

Abstract
OBJECTIVE
Both glucose and triglyceride production are increased in type 2 diabetes and nonalcoholic fatty liver disease (NAFLD). For decades, the leading hypothesis to explain these paradoxical observations has been selective hepatic insulin resistance wherein insulin drives de novo lipogenesis (DNL) while failing to suppress glucose production. Here, we aimed to test this hypothesis in humans.
RESEARCH DESIGN AND METHODS
We recruited obese subjects who met criteria for bariatric surgery with (n = 16) or without (n = 15) NAFLD and assessed 1) insulin-mediated regulation of hepatic and peripheral glucose metabolism using hyperinsulinemic-euglycemic clamps with [6,6-2H2]glucose, 2) fasting and carbohydrate-driven hepatic DNL using deuterated water (2H2O), and 3) hepatocellular insulin signaling in liver biopsy samples collected during bariatric surgery.
RESULTS
Compared with subjects without NAFLD, those with NAFLD demonstrated impaired insulin-mediated suppression of glucose production and attenuated—not increased—glucose-stimulated/high-insulin lipogenesis. Fructose-stimulated/low-insulin lipogenesis was intact. Hepatocellular insulin signaling, assessed for the first time in humans, exhibited a proximal block in insulin-resistant subjects: Signaling was attenuated from the level of the insulin receptor through both glucose and lipogenesis pathways. The carbohydrate-regulated lipogenic transcription factor ChREBP was increased in subjects with NAFLD.
CONCLUSIONS
Acute increases in lipogenesis in humans with NAFLD are not explained by altered molecular regulation of lipogenesis through a paradoxical increase in lipogenic insulin action; rather, increases in lipogenic substrate availability may be the key.
Introduction
The selective hepatic insulin resistance hypothesis posits that increased de novo lipogenesis (DNL) in insulin-resistant individuals is due to paradoxically preserved insulin-mediated activation of lipogenic insulin signaling, with decreased glucoregulatory insulin signaling occurring after a signaling branch point. Many branch points have been proposed (1–3). Perplexingly, the major mechanisms for lipid-mediated hepatic insulin resistance result in defects at the level of the insulin receptor kinase (IRK) because of diacylglycerol accumulation and protein kinase Cε (PKCε) activation (4–8) or at the level of protein kinase B (AKT) because of ceramide accumulation (9,10). These mechanisms suggest proximal insulin resistance, which would be incompatible with pathway-selective (branch point) insulin resistance. Alternative explanations, including substrate-driven and other insulin-independent mechanisms for increased DNL, have also been proposed (11–13). However, the relative contributions of hepatic insulin signaling versus excess nutrient supply to increased lipogenesis in humans with nonalcoholic fatty liver disease (NAFLD) are unknown. Therefore, we performed a comprehensive series of experiments (Supplementary Fig. 1) to determine whether the increased DNL observed in insulin-resistant patients is due to increased insulin-stimulated DNL.
Research Design and Methods
Design
This was a randomized, controlled, single-blinded, multicenter study designed to examine the regulation of hepatic DNL by insulin-mediated and insulin-independent mechanisms in obese control subjects and obese patients with NAFLD (Supplementary Fig. 1). The study was prospectively registered in the Netherlands Trial Registry (www.trialregister.nl: NTR5351).
Subjects
We screened obese patients from the outpatient clinics of two obesity centers in the Amsterdam metropolitan area for NAFLD using proton MRS (1H-MRS) (14). Sixteen obese control subjects without hepatic steatosis and 16 obese patients with NAFLD (defined as liver fat content >5.56% on 1H-MRS [15]) were recruited, but 1 participant did not complete the study because of a cancer diagnosis. Subjects were eligible if they 1) were aged ≥18 years, 2) met the criteria for bariatric surgery in accordance with international guidelines (16), 3) were scheduled to undergo Roux-en-Y gastric bypass surgery, and 4) had stable weight (<5% weight change) for ≥3 months before the study assessments. Exclusion criteria were 1) substance abuse (alcohol >2 units/day, recreational drugs); 2) use of lipid-lowering drugs, exogenous insulin, incretin mimetics, antipsychotics, or antidepressants; 3) childhood-onset obesity; or 4) any somatic disorder except for common obesity-related conditions (e.g., dyslipidemia, hypertension, obstructive sleep apnea). All subjects completed a screening evaluation, including history, physical examination, blood tests, and 1H-MRS. We performed indirect calorimetry using a ventilated hood system (Vmax Encore 29n; CareFusion, San Diego, CA). Body composition was determined by bioelectrical impedance analysis (BF-906; Maltron, Rayleigh, U.K.).
Liver Fat Content
We assessed liver fat content by 1H-MRS, as described (17). This method has high diagnostic accuracy and high precision with low variability for assessment of hepatic steatosis in the context of NAFLD (18). Briefly, single-voxel spectra were obtained during expiration on a 3T Intera magnetic resonance scanner (Philips, Best, the Netherlands). Voxels (27 mm3) were placed in homogeneous liver tissue away from visible vascular and bile structures. Water and fat resonance peaks were integrated using jMRUI software (19) and corrected for T2 relaxation. Liver fat content was calculated as the percentage of liver volume comprising fat (15).
Dietary Intake
After the screening visit, but before the first study procedure, subjects filled in prospective diet records for a minimum of 3 days (mean recorded period 8 days). Subjects were instructed to maintain their usual diet habits. All consumed drinks and foods had to be weighed or documented in standard kitchen measures to allow quantitative estimation of dietary intake. Individual subjects’ records were systematically checked on the day of the first examination to confirm completeness and entered into the Netherlands Nutrition Centre food database (using the online tool available at https://mijn.voedingscentrum.nl) to convert amounts of food into distinct nutrients.
Two-Step Hyperinsulinemic-Euglycemic Clamp
Glucose kinetics and tissue-specific parameters of insulin sensitivity were assessed during a two-step hyperinsulinemic-euglycemic clamp study, as described (20). This experimental protocol allowed us to accurately measure the basal rate of endogenous glucose production (EGP), insulin-mediated suppression of EGP (i.e., hepatic insulin sensitivity), insulin-mediated suppression of plasma nonesterified fatty acid (NEFA) levels (reflecting adipose tissue insulin sensitivity [21]), and the insulin-stimulated Rd of glucose (i.e., peripheral/muscle insulin sensitivity).
Briefly, subjects were admitted to the metabolic research unit after an overnight fast. A primed continuous infusion of the stable isotope-labeled glucose tracer [6,6-2H2]glucose (>99% enriched; Cambridge Isotopes, Andover, MA) was started. Basal EGP was determined after 2 h of tracer equilibration. Next, insulin-mediated suppressions of EGP and circulating NEFAs were assessed after 2 h of low-dose insulin infusion (Actrapid 20 mU ⋅ [m2 body surface area]−1 ⋅ min−1; Novo Nordisk Farma, Alphen aan de Rijn, the Netherlands). Finally, the insulin-stimulated Rd of glucose was assessed after an additional 2 h of high-dose insulin infusion (60 mU ⋅ m−2 ⋅ min−1). During hyperinsulinemia, plasma glucose was maintained constant at 5.0 mmol/L by frequent bedside monitoring of glucose levels and variable infusion of exogenous glucose (enriched with [6,6-2H2]glucose).
Hepatic DNL
To stimulate insulin-mediated and insulin-independent hepatic DNL from monosaccharides, control subjects and patients with NAFLD were randomly assigned (1:1) to glucose or fructose groups. Hepatic DNL was assessed using the incorporation of deuterated water (2H2O) into VLDL triglycerides, as described (22–24). Subjects were administered an oral 2H2O loading dose (2 g/kg) in the evening and fasted overnight. In the morning, they received one of two monosaccharide drinks: glucose (75 g in 300 mL water [25]) or fructose (75 g in 300 mL water). Blood samples were drawn at baseline and for 5 h after monosaccharide ingestion. We found that the optimal time point after carbohydrate ingestion to detect changes in DNL was at 240 min (Supplementary Fig. 2).
Liver Biopsy Collection
We assessed cellular responses to the same monosaccharide in liver biopsy samples obtained during the subjects’ scheduled bariatric surgery. Subjects underwent surgery <2 weeks after the clinical assessments. After an overnight fast and 2 h before the induction of anesthesia, subjects received the same monosaccharide drink (containing either 75 g of glucose or 75 g of fructose). During surgery, an experienced surgeon obtained subcapsular biopsy samples from segment III of the left liver lobe. Administration of the monosaccharide drink (2 h before induction of anesthesia) and biopsy sample collection (at the start of surgery) were carefully timed to minimize variation in the time between monosaccharide challenge and biopsies: median (interquartile range) drink-to-biopsy time was 140 (140–153) or 150 (145–195) min in subjects receiving glucose or fructose, respectively. Biopsy samples were cut and fixed in formaldehyde for histology or snap frozen in liquid nitrogen for other analyses. Subjects were instructed to maintain stable weight through consumption of a weight maintenance diet in the preoperative period.
We also obtained liver biopsy samples taken under fasted/unstimulated conditions from bariatric surgery patients who participated in a previously reported metabolic study (26). We selected age- and BMI-matched historic control subjects with low (≤5.56%, n = 8) or high (>10%, n = 8) liver fat content to compare these groups under fasting conditions.
Liver Histology
Liver histopathology was evaluated by an experienced liver pathologist who was blinded to all subject data and scored in accordance with Nonalcoholic Steatohepatitis Clinical Research Network recommendations (27–29).
Biochemical Analyses
Plasma glucose was determined by the glucose oxidase method using a Biosen C-line Plus glucose analyzer (EKF Diagnostics, Barleben/Magdeburg, Germany). Plasma insulin and cortisol were determined by immunoassay on an Immulite 2000 (Diagnostic Products, Los Angeles, CA), with intra-assay variation of 4–5% and 3–6%, respectively, and interassay variation of 5% and 5–7%, respectively. Plasma glucagon was determined by radioimmunoassay (Linco Research, St. Charles, MO), with intra-assay variation of 4–8% and interassay variation of 6–11%. Plasma NEFAs were determined by enzymatic colorimetric method (NEFA C test kit; Wako Chemicals, Neuss, Germany), with intra-assay variation of 1% and interassay variation of 4–15%. Serum C-peptide was determined by immunoluminometric assay on Attelica Solution (Siemens, Erlangen, Germany), with intra-assay variation of 2.4% and interassay variation of 4.9%. Plasma triglycerides, total cholesterol, and HDL were determined using commercially available assays on a chemistry analyzer (Selectra Pro; ELITechGroup, Dieren, the Netherlands); LDL was calculated using the Friedewald equation. Enrichment of [6,6-2H2]glucose (tracer-to-tracee ratio) in plasma and exogenous glucose infusate was determined by gas chromatography-mass spectrometry (GC-MS), as described (30).
DNL Assessment by GC-MS
VLDL1 and VLDL2 were isolated from 4 mL of fresh plasma by cumulative ultracentrifugation using a discontinuous salt gradient and SW41 rotor in a Beckman L3-50 ultracentrifuge (Palo Alto, CA), as described (31). Fractions were frozen at −80°C until further analysis. Lipids were extracted by vacuum drying and extraction of the pellet in ice-cold chloroform:methanol (2:1). Samples were spotted onto silica gel 60 plates, and thin-layer chromatography was performed with a mobile phase of hexane:diethyl ether:acetic acid (80:20:1). Plates were developed with 0.005% primuline in acetone:water (80:20). Purified samples were collected while absorbed to silica and eluted with diethyl ester. Triglyceride-fatty acids were analyzed by GC-MS (5975CI; Agilent Technologies, Santa Clara, CA) as fatty acid methyl esters following derivatization with methanolic boron trifluoride. The plasma 2H2O pool was assessed by exchange of hydrogens from plasma to acetone in the presence of sodium hydroxide; acetone deuterium enrichment was analyzed by GC-MS.
Protein Phosphorylation
Liver tissues were homogenized in lysis buffer (pH 8.0, 20 mmol/L Tris-HCL, 120 mmol/L KCl, 1 mmol/L EDTA, 2 mmol/L EGTA, 0.1% Triton X-100, 0.5% NP-40, 100 mmol/L NaF) supplemented with protease and phosphatase inhibitor cocktails (Roche, Basel, Switzerland) and caspase-3 inhibitor (1 μg/mL ac-DMQD-CHO; Enzo Life Sciences, Farmingdale, NY). Extracted proteins (20 μg) were separated by 4–12% SDS-PAGE (Invitrogen, Carlsbad, CA) and transferred to polyvinylidene fluoride membranes (Millipore, Burlington, MA) by semidry transfer cell (Bio-Rad, Hercules, CA) for 90 min. After blocking with 3% BSA in Tris-buffered saline with Tween (10 mmol/L Tris, 100 mmol/L NaCl, 0.1% Tween-20), membranes were incubated overnight at 4°C with primary antibodies. Membranes were washed and incubated with secondary antibody (1:1,500; Cell Signaling Technology, Danvers, MA), and immune complexes were detected using enhanced chemiluminescence (Thermo Scientific, Waltham, MA). Immunoblots were quantified by optical densitometry.
Primary antibodies for IRK (insulin receptor β, #3020S), pIRK (Tyr1162, #3918S), AKT (#2920S), pAKT (Ser473; #9271S), glycogen synthase kinase-3β (GSK3B) (#12456S), pGSK3B (Ser9, #5558S), proline-rich AKT substrate of 40 kDa (PRAS40) (#2691S), pPRAS40 (Thr246, #2997S), regulatory-associated protein of mammalian target of rapamycin (RPTOR) (#2280S), and pRPTOR (Ser792, #2083S) were purchased from Cell Signaling Technology. They were prepared at 1:1,000.
Gene Expression
Tissue RNA was extracted using TRIzol Reagent (Invitrogen, Breda, the Netherlands) followed by the NucleoSpin II extraction kit (Macherey-Nagel, Duren, Germany). We synthesized cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics Nederland, Almere, the Netherlands). Tissue mRNA expression was determined by quantitative RT-PCR using the SensiFAST SYBR No-ROX Kit (Bioline, Taunton, MA) on a LightCycler 480 system (Roche Diagnostics Nederland). Primers used for quantitative RT-PCR are listed in Supplementary Table 4. Amplification curves were analyzed using LinRegPCR (v2016.2) (32). Gene expression was normalized to RPLP0 expression.
Calculations
Glucose fluxes (EGP and Rd) were calculated using modified versions of the Steele equations for steady state (basal) or nonsteady state (during insulin infusion) (33,34). These are expressed as absolute flux (μmol ⋅ [kg body weight]−1 ⋅ min−1) as well as relative to basal (percentage suppression or stimulation). Glucose and lipid oxidation rates and resting energy expenditure were calculated from VO2 and VCO2, as described (35,36). The calculation of de novo hepatic palmitate synthesis from isotopic data has been previously described (37–39). Briefly, the fraction of the hepatic palmitate pool newly synthesized during the experiment (F) was calculated as F = ME ÷ (N × p), where molar enrichment (ME) equals m1 + (2 × m2) (where m1 and m2 are the atom percent enrichments [APEs] of singly and doubly deuterium-labeled palmitate; m + 2 APE was corrected for natural abundance from isotopomer distribution of [1-13C]palmitate standard curves); N is the number of exchangeable hydrogen atoms, previously reported to be 22; and p is the plasma APE of deuterium in water.
Statistics
Baseline data were compared by two-sided independent samples t tests or Mann-Whitney U tests, as appropriate. Outcomes in response to monosaccharide ingestion were compared by two-sided t tests (between-group differences) or paired t tests (within-group differences). Correlations were evaluated by simple linear regression. We considered P < 0.05 to be significant. Analyses were performed using SPSS version 25 (IBM Corporation, Armonk, NY) and GraphPad Prism 8 (GraphPad Software, La Jolla, CA) statistical software.
Study Approval
The protocol was approved by the Amsterdam University Medical Center medical ethics committee. All subjects provided written informed consent before inclusion.
Results
We studied morbidly obese subjects scheduled for elective bariatric surgery and recruited 15 obese control subjects without hepatic steatosis as well as 16 obese patients with NAFLD (liver fat content >5.56% [15]). Control subjects and patients with NAFLD did not differ in most baseline clinical and biochemical characteristics, save plasma triglycerides and transaminases (Table 1). Dietary intake did not significantly differ between groups (Supplementary Table 1).
Table 1.
Baseline characteristics of study subjects
| Control | NAFLD | |||||
|---|---|---|---|---|---|---|
| All (n = 15) | Glucose (n = 8) | Fructose (n = 7) | All (n = 16) | Glucose (n = 8) | Fructose (n = 8) | |
| Female sex | 13 (87) | 7 (88) | 6 (86) | 12 (75) | 5 (63) | 7 (88) |
| Age (years) | 43 ± 11 | 37 ± 7 | 49 ± 11 | 49 ± 11 | 50 ± 11A | 48 ± 10 |
| Height (cm) | 171 ± 9 | 173 ± 8 | 169 ± 10 | 172 ± 5 | 173 ± 6 | 171 ± 4 |
| Weight (kg) | 119 ± 18 | 120 ± 18 | 118 ± 18 | 124 ± 15 | 121 ± 13 | 126 ± 17 |
| BMI (kg/m2) | 41 ± 5 | 40 ± 4 | 41 ± 6 | 42 ± 4 | 40 ± 2 | 43 ± 6 |
| Body fat (%) | 47 ± 5 | 46 ± 3 | 47 ± 3 | 48 ± 6 | 45 ± 7 | 51 ± 4 |
| Waist circumference (cm) | 127 ± 14 | 129 ± 15 | 125 ± 13 | 132 ± 10 | 130 ± 8 | 134 ± 13 |
| Systolic blood pressure (mmHg) | 130 ± 19 | 128 ± 19 | 132 ± 20 | 135 ± 15 | 142 ± 13 | 127 ± 15 |
| Diastolic blood pressure (mmHg) | 83 ± 9 | 82 ± 10 | 85 ± 8 | 85 ± 9 | 88 ± 9 | 81 ± 9 |
| Preexisting type 2 diabetes | 1 (7) | 0 (0) | 1 (14) | 4 (25) | 2 (25) | 2 (25) |
| Glucose (mmol/L) | 4.8 ± 0.4 | 4.8 ± 0.2 | 4.8 ± 0.6 | 5.6 ± 1.8 | 6.0 ± 2.4 | 5.1 ± 0.5 |
| Insulin (pmol/L) | 80 ± 26 | 88 ± 21 | 70 ± 29 | 158 ± 78B | 155 ± 59A | 161 ± 98A |
| HbA1c (mmol/mol) | 36 ± 4 | 34 ± 2 | 39 ± 4 | 43 ± 9A | 45 ± 13A | 40 ± 4 |
| Triglycerides (mmol/L) | 1.0 (0.9–1.1) | 0.9 (0.6–1.3) | 1.1 (0.9–1.1) | 1.4 (0.9–1.9)A | 1.8 (1.5–2.6)B | 1.1 (0.7–1.3)C |
| Cholesterol (mmol/L) | 4.9 ± 0.9 | 4.5 ± 0.8 | 5.4 ± 0.9 | 5.1 ± 1.0 | 5.4 ± 1.0 | 4.7 ± 0.9 |
| HDL (mmol/L) | 1.3 ± 0.3 | 1.2 ± 0.3 | 1.4 ± 0.3 | 1.2 ± 0.2 | 1.0 ± 0.2 | 1.2 ± 0.3 |
| LDL (mmol/L) | 3.1 (2.6–3.6) | 2.8 (2.5–3.4) | 3.3 (2.9–4.2) | 3.1 (2.7–3.7) | 3.4 (2.9–3.9) | 2.8 (2.6–3.2) |
| ALT (units/L) | 21 ± 7 | 20 ± 6 | 22 ± 8 | 31 ± 13A | 35 ± 15A | 26 ± 9 |
| AST (units/L) | 20 ± 7 | 20 ± 5 | 19 ± 10 | 40 ± 22B | 50 ± 25A | 29 ± 13 |
| GGT (units/L) | 20 (12–39) | 21 (14–43) | 15 (11–24) | 40 (26–61)B | 42 (26–61)A | 39 (24–63)A |
| C-reactive protein (mg/L) | 6.2 ± 5.4 | 4 ± 3 | 9 ± 6 | 6.1 ± 3.3 | 4 ± 1 | 8 ± 4 |
| Liver fat (%)D | 3.1 (2.4–3.9) | 3.1 (2.2–3.9) | 3.1 (2.4–3.3) | 10.3 (6.2–23.9)B | 16.1 (5.7–27.1)B | 10.3 (7.0–15.2)B |
| Steatosis grade | 0 (0–1) | 0 (0–1) | 0 (0–1) | 1 (1–2)B | 1 (1–2)A | 2 (1–2)B |
| NAFLD activity score | 1 (1–2) | 1 (1–2) | 1 (1–2) | 2 (2–3)B | 2 (2–4)A | 2 (2–3) |
| Fibrosis stage 1 (%)E | 5 (33) | 2 (25) | 3 (43) | 6 (40) | 2 (29) | 4 (50) |
| Fibrosis stage 2 (%) | 10 (67) | 6 (75) | 4 (57) | 9 (60) | 5 (71) | 4 (50) |
Data are n (%), mean ± SD, or median (interquartile range). GGT, γ-glutamyl transferase.
P < 0.05, BP < 0.01 vs. respective control group on t test or Mann-Whitney U test.
P < 0.01 vs. NAFLD/glucose on Mann-Whitney U test.
Assessed by 1H-MRS.
There were no subjects with fibrosis stages 0, 3, or 4 in their subcapsular liver biopsy sample.
Basal and insulin-mediated metabolic fluxes were measured using the two-step hyperinsulinemic-euglycemic clamp method (20). Subjects with NAFLD had elevated fasting plasma insulin concentrations (Table 1). There were no differences in other fasting parameters, basal EGP, and substrate oxidation rates (Supplementary Table 2). Low-dose insulin infusion (step 1 of the clamp) raised plasma insulin to higher levels in subjects with NAFLD, suggesting lower insulin clearance (40). Despite higher plasma insulin concentrations, subjects with NAFLD demonstrated reduced suppression of EGP and reduced suppression of plasma NEFAs during step 1 of the clamp (Fig. 1A and B), reflecting insulin-resistant liver (41) and adipose tissue (21), respectively. The insulin-stimulated Rd of glucose during high-dose insulin infusion (step 2) was lower in subjects with NAFLD (Fig. 1C), indicating systemic/muscle insulin resistance (20).
Figure 1.
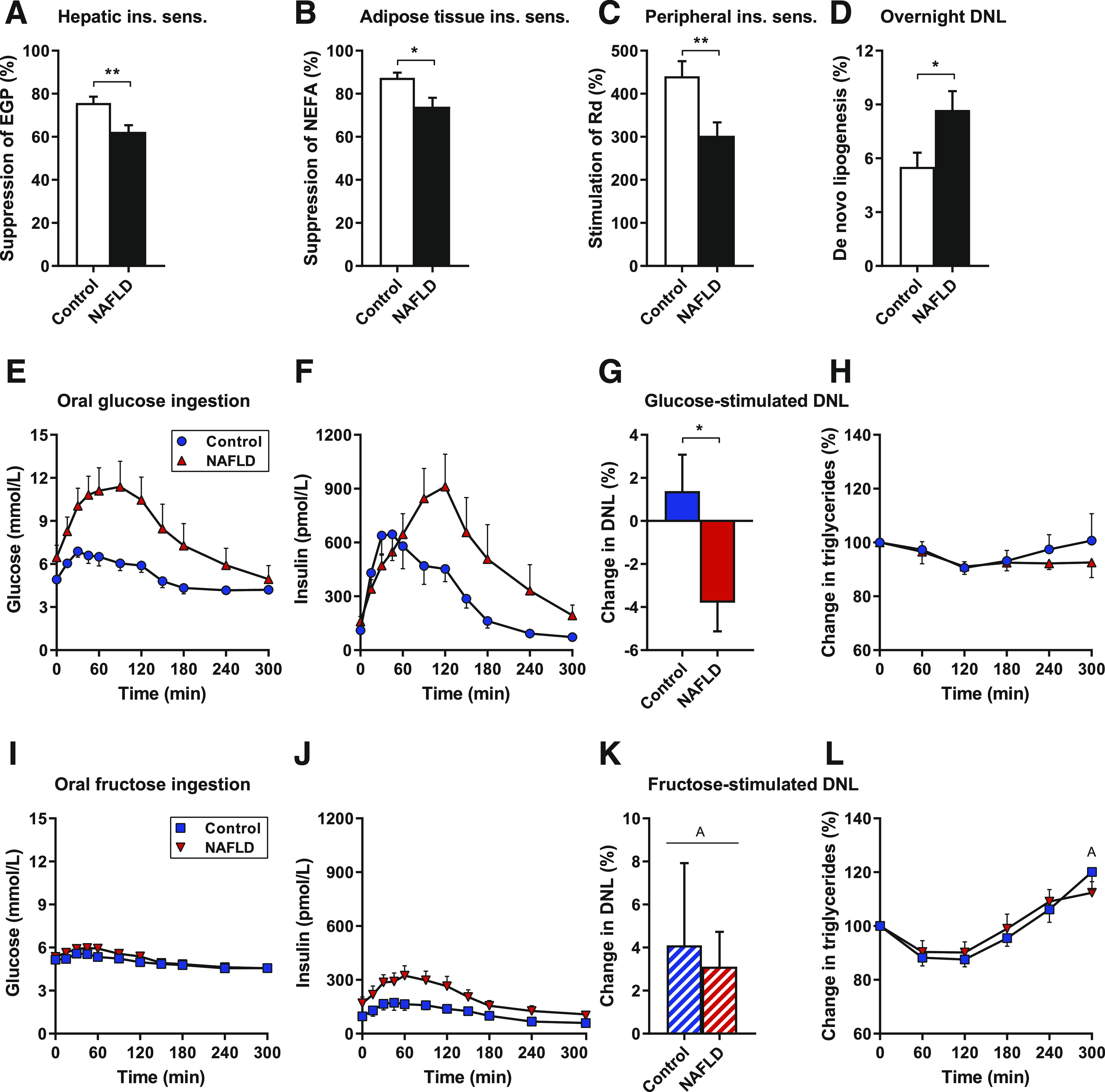
Overnight, glucose-stimulated, and fructose-stimulated DNL in control subjects and subjects with NAFLD. Subjects with NAFLD have hepatic (A), adipose tissue (B), and peripheral (C) insulin resistance (n = 14–16 per group). Overnight DNL is increased in insulin-resistant subjects with NAFLD (n = 14 per group) (D). Oral glucose ingestion produces robust plasma glucose (E) and insulin (F) responses, does not stimulate DNL at 240 min in insulin-resistant subjects with NAFLD (G), and is not associated with increases in circulating triglycerides (H) (n = 8 per group). Oral fructose ingestion produces minimal glucose (I) and insulin (J) responses, stimulates DNL at 240 min in both control subjects and subjects with NAFLD (K), and increases circulating triglycerides (L) (n = 6–8 per group). Data are mean ± SEM. *P < 0.05, **P < 0.01 by t test; AP < 0.05 vs. overnight DNL or basal by paired t test. ins. sens., insulin sensitivity.
Hepatic DNL was assessed using the incorporation of deuterium from 2H2O into VLDL triglycerides (22–24). The rate of DNL after an overnight fast, mostly reflecting the previous evening’s postprandial DNL, was elevated in subjects with NAFLD (Fig. 1D). This finding is consistent with previous studies (42,43) and, in particular, concordant with a recent study demonstrating markedly increased hepatic DNL in obese subjects with NAFLD compared with obese subjects without NAFLD (44).
To evaluate the role of incremental increases in hepatic insulin action on DNL in NAFLD, we assessed hepatic DNL after ingestion of carbohydrates that differentially stimulate insulin secretion. We used oral glucose ingestion to provide lipogenic substrate with elevated plasma insulin concentrations versus oral fructose ingestion as a low-insulin comparator. Subjects were randomly assigned (1:1) to glucose or fructose groups (Table 1). Glucose ingestion rapidly raised plasma glucose concentrations in both groups, but excursions were higher in subjects with NAFLD (area under the curve 1,548 ± 76 vs. 2,474 ± 398, P = 0.04) (Fig. 1E). Glucose potently stimulated postprandial hyperinsulinemia in all subjects (Fig. 1F); the corresponding C-peptide excursions showed that by 120 min, insulin secretion was increased in subjects with NAFLD, demonstrating that the livers of subjects with NAFLD were exposed to excess insulin after glucose ingestion (Supplementary Fig. 3). The pathway-selective insulin signaling hypothesis would predict hepatic DNL to be stimulated to the same (or greater) degree in individuals with NAFLD and greater insulin levels. However, in our insulin-resistant subjects with NAFLD, hepatic DNL at 4 h after glucose ingestion was lower than overnight DNL (P = 0.024 on within-group paired t test), and glucose-stimulated hepatic DNL was greater in control subjects with better insulin sensitivity (Fig. 1G). Circulating triglycerides were unchanged (Fig. 1H).
In contrast, fructose ingestion produced minor changes in plasma glucose and insulin concentrations (Fig. 1I and J) but robustly stimulated hepatic DNL in all subjects (Fig. 1K). Fructose ingestion increased circulating triglycerides (Fig. 1L). Thus, glucose-stimulated/insulin-mediated DNL was not increased in insulin-resistant fatty livers, challenging the concept that increased lipogenesis in human NAFLD is primarily driven by pathway-selective insulin action. Fructose, a substrate that enters the hepatocyte independently of insulin, robustly stimulated hepatic lipogenesis in all subjects.
The view that human hepatic insulin resistance results from a proximal insulin signaling defect has been controversial (45) in part because of difficulties in measuring hepatic insulin signaling in humans. In our study, the administration of monosaccharide solutions just before bariatric surgery afforded us the unique opportunity to safely obtain human liver tissue and examine insulin signaling under conditions of high substrate/high insulin versus high substrate/low insulin. To our knowledge, this is the first study in humans to assess whether cellular hepatic insulin resistance occurs at or downstream of the insulin receptor. We assessed insulin-stimulated hepatocellular phosphorylation events by immunoblotting (Fig. 2A and B). Phosphorylation of IRK, AKT, and GSK3B increased with insulin stimulation (i.e., greater in glucose than fructose ingestion) in control subjects but not in subjects with NAFLD (Fig. 2C–E). Limited by the amount of liver tissue, we were unable to optimize immunoblotting conditions to detect DNL-regulating (cleaved/activated) SREBP1c. As an alternative, we evaluated the lipogenic mammalian target of rapamycin complex 1 (mTORC1) pathway, with its insulin-dependent (through PRAS40) and insulin-independent/AMPK-mediated inputs (through RPTOR) (3). Compared with fructose, glucose ingestion stimulated PRAS40 phosphorylation in control subjects but not in subjects with NAFLD (Fig. 2F). Insulin-independent/AMPK-mediated regulation of mTORC1, as reflected by RPTOR phosphorylation, was unaltered in control subjects and subjects with NAFLD (Fig. 2G). Therefore, these results are consistent with proximal insulin resistance at the level of IRK in subjects with NAFLD. Such a proximal signaling defect would also be consistent with the diacylglycerol-PKCε hypothesis for lipid-mediated hepatic insulin resistance (4–8). Finally, decreased signaling in both glucose and lipogenesis signaling cascades is inconsistent with pathway-selective hepatic insulin resistance hypotheses.
Figure 2.
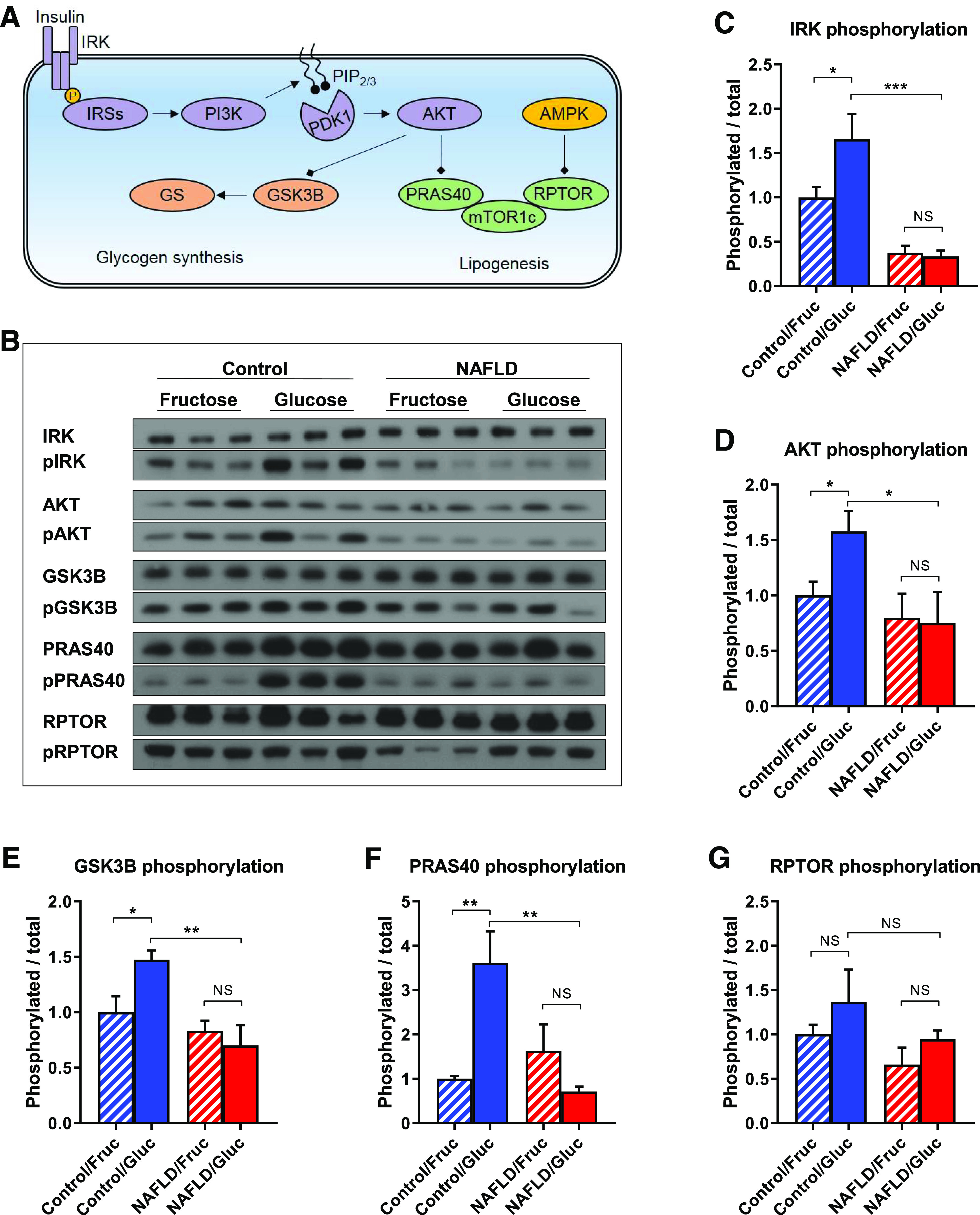
Insulin signaling in both glucose metabolism and lipogenesis pathways is attenuated in subjects with NAFLD. Schematic (A) and representative immunoblots (B) of insulin signaling in the glucose metabolism and lipogenesis pathways. Proximal insulin signaling, as reflected by IRK (C) and AKT (D) phosphorylation, is suppressed in subjects with NAFLD. Downstream insulin signaling in the glucose metabolism pathway (E), as reflected by GSK3B phosphorylation, is suppressed in subjects with NAFLD. Insulin-mediated regulation of mTORC1 (F), as reflected by PRAS40 phosphorylation, is suppressed in subjects with NAFLD. Insulin-independent/AMPK-mediated regulation of mTORC1 (G), as reflected by RPTOR phosphorylation, is unaltered in subjects with NAFLD. Data are mean ± SEM (n = 6 per group). *P < 0.05, **P < 0.01, ***P < 0.001 by t test. Fruc, fructose; Gluc, glucose.
Monosaccharides can act as nutrient regulators of the hepatic lipogenesis transcriptional program (13), particularly through the transcription factor ChREBP. We measured mRNA expression of the predominant transcript ChREBPα and its active isoform ChREBPβ (46): ChREBPα expression was unchanged in any group (Fig. 3A), whereas ChREBPβ was increased to the same degree in all subjects with NAFLD as it was by fructose in control subjects (Fig. 3B). This suggests that the nutrient-ChREBP-lipogenesis program may be constitutively active in NAFLD. We therefore assessed fasting hepatic ChREBP expression in liver biopsy samples from an independent cohort of bariatric surgery patients (26) and compared age- and BMI-matched subjects with low (≤5.56%, n = 8) versus high (>10%, n = 8) liver fat content (Supplementary Table 3). Again, there was no difference in ChREBPα (Fig. 3C) and increased ChREBPβ in subjects with NAFLD (Fig. 3D).
Figure 3.
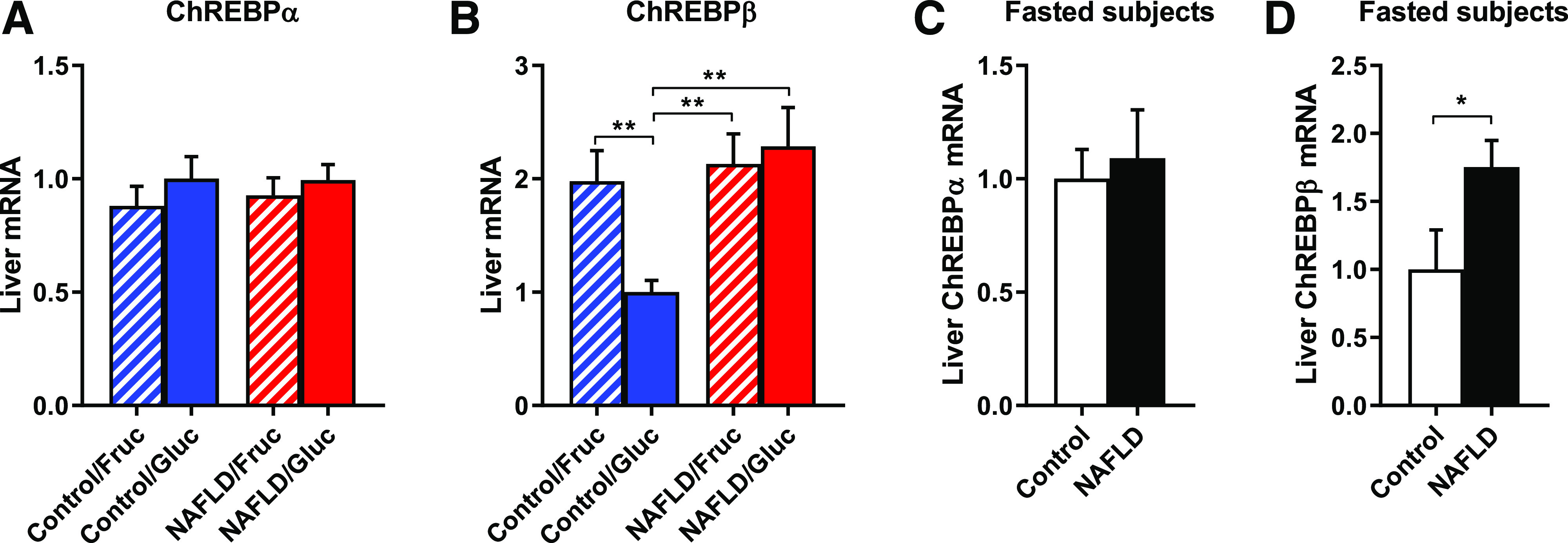
ChREBP may partially explain intact capacity for DNL in the setting of attenuated insulin signaling. Hepatic ChREBPα (A) and ChREBPβ (B) expression after glucose or fructose ingestion. Hepatic ChREBPα (C) and ChREBPβ (D) expression in overnight-fasted obese subjects with or without NAFLD. Data are mean ± SEM (n = 7–8 per group). *P < 0.05, **P < 0.01 by t test. Fruc, fructose; Gluc, glucose.
Conclusions
Taken together, our data provide experimental evidence against the pathway-selective hepatic insulin resistance hypothesis. In humans, hepatic DNL and insulin resistance travel together: Hepatic DNL is increased in patients with NAFLD (42,43) and increased in subjects with peripheral insulin resistance (47,48). More recently, the relationship between hepatic DNL and insulin resistance was shown to hold across a continuum of subjects with varying degrees of insulin sensitivity (44). In that well-conducted study, hepatic DNL was not only inversely related to hepatic and whole-body insulin sensitivity but also directly related to 24-h plasma glucose and insulin concentrations, suggesting that increased glucose and/or insulin concentrations (in the context of insulin resistance) may stimulate hepatic DNL in subjects with NAFLD. Our study extends those findings, benefiting from carbohydrate stimulation testing with stable isotope-labeled tracer analysis and liver biopsy samples taken after carbohydrate stimulation. Using this unique experimental design, we were able to differentiate between DNL driven by alterations in insulin action and DNL driven by other factors (e.g., increased lipogenic substrate). Furthermore, by studying obese patients only, in both the control and NAFLD groups, we are confident that the observed effects are associated with NAFLD and not confounded by group differences in obesity.
Here, we found that subjects with insulin-resistant NAFLD do not display increased hepatic insulin signaling or incremental DNL upon glucose ingestion, despite robust insulin secretion, whereas all subjects demonstrated intact fructose-stimulated DNL under low-insulin conditions. The data indicate that obese individuals with resistance to insulin’s ability to suppress hepatic glucose production also have resistance to insulin’s ability to stimulate hepatic DNL. Moreover, supporting an alternative explanation for increased DNL, we found that hepatic ChREBP was constitutively more active in individuals with NAFLD. Because ChREBP activity is a readout for intrahepatic carbohydrate availability (12), hepatic exposure to lipogenic substrate may largely explain the increased overnight DNL observed in NAFLD, and simple substrate push could explain acute changes in lipogenic flux. In this regard, lipogenic protein expression, and therefore lipogenic capacity, may be maintained by a combination of factors. First, insulin-resistant individuals likely maintain residual insulin-driven expression of lipogenic enzymes through SREBP1c activation; just as EGP in an insulin-resistant individual will suppress in the face of excess circulating insulin, SREBP1c may still be stimulated in the setting of chronic hyperinsulinemia. Second, lipogenic genes are also expressed downstream of substrate-regulated lipogenic transcription factors. To this latter point, monosaccharides have been shown to activate ChREBP and other lipogenic transcription factors (including SREBP1c, liver X receptor, and peroxisome proliferator–activated receptor-γ [13]) by insulin-independent mechanisms. Note that this model likely does not apply to individuals with a complete loss of insulin signaling, such as those with leprechaunism or Rabson-Mendenhall syndrome, because some residual/basal insulin action is likely required to maintain hepatic lipogenic capacity. However, in the usual insulin-resistant patient, no increase in hepatic insulin action need be invoked to explain increased DNL; more likely, it is some combination of excess dietary lipogenic substrate (including not only carbohydrates like fructose but also amino acids, lactate, and citrate) that increases DNL in insulin-resistant individuals.
One of the more surprising findings of this study was that in subjects with NAFLD, the incorporation of de novo synthesized palmitate into VLDL triglycerides appeared to decrease after the administration of a glucose drink. We speculate that this is simply a reflection of how little glucose and insulin are able to stimulate hepatic DNL in patients with NAFLD. The de novo synthesized palmitate observed in the overnight-fasted blood samples largely reflects DNL after the previous night’s dinner meal. In the absence of significant DNL during the course of the experiment, the continued NEFA flux from adipose tissue lipolysis (which was increased in subjects with NAFLD) drives more preformed fatty acids into VLDL triglycerides (11), thereby leading to dilution of the de novo synthesized palmitate pool. This dilution likely explains the apparently negative DNL rates in glucose-stimulated subjects with NAFLD.
Clinically, it will be highly relevant to determine why hepatic ChREBP is constitutively more active in individuals with NAFLD because this may open up new therapeutic avenues. We speculate that multiple sources may contribute to increased intrahepatic carbohydrate availability: 1) dietary carbohydrate (small differences in long-term carbohydrate consumption may not have been picked up by our diet journal analyses), 2) decreased glucose disposal into muscle and adipose tissues whereby circulating glucose may be diverted to and cleared by the liver (12), or 3) increased adipose tissue lipolysis that drives hepatic gluconeogenesis through activation of the pyruvate carboxylase flux by acetyl-CoA (49). Further research is needed to determine the relative contributions of these and other mechanisms.
As a human subjects study addressing a question of fundamental biology, there are several constraints that limit the interpretation of these data. First, our experiments were designed to evaluate hepatic DNL in humans with established NAFLD and insulin resistance, and we cannot specifically address the role of insulin-mediated lipogenesis in the etiology of NAFLD. However, it would be reasonable to suggest that hyperinsulinemia may significantly contribute to the development of hepatic steatosis (i.e., before the development of hepatic insulin resistance). Second, ideally we would compare insulin-stimulated hepatic insulin signaling to the fasted state, but this would have required a second, clinically unnecessary (and thus ethically questionable) liver biopsy. Our current experimental design, comparing glucose versus fructose ingestion, leaves open the theoretical possibility that there are as-yet unexplored differences in fasted-state lipogenesis. However, lipogenesis predominantly occurs postprandially, and hepatic insulin signaling was not significantly activated under high-fructose/low-insulin conditions. It is thus very unlikely that there were meaningful differences in fasting lipogenic capacity. Third, lipogenesis seen in the fructose versus glucose experiments cannot be quantitatively compared directly because carbohydrate entry into glycolysis versus fructolysis cannot be matched. First-pass hepatic extraction of fructose is nearly complete (12), and the amount of carbohydrate in the portal system was not measured. However, the qualitative differences in responses remain valid: Glucose and insulin stimulate DNL in insulin-sensitive subjects while failing to stimulate incremental DNL in insulin-resistant subjects; fructose stimulated DNL equally in insulin-sensitive and insulin-resistant subjects. Finally, we would have preferred the subjects in each group to be matched with respect to every parameter, except those compared in the experiment. However, in light of the limited pool of participants available for this study, it was not possible to match subjects by age, and the control subjects receiving glucose were younger. Such a difference may have introduced some bias. While age per se is unlikely to have altered hepatic metabolism, aging is associated with increased metabolic disease (including type 2 diabetes). Nevertheless, because tissue-specific insulin action and other parameters associated with metabolic health are reported here, we believe that it is unlikely that this imbalance biased our results in a meaningful way.
In conclusion, this series of translational experiments demonstrates that humans with NAFLD have 1) hepatic and extrahepatic insulin resistance, 2) attenuated hepatic insulin signaling at the level of the insulin receptor, 3) reduced glucose-stimulated/insulin-mediated DNL, 4) intact fructose-stimulated DNL under low-insulin conditions, and 5) constitutively elevated hepatic ChREBP expression with increased overnight DNL. Taken together, increased hepatic DNL in obese humans with NAFLD may be better explained by increased substrate availability rather than by an increase in incremental hepatocellular insulin action. In the end, increased DNL in the insulin-resistant patient is no paradox at all.
Article Information
Funding. K.W.t.H. and M.J.S. are supported by the European Commission (FP7-EU 305707). D.F.V. is supported by the National Institutes of Health (R01-DK-124272 and K23-DK-10287). M.N. is supported by ZonMw (VIDI 016.146.327) and Hartstichting (CVON IN-CONTROL). G.I.S. is supported by the National Institutes of Health (R01-DK-113984, R01-DK-119968, and P30-DK-45735).
Duality of Interest. M.J.S. receives unrestricted research funding from Mediq TEFA (Utrecht, the Netherlands). No other potential conflicts of interest relevant to this article were reported.
Author Contributions. K.W.t.H. performed clinical studies. K.W.t.H., D.F.V., D.Z., G.W.C., M.T.A., G.M.D.-T., and M.N. performed and/or supervised laboratory studies. K.W.t.H., D.F.V., G.I.S., J.A.R., and M.J.S. designed the research, interpreted data, and wrote the manuscript. A.J.N. supervised imaging studies. J.V. evaluated liver histopathology. A.D. and B.A.v.W. performed surgeries. All authors contributed to discussions about results and revised the final manuscript. M.J.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract/poster form at the 79th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 7–11 June 2019, and ObesityWeek 2018, Nashville, TN, 11–15 November 2018.
Footnotes
Netherlands trial reg. no. NTR5351, www.trialregisster.nl
K.W.t.H. and D.F.V. are co-first authors.
G.I.S. and M.J.S. are co-senior authors.
This article contains supplementary material online at https://doi.org/10.2337/figshare.13150568.
References
- 1.Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 2000;6:77–86 [PubMed] [Google Scholar]
- 2.Kubota N, Kubota T, Kajiwara E, et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat Commun 2016;7:12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han J, Wang Y. mTORC1 signaling in hepatic lipid metabolism. Protein Cell 2018;9:145–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018;98:2133–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samuel VT, Liu ZX, Wang A, et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 2007;117:739–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen MC, Madiraju AK, Gassaway BM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 2016;126:4361–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ter Horst KW, Gilijamse PW, Versteeg RI, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep 2017;19:1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011;108:16381–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Summers SA, Goodpaster BH. CrossTalk proposal: intramyocellular ceramide accumulation does modulate insulin resistance. J Physiol 2016;594:3167–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summers SA, Garza LA, Zhou H, Birnbaum MJ. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol Cell Biol 1998;18:5457–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vatner DF, Majumdar SK, Kumashiro N, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci U S A 2015;112:1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ter Horst KW, Serlie MJ. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 2017;9:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 2016;126:12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SS, Park SH. Radiologic evaluation of nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:7392–7402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 2005;288:E462–E468 [DOI] [PubMed] [Google Scholar]
- 16.Fried M, Yumuk V, Oppert JM, et al.; European Association for the Study of Obesity; International Federation for the Surgery of Obesity - European Chapter . Interdisciplinary European Guidelines on metabolic and bariatric surgery. Obes Facts 2013;6:449–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Valk F, Hassing C, Visser M, et al. The effect of a diiodothyronine mimetic on insulin sensitivity in male cardiometabolic patients: a double-blind randomized controlled trial. PLoS One 2014;9:e86890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dulai PS, Sirlin CB, Loomba R. MRI and MRE for non-invasive quantitative assessment of hepatic steatosis and fibrosis in NAFLD and NASH: clinical trials to clinical practice. J Hepatol 2016;65:1006–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefan D, Cesare FD, Andrasescu A, et al. Quantitation of magnetic resonance spectroscopy signals: the jMRUI software package. Meas Sci Technol 2009;20:1040355 [Google Scholar]
- 20.ter Horst KW, Gilijamse PW, Koopman KE, et al. Insulin resistance in obesity can be reliably identified from fasting plasma insulin. Int J Obes 2015;39:1703–1709 [DOI] [PubMed] [Google Scholar]
- 21.ter Horst KW, van Galen KA, Gilijamse PW, et al. Methods for quantifying adipose tissue insulin resistance in overweight/obese humans. Int J Obes 2017;41:1288–1294 [DOI] [PubMed] [Google Scholar]
- 22.Diraison F, Yankah V, Letexier D, Dusserre E, Jones P, Beylot M. Differences in the regulation of adipose tissue and liver lipogenesis by carbohydrates in humans. J Lipid Res 2003;44:846–853 [DOI] [PubMed] [Google Scholar]
- 23.Jones PJ Tracing lipogenesis in humans using deuterated water. Can J Physiol Pharmacol 1996;74:755–760 [PubMed] [Google Scholar]
- 24.Murphy EJ Stable isotope methods for the in vivo measurement of lipogenesis and triglyceride metabolism. J Anim Sci 2006;84(Suppl.):E94–E104 [DOI] [PubMed] [Google Scholar]
- 25.American Diabetes Association 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020;43(Suppl. 1):S14–S31 [DOI] [PubMed] [Google Scholar]
- 26.ter Horst KW, Gilijamse PW, Demirkiran A, et al. The FGF21 response to fructose predicts metabolic health and persists after bariatric surgery in obese humans. Mol Metab 2017;6:1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleiner DE, Brunt EM, Van Natta M, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–1321 [DOI] [PubMed] [Google Scholar]
- 28.Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanyal AJ, Brunt EM, Kleiner DE, et al. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011;54:344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ackermans MT, Pereira Arias AM, Bisschop PH, Endert E, Sauerwein HP, Romijn JA. The quantification of gluconeogenesis in healthy men by (2)H2O and [2-(13)C]glycerol yields different results: rates of gluconeogenesis in healthy men measured with (2)H2O are higher than those measured with [2-(13)C]glycerol. J Clin Endocrinol Metab 2001;86:2220–2226 [DOI] [PubMed] [Google Scholar]
- 31.Sondermeijer BM, Battjes S, van Dijk TH, et al. Lactate increases hepatic secretion of VLDL-triglycerides in humans. Atherosclerosis 2013;228:443–450 [DOI] [PubMed] [Google Scholar]
- 32.Ruijter JM, Lorenz P, Tuomi JM, Hecker M, van den Hoff MJ. Fluorescent-increase kinetics of different fluorescent reporters used for qPCR depend on monitoring chemistry, targeted sequence, type of DNA input and PCR efficiency. Mikrochim Acta 2014;181:1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steele R Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci 1959;82:420–430 [DOI] [PubMed] [Google Scholar]
- 34.Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 1987;36:914–924 [DOI] [PubMed] [Google Scholar]
- 35.Frayn KN Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 1983;55:628–634 [DOI] [PubMed] [Google Scholar]
- 36.Ferrannini E The theoretical bases of indirect calorimetry: a review. Metabolism 1988;37:287–301 [DOI] [PubMed] [Google Scholar]
- 37.Diraison F, Pachiaudi C, Beylot M. In vivo measurement of plasma cholesterol and fatty acid synthesis with deuterated water: determination of the average number of deuterium atoms incorporated. Metabolism 1996;45:817–821 [DOI] [PubMed] [Google Scholar]
- 38.Lee WN, Bassilian S, Ajie HO, et al. In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am J Physiol 1994;266:E699–E708 [DOI] [PubMed] [Google Scholar]
- 39.Wadke M, Brunengraber H, Lowenstein JM, Dolhun JJ, Arsenault GP. Fatty acid synthesis by liver perfused with deuterated and tritiated water. Biochemistry 1973;12:2619–2624 [DOI] [PubMed] [Google Scholar]
- 40.Kotronen A, Vehkavaara S, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H. Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab 2007;293:E1709–E1715 [DOI] [PubMed] [Google Scholar]
- 41.ter Horst KW, Gilijamse PW, Ackermans MT, et al. Impaired insulin action in the liver, but not in adipose tissue or muscle, is a distinct metabolic feature of impaired fasting glucose in obese humans. Metabolism 2016;65:757–763 [DOI] [PubMed] [Google Scholar]
- 42.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith GI, Shankaran M, Yoshino M, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 2020;130:1453–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otero YF, Stafford JM, McGuinness OP. Pathway-selective insulin resistance and metabolic disease: the importance of nutrient flux. J Biol Chem 2014;289:20462–20469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herman MA, Peroni OD, Villoria J, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012;484:333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flannery C, Dufour S, Rabøl R, Shulman GI, Petersen KF. Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 2012;61:2711–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabøl R, Petersen KF, Dufour S, Flannery C, Shulman GI. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proc Natl Acad Sci U S A 2011;108:13705–13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perry RJ, Camporez JG, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015;160:745–758 [DOI] [PMC free article] [PubMed] [Google Scholar]