

Abstract
Urbanization represents a fierce driver of phenotypic change, yet the molecular mechanisms underlying observed phenotypic patterns are poorly understood. Epigenetic changes are expected to facilitate more rapid adaption to changing or novel environments, such as our towns and cities, compared with slow changes in gene sequence. A comparison of liver and blood tissue from great tits Parus major originating from an urban and a forest site demonstrated that urbanization is associated with variation in genome‐wide patterns of DNA methylation. Combining reduced representation bisulphite sequencing with transcriptome data, we revealed habitat differences in DNA methylation patterns that suggest a regulated and coordinated response to the urban environment. In the liver, genomic sites that were differentially methylated between urban‐ and forest‐dwelling birds were over‐represented in regulatory regions of the genome and more likely to occur in expressed genes. DNA methylation levels were also inversely correlated with gene expression at transcription start sites. Furthermore, differentially methylated CpG sites, in liver, were over‐represented in pathways involved in (i) steroid biosynthesis, (ii) superoxide metabolism, (iii) secondary alcohol metabolism, (iv) chylomicron remodelling, (v) cholesterol transport, (vi) reactive oxygen species (ROS) metabolic process and (vii) epithelial cell proliferation. This corresponds with earlier studies identifying diet and exposure to ROS as two of the main drivers of divergence between organisms in urban and nonurban environments. Conversely, in blood, sites that were differentially methylated between urban‐ and forest‐dwelling birds were under‐represented in regulatory regions, more likely to occur in nonexpressed genes and not over‐represented in specific biological pathways. It remains to be determined whether diverging patterns of DNA methylation represent adaptive evolutionary responses and whether the conclusions can be more widely attributed to urbanization.
Keywords: birds, DNA methylation, epigenetics, RRBS, urbanization
1. INTRODUCTION
The ability for organisms to adapt quickly is particularly pertinent in the context of current unprecedented rates of global environmental change (Rivkin et al., 2019; Schell, 2018). Urbanization—the process of formation and expansion of towns and cities—is proceeding at a phenomenal rate, across the world (United Nations, 2018), exerting novel selection pressures on free‐living organisms. Persistence of a species within the urban environment likely requires rapid adaptation to the wide range of novel environmental stressors to which animals find themselves subjected (Isaksson, 2015). Organisms living in the urban environment commonly show marked differences in a suite of phenotypic traits, relative to their countryside‐dwelling conspecifics (Cheptou et al., 2008; Chick et al., 2019; Lyons et al., 2017; Salmón et al., 2017; Slabbekoorn & Peet, 2003; Tennessen et al., 2014). However, to what extent variation at the phenotypic level, between urban and nonurban conspecifics, is driven by differences in genetic or nongenetic variation remains unclear.
While the process of genetic adaptation is usually slow and takes several generations to spread within a population, epigenetic modifications—broadly described as alterations to the structure of DNA—occur at a higher rate than changes to the DNA sequence. The epigenome is therefore expected to evolve faster than the genome, and epigenetic changes could be crucial in facilitating rapid adaptation in changing or novel environments, such as the world's growing towns and cities (Bossdorf et al., 2008; Hu et al., 2019; Hu & Barrett, 2017; Jablonka & Raz, 2009; Verhoeven et al., 2016). Indeed, epigenetic divergence has been shown to occur prior to genetic divergence following reproductive isolation in fish (Smith et al., 2016), and high epigenetic diversity, yet low genetic diversity, was found in an introduced population of a passerine bird (Liebl et al., 2013) and among closely related species of Darwin's finches (Skinner et al., 2014). Furthermore, accumulating evidence indicates that epigenetic marks can be induced or removed in response to environmental cues, within the lifetime of an individual (Bollati & Baccarelli, 2010; Gugger et al., 2016; Lea et al., 2016). Despite this suggested reversible plasticity, there is evidence that, if epigenetic modifications occur in early life, phenotypic traits can be permanently affected (Waterland & Jirtle, 2003; Weaver et al., 2004) with consequences for fitness (Rubenstein et al., 2016). Moreover, if environmentally induced epigenetic changes can be stably inherited across multiple generations, epigenetics has the potential to drive the evolutionary trajectory of populations in response to environmental change (Jablonka & Raz, 2009).
DNA methylation, involving the addition of a methyl group (‐CH3) to the 5' carbon of cytosines, is the most widely studied epigenetic mark and plays a key role in regulating gene expression and genome stability (Bird, 2002). Methylation of DNA primarily occurs at cytosine residues within CpG nucleotides, which are concentrated in gene regulatory regions (e.g. promoters and transcription start sites). Changes in DNA methylation can alter expression of associated genes, but the direction of the effect depends upon the genomic location. While methylation at CpG‐rich promoters is often associated with stable repression of gene expression, that is gene silencing, DNA methylation within the gene body has been shown to be positively correlated with gene expression in mammalian tissues (Jones, 2012; Lev Maor et al., 2015). Increasingly, DNA methylation patterns are being shown to be responsive to environmental cues, in wild (Kilvitis et al., 2019; Laubach et al., 2019; Lea et al., 2016) and captive (Jimeno et al., 2019; Viitaniemi et al., 2019) animal populations, suggesting a critical role for DNA methylation as a rapid regulator of phenotypic divergence and adaptation.
Genomic and epigenomic studies offer great potential to understand evolution in cities, yet they remain few and far between (Rivkin et al., 2019; Schell, 2018). While some studies have found genomic signatures of urban adaptation (Harris et al., 2013; Salmón et al., 2020) and consistent differences at the transcriptomic level (Watson et al., 2017), others have found no genetic structure among urban and nonurban congeners (Khimoun et al., 2020). Modifications in DNA methylation have been shown to associate with anthropogenic‐linked factors including feeding on anthropogenic‐provided food in baboons (Lea et al., 2016), exposure to pollution in humans (Baccarelli et al., 2009), and stress resilience in response to handling in tree swallows (Taff et al., 2019). A few studies have specifically investigated epigenetic variation in wild animals in the context of urbanization, revealing varied patterns (Garcia et al., 2019; McNew et al., 2017; Riyahi et al., 2015; Thorson et al., 2019).
In the present study, we investigated variation in genome‐wide DNA methylation of urban‐ and forest‐dwelling great tits Parus major with the aim of elucidating the potential for DNA methylation to mediate well‐established phenotypic differences between the two environments. Urban and nonurban populations of great tits have been shown to differ in respect of oxidative stress physiology (Salmón, Watson, et al., 2018), telomere dynamics (Salmón et al., 2016), immune function (Bailly et al., 2016), breeding performance (Sprau et al., 2017) and survival (Salmón et al., 2017), among other traits. In general, the urban environment appears to be more costly for great tits, but there is evidence for potential adaptation, with upregulation of expression of genes involved in protection against oxidative stress and immune function observed in our urban study population (Watson et al., 2017).
Using reduced representation bisulphite sequencing (RRBS), we characterized CpG methylation patterns in the liver and blood of great tits originating from an urban and a forest environment, and we investigated the functional relevance of observed environmentally induced differences in DNA methylation. Both the avian liver and blood have been shown to be sensitive to urbanization‐associated factors, such as changes in diet (Andersson et al., 2015; Møller et al., 2010) and pollutant exposure (Koivula & Eeva, 2010), and our previous research revealed concordant changes in gene expression in the two tissues, in response to urbanization (Watson et al., 2017). We predict that, if differences in DNA methylation patterns between urban‐ and forest‐dwelling birds represent regulated and coordinated responses associated with urbanization, CpG sites of differential methylation should be over‐represented in (i) regions of the genome regulating gene expression; (ii) regulatory regions associated with expressed, rather than nonexpressed genes; (iii) genomic regions involved in biological pathways relevant to response or adaptation to the urban environment (see below); and, (iv) methylation should correlate with expression of associated genes. Based on our previous findings (Salmón, Stroh et al., 2018; Salmón, Watson, et al., 2018; Watson et al., 2017), we predict that coordinated differences in DNA methylation would be associated with genes involved in immune function, protection against oxidative stress and lipid metabolism.
2. MATERIALS AND METHODS
2.1. Study species and sites
Twelve wild male great tits—six from an urban environment and six from a forest environment—were captured in late winter (February/March 2014) in southern Sweden. Urban birds were captured in urban parkland in Malmö (55°35'N 12°59'E)—the third largest city in Sweden with c. 300,000 inhabitants. Forest birds were captured in Vombs fure (55°39'N 13°33'E), a dense forest surrounded by areas of low human habitation (<5 inhabitants km−2) and located about 36 km east of the urban site. Biometrics were recorded, and a blood sample was collected and immediately transferred to storage at −80°C. Birds were subsequently euthanized, and postmortems were carried out immediately; liver tissue was collected and transferred to storage at −80°C within 5 min of death. All birds were adults: forest birds were all aged as 3 years or older; three of the urban birds were aged 2 years, while the other three were aged as 3 years or older. Due to high adult mortality (61%) and low recruitment rates (7%) at our sites and dispersal of great tit siblings (Dingemanse et al., 2003), genetic relatedness among birds within each habitat is expected to have minimal effect. Furthermore, genetic differentiation between the two sites is very low (FST = 0.0055, (Salmón et al., 2020)), and all birds can be considered to originate from a single population.
2.2. DNA isolation, bisulphite treatment and sequencing
DNA was isolated from 5 µl whole blood and 25 mg liver tissue using NucleoSpin Blood and Tissues kits (Macherey‐Nagel, Germany), respectively, and according to the manufacturer's protocol. Sample quality was assessed using ultraviolet spectrophotometry (NanoDrop, Thermo Scientific) and gel electrophoresis. Average (median ± SD) absorption ratios were 1.95 ± 0.08 (260/280) and 2.57 ± 0.13 (260/230). RRBS library preparation and sequencing were carried out by the SNP&SEQ technology platform (Sweden). RRBS libraries were prepared using 270 ng DNA and an in‐house protocol; DNA was cut using the MspI restriction enzyme and then subjected to bisulphite treatment. Paired‐end (125 bp read length) sequencing was performed using the Illumina HiSeq 2,500 platform with high output mode, v4 chemistry and 12 samples per lane. A PhiX spike‐in of 30% was included.
2.3. Reduced representation bisulphite sequencing sequence alignment and methylation calling
Raw reads are deposited in NCBI's Sequence Read Archive (PRJNA314210, NCBI BioProject, 2017). Raw reads were processed for adapter trimming and removal of low‐quality reads and artificially added bases using Trim Galore! 0.4.4 (Krueger, 2015) with the parameters ‐‐rrbs ‐‐paired ‐‐max_n 1 ‐‐quality 25 ‐‐length 40 ‐‐clip_R2 5. Quality filtering resulted in an average (mean ± SE) of 16.48 ± 2.06 million and 19.94 ± 2.16 million high‐quality paired‐end reads from liver and blood tissues, respectively, per individual. We tested for differences in the total number of CpG sites sequenced (n = 6 birds/habitat; unpaired) using a two‐sample Wilcoxon signed‐rank test in R 3.6.1 (R Core Team, 2013). Trimmed reads were aligned to the great tit genome (Laine et al., 2016) (assembly: Parus_major1.1; RefSeq accession: GCF_001522545.2) using Bismark 0.18.2 (Krueger & Andrews, 2011) in the mode for paired‐end nondirectional RRBS data, including the parameters ‐‐bowtie2 ‐‐fastq ‐X 1,000 ‐‐score_min L,0,‐0.6. An average of 54.1% (liver) and 54.2% (blood) reads aligned uniquely, consistent with other reports (Chatterjee et al., 2012; Yuan et al., 2016). Methylation calling was performed using the bismark_methylation_extractor tool with the parameters ‐‐paired‐end ‐‐no_overlap ‐‐ignore_r2 2. Coverage files were analysed in methylKit (Akalin et al., 2012) to call differentially methylated sites (DMSs) between urban and forest birds, merging reads on both strands of a CpG dinucleotide, including only CpG sites covered in all samples, and discarding bases that had a read coverage lower than ten and with more than the 99.9th percentile coverage in each sample. CpG sites were considered differentially methylated (and thus associated with urbanization) with a methylation difference larger than 25% and a q‐value <0.01 in line with other recent studies (Aluru et al., 2018; McCormick et al., 2017). P‐values were adjusted to q‐values using the SLIM method (Wang et al., 2011). Permutation tests were performed by carrying out the differential methylation analysis, with 1,000 iterations, each time permuting habitat assignments of individuals; the observed differences in DNA methylation were compared to the permutation distribution.
2.4. RNA‐seq alignment
RNA sequencing of the liver and blood transcriptomes from the same 12 individuals was carried out for an earlier study (Watson et al., 2017), and raw reads are deposited in NCBI's Sequence Read Archive (PRJNA314210, NCBI BioProject, 2017). Read quality was first appraised using FastQC 0.11.5 software (Andrews, 2010), and quality trimming and filtering were applied to remove low‐quality reads and adaptor contamination in Trimmomatic (Bolger et al., 2014). Clean reads were aligned to annotated regions of the great tit genome (see above) using HISAT 2.1.0 (Kim et al., 2015) and sorted with SAMtools 1.5 (Li et al., 2009). Counts were made using StringTie 1.3.3 (Pertea et al., 2015) in accordance with the protocol described by Pertea et al. (2016). Genes were classified as being expressed if a read count of ten or higher was found in three or more (≥25%) individuals. This represented a slightly more conservative threshold than used by Lea et al. (2016) with a bigger sample size.
2.5. Genomic locations and gene annotation
In order to evaluate the distribution of sequenced CpG sites throughout the genome, coverage thresholds were relaxed to a minimum of 3 reads in at least one individual. CpG sites were assigned to genomic locations and genes using annotated data from the great tit genome. Sites were assigned to either a) gene body, b) promoter, c) transcription start site (TSS), d) CpG island (CGIs), e) CpG island shore or f) nonregulatory region. Promoters were defined as the region 3 kb upstream from gene start (excluding TSS); TSS regions were defined as the region from 300 bp upstream and 50 bp downstream of gene start; and, CGIs were defined as regions where, over an average of 10 windows of 100 bp and a minimum of 200 bases, the calculated GC content is>=50% and the ratio of observed to expected GC dinucleotides is 0.6, all as per Viitaniemi et al. (2019). CpG island shores were defined as 2 kb regions flanking CpG islands (Irizarry et al., 2009; Rao et al., 2013). Overlap between CpG sites and regulatory regions was determined by intersection using BEDtools 2.27.1 (Quinlan & Hall, 2010), and CpG sites were assigned to one or more specific genes, where overlaps were identified. Prior to the assignment, overlapping regions annotated to the same gene were merged.
2.6. Over‐representation of differentially methylated sites
If changes in DNA methylation associated with urbanization represent coordinated responses to the environment, it is expected that differences in methylation should (i) occur in regions of the genome regulating gene expression; (ii) occur in regulatory regions associated with expressed, rather than nonexpressed genes; (iii) be concentrated in genomic regions involved in related biological pathways; and (iv) relate to changes in gene expression of associated genes. We addressed each of these predictions in turn, as described below. All analyses were performed in R 3.6.1 (R Core Team, 2013) using DMSs as called in the pipeline described above.
Using Fisher's exact test, we tested whether (i) DMSs are over‐represented in functional (i.e. regulatory) regions of the genome and under‐represented in regions with no regulatory function, and (ii) DMSs are more likely to occur in expressed genes. Since RRBS enriches for likely functional areas of the genome, the sequenced CpG sites are expected to be over‐representative of these functional genomic regions. Over‐representation was therefore assessed relative to the background of all sequenced CpG sites. The existence of multiple DMSs in close proximity to one another is more likely to be of functional relevance for gene expression (Lister et al., 2009). We therefore also tested for over‐representation of urbanization‐linked sites in expressed genes, by considering only those DMSs occurring within “differentially methylated regions” (DMRs), defined as the occurrence of three or more urbanization‐linked sites within a 2 kb window, following Lister et al. (2009). For the assessment of over‐representation of DMRs in expressed genes, the background dataset similarly comprised only CpG sites that were located within 2 kb of two or more other CpG sites.
To investigate the functional relevance of differences in DNA methylation, we performed gene over‐representation analyses to test whether (iii) urbanization‐linked CpG sites are over‐represented in or near genes involved in related and relevant biological pathways. Over‐representation analyses were carried out using the BiNGO 3.0.2 plug‐in in Cytoscape 3.2.1, using the great tit gene models as the background. Given the expectation that DMRs are of greater functional relevance, we included only those genes associated with DMSs located within a DMR (defined above) and overlapping with either the gene body, promoter or TSS, using the annotation described above. A full ontology file was created using the annotated great tit genome and the core Gene Ontology (GO) downloaded from www.geneontology.org on 13 September 2018. Hypergeometric tests were employed, and P‐values were corrected using the Benjamini and Hochberg false discovery rate (FDR) (Benjamini & Hochberg, 1995). GO terms with FDR < 0.05 were considered to be significantly over‐represented. GO terms were summarized and redundant terms eliminated, using a clustering algorithm based on semantic similarity in Revigo (Supek et al., 2011), and over‐represented terms were visualized in CirGO (Kuznetsova et al., 2019).
Lastly, we tested whether (iv) DNA methylation at individual DMSs is correlated with gene expression, where DMSs were located in the gene body, TSS or promoter region associated with a gene for which expression levels were also quantified. Linear mixed models were fitted to data on gene expression counts with the covariate of methylation level, the fixed factor of feature (three levels: gene body, TSS or promoter) and the interaction between methylation and feature. A random effect of individual was included, though the estimated variance was zero. Prior to analysis, read expression counts were rlog‐transformed in DESeq2 (Love et al., 2014). Models were fitted to data from liver and blood separately. Pairwise comparisons of slopes were carried out using emtrends() and the Tukey method for P‐value adjustment.
3. RESULTS
3.1. Urbanization influences genome‐wide DNA methylation
DNA methylation was quantified at 6,166,831 and 5,130,570 CpG sites across the great tit genome, in liver and blood, respectively. Average (mean ± SE) read coverage was 8.7 ± 0.04 and 15.7 ± 0.2 across all analysed CpG sites in liver and blood, respectively. There were no consistent differences in the number of CpG sites sequenced in urban birds (n = 6; mean ± SE: liver: 3,645,356 ± 87,634; blood: 3,654,552 ± 42,805), compared with forest birds (n = 6; mean ± SE: liver: 3,513,949 ± 109,839; blood: 3,641,601 ± 38,057) in either liver (W = 22, p = .6) or blood (W = 16, p = .8). A majority of CpG sites were hypomethylated (i.e. median methylation < 50%) in both liver (68.2%) and blood (66.8%). Most CpG sites were methylated at low levels (median ± SD: liver = 11.8 ± 29.4%; blood = 9.09 ± 33.3%), but average methylation ranged from completely unmethylated to constitutively (100%) methylated.
A total of 419 (liver) and 1,144 (blood) CpG sites were differentially methylated between urban and forest birds, and thus from hereon, we refer to these CpG sites and the observed differences in their DNA methylation levels, as being linked to or associated with urbanization (Figure 1). Average (mean ± SE) read coverage at DMSs was 33.3 ± 1.3 and 34.1 ± 0.6 in liver and blood, respectively. Permutation tests revealed that the observed differences in DNA methylation were significantly different to that expected by chance (liver: p = .002; blood: p = .01). Of the differentially methylated sites, in liver, the majority (88.5%) were hypermethylated in urban, compared with forest, birds. In blood, similar proportions of urbanization‐linked CpG sites were hypomethylated (49.3%) and hypermethylated (50.7%) in urban, compared with forest, birds. In liver, 19 DMRs (defined as three or more DMSs within a 2 kb window) were identified, involving 102 DMSs; seventeen (89.5%) DMRs exhibited constitutive hypermethylation (across the constituent DMSs) in urban, compared with forest, birds. A particularly large DMR was identified on the Z chromosome in liver tissue, comprising a total of 36 urbanization‐linked CpG sites within a 300 bp window, but none of the CpG sites were associated with any annotated genes. In blood, 54 DMRs were identified, involving 226 DMSs; eighteen DMRs (33.3%) were constitutively hypomethylated and 14 (25.9%) were constitutively hypermethylated.
Figure 1.
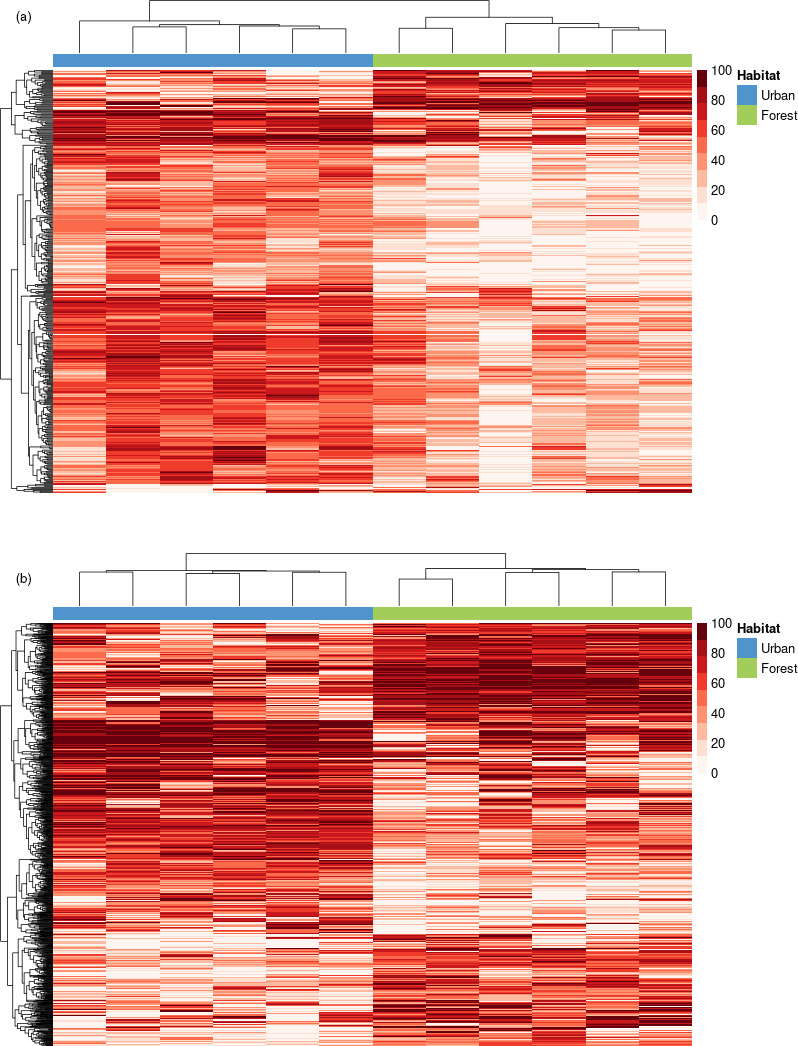
Hierarchical clustering of differentially methylated sites in urban‐ and forest‐dwelling great tits Parus majorin liver (a) and blood (b). Each column represents an individual from either the urban (blue) or forest (green) habitat. Each row represents a CpG site at which DNA methylation levels were significantly different between the two habitats. The colour scale represents percent DNA methylation, with higher methylation indicated by a darker shade of red
3.2. Urbanization‐linked sites are over‐represented in certain genomic regions
In accordance with the fact that RRBS enriches for likely functional areas of the genome, a large proportion of sequenced CpG sites occurred within regulatory genomic regions (Table 1; liver: 88.5%, blood: 91.8%). Overall, urbanization‐associated CpG sites were over‐represented in regulatory regions of the genome, in liver (odds ratio = 1.5, p = .009), but they were under‐represented in regulatory regions in blood (odds ratio = 0.61, p < .001; Table 1; Figure 2). Specifically, DMSs were less likely to occur within a promoter, TSS, gene body (all p < .001) or CGI (blood only: p < .001), but were more likely to be located within CGI shores, the regulatory regions flanking CpG islands in both liver (odds ratio = 1.8, p < .001) and blood (odds ratio = 2.02, p < .001). In liver, there was no difference in the frequency of occurrence of CpG sites in CGIs between the DMS and background data sets (odds ratio = 1.04, p = .7). DMSs were associated with 220 unique genes in liver and 492 unique genes in blood.
Table 1.
Summary of genomic locations of all sequenced CpG sites and differentially methylated sites (DMSs) between urban and forest great tits in liver and blood*
| Total | Promoter | TSS | Gene body | CGI | CGI shore** | Nonregulatory | |
|---|---|---|---|---|---|---|---|
| Liver | |||||||
| All CpG | 6,166,831 | 2,440,376 (39.6%) | 1,508,486 (25.5%) | 3,961,469 (64.2%) | 3,002,199 (48.7%) | 1,316,950 (21.4%) | 711,340 (11.5%) |
| DMSs | 419 | 93 (22.2%) | 31 (7.4%) | 224 (53.5%) | 208 (49.6%) | 137 (32.7%) | 33 (7.9%) |
| Blood | |||||||
| All CpG | 5,130,570 | 2,197,364 (42.8%) | 1,346,853 (26.3%) | 3,339,042 (65.1%) | 2,882,780 (56.2%) | 1,148,229 (22.4%) | 421,862 (8.2%) |
| DMSs | 1,144 | 173 (15.1%) | 69 (6.0%) | 654 (57.2%) | 417 (36.5%) | 422 (36.9%) | 146 (12.8%) |
Coverage thresholds adopted were 3x for all CpG sites and 10x for DMSs.
Abbreviations: CGI, CpG island; TSS, transcription start site.
Note that CpG sites can be associated with one of more genomic features.
CGI shores are defined as 2 kb regions flanking CpG islands.
Figure 2.
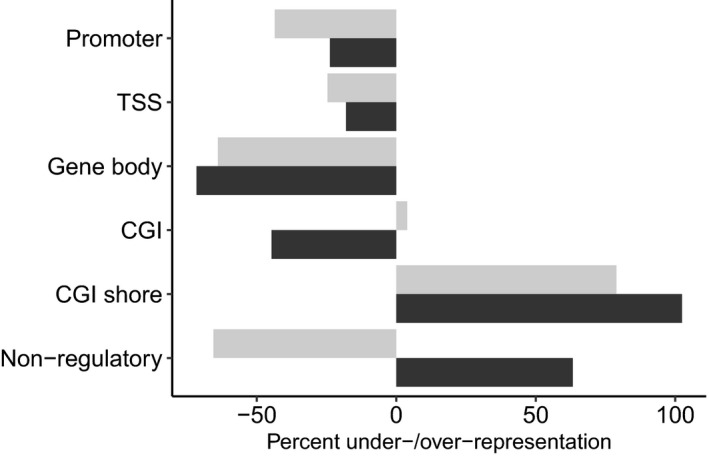
CpG sites associated with urbanization were distributed nonrandomly throughout the genome of great titsParus major. Under‐/over‐representation of urbanization‐associated sites in various regulatory and nonregulatory regions of the genome is illustrated in the liver (grey) and blood (black). Urbanization‐linked differences in DNA methylation were under‐represented in the regulatory regions of promoters, transcription start sites (TSS), gene bodies and CpG islands (CGI; blood only), but were more likely to occur within CGI shores, the regulatory regions flanking CpG islands. Differentially methylated sites were under‐represented in nonregulatory regions in liver, but over‐represented in un‐annotated regions in blood
Urbanization‐linked CpG sites were not more likely to occur in regulatory regions associated with expressed genes, compared with nonexpressed genes, in either liver (odds ratio = 0.99, p = .6) or blood (odds ratio = 0.56, p = 1). In fact, DMSs in blood were more likely (p < .001) to occur in or close to genes that were not expressed (37.5%), compared with the background CpG sites (25.1%). When considering only those differentially methylated sites located within DMRs, in liver, all sites (100%) were located within regulatory regions associated with expressed genes, and this was significantly more so than in the background data set (84.6%; odds ratio = inf.; p < .001). In blood, DMSs located within DMRs were again less likely to be located in regions associated with expressed genes (68.1%), compared with the background (75.0%; odds ratio = 0.71; p = .04).
3.3. Functional relevance of urbanization‐associated sites
Over‐representation analyses revealed that urbanization‐associated regions, in liver, were over‐represented in a few related biological pathways, but there was no such over‐representation among differentially methylated regions in blood. Sixteen genes associated with DMRs in liver and with ontology annotation (a further four genes had no ontology information available) matched to 1,109 GO terms. Of the associated GO terms, 206 terms were significantly over‐represented at an FDR of 0.05. In blood, 26 (out of 34) genes associated with DMRs had available ontology annotation and matched to 1,037 GO terms, but none of the terms were over‐represented at an FDR level of either 0.05 or 0.1. Elimination of redundant GO terms associated with DMRs in liver tissue, using a semantic similarity threshold of 0.4, yielded a subset of 30 representative GO terms, providing a succinct summary of over‐represented pathways (Table 1; Figure 3). The GO terms of highest significance (FDR ≤ 0.002) and lowest dispensability (<0.1) were as follows: (i) steroid biosynthetic process, (ii) superoxide metabolic process, (iii) secondary alcohol metabolic process, (iv) chylomicron remodelling, (v) cholesterol transport, (vi) reactive oxygen species metabolic process and (vii) epithelial cell proliferation.
Figure 3.
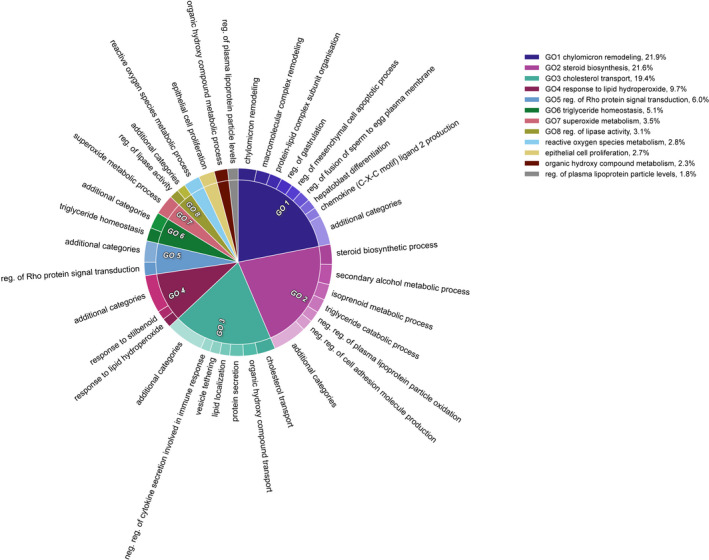
Representation of functional role of genomic regions that are differentially methylated between urban‐ and forest‐dwelling great tits Parus major, in liver tissue. Nonredundant over‐represented gene ontology (GO) terms are grouped by hierarchical clustering. Parent terms are identified in the legend along with their respective proportions, which are directly proportional to statistical significance. GO terms were first summarized based on a semantic similarity of 0.4 using REVIGO and visualized in CirGO. Abbreviations: neg., negative; pos., positive; reg., regulation
We identified seven genes associated with urbanization‐linked CpG sites and whose functions are linked to the most significant over‐represented pathways in the gene ontology network, as highlighted above. These include three genes with key roles in cholesterol transport and metabolism: the apolipoprotein encoded by APOA1 modulates reverse transport of cholesterol from tissues to liver; APOA4 encodes an apolipoprotein that forms a component of chylomicrons and high‐density lipoproteins; and STARD3 encodes a sterol‐binding protein that also mediates cholesterol transport. Another gene, HNF1B, is also linked to cholesterol transport via its involvement in secretion by cells, as well as being linked to epithelial cell proliferation. Farnesyl pyrophosphate synthase, encoded by FDPS, catalyses a pathway upstream of many essential metabolites including sterols and carotenoids. NOX5 and NOXA1 are major sources of superoxide generation and involved in the regulation of redox‐dependent processes. Each of these genes has a cluster of between three and five differentially methylated sites associated with the gene body, promoter and/or TSS region. All, except one, of the involved CpG sites are hypermethylated in urban, compared with forest, birds.
3.4. DNA methylation is linked to gene expression
Significant variation in gene expression was explained by variation in methylation level at associated DMSs, but the slope of the correlation depended on whether an urbanization‐associated CpG site was located in the gene, promoter or TSS region, in both liver (methylation*feature: F 2,4,842 = 2.93, p = .05; Figure 4a) and blood (methylation*feature: F 2,12,738 = 3.32, p = .04; Figure 4b). Post hoc tests confirmed that gene expression was negatively correlated with DNA methylation when a DMS was located in a TSS in liver (slope = −3.1e−2, 95% CI = −4.9e−2‐−1.3e−2) and blood (slope = −1.1e−2, 95% CI = −2.0e−2‐‐3.3e−3), and the slopes were either significantly different, or close to being significantly different, from that of either gene body (liver: p = .07; blood: p = .05) or promoter (liver: p = .05, blood: p = .04) regions. Expression was also negatively correlated with methylation in gene bodies in liver (slope = −9.6e−3, 95% CI = −1.6e−2‐‐2.7e−3). The estimated slopes of the relationship between expression and methylation were not significantly different to zero, when a DMS was located in the promoter in both tissues (liver: slope = −6.6e3, 95% CI = −1.7e−2–3.2e−3; blood: slope = 1.0e−3, 95% CI = −4.7e−3 ––6.7e‐3) and when a DMS was located in a gene body in blood (slope = −9.3e−4, 95% CI = −4.1e−3–2.2e−3). The slopes for gene and promoter regions were not significantly different from one another (liver: p = .9; blood: p = .8). A large amount of residual variation in gene expression levels remained, which was not explained by variation in methylation.
Figure 4.
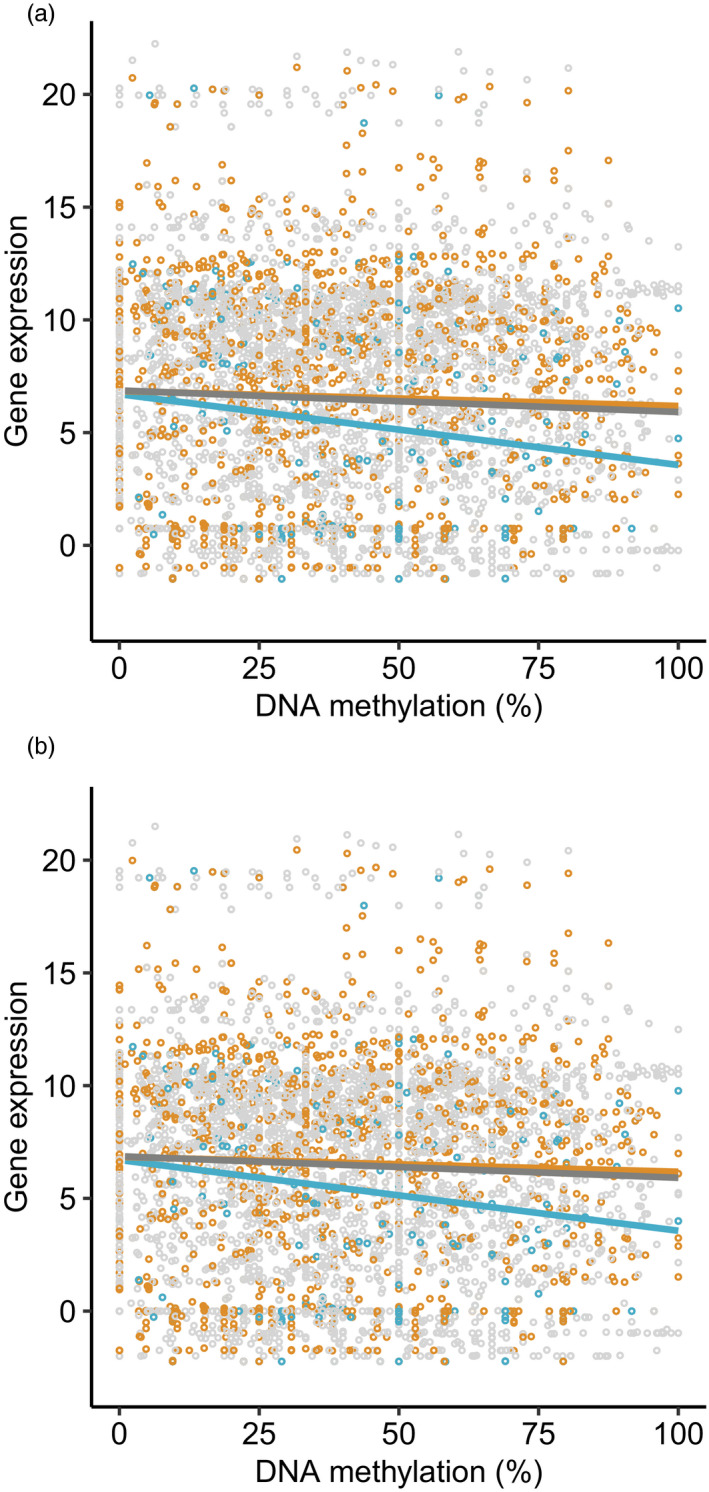
Relationship between gene expression (rlog‐transformed read count) and DNA methylation (%) of urbanization‐associated CpG sites located in gene bodies (grey), promoters (orange) and transcription start sites (blue; TSS) of respective genes in (a) liver and (b) blood of great tits Parus major (n = 12). Lines represent fitted lines from linear models including the interaction between DNA methylation and genomic feature. The slope of the relationship between gene expression and DNA methylation at TSSs was significantly different to zero and different to that of genes or promoters, but there is a large amount of residual variation in expression
4. DISCUSSION
We present one of the first studies to examine DNA methylation patterns in the context of urbanization in vertebrates (Riyahi et al., 2015; McNew et al., 2017; though see Garcia et al., 2019), which provides important advances to our understanding of the role of epigenetic modifications in mediating ecologically relevant phenotypic changes and thereby adaptation. Our results reveal variation in DNA methylation, in both the liver and blood of wild great tits, that corresponds with habitat differences associated with urbanization. Diet and ROS exposure are pinpointed as the possible main drivers of epigenetic divergence between the two populations, based on the biological pathways associated with differences in DNA methylation patterns in the liver. In turn, this suggests that the observed variation in DNA methylation could be driven by variation in food availability and/or pollutant levels, both of which have previously been associated with divergence in DNA methylation patterns (Lea et al., 2016; Romano et al., 2016) and are well understood to characterize key differences between urban and nonurban areas. Furthermore, the results are in accordance with previous studies highlighting marked differences in phenotypic traits, especially those linked to diet and oxidative stress, between urban and nonurban animal populations (Costantini et al., 2014; Isaksson et al., 2007, 2009). However, it is important to acknowledge that, in the absence of replicated sampling sites, we cannot be certain that the results would be reproduced across other populations or that likely causal factors are explicitly a consequence of urbanization rather than other site‐specific differences. Alternatively, variation in DNA methylation could arise as a result of nonadaptive genetic variation between urban and nonurban populations (e.g. drift or ancestry (Husquin et al., 2018)). Further studies are required to determine whether the results can be more widely attributed to urbanization and whether the observed divergent patterns of DNA methylation represent an adaptive evolutionary response.
In line with our predictions associated with a coordinated environmentally induced response, urbanization‐associated CpG sites of differential methylation in liver were (i) over‐represented in regulatory regions of the genome (Figure 2), (ii) more likely to occur in association with expressed genes, (iii) concentrated in relevant biological pathways (Table 2, Figure 3), and (iv) related to gene expression in a manner that was dependent on genomic feature (Figure 4), consistent with other studies (Derks et al., 2016; Jones, 2012). Furthermore, we found almost ubiquitous hypermethylation in the livers of urban, compared with forest, birds, which provides further evidence for a coordinated environmental response (Figure 1). Interestingly, the same pattern of higher levels of DNA methylation in urban birds, relative to rural birds, was also found in two candidate genes linked to personality in great tits (Riyahi et al., 2015).
Table 2.
Functional roles linked to genomic regions that are differentially methylated between urban‐ and forest‐dwelling birds, in liver tissue
| GO ID | GO description | Size | log10 p‐value | Nested terms |
|---|---|---|---|---|
| GO:0006694 | Steroid biosynthetic process | 0.12 | −3.31 | 33 |
| GO:0006801 | Superoxide metabolic process | 0.14 | −3.31 | 9 |
| GO:0034371 | Chylomicron remodelling | 0.00 | −3.00 | 13 |
| GO:1901615 | Organic hydroxy compound metabolic process | 0.83 | −2.20 | 0 |
| GO:0042157 | Lipoprotein metabolic process | 0.21 | −1.46 | 0 |
| GO:0072593 | Reactive oxygen species metabolic process | 0.28 | −2.67 | 0 |
| GO:0030301 | Cholesterol transport | 0.02 | −2.97 | 19 |
| GO:0050673 | Epithelial cell proliferation | 0.07 | −2.62 | 1 |
| GO:0070328 | Triglyceride homeostasis | 0.01 | −2.20 | 7 |
| GO:1902652 | Secondary alcohol metabolic process | 0.10 | −3.31 | 3 |
| GO:0034445 | Negative regulation of plasma lipoprotein particle oxidation | 0.00 | −1.79 | 1 |
| GO:0035023 | Regulation of Rho protein signal transduction | 0.13 | −2.20 | 5 |
| GO:0060191 | Regulation of lipase activity | 0.01 | −1.53 | 5 |
| GO:0097006 | Regulation of plasma lipoprotein particle levels | 0.01 | −1.74 | 0 |
| GO:0099022 | Vesicle tethering | 0.00 | −1.63 | 1 |
| GO:0006982 | Response to lipid hydroperoxide | 0.00 | −1.53 | 2 |
| GO:0071702 | Organic substance transport | 4.98 | −1.35 | 2 |
| GO:0002227 | Innate immune response in mucosa | 0.00 | −1.43 | 5 |
| GO:0034367 | Macromolecular complex remodelling | 0.01 | −2.28 | 0 |
| GO:0060354 | Negative regulation of cell adhesion molecule production | 0.00 | −1.79 | 1 |
| GO:0010470 | Regulation of gastrulation | 0.01 | −1.99 | 55 |
| GO:0015850 | Organic hydroxy compound transport | 0.08 | −2.29 | 1 |
| GO:0061017 | Hepatoblast differentiation | 0.00 | −1.63 | 2 |
| GO:0072567 | Chemokine (C‐X‐C motif) ligand 2 production | 0.00 | −1.53 | 20 |
| GO:0014012 | Peripheral nervous system axon regeneration | 0.00 | −1.43 | 2 |
| GO:0033384 | Geranyl diphosphate biosynthetic process | 0.00 | −1.64 | 1 |
| GO:0071825 | Protein–lipid complex subunit organization | 0.01 | −2.02 | 3 |
| GO:0043012 | Regulation of fusion of sperm to egg plasma membrane | 0.00 | −1.79 | 1 |
| GO:0019433 | Triglyceride catabolic process | 0.01 | −2.29 | 11 |
| GO:0035634 | Response to stilbenoid | 0.00 | −1.47 | 0 |
Significantly (FDR ≤ 0.05) over‐represented gene ontology (GO) terms were summarized based on a semantic similarity of 0.4. Cluster representatives are listed in order of increasing dispensability (i.e. semantic similarity), along with their size, based on the frequency of occurrence in the background GO data (higher frequency = more general terms; lower frequency = more specific terms); adjusted p‐value; and, number of significant GO terms nested within each cluster.
It could be that the urban environment—with its novel stressors or altered resource base—selects for greater phenotypic plasticity, which could be mediated to some extent by DNA methylation. Given the accumulating evidence that conditions experienced during development are crucial in inducing stable phenotypic traits via epigenetic modifications (Jablonka & Raz, 2009; Weaver et al., 2004), it could be that the differences in DNA methylation observed in the present study are established during early life. Indeed, it has been demonstrated that growing up in the urban environment has marked consequences for survival and fitness‐related traits in birds (Salmón et al., 2017; Salmón, Watson, et al. 2018; Sprau et al., 2017), and furthermore, the early life environment has been shown to be a key determinant of DNA methylation patterns in the wild (Laubach et al., 2019; Rubenstein et al., 2016). A key aim for future studies of environmentally induced changes in DNA methylation should be to attempt to disentangle the relative contribution of genetic and environmental variation during the critical period of early life.
While there were differences in DNA methylation patterns in the blood, associated with urbanization, there was little evidence that this reflected a coordinated organismal response to environmental cues: sites of differential methylation between the two habitats were (i) not over‐represented in regulatory regions, (ii) less likely to occur in association with expressed genes, and (iii) not over‐represented in specific biological pathways; however, as in liver, DNA methylation in blood was negatively related to gene expression when DMSs were located in TSS regions. Although other studies have found DNA methylation patterns in blood to reflect environmental and social factors (Jimeno et al., 2019; Lea et al., 2016), tissue‐specific patterns of DNA methylation have been shown in mammals (Christensen et al., 2009) and birds (Siller & Rubenstein, 2019), and it is therefore not expected that all tissues will respond in the same way to environmental cues.
Differentially methylated sites were over‐represented in CGI shores, in both the liver and blood. While it has traditionally been considered that methylation at CGIs themselves is of greatest functional significance, increasing evidence suggests a key role for DNA methylation in the region of CGI shores. It has been recently shown that methylation of DNA in the regions flanking CGIs is more dynamic and variable than that within CGIs themselves, and changes in these regions appear to contribute to cancer development and tissue differentiation (Doi et al., 2009; Irizarry et al., 2009; Rao et al., 2013). Furthermore, Irizarry and colleagues (2009) demonstrated that differential DNA methylation of CGI shores correlates well with tissue‐specific gene expression. The accumulating evidence thus suggests a key involvement for CGI shore methylation in adaptive epigenetic programming. While other studies in the wild have shown environmentally induced differential methylation of DNA to be over‐represented in regions better known to be associated with gene expression (e.g. gene bodies, promoters and TSS) (Gugger et al., 2016; Lea et al., 2016), our observed over‐representation of urbanization‐associated sites in CGI shores could be equally important in regulating gene expression and phenotypic traits. Further research is needed to better understand the role of CGI shore methylation.
The biological pathways with which urbanization‐linked CpG sites were associated, in liver, pinpoint diet and ROS exposure as the two major drivers of the divergence between the urban‐ and forest‐dwelling birds. This is corroborated by previous findings at the transcriptomic level in the liver, where diet and oxidative stress were also identified as key processes underlying differences in gene expression between the same urban and forest populations (Watson et al., 2017). However, transcriptional differences in immune‐related genes, found in that study, were not matched by differences in DNA methylation. Over‐representation of genes, associated with differentially methylated regions, in pathways involved in lipid metabolism and transport suggests differences in energetic demands and/or dietary intake between the urban and forest birds, which could reflect differences in thermoregulatory requirements and resource availability, respectively. Indeed, differences in fatty acids circulating in the plasma have previously been shown in our study populations (Andersson et al., 2015). It is often assumed that urban‐dwelling birds rely more on anthropogenic‐provided foods, which have a higher fat content and different composition of fatty acids (Andersson et al., 2018), compared with naturally available foods (Gavett & Wakeley, 1986; Isaksson et al., 2014). Differential intake of dietary lipids could thus underlie the observed differences in DNA methylation patterns. Since the liver is a major site of lipid biosynthesis and metabolism, and transport of cholesterol and lipoproteins to and from other tissues, we would not necessarily expect to see the same patterns in the blood.
Metabolic processes involving ROS, which include the highly reactive superoxide and peroxides, are essential in the regulation of pro‐oxidants and protection against oxidative stress‐induced damage in the cells of the body. Oxidative stress describes a pathological state that arises when either a surplus of pro‐oxidants or depletion of antioxidants causes an increase in oxidative damage of DNA, lipids and/or proteins. Oxidative stress is considered to be almost ubiquitous in underlying the negative effects of many pollutants and stressors. The urban environment abounds with pro‐oxidant‐generating pollutants, and pollutant‐induced oxidative stress has been demonstrated in urban birds (Fossi et al., 1991; Isaksson, 2010). In turn, increased oxidative stress could be the driver of accelerated erosion of telomeres (Reichert & Stier, 2017), as has been observed in urban bird nestlings (Salmón et al., 2016). However, higher circulating levels of antioxidants (Salmón, Stroh et al. 2018) and elevated expression of several genes involved in metal detoxification and DNA repair (Watson et al., 2017), previously observed in our urban population, could indicate regulated responses to enable birds to cope with living in an environment rich in pro‐oxidants. The over‐representation of differentially methylated regions in genes associated with the metabolism of ROS provides further evidence for the importance of exposure to ROS in shaping the differences between birds living in cities and those living in more benign nonurban environments.
If variation in DNA methylation is mechanistically linked to variation at the phenotypic level, it is expected that there should be effects on regulation of gene expression. However, while often assumed, correlations between DNA methylation and gene expression are commonly not found in practice. It is now becoming increasingly understood that the functional relationship between CpG methylation and gene expression is not consistent across the genome (Lea et al., 2018), or even between CpG sites within a single promoter, as recently demonstrated in birds (Jimeno et al., 2019). Furthermore, additional mechanisms, including non‐CpG methylation and histone acetylation, influence transcription (Derks et al., 2016; Weaver et al., 2004). By using a reduced representation technique, we only sequenced a subsample of the genome's CpG sites. It is therefore perhaps not surprising that our results revealed relatively weak relationships between DNA methylation and gene expression, with large amounts of unexplained variation. This does not mean that there is no functional relevance to the observed variation in DNA methylation patterns, but that the nature and strength of associations are not consistent and/or are not understood in full because the whole methylome has not been sequenced.
Collectively, the observed results suggest that epigenetic mechanisms—specifically DNA methylation—could play a key role in regulating organismal responses to novel environments, such as towns and cities. Observed variation in DNA methylation corresponded with habitat differences associated with urbanization. In liver, CpG sites that were differentially methylated between the two habitats were over‐represented in regulatory regions of the genome, in expressed genes, and in biological pathways that pinpoint diet and oxidative stress as key drivers shaping differences between the urban‐ and forest‐dwelling birds. Conversely, we found little evidence for a coordinated environmental response in DNA methylation patterns in blood. Whether these patterns are consistent across other urban and forest populations, and thus can be more widely attributed to the process of urbanization, remains to be confirmed. An aim for the field of ecological epigenetics is to now further our understanding of the causal mechanisms, and consequences for fitness, of environmentally induced changes in DNA methylation patterns. While we investigated CpG methylation, there is increasing evidence for a potential functional role of non‐CpG methylation (Laine et al., 2016), which demands further research.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
Thanks to Johan Nilsson and Amparo Herrera‐Duenas for assistance in the field; Kees van Oers for providing annotation files; and Hanna Sigeman for advice on processing of bioinformatic data. The SNP&SEQ Technology Platform in Uppsala is part of the National Genomics Infrastructure Sweden and Science for Life Laboratory and is supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. Computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through the Uppsala Multidisciplinary Centre for Advanced Computational Science (UPPMAX) under Project SNIC 2018/8‐221.
Watson H, Powell D, Salmón P, Jacobs A, Isaksson C. Urbanization is associated with modifications in DNA methylation in a small passerine bird. Evol Appl.2021;14:85–98. 10.1111/eva.13160
DATA AVAILABILITY STATEMENT
RRBS and RNA‐seq raw reads are deposited in NCBI’s Sequence Read Archive (BioProject PRJNA314210).
REFERENCES
- Akalin, A. , Kormaksson, M. , Li, S. , Garrett‐Bakelman, F. E. , Figueroa, M. E. , Melnick, A. , & Mason, C. E. (2012). methylKit: A comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biology, 13, R87 10.1186/gb-2012-13-10-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluru, N. , Karchner, S. I. , Krick, K. S. , Zhu, W. , & Liu, J. (2018). Role of DNA methylation in altered gene expression patterns in adult zebrafish (Danio rerio) exposed to 3, 3’, 4, 4’, 5‐pentachlorobiphenyl (PCB 126). Environmental Epigenetics, 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, M. N. , Nilsson, J. , Nilsson, J. Å. , & Isaksson, C. (2018). Diet and ambient temperature interact to shape plasma fatty acid composition, basal metabolic rate and oxidative stress in great tits. Journal of Experimental Biology, 221(Pt 24), jeb186759. [DOI] [PubMed] [Google Scholar]
- Andersson, M. N. , Wang, H.‐L. , Nord, A. , Salmón, P. , & Isaksson, C. (2015). Composition of physiologically important fatty acids in great tits differs between urban and rural populations on a seasonal basis. Frontiers in Ecology and Evolution, 3, 93 10.3389/fevo.2015.00093 [DOI] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Baccarelli, A. , Wright, R. O. , Bollati, V. , Tarantini, L. , Litonjua, A. A. , Suh, H. H. , Zanobetti, A. , Sparrow, D. , Vokonas, P. S. , & Schwartz, J. (2009). Rapid DNA methylation changes after exposure to traffic particles. American Journal of Respiratory and Critical Care Medicine, 179, 572–578. 10.1164/rccm.200807-1097OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly, J. , Scheifler, R. , Belvalette, M. , Garnier, S. , Boissier, E. , Clément‐Demange, V.‐A. , Gète, M. , Leblond, M. , Pasteur, B. , Piget, Q. , Sage, M. , & Faivre, B. (2016). Negative impact of urban habitat on immunity in the great tit Parus major . Oecologia, 182, 1053–1062. 10.1007/s00442-016-3730-2 [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes & Development, 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati, V. , & Baccarelli, A. (2010). Environmental epigenetics. Heredity, 105(1), 105–112. Nature Publishing Group. 10.1038/hdy.2010.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossdorf, O. , Richards, C. L. , & Pigliucci, M. (2008). Epigenetics for ecologists. Ecology Letters, 11, 106–115. [DOI] [PubMed] [Google Scholar]
- Chatterjee, A. , Stockwell, P. A. , Rodger, E. J. , & Morison, I. M. (2012). Comparison of alignment software for genome‐wide bisulphite sequence data. Nucleic Acids Research, 40, e79 10.1093/nar/gks150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheptou, P.‐O. , Carrue, O. , Rouifed, S. , & Cantarel, A. (2008). Rapid evolution of seed dispersal in an urban environment in the weed Crepis sancta . Proceedings of the National Academy of Sciences, 105, 3796–3799. 10.1073/pnas.0708446105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick, L. D. , Strickler, S. A. , Perez, A. , Martin, R. A. , & Diamond, S. E. (2019). Urban heat islands advance the timing of reproduction in a social insect. Journal of Thermal Biology, 80, 119–125. 10.1016/j.jtherbio.2019.01.004 [DOI] [PubMed] [Google Scholar]
- Christensen, B. C. , Houseman, E. A. , Marsit, C. J. , Zheng, S. , Wrensch, M. R. , Wiemels, J. L. , Nelson, H. H. , Karagas, M. R. , Padbury, J. F. , Bueno, R. , Sugarbaker, D. J. , Yeh, R.‐F. , Wiencke, J. K. , & Kelsey, K. T. (2009). Aging and environmental exposures alter tissue‐specific DNA methylation dependent upon CPG island context. PLoS Genetics, 5, e1000602 10.1371/journal.pgen.1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini, D. , Greives, T. J. , Hau, M. , & Partecke, J. (2014). Does urban life change blood oxidative status in birds? Journal of Experimental Biology, 217, 2994–2997. 10.1242/jeb.106450 [DOI] [PubMed] [Google Scholar]
- Derks, M. F. L. , Schachtschneider, K. M. , Madsen, O. , Schijlen, E. , Verhoeven, K. J. F. , & van Oers, K. (2016). Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics, 17, 332 10.1186/s12864-016-2653-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemanse, N. J. , Both, C. , Van Noordwijk, A. J. , Rutten, A. L. , & Drent, P. J. (2003). Natal dispersal and personalities in great tits (Parus major). Proceedings of the Royal Society B‐Biological Sciences, 270, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi, A. , Park, I. H. , Wen, B. , Murakami, P. , Aryee, M. J. , Irizarry, R. , Herb, B. , Ladd‐Acosta, C. , Rho, J. , Loewer, S. , Miller, J. , Schlaeger, T. , Daley, G. Q. , & Feinberg, A. P. (2009). Differential methylation of tissue‐and cancer‐specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Genetics, 41, 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossi, M. C. , Leonzio, C. , Focardi, S. , Lari, L. , & Renzoni, A. (1991). Modulation of mixed‐function oxidase activity in black‐headed gulls living in anthropogenic environments: Biochemical acclimatization or adaptation? Environmental Toxicology and Chemistry, 10, 1179–1188. [Google Scholar]
- Garcia, M. J. , Rodríguez‐Brenes, S. , Kobisk, A. , Adler, L. , Ryan, M. J. , Taylor, R. C. , & Hunter, K. L. (2019). Epigenomic changes in the túngara frog (Physalaemus pustulosus): Possible effects of introduced fungal pathogen and urbanization. Evolutionary Ecology, 33, 671–686. 10.1007/s10682-019-10001-8 [DOI] [Google Scholar]
- Gavett, A. P. , & Wakeley, J. S. (1986). Blood constituents and their relation to diet in urban and rural house sparrows. Condor, 88, 279–284. 10.2307/1368873 [DOI] [Google Scholar]
- Gugger, P. F. , Fitz‐Gibbon, S. , Pellegrini, M. , & Sork, V. L. (2016). Species‐wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Molecular Ecology, 25, 1665–1680. [DOI] [PubMed] [Google Scholar]
- Harris, S. E. , Munshi‐South, J. , Obergfell, C. , & O'Neill, R. (2013). Signatures of rapid evolution in urban and rural transcriptomes of white‐footed mice (Peromyscus leucopus) in the New York Metropolitan Area. PLoS One, 8, e74938 10.1371/journal.pone.0074938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Askary, A. M. , Thurman, T. J. , Spiller, D. A. , Palmer, T. M. , Pringle, R. M. , & Barrett, R. D. H. (2019). The epigenetic signature of colonizing new environments in Anolis lizards . Molecular Biology and Evolution, 36, 2165–2170. 10.1093/molbev/msz133 [DOI] [PubMed] [Google Scholar]
- Hu, J. , & Barrett, R. D. H. (2017). Epigenetics in natural animal populations. Journal of Evolutionary Biology, 30, 1612–1632. 10.1111/jeb.13130 [DOI] [PubMed] [Google Scholar]
- Husquin, L. T. , Rotival, M. , Fagny, M. , Quach, H. , Zidane, N. , McEwen, L. M. , MacIsaac, J. L. , Kobor, M. S. , Aschard, H. , Patin, E. , & Quintana‐Murci, L. (2018). Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Biology, 19, 222 10.1186/s13059-018-1601-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry, R. A. , Ladd‐Acosta, C. , Wen, B. O. , Wu, Z. , Montano, C. , Onyango, P. , Cui, H. , Gabo, K. , Rongione, M. , Webster, M. , Ji, H. , Potash, J. B. , Sabunciyan, S. , & Feinberg, A. P. (2009). The human colon cancer methylome shows similar hypo‐ and hypermethylation at conserved tissue‐specific CpG island shores. Nature Genetics, 41, 178–186. 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson, C. (2010). Pollution and its impact on wild animals: A meta‐analysis on oxidative stress. EcoHealth, 7, 342–350. 10.1007/s10393-010-0345-7 [DOI] [PubMed] [Google Scholar]
- Isaksson, C. (2015). Urbanization, oxidative stress and inflammation: A question of evolving, acclimatizing or coping with urban environmental stress. Functional Ecology, 29, 913–923. [Google Scholar]
- Isaksson, C. , Hanson, M. A. , & Burdge, G. C. (2014). The effects of spatial and temporal ecological variation on fatty acid compositions of wild great tits Parus major . Journal of Avian Biology, 46, 245–253. [Google Scholar]
- Isaksson, C. , Sturve, J. , Almroth, B. C. , & Andersson, S. (2009). The impact of urban environment on oxidative damage (TBARS) and antioxidant systems in lungs and liver of great tits, Parus major. Environmental Research, 109, 46–50. 10.1016/j.envres.2008.10.006 [DOI] [PubMed] [Google Scholar]
- Isaksson, C. , Von Post, M. , & Andersson, S. (2007). Sexual, seasonal, and environmental variation in plasma carotenoids in great tits, Parus major. Biological Journal of the Linnean Society, 92, 521–527. [Google Scholar]
- Jablonka, E. , & Raz, G. (2009). Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. The Quarterly Review of Biology, 84, 131–176. [DOI] [PubMed] [Google Scholar]
- Jimeno, B. , Hau, M. , Gómez‐Díaz, E. , & Verhulst, S. (2019). Developmental conditions modulate DNA methylation at the glucocorticoid receptor gene with cascading effects on expression and corticosterone levels in zebra finches. Scientific Reports, 9, 1–11. 10.1038/s41598-019-52203-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nature Reviews Genetics, 13, 484–492. [DOI] [PubMed] [Google Scholar]
- Khimoun, A. , Doums, C. , Molet, M. , Kaufmann, B. , Peronnet, R. , Eyer, P. A. , Mona, S. (2020). Urbanization without isolation: The absence of genetic structure among cities and forests in the tiny acorn ant Temnothorax nylanderi . Biology Letters, 16, 20190741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilvitis, H. J. , Schrey, A. W. , Ragsdale, A. K. , Berrio, A. , Phelps, S. M. , & Martin, L. B. (2019). DNA methylation predicts immune gene expression in introduced house sparrows (Passer domesticus). Journal of Avian Biology, e01965. [Google Scholar]
- Kim, D. , Langmead, B. , & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12, 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivula, M. J. , & Eeva, T. (2010). Metal‐related oxidative stress in birds. Environmental Pollution, 158, 2359–2370. 10.1016/j.envpol.2010.03.013 [DOI] [PubMed] [Google Scholar]
- Krueger, F. (2015). Trim Galore!: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. www.bioinformatics.babraham.ac.uk/projects/trim_galore/ [Google Scholar]
- Krueger, F. , & Andrews, S. R. (2011). Bismark: A flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics, 27, 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova, I. , Lugmayr, A. , Siira, S. J. , Rackham, O. , & Filipovska, A. (2019). CirGO: An alternative circular way of visualising gene ontology terms. BMC Bioinformatics, 20, 1–7. 10.1186/s12859-019-2671-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, V. N. , Gossmann, T. I. , Schachtschneider, K. M. , Garroway, C. J. , Madsen, O. , Verhoeven, K. J. F. , de Jager, V. , Megens, H.‐J. , Warren, W. C. , Minx, P. , Crooijmans, R. P. M. A. , Corcoran, P. , Sheldon, B. C. , Slate, J. , Zeng, K. , van Oers, K. , Visser, M. E. , & Groenen, M. A. M. (2016). Evolutionary signals of selection on cognition from the great tit genome and methylome. Nature Communications, 7, 10474 10.1038/ncomms10474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach, Z. M. , Faulk, C. D. , Dolinoy, D. C. , Montrose, L. , Jones, T. R. , Ray, D. , Pioon, M. O. , & Holekamp, K. E. (2019). Early life social and ecological determinants of global DNA methylation in wild spotted hyenas. Molecular Ecology, 28(16), 3799–3812. 10.1111/mec.15174 [DOI] [PubMed] [Google Scholar]
- Lea, A. J. , Altmann, J. , Alberts, S. C. , & Tung, J. (2016). Resource base influences genome‐wide DNA methylation levels in wild baboons (Papio cynocephalus). Molecular Ecology, 25, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, A. J. , Vockley, C. M. , Johnston, R. A. , Del Carpio, C. A. , Barreiro, L. B. , Reddy, T. E. , & Tung, J. (2018). Genome‐wide quantification of the effects of DNA methylation on human gene regulation. Elife, 7, 1–27. 10.7554/eLife.37513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev Maor, G. , Yearim, A. , & Ast, G. (2015). The alternative role of DNA methylation in splicing regulation. Trends in Genetics, 31(5), 274–280. 10.1016/j.tig.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebl, A. L. , Schrey, A. W. , Richards, C. L. , & Martin, L. B. (2013). Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integrative and Comparative Biology, 53, 351–358. 10.1093/icb/ict007 [DOI] [PubMed] [Google Scholar]
- Lister, R. , Pelizzola, M. , Dowen, R. H. , Hawkins, R. D. , Hon, G. , Tonti‐Filippini, J. , Nery, J. R. , Lee, L. , Ye, Z. , Ngo, Q.‐M. , Edsall, L. , Antosiewicz‐Bourget, J. , Stewart, R. , Ruotti, V. , Millar, A. H. , Thomson, J. A. , Ren, B. , & Ecker, J. R. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462, 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, J. , Mastromonaco, G. , Edwards, D. B. , & Schulte‐Hostedde, A. I. (2017). Fat and happy in the city: Eastern chipmunks in urban environments. Behavioral Ecology, 28, 1464–1471. 10.1093/beheco/arx109 [DOI] [Google Scholar]
- McCormick, H. , Young, P. E. , Hur, S. S. J. , Booher, K. , Chung, H. , Cropley, J. E. , Giannoulatou, E. , & Suter, C. M. (2017). Isogenic mice exhibit sexually‐dimorphic DNA methylation patterns across multiple tissues. BMC Genomics, 18, 1–9. 10.1186/s12864-017-4350-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew, S. M. , Beck, D. , Sadler‐Riggleman, I. , Knutie, S. A. , Koop, J. A. H. , Clayton, D. H. , & Skinner, M. K. (2017). Epigenetic variation between urban and rural populations of Darwin’s finches. BMC Evolutionary Biology, 17, 1–14. 10.1186/s12862-017-1025-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller, A. P. , Erritzøe, J. , & Karadas, F. (2010). Levels of antioxidants in rural and urban birds and their consequences. Oecologia, 163, 35–45. 10.1007/s00442-009-1525-4 [DOI] [PubMed] [Google Scholar]
- NCBI BioProject (2017). Parus major urban/forest omics. NCBI Sequence Read Archive. [Google Scholar]
- Pertea, M. , Kim, D. , Pertea, G. M. , Leek, J. T. , & Salzberg, S. L. (2016). Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nature Protocols, 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G. M. , Antonescu, C. M. , Chang, T. C. , Mendell, J. T. , & Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology, 33, 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Rao, X. , Evans, J. , Chae, H. , Pilrose, J. , Kim, S. , Yan, P. , Huang, R.‐L. , Lai, H.‐C. , Lin, H. , Liu, Y. , Miller, D. , Rhee, J.‐K. , Huang, Y.‐W. , Gu, F. , Gray, J. W. , Huang, T.‐M. , & Nephew, K. P. (2013). CpG island shore methylation regulates caveolin‐1 expression in breast cancer. Oncogene, 32, 4519–4528. 10.1038/onc.2012.474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert, S. & Stier, A. (2017). Does oxidative stress shorten telomeres in vivo? Biology Letters, 13(5), 20170164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkin, L. R. , Santangelo, J. S. , Alberti, M. , Aronson, M. F. J. , de Keyzer, C. W. , Diamond, S. E. , Fortin, M.‐J. , Frazee, L. J. , Gorton, A. J. , Hendry, A. P. , Liu, Y. , Losos, J. B. , MacIvor, J. S. , Martin, R. A. , McDonnell, M. J. , Miles, L. S. , Munshi‐South, J. , Ness, R. W. , Newman, A. E. M. , … Johnson, M. T. J. (2019). A roadmap for urban evolutionary ecology. Evolutionary Applications, 12(3), 384–398. 10.1111/eva.12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riyahi, S. , Sanchez‐Delgado, M. , Calafell, F. , Monk, D. , & Senar, J. C. (2015). Combined epigenetic and intraspecific variation of the DRD4 and SERT genes influence novelty seeking behavior in great tit Parus major . Epigenetics, 10, 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, A. , De Giorgio, B. , Parolini, M. , Favero, C. , Possenti, C. D. , Iodice, S. , Caprioli, M. , Rubolini, D. , Ambrosini, R. , Gianfranceschi, L. , Saino, N. , & Bollati, V. (2016). Methylation of the circadian Clock gene in the offspring of a free‐living passerine bird increases with maternal and individual exposure to PM10. Environmental Pollution, 220, 29–37. 10.1016/j.envpol.2016.08.060 [DOI] [PubMed] [Google Scholar]
- Rubenstein, D. R. , Skolnik, H. , Berrio, A. , Champagne, F. A. , Phelps, S. , & Solomon, J. (2016). Sex‐specific fitness effects of unpredictable early life conditions are associated with DNA methylation in the avian glucocorticoid receptor. Molecular Ecology, 25, 1714–1728. 10.1111/mec.13483 [DOI] [PubMed] [Google Scholar]
- Salmón, P. , Jacobs, A. , Ahrén, D. , Biard, C. , Dingemanse, N. J. , Dominoni, D. M. , Helm, B. , Lundberg, M. , Senar, J. C. , Sprau, P. , Visser, M. E. , & Isaksson, C. (2020). Repeated genomic signatures of adaptation to urbanisation in a songbird across Europe. bioRxiv. 10.1101/2020.05.05.078568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmón, P. , Nilsson, J. F. , Nord, A. , Bensch, S. , & Isaksson, C. (2016). Urban environment shortens telomere length in nestling great tits, Parus major. Biology Letters, 12, 8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmón, P. , Nilsson, J. F. , Watson, H. , Bensch, S. , & Isaksson, C. (2017). Selective disappearance of great tits with short telomeres in urban areas. Proceedings of the Royal Society B: Biological Sciences, 284, 20171349 10.1098/rspb.2017.1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmón, P. , Stroh, E. , Herrera‐Dueñas, A. , von Post, M. , & Isaksson, C. (2018). Oxidative stress in birds along a NOxand urbanisation gradient: An interspecific approach. Science of the Total Environment, 622–623, 635–643. 10.1016/j.scitotenv.2017.11.354 [DOI] [PubMed] [Google Scholar]
- Salmón, P. , Watson, H. , Nord, A. , & Isaksson, C. (2018). Effects of the urban environment on oxidative stress in early life: Insights from a cross‐fostering experiment. Integrative and Comparative Biology, 58, 986–994. 10.1093/icb/icy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell, C. J. (2018). Urban evolutionary ecology and the potential benefits of implementing genomics. Journal of Heredity, 109, 138–151. 10.1093/jhered/esy001 [DOI] [PubMed] [Google Scholar]
- Siller, S. J. , & Rubenstein, D. R. (2019). A tissue comparison of DNA methylation of the glucocorticoid receptor gene (Nr3c1) in European starlings. Integrative and Comparative Biology, 59, 264–272. 10.1093/icb/icz034 [DOI] [PubMed] [Google Scholar]
- Skinner, M. K. , Gurerrero‐Bosagna, C. , Haque, M. M. , Nilsson, E. E. , Koop, J. A. H. , Knutie, S. A. , & Clayton, D. H. (2014). Epigenetics and the evolution of Darwin’s finches. Genome Biol. Evol., 6, 1972–1989. 10.1093/gbe/evu158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabbekoorn, H. , & Peet, M. (2003). Birds sing at a higher pitch in urban noise. Nature, 424, 267 10.1038/424267a [DOI] [PubMed] [Google Scholar]
- Smith, T. A. , Martin, M. D. , Nguyen, M. , & Mendelson, T. C. (2016). Epigenetic divergence as a potential first step in darter speciation. Molecular Ecology, 25, 1883–1894. 10.1111/mec.13561 [DOI] [PubMed] [Google Scholar]
- Sprau, P. , Mouchet, A. , & Dingemanse, N. J. (2017). Multidimensional environmental predictors of variation in avian forest and city life histories. Behavioral Ecology, 28, 59–68. 10.1093/beheco/arw130 [DOI] [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. , & Šmuc, T. (2011). Revigo summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taff, C. C. , Campagna, L. , & Vitousek, M. N. (2019). Genome‐wide variation in DNA methylation is associated with stress resilience and plumage brightness in a wild bird. Molecular Ecology, 3722–3737. 10.1111/mec.15186 [DOI] [PubMed] [Google Scholar]
- Tennessen, J. B. , Parks, S. E. , & Langkilde, T. (2014). Traffic noise causes physiological stress and impairs breeding migration behaviour in frogs. Conserv. Physiol., 2, 1–8. 10.1093/conphys/cou032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorson, J. L. M. , Smithson, M. , Sadler‐Riggleman, I. , Beck, D. , Dybdahl, M. , & Skinner, M. K. (2019). Regional epigenetic variation in asexual snail populations among urban and rural lakes. Environ. Epigenetics, 5, 1–12. 10.1093/eep/dvz020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations (2018). World Urbanization prospects: The 2018 Revision (ST/ESA/SER.A/420). New York: United Nations. [Google Scholar]
- Verhoeven, K. J. F. , VonHoldt, B. M. , & Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Molecular Ecology, 25, 1631–1638. [DOI] [PubMed] [Google Scholar]
- Viitaniemi, H. M. , Verhagen, I. , Visser, M. E. , Honkela, A. , van Oers, K. , & Husby, A. (2019). Seasonal variation in genome‐wide DNA methylation patterns and the onset of seasonal timing of reproduction in great tits. Genome Biology and Evolution, 11, 970–983. 10.1093/gbe/evz044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. Q. , Tuominen, L. K. , & Tsai, C. J. (2011). SLIM: A sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics, 27, 225–231. 10.1093/bioinformatics/btq650 [DOI] [PubMed] [Google Scholar]
- Waterland, R. A. , & Jirtle, R. L. (2003). Transposable elements: Targets for early nutritional effects on epigenetic gene regulation. Molecular and Cellular Biology, 23, 5293–5300. 10.1128/MCB.23.15.5293-5300.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, H. , Videvall, E. , Andersson, M. N. , & Isaksson, C. (2017). Transcriptome analysis of a wild bird reveals physiological responses to the urban environment. Scientific Reports, 7, 44180 10.1038/srep44180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, I. C. G. , Cervoni, N. , Champagne, F. A. , D'Alessio, A. C. , Sharma, S. , Seckl, J. R. , Dymov, S. , Szyf, M. , & Meaney, M. J. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7, 847–854. 10.1038/nn1276 [DOI] [PubMed] [Google Scholar]
- Yuan, X.‐L. , Gao, N. , Xing, Y. , Zhang, H.‐B. , Zhang, A.‐L. , Liu, J. , He, J.‐L. , Xu, Y. , Lin, W.‐M. , Chen, Z.‐M. , Zhang, H. , Zhang, Z. , & Li, J.‐Q. (2016). Profiling the genome‐wide DNA methylation pattern of porcine ovaries using reduced representation bisulfite sequencing. Scientific Reports, 6, 1–10. 10.1038/srep22138 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RRBS and RNA‐seq raw reads are deposited in NCBI’s Sequence Read Archive (BioProject PRJNA314210).