

Summary
Chordomas are rare spinal tumors addicted to expression of the developmental transcription factor brachyury. In chordomas, brachyury is super-enhancer associated and preferentially downregulated by pharmacologic transcriptional CDK inhibition, leading to cell death. To understand the underlying basis of this sensitivity, we dissect the brachyury transcription regulatory network and compare the consequences of brachyury degradation with transcriptional CDK inhibition. Brachyury defines the chordoma super-enhancer landscape and autoregulates through binding its super-enhancer, and its locus forms a transcriptional condensate. Transcriptional CDK inhibition and brachyury degradation disrupt brachyury autoregulation, leading to loss of its transcriptional condensate and transcriptional program. Compared with transcriptional CDK inhibition, which globally downregulates transcription, leading to cell death, brachyury degradation is much more selective, inducing senescence and sensitizing cells to anti-apoptotic inhibition. These data suggest that brachyury downregulation is a core tenet of transcriptional CDK inhibition and motivates developing strategies to target brachyury and its autoregulatory feedback loop.
Keywords: chordoma, phase separation, transcription, transcription factor, transcriptional inhibition, targeted degradation, brachyury, super-enhancer, cyclin-dependent kinase, transcriptional condensate
Graphical Abstract
Highlights
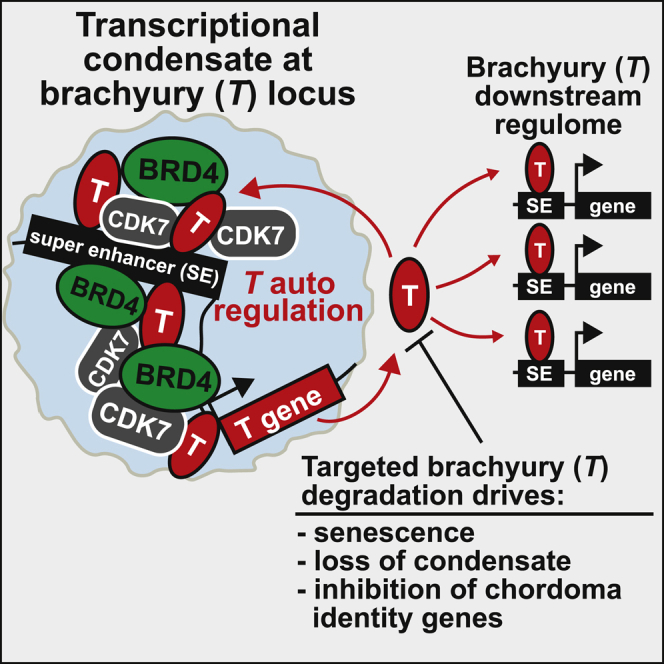
Brachyury defines the chordoma super-enhancer landscape
Brachyury autoregulates through a transcriptional condensate
CDK7/12/13i and brachyury degradation target the brachyury transcriptional condensate
Brachyury degradation inhibits chordoma identity genes and induces senescence
Sheppard et al. map the brachyury regulatory landscape in chordoma and explore its targeting using transcriptional CDK inhibition and targeted brachyury degradation. Brachyury is a highly selective transcriptional regulator of chordoma identity, and they confirm that brachyury targeting is a promising therapeutic strategy.
Introduction
Transcription factor (TF) deregulation is pronounced in pediatric cancers and tumors of lineage-dependent origins.1, 2, 3, 4 In such tumors, TFs that are normally expressed during early development are aberrantly sustained and required to maintain tumor identity.5 Chordoma is a tumor characterized by brachyury, a T-box TF responsible for early mesoderm formation and notochord development.6 Brachyury (encoded by the T/TBXT gene) is not expressed after day 13 in human development and in adults is minimally detected in the pituitary gland, thyroid, and testes.7,8 Chordomas are characterized by brachyury expression, are associated with a germline variant in the protein (G177D), and require brachyury for cell growth.6,9, 10, 11 Efforts to drug brachyury are underway, but as with many TFs, drug discovery is hampered by the absence of ligand-accessible small-molecule binding pockets.12 This has created an unmet clinical need to therapeutically address cancers defined by TF deregulation, such as chordoma.
We and others have observed that inhibitors of the transcription and chromatin machinery downregulate oncogenic master TFs such as brachyury. This was first revealed by selective downregulation of Myc oncogenes by inhibitors of the Bromodomain and Extra-Terminal (BET) family of acetyl-lysine reader transcriptional co-activators.13, 14, 15 Subsequent studies using pharmacological inhibitors of transcriptional cylcin-dependent kinases (CDKs), histone deacetylases (HDACs), and histone acetyltransferases (HATs) have produced similar results in multiple cancers. Examples include the CDK7/12/13 inhibitor THZ1,10,16,17 the HDAC inhibitors vorinostat and romidepsin,18,19 and the p300 inhibitor A-485.20 Treatment of chordoma cells with transcriptional CDK inhibitors like THZ1 leads to preferential downregulation of brachyury and loss of cell viability.10 These data suggest that oncogenic master TFs are acutely sensitive to transcriptional perturbations, which can be exploited therapeutically.
Paralleling the increasing interest in drugging the transcriptional apparatus, we and others have sought to understand the mechanistic basis by which these inhibitors have gene-selective effects. Initial observations noted that many of these oncogenic TFs were adjacent to large enhancers or “super-enhancers” (SEs) that contain disproportionate occupancy of the transcriptional machinery.21, 22, 23 Across multiple cellular contexts, inhibition of transcription selectively downregulates SE-associated gene expression programs.10,17,24 Despite statistical association, the mechanistic differences between SEs and typical enhancers remain poorly understood. Consequently, although SE association can predict selectivity of transcriptional inhibition, it is incomplete in explaining the underlying mechanism.
Emerging studies suggest that SEs achieve high concentrations of the transcriptional machinery by adopting properties of non-membrane-bound phase-separated liquid droplets, called “transcriptional condensates.”25, 26, 27 Weak interactions between the intrinsically disordered regions of transcriptional co-activators nucleate a phase transition and create localized domains of co-activator concentration. Initiation and maintenance of transcriptional condensates require TFs and DNA,28 weak non-covalent interactions mediated by post-translational modifications,29 or signaling events.30 Inhibition of transcription disrupts these condensates,26 and, most recently, antineoplastic drugs have been shown to localize within them,31 providing a potential mechanism for SE target gene selectivity.
Additionally, SE-driven oncogenic master TF regulation is likely autoregulatory. In chordoma, we previously observed brachyury binding its own SE.10 In many different cancer types, networks of oncogenic master TFs form autoregulatory loops via each other’s SEs.32, 33, 34, 35, 36, 37 Disruption of a single oncogenic master TF is often sufficient to collapse this circuitry, leading to cell death or differentiation. We hypothesize that transcriptional inhibition achieves a therapeutic effect by targeting oncogenic TF autoregulation and inducing selective downregulation of these genes. To test this, we developed engineered cell line systems to define brachyury occupancy on the genome using genomic and microscopy approaches and to compare selective brachyury degradation with pharmacological transcriptional CDK inhibition.
Results
Brachyury is a master transcriptional regulator that defines the chordoma SE landscape
To define and modulate brachyury activity, we engineered CH22 and UM-Chor1 human chordoma cell lines to knock out the endogenous T gene and express a degradation-tagged allele, HA-dTAG-T(wild-type [WT] or G177D) (Figures S1A and S1B). Our exogenous T allele contains a hemagglutinin (HA) epitope tag to facilitate brachyury pulldown and detection as well as the dTAG epitope for small-molecule-mediated degradation.38,39 The resulting chordoma cells, with complemented exogenous HA-dTAG-T, CH22, or UM-Chor1 HA-dTAG-T, T−/−, are viable and exhibit growth kinetics similar to their parental lines (Figures S1C and S1D). Genome-wide profiling of WT or G177D-variant brachyury reveals similar binding (Figure S1E) and provides justification for analysis of their pooled binding.
In CH22 and UM-Chor1 HA-dTAG-T, T−/− cells, we define 8,981 and 12,544 distinct brachyury binding sites. Brachyury binding (via HA pull-down) is observed at SEs associated with chordoma pathophysiology, including the keratin (KRT8/KRT18) locus (Figure 1A).40 No binding is observed upon HA pull-down in cells that lack expression of HA (Figure S1F). Ranking brachyury peaks by area-under-curve occupancy, we note a strong association with acetylation of histone H3 on lysine 27 (H3K27ac) and enrichment for binding at SEs (Figures 1B and 1C). De novo motif analysis of top brachyury binding sites reveals enrichment for a series of T-box-like motifs similar to other published profiles of brachyury in vertebrates (Figure 1D).41,42 We were also able to detect a Transcriptional-Enhanced-Associate-Domain (TEAD)-1/Yes-Associated Protein (YAP) motif from the HA-dTAG-brachyury pull-down (Figure 1D). YAP expression is activated by brachyury and can drive cancer stemness in chordomas.43 The TEAD proteins directly interact with YAP and also play a critical role in notochord formation.44 Globally, the majority of brachyury binding sites fall within regions of H3K27ac-marked acetylated chromatin at distal enhancers (Figure 1E). From these data, we find that brachyury predominantly acts as a sequence-specific enhancer binding factor.
Figure 1.

Brachyury is a master transcriptional regulator that defines the chordoma SE landscape
(A) Gene tracks of H3K27ac and HA-dTAG-brachyury in CH22 and UM-Chor1 parental and HA-dTAG-T, T−/− cells at the KRT8/KRT18 loci (units of reads per million per base pair). The SEs are denoted by blue boxes. For CH22, n = 4 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively. For UM-Chor1, n = 3 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively.
(B) Heatmaps showing brachyury (WT and G177D) (red) and H3K27ac (blue) in CH22 and UM-Chor1 parental and HA-dTAG-T, T−/− cells. Each row shows the ± 5 kb centered on the brachyury peak center ranked by average HA-dTAG-brachyury/H3K27ac signal. Color-scaled intensities are in units of reads per million (rpm) per base pair. Brachyury peak overlaps with SEs are noted in black.
(C) Sliding window plot of log2 enrichment for SEs versus HA-dTAG-brachyury peaks ranked by binding (area under curve [AUC]) in CH22 and UM-Chor1 parental and HA-dTAG-T, T−/− cells. Each window is 1,000 brachyury peaks, and each step size is 500 peaks.
(D) De novo motif analysis of brachyury binding from combined analysis of CH22 and UM-Chor1 HA-dTAG-T, T−/− cells.
(E) Pie charts showing genomic distributions of brachyury peaks with active chromatin or genomic features in UM-Chor1 and CH22 parental and HA-dTAG-T, T−/− cells.
(F) Pathway analysis for the combined top CH22- and UM-Chor1 SE-associated genes regulated by brachyury.
(G) Network depiction of the brachyury regulome from combined analysis of CH22 and UM-Chor1 parental and HA-dTAG-T, T−/− cells. Nodes represent SE-associated TFs that regulate or are regulated by brachyury. Edges are bi-directional (solid line) or unidirectional (dotted line). Red edges represent chromatin immunoprecipitation (ChIP)-verified brachyury binding. White nodes show TFs that are upstream regulators of brachyury, blue nodes are upstream and downstream, and gray nodes are downstream only.
Between UM-Chor1 and CH22, we find little overlap of brachyury binding sites and active enhancer regions (Figure S1G). This suggests that, although brachyury is a sequence-specific developmental TF, its regulome is shaped by additional lineage features. This has been observed for other developmental TFs, such as FOXA1, which has distinct binding landscapes in breast versus prostate lineages.45 Many top brachyury-bound genes, such as KRT8/KRT18, are present in both cell lines, suggesting a shared core function for brachyury. Ranking genes by proximal brachyury occupancy, we observe 350 top genes with higher levels of brachyury occupancy by at least an order of magnitude compared with a typical region in both cell lines (Figure S1H). Many of these top brachyury genes are SE associated and strongly enriched for chordoma-associated pathways, including transforming growth factor β (TGF-β) signaling;46 tyrosine kinase signaling, including Epidermal Growth Factor Receptor (EGFR);47 and hypoxia48,49 (Figure 1F).
Master TFs often regulate one another by binding to their proximal enhancers and SEs and forming a TF core regulatory circuitry (CRC) to define cell identity.22 Inferring this CRC in chordoma using our previously established methods,36 we find evidence that brachyury regulates and is regulated by TFs, including MYC, NR3C1, TGFB Induced Factor Homeobox (TGIF), ID1, HoxA, and HoxB (blue nodes, Figure 1G; Figure S1I). Suggestive of upstream regulation, the T proximal SE is also enriched for binding sites for AP-1 TFs (Jun/Fos) and other immediate-early activators (EGR1), which, collectively, are inducers of mesoderm formation and chondrogenic differentiation.50,51 Within this interconnected network, brachyury exhibits the highest overall connectivity, consistent with its overarching role as a chordoma master regulator. Thus, we establish that brachyury is a chordoma master TF that preferentially binds to gene-distal regions of active chromatin, and in particular SEs, where it defines gene programs associated with chordoma identity.
Brachyury autoregulates through an SE transcriptional condensate
Previously, we demonstrated that, in chordoma, the T gene is proximal to a large region of clustered enhancers, typically one or more SEs.10 In particular, a 3′ SE (60 kb downstream), to which brachyury itself was found to bind, was identified in all chordoma cell lines examined and a subset of samples from individuals with chordoma (Figure 2A).10 This autoregulation is maintained in HA-dTAG-T(G177D), T−/− cells. We next investigated whether the 3′ T SE is required to maintain T levels in chordoma. We targeted dCas9 fused to a Krüppel associated box (KRAB) domain (dCas9-KRAB) to the 3′ SE brachyury binding site or the T promoter in parental CH22 cells. Recruitment of this repressive complex via single-guide RNAs (sgRNAs) leads to deposition of repressive histone modifications.52 Silencing of the 3′ SE decreases T mRNA levels equivalent to silencing the T promoter, whereas expression of the control gene MAX remains unaffected (Figure 2B). These results demonstrate that the 3′ proximal SE bound by brachyury contributes to T regulation.
Figure 2.

Brachyury autoregulates through a SE transcriptional condensate
(A) Gene tracks of H3K27ac and HA-dTAG-brachyury (units of reads per million per base pair) at the T locus in UM-Chor1 and CH22 parental and HA-dTAG-T, T−/− cells. The SEs are denoted by blue boxes. The location of sgRNAs used to target dCas9-KRAB are indicated by arrows. For CH22, n = 4 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively. For UM-Chor1, n = 3 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively.
(B) Bar plots depicting T or MAX mRNA levels in CH22 parental cells transduced with a nontargeting sgRNA or sgRNAs targeting the T promoter or the T SE and dCas9-KRAB. Data are expressed as the mean mRNA levels normalized to GAPDH. Error bars denote SD (n = 3 technical replicates).
(C) Line graph showing the log2 fold change of SE-associated TF mRNA levels in parental UM-Chor1 cells with THZ1 (n = 3 biological replicates per time point). T is denoted in red.
(D) Representative FRAP images of HA-EGFP-dTAG-brachyury puncta. The white box indicates the bleached punctum.
(E) Quantification of FRAP data targeting HA-EGFP-dTAG-brachyury. Bleaching occurs at t = 0 s. For the bleached area and unbleached control, fluorescence intensities are plotted relative to a prebleach time point (t = −2 s). Data are plotted as means SD (n = 5 cells).
(F) Top: colocalization between BRD4 and T nascent RNA by immunofluorescence (IF) and nascent RNA fluorescence in situ hybridization (FISH), respectively, in fixed CH22, THA-dTAG-EGFP/+. Bottom: colocalization between BRD4 and GAPDH nascent RNA by IF and nascent RNA FISH in fixed CH22, THA-dTAG-EGFP/+. Separate images of the indicated IF and FISH are shown, along with a merged image.
(G) Zoomed image showing (F) with the calculated Spearman correlation coefficient for BRD4 protein and T or GAPDH nascent RNA colocalization.
(H) Quantification of T nascent RNA and BRD4 protein colocalization compared with GAPDH nascent RNA and BRD4 protein colocalization. Cells from two biological populations were prepared and imaged in parallel. Spearman correlation coefficients between T or GAPDH nascent RNA and with BRD4 protein signal are plotted. ∗∗∗∗p < 0.0001, derived from a two-tailed, unpaired t test.
It has been observed that the CDK7/12/13 inhibitor THZ1 can downregulate SE-driven brachyury expression but that exogenous T, dissociated from its endogenous regulatory elements, is less susceptible to downregulation.10 To confirm that transcriptional CDK inhibition selectively targets SE-mediated regulation of T in our engineered cell line system, we profiled CH22 cells that express an HA-dTAG-T(G177D) allele in the context of endogenous T (T+/+) and treated them with a panel of pharmacological transcriptional inhibitors. It is important to note that CH22 cells are heterozygous for the G177D brachyury variant. We find that all compounds targeting both members of transcriptional elongation (CDK9, CDK12, and CDK13) and transcriptional initiation (CDK7) show selective downregulation of endogenous versus exogenous T mRNA and brachyury protein (Figures S2A–S2C). The brachyury target MYC is also downregulated with THZ1 treatment (Figure S2D). In UM-Chor1 cells, pharmacological transcriptional CDK inhibition using THZ1 is able to preferentially downregulate T compared with all other SE-associated TFs in terms of timing and magnitude (Figure 2C). These observations support our model that brachyury autoregulates through a 3′ SE and provide additional evidence that this SE is critical for preferential targeting via transcriptional inhibition using several pharmacological agents.
Because master TF-bound SEs have been shown to adopt properties of liquid-like transcriptional condensates,25,27,29 we investigated whether the same applied to the T locus. The brachyury protein encodes an intrinsically disordered domain required for activation of transcription.53 Thus, brachyury has been hypothesized to undergo phase separation. To visualize brachyury, we engineered CH22 cells with an HA-dTAG-EGFP epitope endogenously inserted into the N terminus of T (Figure S2E). Live imaging of CH22, T HA-dTAG-EGFP/+ cells shows clear formation of brachyury puncta in the nucleus (Figure 2D; Figure S2F). Furthermore, our engineered cell lines, CH22 HA-dTAG-T (WT), T−/− and CH22 HA-dTAG-T(G117D), T−/−, reveal that WT and G177D brachyury are able to form puncta (Figure S2G). Puncta are no longer visible upon immediate treatment with 1,6-hexanediol, a small-molecule aliphatic alcohol used to differentiate liquid from solid-like condensates27,54 (Figure S2H). Liquid-like condensates also exhibit rapid exchange kinetics, which can be validated through fluorescence recovery after photobleaching (FRAP).55, 56, 57 In live cells, we find that brachyury puncta are rapidly turned over in seconds (Figures 2D and 2E), consistent with brachyury forming a liquid-like condensate in the nucleus.
Transcriptional condensates also exhibit high concentrations of the transcriptional machinery and transcriptional co-activators, including the BET-bromodomain BRD4.23,27 Consistent with SE regulation of T through an autoregulatory transcriptional condensate, we observe brachyury and BRD4 condensates at the T locus (Figures 2F and 2G; Figure S2I). When comparing SE-associated T with the non-SE-associated GAPDH housekeeping gene (Figures 2F and 2G; Figure S2I), we see increased colocalization between T RNA and BRD4 condensates (Figure 2H). There is also increased colocalization between brachyury condensates and T RNA compared with GAPDH (Figure S2J) Thus, throughout the nucleus, brachyury is found at transcriptional condensates, including at the T locus in particular, where BRD4 is consistently co-localized. These results support our hypothesis that the T locus adopts properties consistent with a transcriptional condensate.
Transcriptional CDK inhibition-induced apoptosis is associated with brachyury downregulation
Inhibiting components of transcription and splicing disrupts the respective protein condensates, including BRD4 and SRSF2 condensates, suggesting one mechanism by which pharmacologic transcriptional CDK inhibitors kill cancerous cells.26,29 In chordoma, transcriptional CDK inhibitors cause not only preferential downregulation of brachyury but also induction of apoptosis10 (Figure S3A). Moreover, previous work using a non-dTAG, open reading frame (ORF)-based T overexpression system has shown that exogenous T, lacking its endogenous regulatory elements, is less susceptible to THZ1-induced downregulation and can partially rescue cellular sensitivity to THZ1.10 We thus tested whether transcriptional CDK inhibition-induced apoptosis is associated with brachyury downregulation in our engineered dTAG/degron system. We first confirmed the ability of transcriptional CDK inhibitors to downregulate endogenous brachyury in parental and engineered CH22 HA-dTAG-T, T+/+ cells (Figure 3A; Figure S3B). At the protein level, the exogenous brachyury is not downregulated by THZ1 (Figure 3B).
Figure 3.

Transcriptional CDK inhibition-induced apoptosis is associated with brachyury downregulation
(A) Schematic depicting engineered chordoma cell lines.
(B) Immunoblot of brachyury protein levels in CH22 parental and HA-dTAG-T, T+/+ cells with THZ1 (n = 2, one experiment is shown).
(C) Immunoblots of HA-dTAG-brachyury expression in CH22 HA-dTAG-T, T−/− cells with concentrations of degron for 8 h (left) and 1 μM degron over time (right) (n = 1).
(D) Cell viability of HA-dTAG-T, T+/+ or parental cells treated with 15 nM THZ1 or 1 μM degron + 15nM THZ1 for 6 days. Data are plotted as the mean fraction of cell viability relative to DMSO (n = 8 biological replicates). Error bars denote SD. ∗∗∗∗p < 0.0001, derived from a two-tailed, unpaired t test.
(E) Representative images of CH22, THA-dTAG-EGFP/+ with degron or THZ1.
(F) Left: quantification of live-cell imaging of CH22, THA-dTAG-EGFP/+ with DMSO, THZ1 or degron (n = 3 biological replicate populations per treatment). The integral EGFP signal is normalized to t = 0. Right: boxplots comparing median EGFP levels in apoptotic and nonapoptotic individual cells (n = 21). ∗∗∗p < 0.001, derived from a two-tailed, unpaired t test.
We then returned to CH22 cell lines expressing exogenous HA-dTAG-T(G177D) with endogenous T knockout (T−/−) (Figures S1B and S3B), hereafter referred to as HA-dTAG-T, T−/− cells. Upon addition of degron, exogenous HA-dTAG-brachyury is degraded rapidly in a time- and concentration-dependent manner (Figure S3C; Figure 3C). Degron treatment of CH22 HA-dTAG-T, T−/− cells halts chordoma cell growth and phenocopies the morphological effect of shRNA knockdown of T9, confirming brachyury dependence (Figures S3D and S3E). Cells expressing exogenous HA-dTAG-brachyury and endogenous brachyury are less sensitive to THZ1 compared with parental controls, and sensitivity is also restored with degron and removal of HA-dTAG-brachyury (Figure 3D). Degron treatment exhibits minimal toxicity to WT cells (Figure S3F).
We then performed live imaging of CH22, T HA-dTAG-EGFP/+ to compare the kinetics of THZ1-mediated brachyury downregulation with direct degradation (Figures 3E and 3F). Although downregulation of brachyury by THZ1 is evident between 8–23 h (Figures 2C, 3E, and 3F), degradation occurs within the first hour and is nearly complete by 8 h (Figures 3C, 3E, and 3F). This suggests that targeting brachyury at the protein level is kinetically favorable, given the longer half-life of brachyury protein. Furthermore, brachyury downregulation precedes THZ1-mediated cell death (Video S1). In cells that undergo cell death (Figures 3E and 3F), we observe lower basal levels of brachyury (Figure 3F). Consistent with previous findings,10 these data suggest that brachyury downregulation contributes to THZ1-induced cell death and that overexpression can partially abrogate these effects.
Each frame represents 5 minutes and filming occurred over 23 hours. When CH22, THA-dTAG-EGFP/+ are treated with 500nM THZ1, individual cells with lower brachyury expression levels are primed to undergo apoptosis and detach from their surface.
Brachyury is a highly selective transcriptional regulator
To dissect functional differences between THZ1 and brachyury degradation, we performed kinetic gene expression profiling in CH22 HA-dTAG-T, T−/− chordoma cells. To capture the overall effects of both perturbations on the mRNA pool as well as the immediate effects on nascent transcription, we also performed thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq), a metabolic labeling technique that measures changes in nascent transcription.58 Consistent with transcriptional CDK inhibition acting on a global scale, 8-h THZ1 treatment results in marked global downregulation of transcription, as revealed by nascent transcription profiling (Figure 4A).
Figure 4.

Brachyury is a highly selective transcriptional regulator
(A) Boxplots depicting log2 fold changes in steady state mRNA (left) or mean percentage of nascent mRNA reads (right) in CH22 HA-dTAG-T, T−/− cells with DMSO, degron, or THZ1 (n = 3 biological replicates per treatment).
(B) Bar plots depicting mean nascent mRNA levels for KRT18, MCL1, or GAPDH in CH22 HA-dTAG-T, T−/− cells with DMSO, degron, or THZ1. Error bars denote SD (n = 3 biological replicates per treatment). The p values were derived from a two-tailed, unpaired t test.
(C) Bar plots showing mean total mRNA levels for KRT18, MCL1, or GAPDH in CH22 HA-dTAG-T, T−/− cells with DMSO, degron, or THZ1. Error bars denote SD (n = 3 biological replicates per treatment).
(D) Left: scatterplot of percent nascent mRNA reads for all active genes with degron versus DMSO in CH22 HA-dTAG-T, T−/− cells (n = 3 biological replicates per treatment). The top brachyury-bound, SE-associated genes are shown in red. Right: boxplot comparing the log2 fold change in nascent mRNA for the top brachyury-bound SE-associated genes (red); the top brachyury-bound, non-SE-associated genes (blue); and other, non-brachyury-bound, active genes (gray) with degron. The p values were derived from a two-tailed, unpaired t test.
(E) Left: scatterplot of percent nascent mRNA reads for all active genes with THZ1 versus DMSO in CH22 HA-dTAG-T, T−/− cells (n = 3 biological replicates per treatment). The top brachyury-bound, SE-associated genes are shown in red. Right: boxplot comparing the log2 fold change in nascent mRNA for the top brachyury-bound SE-associated genes (red); the top brachyury-bound, non-SE-associated genes (blue); and all other, non-brachyury-regulated, active genes (gray) with THZ1. The p values were derived from a two-tailed, unpaired t test.
(F) GSEA plots of the top, brachyury-bound SE-associated genes with THZ1 or degron defined by leading edge analysis in CH22 HA-dTAG-T, T−/− cells. Genes are ranked from left to right by the log2 fold change in nascent transcription.
At KRT18, a prominent brachyury-bound and SE-associated gene, brachyury degradation downregulates nascent transcription (Figure 4B). At MCL1 and GAPDH, which lack brachyury binding, degradation has little effect. In contrast, nascent transcription at all three genes is downregulated strongly by THZ1. At 8 h, changes in nascent transcription are not yet reflected in total mRNA levels (Figure 4C), with the exception of short-half-life genes like MCL1.59 Across all active genes, top brachyury target genes that are SE associated (defined in Figure 1) show selective downregulation of nascent transcription upon brachyury degradation (Figure 4D). Degradation also results in modest downregulation of select non-brachyury-bound active genes, which, we hypothesize, is due to secondary effects and loss of cellular fitness. Preferential downregulation of brachyury-bound SE genes is also captured by leading-edge enrichment of top brachyury targets (Figure 4F). Brachyury degradation is highly significant (false discovery rate [FDR] 0.022), whereas THZ1 shows no enrichment (FDR = 0.4). Because the exogenous HA-dTAG-brachyury expressed in these cells is not SE associated or selectively downregulated by THZ1, no preferential downregulation of the top SE-associated, brachyury-bound target genes is expected or observed with THZ1 (Figures 4E and 4F).
Traditional RNA sequencing (RNA-seq) in CH22-derived cells also captures the selective downregulation of top brachyury target genes upon degradation, but not treatment with low concentrations of THZ1, at 8 and 24 h (Figure S4A). In UM-Chor1 parental cells, at higher THZ1 concentrations that selectively downregulate brachyury, there is preferential depletion of top brachyury SE-associated target genes by 24 h (likely as a secondary consequence of brachyury loss) (Figure S4B). In CH22 HA-dTAG-T, T−/− cells, genes significantly downregulated upon brachyury degradation are enriched for top SE-associated, brachyury-bound target genes (p = 4.568e–14, two-sided Fisher’s exact test) (Figure S4C). However, overall, only a small number of genes are affected when comparing brachyury degradation with controls (Figure S4D). In contrast, THZ1 results in many thousands of differentially expressed genes at 8 and 24 h (Figures S4C and S4D). A small number of genes are upregulated with THZ1 and brachyury degradation. Examining the gene signatures from each treatment reveals no overlap (Tables S1 and S2). We conclude that brachyury is a highly selective regulator of gene expression and that its degradation downregulates key chordoma identity targets.
THZ1 and brachyury degradation converge on disrupting the T transcriptional condensate
From our transcriptional profiling, we conclude that small-molecule transcriptional CDK inhibition selectively downregulates endogenous T expression but also has global effects on nascent transcription. Brachyury degradation alone is highly specific and primarily affects the chordoma SE-associated gene expression program. Considering the mechanism by which pharmacological transcriptional CDK inhibition targets brachyury, we hypothesize that these agents are capable of disrupting transcriptional condensates. Given the importance of brachyury autoregulation, THZ1-mediated inhibition of this cycle via disruption of the transcriptional condensate is likely to have negative feedback on T expression.
In CH22, THA-dTAG-EGFP/+, 24-h THZ1 treatment results in lower levels of brachyury protein and partial depletion of brachyury condensates in the nucleus (Figure 5A; Figure S5A). At this same time point, brachyury degradation using the dTAG/degron system is nearly complete, and few remaining brachyury condensates are observed. Neither degron nor THZ1 alter the levels of BRD4 protein (Figure S5B), but THZ1 treatment increases the total number of BRD4 condensates per nucleus, whereas brachyury degradation has no significant effect (Figure S5C). Specifically inspecting the T locus, we find that THZ1 and brachyury degradation decrease co-localization of BRD4 condensates at T (Figures 5C–5F; Figures S5D and S5E). This evidence, showing that brachyury degradation alone is capable of disrupting its transcriptional condensate, further highlights the importance of brachyury autoregulation in chordoma. Examining a second SE-associated, non-brachyury-regulated locus, MCL1, we see that THZ1 can disrupt the MCL1 transcriptional condensate, whereas brachyury degradation has no significant effect (Figures S5F and S5G). These data confirm that, despite inducing highly divergent transcriptional responses, pharmacological transcriptional CDK inhibition and brachyury degradation disrupt the T transcriptional condensate.
Figure 5.

THZ1 and brachyury degradation converge on disrupting the T transcriptional condensate
(A) Left: brachyury IF in CH22, THA-dTAG-EGFP/+ with 500 nM THZ1 or DMSO. Right: quantification of brachyury condensates per nucleus. Error bars represent SD. ∗∗∗p < 0.001, derived from a two-tailed, unpaired t test (n = 1 biological replicate).
(B) Left: brachyury IF in CH22 HA-dTAG-T, T−/− cells with 1 μM degron or DMSO. Right: quantification of brachyury condensates per nucleus. Error bars represent SD. ∗∗∗p < 0.001, derived from a two-tailed, unpaired t test (n = 1 biological replicate).
(C) Colocalization between BRD4 and the T DNA locus by IF and DNA FISH, respectively, in fixed CH22, THA-dTAG-EGFP/+ with 500 nM THZ1 or DMSO (24 h). Separate images of the indicated IF and FISH are shown, along with an image showing the merged channels and the Spearman correlation coefficient for BRD4 protein and T colocalization.
(D) Quantification of the colocalization of T DNA FISH with BRD4 protein with DMSO or 500 nM THZ1 (24 h) in CH22, THA-dTAG-EGFP/+. Spearman correlation coefficients between the T DNA FISH signal with the BRD4 protein signal are plotted. Three biological replicates were treated and imaged in parallel. ∗∗p < 0.01, derived from a two-tailed, unpaired t test.
(E) Colocalization between BRD4 and the T DNA locus by IF and DNA-FISH, respectively, in fixed CH22 HA-dTAG-T, T−/− cells with 1 μM degron or DMSO (24 h). Separate images of the indicated IF and FISH are shown, along with an image showing the merged channels and the Spearman correlation coefficient BRD4 protein and T colocalization.
(F) Quantification of the colocalization of T DNA FISH with BRD4 protein with DMSO or 1 μM degron (24 h) in CH22 HA-dTAG-T, T−/−. Spearman correlation coefficients between the T DNA FISH signal with the BRD4 protein signal are plotted. Three biological replicates were treated and imaged in parallel. ∗∗∗∗p < 0.0001, derived from a two-tailed, unpaired t test.
Brachyury degradation induces senescence and sensitizes chordoma cells to anti-apoptotic inhibitors
Given the evident pleiotropic effects of transcriptional CDK inhibition, our data suggest direct brachyury inhibition as a targeted approach to disrupt chordoma identity. Like the CH22 HA-dTAG-T, T−/− engineered chordoma cell line, UM-Chor1 HA-dTAG-T, T−/− chordoma cells arrest upon brachyury degradation (Figure S6A). To assess long-term effects of brachyury degradation, we treated CH22 HA-dTAG-T, T−/− cells for 10 days with degron followed by washout. We find that brachyury degradation halts chordoma cell proliferation and that this is not rescued upon degron washout and re-expression of brachyury (Figures 6A and 6B). These data suggest that, upon brachyury loss, cells undergo an irreversible change in cell state, and indeed we observe a strong increase in cellular senescence, as measured by senescence-associated β-galactosidase (Figure 6C).
Figure 6.

Brachyury degradation induces senescence and sensitizes chordoma cells to anti-apoptotic inhibitors
(A) Fold change (relative to t = 0) of cell growth with DMSO or 1 μM degron in CH22 HA-dTAG-T, T−/− cells. At t = 10 days, degron- and DMSO-treated media were replaced with normal medium. Error bars denote SD (n = 3 biological replicates).
(B) Immunoblot of brachyury levels in CH22 HA-dTAG-T, T−/− cells with DMSO (t = 0), 1 μM degron (t = 0), DMSO washout (t = 18), and degron washout (t = 18) (n = 1).
(C) Percentage of SA-β-galactosidase (β-gal)-positive CH22 HA-dTAG-T, T−/− cells treated with DMSO or degron for 6 days. ∗∗p < 0.00, derived from a two-tailed, unpaired t test (n = 3 biological replicates).
(D) Gene set enrichment analysis (GSEA) in CH22 HA-dTAG-T, T−/− cells with 1 μM degron or CH22 parental cells with 60 nM THZ1 (n = 3 biological replicates each). Gene sets are from the Molecular Signatures Database.
(E) Immunoblots validating MCL1 levels and cleaved PARP levels in CH22 HA-dTAG-T, T−/− cells with THZ1 or degron (n = 2 biological replicates, one experiment shown).
(F) Gene tracks of H3K27ac and HA-dTAG-brachyury (units of reads per million per base pair) at the MCL1 locus in CH22 parental and HA-dTAG-T, T−/− cells (n = 4 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively).
(G) Cell viability of CH22 HA-dTAG-T, T−/− cells with maritoclax, degron, or maritoclax + degron for 6 days. Data are plotted as the mean fraction of cell viability relative to DMSO-treated cells (n = 5 biological replicates). Error bars denote SD. ∗p < 0.05, derived from a two-tailed, unpaired t test.
(H) Caspase-3/7 levels in CH22 HA-dTAG-T, T−/− cells with maritoclax, degron, or maritoclax + degron for 3 days. Caspase-3/7 levels and cell viability were measured in parallel. Data are plotted as the normalized mean caspase-3/7 levels relative to DMSO-treated cells, normalized again to cell viability for each treatment. Error bars denote SD (n = 5 biological replicates). ∗p < 0.05, derived from a two-tailed, unpaired t test.
(I) Gene tracks of H3K27ac and HA-dTAG-brachyury (units of reads per million per base pair) at the BCL-xL locus in CH22 parental and HA-dTAG-T, T−/− cells (n = 4 biological replicates for H3K27ac and HA-dTAG-brachyury, respectively).
(J) Cell viability of CH22 HA-dTAG-T, T−/− cells with navitoclax, degron, or navitoclax + degron for 6 days. Data are plotted as the mean fraction of cell viability relative to DMSO-treated cells. (n = 5 biological replicates). Error bars denote SD. ∗∗p < 0.01, derived from a two-tailed, unpaired t test.
(K) Caspase-3/7 levels in CH22 HA-dTAG-T, T−/− cells with navitoclax, degron, or navitoclax + degron for 3 days. Caspase-3/7 levels and cell viability were measured in parallel. Data are plotted as the normalized mean caspase-3/7 level relative to DMSO-treated cells, normalized again to the cell viability for each treatment (n = 5 biological replicates). Error bars denote SD. ∗∗p < 0.01, derived from a two-tailed, unpaired t test.
The irreversible consequences of brachyury degradation outlined above validate direct brachyury targeting as an attractive therapeutic strategy, especially when considering its limited tissue expression. However, the failure to kill cells through brachyury depletion (Figure S6B) raises the possibility of adaptive resistance and contrasts pharmacologic transcriptional CDK inhibition (Figure S3C). Gene expression profiling of chordoma cells following THZ1 treatment (24 h) versus long-term brachyury degradation (6 days) (Figure S6C) reveals two distinct gene expression signatures: apoptosis with THZ1 treatment and cellular arrest with brachyury degradation (Figure 6D). These findings were confirmed with downregulation of MCL1, an inhibitor of apoptosis, and cleavage of poly(ADP-ribose) polymerase (PARP), a target of caspase-1, following THZ1 treatment but not with brachyury degradation (Figure 6E). After 6 days of brachyury degradation, we also find that the top brachyury non-SE-associated genes are upregulated relative to those that are SE associated (Figure S6D). We hypothesize that this finding reflects an added role of brachyury as a transcriptional repressor, which has been validated in previous reporter assays.53 These data suggest that brachyury degradation leads to irreversible reprogramming of gene expression to adopt a senescent cell state in chordoma.
We then aimed to identify targets in combination with brachyury degradation that could achieve chordoma cell killing. Anti-apoptotic regulators feature prominently among highly transcribed short half-life genes that are downregulated by transcriptional CDK inhibition (Figure 4).16,60,61 In chordoma, MCL1 and BCL2L1 (BCL-xL) are highly transcribed and strongly downregulated by THZ1. MCL1 and BCL-xL, members of the BCL-2 family of anti-apoptotic proteins, are often overexpressed in drug-resistant cancers.62 Neither MCL1 nor BCL-xL are downregulated by brachyury degradation, which is consistent with the absence of brachyury binding at their loci (Figures 6F and 6I). There are promising selective inhibitors of MCL1 and BCL-xL.63,64
We demonstrate that brachyury degradation sensitizes chordoma cells to anti-apoptotic protein inhibitors, maritoclax (inhibiting MCL1) and navitoclax (inhibiting BCL-xL) (Figure S6E), as measured by caspase-3/7 levels (Figures 6G, 6H, 6J, and 6K). These combined treatments also decrease chordoma cell viability. As a negative control, we used venetoclax, an anti-apoptotic inhibitor of BCL2 that is not expressed in chordoma (Figure S6F). Combination of navitoclax or maritoclax with brachyury degradation does not increase cellular senescence (Figure S6G). From these data, we conclude that brachyury degradation has irreversible effects on chordoma cell growth, induces senescence alone, but can also sensitize cells to apoptosis in combination with inhibitors of MCL1 and BCL-xL.
Discussion
In this study, we show that brachyury defines the chordoma SE landscape and that a 3′ SE binding site at the brachyury (T) locus is required for its autoregulation. High-resolution microscopy in chordoma cells reveals that the T SE forms an autoregulatory transcriptional condensate that is disrupted by pharmacological transcriptional CDK inhibition and brachyury degradation. CDK inhibition induces apoptosis, and brachyury degradation promotes senescence and sensitizes chordoma cells to anti-apoptotic inhibitors. We explain this difference by showing that degradation results in selective transcriptional reprogramming. In contrast, transcriptional CDK inhibition globally downregulates brachyury and short-half-life genes. Given these findings, we posit that the autoregulatory capacity of driver master TFs such as brachyury may underlie the unique sensitivity to transcriptional inhibition.
Positive autoregulatory feedback loops are attractive therapeutic targets because they provide a clear mechanistic explanation for how general perturbation can achieve a selective effect and a therapeutic index for cancer cells.65 Whether this mechanism extends to other tumors remains to be studied. Chordoma, a rare and understudied cancer, proved to be an ideal model system, given that brachyury is the single oncogenic master TF.
Although pharmacologic transcriptional CDK inhibition is one route to target brachyury autoregulation, the global effects must be properly considered as sources of on-target toxicity. Because transcriptional CDKs are core components of transcription, their knockout is lethal in most cell types.66,67 Pharmacological dosing of these inhibitors should be designed with the understanding that the therapeutic index likely arises from their ability to disrupt transcriptional condensates and oncogenic TF autoregulation, specifically at lower concentrations than those that globally downregulate transcription. Because a compound like THZ1 inhibits multiple CDKs, including those involved in transcription and the cell cycle, there will be many non-brachyury-selective effects even when there is a preference for downregulating SE-associated genes. Subsequently, there are differing phenotypes associated with THZ1 versus brachyury degradation.
Our also work provides proof of concept that direct pharmacological targeting of brachyury irreversibly halts tumor cell growth and sensitizes chordoma cells to anti-apoptotic inhibitors. Because brachyury is a highly selective transcriptional regulator and minimally expressed in human adult tissue, its targeting is likely to have a better therapeutic index than transcriptional CDK inhibition. Furthermore, an emerging interest in applying senolytics,68 which selectively kill senescent cells, to cancer is highly applicable to chordoma. If brachyury inhibition could be combined with a senolytic agent, then there is potential to kill reprogrammed senescent cells that pose a threat of adaptive resistance. Although TFs have been historically considered undruggable, emerging technologies for targeted degradation are challenging this label. Given the centrality of brachyury in chordoma and the lack of an effective targeted therapy, our work further supports pharmacological transcriptional CDK inhibition as an immediately actionable therapeutic strategy but also motivates future efforts to drug brachyury directly.
Limitations of study
First, we did not evaluate the presence of the brachyury transcriptional condensate in vivo and its role in promoting chordoma tumorigenesis. It will also be important to examine brachyury-degraded cells at a longer time point to determine whether the cells maintain a senescent state after re-expression. Finally, we aim to validate the combination of anti-apoptotic inhibitors with brachyury degradation in vivo to understand their therapeutic potential.
STAR★methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Acetyl-Histone H3 (Lys27) Rabbit mAb D5E4 | Cell Signaling | Cat # 8173; RRID:AB_10949503 |
| HA-Tag Rabbit mAb C29F4 | Cell Signaling | Cat # 3724; RRID:AB_1549585 |
| Anti-BRD4 Rabbit mAb 128874 | abcam | Cat # EPR5150(2); RRID:AB_2868517 |
| Brachyury Rabbit mAb D2Z3J | Cell Signaling | Cat # 81694; RRID:AB_2799983 |
| Anti-GFP Mouse mAb 1218 | abcam | Cat # 9F9.F9; RRID:AB_298911 |
| c-Myc Rabbit Antibody | Cell Signaling | Cat # 9402; RRID:AB_2151827 |
| PARP Rabbit Antibody | Cell Signaling | Cat # 9542; RRID:AB_2160739 |
| Mcl-1 Rabbit mAb D2W9E | Cell Signaling | Cat # 94296; RRID:AB_2722740 |
| GAPDH Mouse mAb D4C6R | Cell Signaling | Cat # 97166; RRID:AB_2756824 |
| Chemicals, peptides, and recombinant proteins | ||
| X-Gal | Sigma | Cat # 3117073001 |
| 4-Thiouridine | Sigma | Cat # T4509 |
| Iodoacetamide | Sigma | Cat # I1149 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat # G7570 |
| Caspase-Glo® 3/7 Assay System | Promega | Cat # G8090 |
| NEBNext® Ultra II Directional RNA Library Prep Kit for Illumina® | NEB | Cat # E7760 |
| NEBNext® Library Quant Kit for Illumina® | NEB | Cat # E7630 |
| Magnetic mRNA Isolation Kit | NEB | Cat # S1550 |
| QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina | Lexogen | Cat # 015.24 |
| Fast SYBR Green Master Mix | ThermoFisher | Cat # 4385610 |
| Deposited data | ||
| RNA-seq | This paper | GSE153972 |
| UM-Chor1 Parental H3K27ac ChIP-seq | Sharifnia et al.10 | GSE109794 |
| ChIP-seq | This paper | GSE153971 |
| Experimental models: cell lines | ||
| UM-Chor1 | ATCC | Cat # CRL-3270 |
| CH22 | Chordoma Foundation | N/A |
| 293T | ATCC | Cat # CRL-3216 |
| Oligonucleotides | ||
| HDR Templates (Table S3) | Twist Bioscience | N/A |
| Custom synthetic sgRNA (sgRNA_T_n_term): TTTCCCGCGCTCTCGGTGCC | Synthego | N/A |
| Custom oligonucleotides (Table S6) | IDT | N/A |
| Recombinant DNA | ||
| Phage-PGK-Flag-HA-FKBP-T(G177D)-DEST | This paper | N/A |
| pLV hUbC-dCas9-KRAB-T2a-Puro | Addgene | Cat # 71236 |
| Lenti-Cas9-Blast | Addgene | Cat # 52962 |
| pLKO5.sgRAN.EGS.tRFP | Addgene | Cat # 57823 |
| Phage-PGK-Flag-HA-FKBP-T(WT)-DEST | This paper | N/A |
| Software and algorithms | ||
| Graphpad Prism 8.0 | Graphpad Software | https://www.graphpad.com:443/ |
| Fiji image processing package | Schindelin et al.69 | https://fiji.sc/ |
| Genialis Data Analysis Software | Genialis | https://www.genialis.com |
| ROSE2 | Brown et al.70 | https://bitbucket.org/young_computation/rose |
| mSigDB | Liberzon et al.71; Subramanian et al.72 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| Core regulatory circuitry mapper | Lin et al.36 | https://github.com/linlabcode/CRC |
| SlamDunk | Neumann et al., 201973 | https://t-neumann.github.io/slamdunk/index.html |
| HiSat2 | Kim et al., 201574 | https://ccb.jhu.edu/software/hisat2/index.shtml |
| Cufflinks | Trapnell et al., 201275 | http://cole-trapnell-lab.github.io/cufflinks/ |
| BWA ALN | Li and Durbin, 200976 | http://bio-bwa.sourceforge.net/ |
| MACS2 | Zhang et al., 200877 | https://github.com/macs3-project/MACS |
| Other | ||
| T RNA FISH probe | Stellaris | N/A |
| GAPDH RNA FISH Probe | Stellaris | N/A |
| T DNA FISH Probe | Empire Genomics | RP11-5E5 |
| MCL1 DNA FISH Probe | Empire Genomics | RP11-663F24 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles Y. Lin (charles.y.lin@bcm.edu).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact author. This includes plasmids and engineered cell lines generated in this study.
Data and code availability
Sequencing datasets generated in this study are summarized in Table S5. All raw data are available on NCBI GEO. The superseries accession number for new data reported in this paper is GSE153973. Specifically, RNA-seq data is available under accession number GSE153972 and ChIP-seq data under accession number GSE153971. All custom scripts associated with this study are deposited on github.com at: https://github.com/linlabcode/chordoma_code/.
Experimental model and subject details
Cell lines
UM-Chor1 and CH22 chordoma cell lines were obtained from the Chordoma Foundation. HEK293T cells were provided by the laboratory of Dr. Daisuke Nakada at Baylor College of Medicine. Cells were tested and were negative for mycoplasma and STR profiling for cell line authentication was performed at MD Anderson Cancer Center.
Cell culture conditions
UM-Chor1 chordoma cells were maintained in IMDM/RPMI (4:1) + 10% fetal bovine serum (FBS). CH22 chordoma cells were maintained in RPMI media + 10% FBS. All cell lines were maintained on tissue-culture treated plates.
Method details
Lentiviral vectors and cell line engineering
Lenti-Cas9-Blast (Addgene #52962) was used for Cas9 expression. For T sgRNA expression, the spacer sequence for T-targeting (sg_T_knockout: CCCTGAGACCCAGTTCATAG) was cloned into the Bsmb1-digested pLKO5.sgRAN.EGS.tRFP lentiviral backbone (Addgene #57823). For the dTAG-T lentiviral expression vector, the brachyury ORF was generously provided as a gift from Dr. Claudia Palena. Using the Gateway cloning strategy, this ORF was bp cloned into pDONR_223 and subjected to site directional mutagenesis to introduce the G177D variant and a silent mutation in the PAM site of sg_T (amino acid 194 - C to T at position 3 of alanine). This mutagenized T_pDONR_223 was then LR cloned into the destination vector Phage-PGK-Flag-HA-FKBP-DEST, a generous gift from Dr. Kristen Karlin at Baylor College of Medicine. Lentivirus was produced by transfection of 293T packaging cells with four plasmids (HDM-tat, HDM-VSV, HDM-Hypm2(Gag), and PRC-CMV-RaII(Pol)) and Mirus Bio TransIT-LT1 Transfection Reagent (Mirus 2304). Virus-containing supernatant containing Lenti-Cas9-Blast, sg_T, or Phage-PGK-Flag-HA-FKBP-T-DEST was collected 48- and 72-hours post transfection and concentrated 100-fold with Lenti-X Concentrator (Clontech 631232). UM-Chor1 and CH22 chordoma lines were transduced with Phage-PGK-Flag-HA-FKBP-T(WT or G177D)-DEST virus alone (to generate HA-dTAG-T, T+/+ cells) or in combination with Lenti-Cas9-Blast and sg_T virus (to generate HA-dTAG-T, T−/− cells) with 8μg/mL polybrene (Sigma TR-1003). The polyclonal population of HA-dTAG-T, T+/+ cells was used for subsequent experiments while clonal populations of HA-dTAG-T, T−/− were used unless specifically noted.
ChIPmentation
ChIPmentation was carried out as previously described,78 with minor adaptions. Cells were washed once with PBS and fixed with 11% formaldehyde fixation solution (1M HEPES-KOH, pH 7.5, 5M NaCl, 0.5M EDTA, pH 8.0, 0.5M EGTA, pH 8.0, and 37% formaldehyde and H20) in 10 mL PBS for 10 min at room temperature. Glycine was added at a final concentration of 2.5M to stop the reaction. Cells were collected at 500 g for 10 min at 4C (subsequent work was performed on ice and used cool buffers and solutions unless otherwise specified) and washed twice with up to 0.5mL ice-cold PBS supplemented with 1X Halt Protease Inhibitor Cocktail Solution (ThermoFisher 78446). The pellet was lysed in sonication buffer (10mM Tris-HCl pH 8.0, 2mM EDTA pH 8.0, 0.25% SDS, 1X Halt Protease Inhibitor Cocktail Solution) and sonicated with a Covaris LE220 Ultrasonicator for 8 minutes (CH22 parental and engineered chordoma cells) or 4 minutes (UM-Chor1 parental and engineered chordoma cells) in a microtube until the size of fragments was in the range of 200–700 base pairs. Lysates were transferred to new tube and diluted 1:1.5 with equilibration buffer (10mM Tris, 233mM NaCl, 1.66% Triton X-100, 1.66% sodium deoxycholate, 1 mM EDTA, 1X Halt Protease Inhibitor Cocktail Solution). Lysates were centrifuged at full speed for 10 min at 4C and the supernatant containing the sonicated chromatin was transferred to a new tube. 10L of chromatin was removed for the whole cell extract (WCE). The antibody was added to the lysates and incubated on a rotator overnight at 4C. The following antibodies were used: H3K27ac (Cell signaling D5E4), and HA (Cell signaling C29F4) (1μg per one million cells per immunoprecipitation). Magnetic Protein A beads (ThermoFisher 10001D) (10μL per IP) were washed twice and blocked overnight in 1mL of PBS supplemented with 0.1% BSA. Blocked beads were then added to immunoprecipitated lysates and incubated for 2hrs on a rotator at 4C. Beads were washed subsequently with RIPA-LS (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0,140 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate and 1X Halt Protease Inhibitor Cocktail Solution) (twice), RIPA-HS (10mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate and 1X Halt Protease Inhibitor Cocktail Solution) (twice) and RIPA-LiCl (10mM Tris-HCl pH 8.0, 1mM EDTA pH 8.0, 250 mM LiCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% NP40 and 1X Halt Protease Inhibitor Cocktail Solution) (twice). Beads were washed with cold 10mM Tris-HCl pH 8.0, to remove detergent, salts and EDTA. Beads were washed once more with cold 10mM Tris-HCl pH 8.0 and the whole reaction including beads was transferred to a new tube and then placed on a magnet to remove supernatant to decrease background. Beads were then resuspended in 25μL of the tagmentation reaction mix (2X Tagment DNA buffer (Illumina) and water containing 1μL Tagment DNA Enzyme (Illumina)) and incubated at 37C for 7 min in a thermocycler. The beads were washed with RIPA-LS (twice) and with cold TE pH 8.0 (twice) before removing the supernatant. Beads were then incubated with 50mL elution buffer (0.5% SDS, 300mM NaCl, 5mM EDTA and 10mM Tris-HCl pH 8.0) containing 2μL of Proteinase K (ThermoFisher AM2548) for 1h at 55C and 10h at 65C, to revert formaldehyde crosslinking. 10μL of WCE was also de-crosslinked with elution buffer and Proteinase K in a 50μL reaction. Post de-crosslinking, supernatants were transferred to a new tube. Antibody-treated samples were de-crosslinked a second time for 1 hour at 55C in 19μL of ChIP elution buffer and 1μL Proteinase K and then purified with the Zymo DNA Clean and Concentrator (−5) kit (Zymo D4013) and eluted in 27μL of water. WCE DNA was purified with Zymo DNA Clean and Concentrator (−5) kit (Zymo D4013) and eluted in 12.5μL of water before preforming the tagmentation reaction described above and purifying the DNA again (eluting in 27μL of water). Enrichment of the libraries was performed in a 50μL reaction using 0.75mM primers, 25mL NEBNext High-Fidelity 2X PCR MasterMix (M0541) and 20μL of the purified library. Libraries were amplified for N+2 cycles, where N is equal to the rounded-up CT value determined in a test qPCR reaction with 4μL of the library. Enriched libraries were size selected and purified using SPRI AMPure XP beads (Beckman A63881) at a beads-to-sample ratio of 0.65:1 to recover libraries with a fragment length of 200–400 bp. ChIPmentation libraries were run on an Illumina Nextseq 500 instrument (single-end 75 bp reads).
CRISPRi of the T promoter and SEs
Protospacer sequences targeting either the T promoter, T SE autoregulatory site, and non-targeting control were individually cloned into plv hu6-sgRNA hUbC-dCas9-KRAB-T2a-Puro, a gift from Charles Gersbach (addgene #71236) (See Table S6 for sequences). Plasmids were sequence verified and co-transfected with lentiviral packaging plasmids into 293T cells to make virus as previously described above. Concentrated virus was used to infect CH22 chordoma cells as previously described above. One-week post infection, RNA was collected for cDNA and subsequent qPCR.
RT-qPCR
For all RT-qPCR, 250,000 cells were collected per sample. RNA extraction was performed using the RNA Clean and Concentrator-5 kit (Zymo R1017). Total RNA was reverse transcribed to cDNA using the SuperScript VILO cDNA Synthesis Kit (ThermoFisher 11756050). cDNA was used for qPCR using the Fast SYBR Green Master Mix (ThermoFisher 4385612) and uniquely designed gene expression probes (IDT), including a GAPDH reference gene, in three technical replicates per reaction. For Figure 2B, the Endogenous_T- Fwd/Rev, Max-Fwd/Rev, and GAPDH-Fwd/Rev probe sequences were used (Table S6). For Figure S2A, the Endogenous_T-Fwd/Rev, Exogenous_T- Fwd/Rev and GAPDH-Fwd/Rev probe seuqennces were used (Table S6).
CRISPR-mediated endogenous tagging
Templates for homology directed repair (HDR) were ordered as linear DNA fragment pools from Twist Biosciences (Table S3). HDR templates were designed with 400bp homology arms, with the left homology arm designed from one base pair upstream of the brachyury start and the right homology arm to include and continue downstream of the brachyury start codon. Templates were PCR amplified with KAPA HiFi HotStart ReadyMix and TWIST_F and TWIST_R primers. PCR products were purified using the Zymo DNA Clean and Concentrator-25 (Zymo D4005) kit following manufacturer’s instructions. Protospacer sequences for tagging the N terminus of brachyury were identified using the Zhang Lab sgRNA design tool (former). An sgRNA for endogenous tagging as ordered as a synthetic RNA from synthego (sgRNA_T_n_term). To obtain Cas9-sgRNA RNPs, 1μg of synthetic sgRNA was incubated with 1.5μg Cas9 (IDT 1081058) for 30 min at room temperature. 350,000 CH22 chordoma cells were electroporated in Buffer R (ThermoFisher) with 500ng of HDR template using the Neon Transfection System (ThermoFisher MPK1025). Electroporation conditions for CH22 cells were 1230 V, 10 ms, 4 pulses. Once the cells recovered, EGFP positive cells were sorted for using flow cytometry.
Live cell imaging of brachyury puncta
CH22, THA-dTAG-EGFP/+ chordoma cells were grown on glass coverslips coated with Poly-D-lysine solution (ThermoFisher A3890401). Before imaging, cell culture media was replaced with phenol red-free RPMI media + 10% FBS and imaged using the Airyscan detector on an LSM880 confocal microscope (Zeiss, Thornwood, NY). Cells were imaged on a heated stage (37°C) and supplemented with warmed (37°C), humidified air. Additionally, the microscope was enclosed in an incubation chamber heated to 37°C. ZEN black edition version 2.3 (Zeiss, Thornwood NY) was used for acquisition. Images were acquired with the Airyscan detector in super-resolution (SR) mode with a Plan-Apochromat 63x/1.4 oil objective. Raw Airyscan images were processed using ZEN 2.3 (Zeiss, Thornwood NY).
Fluorescence Recovery After Photobleaching (FRAP)
FRAP was performed on the LSM880 Airyscan microscope with 488nm laser (Harvard University). Bleaching was performed over a rbleach ≈1μm using 100% laser power. Images were collected every two seconds.
1,6-hexanediol treatment for live imaging
CH22, THA-dTAG-EGFP/+ chordoma cells were grown on glass plates in 1mL of complete RPMI media and cells were imaged every 2 s. After the second acquisition, 1mL of 3% 1,6-hexanediol was added on the plate for 10 s (2 s per acquisition).
Immunofluorescence
Immunofluorescence was performed as previously described with some modifications.27 70,000 cells were grown on coated glass and fixed in 4% paraformaldehyde, PFA, (ThermoFisher 28906) in PBS for 10min at RT. After three washes in PBS for 5min, cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min at RT and washed with one quick wash of PBS, followed by two 5min washes at RT. Cells were blocked with 4% IgG-free Bovine Serum Albumin (Jackson ImmunoResearch 001-000-161) (BSA) for 30mins at RT and incubated with primary antibodies (anti-BRD4 Abcam ab128874 1:500 dilution, anti-Brachyury Cell Signaling D2Z3J 1:1600 dilution, anti-GFP Abcam ab1218 1:3000 dilution) in 4% IgG-free BSA overnight at RT. After three 5min washes in PBS, secondary antibodies (Goat anti-Rabbit IgG Alexa Fluor 647 ThermoFisher A21244 1:500 dilution, Goat anti-Mouse IgG Alexa Fluor 488 Invitrogen A11029 1:500 dilution) were incubated for one hour in the dark. Cells were washed three times with PBS (5mins each) and Hoechst solution (ThermoFisher H3569) was used to stain nuclei for 5min. Glass coverslips were mounted onto slides with Vectashield (VWR, 101098-042). Coverslips were sealed with transparent nail polish (Electron Microscopy Science Nm, 72180) and stored at 4°C. Images were taken using a DeltaVision Live Deconvolution Microscope with an Olympus PlanApo 100x/1.4 objective and a 1.9k x 1.9x sCMOS camera. Images were acquired with 0.2μm z-steps, deconvolved using quantitative restorative algorithms in SoftWorx 7.0 and post-processed using Fiji Is Just ImageJ (Fiji).69
RNA FISH combined with immunofluorescence
Immunofluorescence was performed in an RNase-free environment as previously described and RNA FISH performed as previously described with some modifcations27. Cells were blocked with 4% IgG-free BSA for 10 minutes at RT and primary antibodies were diluted in RNase-free PBS. After immunofluorescence, cells were fixed with 4% PFA in RNase-free PBS for 10 min at RT. Prior to probe hybridization, cells were washed once with 20% 6 Stellaris RNA FISH Wash Buffer A (Biosearch Technologies, Inc., SMF-WA1-60) and 10% Deionized Formamide (EMD Millipore, S4117) in RNase-free water for 5 min at RT. Cells were hybridized with 90% Stellaris RNA FISH Hybridization Buffer (Biosearch Technologies, SMF-HB1-10), 10% Deionized Formamide, and 4μL of 12.5μM Stellaris RNA FISH probes designed to hybridize introns of the transcripts T or GAPDH, respectively. Hybridization was performed overnight at 37°C. The next morning, coverslips were washed with 20% 6 Stellaris RNA FISH Wash Buffer A (Biosearch Technologies, Inc., SMF-WA1-60) and 10% Deionized Formamide (EMD Millipore, S4117) in RNase-free water for 5 min at RT 30 min at 37°C and nuclei were stained with Hoechst (ThermoFisher H3569) in 20% Stellaris RNA FISH Wash Buffer A (Biosearch Technologies, Inc., SMF-WA1-60) and 10% Deionized Formamide (EMD Millipore, S4117) in RNase-free water for 5 min at RT. Cells were then washed with Stellaris RNA FISH Wash Buffer B (Biosearch Technologies, SMF-WB1-20) at RT for five minutes. Glass coverslips were mounted onto slides with Vectashield (VWR, 101098-042). Coverslips were sealed with transparent nail polish (Electron Microscopy Science Nm, 72180). Images were taken using a DeltaVision Live Deconvolution Microscope with an Olympus PlanApo 100x/1.4 objective and a 1.9k x 1.9x sCMOS camera. Images were acquired with 0.2μm z-steps, deconvolved using quantitative restorative algorithms in SoftWorx 7.0 and post-processed using Fiji Is Just ImageJ (Fiji).
Molecule synthesis
Degron (dTAG-47) was synthesized as previously described.39
Immunoblotting
For each sample, 200,000 cells were lysed in RIPA Lysis and Extraction Buffer (ThermoFisher 89901) with protease and phosphatase inhibitors (ThermoFisher 78440). Lysates were incubated on ice for 30 minutes and then centrifuged for 20 minutes at 1600 g and 4C. The protein concentrations of the resulting supernatants were quantified using a BCA protein assay kit (Pierce, 23228), and then denatured for 10 minutes at 99C. Protein samples were run on NuPAGE 4%–12% Bis-Tris Protein gels. Protein was then transferred to iBlot Transfer Stack PVDF membranes (ThermoFisher 1B401001). Membranes were blocked in Odyssey blocking buffer (LI-COR) for one hour at room temperature. Membranes were then probed with the indicated primary antibody (Brachyury: Cell Signaling D2Z3J, HA: Cell signaling #3724, MYC: Cell Signaling #9402, BRD4: Abcam #128874, PARP: Cell Signaling #9542, MCL1 Cell Signaling #94296 and GAPDH: Cell Signaling #97166) (each diluted 1:1000) in Odyssey Blocking Buffer at 4°C overnight. Membranes were then washed for 5 minutes with TBST, three times. Membranes were probed with secondary antibodies (LI-COR #926-32213 and #926-68072) (each diluted 1:10,000) for 1 hour at room temperature and washed TBST for 5 minutes, three times. Membranes were imaged, and if applicable also quantified, with the Odyssey Imaging System (LI-COR Biosciences).
Live cell imaging of CH22, THA-dTAG-EGFP/+
CH22, THA-dTAG-EGFP/+ chordoma cells were seeded overnight in in 6-well tissue culture-treated plates at a density of 100,000 cells per well. The following day, media was replaced with phenol-red-free growth media containing either DMSO, THZ1 or degron at the final indicated concentration. Each well was imaged in 9 fields every five minutes for a total of 23 hours using the IncuCyte S3 Live-Cell Analysis system.
Caspase-3/7 apoptotic assay
The day before addition of compound, 1000 cells were seeded overnight in Pierce 96-well Polystyrene Plates (white, opaque) (ThermoFisher 15402). The following day, as pertaining to the experiment, the compound of interest or DMSO was added to wells. Cells were incubated at 37°C, and caspase-3/7 activity was measured 3- or 6-days post addition of compound using the Caspase-Glo 3/7 reagent (Promega G89090). In parallel, cell number was measured with the same DMSO or compound treatment using the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7570). To calculate caspase-3/7 activity, luminescence values from compound-treated cells were double normalized to both cell number and DMSO controls.
RNA-seq
Traditional RNA-sequencing experiments with either THZ1 or degron treatment in UM-Chor1 chordoma cells (Figure 2) and CH22 parental or HA-dTAG-T, T−/− chordoma cells (Figures 4, S4, and 6) was performed by seeding 350,000 cells per well in 6 well tissue culture-treated plates the night before addition of compound. The following day, media was replaced with growth media containing compound at the indicated final concentration (see respective figures). Compound-treated UM-Chor1 chordoma cells were collected at 0, 4, 8, 12, and 24 hours after compound treatment. CH22 parental or HA-dTAG-T, T−/− chordoma cells were collected at 0, 4, 8, 24 hours or 6 days after compound treatment. RNA extraction was performed using the RNA Clean and Concentrator-5 kit (Zymo R1017). ERCC RNA Spike-In Control Mixes (ThermoFisher 4456740) were added to each sample to allow for cell count normalization of gene expression. Sequencing libraries were prepared using the NEB Ultra II Directional Library Prep Kit for Illumina (NEB E7760L) and libraries were sequenced using the Illumina Nextseq 500 instrument with single end, 75bp reads.
SLAM-seq
SLAM-sequencing protocols were followed as previously described.58 In brief, either parental CH22 and HA-dTAG-T, T−/− CH22 chordoma cells were seeded the night before addition of compound in 6-cm tissue culture-treated dishes at 750,000 cells per dish. The following morning, media was replaced to contain DMSO or THZ1 (60nM) or degron at (1μM). One hour later, media was replaced with DMSO or compound containing media and s4U (Sigma T4509) to a final concentration of 750 μM. Media containing compound and s4U were exchanged every 3 hours for the duration of the pulse, before collecting the cells at 8 hours post initial addition of compound. Cells were harvested by total RNA extraction using TRIzol (with 0.1mM DTT during isopropanol precipitation). RNA was resuspended in 1mM DTT and ERCC RNA Spike-In Control Mixes (ThermoFisher 4456740) were added to each sample to allow for cell count normalization of gene expression. 5μg of total RNA was treated with 10mM iodoacetamide (Sigma I1149) under denaturing conditions, ethanol precipitated and purified, and subjected to Quant-seq 3′ end mRNA library preparation for Illumina (Lexogen 015). Libraries were sequenced using the Illumina Nextseq 500 instrument with single-end, 75bp reads.
DNA FISH combined with immunofluorescence
Immunofluorescence was performed as previously described and DNA FISH performed as previously described with some modifcations.27 After incubating the cells with the secondary antibodies, cells were washed three times in PBS for 5min at RT, fixed with 4% PFA in PBS for 10min and washed three times in PBS. Cells were incubated in 70% ethanol, 85% ethanol and then 100% ethanol for 1min at RT, each. Probe hybridization mixture was made mixing 7μl of FISH Hybridization Buffer (Agilent G9400A), 1μl of FISH probes (T: Empire Genomics, RP11-5E5 BAC clone CHR6: 166,463,032-166,602,569 on build hg19, MCL1: Empire Genomics RP11-663F24 BAC clone Chr1: 150,493,599-150,684,687) and 2μl of water. 5μl of mixture was added on a slide and coverslips were placed on top, face down, and sealed using rubber cement. Genomic DNA and probes were denatured at 80°C for 5 min and slides were incubated at 37C in the dark O/N. The following morning, the coverslip was removed from the slide and incubated in pre-warmed Wash buffer 1 (Agilent, G9401A) at 73°C for 2 min and in Wash Buffer 2 (Agilent, G9402A) for 1 min at RT. Slides were air-dried and stained with Hoechst (ThermoFisher H3569) for 5 min at RT. Coverslips were washed three times in PBS (each wash 5 minutes), and mounted onto slides with Vectashield (VWR, 101098-042). Coverslips were sealed with transparent nail polish (Electron Microscopy Science Nm, 72180). Images were taken using a DeltaVision Live Deconvolution Microscope with an Olympus PlanApo 100x/1.4 objective and a 1.9k x 1.9x sCMOS camera. Images were acquired with 0.2μm z-steps, deconvolved using quantitative restorative algorithms in SoftWorx 7.0 and post-processed using Fiji Is Just ImageJ (Fiji).
Senescence staining
CH22 HA-dTAG-T, T−/− polyclonal chordoma cells were seeded the night before the addition of compound in tissue culture-treated 12-well dishes at 30,000 cells per well. The following day, media was replaced with media containing DMSO or degron, maritoclax, navitoclax, maritoclax+degron, or navitoclax + degron. Cells were incubated for 6 days at 37°C with media changes every 2 days. On day 6, the media was removed, cells were washed with PBS, and fixed in 0.5% glutaraldehyde solution (in PBS pH7.4) for 15 minutes at room temperature. Following three washes with PBS (each five minutes at room temperature), the cells were washed twice for 5 minutes with PBS/MgCl2 pH 6.0 at room temperature. X-Gal staining was prepared (0.2M K3Fe(CN)6, 0.2M K4Fe(CN)2•3H2O, X-Gal stock (40X, Sigma 3117073001), in PBS/MgCl2 pH 6.0) and added to all wells. The cells were incubated at 37°C overnight. The following morning, the cells were washed with PBS for five minutes at room temperature, three times. Pictures were acquired on the Cyation 5 Cell Imaging Multi-Mode Reader. The number of SA-β-Gal positive cells were divided by the total number of cells to generate the percentage of SA-β-Gal positive cells with either DMSO or degron treatment.
Compound viability assays
The day before addition of compound, 1000 cells were seeded overnight in Pierce 96-well Polystyrene Plates (white, opaque) (ThermoFisher 15402). The following day, as pertaining to the experiment, the compound or DMSO was added to wells (50μM navitoclax, 50μM maritoclax, 10μM venetoclax, or each compound in combination with 1μM degron). Each compound was serially diluted by a factor of 1:3 to test a total of nine concentrations in triplicate. Cells were incubated at 37°C for 3 days and cell viability was assayed using CellTiter-Glo (Promega G7570). Dose response curves and EC50 calculations were generated using GraphPad Prism (v.6).
Quantification and statistical analysis
Analysis of RT-qPCR data
RT-qPCR data were analyzed using the CT method. The CT values were calculated by subtraction of the average reference-gene (GAPDH) CT values from average target-gene CT values (three replicates each). CT values were calculated by subtracting CT values for the “reference” samples (non-targeting sgRNA-infected [Figure 2B] or DMSO-treated [Figure S2A]) from the CT of the “treated” samples (targeting sgRNA-infected [Figure 2B] or compound-treated [Figure S2A]). The change in mRNA levels was calculated as 2ˆ-CT.
FRAP Analysis
Fluorescence intensity was measured using Fiji.69 Values are reported relative to pre-bleaching time points.
Quantifying brachyury puncta
The number of puncta from immunofluorescent experiments were calculated with the “Objects Counter3D” Plugin in FIJI. For each image analyzed, the “threshold” parameter was manually set such that the individual puncta could be considered as separate objects.
Colocalization analysis (RNA/DNA FISH)
RNA/DNA FISH foci were visually identified. Spearman correlation coefficients were calculated as a quantitative metric to determine colocalization between protein and RNA/DNA FISH foci. Spearman correlation coefficients were calculated between DNA/RNA foci and IF signal using Fiji’s Coloc2 plugin through the z-tack in a region of interest encompassing the RNA or DNA foci.
Analysis of CH22, THA-dTAG-EGFP/+ live cell imaging data
Whole field analysis
To measure change in HA-dTAG-EGFP-brachyury expression over time, the integral EGFP pixel intensity per well was measured at each time point. Background signal was first reduced by top hat filtering and EGFP positive signal was then determined by global thresholding. Size filtering was then applied to keep cell-sized objects, and the resulting mask was used to measure the integral EFP intensity on the original image.
Individual Cell tracking
For cell tracking, brightfield and EGFP images were first registered using the first frame as a reference. Nuclear segmentation was done on the EGFP images by global thresholding after background removal and denoising, and the mean EGFP intensity was recorded for each object. For track detection, two consecutive nuclear masks were superimposed, and overlapping objects between frames were assigned to the same single track. Track errors, discontinuities or ambiguities were manually corrected after visual inspection. Image processing and analysis pipelines were developed in MATLAB R2016a.
Quantifying BRD4 puncta
Images (16bit TIFF grayscale) were analyzed using CellProfiler (cellprofiler.org). Briefly, nuclei were segmented using the DAPI channel as a reference, adjacent nuclei were separated by shape and intensity and nuclei touching the borders of the image were discarded. The BRD4 channel was first background corrected, then we applied the “EnhanceOrSuppressFeatures” step to enhance speckles with a radius of 5 pixels. Then we segmented the speckles (“puncta”) setting a rage of size from 3 to 50 pixels. Finally, every speckle was related to a nucleus and feature extracted from each speckle.
Genomic coordinates and gene annotation
All coordinates and gene annotations in this study were based on human reference genome assembly HG19, GRCh37 (https://www.ncbi.nlm.nih.gov/assembly/2758/) and RefSeq genes. The genome and transcriptome gene gtf were obtained from (http://ftp.illumina.com/Homo_sapiens/UCSC/hg19/Homo_sapiens_UCSC_hg19.tar.gz). The HG19 NCBI RefSeq gene table was downloaded directly from the UCSC genome browser from their table browser utility (http://genome.ucsc.edu/cgi-bin/hgTables). The HG19 ENCODE blacklist was used to filter genomic regions and was obtained from (https://sites.google.com/site/anshulkundaje/projects/blacklists).
ChIPmentation processing and quality control
ChIPmentation datasets were aligned to the human HG19 genome using BWA ALN. MACS2 (Model-based Analysis of ChIP-seq) was used to identify enriched regions relative to background. A P value threshold of enrichment of 1e-5 was used for all datasets. All processed ChIPmentation datasets adhered to the ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia.79 This included at least 20 million mapped sequencing reads per sample. Each sequenced sample had an NSC (normalized strand coefficient) > 1.05 and RSC (relative strand correlation) > 0.8.
Calculating ChIPmentation density
We calculated the normalized read density of ChIPmentation datasets in any genomic region using the Bamliquidator (version 1.0) (https://github.com/BradnerLab/pipeline/wiki/bamliquidator). Briefly, reads aligning to the region were extended to 200bp and the density of reads per base pair (bp) was calculated. The density of reads in each region was normalized to the total number of million mapped reads producing read density in units of reads per million mapped reads per bp (rpm/bp). The area under curve (AUC) of read occupancy of a given region was reported simply as the number of reads divided by the total number of million mapped reads producing an AUC measurement in total reads per million (total rpm).
Defining ChIPmentation binding sites
UM-Chor1 and CH22 ChIPmentation peaks were called using MACS280 using a p value threshold of 1e-5 and all other parameters set to default. For brachyury pulldowns using HA, we filtered out known background HA-peaks using peaks called from HA pulldowns in human cells not expressing HA-tagged proteins (datasets from GSM4025790 and GSM4025791 on https://www.ncbi.nlm.nih.gov/geo/) as well as CH22 and UM-Chor1 parental cells that lack an HA tag.
Comparing brachyury binding landscapes
ChIPmentation peaks from HA-dTAG-T(WT), T−/− and HA-dTAG-T(G177D), T−/− chordoma cells were compared to assess similarities of binding landscapes. For each dataset we generated pairwise scatterplots of AUC and determined spearman correlations (Figure S1E).
Mapping enhancers and SEs in chordoma cells
Per the original definition of SEs, a small number of cis-regulatory enhancers in the genome display disproportionate or “asymmetric” occupancy for a factor. This asymmetry is defined graphically by the tangent between the ranked occupancy plot at a line with the same slope as the diagonal. This definition robustly identifies a small number of regions that typically have an order of magnitude higher occupancy than the median or typical region. Enhancers and SEs were mapped for H3K27ac datasets in either CH22 (n = 4), UM-Chor1 (n = 3), or the combination of CH22 and UM-Chor1 (n = 7) using ROSE2_META (https://github.com/linlabcode/pipeline/blob/master/ROSE2_META.py). In each cell line, H3K27ac datasets from both parental and HA-dTAG-T expressing cells were aggregated and average H3K27ac signal (background-subtracted AUC) was quantified at the union of all binding sites. Binding sites contained within the ± 2.5kb region surrounding transcription start sites (TSSs) were excluded. The cutoff delineating typical from SEs was defined as originally described.21
Plotting brachyury occupancy versus SEs
To assess the statistical enrichment of SEs overlapping top brachyury peaks, we ranked all brachyury peaks in either CH22 or UM-Chor1 by average signal (background subtracted AUC). Using a sliding bin approach with 1,000 peaks per bin sliding in increments of 500 peaks, we assessed the enrichment of SE binding as the log2 ratio of observed over expected where expected represents the overall frequency of SE overlap with brachyury binding sites.
Motif analysis
Genomic sequence was extracted for the top 1,000 brachyury peaks as ranked by average signal (by AUC) for brachyury peaks in CH22, UM-Chor1, and the union of both CH22 and UM-Chor1. To identify de novo brachyury sequence binding motifs, we used MEME-chip81 to find the top 5 motifs (using the option -meme-nmotifs 5). To connect de novo motifs to known transcription factor binding motifs, we also supplied an annotated table of transcription factor position weight matrices drawn from TRANSFAC82 and the Jolma 2013 study83 available at (https://github.com/linlabcode/CRC/blob/master/crc/annotation/VertebratePWMs.txt). Motif diagrams of the top 5 motifs along with their associated known binding transcription factors are presented in Figure 1F.
Overlapping brachyury with genomic features
Brachyury peaks in either CH22 or UM-Chor1 were overlapped with aggregate H3K27ac peaks defined using MACS2 in their respective cell lines. For genomic features, enhancers and SEs were defined as above. Transcription start sites were defined using the ± 1,000 bp window flanking annotated gene start sites. Active TSSs were defined as those overlapping a H3K27ac peak in their respective cell lines.
Defining top brachyury-bound SE genes
We ranked all expressed genes in CH22 or UM-Chor1 (FPKM > 10 in RNA-seq data) by their total brachyury proximal occupancy (total AUC in the ± 100kb window flanking the TSS). To separate top brachyury genes, we used the same graphical cutoff from the ROSE2 SE algorithm which identifies the tangent of the intersect between the rank curve and the diagonal. These genes were further filtered for proximal association to a SE (in the same ± 100kb window) to define the top brachyury SE-associated genes for CH22, UM-Chor1, or the combination the two cell lines.
To define the functional categories associated with these top brachyury SE genes, we used the broad mSigDB tool71,72 (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) and performed overlap with the C2 collection of curated gene signatures. The top 5 most significant signatures and their associated significance are shown in Figure 1E.
Identifying shared brachyury-regulated genes
Brachyury signal proximal to active genes was aggregated in each cell line and plotted between CH22 and UM-Chor1 (Figure S1G). Top brachyury bound SE-associated in either CH22, UM-Chor1, or the combination of both lines are annotated.
Inferring brachyury core regulatory circuitry
To understand how the T gene is regulated and how brachyury regulates itself and other transcription factors, we mapped the transcription factor core regulatory circuitry in combined CH22 and UM-Chor1 cells. Here we used our previously established core regulatory circuitry mapper36 (https://github.com/linlabcode/CRC). This approach first identified all SE proximally associated (+/− 100kb from transcription start site) TFs. SEs were assigned to their proximal transcription factors and within each SE, putative nucleosome free regions were identified by determining “valleys” between H3K27ac peaks. Transcription factor binding motifs were identified in these valleys using FIMO.81 This enabled us to map how each SE-associated TF regulated other SE-associated TFs by the inferred binding at proximal SEs. The transcription factor core regulatory circuitry is shown in Figure 1G. Interactions that are bidirectional are drawn in solid line. Interactions that are uni-directional are drawn in dotted line. Transcription factors that bind to the T SE, but themselves are not bound by brachyury are shown in white. Those with bidirectional binding are shown in blue. TFs bound by brachyury only are shown in gray. Brachyury interactions that are supported by brachyury ChIPmentation data are shown in red.
RNA-seq processing
RNA-sequencing data was processed using the Genialis platform (https://www.genialis.com). In brief, reads were first preprocessed by BBDuk to remove adaptors. Preprocessed reads were aligned to HG19 by HISAT2,84 per sample quantification was performed with Cuffquant, and Cuffnorm was used to estimate gene abundance.85 FPKM values were normalized to ERCC spike-in RNAs as described previously.86
Plotting SE-associated TFs in UM-Chor1
SE-associated TFs were identified as previously described.10 In brief, SEs and typical and enhancers were defined by H3K27ac signal and mapped using the ROSE2 software package.70 Discrete H3K27ac regions were defined and a stitching parameter consolidated proximal peaks. All enhancers were ranked according to increasing H3K27ac, and a 50kb proximity window was used to assigned enhancers to target genes. Of all the identified SE-associated genes, genes that were annotated as TFs in UniProt87 (115) were obtained and filtered for expression > 10 FPKM and for the subsequent line plot.
SLAM-seq data processing
SLAM-seq data were processed as in the original SLAM-seq publication using the SlamDunk pipeline (https://t-neumann.github.io/slamdunk/index.html) with the following parameters (−5 12 -n 100 -t 32 -m -rl 75). Given the sparsity of SLAM-seq nascent converted reads (typically 3%–5% of total reads), we applied a more stringent cutoff for actively transcribed genes. We used genes in the top quartile by total counts per million (cpm). We next intersected these genes with those that have evidence of nascent reads in all samples. Finally, this list was intersected with expressed genes as detected by RNA-seq (FPKM > 1). This resulted in 6,959 genes used for subsequent analysis. For all analysis we either compared total counts per million (cpm) which is a representation of total mRNA abundance or conversion fraction (presented as % nascent reads) which is a measure of the fraction of reads that have a converted C- > T (indicative of metabolic labeling and therefore nascent transcription).
For individual genes, total counts per million (cpm) or % nascent reads were plotted as the mean and standard error of the mean (Figures 4B and 4C). For comparisons of active genes or top brachyury SE genes, a two-tailed t test was utilized to assess statistical significance (Figures 4D and 4E).
To assess the leading-edge enrichment of these genes in either the context of brachyury degradation or THZ1 treatment, we first converted the CH22 top brachyury SE genes into a gene set. We next utilized GSEA. GSEA was called as follows with custom inputs denoted in brackets [ ]:
java -Xmx4000m -cp ./gsea2-3.0_beta_2.jar xtools.gsea.Gsea -res [GCT] -cls [CLS] -gmx [TOP_BRACHYURY_SE_GENES.gmt] -collapse false -mode Max_probe -norm meandiv -nperm 1000 -permute gene_set -rnd_type no_balance -scoring_scheme weighted -rpt_label [ANALYSIS_NAME] -metric log2_Ratio_of_Classes -sort real -order descending -include_only_symbols true -make_sets true -median false -num 100 -plot_top_x 20 -rnd_seed timestamp -save_rnd_lists false -set_max 500 -set_min 15 -zip_report false -out [OUTPUT_FOLDER] -gui false
The enrichment plot is shown in Figure 4F.
RNA-seq analysis
For CH22 and UM-Chor1 RNA-seq analysis, active genes were defined as those expressed with an average > 10 FPKM in at least one sample and for which an average expression > 0 was detected in all groups. The significance of differences between groups of genes was assessed using a two-tailed t test (Figures S4A and S4B). Differential genes in CH22 were defined by having a > 1.5-fold change in either direction and a p value for significance < 0.05 as defined by a two-tailed t test. Figure S4C shows a heatmap of differential genes comparing brachyury degradation at 8hr or 24hr to the 0hr control. Figure S4D shows the log2 fold change and p value at 8 hours for either THZ1 treatment or brachyury degradation for all expressed genes.
GSEA for long-term brachyury degradation
Gene set enrichment analysis is a computational platform developed by the Broad institute (https://www.gsea-msigdb.org/gsea/index.jsp).72,88 A Gene Cluster Text file (.gct), a Categorical Class file (.cls), and a Gene Matrix Transposed file (.gmt) are required for this analysis. The Gene Cluster Text file was generated from normalized FPKM expression data. Phenotypic data comprised of number and classification of samples was input for the Categorical Class file. Finally, the C2 all curated gene set from the GSEA website was used to create the Gene Matrix Transposed file. Parameters were kept at their defaults.
Acknowledgments
Imaging for this project was in part supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (DK56338 and CA125123), CPRIT (RP150578 and RP170719), the Dan L. Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. C.Y.L. is a CPRIT Scholar for Cancer Research and is supported by CPRIT (RR150093). C.Y.L. and H.E.S. are supported by the Chordoma Foundation, and H.E.S. is also supported by the NIH (1F31CA236130-01). A.D. is supported by the Hope Funds for Cancer Research. T.S., M.J.W., P.A.C., and S.L.S. are supported in part by the NCI Cancer Target Discovery and Development (CTD2) Network (U01CA176152/U01CA217848 to S.L.S.). In addition to being supported by the NCI, T.S. is supported by a grant from the Chordoma Foundation. B.N. is supported by American Cancer Society postdoctoral fellowship PF-17-010-01-CDD. B.N. and N.S.G. are supported by the Katherine L. and Steven C. Pinard Research Fund. P.W. is supported by Cancer Research UK (CRUK) (C2739/A22897) and is a CRUK Life Fellow. We thank Dennis L. Buckley for his work relating to dTAG-47 reagents.
Author contributions
H.E.S. and C.Y.L. designed the study. H.E.S., C.Y.L., T.S., M.J.W., P.A.C., S.L.S., and R.A.Y. supervised the execution of experiments and data analysis. B.N. and N.S.G. provided dTAG-47 reagents. H.E.S., A.D., H.L.J., W.D.P., and M.H.S. performed experiments. H.E.S., P.W., J.D., F.S., W.D.P., and C.Y.L. performed data analysis and interpretation. J.S., P.C., and J.L. provided the CH22 cell line via the Chordoma Foundation. H.E.S. and C.Y.L. wrote the original draft of the manuscript. All authors contributed to reviewing and editing the manuscript.
Declaration of interests
C.Y.L. is an inventor of intellectual property licensed to Syros Pharmaceuticals, and is the Vice President of Biology and a shareholder of Kronos Bio, Inc. S.L.S. is a shareholder and serves on the Board of Directors of Jnana Therapeutics; is a shareholder of Forma Therapeutics and Decibel Therapeutics; is a shareholder and advises Kojin Therapeutics, Kisbee Therapeutics, Exo Therapeutics, and Eikonizo Therapeutics; serves on the Scientific Advisory Boards of Eisai Co., Ltd., Ono Pharma Foundation, Biogen, Inc., and F-Prime Capital Partners and the Board of Advisers of the Genomics Institute of the Novartis Research Foundation; and is a Novartis Faculty Scholar. M.J.W. is an employee and equity holder of Kojin Therapeutics. P.A.C. is an advisor to Pfizer, Inc. and nference, Inc. R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics, and Dewpoint Therapeutics. P.W. is a current employee of The Institute of Cancer Research, which has a Rewards to Inventors scheme and has a commercial interest in development of inhibitors of the WNT pathway, CDK8/19, and other CDKs, with intellectual property licensed to Merck and Cyclacel Pharmaceuticals. P.W. is also a consultant for Astex Pharmaceuticals, CV6 Therapeutics, Nextechinvest, Nuevolution, Black Diamond, and STORM Therapeutics and holds equity in Chroma Therapeutics, Nextech, Black Diamond, and STORM. B.N. is an inventor on patent applications related to the dTAG system (WO/2017/024318, WO/2017/024319, WO/2018/148443, and WO/2018/148440). N.S.G. is a Scientific Founder, member of the Scientific Advisory Board (SAB), and equity holder in C4 Therapeutics, Syros, Soltego (board member), B2S, Allorion, and Inception. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voroni, Arbella, Deerfield, and Sanofi.
Published: January 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100188.
Supplemental information
References
- 1.Gonda T.J., Ramsay R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer. 2015;15:686–694. doi: 10.1038/nrc4018. [DOI] [PubMed] [Google Scholar]
- 2.Bradner J.E., Hnisz D., Young R.A. Transcriptional Addiction in Cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEvoy J., Flores-Otero J., Zhang J., Nemeth K., Brennan R., Bradley C., Krafcik F., Rodriguez-Galindo C., Wilson M., Xiong S. Coexpression of normally incompatible developmental pathways in retinoblastoma genesis. Cancer Cell. 2011;20:260–275. doi: 10.1016/j.ccr.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart E., McEvoy J., Wang H., Chen X., Honnell V., Ocarz M., Gordon B., Dapper J., Blankenship K., Yang Y., St. Jude Children’s Research Hospital – Washington University Pediatric Cancer Genome Project Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell. 2018;34:411–426.e19. doi: 10.1016/j.ccell.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanda T., Lawton L.N., Barrasa M.I., Fan Z.P., Kohlhammer H., Gutierrez A., Ma W., Tatarek J., Ahn Y., Kelliher M.A. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vujovic S., Henderson S., Presneau N., Odell E., Jacques T.S., Tirabosco R., Boshoff C., Flanagan A.M. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J. Pathol. 2006;209:157–165. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson D.G., Bhatt S., Herrmann B.G. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature. 1990;343:657–659. doi: 10.1038/343657a0. [DOI] [PubMed] [Google Scholar]
- 8.Roselli M., Fernando R.I., Guadagni F., Spila A., Alessandroni J., Palmirotta R., Costarelli L., Litzinger M., Hamilton D., Huang B. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin. Cancer Res. 2012;18:3868–3879. doi: 10.1158/1078-0432.CCR-11-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Presneau N., Shalaby A., Ye H., Pillay N., Halai D., Idowu B., Tirabosco R., Whitwell D., Jacques T.S., Kindblom L.G. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J. Pathol. 2011;223:327–335. doi: 10.1002/path.2816. [DOI] [PubMed] [Google Scholar]
- 10.Sharifnia T., Wawer M.J., Chen T., Huang Q.Y., Weir B.A., Sizemore A., Lawlor M.A., Goodale A., Cowley G.S., Vazquez F. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019;25:292–300. doi: 10.1038/s41591-018-0312-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pillay N., Plagnol V., Tarpey P.S., Lobo S.B., Presneau N., Szuhai K., Halai D., Berisha F., Cannon S.R., Mead S. A common single-nucleotide variant in T is strongly associated with chordoma. Nat. Genet. 2012;44:1185–1187. doi: 10.1038/ng.2419. [DOI] [PubMed] [Google Scholar]
- 12.Dang C.V., Reddy E.P., Shokat K.M., Soucek L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer. 2017;17:502–508. doi: 10.1038/nrc.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delmore J.E., Issa G.C., Lemieux M.E., Rahl P.B., Shi J., Jacobs H.M., Kastritis E., Gilpatrick T., Paranal R.M., Qi J. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson M.A., Gudgin E.J., Horton S.J., Giotopoulos G., Meduri E., Robson S., Cannizzaro E., Osaki H., Wiese M., Putwain S. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28:311–320. doi: 10.1038/leu.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puissant A., Frumm S.M., Alexe G., Bassil C.F., Qi J., Chanthery Y.H., Nekritz E.A., Zeid R., Gustafson W.C., Greninger P. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3:308–323. doi: 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwiatkowski N., Zhang T., Rahl P.B., Abraham B.J., Reddy J., Ficarro S.B., Dastur A., Amzallag A., Ramaswamy S., Tesar B. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chipumuro E., Marco E., Christensen C.L., Kwiatkowski N., Zhang T., Hatheway C.M., Abraham B.J., Sharma B., Yeung C., Altabef A. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwamoto M., Friedman E.J., Sandhu P., Agrawal N.G., Rubin E.H., Wagner J.A. Clinical pharmacology profile of vorinostat, a histone deacetylase inhibitor. Cancer Chemother. Pharmacol. 2013;72:493–508. doi: 10.1007/s00280-013-2220-z. [DOI] [PubMed] [Google Scholar]
- 19.VanderMolen K.M., McCulloch W., Pearce C.J., Oberlies N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. (Tokyo) 2011;64:525–531. doi: 10.1038/ja.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lasko L.M., Jakob C.G., Edalji R.P., Qiu W., Montgomery D., Digiammarino E.L., Hansen T.M., Risi R.M., Frey R., Manaves V. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–132. doi: 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eliades P., Abraham B.J., Ji Z., Miller D.M., Christensen C.L., Kwiatkowski N., Kumar R., Njauw C.N., Taylor M., Miao B. High MITF Expression Is Associated with Super-Enhancers and Suppressed by CDK7 Inhibition in Melanoma. J. Invest. Dermatol. 2018;138:1582–1590. doi: 10.1016/j.jid.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boija A., Klein I.A., Sabari B.R., Dall’Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 2018;175:1842–1855.e16. doi: 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho W.K., Spille J.H., Hecht M., Lee C., Li C., Grube V., Cisse I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361:412–415. doi: 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shrinivas K., Sabari B.R., Coffey E.L., Klein I.A., Boija A., Zamudio A.V., Schuijers J., Hannett N.M., Sharp P.A., Young R.A., Chakraborty A.K. Enhancer Features that Drive Formation of Transcriptional Condensates. Mol. Cell. 2019;75:549–561.e7. doi: 10.1016/j.molcel.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y.E., Manteiga J.C., Henninger J.E., Sabari B.R., Dall’Agnese A., Hannett N.M., Spille J.H., Afeyan L.K., Zamudio A.V., Shrinivas K. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature. 2019;572:543–548. doi: 10.1038/s41586-019-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamudio A.V., Dall’Agnese A., Henninger J.E., Manteiga J.C., Afeyan L.K., Hannett N.M., Coffey E.L., Li C.H., Oksuz O., Sabari B.R. Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol. Cell. 2019;76:753–766.e6. doi: 10.1016/j.molcel.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein I.A., Boija A., Afeyan L.K., Hawken S.W., Fan M., Dall’Agnese A., Oksuz O., Henninger J.E., Shrinivas K., Sabari B.R. Partitioning of cancer therapeutics in nuclear condensates. Science. 2020;368:1386–1392. doi: 10.1126/science.aaz4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durbin A.D., Zimmerman M.W., Dharia N.V., Abraham B.J., Iniguez A.B., Weichert-Leahey N., He S., Krill-Burger J.M., Root D.E., Vazquez F. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018;50:1240–1246. doi: 10.1038/s41588-018-0191-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gryder B.E., Yohe M.E., Chou H.C., Zhang X., Marques J., Wachtel M., Schaefer B., Sen N., Song Y., Gualtieri A. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017;7:884–899. doi: 10.1158/2159-8290.CD-16-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaffer A.L., Emre N.C., Lamy L., Ngo V.N., Wright G., Xiao W., Powell J., Dave S., Yu X., Zhao H. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsueh Y.S., Chang H.H., Shan Y.S., Sun H.S., Fletcher J.A., Li C.F., Chen L.T. Nuclear KIT induces a NFKBIB-RELA-KIT autoregulatory loop in imatinib-resistant gastrointestinal stromal tumors. Oncogene. 2019;38:6550–6565. doi: 10.1038/s41388-019-0900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin C.Y., Erkek S., Tong Y., Yin L., Federation A.J., Zapatka M., Haldipur P., Kawauchi D., Risch T., Warnatz H.J. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530:57–62. doi: 10.1038/nature16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ott C.J., Federation A.J., Schwartz L.S., Kasar S., Klitgaard J.L., Lenci R., Li Q., Lawlor M., Fernandes S.M., Souza A. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell. 2018;34:982–995.e7. doi: 10.1016/j.ccell.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H.T., Seo H.S., Zhang T., Wang Y., Jiang B., Li Q., Buckley D.L., Nabet B., Roberts J.M., Paulk J. MELK is not necessary for the proliferation of basal-like breast cancer cells. eLife. 2017;6:e26693. doi: 10.7554/eLife.26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naka T., Iwamoto Y., Shinohara N., Chuman H., Fukui M., Tsuneyoshi M. Cytokeratin subtyping in chordomas and the fetal notochord: an immunohistochemical analysis of aberrant expression. Mod. Pathol. 1997;10:545–551. [PubMed] [Google Scholar]
- 41.Faial T., Bernardo A.S., Mendjan S., Diamanti E., Ortmann D., Gentsch G.E., Mascetti V.L., Trotter M.W., Smith J.C., Pedersen R.A. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development. 2015;142:2121–2135. doi: 10.1242/dev.117838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebé-Pedrós A., Ariza-Cosano A., Weirauch M.T., Leininger S., Yang A., Torruella G., Adamski M., Adamska M., Hughes T.R., Gómez-Skarmeta J.L., Ruiz-Trillo I. Early evolution of the T-box transcription factor family. Proc. Natl. Acad. Sci. USA. 2013;110:16050–16055. doi: 10.1073/pnas.1309748110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah S.R., David J.M., Tippens N.D., Mohyeldin A., Martinez-Gutierrez J.C., Ganaha S., Schiapparelli P., Hamilton D.H., Palena C., Levchenko A., Quiñones-Hinojosa A. Brachyury-YAP Regulatory Axis Drives Stemness and Growth in Cancer. Cell Rep. 2017;21:495–507. doi: 10.1016/j.celrep.2017.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sawada A., Kiyonari H., Ukita K., Nishioka N., Imuta Y., Sasaki H. Redundant roles of Tead1 and Tead2 in notochord development and the regulation of cell proliferation and survival. Mol. Cell. Biol. 2008;28:3177–3189. doi: 10.1128/MCB.01759-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lupien M., Eeckhoute J., Meyer C.A., Wang Q., Zhang Y., Li W., Carroll J.S., Liu X.S., Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malgulwar P.B., Pathak P., Singh M., Kale S.S., Suri V., Sarkar C., Sharma M.C. Downregulation of SMARCB1/INI1 expression in pediatric chordomas correlates with upregulation of miR-671-5p and miR-193a-5p expressions. Brain Tumor Pathol. 2017;34:155–159. doi: 10.1007/s10014-017-0295-7. [DOI] [PubMed] [Google Scholar]
- 47.Shalaby A., Presneau N., Ye H., Halai D., Berisha F., Idowu B., Leithner A., Liegl B., Briggs T.R., Bacsi K. The role of epidermal growth factor receptor in chordoma pathogenesis: a potential therapeutic target. J. Pathol. 2011;223:336–346. doi: 10.1002/path.2818. [DOI] [PubMed] [Google Scholar]
- 48.Lee D.H., Zhang Y., Kassam A.B., Park M.J., Gardner P., Prevedello D., Henry S., Horbinski C., Beumer J.H., Tawbi H. Combined PDGFR and HDAC Inhibition Overcomes PTEN Disruption in Chordoma. PLoS ONE. 2015;10:e0134426. doi: 10.1371/journal.pone.0134426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X., Ji Z., Ma Y., Qiu X., Fan Q., Ma B. Expression of hypoxia-inducible factor-1α, vascular endothelial growth factor and matrix metalloproteinase-2 in sacral chordomas. Oncol. Lett. 2012;3:1268–1274. doi: 10.3892/ol.2012.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J., Lin J.J., Xu R.H., Kung H.F. Mesoderm induction by heterodimeric AP-1 (c-Jun and c-Fos) and its involvement in mesoderm formation through the embryonic fibroblast growth factor/Xbra autocatalytic loop during the early development of Xenopus embryos. J. Biol. Chem. 1998;273:1542–1550. doi: 10.1074/jbc.273.3.1542. [DOI] [PubMed] [Google Scholar]
- 51.Smith J.C., Price B.M., Green J.B., Weigel D., Herrmann B.G. Expression of a Xenopus homolog of Brachyury (T) is an immediate-early response to mesoderm induction. Cell. 1991;67:79–87. doi: 10.1016/0092-8674(91)90573-h. [DOI] [PubMed] [Google Scholar]
- 52.Thakore P.I., D’Ippolito A.M., Song L., Safi A., Shivakumar N.K., Kabadi A.M., Reddy T.E., Crawford G.E., Gersbach C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kispert A., Koschorz B., Herrmann B.G. The T protein encoded by Brachyury is a tissue-specific transcription factor. EMBO J. 1995;14:4763–4772. doi: 10.1002/j.1460-2075.1995.tb00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kroschwald S., Maharana S., Simon A. Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. 2017 doi: 10.19185/matters.201702000010. Published online May 22, 2017. [DOI] [Google Scholar]
- 55.Hyman A.A., Weber C.A., Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- 56.Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shin Y., Brangwynne C.P. Liquid phase condensation in cell physiology and disease. Science. 2017;357:eaaf4382. doi: 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- 58.Herzog V.A., Reichholf B., Neumann T., Rescheneder P., Bhat P., Burkard T.R., Wlotzka W., von Haeseler A., Zuber J., Ameres S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods. 2017;14:1198–1204. doi: 10.1038/nmeth.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang T., Buchan H.L., Townsend K.J., Craig R.W. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J. Cell. Physiol. 1996;166:523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 60.Wittmann S., Bali P., Donapaty S., Nimmanapalli R., Guo F., Yamaguchi H., Huang M., Jove R., Wang H.G., Bhalla K. Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer Res. 2003;63:93–99. [PubMed] [Google Scholar]
- 61.Desai B.M., Villanueva J., Nguyen T.T., Lioni M., Xiao M., Kong J., Krepler C., Vultur A., Flaherty K.T., Nathanson K.L. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS ONE. 2013;8:e59588. doi: 10.1371/journal.pone.0059588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiang W., Yang C.Y., Bai L. MCL-1 inhibition in cancer treatment. OncoTargets Ther. 2018;11:7301–7314. doi: 10.2147/OTT.S146228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doi K., Li R., Sung S.S., Wu H., Liu Y., Manieri W., Krishnegowda G., Awwad A., Dewey A., Liu X. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012;287:10224–10235. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J., Jin S., Abraham V., Huang X., Liu B., Mitten M.J., Nimmer P., Lin X., Smith M., Shen Y. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011;10:2340–2349. doi: 10.1158/1535-7163.MCT-11-0415. [DOI] [PubMed] [Google Scholar]
- 65.Su S., Liu Q., Chen J., Chen J., Chen F., He C., Huang D., Wu W., Lin L., Huang W. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605–620. doi: 10.1016/j.ccr.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 66.Ganuza M., Sáiz-Ladera C., Cañamero M., Gómez G., Schneider R., Blasco M.A., Pisano D., Paramio J.M., Santamaría D., Barbacid M. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J. 2012;31:2498–2510. doi: 10.1038/emboj.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lim S., Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 68.Wang B., Kohli J., Demaria M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer. 2020;6:838–857. doi: 10.1016/j.trecan.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 69.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown J.D., Lin C.Y., Duan Q., Griffin G., Federation A., Paranal R.M., Bair S., Newton G., Lichtman A., Kung A. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann T., Herzog V.A., Muhar M., von Haeseler A., Zuber J., Ameres S.L. Quantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets. BMC Bioinformatics. 2019;20:258. doi: 10.1186/s12859-019-2849-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmidl C., Rendeiro A.F., Sheffield N.C., Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods. 2015;12:963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol. Biol. 2014;1150:81–95. doi: 10.1007/978-1-4939-0512-6_4. [DOI] [PubMed] [Google Scholar]
- 81.Grant C.E., Bailey T.L., Noble W.S. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 84.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lovén J., Orlando D.A., Sigova A.A., Lin C.Y., Rahl P.B., Burge C.B., Levens D.L., Lee T.I., Young R.A. Revisiting global gene expression analysis. Cell. 2012;151:476–482. doi: 10.1016/j.cell.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Each frame represents 5 minutes and filming occurred over 23 hours. When CH22, THA-dTAG-EGFP/+ are treated with 500nM THZ1, individual cells with lower brachyury expression levels are primed to undergo apoptosis and detach from their surface.
Data Availability Statement
Sequencing datasets generated in this study are summarized in Table S5. All raw data are available on NCBI GEO. The superseries accession number for new data reported in this paper is GSE153973. Specifically, RNA-seq data is available under accession number GSE153972 and ChIP-seq data under accession number GSE153971. All custom scripts associated with this study are deposited on github.com at: https://github.com/linlabcode/chordoma_code/.