

Abstract
tRNA-derived fragments (tRFs) and tRNA halves (tiRNAs) are originated from the specific cleavage of endogenous tRNAs or their precursors and regulate gene expression when the cells are in stressful circumstances. Here, we replicated the rat common carotid artery (CCA) intimal hyperplasia model and investigated the expression of tRFs/tiRNAs in the artery. The normal and the balloon-injured rat CCAs were subjected to small RNA sequencing, and then the differentially expressed tRFs/tiRNAs were identified and analyzed. The expression profiles of tRFs/tiRNAs in the healthy and injured CCAs were remarkably different. tRNAGlnCTG-derived fragments (tRFGlnCTG) were found to be overexpressed with a high abundance in the injured CCA. In in vitro experiments, the synthetic tRFGlnCTG mimetics elevated the proliferation and migration of rat vascular smooth muscle cells (VSMCs). Through bioinformatics analysis and an overexpression experiment, tRFGlnCTG was found to negatively regulate the expression of FAS cell surface death receptor (FAS). This study revealed that tRFGlnCTG is a crucial regulator in promoting VSMC proliferation. The investigation of the roles of tRFs/tiRNAs is of significance for understanding the mechanism, diagnosis, and treatment of intimal hyperplasia.
Keywords: tRNA-derived fragments, intimal hyperplasia, vascular smooth muscle cell, proliferation, FAS receptor
Graphical abstract
tRNA-derived fragments (tRFs) originated from the specific cleavage of tRNAs and regulate gene expression when the cells are in stressful circumstances. We described that tRFGlnCTG was highly expressed in the balloon-injured rat carotid artery. tRFGlnCTG was found to positively regulate VSMC proliferation via the FAS gene.
Introduction
Transfer RNAs (tRNAs) are molecules that assemble amino acids into proteins. In recent years, studies have found that when cells are under environmental stress, such as starvation, oxidative stress, or hypoxia, endogenous tRNAs are prone to be cleaved into small fragments at the specific sites.1 The half molecules formed by the cleavage on the tRNA anticodon loop are called tRNA halves (tiRNAs). The small fragments of tRNA are called tRNA-derived fragments (tRFs). tRFs are approximately 16∼18 nt in length and are derived from mature or precursor tRNA. According to their corresponding position on the parent tRNA, they can be further classified into tRF-5, which corresponds to the 5′ end of mature tRNA and is produced by cleaving the tRNA on the D-loop; tRF-3, which corresponds to the 3′ end of mature tRNA, is produced by cutting the tRNA on the T-loop, and contains the common carotid artery (CCA) sequence; tRF-1, which is derived from the 3′ tail of the precursor tRNA and includes a poly U sequence at the 3′ end; and i-tRF, which is derived from the internal region of mature tRNA (Figure S1A). Unlike intact tRNAs, tRFs/tiRNAs perform another function: if their nucleic acid sequences are complementary to messenger RNAs (mRNAs), then they interfere with the production of proteins. tRFs/tiRNAs play essential roles in biological regulation and are related to diverse diseases. tiRNA or tRFs are found to be involved in degenerative neural diseases,2, 3, 4 acquired metabolic disorders,5,6 stress injuries,7,8 and cancers.9,10 Their action mechanisms include gene silencing,11 affecting protein translation,12 competitive binding to essential proteins,9,13 and so on.
Intimal hyperplasia is a fundamental pathological change in cardiovascular diseases, such as hypertension and atherosclerosis. It also occurs after vascular surgeries, such as arterial bypass, balloon dilatation and stenting, and arteriovenous fistula. Neointima formation is a crucial cause of vascular stenosis, leading to the failure of revascularization surgery or aggravation of the original disease. There are a variety of cells involved in the formation of neointima. After the endothelium injury, platelets accumulate on the sites where endothelial cells (ECs) fall off, triggering inflammation. Then vascular smooth muscle cells (VSMCs) or adventitial fibroblasts (AFs) accumulate under the intima, hyperproliferating and synthesizing excessive extracellular matrix. In the end, ECs regenerate and cover the intima, reshaping the protective barrier of the intima. In the process of neointima formation, the migration and proliferation of VSMCs play pivotal roles.
We hypothesized that as a stress condition, the vascular injury might regulate the production of tiRNA or tRF, which have specific biological effects. In this study, we replicated the rat carotid artery intimal hyperplasia model by balloon injury (Figure 1A) and performed a high-throughput deep RNA sequencing. tiRNA and tRFs were identified and analyzed. One tRF, the tRNAGlnCTG-derived fragment (tRFGlnCTG), was found to be upregulated in the injured arteries. RNA fluorescent in situ hybridization (RNA-FISH) demonstrated that tRFGlnCTG was expressed in the cytoplasm of cells from the tunica media and the neointima. The synthesized tRFGlnCTG mimetics promoted VSMC proliferation and migration. In contrast, the inhibitors of tRFGlnCTG suppressed the migration and proliferation of VSMCs. Bioinformatics analysis suggested that tRFGlnCTG may modulate genes involved in cell growth and death. We identified its target genes as FAS cell surface death receptor (FAS). Our study provides new insights for further understanding of intimal hyperplasia mechanism and searching for gene-therapy targets.
Figure 1.

Deep sequencing showed the expression of tRFs and tiRNAs in the rat carotid artery
(A) The schema shows the position of injury to the rat common carotid artery (CCA). (B) Neointima (indicated by a dotted line) appeared in the injured artery 14 days after surgery. The ratio of the intima/media area markedly increased in the injured artery (n = 5). (C) The expression of tRFs and tiRNA in the CCA was detected by deep sequencing. The Venn plot showed the tRF and tiRNA numbers, which were expressed in both of the injured and control CCAs and also indicated the number of specific expression tRFs and tiRNAs. (D) Scatterplot and volcano plot of tRFs and tiRNAs between the healthy and the injured CCAs. tRFs and tiRNAs above the top line (red dots, upregulation) or below the bottom line (blue dots, downregulation) indicate a more than 2.0-fold change between two compared groups. Brown dots show the tRFs and tiRNAs without differential expression. (E) Heatmap of gene expression obtained from the control and the injured CCAs; it includes the 500 tRFs and tiRNAs that have the most significant coefficient of variation (CV) based on tag counts per million of total aligned tRNA reads (TPM). Blue represented an expression level below the mean. Red represented an expression level above the mean (n = 3). Data are expressed as means ± SEM. ∗∗p < 0.01.
Results
The expression profiles of tiRNAs and tRFs were markedly different between the healthy and injured rat CCAs
On day 14 after the balloon injury, the rat CCAs were harvested. Histological sections showed the neointima formed in the carotid artery with a marked increase in the intimal/medial area ratio (Figure 1B). The intimal hyperplasia model was successfully made. Small RNA fragments consistent with pre-tRNA and mature-tRNA sequences were screened from the sequencing results for differential expression analysis between two groups.
The expression profiles of tiRNAs and tRFs in the healthy and injured CCA were markedly different. A total of 1,131 tiRNAs and tRFs were identified, of which 283 were only expressed in normal CCAs, and 87 were exclusively expressed in injured CCAs (Figure 1C). Fragments with expression difference >2.0-fold between the two groups and p values <0.05 were defined as the differentially expressed tiRNAs or tRFs (Figures 1D and 1E). The tiRNAs and tRFs expressed in arteries were mainly tiRNA-5, tRF-5, and i-tRF. Other types, such as tRF-3, tRF-1, and tiRNA-3, just occupied a minimum proportion. The proportion of tRF-5 was higher in the injured arteries than in the healthy arteries (Figure 2A). The source tRNA of tiRNAs and tRFs was also different in the healthy arteries and injured arteries (Figure 2B). There were 14 statistically differentially expressed tiRNAs and tRFs between the healthy and injured arteries (Figure 2C). Nine of them were upregulated, and the other five were downregulated in the injured arteries. In the upregulated tRFs, tRFGlnCTG had a high expression abundance (with high tag counts per million of total aligned tRNA reads [TPM] values) in carotid arteries.
Figure 2.

The analysis of the expression of tRFs and tiRNAs in control and injured CCA
(A) Pie plots demonstrated tRF and tiRNA populations in control and injured rat carotid artery. (B) Stacked plots for all subtypes of tRF and tiRNA clustering by the anticodon of the tRNAs. The x axes represent the tRNAs with the same anticodon, and the y axes show the number of all subtype tRFs and tiRNAs derived from the same anticodon tRNA. (C) The gene ID, tRNA origin, type, fold changes, and p value of the differentially expressed tRFs and tiRNAs were given (n = 3). The red box marks tDR-010323 (tRFGlnCTG), which is highly expressed in the rat carotid artery.
tRFGlnCTG was highly expressed in the neointima and also in platelet-derived growth factor (PDGF)-BB- or transforming growth factor (TGF)-β1-induced VSMCs
The results of deep sequencing showed that the expression of tRFGlnCTG was increased in injured arteries (Figure 3A). According to its sequence, tRFGlnCTG was derived from the internal area of tRNAGlnCTG (Figure 3B), so-called i-tRFs. We utilized quantitative real-time PCR to validate its expression in healthy and injured arteries. Its increased expression in injured arteries was confirmed (Figure 3C).
Figure 3.

The expression of tRFGlnCTG in the rat CCAs
(A) Small RNA sequencing demonstrated that tRFGlnCTG was highly expressed in injured CCA (n = 3). (B) tRFGlnCTG is derived from the internal area of mature tRNAGlnCTG. The schema shows its sequence. (C) The real-time qPCR primers were designed according to the sequences of tRFGlnCTG and the adaptors to validate its expression in arteries. PCR confirmed that the expression of tRFGlnCTG was elevated in the injured CCA. (D) RNA in situ fluorescence demonstrated that tRFGlnCTG localized in the cytoplasm of VSMCs from tunica media and neointima. It was highly expressed in the neointima (n = 5). (E) Recombinant PDGF-BB and TGF-β1 were used to stimulate cultured VSMCs, and both elevated the expression of tRFGlnCTG (n = 5). Data are expressed as means ± SEM. ∗p < 0.05, ∗∗p < 0.01.
RNA-FISH demonstrated that tRFGlnCTG were localized in the cytoplasm of VSMCs from the tunica media and in the cytoplasm of cells from the neointima. In the neointima, the expression of tRFGlnCTG was markedly elevated (Figure 3D).
VSMCs from rat thoracic aorta were stimulated with recombinant proteins of PDGF-BB (rPDGF-BB) and TGF-β1 (rTGF-β1) in vitro. Both rPDGF-BB and rTGF-β1 resulted in increased expression of tRFGlnCTG (Figure 3E).
tRFGlnCTG promoted the proliferation and viability of VSMCs
We investigated the biological effects of tRFGlnCTG by transfecting the synthetic RNA mimetics or inhibitors into rat VSMCs (Figure 4A).
Figure 4.

Synthetic tRFGlnCTG mimetics enhanced the proliferation and migration of the cultured rat VSMCs, whereas the inhibitor reduced the proliferation and migration
(A) The transfection efficacy of tRFGlnCTG mimetics and inhibitors was validated by qPCR. (B) Cell counting kit-8 (CCK-8) assay showed that the transfection of tRFGlnCTG mimetics increased DNA synthesis in VSMCs, and the inhibitors reduced it. (C) EdU incorporation assay demonstrated that the tRFGlnCTG mimetics elevated the cell viability, and the inhibitors weakened it. (D) Transwell assay revealed that tRFGlnCTG mimetics induced the migration of VSMCs, and the inhibitors reduced it. (E) Wound-closure assay showed that mimetics accelerated the wound-healing rate, and the inhibitors declined it (n = 5). Data are expressed as means ± SEM. ∗p < 0.05, ∗∗p < 0.01.
The Cell Counting Kit-8 (CCK-8) assay showed that the mimetics increased, and the inhibitors decreased the cell viability (Figure 4B).
For the 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay, 50 nM of synthetic tRFGlnCTG mimetics or inhibitors were transfected into VSMCs by Lipofectamine 3000 for 24 h. Then EdU was added to the culture medium. After another 24 h, the number of proliferating cells was counted under a fluorescence microscope, and the cell proliferation rate was calculated. The proliferation of VSMCs transfected with tRFGlnCTG mimetics was increased. On the contrary, tRFGlnCTG inhibitors reduced the number of proliferating cells (Figure 4C).
tRFGlnCTG enhanced the migration of VSMCs
The pretreatment of VSMCs was identical to the cell proliferation assay. Cell motility was assessed by the Transwell and scratch wound-closure assay. The transfection of tRFGlnCTG mimetics significantly increased the migration (Figure 4D) and the wound-closure velocity of VSMCs (Figure 4E). On the other hand, the inhibitors of tRFGlnCTG alleviate cell migration.
Target gene prediction of tRFGlnCTG
In the R environment, TargetScan and miRanda were used to predict the possible target genes of tRFGlnCTG. We performed functional enrichment and pathway prediction of the assumed genes by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (Figure 5A). The genes were functionally clustered, and genes related to cell growth, proliferation, death, and movement were paid attention. By querying the results of the GEO (GEO: GSE48279), we found that some of the assumed genes were downregulated in intimal hyperplasia and were previously reported to be involved in vascular biology. These target genes may be involved in the regulation of vascular cell death and movement.
Figure 5.

Bioinformatics analysis of tRFGlnCTG
(A) The target genes of tRFGlnCTG were predicted by TargetScan and miRanda. Gene Ontology (GO) and KEGG pathway analysis were performed on these target genes. Here is the number of target genes involved in related biological processes, molecular functions, and pathways. (B) Ingenuity Pathway Analysis (IPA) was used to analyze further the biological processes and signaling pathways of potential target genes. AFT6, FAS, IL-10, MAPK1, and SPRY2 (all highlighted in blue) are located at the predicted protein-protein interaction network hubs.
The biological processes and signaling pathways of the potential target genes of TRFGlnCTG were analyzed with Ingenuity Pathway Analysis (IPA) software (QIAGEN Bioinformatics, Hilden, Germany https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis,content). In the forecasted protein-protein interact networks, ATF6 (activating transcription factor 6), FAS, IL-10 (interleukin 10), MAPK1 (mitogen-activated protein kinase 1), and SPRY2 (Sprouty homolog 2) were located at the hubs of the network (Figure 5B).
FAS was the target gene of tRFGlnCTG in VSMCs
Bioinformatic analysis forecasted the potential binding sites of tRFGlnCTG with AFT6, FAS, IL-10, MAPK1, and SPRY2 (Figure 6A). These genes have been reported to modulate the growth of vascular cells. The transfection of tRFGlnCTG mimetics to rat VSMCs showed that the level of FAS was decreased, whereas the inhibitors of tRFGlnCTG elevated the level of FAS (Figure 6B). RNAhybrid 2.2 predicted 3 potential binding sites between tRFGlnCTG and FAS mRNA. The binding site with the smallest minimum free energy (MFE) value (−21.4 kcal/mol) (Figure 7A) was consistent with the results predicted by TargetScan and miRanda. We chose this site for the construction of the FAS 3′ UTR luciferase reporter vector. The luciferase reporter assay revealed that tRFGlnCTG mimetics inhibited the expression of FAS mRNA (Figure 7B).
Figure 6.

FAS was the target gene of tRFGlnCTG in VSMCs
(A) AFT6, FAS, IL-10, MAPK1, and SPRY2 have been reported to regulate the growth of vascular cells. The possible bindings sites between them and tRFGlnCTG and their functions in vascular biology are listed in the table. (B) Rat VSMCs were transfected with tRFGlnCTG mimetics or inhibitors, and the levels of AFT6, FAS, IL-10, MAPK1, and SPRY2 were detected. tRFGlnCTG mimetics decreased the level of FAS, and the inhibitors increase it (n = 7). Data are expressed as means ± SEM. ∗p < 0.05, ∗∗p < 0.01.
Figure 7.

tRFGlnCTG regulated VSMC proliferation via FAS
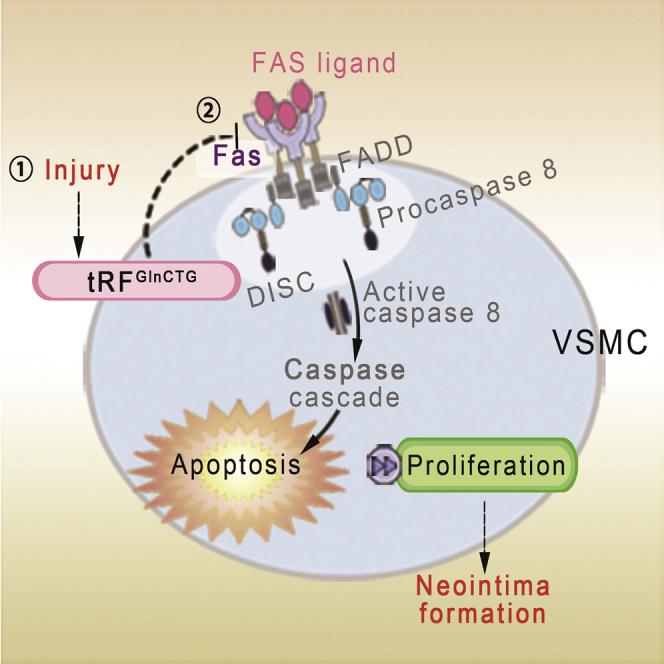
(A) The potential bindings sites of FAS mRNA and tRFGlnCTG were analyzed by RNAhybrid 2.2. The minimum free energy (MFE) of the binding was labeled on the digraph. The binding site with the smallest MFE value (−21.4 kcal/mol) was consistent with the results predicted by TargetScan and miRanda. We chose this site for the next experiment. (B) Luciferase reporter assay demonstrated that tRFGlnCTG reduced the expression of wild-type FAS mRNA. On the contrary, tRFGlnCTG showed no significant effect on the expression of mutant FAS mRNA. (C) The efficiency of overexpression of FAS in rat VSMC by the lentiviral vector. (D) FAS overexpression reduced the proliferation of VSMCs upregulated by tRFGlnCTG (n = 3). Data are expressed as means ± SEM. ∗∗p < 0.01. (E) Schematic diagram of the mechanism of tRFGlnCTG involved in intimal hyperplasia. FAS binds to the FAS ligand and forms a death complex, which activates the caspase cascade and causes cell death. tRFGlnCTG attenuates apoptosis by inhibiting FAS, thereby promoting intimal hyperplasia.
FAS overexpression (FAS-OE) abolished the effect of tRFGlnCTG
To validate the causal link between tRFGlnCTG and FAS, a recombinant lentiviral vector carrying the rat FAS gene was transfected to rat VSMCs, the control group treated with an empty vector. Western blotting confirmed the overexpression of FAS in VSMCs (Figure 7C). Then, the proliferation of FAS-OE VSMCs was investigated under tRFGlnCTG overexpression. FAS-OE VSMCs showed decreased proliferation compared with the control group (Figure 7D). Thus, the results indicated that the overexpression of FAS in VSMCs eliminated the upregulation of proliferation by tRFGlnCTG.
Discussion
Studies have shown that in many species, under certain stressful conditions, such as starvation or hypoxia, endogenous tRNAs or their precursors are prone to be cleaved to produce specific small RNA fragments. Such RNA fragments are a class of gene-expression regulators, and their mechanisms of action are diverse. Some inhibit mRNA translation by microRNAs (miRNAs) manners.10,14,15 Some serve as reverse transcription primers for the retroviral RNA genome.16,17 Some are involved in the assembly of precursor rRNA cleavage complexes.18 Others inhibit protein translation.19 tRFs/tiRNAs play crucial roles in diseases. For example, CU1276, a tRF from human mature B cells, suppresses proliferation and regulates the molecular response to DNA damage, and it is absent in germinal center-derived lymphomas.10 tRFs originated from tRNAGlu, tRNAAsp, tRNAGly, and tRNATyr suppress breast cancer metastasis by competitively binding to YBX1, the stabilizer of pro-oncogenic transcripts.9
Vascular injury induces excessive proliferation of the intima, leading to stenosis or even occlusion of blood vessels. We speculate that intimal injury, as a stress condition, may cause changes in tiRNA and tRF expression. The small RNA sequencing to the healthy and injured rat CCA demonstrated that the types and expression levels of tRNA-derived small fragments markedly changed in intimal hyperplasia. A total of 14 differentially expressed tiRNAs and tRFs were identified. Among them, we investigated the regulation of tRFGlnCTG on the biological behavior of VSMCs. Synthetic tRFGlnCTG mimetics promoted the proliferation and migration of VSMCs in vitro. On the contrary, inhibitors of tRFGlnCTG weaken VSMC proliferation and migration. Previous studies have also found that tRFs can negatively regulate cell death or apoptosis, promoting cell proliferation.13,20,21 The change of VSMC biological behavior is pivotal in the process of neointima formation. Therefore, tRFGlnCTG may be essential for the diagnosis and treatment of intimal hyperplasia.
tRNAs are one of the main sources of natural small noncoding RNAs. By bioinformatic tools, we predicted possible target genes of tRFGlnCTG. ATF6,22, 23, 24 FAS,25, 26, 27, 28, 29 IL-10,30, 31, 32 MAPK1,33,34 and SPRY235, 36, 37, 38 have been reported to regulate vascular cells’ growth. We confirmed that tRFGlnCTG decreased the level of FAS. Furthermore, the overexpression of FAS reduced the proliferation of VSMCs upregulated by tRFGlnCTG. Studies have shown that the decrease in FAS is one of the critical reasons for intimal hyperplasia. FAS binds to the FAS ligand and forms a death complex, reducing inflammatory cells’ infiltration and the proliferation of smooth muscle cells.26,27 Intimal hyperplasia can be alleviated by local overexpression of the FAS ligand in the arteries injured by the balloon.25 ECs are relatively resistant to the FAS mechanism. Overexpression of the FAS ligand in transplanted ECs attenuates intimal hyperplasia.28 The combination of the overexpressing FAS ligand and nitric oxide suppresses smooth muscle growth without affecting endothelium repairment.29 Our study found that vascular injury induces increased expression of tRFGlnCTG, which promotes neointimal formation by inhibiting FAS (Figure 7E). The tRFGlnCTG-FAS pathway may be one of the mechanisms leading to intimal hyperplasia after arterial injury. The utilization of tRFGlnCTG inhibitors might be a feasible strategy to alleviate neointima formation.
Our understanding of the correlation between tRFs/tiRNAs and vascular biology is still in the preliminary stage. Although we have predicted the possible targets for tRFGlnCTG, the relationship between tRFGlnCTG and neointima formation requires more experiments to determine. Besides, studies of other differentially expressed tRFs and tiRNAs, especially those that are downregulated in intimal hyperplasia, have not been conducted. We hope that shortly, knowledge of the modulation mechanisms of tiRNA and tRFs on vascular biology will be developed and enriched and will provide more tools for the diagnosis and treatment of cardiovascular diseases.
Materials and methods
Intima hyperplasia model of rat CCA
The animal protocol conformed to the Animal Management Rules of China (documentation 55, 2201, Ministry of Health, China) and the recommendations in the 8th edition of the Guide for the Care and Use of Laboratory Animals of the NIH (NIH revised 2011). All experiments were approved by the Southwest Medical University Laboratory Animal Care and Use Committee. 8-week-old healthy male Sprague-Dawley rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (300 mg/kg), as described in the previous study.39 A 2F Fogarty catheter (Edward Lifesciences, Irvine, CA, USA) was inserted into the left CCA of the rat, dragged, and rotated. The right CCA of the rat was dissected, but not invaded, as a sham control. The rats were sacrificed under general anesthesia 14 days after surgery. The bilateral CCAs were harvested, sectioned, and subjected to a small RNA sequencing.
Small RNA sequencing
Total RNA of arteries was extracted by the TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) method. First, the samples were pretreated to remove the modifications, including 3′-aminoacyl, 2′,3′-cyclic phosphate, 5′-OH, m1A, and m3C. With the use of RT primers and amplification primers, cDNA was synthesized and amplified. Second, 135−160 bp PCR-amplified fragments (corresponding to a small RNA size of 15∼40 nt) were extracted and purified from the polyacrylamide gel electrophoresis (PAGE) gel. Finally, the completed libraries were quantified by a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). The diluted library was sequenced on a NextSeq 500 system (Illumina, San Diego, CA, USA).
The identification and analysis of tiRNA and tRFs
The Illumina chastity filter analyzed the raw sequencing reads data. The sequence of the 5′ and 3′ adaptors was removed from the small RNA data. The trimmed reads conformed to tRNAs were processed to study further. The other small RNAs, including mRNA, rRNA, small nuclear RNA (snRNA), Piwi-interacting RNA (piRNA), small nucleolar RNA (snoRNA), and miRNA, were excluded. Via R software, the differentially expressed tRFs or tiRNAs were hierarchical clustered and made scatterplots.
Quantitative real-time PCR
RNA modifications, including terminal and methylations, were removed using the rtStar tRF&tiRNA Pretreatment Kit (Arraystar, Rockville, MD, USA), according to the manufacturer’s instructions. Total RNA of each sample was sequentially ligated to 3′ and 5′ small RNA adaptors, and each adaptor was 60 nt. Therefore, the length of the amplified cDNA is about 134−160 bp (120 nt + 15−40 nt) (Figure S1B). cDNA was synthesized and amplified using Illumina’s proprietary RT primers and amplification primers. With the use of the rtStar First-Strand cDNA Synthesis Kit (Arraystar), cDNA was synthesized and amplified. Stem-loop primers were added into the RT mixtures, and the reaction mixtures were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 60°C for 60 s in a ViiA 7 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). U6 was used as an internal control. The 2–ΔΔCt methods were used for the analysis of the gene expression.
Rat thoracic aorta VSMC culture
As mentioned in the previous study,40 the rat thoracic aorta was dissected under the aseptic circumstance. First, the tunica adventitia was removed. Subsequently, the ECs on the intima were digested using 0.1% type I collagenase. Finally, the tunica media were cut into small pieces and immersed in DMEM containing 15 mM HEPES, 100 U/mL penicillin, 100 mg/mL streptomycin (Sigma-Aldrich, St. Louis, MO, USA), and 10% newborn bovine serum (Gibco). VSMCs were identified by immunocytochemistry using the anti-alpha-smooth muscle actin (α-SMA) antibody (Cell Signaling Technology, Danvers, MA, USA). Passages 4−7 VSMCs were used for subsequent experiments.
PDGF-BB and TGF-β1 stimulation on VSMCs
VSMCs were cultivated on 6-well plates at a density of 2 × 105 cells with DMEM containing 10% neonatal bovine serum. 25 ng/mL rPDGF-BB (R&D Systems, MN, USA) or 1 ng/mL rTGF-β1 (R&D Systems) was added into the culture medium. The expression of tRFGlnCTG in VSCMs was detected after 24 h by qPCR.
Transfection of RNA mimetics or inhibitors
The exogenous tRF mimetics and inhibitors were synthesized (GenePharma, Shanghai, China.). VSMCs were seeded at a density of 2 × 105 cells/mL and cultured in 6-well plates for 24 h. RNA mimetics or inhibitors were transfected into VSMCs at a final concentration at 50 nM using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s protocol.
Cell viability and proliferation assay
VSMCs were transfected with tRF mimetics or inhibitors and then seeded at 5 × 104 cells/mL in a 96-well plate for 48 h. 10 μL of CCK-8 (Dojindo, Kumamoto, Japan) solution was added to the VSMCs, and the cells were incubated at 37 °C. The absorbance was measured at 450 nm using an iMark microplate reader (Bio-Rad, Hercules, CA, USA).
VSMCs were cultured in a 96-well plate for 8 h. 50 μM EdU was added into the culture medium for 2 h. Then the staining solution was added for 30 min in the dark. The proliferation cells were observed using a fluorescence microscope (Olympus BX-51).
Cell migration assay
After transfected with tRF mimetics or inhibitors, VSMCs were seeded at a density of 2 × 105 cells/mL in 24-well plates and cultivated 48 h to the confluence. A 200 μL pipette tip was used to scratch the cell layer to form a wound. The cells were photographed under a BH-2 optical microscope (Olympus, Tokyo, Japan) at 0 h and 24 h after wounding.
A Costar Transwell chamber (pore size of 8 μm; Corning Life Sciences, Tewksbury, MA, USA) containing serum-free DMEM was used to culture VSMCs. The chamber was immersed in the DMEM with 20% fetal bovine serum for 8 h. The unmigrated cells on the inner side of the chamber were removed using a cotton bar. The migrated cells on the chamber’s outer side were dyed with crystal violet and counted using an optical microscope.
Bioinformatical analysis
In the R environment, TargetScan and miRanda databases are applied to predict the potential target genes and sites of tRFGlnCTG. The Database for Annotation, Visualization and Integrated Discovery (DAVID) version (v.)6.8 was used to perform the functional clustering of target genes. KEGG pathway was used to analyze the signaling pathways in which target genes may participate. RNAhybrid 2.2 (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/submission.html) was used to analyze the potential binding sites and their MFE between tRNAGlnCTG and the target gene.
Western blotting
The rat CCAs or cultured cells’ total protein were abstracted using radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China). Proteins were isolated by PAGE and transferred to a nitrocellulose membrane (GE Healthcare Life Sciences, PA, USA). The target proteins were identified with ATF6, FAS, IL-10, MAPK1, and SPRY2 antibodies (Absin, Shanghai, China).
Luciferase reporter assay
1.0 × 104 HEK293T cells were cultured in a 96-well plate. tRFGlnCTG mimetics or nontarget control were diluted in 5 μL of Opti-MEM medium (Beyotime, China). FAS 3′ UTR dual reporter vector (or mutation vector) and 0.25 μL of Lipo6000 transfection reagent (Beyotime, China) were diluted in 5 μL of Opti-MEM medium. The two solutions were mixed gently and added to the cells. The transfection concentration of mimetics was 50 nM. The plasmid concentration was 50 ng/well. After 48 h of transfection, the medium was removed, and luciferase reagent (Promega, Fitchburg, WI, USA) was added. The fluorescence value was measured with a GloMax 96 spectrophotometer (Promega).
Overexpression of FAS
Lentiviral vector overexpressing recombinant FAS (rFAS-OE-LV) encoding FAS (GenBank: NM_139194.2) was obtained by cloning the coding region of FAS from rat VSMC cDNA into lentiviral vector GV492 (GeneChem, Shanghai, China). VSMCs were infected with FAS-OE-LV particles, and the efficacy of transfection was investigated by western blotting.
Statistical analysis
If there is no specific remark in all of the experiments, then the replicates used in each experimental group are 5 (n = 5). The statistical analysis was processed with Prism 7 (GraphPad, San Diego, CA, USA). Two-tailed Student’s t test was performed for pairwise comparison, and a one-way ANOVA was performed for multiple comparisons. Bonferroni corrected p <0.05 was considered statistically significant. The results were expressed as the mean ± standard error of the mean (SEM).
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant numbers 82070288 and 31900813); Science & Technology Department of Sichuan Province (grant numbers 20ZDYF2108 and 2019YJ0409); Office of Science and Technology and Intellectual Property of Luzhou (grant number: 2019LZXNYDJ29); Talent Development Project of The Affiliated Hospital of Southwest Medical University; and Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province, Southwest Medical University (grant number xtcx2019-18).
Author contributions
J.J. designed the research and wrote and reviewed the manuscript. J.J. and R.J. analyzed the data. X.-L.Z., T.L., Y.C., Q.-P.Y., X.L., Y.L., Y.-Y.G., and J.-J.D. performed the research.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.010.
Contributor Information
Rui Jiang, Email: jiangrui@126.com.
Jun Jiang, Email: jiangjun@swmu.edu.cn.
Supplemental information
References
- 1.Emara M.M., Ivanov P., Hickman T., Dawra N., Tisdale S., Kedersha N., Hu G.F., Anderson P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. J. Biol. Chem. 2010;285:10959–10968. doi: 10.1074/jbc.M109.077560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanada T., Weitzer S., Mair B., Bernreuther C., Wainger B.J., Ichida J., Hanada R., Orthofer M., Cronin S.J., Komnenovic V. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenway M.J., Andersen P.M., Russ C., Ennis S., Cashman S., Donaghy C., Patterson V., Swingler R., Kieran D., Prehn J. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 4.van Es M.A., Schelhaas H.J., van Vught P.W., Ticozzi N., Andersen P.M., Groen E.J., Schulte C., Blauw H.M., Koppers M., Diekstra F.P. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann. Neurol. 2011;70:964–973. doi: 10.1002/ana.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Q., Yan M., Cao Z., Li X., Zhang Y., Shi J., Feng G.H., Peng H., Zhang X., Zhang Y. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351:397–400. doi: 10.1126/science.aad7977. [DOI] [PubMed] [Google Scholar]
- 6.Sharma U., Conine C.C., Shea J.M., Boskovic A., Derr A.G., Bing X.Y., Belleannee C., Kucukural A., Serra R.W., Sun F. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishima E., Inoue C., Saigusa D., Inoue R., Ito K., Suzuki Y., Jinno D., Tsukui Y., Akamatsu Y., Araki M. Conformational change in transfer RNA is an early indicator of acute cellular damage. J. Am. Soc. Nephrol. 2014;25:2316–2326. doi: 10.1681/ASN.2013091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Q., Lee I., Ren J., Ajay S.S., Lee Y.S., Bao X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol. Ther. 2013;21:368–379. doi: 10.1038/mt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodarzi H., Liu X., Nguyen H.C., Zhang S., Fish L., Tavazoie S.F. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell. 2015;161:790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maute R.L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA. 2013;110:1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatesh T., Suresh P.S., Tsutsumi R. tRFs: miRNAs in disguise. Gene. 2016;579:133–138. doi: 10.1016/j.gene.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 12.Sobala A., Hutvagner G. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10:553–563. doi: 10.4161/rna.24285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saikia M., Jobava R., Parisien M., Putnam A., Krokowski D., Gao X.H., Guan B.J., Yuan Y., Jankowsky E., Feng Z. Angiogenin-cleaved tRNA halves interact with cytochrome c, protecting cells from apoptosis during osmotic stress. Mol. Cell. Biol. 2014;34:2450–2463. doi: 10.1128/MCB.00136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schopman N.C., Heynen S., Haasnoot J., Berkhout B. A miRNA-tRNA mix-up: tRNA origin of proposed miRNA. RNA Biol. 2010;7:573–576. doi: 10.4161/rna.7.5.13141. [DOI] [PubMed] [Google Scholar]
- 16.Morris S., Leis J. Changes in Rous sarcoma virus RNA secondary structure near the primer binding site upon tRNATrp primer annealing. J. Virol. 1999;73:6307–6318. doi: 10.1128/jvi.73.8.6307-6318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggero K., Guffanti A., Corradin A., Sharma V.K., De Bellis G., Corti G., Grassi A., Zanovello P., Bronte V., Ciminale V., D’Agostino D.M. Small noncoding RNAs in cells transformed by human T-cell leukemia virus type 1: a role for a tRNA fragment as a primer for reverse transcriptase. J. Virol. 2014;88:3612–3622. doi: 10.1128/JVI.02823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couvillion M.T., Bounova G., Purdom E., Speed T.P., Collins K. A Tetrahymena Piwi bound to mature tRNA 3′ fragments activates the exonuclease Xrn2 for RNA processing in the nucleus. Mol. Cell. 2012;48:509–520. doi: 10.1016/j.molcel.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanov P., Emara M.M., Villen J., Gygi S.P., Anderson P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell. 2011;43:613–623. doi: 10.1016/j.molcel.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang X., Wang X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 21.Mei Y., Yong J., Liu H., Shi Y., Meinkoth J., Dreyfuss G., Yang X. tRNA binds to cytochrome c and inhibits caspase activation. Mol. Cell. 2010;37:668–678. doi: 10.1016/j.molcel.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X., Karamariti E., Simpson R., Wang W., Xu Q. Dickkopf Homolog 3 Induces Stem Cell Differentiation into Smooth Muscle Lineage via ATF6 Signalling. J. Biol. Chem. 2015;290:19844–19852. doi: 10.1074/jbc.M115.641415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karamariti E., Zhai C., Yu B., Qiao L., Wang Z., Potter C.M.F., Wong M.M., Simpson R.M.L., Zhang Z., Wang X. DKK3 (Dickkopf 3) Alters Atherosclerotic Plaque Phenotype Involving Vascular Progenitor and Fibroblast Differentiation Into Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2018;38:425–437. doi: 10.1161/ATVBAHA.117.310079. [DOI] [PubMed] [Google Scholar]
- 24.Karali E., Bellou S., Stellas D., Klinakis A., Murphy C., Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell. 2014;54:559–572. doi: 10.1016/j.molcel.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 25.Luo Z., Sata M., Nguyen T., Kaplan J.M., Akita G.Y., Walsh K. Adenovirus-mediated delivery of fas ligand inhibits intimal hyperplasia after balloon injury in immunologically primed animals. Circulation. 1999;99:1776–1779. doi: 10.1161/01.cir.99.14.1776. [DOI] [PubMed] [Google Scholar]
- 26.Mano T., Luo Z., Suhara T., Smith R.C., Esser S., Walsh K. Expression of wild-type and noncleavable Fas ligand by tetracycline-regulated adenoviral vectors to limit intimal hyperplasia in vascular lesions. Hum. Gene Ther. 2000;11:1625–1635. doi: 10.1089/10430340050111287. [DOI] [PubMed] [Google Scholar]
- 27.Jiang C., Yang Y.F., Cheng S.H. Fas ligand gene therapy for vascular intimal hyperplasia. Curr. Gene Ther. 2004;4:33–39. doi: 10.2174/1566523044578022. [DOI] [PubMed] [Google Scholar]
- 28.Sata M., Luo Z., Walsh K. Fas ligand overexpression on allograft endothelium inhibits inflammatory cell infiltration and transplant-associated intimal hyperplasia. J. Immunol. 2001;166:6964–6971. doi: 10.4049/jimmunol.166.11.6964. [DOI] [PubMed] [Google Scholar]
- 29.Kural M.H., Wang J., Gui L., Yuan Y., Li G., Leiby K.L., Quijano E., Tellides G., Saltzman W.M., Niklason L.E. Fas ligand and nitric oxide combination to control smooth muscle growth while sparing endothelium. Biomaterials. 2019;212:28–38. doi: 10.1016/j.biomaterials.2019.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazighi M., Pellé A., Gonzalez W., Mtairag E.M., Philippe M., Hénin D., Michel J.-B., Feldman L.J. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H866–H871. doi: 10.1152/ajpheart.00918.2003. [DOI] [PubMed] [Google Scholar]
- 31.Verma S.K., Garikipati V.N., Krishnamurthy P., Khan M., Thorne T., Qin G., Losordo D.W., Kishore R. IL-10 Accelerates Re-Endothelialization and Inhibits Post-Injury Intimal Hyperplasia following Carotid Artery Denudation. PLoS ONE. 2016;11:e0147615. doi: 10.1371/journal.pone.0147615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldman L.J., Aguirre L., Ziol M., Bridou J.P., Nevo N., Michel J.B., Steg P.G. Interleukin-10 inhibits intimal hyperplasia after angioplasty or stent implantation in hypercholesterolemic rabbits. Circulation. 2000;101:908–916. doi: 10.1161/01.cir.101.8.908. [DOI] [PubMed] [Google Scholar]
- 33.Pintucci G., Saunders P.C., Gulkarov I., Sharony R., Kadian-Dodov D.L., Bohmann K., Baumann F.G., Galloway A.C., Mignatti P. Anti-proliferative and anti-inflammatory effects of topical MAPK inhibition in arterialized vein grafts. FASEB J. 2006;20:398–400. doi: 10.1096/fj.05-4114fje. [DOI] [PubMed] [Google Scholar]
- 34.Evans B.C., Hocking K.M., Osgood M.J., Voskresensky I., Dmowska J., Kilchrist K.V., Brophy C.M., Duvall C.L. MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia. Sci. Transl. Med. 2015;7:291ra95. doi: 10.1126/scitranslmed.aaa4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taniguchi K., Sasaki K., Watari K., Yasukawa H., Imaizumi T., Ayada T., Okamoto F., Ishizaki T., Kato R., Kohno R. Suppression of Sproutys has a therapeutic effect for a mouse model of ischemia by enhancing angiogenesis. PLoS ONE. 2009;4:e5467. doi: 10.1371/journal.pone.0005467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wietecha M.S., Chen L., Ranzer M.J., Anderson K., Ying C., Patel T.B., DiPietro L.A. Sprouty2 downregulates angiogenesis during mouse skin wound healing. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H459–H467. doi: 10.1152/ajpheart.00244.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biyashev D., Veliceasa D., Topczewski J., Topczewska J.M., Mizgirev I., Vinokour E., Reddi A.L., Licht J.D., Revskoy S.Y., Volpert O.V. miR-27b controls venous specification and tip cell fate. Blood. 2012;119:2679–2687. doi: 10.1182/blood-2011-07-370635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C., Chaturvedi D., Jaggar L., Magnuson D., Lee J.M., Patel T.B. Regulation of vascular smooth muscle cell proliferation and migration by human sprouty 2. Arterioscler. Thromb. Vasc. Biol. 2005;25:533–538. doi: 10.1161/01.ATV.0000155461.50450.5a. [DOI] [PubMed] [Google Scholar]
- 39.Xu J.Y., Chang N.B., Rong Z.H., Li T., Xiao L., Yao Q.P., Jiang R., Jiang J. circDiaph3 regulates rat vascular smooth muscle cell differentiation, proliferation, and migration. FASEB J. 2019;33:2659–2668. doi: 10.1096/fj.201800243RRR. [DOI] [PubMed] [Google Scholar]
- 40.Xu J.Y., Chang N.B., Li T., Jiang R., Sun X.L., He Y.Z., Jiang J. Endothelial Cells Inhibit the Angiotensin II Induced Phenotypic Modulation of Rat Vascular Adventitial Fibroblasts. J. Cell. Biochem. 2017;118:1921–1927. doi: 10.1002/jcb.25941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.