium channel Kir4.1 (Kir4.1 KO). Control mice received 0.5 μl of AAV2/5 GfaABC1D-tdTomato together with RiboTag AAV. To silence astrocyte intracellular Ca2+ signalling, we used two strategies: CalEx AAV together with RiboTag AAV were microinjected into the dorsal striatum of C57BL/6NTac mice to attenuate astrocyte Ca2+ signalling in adulthood. Control mice received 0.5 μl of AAV2/5 GfaABC1D-tdTomato together with RiboTag AAV. Transgenic Ip3r2 knockout mice (IP3R2 KO) were used to silence astrocyte Ca2+ signalling throughout the development. Age-matched WT littermates were used as controls. The fourth group evaluated astrocytes after activating specific astrocyte G-protein coupled receptor (GPCR) pathways (Gi, Gq and Gs) using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs). AAV2/5 GfaABC1D-hM3Dq-mCherry, AAV2/5 GfaABC1D-hM4Di-mCherry or AAV2/5 GfaABC1D-rM3Ds-mCherry was co-injected with RiboTag AAV into the striatum of C57BL/6NTac mice. A selective synthetic agonist CNO (1 mg/kg body weight) for DREADDs was given by i.p. injection 2-6 hours before tissue harvest. Control mice received i.p. injection of vehicle (saline) 2-6 hours before tissue harvest. All EPs were validated either in previously published studies or in the current study (Table S1 and Figure S2).
Striatal astrocyte RNA-sequencing (RNA-seq) and analysis
To exact RNA from striatal astrocytes, RiboTag AAV was microinjected bilaterally into the dorsal striatum of mice. Two to three weeks after the RiboTag AAV injection, RNA extraction was performed as previously described (Diaz-Castro et al., 2019; Nagai et al., 2019; Yu et al., 2018). Briefly, striatal tissues were dissected and homogenized in ice-cold homogenization buffer. RNA was extracted from 10-20% of homogenate after centrifugation as input sample, which contained RNA from all cell types in the striatum (Qiagen Rneasy Plus Micro #74034). The remaining homogenate was incubated with mouse anti-HA antibody (1:250; Covance, #MMS-101R) for 4 hours at 4 °C followed by the addition of magnetic beads (Invitrogen, Dynabeads #110.04D) for overnight incubation at 4°C. RNA was purified from the immunoprecipitation (IP) sample, which contained astrocyte-enriched RNA (Qiagen RNeasy Plus Micro #74034).
RNA concentration and quality were assessed with Agilent 2100 Bioanalyzer. RNA samples with RNA integrity number (RIN) greater than 7 were used for multiplexed library preparation with Nugen Ovation RNA-Seq System V2. For each experiment, all samples were multiplexed into a single pool in order to avoid batch effects (Auer and Doerge, 2010), and sequencing was performed on Illumina NextSeq 4000 for 2 × 75 yielding at least 45 million reads per sample. Demultiplexing was performed with Illumina Bcl2fastq2 v 2.17 program. Reads were aligned to the mouse mm10 reference genome using the STAR spliced read aligner (Dobin et al., 2013) with default parameters and fragment counts were derived using HTS-seq program. Approximately 70% of the reads mapped uniquely to the mouse genome and were used for subsequent analyses. Differential gene expression analysis was performed with Bioconductor packages edgeR (Law et al., 2014; Robinson et al., 2010) with false discovery rate (FDR) threshold < 0.05 (http://www.bioconductor.org). Lowly expressed genes that had CPM > 3 in at least 4 samples were filtered out. In addition, we have applied FPKM >1 as an additional threshold to exclude low expression genes for the analyses of DEG numbers, canonical pathways and upstream regulators. This value was chosen based on previously published literature (Hebenstreit et al., 2011; Uhlen et al., 2017). For the top 20 gene list, we used FPKM > 5 as a threshold to select genes with higher expression levels. Differentially expressed genes (DEGs) that were more than two-fold higher in the IP samples than the input samples were designated as astrocyte-enriched DEGs. Weighted gene co-expression network analysis (WGCNA) was performed using an R package of WGCNA for finding modules of highly correlated genes. The gene ontology (GO) enrichment analysis was performed using PANTHER overrepresentation test (GO ontology database released 2020-01-01) with FDR < 0.05, with allMus musculus genes used as the reference. Significantly associated canonical pathways and upstream regulators (P < 0.05) were identified by Ingenuity Pathway Analysis (IPA) using DEGs with thresholds FPKM > 1 and FDR < 0.05. All RNA-seq data have been deposited within the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo) with accession numbers provided in Table S1 and Key Resources Table.
We comment briefly on the use of the Rpl22-HA RiboTag method in our evaluations. First, from the immunohistochemistry analysis that we performed previously (Diaz-Castro et al., 2019; Nagai et al., 2019; Yu et al., 2018), Rpl22-HA was expressed in ~ 85% of the S100β+ astrocytes in the dorsal striatum. The expression of Rpl22-HA was consistent and uniform across astrocytes, ages and mouse models. It was not expressed in neurons. Second, we have identified subpopulations of striatal astrocytes from our scRNA-seq using droplet based methods (i.e. not dependent on RiboTag). Gfap has relatively low and comparable expression level across all astrocyte subpopulations, and is not a marker gene for any astrocyte subpopulation: these data recall RiboTag methods for Gfap and more broadly for astrocyte markers (Chai et al., 2017). Thus, the finding we can observe similar astrocyte gene expression with scRNA-seq and RiboTag methods provides assurance for the methodology. Furthermore, based on the available data it is very unlikely that GfaABC1D promoter activity biases towards any particular astrocyte population, as Gfap is equivalently low across astrocyte populations. Third, it is conceivable that experimental perturbations may affect the translational efficiency or ribosome association in disease settings. However, it is highly unlikely that HA-tagged ribosomes discriminate transcripts based on their relative abundance. To avoid potential technical bias, we normalized the concentration and amount of total RNA for sequencing across all samples, which have high RNA integrity with RIN > 7.0. In addition, we filtered out lowly expressed genes in the subsequent analysis and applied several expression thresholds for fair comparisons. Fourth, we have previously used the RiboTag approach in the context of R6/2 and Q175 HD-model mice. Reassuringly, these same mice at the same ages have recently been reassessed using a different BAC-Trap method (Lee et al., 2020). The findings in that study, with an independent method, were remarkably similar to our past work with RiboTag (Diaz-Castro et al., 2019). Fifth, when we first reported the RiboTag method to assess astrocyte-specific gene expression (Srinivasan et al., 2016), we performed detailed analyses by comparing our cortical data to cortical astrocyte gene expression data gathered with FACS isolated cells (Zhang et al., 2014). Specifically, we determined the 4727 transcripts enriched in astrocytes in either dataset and used the rank-rank hypergeometric overlap (RRHO) method to compare their relative rank according to FPKM percentile (across all 4727 genes). Most transcripts were significantly clustered along the diagonal, which indicated similarity in rank between RiboTag and FACS methods (Srinivasan et al., 2016). These published validations of the method also provide assurance that the RiboTag method works reliably. In summary then, with the data that are available for us to candidly assess, we suggest that the RiboTag method can sample astrocyte-specific gene expression reliably. Nonetheless, our data should not be over interpreted and they should be appraised appropriately and considered with the aforementioned caveats in mind in future work. Our data also do not obviate the need for future controls by others on a case-by-case basis when they use the methods we report. To aid further assessments, we have made all the RNA-seq data openly available (Key Resources Table).
Striatal single cell RNA-seq (scRNA-seq) and analysis
The procedure of Act-seq (Wu et al., 2017) was optimized to improve the enrichment of glial cells and scRNA-seq was performed to profile the whole striatum of adult mice. Male mice at 8-9 weeks old of age were anesthetized and decapitated. The brain was immediately dissected out and was sectioned on a vibratome (Microslicer DTK-Zero 1; Ted Pella, Inc.) into 400 pm slices in ice-cold artificial cerebrospinal fluid (ACSF) (124 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 24 mM NHCO3, 5 mM HEPES, 13 mM glucose, 2 mM MgSO4, and 2 mM CaCl2, pH adjusted to 7.3-7.4) oxygenated with 95% O2/5% CO2. The slices containing the striatum were immediately transferred to an oxygenated recovery solution (93 mM N-methyl-D-glucamine, 2.5 mM KCl, 1.2 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 10 mM MgSO4, 0.5 mM CaCl2, 5 mM sodium ascorbate, 2 mM thiourea, and 3 mM sodium pyruvate with a pH of 7.3-7.4) for 15 min on ice. The striatum was dissected out under a dissecting microscope in ice-cold ACSF and cut into small pieces (< 1 mm in all dimensions). Tissue was then transferred to a petri dish for digestion with ACSF containing 1 mg/ml pronase (Sigma-Aldrich, P6911) and incubated at 34 °C for 30 min. The digestion solution was replaced with ice-cold oxygenated ACSF containing 1% fetal bovine serum. The tissue was dissociated sequentially by gentle trituration through glass pipettes with polished tip openings of 500 μm, 300 μm and 150 μm diameter. Actinomycin D (Sigma-Aldrich, A1410) was added to the recovery solution at 45 μM, the pronase solution at 45 μM and trituration solution at 3 μM to prevent stress-induced transcriptional alterations. To increase the yield of glial cells, filters with various pore sizes (70 μm, 40 μm and 20 μm) were tested and 20 μm filter gave the highest yield and therefore was chosen. The dissociated cells were filtered through a 20 μm filter and washed with ice-cold ACSF. To remove myelin, cell pellet was resuspended in PBS and processed with debris removal kit (Miltenyi Biotec, 130-109-398). Cell density was counted and isolated cells were diluted to 1000 cells/μl and processed with 10X Genomics platform within 10 min. Single cell libraries were generated and sequenced on the Illumina NextSeq500 sequencer.
Sequence reads were processed and aligned to the mouse genome (mm10) using CellRanger 3.0. Striatal cells with > 300 genes and genes expressed in > 3 cells were used for the subsequent analysis in Python. Principal component analysis (PCA) was performed on the expression data matrix using Arpack wrapper function in the Scanpy package (Wolf et al., 2018). PCs 1-50 were used for generating t-distributed stochastic neighbor embedding (tSNE) plots. Eleven transcriptomic clusters were identified from total 20912 striatal cells with a Louvain-Jaccard graph clustering algorithm with resolution set to 0.1 and were then annotated based on the expression of cell lineage marker genes (Saunders et al., 2018). For the subclustering of astrocytes, cells that were classified as astrocytes in the major cell type analysis were used (3244 astrocytes) and 5 astrocyte subpopulations were identified with resolution set to 0.18. Marker genes of oligodendrocytes, astrocytes, microglia and neurons from the human striatum were selected from Kelly et al. (Kelley et al., 2018) ranked by the fidelity scores. Human genes associated with basal ganglia disorders were based on the database of Phenopedia (Yu et al., 2010) and then mapped onto 11 major cell clusters based on the top 1000 marker genes identified from the mouse striatal scRNA-seq. scRNA-seq data have been deposited within the GEO repository (www.ncbi.nlm.nih.gov/geo) with accession number GSE156628.
Drug administration in vivo
AAV2/5 GfaABC1D-hMADi-mCherry was microinjected into the dorsal striatum of R6/2 or NCAR mice at 4 weeks old. Two weeks after the microinjection, 1 mg/kg clozapine N-oxide (CNO; Tocris Bioscience, 4936) was administered to mice by i.p. injection every other day for 3-6 weeks to chronically activate hM4Di in dorsal striatal astrocytes in vivo. Behavioral tests, brain slice experiments and immunohistochemistry were performed two hours after the last CNO administration. In some cases, 100 mg/kg Gabapentin (Tocris Bioscience, 0806) was administered by i.p. injection to mice 1 hour before every CNO injection. Previous work shows that the effects of striatal astrocyte hM4Di DREADD activation in mice are reversible (Nagai et al., 2019), which is consistent with prior assessments in other cells (Alexander et al., 2009). However, the reversibility experiment could not be performed in R6/2 mice for the present study, because by ~12 weeks the mice are sick from the disease in other parts of the body (recall we microinject AAV for hM4Di only into the striatum, whereas the disease affects other parts of the body as well). Hence, the humane endpoint considerations of our experiments mandate that the mice be euthanized before they succumb to disease throughout the body.
Immunohistochemistry (IHC) and analysis
Frozen sections:
For transcardial perfusion, mice were anesthetized with 5% isoflurane and once all reflexes subsided, the abdominal cavity was opened and heparin (50 units) was injected into the left ventricle to prevent blood coagulate. The animal was perfused with 20 ml ice cold 0.1 M phosphate buffered saline (PBS) followed by 60 ml 10% buffered formalin (Fisher #SF100-20). After gentle removal from the skull, the brain was post-fixed in 10% buffered formalin overnight at 4°C. The tissue was cryoprotected in 30% sucrose (0.1M PBS) and serial 40 μm coronal sections were prepared using a cryostat microtome (Leica) at −20°C and processed for immunohistochemistry. Sections were incubated with agitation in primary antibodies diluted in 0.1 M PBS with 0.5% Triton-X 100 overnight at 4°C. The following primary antibodies were used: mouse anti-S100β (1:1,000; Sigma, S2532), mouse anti-NeuN (1:1,000; Millipore, MAB377), chicken anti-GFP (1:1,000; Abcam, ab13970), mouse anti-tyrosine hydroxylase (1:1,000; Immunostar, 22941); rabbit anti-SAPAP3 (1: 600; a gift from Dr. Feng’s laboratory at MIT); rabbit anti-Kir4.1 (1:1,000; Alomone, APC-035), rabbit anti-DARPP-32 (1:200; Abcam, ab40801) or rabbit anti-cFos (1:1,000; Millipore, ABE457). The sections were then washed 3 times in 0.1 M PBS for 10 min each before incubation at room temperature for 2 hr with secondary antibodies diluted in 0.1 M PBS. Alexa conjugated (ThermoFisher Scientific) secondary antibodies were used at 1:1000 dilution except streptavidin conjugated Alexa 647 at 1:250 dilution. Fluorescent images were taken using UplanSApo 20X 0.85 NA, UplanFL 40X 1.30 NA oil immersion or PlanApo N 60X 1.45 NA oil immersion objective lens on a confocal laser-scanning microscope (FV10-ASW; Olympus). Laser settings were kept the same within each experiment. Images represent maximum intensity projections of optical sections with a step size of 1.0 μm.
Images were processed with ImageJ. Cell counting was done on maximum intensity projections using the Cell Counter plugin; only cells with soma completely within the region of interest (ROI) were counted. For the analysis of astrocyte territory size, the images of Lck-GFP-expressing astrocytes were thresholded to remove background signals. Images were then converted to a binary format in which pixels above the threshold were counted as 1 and pixels with signal at the level of background or lower were counted as 0. Astrocyte territory sizes were estimated by measuring the area of a ROI that surrounded the thresholded fluorescence profile of astrocytes. For analysis of the size of the striatum and the lateral ventricle, coronal serial cryosections with 40 μm step size were prepared. Measurements were made by tracing the boundaries of the striatum and lateral ventricles using ImageJ to calibrate the area in square millimeters for each section. Landmarks used to define the borders of the striatum were the corpus callosum on the dorsal and lateral sides, the lateral ventricle on the medial side, and the anterior commissure and internal capsule on the ventral side. The section at + 0.9 mm AP from bregma from each mouse was subjected to the comparisons between experimental groups.
Acute sections:
Fresh brain slices (300 μm) were placed into 10% buffered formalin overnight at 4°C and processed for IHC. Sections were washed 3 times in 0.1 M PBS with 2% Triton-X 100 for 5 min each, and then incubated in a blocking solution containing 10% NGS in 0.1 M PBS with 1% Triton-X 100 for 1 hour at room temperature with agitation. Sections were then incubated with agitation in primary antibodies diluted in 0.1 M PBS with 0.4% Triton-X 100 for 3 days at 4°C. The primary antibody was guinea pig anti-vGluT1 (1:2000; Synaptic Systems 135302). Sections were washed 3 times in 0.1 M PBS with 0.4% Triton-X 100 for 10 min each before incubation 3 days at 4°C with streptavidin conjugated Alexa 647 (1:250) diluted in 0.1 M PBS with 0.4% Triton-X 100. The sections were rinsed 3 times in 0.1 M PBS for 10 min each before being mounted on microscope slides in fluoromount-G. Images were obtained in the same way as IHC for frozen sections except a step size of 0.33 μm.
Spine density was quantified in cells that had been characterized by electrophysiology by whole-cell current-clamp recording (Methods described in a following section) and filled intracellularly with biocytin (1 mg/ml) via the patch pipette. Based on the electrophysiological properties of the cells (membrane resistance, membrane potential and action potential firing), we only analyzed neurons that were classified as MSNs. For quantification of spine density, we only analyzed spines on secondary dendritic shafts that are parallel to the imaging plane to minimize the possibility of rotational artifacts. The primary dendrite of MSNs does not have dendritic spines. Spine density was calculated by dividing the number of spines by the length of the dendritic segment. For quantification of spine head size, a line ROI across the maximum diameter of the spine was made and a profile that has a single peak and is closer to a Gaussian curve was obtained. Full-Width Half-Maximum of that was defined as a spine head size to avoid the point spread function. For counting the number of vGluT1-positive synapse, only spines that are off from optical plane were analyzed. As described previously (Nagai et al., 2019), a line ROI was made over MSN spine and vGluT1 puncta that is closest to the spine. FWHM of each profile was measured. A MSN spine was recognized as forming vGluT1-positive synapse when each FWHM is overlapped, while recognized as not forming vGluT1-positive synapse when there is a gap between each FWHM. Analysis of MSN morphology was performed by the Sholl method of concentric circles using an ImageJ plugin essentially as described previously (Reeves et al., 2011). Each cell was thus analyzed by selecting the center of its soma and then performing the Sholl analysis, which counts the number of intersections at circles of increasing radii from the center with 20 μm step size.
Dual in situ hybridization (ISH) with RNAscope and IHC
Cryosections were prepared as described above and stored at −80°C. ISH was performed using Multiplex RNAscope (ACDBio 320851). Sections were washed at least for 15 min with 0.1 M PBS, and then incubated in 1X Target Retrieval Reagents (ACDBio 322000) for 5 min at 99-100°C. After washing with ddH2O twice for 1 min each, they were dehydrated with 100% ethanol for 2 min and dried at RT. Then, the sections were incubated with Protease Pretreat-4 solution (ACDBio 322340) for 30 min at 40°C. The slides were washed with ddH2O twice for 1 min each and then incubated with probe Mm-Thbs1-C3 (ACDBio 457891-C3) for 2 hours at 40°C. The sections were incubated in AMP 1-FL for 30 min, AMP2-FL for 15 min, AMP3-FL for 30 min and AMP4-FL for 15 min at 40°C with washing in 1X Wash Buffer (ACDBio 310091) twice for 2 min each prior to the first incubation and in between incubations. All the incubations at 40°C were performed in the HybEZ™ Hybridization System (ACDBio 310010). Slices were washed in 0.1 M PBS three times for 10 min each, followed by IHC that was performed as described above except with antibody dilutions: chicken anti-GFP (Abcam ab13970) was used at 1:250 dilution to stain Lck-GCaMP. Images were obtained in the same way as IHC described above except a step size of 0.8 μm. Images were processed with ImageJ (NIH). Astrocyte somata were demarcated based on GFP signal, and number of puncta and intensity of probe signals within somata were measured.
Acute brain slice preparation
Mice were transferred from the vivarium to the laboratory during the light cycle between 8 am and 10 am. Brain slices were prepared 30 min to 1 hour afterwards and were used for experiments within 8 h of slicing (mostly within 6 hours). Sagittal striatal slices (300 μm thick) were prepared from 11-12 week old R6/2 or NCAR mice for imaging and electrophysiological recordings in ice-cold sucrose cutting solution (30 mM NaCl, 4.5 mM KCl, 1.2 mM NaH2PO4, 26 mM NaHCO3, 10 mM D-glucose, 194 mM sucrose and 1 mM MgCl2) using a vibratome (DSK Zero1 Microslicer; Ted Pella, Inc.). Slices were then incubated in artificial cerebrospinal fluid (ACSF) (124 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, 10 mM D-glucose, and 2.0 mM CaCl2) for 30 min at 32°C and 1 h at room temperature before recording. All the solutions were oxygenated with 95% O2/5% CO2.
Intracellular Ca2+ imaging of striatal astrocytes and analysis
Striatal slice preparation was performed as described above. Striatal slices were maintained in oxygenated ACSF (124 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, 10 mM D-glucose, and 2.0 mM CaCl2) through a perfusion system. Astrocytes for all the experiments were imaged using a confocal microscope (Fluoview 1200; Olympus) with a 40× water-immersion objective lens with a numerical aperture (NA) of 0.8 and at a digital zoom of two to three. We used the 488 nm line of an Argon laser, with the intensity adjusted to 9% of the maximum output of 10 mW. Astrocytes were chosen typically ~20 to ~30 μm below the slice surface and scanned at 1 frame per second for imaging sessions.
Analyses of time-lapse image series were performed using ImageJ (NIH). XY drift was corrected using a custom plugin in ImageJ. Time traces of fluorescence intensity were extracted from the ROIs and converted to dF/F values. For analyzing spontaneous Ca2+ signaling, ROIs were defined in normal aCSF (control). Using Origin 2016 (Origin Lab Corp), Ca2+ events were manually marked. Event amplitudes, half width, event frequency per ROI per min, the integrated area-under-the-curve (AUC) of dF/F traces were measured. Events were identified based on amplitudes that were at least 2-fold above the baseline noise of the dF/F trace.
Electrophysiological recordings in the striatal slices
Electrophysiological recordings were performed using standard methods as described below. Slices were placed in the recording chamber and continuously perfused with 95% O2 and 5% CO2 bubbled normal ACSF. pCLAMP10.4 software and a Multi-Clamp 700B amplifier was used for electrophysiology (Molecular Devices). Whole-cell patch-clamp recordings were made from medium spiny neurons (MSNs) in the dorsolateral striatum using patch pipettes with a typical resistance of 5-6 MΩ. MSNs were morphologically and electrophysiologically identified. The intracellular solution for MSN EPSCs recordings comprised the following (in mM): 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 0.3 Na-GTP, 2 Mg-ATP, 10 TEA-Cl, with pH adjusted to 7.3 with CsOH. The intracellular solution for other experiments comprised the following (in mM): 135 K gluconate, 5 KCl, 0.5 CaCl2, 5 HEPES, 5 EGTA, 2 Mg-ATP and 0.3 Na-GTP, pH 7.3 adjusted with KOH. To assess evoked EPSCs, electrical field stimulation (EFS) was achieved using a bipolar matrix electrode (FHC) that was placed on the dorsolateral corpus callosum to evoke glutamate release from the cortico-striatal pathway. The MSNs to be assessed were typically located ~300-400 μm away from the stimulation site to avoid the EFS-evoked astrocyte calcium increase that occurs nearby the stimulating electrode. We could not determine full stimulation-response curves for NMDA EPSCs, because clamping the cells at +40 mV for the prolonged periods needed for such assessments decreased the quality of whole-cell recording. However, we evaluated AMPA and NMDA EPSCs equivalently when stimulation intensities were set to 200 μA that approximately evoke responses at 50% maximal amplitude of the AMPA EPSCs. To isolate the AMPAR- and NMDAR-mediated evoked EPSCs, MSNs were voltage-clamped at −70 mV or +40 mV in the presence of 10 μM bicuculline (Tocris Bioscience 0131). Paired pulses were delivered at 50 ms inter-pulse intervals. The AMPAR-mediated EPSC was measured at the peak amplitude of the EPSC at −70 mV, while the amplitude of the EPSC 50 ms after stimulation at +40 mV was used to estimate the NMDAR-mediated component. To isolate mEPSCs, MSNs were voltage-clamped at −70 mV and pre-incubated with 10 μM bicuculline and 300 nM tetrodotoxin (TTX, Cayman Chemical 14964) for 5 min before recording. In some cases, 1 mg/ml biocytin (Tocris, 3349) was added to the intracellular solution to subsequently visualize patched neuron. All recordings were performed at room temperature, using pCLAMP10.4 (Axon Instruments, Molecular Devices) and a MultiClamp 700B amplifier (Axon Instruments, Molecular Devices). Cells with Ra that exceeded 20 MΩ were excluded from analysis. Analysis was performed using ClampFit 10.4 software.
Behavioral tests
Behavioral tests were performed during the light cycle between 12:00 pm and 6:00 pm. Only male mice were used in behavioral tests because of gender-dependent differences known for striatal physiology. All the experimental mice were transferred to the behavior testing room at least 30 min before the tests to acclimatize to the environment and to reduce stress. Temperature and humidity of the experimental rooms were kept at 23 ± 2°C and 55 ± 5%, respectively. Background noise (65 ± 2 dB) was generated by white noise generator (San Diego Instruments).
Sucrose preference test:
Two days prior to LPS administration, mice at 9 weeks old were provided with ad libitum access to two regular bottles of water for 24 h in the home cage. On the following day, one bottle of water was replaced with 1% (wt/vol) sucrose water and mice were given continuous exposure to both bottles for 24h. Then mice received either LPS (5 mg/kg body weight) or PBS vehicle through i.p. injection and were given one bottle of water and one bottle of 1% sucrose water for 24h. The bottles were weighed every day to measure the amounts of liquid consumption. The preference ratio was calculated as a ratio of sucrose water intake and regular water intake over 24 h testing period.
Open field test:
The open field chamber consisted of a square arena (28.7 × 30 cm) enclosed by walls made of translucent polyethylene (15 cm tall). The brightness of the experimental room was kept < 10 lux. Locomotor activity of mice at 10 weeks old was then recorded for 30 min using an infrared camera located above the open field chamber. Recording camera was connected to a computer operating an automated video tracking software ANY-maze from Stoelting.
Self-grooming behavior:
Mice were placed individually into plastic cylinders (15 cm in diameter and 35 cm tall) and allowed to habituate for 20 min. The brightness of the experimental room was kept < 10 lux. Rearing behavior was recorded for 10 min. The number of rearing bouts was counted, in which mice support their weight freely on its hind legs without using its tail or forepaws. A timer was used to assess the cumulative time spent in self-grooming behavior, which included paw licking, unilateral and bilateral strokes around the nose, mouth, and face, paw movement over the head and behind ears, body fur licking, body scratching with hind paws, tail licking, and genital cleaning. The number of self-grooming bouts was also counted. Separate grooming bouts were considered when the pause was more than 5 s or behaviors other than self-grooming occurred. Self-grooming microstructure was not assessed.
Forelimb grip test:
Mice at 10 weeks old were used in this test. Forelimb strength was measured using a customized grip strength meter (Chatillon Digital force gauge) with an adjustable angel wire mesh grip (San Diego Instruments). Both the grip strength meter and the mesh grip were positioned horizontally. Each mouse was held by the base of the tail and lowered to grasp the mesh with both forelimbs. A steady horizontal force was then applied to extend the mouse away from the meter until it relinquished its grip on the mesh. Maximum pulling force required to separate the mouse from the mesh grip was recorded by the meter. Each mouse was tested in five trials with a 5 min intertrial interval and average maximum pulling force was used to assess the muscle strength.
Hind limb footprint test:
Mice at 10 weeks old were used in this test. A one-meter long runway (8 cm wide) was lined with paper. Each mouse with hind paws painted with non-toxic ink was placed at an open end of the runway and allowed to walk to the other end with a darkened box. For the gait analysis, stride length and width were measured and averaged for both left and right hind limbs over 5 steps.
Hind limb clasping test:
Mice at 10-11 weeks old were used in this test. Each mouse was suspended by holding the tail. The experiments were conducted for 60 s. Time taken for the mouse to move both limbs close to the body and for both paws to clasp was recorded. The mice that did not clasp in this time period were given a data value of 60 s.
Behavioral and deficit scores:
We evaluated HD-related behavioral phenotypes in three groups: the first group comprised control NCAR mice that received control AAVs (“NCAR + AAV”); the second group comprised R6/2 mice that also received control AAVs (“R6/2 + AAV”); and the third group comprised R6/2 mice received hM4Di AAVs (“R6/2 + hM4Di”). All groups were identically treated with CNO to control for off-target effects, which were not observed. Seven behavioral parameters were assessed: (i) the distance travelled by the mice over 30 min in the open field test, (ii) the frequency of rearing in an open field chamber, (iii) the grip strength of forelimbs, (iv) the stride length, (v) the stride width of hind limbs, (vi) the latency to clasp hind limbs upon tail suspension, and (vii) the duration of self-grooming. Behavioral score (x) for each test parameter was calculated as the normalized difference between the mean value of an experimental group (y) and the mean value of the control NCAR + AAV group (z): . Aggregate behavioral deficit score was calculated as the sum of the absolute values of behavioral scores from all seven parameters.
GPCR-G protein coupling analysis
Genes identified in IP samples from R6/2 and NCAR mice at 3 months old (FPKM > 0) (Diaz-Castro et al., 2019) were used to search for all GPCR genes annotated in GPCRdb (Pándy-Szekeres et al., 2018a). Coupling information of their primary and secondary G-protein signaling transducers was obtained using the recently implemented GPCR-G protein couplings in the GPCRdb database that merge information on receptor coupling partners (Flock et al., 2017; Harding et al., 2018; Inoue et al., 2019). Relative enrichment in astrocytes vs. all cells in the striatum and relative enrichment in striatal astrocytes vs. hippocampal astrocytes of the identified GPCR genes were obtained from a previously published data set (Chai et al., 2017).
Quantitation and statistical analysis
Sample sizes were based on previously published work (Chai et al., 2017; Nagai et al., 2019; Yu et al., 2018). Raw replicate values for all experiments are provided in Excel file S4. The results of statistical comparisons, n numbers and P values are shown in the figure panels or figure legends with the average data. N is defined as the numbers of cells, sections or mice throughout on a case-by-case basis; the unit of analysis is stated in the text or each figure legend. Full details of n numbers, precise P values, statistical tests are reported in Excel file S3. When the average data are reported in the text, the statistics are reported there. Statistical tests were run in OriginPro 2016 or GraphPad Prism 8. Summary data are presented as mean ± SEM along with the individual data points. Note that in some of the graphs the bars representing the SEM in figure panels are smaller than the symbols used to represent the mean. For most statistical analyses we used non-parametric tests due to the relatively small sample size throughout the study: two-tailed Mann–Whitney test for the comparisons between two groups and Kruskal-Wallis one-way ANOVA test followed by Dunn’s post-hoc test for the comparisons between more than two groups with significance declared at P < 0.05. The repeated measures two-way ANOVA test was used to compare means across variables that are based on repeated observations (Figure S6B and S6E). In the figures, P values were stated by asterisk(s): *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. A statistical FDR value < 0.05 was used for all RNA-seq analyses. All mice were assigned to particular experimental groups at random. No data points were excluded from any experiment.
Supplementary Material
Excel File S1: Data reporting the IP DEGs (FDR < 0.05, FPKM > 1) for all the astrocyte-specific RNA-seq projects (related to Figure 1).
Excel File S2: Data reporting genes that belong to each of the WGCNA modules (related to Figure 1).
Excel File S3: Data reporting statistical analyses, n numbers and relevant parameters (related to Figures 3–5, 7 and Figure S6).
Excel File S5: Data reporting the GPCRs identified from RNA-seq of NCAR and R6/2 (related to Figure 8).
Highlights.
Striatal astrocytes display context-specific molecular responses
Astrocytic HD molecular signatures are reciprocally regulated by Gi-GPCR activation
Activation of astrocyte Gi-GPCR signaling in vivo corrects several HD-phenotypes
Astrocyte responses can be used for phenotypic benefit in a brain disorder model
Acknowledgements
We thank the UCLA Neuroscience Genomics Core (www.semel.ucla.edu/ungc) and Fuying Gao for assistance with RNA-seq and analysis. Thanks to Ye Emily Wu for guidance on scRNA-seq. Thanks to Guoping Feng for SAPAP3 antibody. Thanks to Blanca Diaz Castro, Vahri Beaumont and Ignacio Munoz-Sanjuan for discussions. This work was supported by the National Institutes of Health (NS111583 and MH104069), CHDI Inc, by an Allen Distinguished Investigator Award through The Paul G. Allen Frontiers Group, and by the Ressler Family Foundation (to BSK). XY was partly supported by the American Heart Association (16POST27260256). JN was partly supported by a JSPS Overseas Research Fellowship (H28-729) and the Uehara Memorial Foundation Overseas Postdoctoral Research Fellowship (201730082). We acknowledge the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691 to GC) and the Genetics, Genomics and Informatics Core of the Semel Institute of Neuroscience at UCLA (U54HD087101-01; Eunice Kennedy Shriver National Institute of Child Health and Human Development). MM-S and MMB acknowledge the MRC (MC_U105185859) for support. MM-S is a Wolfson College Junior Research Fellow and has been supported by a Federation of European Biochemical Societies Long-Term Fellowship and a Marie Sklodowska-Curie Individual Fellowship from the European Union’s Horizon 2020 research and innovation programme (No 832620). MMB is a Lister Institute Fellow and is supported by the ERC (ERC-COG-2015-682414) and by ALSAC. JSS is supported by the National Science Foundation Graduate Research Fellowship Program (2017238856).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Agus F, Crespo D, Myers RH, and Labadorf A (2019). The caudate nucleus undergoes dramatic and unique transcriptional changes in human prodromal Huntington’s disease brain. BMC Med Genomics October 16;12(1):137. doi: 10.1186/s12920-019-0581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, Adorjan I, Menon V, and Goldman JE (2020). Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun February 18;8(1):19. doi: 10.1186/s40478-020-0880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves de Lima K, Rustenhoven J, and Kipnis J (2020). Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu Rev Immunol 38, 597–620. [DOI] [PubMed] [Google Scholar]
- Auer PL, and Doerge RW (2010). Statistical design and analysis of RNA sequencing data. Genetics 185, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, et al. (2015). Huntington disease. Nat Rev Dis Primers April 23;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, and Martin JB (1986). Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 321, 168–171. [DOI] [PubMed] [Google Scholar]
- Bensadoun JC, de Almeida LP, Dreano M, Aebischer P, and Deglon N (2001). Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in the quinolinic acid rat model of Huntington’s syndrome. Eur J Neurosci 14, 1753–1761. [DOI] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, and Li S (2009). Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A 106, 22480–22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S, and Li XJ (2010). Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem 285, 10653–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandre EP, Rico-Villademoros F, and Slim M (2016). Alpha2delta ligands, gabapentin, pregabalin and mirogabalin: a review of their clinical pharmacology and therapeutic use. Expert Rev Neurother 16, 1263–1277. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Cummings DM, André VM, Holley SM, and Levine MS (2010). Genetic mouse models of Huntington’s disease: focus on electrophysiological mechanisms. ASN Neuro April 7;2(2):e00033. doi: 10.1042/AN20090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whittelegge JP, et al. (2017). Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological and functional evidence. Neuron 95, 531–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, and Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Colpo GD, Stimming EF, Rocha NP, and Teixeira AL (2017). Promises and pitfalls of immune-based strategies for Huntington’s disease. Neural Regen Res 12, 1422–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Jiang X, Taylor A, and Mennerick S (2012). Astrocyte-derived thrombospondins mediate the development of hippocampal presynaptic plasticity in vitro. J Neurosci 32, 13100–13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Mesquita S, Fu Z, and Kipnis J (2018). The Meningeal Lymphatic System: A New Player in Neurophysiology. Neuron 100, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Castro B, Gangwani M, Yu X, Coppola G, and Khakh BS (2019). Astrocyte molecular signatures in Huntington’s disease. Sci Transl Med 11(514). pii: eaaw8546. doi: 10.1126/scitranslmed.aaw8546. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594, 5369–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. (2009). Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito S, Bianchini S, Baggi E, Fattizzo M, and Rigante D (2014). Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: an overview. Eur J Clin Microbiol Infect Dis 33, 2105–2109. [DOI] [PubMed] [Google Scholar]
- Flock T, Hauser AS, Lund N, Gloriam DE, Balaji S, and Babu MM (2017). Selectivity determinants of GPCR-G-protein binding. Nature 545, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Grafton ST (2015). The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol August 3;7(8):a021691. doi: 10.1101/cshperspect.a021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nature Neuroscience 22, 2087–2097. [DOI] [PubMed] [Google Scholar]
- Haim LB, and Rowitch DH (2017). Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, Gray AJG, Bruce L, Alexander SPH, Anderton S, et al. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46, D1091–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haustein MD, Kracun S, Lu XH, Shih T, Jackson-Weaver O, Tong X, Xu J, Yang XW, O’Dell TJ, Marvin JS, et al. (2014). Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 82, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, and Teichmann SA (2011). RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol June 7;7:497. doi: 10.1038/msb.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 15, 965–977. [DOI] [PubMed] [Google Scholar]
- Inoue A, Raimondi F, Kadji FMN, Singh G, Kishi T, Uwamizu A, Ono Y, Shinjo Y, Ishida S, Arang N, et al. (2019). Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 177, 1933–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A, Ao Y, Farkhondeh V, Johnsonbaugh H, Burda J, et al. (2018). Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proceedings of the National Academy of Sciences 115, E302–E309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Lewis V, and Przedborski S (2007). Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Tong X, Looger LL, and Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan WT, Fernandez DC, Shumway LJ, Zhao H, and Hattar S (2017). Eye-Drops for Activation of DREADDs. Front Neural Circuits November 23;11:93. doi: 10.3389/fncir.2017.00093. eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley KW, Nakao-Inoue H, Molofsky AV, and Oldham MC (2018). Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci 21, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS (2019). Astrocyte-Neuron Interactions in the Striatum: Insights on Identity, Form, and Function. Trends Neurosci 42, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, and Grantyn R (2017). Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, and Deneen B (2019). The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci 42, 187–207. [DOI] [PubMed] [Google Scholar]
- Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, Inada H, Roh SE, Kim SJ, Lee G, et al. (2016). Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest 126, 1983–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadorf A, Hoss AG, Lagomarsino V, Latourelle JC, Hadzi TC, Bregu J, MacDonald ME, Gusella JF, Chen JF, Akbarian S, et al. (2015). RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS One December 4;10(12):e0143563. doi: 10.1371/journal.pone.0143563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, et al. (2020). Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron Jul 15:S0896-6273(20)30475-X. doi: 10.1016/j.neuron.2020.06.021. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Mϋnch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb A, Weston M, and Kullmann DM (2019). Designer receptor technology for the treatment of epilepsy. EBioMedicine 43, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai J, Rajbhandari AK, Gangwani MR, Hachisuka A, Coppola G, Masmanidis SC, Fanselow MS, and Khakh BS (2019). Hyperactivity with Disrupted Attention by Activation of an Astrocyte Synaptogenic Cue. Cell 177, 1280–1292 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris GT, and Kipnis J (2019). Immune cells and CNS physiology: Microglia and beyond. J Exp Med 216, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwaobi SE, Cuddapah VA, Patterson KC, Randolph AC, and Olsen ML (2016). The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol 132, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octeau JC, Chai H, Jiang R, Bonanno SL, Martin KC, and Khakh BS (2018). An Optical Neuron-Astrocyte Proximity Assay at Synaptic Distance Scales. Neuron 98, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsoe K, Hauser AS, Bojarski AJ, and Gloriam DE. (2018a). GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res 46, D440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpse K, Hauser AS, Bojarski AJ, and Gloriam DE (2018b). GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res 46, D440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AM, Shigetomi E, and Khakh BS (2011). Bulk loading of calcium indicator dyes to study astrocyte physiology: key limitations and improvements using morphological maps. J Neurosci 31, 9353–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, et al. (2018). Thrombospondin receptor α2δ-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J Cell Biol 217, 3747–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch NP, Ribeiro FM, Furr-Stimming E, and Teixeira AL (2016). Neuroimmunology of Huntington’s Disease: Revisiting Evidence from Human Studies. Mediators Inflamm 2016:8653132. doi: 10.1155/2016/8653132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues FB, Byrne LM, McColgan P, Robertson N, Tabrizi SJ, Zetterberg H, and Wild EJ (2016). Cerebrospinal Fluid Inflammatory Biomarkers Reflect Clinical Severity in Huntington’s Disease. PLoS One September 22;11(9):e0163479. doi: 10.1371/journal.pone.0163479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues FB, Byrne LM, and Wild EJ (2018). Biofluid Biomarkers in Huntington’s Disease. Methods Mol Biol 1780, 329–396. [DOI] [PubMed] [Google Scholar]
- Roth BL (2016). DREADDs for Neuroscientists. Neuron 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, and Khakh BS (2013). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, Wang C, Jacob F, Wu K, Traglia M, et al. (2019). Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nature Genetics 51, 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, and Khakh BS (2015). Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci 18, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu T-Y, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS (2016). New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron 92, 1181–1195: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, and Zlokovic BV (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science August 18;357(6352):eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Urban DJ, and Roth BL (2015). DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu Rev Pharmacol Toxicol 55, 399–417. [DOI] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP (2007). Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115, 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JM, Lu J, Rodriguiz RM, Trotta NC, Peca J, Ding JD, Feliciano C, Chen M, Adams JP, Luo J, et al. (2007). Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 448, 894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz MH, Pineda SS, Lee H, Kulicke R, Kellis M, and Heiman M (2020). Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol Neurodegener May 24;15(1):29. doi: 10.1186/s13024-020-00379-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston M, Kaserer T, Wu A, Mouravlev A, Carpenter JC, Snowball A, Knauss S, von Schimmelmann M, During MJ, Lignani G, et al. (2019). Olanzapine: A potent agonist at the hM4D(Gi) DREADD amenable to clinical translation of chemogenetics. Sci Adv April 17;5(4):eaaw1567. doi: 10.1126/sciadv.aaw1567. eCollection 2019 Apr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton DK, and Stevens B (2020). The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol Dis Sep;143:104963. doi: 10.1016/j.nbd.2020.104963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19(1):15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, and Hong W (2017). Detecting Activated Cell Populations Using Single-Cell RNA-Seq. Neuron 96, 313–329. [DOI] [PubMed] [Google Scholar]
- Yang P, Wen H, Ou S, Cui J, and Fan D (2012). IL-6 promotes regeneration and functional recovery after cortical spinal tract injury by reactivating intrinsic growth program of neurons and enhancing synapse formation. Exp Neurol 236, 19–27. [DOI] [PubMed] [Google Scholar]
- Yu K, Nagai J, and Khakh BS (2020). Improved tools to study astrocytes. Nat Rev Neurosci 21, 121–138. [DOI] [PubMed] [Google Scholar]
- Yu W, Clyne M, Khoury MJ, and Gwinn M (2010). Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 26, 145–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, and Khakh BS (2018). Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, and Barres BA (2012). Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond RE (2012). Cytokines that promote nerve regeneration. Exp Neurol 238, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel File S1: Data reporting the IP DEGs (FDR < 0.05, FPKM > 1) for all the astrocyte-specific RNA-seq projects (related to Figure 1).
Excel File S2: Data reporting genes that belong to each of the WGCNA modules (related to Figure 1).
Excel File S3: Data reporting statistical analyses, n numbers and relevant parameters (related to Figures 3–5, 7 and Figure S6).
Excel File S5: Data reporting the GPCRs identified from RNA-seq of NCAR and R6/2 (related to Figure 8).
Data Availability Statement
All data and codes are available upon request from the Lead Contact. Excel file S4 reports raw replicates used for the figures. RNA-seq data are deposited at the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo) with accession numbers provided in Table S1 and Key Resources Table. Lists of DEGs for each EPs are provided in Excel file S1.