

Abstract
The nuclear receptor-binding SET domain (NSD) protein family encoding histone lysine methyltransferases is involved in cancer progression. However, the role of NSDs in esophageal squamous cell carcinoma (ESCC) remains unclear. Here we examined the expression of NSDs in cisplatin-resistant and parental ESCC cells and revealed the upregulation of NSD2 in cisplatin-resistant cells. Ectopic expression of NSD2 increased cisplatin resistance and attenuated cisplatin-induced apoptosis. Colony formation assay indicated that NSD2 overexpression enhanced long-term survival of ESCC cells after treatment with cisplatin. In contrast, knockdown of NSD2 inhibited ESCC cell proliferation and sensitized ESCC cells to cisplatin. Depletion of NSD2 augmented the cytotoxic effect of cisplatin on EC109 xenograft tumors. NSD2 stimulated long non-coding RNA MACC1-AS1 in ESCC cells. Knockdown of MACC1-AS1 impaired NSD2-induced cisplatin resistance. Moreover, MACC1-AS1 overexpression promoted ESCC cell proliferation and cisplatin resistance. Clinically, MACC1-AS1 was upregulated in ESCC relative to adjacent noncancerous tissues. High MACC1-AS1 levels were significantly associated with reduced overall survival of ESCC patients. There was a positive correlation between MACC1-AS1 and NSD2 expression in ESCC specimens. Taken together, MACC1-AS1 induced by NSD2 mediates resistance to cisplatin in ESCC and may represent a novel target to improve cisplatin-based chemotherapy.
Keywords: apoptosis, cisplatin resistance, esophageal cancer, MACC1-AS1, NSD2
Graphical Abstract
NSD2 promotes cisplatin resistance in ESCC cells. MACC1-AS1 mediates NSD2-induced cisplatin resistance. MACC1-AS1 may represent a novel target to improve cisplatin-based chemotherapy against ESCC.
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most frequently diagnosed cancers, especially in Asian countries including China.1,2 Although surgery remains the primary treatment for ESCC, chemotherapy is suggested to improve therapeutic outcomes.3,4 Cisplatin is a widely used chemotherapeutic agent for ESCC.5 However, chemotherapeutic response varies greatly among patients with ESCC. The development of drug resistance limits chemotherapeutic efficacy. Therefore, exploring the molecular mechanism of chemoresistance is of significance in treating ESCC.
The nuclear receptor-binding SET domain (NSD) protein family encoding histone lysine methyltransferases comprises three members, i.e., NSD1, NSD2 (MMSET/WHSC1), and NSD3 (WHSC1L1). These NSD enzymes direct methylation of histone H3 lysine 36 (H3K36) and contributes to active transcription.6 Dysregulation of NSDs has been reported in many cancers, suggesting their involvement in cancer progression.6,7 NSD1 is epigenetically silenced via promoter methylation and induces tumor-suppressive effects in clear cell renal cell carcinoma and glioma.8,9 In contrast to NSD1, NSD2 is frequently upregulated in malignant lesions such as prostate cancer and osteosarcoma.10,11 Knockdown of NSD2 suppresses prostate cancer metastasis and improves osteosarcoma sensitivity to cisplatin.10,11 Upregulation of NSD3 as a result of genomic amplification enhances breast cancer initiation and metastasis.12 These studies suggest differential regulation of NSD family members in cancers. Despite these findings, little is known about the expression and function of NSDs in ESCC.
Long non-coding RNAs (lncRNAs) are a class of noncoding transcripts of longer than 200 nucleotides. lncRNAs can exert their biological effects through interplay with protein or other RNA molecules.13,14 To date, many lncRNAs have been found to be aberrantly expressed and exhibit diagnostic and prognostic potential in cancers.13, 14, 15, 16 Accumulating evidence indicates that lncRNAs play a pivotal role in various aspects of tumor biology, such as proliferation, invasion, and survival.15,16 For instance, lncRNA MACC1-AS1, which is induced in gastric cancer cells by mesenchymal stem cells, has the ability to enhance stemness and chemoresistance.16 MACC1-AS1 is also upregulated in pancreatic cancer and participates in cancer growth and metastasis.17 lncRNA FOXD2-AS1 has been shown to induce cisplatin resistance in non-small cell lung cancer and ESCC.18,19 These studies suggest lncRNAs as an important regulator of cancer cell chemoresistance.
In the present study, we revealed the induction of NSD2 in cisplatin-resistant ESCC cells and explored the role of NSD2 in the regulation of ESCC cisplatin resistance. We further identified the key lncRNA(s) that mediates the function of NSD2. In addition, the expression and function of the lncRNA(s) in ESCC were investigated.
Results
NSD2 is stimulated in cisplatin-resistant ESCC cells
To determine whether the NSD family is related to cisplatin resistance of ESCC cells, we examined the expression of NSD1-3 in cisplatin-resistant and parental KYSE150 cells. Cisplatin-resistant KYSE150 cells had a 6.5-fold increase in the half maximal inhibitory concentration (IC50) for cisplatin relative to parental control cells (Figure 1A). Of note, NSD2 was remarkably upregulated in cisplatin-resistant KYSE150 cells (Figures 1B and 1C). In contrast, both NSD1 and NSD3 did not significantly differ between cisplatin-resistant and parental KYSE150 cells. The upregulation of NSD2, but not NSD1 or NSD3, was also detected in cisplatin-resistant KYSE30 cells (Figures 1D–1F). These results suggest an implication of NSD2 in ESCC cisplatin resistance.
Figure 1.

NSD2 is stimulated in cisplatin-resistant ESCC cells
(A) Cisplatin-resistant and parental KYSE150 cells were treated with different concentrations of cisplatin for 72 h, and cell viability was measured. Bar graphs show the cisplatin IC50 values. (B and C) Measurement of the (B) mRNA and (C) protein levels of NSDs in cisplatin-resistant and parental KYSE150 cells. (D) Measurement of the cisplatin IC50 values in cisplatin-resistant and parental KYSE30 cells. (E) Detection of indicated transcripts in cisplatin-resistant and parental KYSE30 cells by real-time PCR analysis. (F) Detection of NSD2 protein expression in cisplatin-resistant and parental KYSE30 cells. ∗p < 0.05. ns indicates no significance.
NSD2 upregulation leads to cisplatin resistance in ESCC cells
To investigate the role of NSD2 in regulating the response of ESCC cells to cisplatin, we ectopically expressed NSD2 in both KYSE150 and KYSE30 cells (Figure 2A). Consistent with the elevation of NSD2, global H3K36me2 levels were increased in NSD2-overexpressing ESCC cells (Figure 2A). TGFA, PAK1, and MET have been identified as direct target genes of NSD2.20 As expected, TGFA, PAK1, and MET were induced by NSD2 overexpression (Figure S1). Next, we evaluated the effect of NSD2 overexpression on cisplatin sensitivity. After treatment with different concentrations of cisplatin for 72 h, cell viability was measured. The results showed that NSD2 overexpression rendered ESCC cells more resistant to cisplatin (Figure 2B). To exclude the possibility that the NSD2-mediated cisplatin resistance is a result of increased proliferation, we performed EdU proliferation assays. The cancer cell proliferation capacity was not affected by NSD2 overexpression (Figure 2C). Interestingly, analysis of apoptosis by Annexin V/propidium iodide (PI) staining revealed that cisplatin-induced apoptosis was attenuated by overexpression of NSD2 (Figure 2D), suggesting a pro-survival role for NSD2. Colony formation assay further confirmed that NSD2 overexpression augmented long-term survival of ESCC cells after treatment with cisplatin (Figure 2E). In contrast, overexpression of NSD2 did not affect the migration capacity of KYSE150 and KYSE30 cells, as determined by Transwell migration assay (Figure S2). Taken together, NSD2 upregulation prevents cisplatin-induced apoptosis and contributes to cisplatin resistance in ESCC cells.
Figure 2.

NSD2 upregulation leads to cisplatin resistance in ESCC cells
(A) Western blot analysis of NSD2 and H3K36me2 protein levels in ESCC cells transfected with empty vector or NSD2-expressing plasmids. (B) Bar graphs show the cisplatin IC50 values in ESCC cells transfected with empty vector or NSD2-expressing plasmids. (C) Cell proliferation was determined by EdU incorporation assay after treatment with or without cisplatin for 72 h. Left, representative merged images of EdU incorporation (red) and nuclear staining (blue). Scale bar, 50 μm. Right, quantification of EdU-positive cells. ∗p < 0.05. ns indicates no significance. (D) Measurement of apoptosis by Annexin V/PI staining in the transfected cells after 10 μM cisplatin treatment. (E) Colony formation assay. NSD2 overexpression enhanced long-term survival of ESCC cells after treatment with 10 μM cisplatin. Right, quantitative analysis of colonies. ∗p < 0.05.
NSD2 depletion increases cisplatin sensitivity in ESCC cells
Next, we checked whether knockdown of NSD2 can enhance the cytotoxic effects of cisplatin on ESCC cells. Since EC109 cells had abundant expression of endogenous NSD2 (Figure 3A), this ESCC cell line was used in NSD2 knockdown experiments. qRT-PCR analysis confirmed the depletion of NSD2 in EC109 cells by transfecting with NSD2-targeting short hairpin RNAs (shRNAs) (Figure 3B). Western blot analysis confirmed the reduction of NSD2 protein and H3K36me2 levels in EC109 cells transfected with NSD2 shRNAs (Figure 3C). Knockdown of NSD2 inhibited the proliferation of EC109 cells (Figure 3D). Most interestingly, silencing of NSD2 significantly increased the sensitivity of EC109 cells to cisplatin, as evidenced by a reduction in the IC50 (Figure 3E). Furthermore, NSD2 depletion significantly increased EC109 cell apoptosis in response to cisplatin treatment (Figure 3F). We also determined whether knockdown of NSD2 is sufficient to re-sensitize cisplatin-resistant ESCC cells. As shown in Figures 3G and 3H, silencing of NSD2 re-sensitized resistant KYSE150 and KYSE30 cells to cisplatin, compared to control shRNA groups (p < 0.05).
Figure 3.

NSD2 depletion increases cisplatin sensitivity in ESCC cells in vitro
(A) Western blot analysis of NSD2 protein levels in ESCC cell lines and HEsEpiC cells. (B) Analysis of NSD2 mRNA levels in EC109 cells transfected with control shRNA (shCtrl) or NSD2-targeting shRNA (shNSD2). (C) Western blot analysis of NSD2 and H3K36me2 protein levels in EC109 cells transfected with shCtrl or shNSD2. (D) Silencing of NSD2 suppressed the proliferation of EC109 cells. (E) Measurement of the cisplatin IC50 values in EC109 cells transfected with shCtrl or shNSD2. (F) Detection of apoptosis in EC109 cells transfected with shCtrl or shNSD2 after treatment with 10 μM cisplatin. (G) Measurement of NSD2 mRNA levels in resistant KYSE150 and KYSE30 cells transfected with shCtrl or shNSD2. (H) Measurement of the cisplatin IC50 values in resistant KYSE150 and KYSE30 cells transfected with shCtrl or shNSD2. ∗p < 0.05.
To validate the effect of NSD2 knockdown on the cisplatin resistance of ESCC cells in vivo, we performed xenograft experiments in nude mice. We found that EC109 cells with depletion of NSD2 were more sensitive to cisplatin treatment compared to those transfected with control shRNA (Figures 4A–4C). The terminal dideoxynucleotidetransferase (TdT)-mediated X-dUTP nick and end labeling (TUNEL) staining was performed to assess apoptosis. The results showed that NSD2 depletion was associated with increased apoptosis in cisplatin-treated xenograft tumors (Figure 4D). Similar to that in EC109 cells, depletion of NSD2 restored the sensitivity to cisplatin in cisplatin-resistant KYSE30 cells (Figures 4E–4H). Collectively, these observations suggest NSD2 as a potential target for improving cisplatin sensitivity in ESCC cells.
Figure 4.

NSD2 depletion increases cisplatin sensitivity in ESCC cells in vivo
ESCC cells stably transfected with shCtrl or shNSD2 were injected into nude mice. In the drug treatment groups, 3 mg/kg cisplatin was injected. (A and E) Representative images of subcutaneous xenograft tumors from 2 mice of each group. (B and F) Tumor growth curves were plotted. (C and G) Quantitative analysis of the weight of xenograft tumors. (D and H) TUNEL staining showed that NSD2 depletion increased cisplatin-induced apoptosis in xenograft tumors. ∗p < 0.05.
NSD2-induced cisplatin resistance requires the upregulation of MACC1-AS1 and FOXD2-AS1
Because lncRNAs play critical roles in various biological processes,13, 14, 15, 16 we attempted to identify the key lncRNA(s) that mediates NSD2-induced cisplatin resistance. To this end, we analyzed the expression of cancer-related lncRNAs between NSD2-overexpressing and control KYSE150 cells using a functional lncRNA PCR array. This PCR array can simultaneously examine 84 lncRNAs. Among the 84 lncRNAs tested, 3 lncRNAs (i.e., MACC1-AS1, FOXD2-AS1, and DANCR) showed a >2-fold change in expression levels (Figure 5A). Chromatin immunoprecipitation (ChIP) assay revealed that the levels of H3K36me2 were significantly increased at the promoters of MACC1-AS1, FOXD2-AS1, and DANCR in NSD2-overexpressing cells (Figure 5B). To validate whether these lncRNAs are required for NSD2-induced cisplatin resistance, we knocked down their expression using shRNA technology (Figure 5C). In response to cisplatin treatment, NSD2-induced cisplatin resistance was significantly impaired when MACC1-AS1 or FOXD2-AS1 was depleted (Figure 5D). However, depletion of DANCR failed to reverse NSD2-induced cisplatin resistance. Knockdown of MACC1-AS1 or FOXD2-AS1 also re-sensitized NSD2-overexpressing KYSE30 cells to cisplatin (Figures 5E and 5F). Moreover, double knockdown of MACC1-AS1 and FOXD2-AS1 resulted in increased sensitivity to cisplatin (Figure 5F). Next, we tested whether knockdown of MACC1-AS1 and FOXD2-AS1 can reverse cisplatin resistance in cisplatin-resistant ESCC cells. As expected, depletion of either MACC1-AS1 or FOXD2-AS1 increased cisplatin sensitivity in cisplatin-resistant KYSE150 and KYSE30 cells (Figure 5G). Knockdown of both MACC1-AS1 and FOXD2-AS1 produced an additive effect on cisplatin sensitivity. Taken together, these findings suggest that both MACC1-AS1 and FOXD2-AS1 play an essential role in mediating NSD2-induced cisplatin resistance.
Figure 5.

NSD2-induced cisplatin resistance requires the upregulation of MACC1-AS1 and FOXD2-AS1
(A) Quantitative real-time PCR analysis of MACC1-AS1, FOXD2-AS1, and DANCR levels in NSD2-overexpressing cells. (B) ChIP analysis of the enrichment of H3K36me2 at the promoter regions of indicated genes in control vector and NSD2 overexpression KYSE150 cells. (C) Quantitative real-time PCR analysis of MACC1-AS1, FOXD2-AS1, and DANCR levels after transfection with indicated shRNAs. (D) Measurement of the cisplatin IC50 values in KYSE150 cells transfected with indicated constructs. (E) Quantitative real-time PCR analysis of MACC1-AS1 and FOXD2-AS1 levels in KYSE30 cells transfected with indicated shRNAs. (F) Measurement of the cisplatin IC50 values in KYSE30 cells transfected with indicated constructs. (G) Measurement of the cisplatin IC50 values in cisplatin-resistant KYSE150 and KYSE30 cells transfected with indicated constructs. ∗p < 0.05. ns indicates no significance.
MACC1-AS1 promotes ESCC cell growth and cisplatin resistance
A previous study has indicated that FOXD2-AS1 is involved in the growth, invasion, and cisplatin resistance of ESCC cells.19 Yet, the expression and function of MACC1-AS1 in ESCC is still unclear. Therefore, in this work we focused on the role of MACC1-AS1 in ESCC progression. We observed that the level of MACC1-AS1 was significantly greater in ESCC cell lines than that in HEsEpiC cells (Figure 6A). Knockdown of MACC1-AS1 (Figure 6B) inhibited cell proliferation (Figure 6C) and increased cisplatin sensitivity (Figure 6D) in EC109 cells. Conversely, ectopic expression of MACC1-AS1 (Figure 6E) enhanced EC109 cell proliferation (Figure 6F) and cisplatin resistance (Figure 6G). These results collectively indicate MACC1-AS1 as an oncogene in ESCC.
Figure 6.

MACC1-AS1 promotes ESCC cell growth and cisplatin resistance
(A) Quantitative real-time PCR analysis of MACC1-AS1 in ESCC cell lines and HEsEpiC cells. (B) Quantitative real-time PCR analysis of MACC1-AS1 after transfection with indicated shRNAs. (C) Silencing of MACC1-AS1 suppressed the proliferation of EC109 cells. (D) Measurement of the cisplatin IC50 values in EC109 cells transfected with shCtrl or shMACC1-AS1. (E) Quantitative real-time PCR analysis of MACC1-AS1 after transfection with indicated constructs. (F) Overexpression of MACC1-AS1 promoted the proliferation of EC109 cells. (G) Measurement of the cisplatin IC50 values in EC109 cells transfected with indicated constructs. ∗p < 0.05.
Clinical significance of NSD2 and MACC1-AS1 in ESCC
Next, we examined the expression of NSD2 mRNA and MACC1-AS1 in 70 pairs of ESCC and normal esophageal tissue samples by quantitative real-time PCR analysis. The results showed that NSD2 mRNA (p = 0.0097) and MACC1-AS1 (p = 0.0001) expression was remarkably upregulated in ESCC tissues relative to adjacent noncancerous tissues (Figures 7A and 7B). Immunohistochemistry for NSD2 was also performed in the 70 ESCC cases and normal tissues. The results confirmed that NSD2 immunostaining score was higher in ESCC tissues than that in normal tissues (Figure 7C). High MACC1-AS1 levels were significantly associated with advanced tumor stage (p = 0.0003) and lymph node metastasis in ESCC (p = 0.0323; Table 1). Similarly, there was a significant correlation between high NSD2 expression and advanced tumor stage of ESCC (p = 0.0168; Table 1). We further analyzed the prognostic significance of MACC1-AS1 expression using The Cancer Genome Atlas (TCGA) database. Kaplan-Meier analysis showed that ESCC patients with high MACC1-AS1 expression in tumors had worse overall survival compared to those with low MACC1-AS1 expression (p = 0.0057; Figure 7D). These results suggest that MACC1-AS1 is a potential indicator for poor prognosis in ESCC patients. We also examined the correlation between MACC1-AS1 and NSD2 expression in the 70 ESCC cases. Interestingly, there was a positive correlation between MACC1-AS1 and NSD2 mRNA expression (r = 0.3477, p = 0.0032; Figure 7E).
Figure 7.

Clinical significance of NSD2 and MACC1-AS1 in ESCC
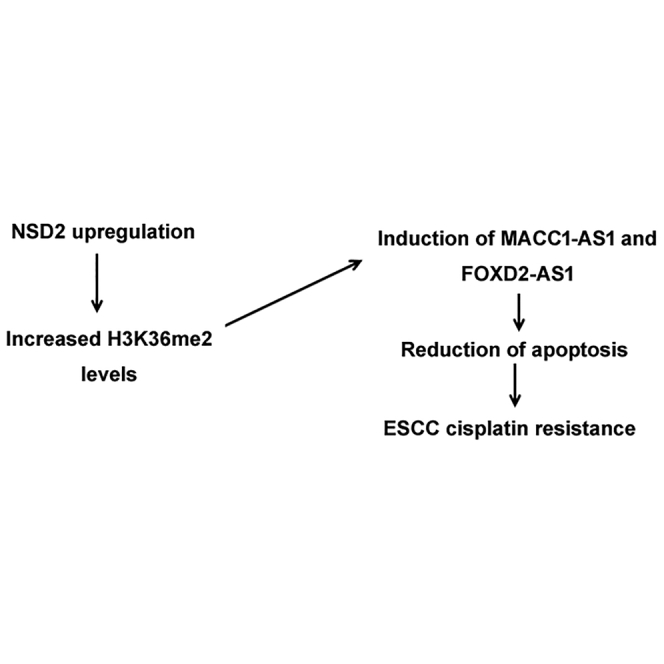
(A and B) Quantitative real-time PCR analysis of (A) MACC1-AS1 and (B) NSD2 in 70 pairs of ESCC and normal esophageal tissue samples. (C) Immunohistochemistry for NSD2 in 70 ESCC cases and normal tissues. Left, representative images of NSD2 staining. Scale bar, 40 μm. Right, quantitative evaluation of NSD2 staining results. (D) Survival analysis of ESCC patients from TCGA dataset indicated that ESCC patients with high MACC1-AS1 expression in tumors had worse overall survival compared to those with low MACC1-AS1 expression. (E) Analysis of the correlation between MACC1-AS1 and NSD2 expression in ESCC tissues (n = 70). (F) Schematic model showing that NSD2 promotes the expression of MACC1-AS1 and FOXD2-AS1, enhancing ESCC cell survival and cisplatin resistance.
Table 1.
Correlation of MACC1-AS1 expression with clinicopathological parameters in 70 ESCC patients
| Parameter | MACC1-AS1 expression |
p | NSD2 mRNA expression |
p | ||
|---|---|---|---|---|---|---|
| Low (n = 34) | High (n = 36) | Low (n = 28) | High (n = 42) | |||
| Gender | ||||||
| Male | 30 | 32 | 0.9315 | 25 | 37 | 0.8781 |
| Female | 4 | 4 | 3 | 5 | ||
| Age, years | ||||||
| ≤60 | 13 | 13 | 0.8541 | 10 | 16 | 0.8399 |
| >60 | 21 | 23 | 18 | 26 | ||
| TNM stage | ||||||
| I-II | 21 | 7 | 0.0003 | 16 | 12 | 0.0168 |
| III-IV | 13 | 29 | 12 | 30 | ||
| Lymph node metastasis | ||||||
| Negative | 19 | 11 | 0.0323 | 10 | 20 | 0.3241 |
| Positive | 15 | 25 | 18 | 22 | ||
Discussion
In this study, we show that NSD2, but not NSD1 or NSD3, is induced in cisplatin-resistant ESCC cells, suggesting an involvement of NSD2 in ESCC chemoresistance. Previous studies have reported that NSD2 drives oncogenic cascades in several malignancies including prostate cancer,10 myeloma,21 neuroblastoma,22 and osteosarcoma.11 Consistently, we demonstrate that NSD2 also plays an oncogenic role in ESCC, and its silencing restrains ESCC cell proliferation and enhances apoptosis. Moreover, NSD2 overexpression induces cisplatin resistance and attenuates cisplatin-induced apoptosis in ESCC cells. Using a mouse xenograft model, we validate the increased sensitivity to cisplatin in NSD2-depleted xenograft tumors. Taken together, NSD2 plays an essential role in the growth and cisplatin resistance of ESCC cells. However, it should be noted that the IC50 of cisplatin in EC109 cells is comparable to parental KYSE150 and KYSE30 cells despite high variation in NSD2 expression levels. This finding suggests that NSD2 is not the sole determinant of ESCC cell response to cisplatin. The ability of NSD2 to modulate cisplatin sensitivity may be affected by genetic contexts.
It has been documented that NSD2 facilitates DNA damage repair in multiple myeloma cells, thus conferring resistance to chemotherapeutic agents.23 In osteosarcoma, NSD2 regulates cisplatin sensitivity through the ERK and AKT pathways.11 NSD2 can induce gene expression through its ability to promote H3K36 methylation.6,24 Huang et al.24 reported that NSD2 overexpression leads to H3K36 dimethylation at the promoter regions of multiple cancer-related genes including BACE2, CA2, and IGF2BP2, contributing to their upregulation. Our data show that NSD2 overexpression causes a marked increase in global H3K36me2 levels in ESCC cells, which is accompanied by induction of multiple target genes including TGFA, PAK1, and MET. These finding suggest that NSD2 may epigenetically regulate many genes in ESCC cells. Since several lncRNAs such as LINC01234, CCAT1, and linc00173 are capable of modulating chemosensitivity of cancer cells,25, 26, 27 here we sought to identify the key lncRNA(s) involved in the function of NSD2. Intriguingly, we find that NSD2 overexpression stimulates the expression of MACC1-AS1 and FOXD2-AS1 in ESCC cells. Knockdown of MACC1-AS1 or FOXD2-AS1 significantly reverses NSD2-dependent cisplatin resistance of ESCC cells. Double knockdown of MACC1-AS1 and FOXD2-AS1 causes more sensitivity to cisplatin NSD2-overexpressing ESCC cells. In contrast, knockdown of another lncRNA, DANCR, fails to overcome NSD2-induced cisplatin resistance. Therefore, we suggest that both MACC1-AS1 and FOXD2-AS1 play a nonredundant role in mediating NSD2-induced cisplatin resistance. These findings encourage us to conduct genome-wide experiments in future work to identify more NSD2 downstream target genes.
It has been previously reported that FOXD2-AS1 is upregulated and serves as a poor prognostic factor in ESCC.28 Another study further indicates that FOXD2-AS1 overexpression promotes ESCC cell growth and invasion and attenuates cisplatin-induced apoptosis.19 These studies are consistent with our findings that FOXD2-AS1 contributes to cisplatin resistance induced by NSD2. Although the function of FOXD2-AS1 in ESCC has been uncovered, few studies have addressed the effect of MACC1-AS1 on ESCC cells. In the current study, we show that MACC1-AS1 is expressed at greater levels in ESCC tissues and cells than nonmalignant controls. Clinically, high MACC1-AS1 levels are associated with advanced tumor stage and reduced overall survival in ESCC patients. Moreover, a positive correlation between MACC1-AS1 and NSD2 expression was observed in ESCC tissues. Biologically, silencing of MACC1-AS1 blocks ESCC cell proliferation and increases cisplatin sensitivity. Our data provide the first evidence that MACC1-AS1 plays an oncogenic role in ESCC and represents a potential target to overcome cisplatin resistance. MACC1-AS1 also functions as an oncogene in several other malignancies such as gastric cancer16,29 and pancreatic cancer17. Qi et al.17 reported that MACC1-AS1 is capable of enhancing pancreatic cancer growth and metastasis through the PAX8/NOTCH1 signaling pathway. Zhao et al.29 reported that MACC1-AS1 promotes gastric cancer cell proliferation and enhances cell survival under metabolic stress through stabilization of MACC1 mRNA. However, the detailed mechanism for MACC1-AS1-mediated aggressive phenotype in ESCC remains to be clarified in future work.
In conclusion, NSD2 is stimulated in cisplatin-resistant ESCC cells and required for the development of cisplatin resistance in ESCC. MACC1-AS1 is upregulated by NSD2 and contributes to ESCC growth and cisplatin resistance (Figure 7F). Upregulation of MACC1-AS1 is associated with advanced tumor stage and poor prognosis in ESCC. Therefore, MACC1-AS1 represents a promising target to improve chemotherapy against ESCC.
Materials and methods
Cell culture
Three ESCC cell lines (KYSE150, KYSE30, and EC109) were obtained from Shanghai Institutes for Biological Sciences (Shanghai, China). They were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA) in a humidified atmosphere of 5% CO2 at 37°C. HEsEpiC esophageal epithelial cells were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in epithelial cell medium-2 (ScienCell Research Laboratories).
Cisplatin treatment
Cisplatin-resistant ESCC cell lines were established from parental cells through exposure to increasing concentrations of cisplatin, as described previously.30 Briefly, parental KYSE150 and KYSE30 cells were cultured in the presence of cisplatin (0.2–4 μM; Sigma-Aldrich) over a period of 3 months. For determination of cisplatin IC50 values, ESCC cells were exposed to 0–100 μM cisplatin for 72 h, and viable cells were determined using the MTT Cell Proliferation Kit I (Sigma-Aldrich). For apoptosis and colony formation assays, 10 μM cisplatin was used.
Quantitative real-time PCR analysis
Total RNA was isolated using TRIzol Reagent (Invitrogen) and reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (QIAGEN, Hilden, Germany). Quantitative real-time PCR assays were performed using a SYBR Green PCR Kit (QIAGEN). PCR primers are summarized in Table S1. The relative gene expression levels were determined by the 2−ΔΔCt method.31 GAPDH was used as a normalization control.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay buffer supplemented with a protease inhibitor cocktail (Abcam, Cambridge, UK). Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of total protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Anti-NSD1 (sc-130470; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-NSD2 (ab137429; Abcam), anti-NSD3 (ab137430; Abcam), anti-H3K36me2 (ab176921, Abcam), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (ab181602; Abcam) antibodies were used as the primary antibodies. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h and visualized by enhanced chemiluminescence (ECL) reagent (Millipore, Billerica, MA, USA).
Plasmids and transfections
The full-length sequences of NSD2 (NM_133330.2) and MACC1-AS1 (NR_046756.1) were cloned to pcDNA3.1(+) expression vector. The shRNAs targeting NSD2, MACC1-AS1, FOXD2-AS1, and DANCR, which were cloned to the pLKO.1 vector, were obtained from Yangzhou Oncogene Biotechnology (Yangzhou, China). All the plasmids were validated by sequencing. Cell transfection was performed using Lipofectamine 3000 transfection reagent (Invitrogen) as per the manufacturer’s protocol. For generation of NSD2-depleted EC109 cell lines, the EC109 cells were transfected with control shRNA (shCtrl) or NSD2-targeting shRNA (shNSD2) and selected in the presence of 2 μg/mL puromycin (Thermo Fisher Scientific).
Apoptosis analysis
Cells were exposed to cisplatin for 72 h, and apoptosis was detected using the Annexin V-fluorescein isothiocyanate (FITC) Assay Kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s protocol. Briefly, cells were stained with Annexin V and PI and analyzed using a FACSCalibur flow cytometer.
Colony formation assay
Colony formation assay was performed as described previously.32 Briefly, ESCC cells (600 per well) were cultured in the presence of 10 μΜ cisplatin for 48 h. After drug treatment, the cells were cultured in fresh media supplemented with 10% FBS for additional 2 weeks. The colonies were stained with 0.25% crystal violet for 30 min and counted under a microscope.
Cell proliferation assay
After transfection with indicated constructs, cells were seeded into 24-well plates (0.5 × 105 cells/well) and cultured for 24–72 h. Cells were directly counted with a hemocytometer, and cell proliferation curves were plotted. Cell proliferation was also evaluated by EdU incorporation. Briefly, cells were seeded into 24-well plates in triplicate and treated with or without 10 μM cisplatin. After 72 h, cells were incubated with 10 μM EdU reagent (BeyoClick EdU-594, Beyotime, Haimen, China) for 2 h at 37°C. Nuclei were stained with Hoechst 33342 (Beyotime) for 30 min. The stained cells were photographed under an inverted fluorescent microscope. The percentage of EdU-positive cells was determined.
Animal studies
Animal studies were performed as described previously.33,34 The procedure involving animals was approved by the Institutional Animal Care and Use Committee of Zhengzhou University (Zhengzhou, China). EC109 or cisplatin-resistant KYSE30 cells stably transfected with control shRNA or NSD2-targeting shRNA were subcutaneously injected into 5-week-old, male nude mice (2 × 106 cells/mouse). Cisplatin (3 mg/kg) was intraperitoneally administered to nude mice 10 days after cell injection. Cisplatin was given twice every week for 2 weeks. Each group had 4 mice. Tumor volume was measured every week, and growth curves were plotted. At 4 weeks after cell injection, the mice were sacrificed using CO2 inhalation. The xenograft tumors were excised and weighed. For analysis of apoptosis, sections of the tumor samples were stained using the TUNEL procedure, as described previously.35 The percentage of TUNEL-positive cells was determined under a microscope.
Patient samples
Tumor specimens and paired noncancerous tissues were collected from 70 patients with ESCC who underwent resection between July 2017 and July 2018. No patients received anticancer treatment before operation. Tissue samples were snap-frozen in liquid nitrogen and stored at −80 °C until RNA analysis. Written informed consent for research was given by all the patients. This study was approved by the Ethics Committee of Zhengzhou University.
Immunohistochemistry
ESCC and normal tissue samples were subjected to immunohistochemistry using anti-NSD2 antibody (Abcam; 1:100 dilution). The staining results were evaluated by experienced pathologists in a blind manner. The staining intensity was scored as 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). The extent of staining was scored according to the percentage of positive cells, i.e., 0 (<5%), 1 (5%–25%), 2 (26%–50%), 3 (51%–75%), and 4 (>76%). NSD2 staining score was calculated by multiplying the staining intensity and staining percentage score.
ChIP assay
ChIP assay was performed using the Magna ChIP Kit (Merk-Millipore, Billerica, MA, USA) following the manufacturer’s instructions. In brief, cells were fixed in 1% formaldehyde and lysed in lysis buffer. The lysates were sonicated for 15 min to yield DNA fragments of ∼500 bp in size. ChIP experiments were performed using anti-H3K36me2 (Abcam) or control isotype immunoglobulin G (IgG) (Abcam). ChIP DNA was subjected to quantitative real-time PCR analysis using specific primers (Table S1).
Statistics
The data are expressed as mean ± standard deviation and were analyzed by the Student’s t test or one-way analysis of variance followed by Tukey’s post hoc tests. The Mann-Whitney U test was used to determine the differences in MACC1-AS1 expression between tumor samples and adjacent noncancerous tissues. The associations of MACC1-AS1 expression with clinicopathological parameters of ESCC were analyzed by the chi-square test. Pearson’s correlation analysis was performed to determine the correlation between MACC1-AS1 and NSD2 expression. Kaplan-Meier survival plots and log-rank statistics were used to evaluate the overall survival of ESCC patients based on TCGA data using the ENCORI Pan-Cancer Analysis Platform (http://starbase.sysu.edu.cn/panCancer.php). Differences were considered significant when p <0.05.
Acknowledgments
This study was supported by grants from the National Key Research and Development Program of China (grant number 2017YFC0909900), the National Natural Science Foundation of China (grant number 31670895 and 82002433), the Science and Technology Project of Henan Provincial Department of Education (grant number 18A320044 and 21A320036), the Henan Province Medical Science and Technology Research Project Joint Construction Project (grant number LHGJ20190003 and LHGJ20190055), and the Natural Science Foundation of Henan Province of China (grant number 202300410460).
Author contributions
W.X., J.Z., and Q.K. conceived the project and designed the experiments. W.X., Z.S., L.L., Y.Z., and D.Y. performed the experiments and analyzed the data. W.X. wrote the manuscript. All authors approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.007.
Contributor Information
Quancheng Kan, Email: kanqc@zzu.edu.cn.
Jie Zhao, Email: jiezhaoz2016@163.com.
Supplemental information
References
- 1.Yang C.S., Chen X., Tu S. Etiology and Prevention of Esophageal Cancer. Gastrointest. Tumors. 2016;3:3–16. doi: 10.1159/000443155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin D.C., Wang M.R., Koeffler H.P. Genomic and epigenomic aberrations in esophageal squamous cell carcinoma and implications for patients. Gastroenterology. 2018;154:374–389. doi: 10.1053/j.gastro.2017.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Y., Han L., Zhang W., Shan L., Wang Y., Song P., Peng C., Zhao X. Preoperative chemotherapy compared with postoperative adjuvant chemotherapy for squamous cell carcinoma of the thoracic oesophagus with the detection of circulating tumour cells randomized controlled trial. Int. J. Surg. 2020;73:1–8. doi: 10.1016/j.ijsu.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Mokdad A.A., Yopp A.C., Polanco P.M., Mansour J.C., Reznik S.I., Heitjan D.F., Choti M.A., Minter R.R., Wang S.C., Porembka M.R. Adjuvant Chemotherapy vs Postoperative Observation Following Preoperative Chemoradiotherapy and Resection in Gastroesophageal Cancer: A Propensity Score-Matched Analysis. JAMA Oncol. 2018;4:31–38. doi: 10.1001/jamaoncol.2017.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B., Li Q., Li Q., Qiu B., Xi M., Liu M., Hu Y., Zhu Y. Weekly Chemotherapy of 5-Fluorouracil plus Cisplatin Concurrent with Radiotherapy for Esophageal Squamous Cell Carcinoma Patients with Postoperative Locoregional Recurrence: Results from a Phase II Study. Oncologist. 2020;25:308–e625. doi: 10.1634/theoncologist.2019-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett R.L., Swaroop A., Troche C., Licht J.D. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb. Perspect. Med. 2017;7:a026708. doi: 10.1101/cshperspect.a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papillon-Cavanagh S., Lu C., Gayden T., Mikael L.G., Bechet D., Karamboulas C., Ailles L., Karamchandani J., Marchione D.M., Garcia B.A. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017;49:180–185. doi: 10.1038/ng.3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su X., Zhang J., Mouawad R., Compérat E., Rouprêt M., Allanic F., Parra J., Bitker M.O., Thompson E.J., Gowrishankar B. NSD1 Inactivation and SETD2 Mutation Drive a Convergence toward Loss of Function of H3K36 Writers in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017;77:4835–4845. doi: 10.1158/0008-5472.CAN-17-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berdasco M., Ropero S., Setien F., Fraga M.F., Lapunzina P., Losson R., Alaminos M., Cheung N.K., Rahman N., Esteller M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. USA. 2009;106:21830–21835. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aytes A., Giacobbe A., Mitrofanova A., Ruggero K., Cyrta J., Arriaga J., Palomero L., Farran-Matas S., Rubin M.A., Shen M.M. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat. Commun. 2018;9:5201. doi: 10.1038/s41467-018-07511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He C., Liu C., Wang L., Sun Y., Jiang Y., Hao Y. Histone methyltransferase NSD2 regulates apoptosis and chemosensitivity in osteosarcoma. Cell Death Dis. 2019;10:65. doi: 10.1038/s41419-019-1347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong G.Y., Park M.K., Choi H.J., An H.W., Park Y.U., Choi H.J., Park J., Kim H.Y., Son T., Lee H., Min K.W., Oh Y.H., Lee J.Y., Kong G. NSD3-induced methylation of H3K36 activates NOTCH signaling to drive breast tumor initiation and metastatic progression. Cancer Res. 2020;81:1–14. doi: 10.1158/0008-5472.CAN-20-0360. [DOI] [PubMed] [Google Scholar]
- 13.Chen X., Wang Z., Tong F., Dong X., Wu G., Zhang R. lncRNA UCA1 Promotes Gefitinib Resistance as a ceRNA to Target FOSL2 by Sponging miR-143 in Non-small Cell Lung Cancer. Mol. Ther. Nucleic Acids. 2020;19:643–653. doi: 10.1016/j.omtn.2019.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Dong H.T., Liu Q., Zhao T., Yao F., Xu Y., Chen B., Wu Y., Zheng X., Jin F., Li J., Xing P. Long Non-coding RNA LOXL1-AS1 Drives Breast Cancer Invasion and Metastasis by Antagonizing miR-708-5p Expression and Activity. Mol. Ther. Nucleic Acids. 2020;19:696–705. doi: 10.1016/j.omtn.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang P., Yang Z., Ye T., Shao F., Li J., Sun N., He J. lncTUG1/miR-144-3p affect the radiosensitivity of esophageal squamous cell carcinoma by competitively regulating c-MET. J. Exp. Clin. Cancer Res. 2020;39:7. doi: 10.1186/s13046-019-1519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He W., Liang B., Wang C., Li S., Zhao Y., Huang Q., Liu Z., Yao Z., Wu Q., Liao W. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene. 2019;38:4637–4654. doi: 10.1038/s41388-019-0747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi C., Xiaofeng C., Dongen L., Liang Y., Liping X., Yue H., Jianshuai J. Long non-coding RNA MACC1-AS1 promoted pancreatic carcinoma progression through activation of PAX8/NOTCH1 signaling pathway. J. Exp. Clin. Cancer Res. 2019;38:344. doi: 10.1186/s13046-019-1332-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Ge P., Cao L., Yao Y.J., Jing R.J., Wang W., Li H.J. lncRNA FOXD2-AS1 confers cisplatin resistance of non-small-cell lung cancer via regulation of miR185-5p-SIX1 axis. OncoTargets Ther. 2019;12:6105–6117. doi: 10.2147/OTT.S197454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H., Zhang J., Luo X., Zeng M., Xu L., Zhang Q., Liu H., Guo J., Xu L. Overexpression of the long non-coding RNA FOXD2-AS1 promotes cisplatin resistance in esophageal squamous cell carcinoma through the miR-195-Akt-mTOR axis. Oncol. Res. 2020;28:65–73. doi: 10.3727/096504019X15656904013079. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Kuo A.J., Cheung P., Chen K., Zee B.M., Kioi M., Lauring J., Xi Y., Park B.H., Shi X., Garcia B.A. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Min D.J., Ezponda T., Kim M.K., Will C.M., Martinez-Garcia E., Popovic R., Basrur V., Elenitoba-Johnson K.S., Licht J.D. MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia. 2013;27:686–694. doi: 10.1038/leu.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudlebusch H.R., Skotte J., Santoni-Rugiu E., Zimling Z.G., Lees M.J., Simon R., Sauter G., Rota R., De Ioris M.A., Quarto M. MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res. 2011;71:4226–4235. doi: 10.1158/0008-5472.CAN-10-3810. [DOI] [PubMed] [Google Scholar]
- 23.Shah M.Y., Martinez-Garcia E., Phillip J.M., Chambliss A.B., Popovic R., Ezponda T., Small E.C., Will C., Phillip M.P., Neri P. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene. 2016;35:5905–5915. doi: 10.1038/onc.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Z., Wu H., Chuai S., Xu F., Yan F., Englund N., Wang Z., Zhang H., Fang M., Wang Y. NSD2 is recruited through its PHD domain to oncogenic gene loci to drive multiple myeloma. Cancer Res. 2013;73:6277–6288. doi: 10.1158/0008-5472.CAN-13-1000. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y., Zhao H., Li H., Feng X., Tang H., Qiu C., Zhang J., Fu B. LINC01234/MicroRNA-31-5p/MAGEA3 Axis Mediates the Proliferation and Chemoresistance of Hepatocellular Carcinoma Cells. Mol. Ther. Nucleic Acids. 2020;19:168–178. doi: 10.1016/j.omtn.2019.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Hu M., Zhang Q., Tian X.H., Wang J.L., Niu Y.X., Li G. lncRNA CCAT1 is a biomarker for the proliferation and drug resistance of esophageal cancer via the miR-143/PLK1/BUBR1 axis. Mol. Carcinog. 2019;58:2207–2217. doi: 10.1002/mc.23109. [DOI] [PubMed] [Google Scholar]
- 27.Zeng F., Wang Q., Wang S., Liang S., Huang W., Guo Y., Peng J., Li M., Zhu W., Guo L. Linc00173 promotes chemoresistance and progression of small cell lung cancer by sponging miR-218 to regulate Etk expression. Oncogene. 2020;39:293–307. doi: 10.1038/s41388-019-0984-2. [DOI] [PubMed] [Google Scholar]
- 28.Bao J., Zhou C., Zhang J., Mo J., Ye Q., He J., Diao J. Upregulation of the long noncoding RNA FOXD2-AS1 predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Biomark. 2018;21:527–533. doi: 10.3233/CBM-170260. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y., Liu Y., Lin L., Huang Q., He W., Zhang S., Dong S., Wen Z., Rao J., Liao W., Shi M. The lncRNA MACC1-AS1 promotes gastric cancer cell metabolic plasticity via AMPK/Lin28 mediated mRNA stability of MACC1. Mol. Cancer. 2018;17:69. doi: 10.1186/s12943-018-0820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimura K., Miyata H., Tanaka K., Hamano R., Takahashi T., Kurokawa Y., Yamasaki M., Nakajima K., Takiguchi S., Mori M., Doki Y. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin. Cancer Res. 2012;18:5144–5153. doi: 10.1158/1078-0432.CCR-12-0701. [DOI] [PubMed] [Google Scholar]
- 31.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y.H., Huang Y.F., Chen C.C., Chou C.Y. Akt inhibitor SC66 promotes cell sensitivity to cisplatin in chemoresistant ovarian cancer cells through inhibition of COL11A1 expression. Cell Death Dis. 2019;10:322. doi: 10.1038/s41419-019-1555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y., Zhou L., Huang J., Liu F., Yu J., Zhan Q., Zhang L., Zhao X. Role of Smac in determining the chemotherapeutic response of esophageal squamous cell carcinoma. Clin. Cancer Res. 2011;17:5412–5422. doi: 10.1158/1078-0432.CCR-11-0426. [DOI] [PubMed] [Google Scholar]
- 34.Xu J., Zhu C., Yu Y., Wu W., Cao J., Li Z., Dai J., Wang C., Tang Y., Zhu Q. Systematic cancer-testis gene expression analysis identified CDCA5 as a potential therapeutic target in esophageal squamous cell carcinoma. EBioMedicine. 2019;46:54–65. doi: 10.1016/j.ebiom.2019.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon K.W., Byun S., Kwon E., Hwang S.Y., Chu K., Hiraki M., Jo S.H., Weins A., Hakroush S., Cebulla A. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science. 2015;349:1261669. doi: 10.1126/science.1261669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.