

SUMMARY
During chronic infection, the inflammatory cytokine interferon gamma (IFNγ) damages hematopoietic stem cells (HSCs) by disrupting quiescence and promoting excessive terminal differentiation. However, the mechanism by which IFNγ hinders HSC quiescence remains undefined. Using intravital 3-dimensional microscopy, we find that IFNγ disrupts the normally close interaction between HSCs and CXCL12-abundant reticular (CAR) cells in the HSC niche. IFNγ stimulation increases expression of the cell surface protein BST2, which we find is required for IFNγ-dependent HSC relocalization and activation. IFNγ stimulation of HSCs increases their E-selectin binding by BST2 and homing to the bone marrow, which depends on E-selectin binding. Upon chronic infection, HSCs from mice lacking BST2 are more quiescent and more resistant to depletion than HSCs from wild-type mice. Overall, this study defines a critical mechanism by which IFNγ promotes niche relocalization and activation in response to inflammatory stimulation and identifies BST2 as a key regulator of HSC quiescence.
Graphical Abstract
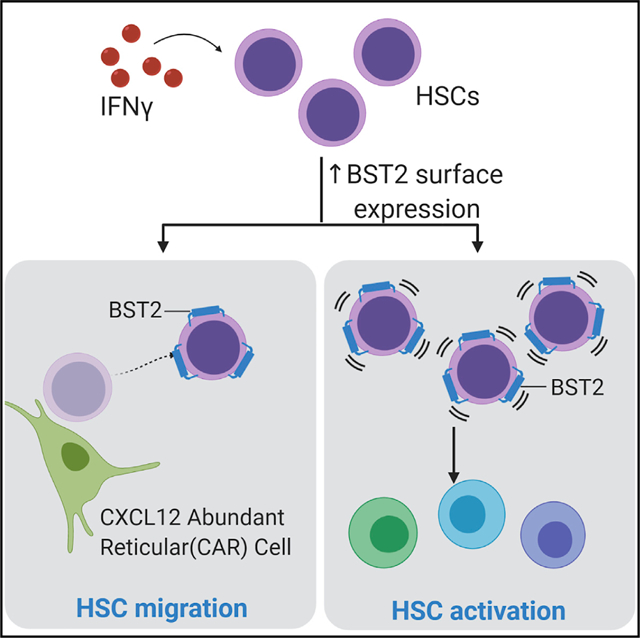
In Brief
Florez et al. identify BST2 as a surface protein, induced by interferon gamma on hematopoietic stem cells, that is required for their relocalization and cell cycle activation in response to infection. BST2 may become an important target for enhancing stem cell homing and persistence in the face of inflammatory stress.
INTRODUCTION
The inflammatory cytokine interferon gamma (IFNγ) exerts powerful effects on hematopoietic stem and progenitor cells (HSPCs) (King and Goodell, 2011; Morales-Mantilla and King, 2018; Pietras, 2017). Patients with aplastic anemia, a disease characterized by loss of HSPCs and pancytopenia, have high levels of IFNγ in their serum (Nisticò and Young, 1994; Young and Maciejewski, 1997). Similarly, infectious diseases such as hepatitis C and chronic mycobacterial infections that induce IFNγ-mediated immune responses are associated with impaired hematopoiesis (Achi et al., 2013; Ramos-Casals et al., 2003; Scadden et al., 1989). However, the exact mechanisms by which IFNγ damages hematopoiesis remain poorly defined.
In a recent study, we used a mouse model of chronic Mycobacterium avium infection to evaluate the mechanisms of stem cell loss during a chronic IFNγ-mediated immune response (Matatall et al., 2016). We found that blood counts decline and HSCs are significantly depleted during chronic infection but not through apoptosis or peripheral mobilization. Rather, chronic infection activates HSCs to divide by an IFNγ-dependent mechanism (Baldridge et al., 2010; MacNamara et al., 2011), and this division occurs preferentially among myeloid-biased HSCs, which express higher levels of IFNγ receptor (Ifngr1) (Matatall et al., 2014). Increased cell division translated to a net loss of HSCs because the divisions predominantly resulted in differentiation rather than self-renewal events. In summary, persistent IFNγ exposure contributes to the loss of HSCs by triggering their division and excessive terminal differentiation.
Impaired HSC quiescence has also been observed upon IFNα, interleukin-1 (IL-1), lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), and IL-6 stimulation in both mice and humans (Essers et al., 2009; King et al., 2015; Pietras et al., 2016; Schürch et al., 2014; Takizawa et al., 2017; Yamashita and Passegué, 2019). The dynamic nature of HSCs and the complexity of their microenvironment challenge our understanding of how chronic infections and inflammatory mediators induce the activation, differentiation, and loss of HSCs.
Interactions between HSCs and their bone marrow (BM) niche are an important determinant of HSC quiescence. CXCL12-abundant reticular (CAR) cells are BM perivascular stromal cells critical for the maintenance of HSC populations (Calvi and Link, 2015). Deletion of CXCL12 or its receptor CXCR4 or depletion of CAR cells leads to loss of HSC quiescence and depletion of HSCs over time (Nie et al., 2008; Sugiyama et al., 2006; Tzeng et al., 2011). Although the production of CXCL12 by early mesenchymal progenitors (a.k.a. perivascular stromal cells) and their interaction with HSCs have been reported to be particularly important for HSC maintenance (Ding and Morrison, 2013; Greenbaum et al., 2013), how chronic infections and inflammatory cytokines affect these interactions in vivo remains elusive.
To evaluate the fundamental question of how IFNγ affects HSCs within the BM niche and HSC niche interactions, we used intravital imaging of HSCs in CXCL12-GFP knockin animals. We report that a surface-expressed protein called BST2 facilitates displacement of HSCs from proximity with quiescence-enforcing CAR cells and further demonstrate that BST2 is required for cell cycle activation of HSCs upon IFNγ stimulation.
RESULTS
IFNγ Increases the Distance between HSCs and CAR Cells by a Cell Autonomous Process
To evaluate how IFNγ affects the interactions between HSC and CAR cells, we conducted sequential intravital imaging of HSCs in CXCL12-GFP transgenic mice with or without IFNγ treatment and measured the distance between HSCs and CAR cells in living animals. Briefly, HSCs (lineage Sca1+ cKit+ CD150+ CD48−) were sorted from wild-type (WT) C57BL/6 mice and stained with cell-tracker 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR) dye. The labeled HSCs were then transplanted into irradiated CXC12-GFP mice, and the calvaria of the recipient mice were imaged 24 h later to confirm homing into the BM. Consistent with previous studies, HSCs were located in close proximity to the CAR cells but were undetectable in the CAR cell-free BM cavity (Figure S1A). The same mice were then injected with either recombinant IFNγ, TGF-β, or PBS control. In vivo imaging of the mice 24 h later revealed that HSCs from IFNγ-treated mice were significantly further away from CAR cells compared to those treated with PBS alone (Figures 1A and S1A). The difference in distance was several microns, similar to niche distancing effects that have been observed in other models of HSC activation (Christodoulou et al., 2020). In contrast, the HSCs remained in close proximity to the CAR cells when recipient CXCL12-GFP mice were treated with 100 ng TGF-β, which is not known to induce HSC proliferation at this dose (Figure 1A).
Figure 1. IFNγ Induces HSC Relocalization Away from CAR Cells.
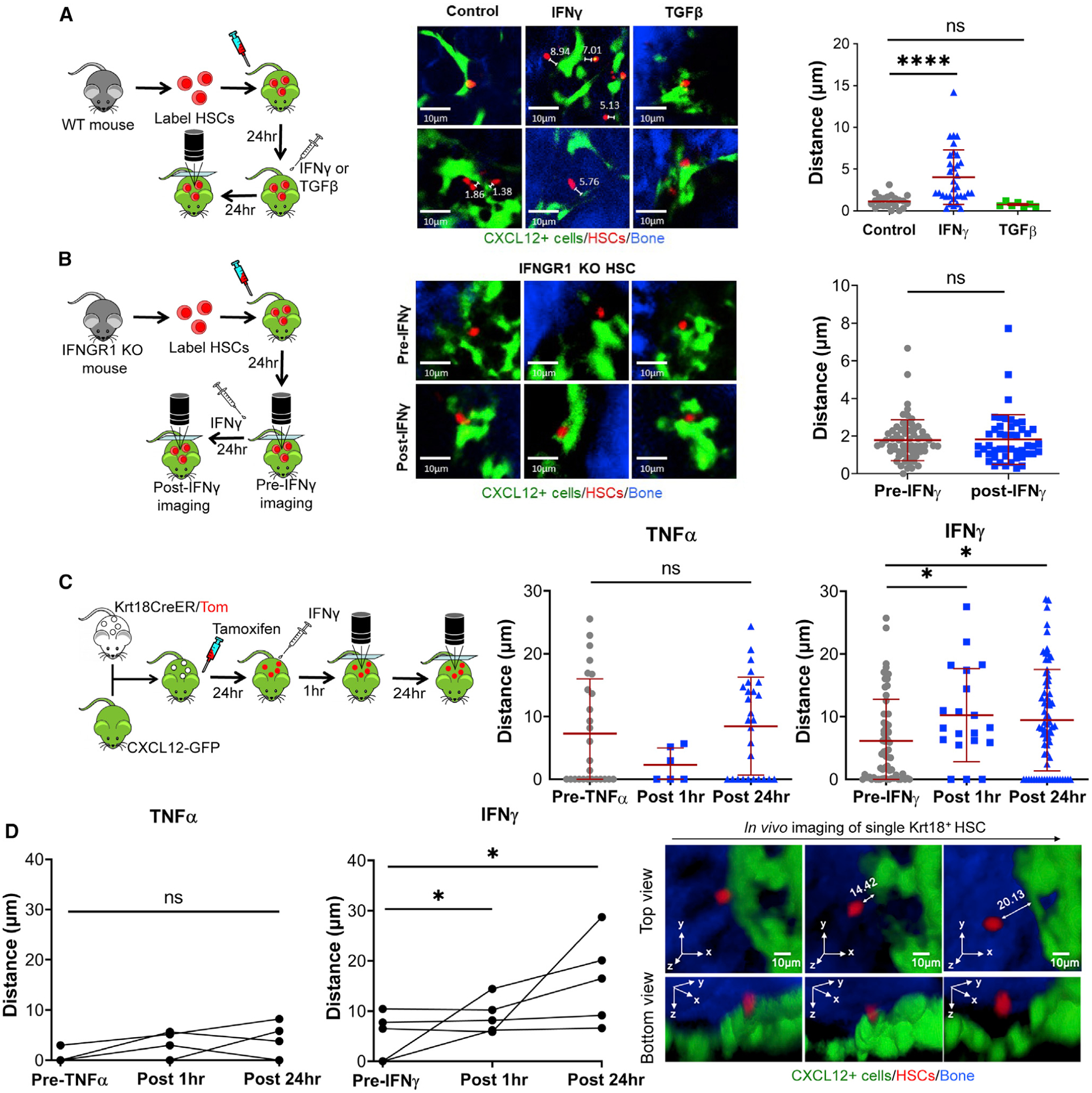
(A and B) Intravital imaging of the calvarium of CXCL12-GFP mice transplanted with CMTMR-stained HSCs. Representative images show HSCs in red (CMTMR), CAR cells in green (GFP), and bone in blue. For quantification, each point represents a single CMTMR-stained HSC.
(A) Recipient CXCL12-GFP mice were treated with IFNγ or TGF-β following transplant, and mice were imaged 24 h after treatment. Distances shown are in μm. Mean distances: control, 1.12 μm, n = 22; TGF-β, 0.74 μm, n = 7; IFNγ, 4.03 μm, n = 33. Data are compiled from 4 independent experiments.
(B) HSCs were isolated from Ifngr1−/− mice. Recipient CXCL12-GFP mice were imaged before (pre-IFNγ) and after treatment with IFNγ (post-IFNγ) following transplant of labeled HSCs. n = 48–66. Mean distances: control, 1.78 μm, n = 48; IFNγ, 1.82 μm, n = 66. Data are representative of 2 independent experiments.
(C) Intravital imaging of the calvarium of Krt18/Tom/CXCL12-GFP mice 24 h after a single dose of tamoxifen (50 mg/kg) and imaging of Krt18+ HSCs after IFNγ or TNF-α treatment. n = 6–71. Mean distances: pre-TNFα, 7.28 μm, n = 26; post-TNFα 1 h, 2.30 μm, n = 6; post-TNF-α 24 h, 8.45 μm, n = 27; pre-IFNγ, 6.16 μm, n = 64; post-IFNγ 1 h, 10.24 μm, n = 20; post-IFNγ 24 h, 9.30 μm, n = 71. Data are representative of 2 independent experiments.
(D) Change in distance of the same Krt18+ HSCs at the indicated time after TNF-α or IFNγ treatment. Representative 3-dimensional images of a single Krt18+ HSC tracking at 24 h after tamoxifen (pre) or 1 h and 24 h after IFNγ treatment. Scale bars are 10μm; A–C error bars are presented as mean ± SD;*p < 0.05, ****p <0.0001; ns, not significant by Mann-Whitney test or Kruskal-Wallis test.
To ascertain whether relocalization effects are cell autonomous, we transplanted HSCs from an Ifngr1-deficient (Ifngr1−/−) mouse into CXCL12-GFP mice. In contrast to WT HSCs, consecutive in vivo imaging of Ifngr1−/− HSCs before and 24 h after IFNγ-treatment revealed that these HSCs were not displaced from the CAR cells in response to IFNγ (Figure 1B). These results indicate that IFNγ acts to distance HSCs from quiescence-enforcing CAR cells by a cell autonomous mechanism.
Additionally, we treated donor mice with recombinant IFNγ or PBS control 24 h prior to harvesting the HSCs. These HSCs were then stained and injected to CXCL12-GFP recipient mice as described above. HSCs from mice that had been treated with recombinant murine IFNγ for 24 h prior to harvest were located slightly further away from CAR cells compared to control HSCs, although the difference was not statistically significant (Figure S1B). These data support a cell autonomous effect of IFNγ on HSCs but indicate that the effects of IFNγ on HSC localization may be transient.
To test whether the distancing phenomenon is specific to IFNγ, we transplanted WT CMTMR-labeled HSCs into a CXCL12-GFP recipient and treated the mice with a single intraperitoneal (i.p.) dose of polyinositic/polycytidylic acid (PIPC) to induce an IFNα response. Twenty-four h after PIPC induction, there was a trend toward increased distance between HSCs and CAR cells (Figure S1C), similar to the effects of IFNγ and consistent with prior reports of niche relocalization upon IFNα stimulation (Kunisaki et al., 2013).
To assess the specificity of the IFNγ-dependent relocalization response in the HSC compartment, we conducted a control experiment with CMTMR-stained CD19+ B cells, which also depend on CXCL12 signaling (Aurrand-Lions and Mancini, 2018). As shown in Figure S1D, the distances between B cells and CAR cells varied more widely than the distances between HSCs and CAR cells. Furthermore, there was no detectable change in the median distances between B cells and CAR cells after IFNγ treatment (Figure S1E), supporting that its distancing effect is specific to the HSC compartment.
To exclude the impact of toxic effects of irradiation on distancing of HSCs from CAR cells after IFNγ stimulation, we generated trigenic Krt18-CreER+Rosa26-Tomato+CXCL12-GFP+ (Krt18/Tomato/CXCL12-GFP) mice. Consistent with a previous report (Chapple et al., 2018), we confirmed that a single tamoxifen treatment of Krt18-CreER+Rosa26-Tomato+ mice induced selective labeling of CD48−CD150+ HSCs (~72%) and CD48+CD150+ HSPCs (~22%), whereas there was no detectable labeling in other populations (HPC1, HPC2, and multipotent progenitors [MPPs]) (Figure S1F). To measure the distance between endogenous steady-state HSCs and CAR cells, we conducted intravital 3-dimensional imaging of Krt18/Tomato/ CXCL12-GFP mice at 24 h after a single dose of tamoxifen treatment. The mice were then retroorbitally injected with IFNγ or TNF-α, another proinflammatory cytokine that promotes HSC cycling (Yamashita and Passegué, 2019). Notably, in vivo tracking of the same HSCs at 1 h and 24 h after IFNγ treatment revealed that the majority of Krt18-Tomato+ HSCs migrated away from the CAR cells, and the effects of TNF-α treatment were similar but did not reach statistical significance (Figures 1C and 1D). These results indicate that IFNγ provides a powerful stimulus to endogenous HSCs and induces their distancing away from CAR cells even in the absence of irradiation.
IFNγ Exposure Increases BST2 Expression on HSCs
In order to determine whether impaired CXCL12 sensing contributed to IFNγ-dependent distancing of HSCs from CAR cells, we measured expression of the CXCL12 receptor CXCR4 on HSCs in the presence and absence of IFNγ. We found no change, either by mRNA or protein expression, in CXCR4 expression upon IFNγ treatment (Figures 2A and 2B). We further conducted functional studies to test migration of HSPCs toward CXCL12 following IFNγ treatment in a transwell migration assay. Again, we saw no difference in the ability of HSCs to migrate toward CXCL12 upon IFNγ stimulation (Figure 2C). Together, these data suggest that IFNγ does not impair the ability of the HSCs to respond to CXCL12.
Figure 2. IFNγ Induces HSCs to Upregulate BST2.
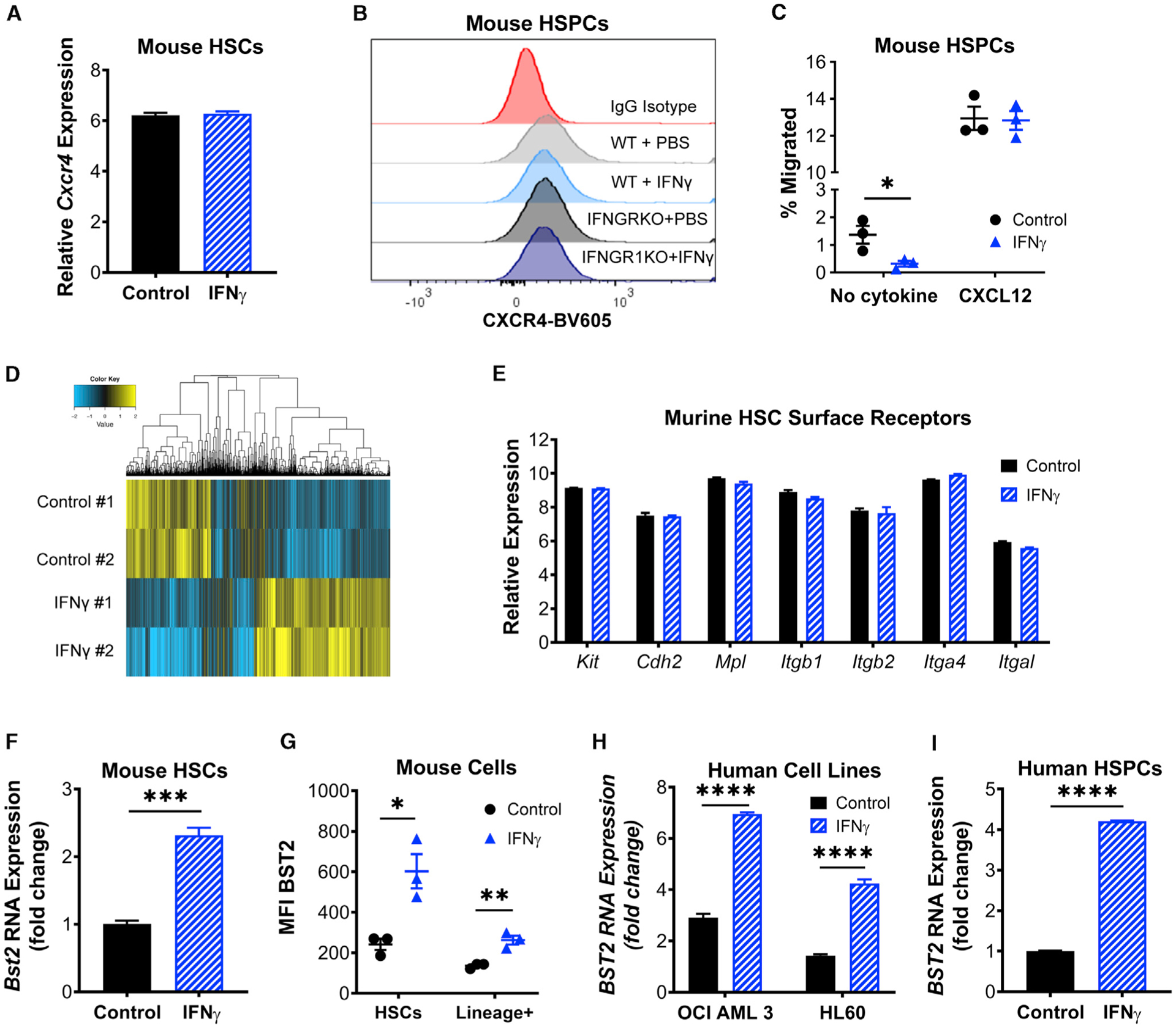
(A) RNA expression level of Cxcr4 in HSCs isolated from control or IFNγ-treated mice, shown as relative expression.
(B) CXCR4 protein expression in HSPCs (cKIT+ cells) isolated from wild-type (WT) or IFNGRKO mice that were treated with PBS or IFNγ. Plot shows the mean fluorescence intensity (MFI) of antibody-stained cells.
(C) CXCL12 transwell migration assay of cKit+ cells isolated from control or IFNγ-treated mice. Shown are the number of cells that migrated as a percentage of the total cells in both upper and lower chambers. Performed in triplicate; data are representative of 2 independent experiments.
(D) Heatmap of differentially expressed genes from expression profiling of HSCs from control or 24-h IFNγ-treated mice. Fold change >1.5; p < 0.1.
(E) RNA expression level of HSC surface markers Kit, Cdh2, Mpl, Itgb1, Itgb2, Itga4, and Itga1 in HSCs isolated from control or IFNγ-treated mice, shown as relative expression.
(F) RNA expression level of Bst2 in HSCs isolated from control or IFNγ-treated mice, shown as fold change over control. Experiment was performed once in triplicate.
(G) Protein expression level of BST2 on HSCs and isolated from control or IFNγ-treated mice. Data are shown as the MFI for surface BST2. Performed in triplicate; representative of 2 independent experiments.
(H) RNA expression level of BST2 in control or IFNγ-treated AML cell lines OCI-AML3 and HL60, shown as fold change over control. Experiment was performed once in triplicate.
(I) RNA expression level of BST2 in control or 20-h IFNγ-treated CD34+ HSPCs isolated from frozen cord blood, shown as fold change over control. Performed in triplicate; data are representative of 2 independent experiments.Error bars represent mean ± SEM; *p < 0.05, ** p<0.01, ***p < 0.001, ****p < 0.0001; ns, not significant by Student’s t-test.
In order to investigate other potential mechanisms for HSC relocalization, we analyzed microarray data of HSCs from control or IFNγ-treated mice and looked for cell surface proteins whose expression changed with 24-h IFNγ treatment (Figure 2D). Consistent with a prior report (Umemoto et al., 2017), we found no IFNγ-dependent changes in the expression levels of common HSC receptors thought to be important for maintaining HSC quiescence, including cKit, Cdh2, Mpl, Itgb1, Itbg2, Itga4, and Itga1 (Figure 2E). Because M. avium infection also triggers HSC proliferation by an IFNγ-dependent mechanism, we cross-referenced the microarray data with previously reported RNA sequencing data from control and M. avium-infected animals (Matatall et al., 2016) to find surface-expressed proteins with altered expression under both conditions. From the short list of upregulated proteins, we were able to validate increased expression of only one surface protein, BST2, also known as tetherin. Bst2 mRNA expression was increased in HSCs by 2-fold upon IFNγ treatment (Figure 2F), and BST2 protein levels were increased significantly in HSCs upon IFNγ treatment, as confirmed by fluorescence-associated cell sorting (FACS) (Figure 2G). We verified that protein expression was elevated upon IFNγ treatment in WT but not Ifngr1−/− host animals (Figure S2A). Of note, the increase in BST2 protein expression on HSCs was higher than on terminally differentiated cells of the mouse BM (Figure 2G). BST2 expression was also induced by IFNγ in two human acute myelogenous leukemia (AML) cell lines, OCI AML3 and HL60 (Figures 2H and S2B), and in primary human CD34+ HSPCs (Figure 2I).
BST2 Is Required for IFNγ-Dependent HSC Niche Relocalization
In order to determine whether BST2 is required to displace HSCs from CAR cells after in vivo IFNγ stimulation, we conducted intravital imaging of WT versus Bst2−/− HSCs in CXCL12-GFP transgenic mice before and after IFNγ treatment. Most WT cells were displaced from CAR cells after IFNγ treatment, whereas there was no change in distance of the Bst2−/− HSCs under the same conditions (Figures 3A, 3B, and S3A). These data indicate that BST2 is required for the displacement of HSCs from the CAR cell niche elicited by IFNγ exposure.
Figure 3. Loss of BST2 Prevents IFNγ-Mediated Relocalization.
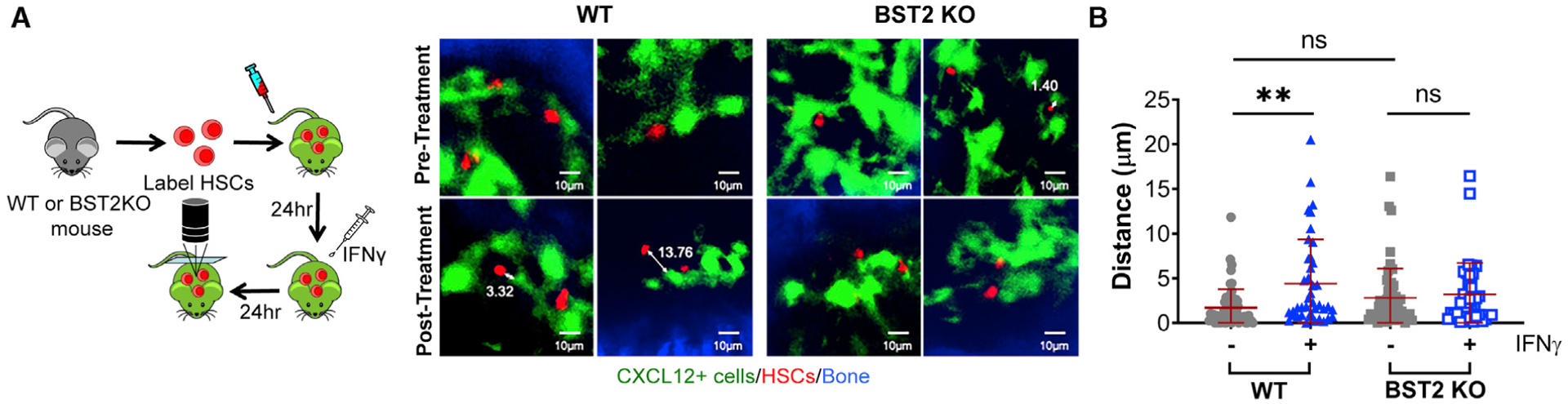
(A and B) Representative imaging of WT versus Bst2−/− HSCs before or 24 h after systemic treatment with IFNγ. Data are representative of 2 independent experiments with total n = 39–61 cells per group and were analyzed by Kruskal-Wallace one-way analysis of variance. Mean distances: WT control, 1.72 μm, n = 61; WT IFNγ, 4.40 μm, n = 41; BST2 knockout (KO) control, 2.80 μm, n = 56; BST2 KO IFNγ, 3.18 μm, n = 39. **p < 0.01. Scale bars are 10μm; ns, not significant; error bars represent mean ± SD.
Because we also noted distancing of HSCs from CAR cells in response to IFNα, we tested whether this displacement was dependent on BST2. Similar to the results of IFNγ treatment, we found that there was no change in the distance of Bst2−/− HSCs after PIPC treatment (Figure S3B). These results suggest that niche relocalization in response to either IFNγ or IFNα stimulation requires BST2.
IFNγ-Induced BST2 Facilitates E-Selectin Binding and Homing in Human and Murine HSCs
BST2 is an IFNγ-induced cell surface protein that contributes to immunity by preventing the budding of viruses (Liberatore and Bieniasz, 2011) and has also been shown to facilitate endothelial binding of monocytes and lung epithelial progenitor cells (Lee et al., 2018; Yoo et al., 2011). Previous reports indicate that BST2 is a non-canonical E-selectin ligand (Julien et al., 2011). E-selectin has been characterized as a marker of vascular endothelial cells in an “activated” HSC niche, as mice lacking E-selectin have increased HSC quiescence (Winkler et al., 2012). However, prior studies indicated that canonical E-selectin ligands such as PSGL-1 and CD44 were not involved in this process and the HSC ligand for E-selectin has heretofore not been identified (Winkler et al., 2012).
To determine if IFNγ affects E-selectin binding, we conducted in vitro binding assays by using cKit-enriched HSPCs. We found that HSPCs isolated from IFNγ-treated mice indeed had increased E-selectin binding compared to control HSPCs by using both static and flow cytometric binding assays (Figures 4A and S4A). Meanwhile, IFNγ-treated HSPCs did not show increased binding to P-selectin, a closely related selectin (Figure S4B).
Figure 4. IFNγ Increases HSPC Binding to E-Selectin by BST2.
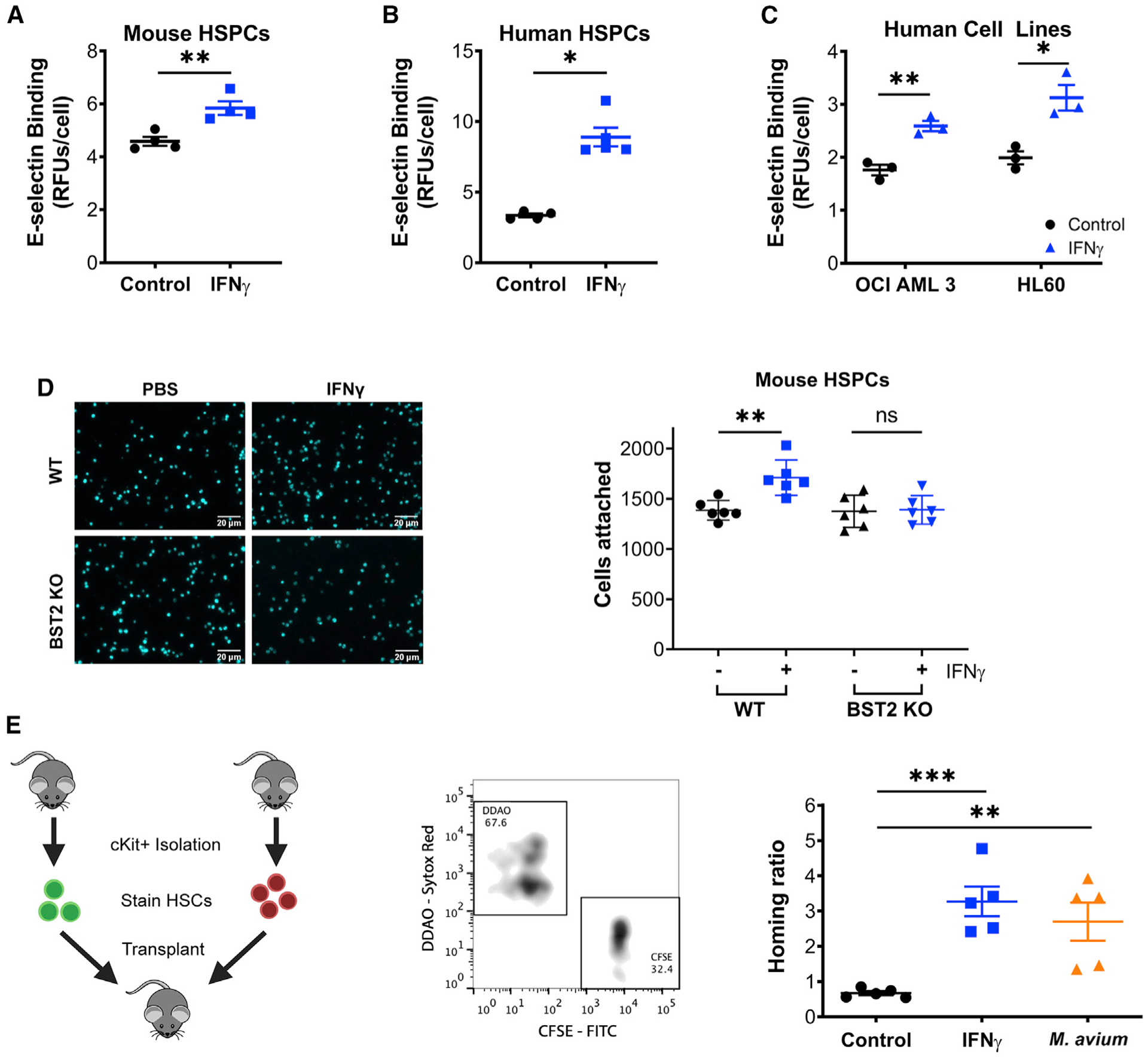
(A) In vitro E-selectin adhesion plate assay of cKit+ progenitors isolated from control or IFNγ-treated mice. Performed in triplicate; representative of 3 independent experiments.
(B) In vitro E-selectin adhesion plate assay of control or IFNγ-treated human CD34+ HSPCs. n = 5; representative of 2 independent experiments.
(C) In vitro E-selectin adhesion plate assay of control or IFNγ-treated AML cell lines OCI-AML3 and HL60. n = 3; representative of 3 independent experiments. Data shown in (A)–(C) are normalized to background and reported in relative fluorescent units/cell.
(D) In vitro E-selectin adhesion plate assay of cKit+ progenitors isolated from control or IFNγ-treated Bst2−/− mice, with representative images and aggregate data. Each data point represents the average cell count of 4 high-powered fields of a separate well. Scale bars are 20μm. Data are representative of 2 independent experiments.
(E) Homing assays in which test marrow (carboxyfluorescein succinimidyl ester [CFSE] stained) was mixed with normal marrow (9H-(1,3-Dichloro-9,9-Dimethylacridin-2-One-7-yl [DDAO] stained) before injection into recipient mice. Bone marrow was isolated 17 h later, and the ratio between the test and normal marrow was compared to the input ratio; shown as the homing ratio. Test marrow was from control, 24-h IFNγ or 4-week M. avium-treated mice. n = 5 per group. Data are representative of 2 independent experiments. Error bars represent mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001.
To test the relevance of these findings in human cells, we treated both primary human CD34+ HSPCs and AML cell lines OCI AML3 and HL60 with IFNγ in vitro and again found increased E-selectin binding following IFNγ treatment (Figures 4B and 4C). IFNγ treatment did not change RNA expression of canonical E-selectin ligands Gig1, Selplg, and Cd44 in mouse HSCs, indicating that a non-canonical ligand is likely involved (Figure S4C). Together, these data show that IFNγ promotes E-selectin binding by HSCs, and they suggest that IFNγ may induce the relocalization of HSCs from the niche, possibly by inducing a higher binding affinity for E-selectin+ endothelial cells.
To test whether BST2 is required for IFNγ-induced HSC binding to E-selectin, we conducted E-selectin binding assays for WT versus Bst2−/− HSCs in the presence and absence of IFNγ. The binding of WT but not Bst2−/− murine HSCs to E-selectin increased in the presence of IFNγ (Figure 4D). We further used CRISPR-Cas9 gene editing with a combination of four single-guide RNAs to disrupt BST2 in primary human CD34+ cells (Figure S4D). We confirmed efficient gene editing and protein reduction of BST2 by PCR and flow cytometry, respectively (Figures S4E and S4F). Using in vitro binding assays, we found that loss of BST2 significantly reduced the binding capacity of primary HSPCs for E-selectin (Figure S4G). Loss of BST2 also decreased the IFNγ-induced E-selectin binding capacity of these cells, although it was not completely ablated (Figure S4H). Collectively, these studies suggest that BST2 is a critical mediator of IFNγ-induced binding of HSCs to E-selectin.
E-selectin serves as an important mediator of vascular attachment and transendothelial migration of leukocytes and other hematopoietic cells. Adhesion through E-selectin allows cells to transition from rolling to “spreading activation” during this process (van Buul and Hordijk, 2004). Because transendothelial migration is a critical component of HSC homing, we assessed the effect of IFNγ stimulation on HSC homing. By comparing the relative efficiency of CFSE-stained test cells versus DDAO-stained control cells to BM, we found that cKit+ test cells from IFNγ-treated animals homed to the BM with greater efficiency than untreated cKit+ test cells. Similarly, cKit+ test cells from M. avium-infected animals showed increased homing efficiency compared to untreated cells (Figure 4E). Conversely, cKit+ cells from Ifngr1−/− animals had a decreased homing efficiency (Figure S4I). To confirm these findings, we conducted limiting dilution transplants of BM from WT versus Ifngr1−/− mice, as these studies do not depend on surface marker expression to measure stem cell content and homing. Consistent with a defect in homing, BM cells from Ifngr1−/− mice showed reduced long-term engraftment compared to those from WT mice (Figures S4J and S4K). Moreover, engraftment was identical if the BM was transplanted intrafemorally (Figure S4L), indicating no defect in stem cell viability and function. Collectively, these studies indicate that IFNγ signaling promotes E-selectin binding through BST2 and contributes to appropriate HSC homing, possibly through E-selectin-mediated transendothelial migration.
BST2 Is Required for IFNγ-Dependent HSC Activation
To test the functional role of BST2 in HSC activation during infection, we infected WT or Bst2−/− mice with M. avium. At 1 month of infection, WT HSC quiescence was impaired, as evidenced by a decreased percentage of cells in G0 based on Hoechst/pyronin Y staining (Figure 5A). In contrast, Bst2−/− HSCs remained quiescent (~83% in G0) even in the setting of M. avium infection (Figure 5A). We confirmed that the infection was robust in both the WT and Bst2−/− mice, as shown by increased spleen weight and size (Figures S5A and S5B). These findings indicate that BST2 is essential for the activation of HSCs during chronic M. avium infection.
Figure 5. BST2 Regulates HSC Quiescence and Maintenance.
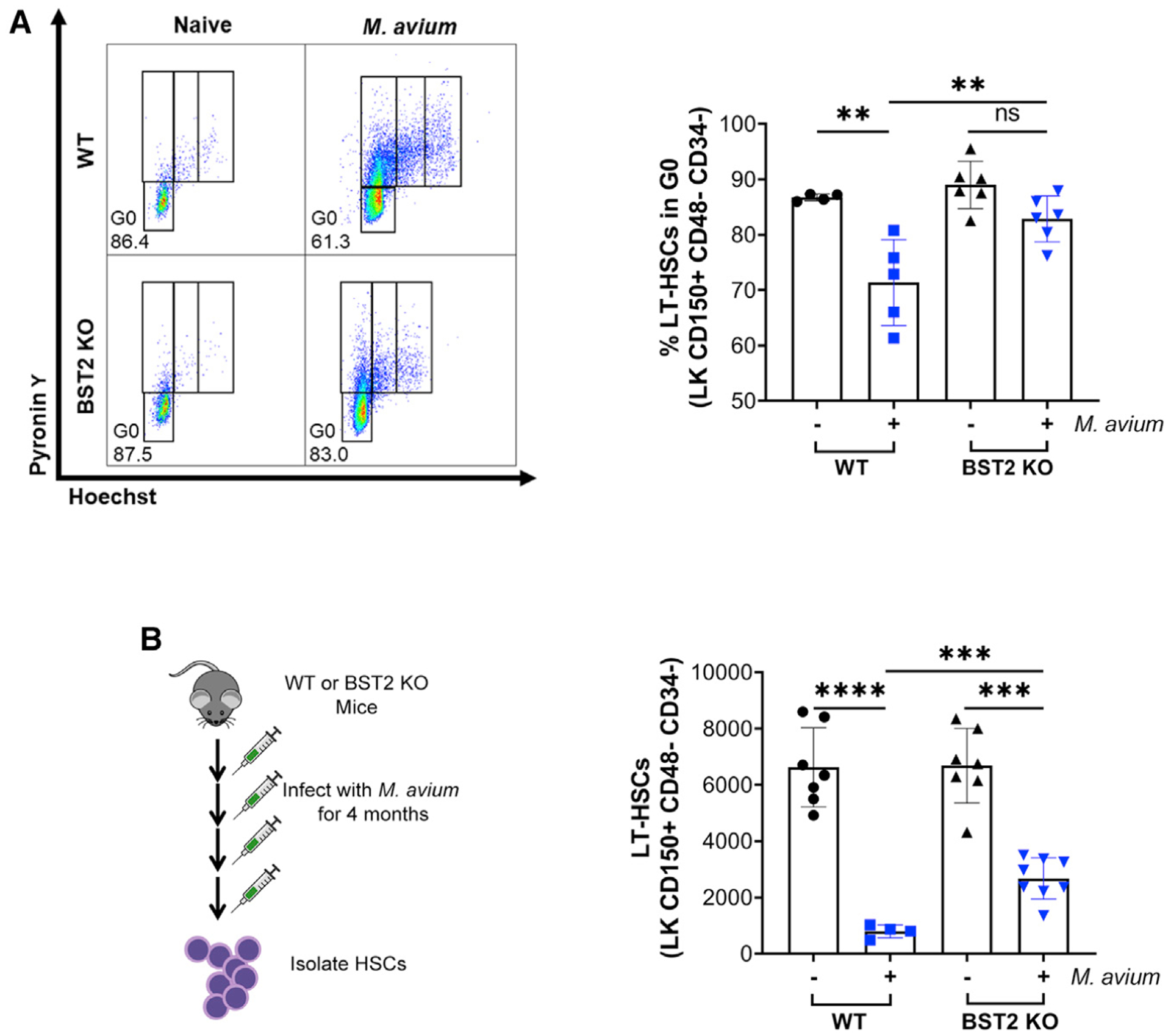
(A) Representative flow plots of cell cycle analysis of HSCs using Hoechst pyronin Y. Bottom left gating indicates G0. Hoechst pyronin Y staining of HSCs isolated from WT or Bst2−/− naive or M. avium-infected mice. Shown as % of total HSCs (LK CD150+ CD48− CD34−) in G0.
(B) LT-HSCs (LK CD150+ CD48− CD34−) per bone after 4 months of M. avium infection. *p <0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± SD.
Because IFNγ-mediated HSC activation and differentiation eventually result in HSC depletion after repeated M. avium infection, we hypothesized that Bst2−/− HSCs should be relatively protected from exhaustion during chronic infection. Therefore, we repeatedly infected WT and Bst2−/− mice with M. avium over the span of 4 months and measured HSC numbers at the end of this chronic infection. As expected, after 4 months of infection, WT HSCs were nearly completely depleted, whereas Bst2−/− HSCs were more preserved, with over 30% remaining compared to the uninfected group (Figures 5B and S5C). A similar pattern was noted for the MPP3 population (Figures S5D–S5G). These findings indicate that BST2 is critical for IFNγ-mediated HSC activation and exhaustion of the HSC compartment in the setting of chronic infection.
BST2 Regulates HSC Quiescence and Maintenance
Because baseline IFNγ has been shown to affect HSC quiescence (Baldridge et al., 2010), we assessed the quiescence and maintenance of HSCs in Bst2−/− mice. Bromodeoxyuridine (BrdU) incorporation studies revealed that the HSCs from Bst2−/− mice were significantly more quiescent than HSCs of WT mice (Figures 6A and S6A), consistent with a role for BST2 in HSC cell cycle activation. Reduced BrdU incorporation was also noted in several classes of MPPs (Figures S6B–S6D). Furthermore, the overall number of HSCs was greater in Bst2−/− mice, both as a percentage of total BM cells and in absolute number (Figures 6B and 6C). The percentage of MPP1s and MPP2s were unchanged, whereas the percentage of MPP3s and MPP4s was higher in Bst2−/− mice than in WT mice (Figures S6E–S6G). Consistent with a greater number of HSCs in the BM, transplantation of whole BM from Bst2−/− mice into lethally irradiated WT recipients resulted in enhanced engraftment across all lineages compared to WT BM (Figures 6D and S6H–S6J). However, transplantation of an identical number of Bst2−/− versus WT HSCs resulted in equal levels of engraftment (Figure 6E), suggesting that the increased engraftment seen in the whole BM transplants was due to a greater number of HSCs rather than a cell autonomous advantage of Bst2−/− HSCs. Together, these data suggest a role for BST2 in HSC maintenance even during homeostatic conditions.
Figure 6. Loss of BST2 Reduces HSC Proliferation and Increases HSC Numbers.
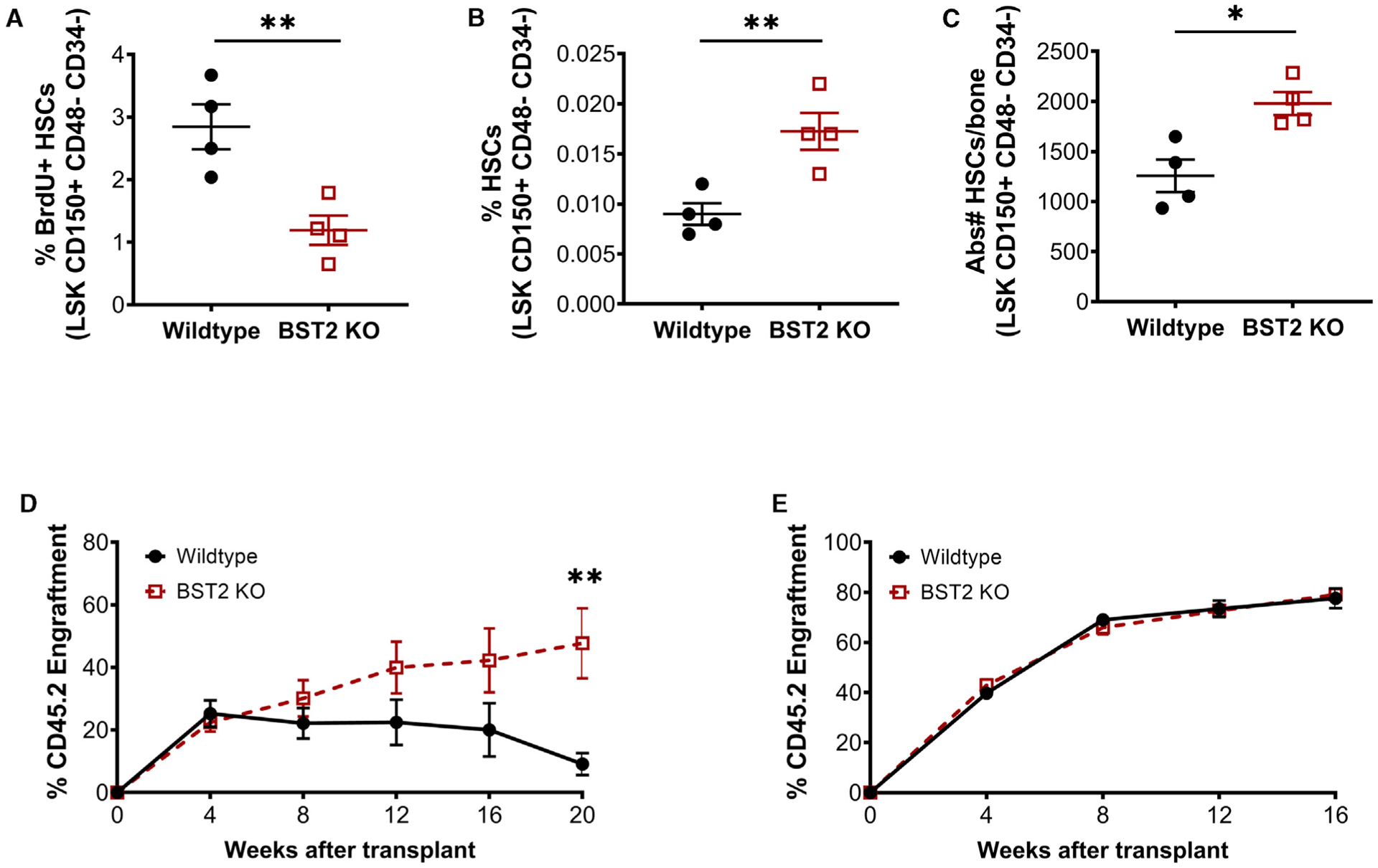
(A) BrdU+ HSCs isolated from WT or Bst2−/− mice. Shown as % of total HSCs (LSK CD150+ CD48− CD34−). n = 4; data are representative of 2 independent experiments.
(B and C) The number of HSCs in WT or Bst2−/− mice.
(B) The % of live WBM cells.
(C) Absolute number per bone. n = 4; Data are representative of 4 independent experiments.
(D) Total peripheral blood engraftment following transplant of 2 × 105 CD45.2 WT or Bst2−/− WBM cells mixed with 2 × 105 rescue marrow (CD45.1). n = 10.
(E) Total peripheral blood engraftment following transplant of 300 CD45.2 WT or Bst2−/− HSCs mixed with 2 × 105 rescue marrow (CD45.1). n = 10. Data are presented as mean ± SEM; *p < 0.05, **p < 0.01.
DISCUSSION
Here, we demonstrated that IFNγ displaces HSCs from quiescence-enforcing CAR cells in the niche. Furthermore, we found that the glycoprotein BST2 is upregulated on the surface of HSCs upon IFNγ treatment and is required for IFNγ-dependent displacement of HSCs from CAR cells. Although IFNγ exposure does not interfere with normal CXCL12-CXCR4 interactions, induction of BST2 appears to be sufficient to overpower this chemoattractive relationship, resulting in displacement of HSCs away from CAR cells. Furthermore, we found that BST2 is required for the activation of HSC cell cycle activity and HSC exhaustion during chronic infection. Conversely, HSCs from Bst2−/− mice were increased in abundance and hyperquiescent, highlighting the role of BST2 in stem cell regulation even under steady-state conditions.
Our finding that IFNγ stimulation resulted in distancing of HSCs from CAR cells mirrors findings of Kunisaki et al. (2013), who reported that polyinositol:polycytidylic-acid-induced IFNα stimulation increased the distance between HSCs and the quiescence-enforcing arteriolar niche. Recent live imaging studies support this dynamic localization by showing that activated HSCs were closer to the vasculature, with an average change in distance of ~1 μm (Christodoulou et al., 2020). Given prior reports that indicate that HSCs are normally in direct contact with vascular endothelial cells (Chen et al., 2016), even small changes in mean distance between HSCs and niche cells (1.12 μm to ~4 μm after IFNγ stimulation; Figure 1A) may have significant functional consequences.
HSC-niche interactions are an important determinant of HSC quiescence (Calvi and Link, 2015; Cordeiro Gomes et al., 2016). E-selectin is expressed in specialized locations within the BM and on small venule endothelial cells (Runnels et al., 2006). Winkler et al. (2012) showed that HSCs are more quiescent in the absence of E-selectin, suggesting that E-selectin preferentially marks an activated portion of the HSC niche. Indeed, our data indicate IFNγ increases the binding efficiency of HSCs for E-selectin but not to the closely related P-selectin, which does not play a role in HSC activation. Despite testing many of the canonical ligands for E-selectin binding, Winkler et al. (2012) never identified the ligand for E-selectin binding in the HSC niche. Our data provide evidence that BST2 is that ligand, as loss of BST2 in both murine and primary human cells significantly reduced binding to E-selectin. Furthermore, we found that HSCs lacking BST2 phenocopy the hyperquiescent phenotype of E-selectin KO HSCs. Based on these studies and our findings, we speculate that BST2 activates HSCs in part by promoting their relocalization toward an activated perisinusoidal niche.
Of note, IFNα treatment also induces cell proliferation in HSCs and has been reported to upregulate BST2 (Bujanover et al., 2018). Thus, the mechanism we report here may be relevant to a number of inflammatory conditions beyond IFNγ exposure. Our methods do not provide the resolution of recent confocal microscopy studies (Kokkaliaris et al., 2020), and they do not allow us to comment on positioning of HSCs relative to various structures of the BM such as endosteum, which was recently reported to be important for maintaining reserve HSCs (Zhao et al., 2019). Future studies should assess how IFNγ exposure alters the interactions of HSCs with other niche cell types such as osteoblasts or macrophages.
Baseline E-selectin binding of primary human CD34+ cells was lower in BST2 mutant cells than in controls, but increased E-selectin binding upon IFNγ stimulation remained intact even in the mutants, suggesting that BST2 may not be the only ligand capable of mediating this interaction. It is important to note, however, that the human cells in these binding experiments were subject to the high stress conditions of IFNγ treatment in vitro, possibly inducing alternative binding mechanisms. Further in vivo studies are necessary to address the involvement of BST2 in IFNγ-induced E-selectin binding.
Given the link between IFNγ-induced BST2 expression and E-selectin binding and prior literature showing that E-selectin binding is a critical contributor to stem cell homing and engraftment (Krause et al., 2014), we hypothesized that IFNγ signaling may affect transendothelial migration and, thus, homing of HSCs. Indeed, we show that infections that induce IFNγ, as well as recombinant IFNγ alone, promote increased homing of HSCs to the BM from the peripheral circulation. Although others have speculated that infections can promote trafficking of HSPCs for immunosurveillance (Massberg et al., 2007), our prior data indicate that IFNγ is not a prime mediator of such migration (Baldridge et al., 2010; Matatall et al., 2014). These findings will be useful for interpreting BM transplant studies, as inflammatory cues may affect overall engraftment independent of intrinsic HSC function. The beneficial effects of IFNγ in HSC homing are likely offset by the negative effects in self-renewal, and further exploration of how IFNγ promotes homing of HSCs is worthy of pursuit. For example, these findings could have important implications for HSC transplant patients, especially those with inborn errors of immunity (Roesler et al., 2004).
Aside from BST2’s role in HSC localization in the niche, we further found that BST2 is required for HSC cell cycle activation and depletion in the setting of chronic infection. Bst2−/− HSCs were uniformly more quiescent than WT HSCs even at baseline, suggesting that BST2 may have a cell autonomous role in regulating HSC quiescence. Indeed, BST2 has been reported to trigger nuclear factor κB (NF-κB) signaling in cancer cells (Liu et al., 2018). Whether BST2 activates NF-κB or other signaling pathways in HSCs will be the subject of future studies. In light of the above findings, modulating BST2 expression on HSCs may represent a previously unrecognized route to modify HSC homing, activation, and/or persistence in the setting of chronic inflammation. Recently, CCL2+ and HDC+ HSCs have been shown to be especially responsive to stress stimuli such as injury after myocardial infarction or LPS administration, respectively (Chen et al., 2017; Dutta et al., 2015). Whether these phenotypes also correlate with BST2 upregulation remains unknown. As we found that BST2 expression correlates with poor survival in patients with AML (LinkedOmics; Figure S6K), blocking BST2 binding may prove to be a strategy to limit leukemic stem cell proliferation. Further investigation of the mechanisms by which BST2 affects HSCs may thus have wide-ranging future implications for HSC biology and medicine.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, KatherineY. King (kyk@bcm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The microarray data generated during this study are available at https://data.mendeley.com/datasets/mhc8b4yn9d/1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Wild-type C57BL/6 (CD45.2) and C57BL/6.SJL (CD45.1) mice 6–18 weeks of age were used, with equal numbers of males and females. C57BL/6 Ifngr1−/− (Stock #3288) and Bst2−/− mice (Stock #029892) (Liberatore and Bieniasz, 2011) were obtained from Jackson Laboratories (Bar Harbor, ME [14]). CXCL12-GFP (Mignone et al., 2004) (C57/BL6 background) mice were kindly provided by Dr. Takashi Nagasawa. Krt18-CreERT Rosa26-tdTomato+ reporter mice were generously provided by Dr. Daisuke Nakada (Chapple et al., 2018) and crossed with CXCL12-GFP transgenic mice to create trigenic Krt18-CreER+Rosa26-Tomato+CXCL12-GFP+ (Krt18/Tomato/CXCL12-GFP) mice. All mice were maintained at an AALAC-accredited, specific-pathogen-free animal facility at Baylor College of Medicine under the approved protocol AN-4802. Genotypes were confirmed by PCR. Experiments were always done at the same time of the day, beginning at 7 am, with the control and test samples compared at the same time of the day.
Microbial infections
Mice were infected with 2×106 colony-forming units of Mycobacterium avium IV as described (Feng et al., 2008). M. avium was detected by growth on Middlebrook agar and by PCR (Park et al., 2000). For chronic infections, mice were infected every month for four months.
Cell lines
HL-60 and OCI AML3 cell lines were provided by the Goodell laboratory. Both cell lines were cultured in RPMI 10% FBS supplemented with 2mM L-glutamate and penicillin/streptomycin.
Primary cell cultures
Human CD34+ cells were isolated from Buffy coat samples or frozen umbilical cord blood samples by enrichment using anti-CD34 magnetic microbeads (Macs, Miltenyi). CD34+ cord blood cells were expanded in StemSpan SFEM (Stem Cell Technologies) supplemented with 100ng/ml rhFLT-3L, 100ng/ml rhSCF, 100ng/ml rhTPO and 20ng/ml IL-3 (Preprotech). rhIFNγ (1000 U/mL) was added to media for 18hrs before harvest.
METHOD DETAILS
Limiting dilution transplants
Limiting dilution experiments were carried out by transplanting 1×104, 8×104, or 2×105 CD45.2 WBM cells from WT or Ifngr1−/− mice along with 2×105 CD45.1 rescue marrow into lethally irradiated recipient mice. Ten mice were transplanted per group. The CD45.2 WBM engraftment was determined 28 weeks post-transplant. Stem cell frequency was determined using Extreme Limiting Dilution Analysis (Hu and Smyth, 2009). Any transplanted mouse with total engraftment below 1% or less than 1% contribution to any lineage was considered a non-responder.
Bst2−/− Transplants
Bst2−/− WBM transplants were carried out by transplanting 2×105 WBM cells from wild-type or Bst2−/− CD45.2 mice along with 2×105 CD45.1 rescue marrow into lethally irradiated recipient mice. Bst2−/− HSC transplants were carried out by transplanting 300 sorted HSCs (LSK CD150+ CD48− CD34−) from wild-type or Bst2−/− CD45.2 mice along with 2×105 CD45.1 rescue marrow into lethally irradiated recipient mice. CD45.2 peripheral blood engraftment was assessed at 4, 8, 12, 16, and 20 weeks post-transplant. CD45.2 WBM engraftment was determined at 20 weeks post-transplant.
Homing assays
For homing studies, cKit+ cells were isolated from control or test animals by magnetic bead enrichment using CD117 beads (Automacs, Miltenyi Biotech). Control cells were stained with 2mM DDAO (Molecular Probes) and test cells were stained with 2 mM CFSE (Molecular Probes). Cells were admixed and ~1 × 105 cells of each kind were injected intravenously into lethally irradiated animals. 17 h later, the bones were harvested and the ratio of CFSE:DDAO positive cells in bone marrow was measured by flow cytometry.
Intrafemoral transplants
Whole bone marrow cells (2×105) from 12 week old WT or Ifngr1−/− mice were admixed with an equal number of WBM cells from WT C57BL/6.SJL (CD45.1) mice and injected in a total volume of 50 uL directly into the femur of each recipient animal. Five animals were used per cohort and engraftment was assessed monthly for twelve weeks.
Intravital imaging of cell-niche interactions
For live in vivo imaging of HSC and niche interaction, CXCL12-GFP knockin mice were lethally irradiated with 9.5 Gy the day of HSC transplantation. Sorted HSCs or B cells from indicated mice were stained with 1 uM of CMTMR for 30 min and transplanted intravenously into CXCL12-GFP knockin mice. 24 h later, mice were treated with 100 μg rIFNγ, 100 ng TGFβ, TNFα or 300 μg IFNα and prepared for visualization under a customized two-photon and confocal hybrid microscope (Leica TCS SP8MP with DM6000CFS) specifically designed for live animal imaging, as described in previous report (Park et al., 2012). The mice were then mounted on a 3-D axis motorized stage (Anaheim Automation Anaheim, CA), and the calvarial bone was scanned for second harmonic generation (SHG from bone collagens by femto-second titanium:sapphire laser pulses: 880 nm) to identify bone and bone marrow structures and the intersection of the sagittal and coronal sutures. CXCL12-GFP-expressing cells (488 nm excitation, 505–545 nm detection) and CMTMR+ HSCs (541 nm excitation, 590–620 nm detection) were simultaneously imaged using confocal spectral fluorescence. All images were recorded with their distances to the intersection of the sagittal and coronal sutures to define their precise location. Each image was recorded by Z stacks with 50–100 μm depth from the bone surface at a 2-μm interval. A PCI-based image capture board (Snapper, Active Silicon) was used to acquire up to three channels simultaneously using the Leica Application Suite software (Version 3.3). 3-D Images and the distance between HSCs and niche cells were measured using the Leica Application Suite software.
RNA purification and realtime PCR
RNA was isolated from HSCs (Lin− cKit+ Sca1+ CD150+ CD48− CD34−) from control, 24hr IFNγ-treated, or 4-week M. avium-infected wild-type and Ifngr1−/− mice using RNeasy Micro Plus (QIAGEN). Real-time PCR was performed using iTaq Universal SYBR Green SupermixBR Green Master Mix (BioRad). All data were normalized to an 18 s endogenous control.
PRIMER SEQUENCES
| Gene | Primer | Sequence |
|---|---|---|
| Bst2 | Forward | TCAGGAGTCCCTGGAGAAGAA |
| Bst2 | Reverse | AG AAGTCTCCTTTTG G ATCCTCAG |
Flow cytometry
Whole bone marrow cells were isolated from femurs and tibias. Following RBC lysis, cells were co-stained for BST2 and HSCs markers. Unless otherwise stated, HSCs were defined as Lineage− cKit+ Sca1+, CD150+, CD48−, CD34−. Cells were suspended at a concentration of 108 cells/mL and incubated in 4°C for 15 min with the desired antibodies. See Key Resources Table for a list of antibodies used.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD45.1 (PB conjugated, clone A20) | Biolegend | Cat#110721; RRID: AB_492867 |
| Anti-Mouse CD45.1 (APC conjugated, clone A20) | eBioscience | Cat#17–0453-81; RRID: AB_469397 |
| Anti-Mouse CD45.2 (BV 605 conjugated, clone 104) | Biolegend | Cat#109841; RRID: AB_2563485 |
| Anti-Mouse CD45.2 (PE conjugated, clone A20) | eBioscience | Cat#12–0453-81; RRID: AB_465674 |
| Anti-Mouse c-kit/CD117 (APC-Cy7 conjugated, clone 2B8) | eBioscience | Cat#25–1171-82; RRID: AB_469644 |
| Anti-Mouse CD4 (PE-Cy5 conjugated, clone GK 1.5) | eBioscience | Cat#15–0041-82; RRID: AB_468695 |
| Anti-Mouse CD4 (FITC conjugated, clone GK1.5) | eBioscience | Cat#11–0041-82; RRID: AB_464892 |
| Anti-Mouse CDS (PE-Cy5 conjugated, clone 53–6.7) | eBioscience | Cat#15–0081-82; RRID: AB_468706 |
| Anti-Mouse CDS (FITC conjugated, clone 53–6.7) | eBioscience | Cat#11 −0081 −82; RRID: AB_464915 |
| Anti-Mouse CD45R/B220 (PE-Cy5 conjugated, clone RA3–682) | eBioscience | Cat#15–0452-82; RRID: AB_468755 |
| Anti-Mouse CD45R/B220 (PE-Cy7 conjugated, clone RA3–6B2) | eBioscience | Cat#25–0452-82; RRID: AB_469627 |
| Anti- Mouse/Human CD45R/B220 (FITC conjugated, clone RA3–6B2) | eBioscience | Cat#11–0452-82; RRID: AB_465054 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy5 conjugated, clone RB6–8C5) | eBioscience | Cat#15–5931-82; RRID: AB_468813 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy7 conjugated, clone RB6–8C5) | eBioscience | Cat#25–5931-82; RRID: AB_469663 |
| Anti-Mouse CD11b/Mac1 (PE-Cy5 conjugated, clone M1/70) | eBioscience | Cat#15–0112-82; RRID: AB_468714 |
| Anti-Mouse CD11b/Mac1 (PE-Cy7 conjugated, M1/70) | eBioscience | Cat#25–0112-81; RRID: AB_469587 |
| Anti-Mouse Ter119 (PE-Cy5 conjugated, clone M1/70) | eBioscience | Cat#15–5921-82; RRID: AB_468810 |
| Anti-Mouse CD150/SLAM (PE-Cy7 conjugated, clone TC15–12F12.2) | Biolegend | Cat#115914; RRID: AB_439797 |
| Anti-Mouse CD48 (APC conjugated, clone HM48–1) | eBioscience | Cat#17–0481-82; RRID: AB_469408 |
| Anti-Mouse CD34 (FITC conjugated, clone RAM 34) | eBioscience | Cat#11–0341-82; RRID: AB_465021 |
| Anti-Mouse Flk2/Flt3/CD135 (PE conjugated, clone A2F10) | eBioscience | Cat#12–1351-82; RRID: AB_465859 |
| Anti-Mouse Annexin V (PE conjugated) | BD PharMingen | Cat#556422; RRID: AB_2108590 |
| Anti-Human CD34 (PB conjugated, clone RAM 34) | eBioscience | Cat# 48–0341-82; RRID: AB_2043837 |
| DAPI | Life technologies | Cat#D1306; RRID: AB_2629482 |
| Anti-E-selectin Rat monoclonal anti body (clone 9A9) | Bio X cell | Cat# BE0294; RRID: AB_2687816 |
| Purified Rat lgG1 lambda isotype control (cloneA110–1) | BD | Cat#553993; RRID: AB_395190 |
| Anti-Mouse CD184 (BV605 conjugated, clone 2B11) | BD | Cat#740378; RRID: AB_2740109 |
| Anti-Mouse lgG2b,k (BV605 conjugated, clone R35–38) | BD | Cat#563145; RRID: N/A |
| Anti-Human CD317 (PE conjugated, clone 26F8) | eBioscience | Cat#12–3179-42; RRID: ABJ0596640 |
| Anti-Mouse CD317(Biotin conjugated, clone eBio927) | eBioscience | Cat#13–3172-82; RRID: AB_763415 |
| Bacterial and Virus Strains | ||
| Mycobacterium avium | N/A | strain SmT2151 |
| Biological Samples | ||
| Human Buffy coat samples | Gulf Coast Regional Blood Center | https://www.gulfcoastconsortia.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IFNy | eBioscience | Cat#BMS326 |
| Recombinant Human IFNy | RD Biosystems | Cat#285-IF-100 |
| Recombinant Human Flt3-Ligand | Preprotech | Cat#300–19 |
| Recombinant Human IL-3 | Preprotech | Cat#2 00–03 |
| Recombinant Human SCF | Preprotech | Cat#300–07 |
| Recombinant Human TPO | Preprotech | Cat#300–18 |
| Recombinant Mouse TGFp | R&D Biosystems | Cat#7346-B2/CF |
| Mouse E-selectin | R&D Biosystems | Cat#575-ES100 |
| Mouse P-selectin | R&D Biosystems | Cat#737-PS-050 |
| Human E-selectin | R&D Biosystems | Cat#724-ES-100 |
| Mouse CD117 Micro Beads | MACS Miltenyi Biotec | Cat#130–091 −224 |
| Human CD34 Micro Beads | MACS Miltenyi Biotec | Cat#130–046-702 |
| CFSE | Life Technologies | C34554 |
| DDAO | Life Technologies | C34553 |
| Recombinant human TNFa protein | Fisher | PHC3016 |
| Tamoxifen | SantaCruz Biotechnology | CAS 10540–29-1 |
| Critical Commercial Assays | ||
| NucleoSpin RNA Plus XS kit | Macherey-Nagel | Cat# 740990 |
| SMARTer® Stranded Total RNA-Seq Kit i/2 - Pico Input Mammalian | Takara | Cat# 634411 |
| RNAqueous Kit | Ambion | Cat#AM1912 |
| Superscript III First-Strand Synthesis Supermix | Invitrogen | Cat#18080051 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#172–5121 |
| BrdU Flow Kit (FITC conjugated) | BD | Cat# 559619 |
| Deposited Data | ||
| https://data.mendeley.com/datasets/mhc8b4yn9d/1 | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| CD45.1 mice | Baylor College of Medicine | JAX Stock#0066584 |
| CD45.2 mice | Baylor College of Medicine | Stock#14170 |
| Bst2−/−mice | Jackson Labs | Stock#029892 |
| Ifngr1−/− mice | Jackson Labs | Stock#025545 |
| CXCL12GFP/+ | (Toshiaki et al., 2003) | N/A |
| Krt18-CreER (Tg(KRT18-cre/ERT)23Blpn/J | Dr. Nakada Daisuke, Baylor College of Medicine | N/A |
| B6.Cg-Gt(ROSA)26Sor1m14(CAG”1dToma1o)Hz7 J | Jackson Labs | JAX:007914 |
| Oligonucleotides | ||
| sgRNA: BST2+17403843 - UGC GGUACAGAUGGCAAAUC | Synth ego | N/A |
| sgRNA: BST2–17403817 - GUA CCG CAG AAGAG AAAACC | Synth ego | N/A |
| sgRNA: BST2+17403810 - CGA UUCUCACGCUUAAGACC | Synth ego | N/A |
| sgRNA: BST2+17403763 - CGC A.GCGGAGCUGGAGUCCU | Synth ego | N/A |
| Forward Primer 1: GATCAAG G G AATGTTCAAGCG AAA | Integrated DNA Technologies | N/A |
| Reverse Primer 1: AGGATCT CCTTTGCTCCCAAAATC | Integrated DNA Technologies | N/A |
| Forward Primer 2: CATCCTT CTCACTG G ATTCTCCC | Integrated DNA Technologies | N/A |
| Reverse Primer 2: ATTAAAC CATAAG CTTCAG GACG C | Integrated DNA Technologies | N/A |
| Forward sequencing primer 1: CTGCAGCCTCT CTCTCTAGACTT | Integrated DNA Technologies | N/A |
| Forward sequencing primer 2: CTGCAGCCTCT CTCTCTAGACTTC | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| Prism | N/A | N/A |
| Flow Jo | N/A | N/A |
| Fiji | N/A | N/A |
| Leica Application Suite software (Version 3.3), Leica Microsystems | N/A | N/A |
| Other | ||
| StemSpan SFEM | StemCell Technologies, Inc. | Cat#9600 |
CXCR4 expression analysis
Wild-type and Ifngr−/− mice were treated with either PBS (control) or IFNγ 24 h prior to bone marrow harvest. WBM was RBC lysed and cKit+ cells were isolated from control or test animals by magnetic bead enrichment using CD117 beads. CXCR4 protein expression was determined by flow cytometry.
Selectin binding assays
For the plate binding assays 96-well plates were coated with E-selectin or P-selectin (R&D Systems) at 3μg/mL for 16hrs at 4°C. Unbound selectin was removed and plates were blocked for 1hr at room temperature with 1% BSA. Plates were then washed 4 times with PBS + 1% BSA and used immediately. WBM was harvested from wild-type or BST2 KO mice 18hrs post IFNγ treatment. WBM was then RBC lysed and enriched for cKit+ cells using CD117 MicroBeads (Miltenyi-Biotec) and an AutoMACS Pro Separator. cKit-enriched cells were then stained at 1×106 cells/mL with 5μM CFSE for 20mins at 37°C. Stained cells were then washed and resus-pended at 2×105 cells/mL in media containing 1mM CaCl2 or 5mM EDTA. 100μl of this cell suspension was plated per well on the selectin coated 96-well plates. Cells were sedimented at 1000rpm for 5 mins followed by incubation at 4°C for 30mins. Plates were then washed 4 times before lysing the cells with 1% SDS. Fluorescent intensity was measured at an excitation of 492 and emission of 517 using a BioTek Cytation5 Imaging Reader. Similarly, E-selectin assays were also performed by staining cKit-enriched 1×106 cells/mL cells with 2 μM DDAO, resuspending at 2×105 cells/mL in media containing 1mM CaCl2 or 5mM EDTA and plating 100 μl on 96 well plates. Cells were sedimented at 1000rpm for 5 mins followed by incubation at 4°C for 30mins. Plates were then washed 4 times and wells were imaged using Echo Revolve florescent microscope. Cells were counted using Fiji software.
For the flow cytometry binding assay cKit+ progenitor cells were enriched from WBM using CD117 MicroBeads (Miltenyi-Biotec) and an AutoMACS Pro Separator. cKit+ cells were then stained for HSPC markers (Lineage, cKit, CD150 and CD48). Recombinant E-selectin (E-Selectin/CD62E Fc Chimera Protein) was pre-incubated with human IgG-FITC for 1hr at 4°C. 2×105 stained cKit+ cells were then incubated with 3μg of FITC-tagged E-selectin for 20mins at 4°C in PBS + 0.2% BSA + 1mM CaCl2. Cells were then washed and analyzed by flow cytometry.
Transwell migration assay
WBM was isolated from mice 18hrs post IFNγ-treatment. WBM was then RBC lysed and enriched for cKit+ cells using CD117 MicroBeads (Miltenyi-Biotec) and an AutoMACS Pro Separator. cKit+ cells were then plated in the top chamber of a transwell plate (5μm pore, 24 well) at 1×106 cells/well. Recombinant mCXCL12 (Peprotech) at 20ng/mL was added to the bottom chamber of the well and cells were allowed to migrate for 2hrs at 37°C. Cells were then harvested from both the top and bottom chambers. % cells migrated was calculated as the number of the cells in the lower chamber compared to the total cells. Flow cytometry was performed to confirm that the % HSCs in the upper and lower chambers were the same.
CRISPR deletion
BST2 was edited in the human cell line HL60 and in CD34+ HSPCs isolated from adult peripheral blood. Four separate single guide RNAs (sgRNAs) were designed using CRISPRSCAN software (Moreno-Mateos et al., 2015) and cloned by PCR into pKLV-U6gRNAEF(BbsI)-PGKpuro2ABFP (Ochiai, 2015). The 4 forward primers used were of sequence: TTAATACGACTCACTATAGG + 18N+GTTTAAGAGCTATGCTGGAAACAGC and the common reverse primer was AGCACCGACTCGGTGCCACT. Guide sequences are listed in the Key Resources Table. Single guide RNAs were used at concentration 1 μg/μl, mixed with 1 μg Cas9 protein (PNA Bio) and transduced into CD34+ cells by electroporation.
Gene expression profiling
Expression profiling of HSCs from control or 24hr IFNγ-treated mice was performed using Agilent’s SurePrint G3 Mouse Exon 4×180K Microarray. The Bioconductor package limma was used to analyze the microarray data. The data were background corrected by ‘normexp’ method with an offset of 16 added to the intensities before log2-transforming and then quantile normalized. Probes without gene description were filtered out before hypothesis testing. Moderated t-statistics were used to test if genes were differentially expressed between the groups of interest and Benjamini-Hochberg method was used to estimate false discovery rate (FDR). Probes with p value < 0.1 and fold change > 1.5 was considered top differentially expressed genes.
BrdU incorporation
Mice were injected with 1mg BrdU (10 mg/mL in PBS) per 6g body weight by IP 24 h before euthanasia. Bone marrow was stained using the BrdU flow kit (BD) and anti-BrdU FITC antibody. The BrdU staining of whole bone marrow was used as the “positive” control.
Hoechst Pyronin Y
Mice were infected with 2×106 colony-forming units M. avium for one month prior to WBM harvest. WBM was then RBC lysed and washed with PBS. Cells were pelleted and fixed with 70% ethanol added dropwise with gentle mixing. Cells were incubated on ice for > 2 h. Afterward, cells were washed twice with media followed by staining with 5 μM Hoechst and 10 μM pyronin Y for 1 h on ice. Cells were washed and subsequently stained for LT-HSCs (Lineage, cKit, CD150, CD48, CD34) and analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mean values ± SEM are shown. Identification of outliers (ROUT method, Q = 5%), normality and lognormality tests were performed to determine the proper use of parametric and nonparametric tests for analysis. Generally, Student’s t test, one-way ANOVA or two-way ANOVA were used for comparisons (GraphPad Prism Version 5.0).
Supplementary Material
Highlights.
Interferon gamma displaces hematopoietic stem cells in the bone marrow niche
Stem cell relocalization is mediated by BST2, an E-selectin ligand
Bst2−/− stem cells are hyperquiescent and resist depletion upon chronic infection
ACKNOWLEDGMENTS
The authors would like to thank T. Horton for sharing research equipment, D. Nakada for sharing Krt18 reporter mice, F. Lam for assistance with binding assays, and C. Gillespie for critical reading of the manuscript. This project depended on the support of Joel Sederstrom and the BCM Cytometry and Cell Sorting Core with funding from the NIH (NCRR grant S10RR024574, NIAID grant AI036211, and NCI grant P30CA125123), NIH grant S10 OD020066, and the Dan L. Duncan Cancer Center, as well as help from Lisa White and the genomic and RNA profiling core at Baylor College of Medicine, with funding from the NIH (NIDDK-DK56338 and NCI-CA125123). This project was also supported by the BCM Integrated Microscopy Core with funding from the NIH (HD007495, DK56338, and CA125123), the Dan L. Duncan Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. K.Y.K., M.A.F., and K.A.M. were supported by the NIH grants R01HL136333 and R01HL134880 (K.Y.K.) and T32DK060445 (K.A.M. and M.A.F.), the Aplastic Anemia and MDS International Foundation Liviya Anderson Award (K.Y.K.), and a March of Dimes Basil O’Connor Starter Scholar Award (K.Y.K.). R.J. and M.K. were supported by the Polish National Science Centre grants 2016/23/D/ST7/03665 and 2018/29/B/ST7/02550, respectively. D.P., L.O., and Y.J. were supported by the NIH grants R01AR072018 (D.P., Y.J., andL.O.), R01CA221946 (D.P. and Y.J.), and R21AG064345 (D.P.).
AML survival data are based upon data generated by the TCGA Research Network: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108530.
A video abstract is available at https://doi.org/10.1016/j.celrep.2020.108530#mmc3.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Achi HV, Ahui BJ, Anon JC, Kouassi BA, Dje-Bi H, and Kininlman H (2013). [Pancytopenia: a severe complication of miliary tuberculosis]. Rev. Mal. Respir 30, 33–37. [DOI] [PubMed] [Google Scholar]
- Aurrand-Lions M, and Mancini SJC (2018). Murine Bone Marrow Niches from Hematopoietic Stem Cells to B Cells. Int. J. Mol. Sci 19, 2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujanover N, Goldstein O, Greenshpan Y, Turgeman H, Klainberger A, Scharff Y, and Gazit R (2018). Identification of immune-activated hematopoietic stem cells. Leukemia 32, 2016–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, and Link DC (2015). The hematopoietic stem cell niche in homeostasis and disease. Blood 126, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple RH, Tseng YJ, Hu T, Kitano A, Takeichi M, Hoegenauer KA, and Nakada D (2018). Lineage tracing of murine adult hematopoietic stem cells reveals active contribution to steady-state hematopoiesis. Blood Adv 2, 1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, Seita J, Sahoo D, Nakauchi H, and Weissman IL (2016). Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature 530, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Deng H, Churchill MJ, Luchsinger LL, Du X, Chu TH, Friedman RA, Middelhoff M, Ding H, Tailor YH, et al. (2017). Bone Marrow Myeloid Cells Regulate Myeloid-Biased Hematopoietic Stem Cells via a Histamine-Dependent Feedback Loop. Cell Stem Cell 21, 747–760.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou C, Spencer JA, Yeh SA, Turcotte R, Kokkaliaris KD, Panero R, Ramos A, Guo G, Seyedhassantehrani N, Esipova TV, et al. (2020). Live-animal imaging of native haematopoietic stem and progenitor cells. Nature 578, 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, Tani-Ichi S, Schlenner S, Richie E, Rodewald H-R, et al. (2016). Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 45, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, and Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P, Sager HB, Stengel KR, Naxerova K, Courties G, Saez B, Silberstein L, Heidt T, Sebas M, Sun Y, et al. (2015). Myocardial Infarction Activates CCR2(+) Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 16, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFNαlpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Feng CG, Zheng L, Jankovic D, Báfica A, Cannons JL, Watford WT, Chaussabel D, Hieny S, Caspar P, Schwartzberg PL, et al. (2008). The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-γ-induced cell death. Nat. Immunol 9, 1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A, Hsu Y-MS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays.J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Julien S, Ivetic A, Grigoriadis A, QiZe D, Burford B, Sproviero D, Picco G, Gillett C, Papp SL, Schaffer L, et al. (2011). Selectin ligand sialyl-Lewis × antigen drives metastasis of hormone-dependent breast cancers. Cancer Res 71, 7683–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, and Goodell MA (2011). Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol 11, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, Matatall KA, Shen C-C, Goodell MA, Swierczek SI, and Prchal JT (2015). Comparative long-term effects of interferon α and hydroxyurea on human hematopoietic progenitor cells. Exp. Hematol 43, 912–918.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkaliaris KD, Kunz L, Cabezas-Wallscheid N, Christodoulou C, Renders S, Camargo F, Trumpp A, Scadden DT, and Schroeder T (2020). Adult blood stem cell localization reflects the abundance of reported bone marrow niche cell types and their combinations. Blood 136, 2296–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DS, Lazarides K, Lewis JB, von Andrian UH, and Van Etten RA (2014). Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 123, 1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BNR, Chang HK, Son YS, Lee D, Kwon SM, Kim PH, and Cho JY (2018). IFN-γ enhances the wound healing effect of late EPCs (LEPCs) via BST2-mediated adhesion to endothelial cells. FEBS Lett 592, 1705–1715. [DOI] [PubMed] [Google Scholar]
- Liberatore RA, and Bieniasz PD (2011). Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc. Natl. Acad. Sci. USA 108, 18097–18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Cao Y, Guan Y, and Zheng C (2018). BST2 promotes cell proliferation, migration and induces NF-κB activation in gastric cancer. Biotechnol. Lett 40, 1015–1027. [DOI] [PubMed] [Google Scholar]
- MacNamara KC, Jones M, Martin O, and Winslow GM (2011). Transient activation of hematopoietic stem and progenitor cells by IFNγ during acute bacterial infection. PLoS One 6, e28669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, and von Andrian UH (2007). Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 131, 994–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Shen C-C, Challen GA, and King KY (2014). Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells 32, 3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, and King KY (2016). Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep 17, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, and Enikolopov G (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. J. Comp. Neurol 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Morales-Mantilla DE, and King KY (2018). The Role of Interferon-Gamma in Hematopoietic Stem Cell Development, Homeostasis, and Disease. Curr. Stem Cell Rep 4, 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ, et al. (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12 (10), 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Han Y-C, and Zou Y-R (2008). CXCR4 is required for the quiescence of primitive hematopoietic cells. J. Exp. Med 205, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisticò A, and Young NS (1994). gamma-Interferon gene expression in the bone marrow of patients with aplastic anemia. Ann. Intern. Med 120, 463–469. [DOI] [PubMed] [Google Scholar]
- Ochiai H (2015). Single-Base Pair Genome Editing in Human Cells by Using Site-Specific Endonucleases. Int. J. Mol. Sci 16 (9), 21128–21137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Jang H, Kim C, Chung B, Chang CL, Park SK, and Song S (2000). Detection and identification of mycobacteria by amplification of the internal transcribed spacer regions with genus- and species-specific PCR primers. J. Clin. Microbiol 38, 4080–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, and Scadden DT (2012). Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM (2017). Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood 130, 1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner J-M, Will B, et al. (2016). Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol 18, 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Casals M, García-Carrasco M, López-Medrano F, Trejo O, Forns X, López-Guillermo A, Muñoz C, Ingelmo M, and Font J (2003). Severe autoimmune cytopenias in treatment-naive hepatitis C virus infection: clinical description of 35 cases. Medicine (Baltimore) 82, 87–96. [DOI] [PubMed] [Google Scholar]
- Roesler J, Horwitz ME, Picard C, Bordigoni P, Davies G, Koscielniak E, Levin M, Veys P, Reuter U, Schulz A, et al. (2004). Hematopoietic stem cell transplantation for complete IFN-gamma receptor 1 deficiency: a multi-institutional survey. J. Pediatr 145, 806–812. [DOI] [PubMed] [Google Scholar]
- Runnels JM, Zamiri P, Spencer JA, Veilleux I, Wei X, Bogdanov A, and Lin CP (2006). Imaging molecular expression on vascular endothelial cells by in vivo immunofluorescence microscopy. Mol. Imaging 5, 31–40. [PMC free article] [PubMed] [Google Scholar]
- Scadden DT, Zon LI, and Groopman JE (1989). Pathophysiology and management of HIV-associated hematologic disorders. Blood 74, 1455–1463. [PubMed] [Google Scholar]
- Schürch CM, Riether C, and Ochsenbein AF (2014). Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 14, 460–472. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, and Nagasawa T (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. [DOI] [PubMed] [Google Scholar]
- Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, Ahuja AK, Lopes M, Hausmann A, Hardt W-D, et al. (2017). Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 21, 225–240.e5. [DOI] [PubMed] [Google Scholar]
- Toshiaki Ara, Koji Tokoyoda, Tatsuki Sugiyama, Takeshi Egawa, Kenji Kawabata, and Takashi Nagasawa (2003). Long-Term Hematopoietic Stem Cells Require Stromal Cell-Derived Factor-1 for Colonizing Bone Marrow during Ontogeny. Immunity 19, 257–267. [DOI] [PubMed] [Google Scholar]
- Tzeng YS, Li H, Kang YL, Chen WC, Cheng WC, and Lai DM (2011). Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 117, 429–439. [DOI] [PubMed] [Google Scholar]
- Umemoto T, Matsuzaki Y, Shiratsuchi Y, Hashimoto M, Yoshimoto T, Nakamura-Ishizu A, Petrich B, Yamato M, and Suda T (2017). Integrin αvβ3 enhances the suppressive effect of interferon-γ on hematopoietic stem cells. EMBO J 36, 2390–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul JD, and Hordijk PL (2004). Signaling in leukocyte transendothelial migration. Arterioscler. Thromb. Vasc. Biol 24, 824–833. [DOI] [PubMed] [Google Scholar]
- Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, Magnani JL, and Lévesque J-P (2012). Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med 18, 1651–1657. [DOI] [PubMed] [Google Scholar]
- Yamashita M, and Passegué E (2019). TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 25, 357–372.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H, Park SH, Ye SK, and Kim M (2011). IFN-γ-induced BST2 mediates monocyte adhesion to human endothelial cells. Cell. Immunol 267, 23–29. [DOI] [PubMed] [Google Scholar]
- Young NS, and Maciejewski J (1997). The pathophysiology of acquired aplastic anemia. N. Engl. J. Med 336, 1365–1372. [DOI] [PubMed] [Google Scholar]
- Zhao M, Tao F, Venkatraman A, Li Z, Smith SE, Unruh J, Chen S, Ward C, Qian P, Perry JM, et al. (2019). N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep 26, 652–669.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data generated during this study are available at https://data.mendeley.com/datasets/mhc8b4yn9d/1