

Abstract
G protein‐coupled receptors (GPCRs) are key players in mediating signal transduction across the cell membrane. However, due to their intrinsic instability, many GPCRs are not suitable for structural investigations. Various approaches have been developed in recent years to remedy this situation, ranging from the use of more native membrane mimetics to protein‐stabilization methods. The latter approach typically results in GPCRs that contain various numbers of mutations. However, probing the functionality of such variants by in vitro and in vivo assays is often time consuming. In addition, to validate the suitability of such GPCRs for structural investigations, an assessment of their conformation state is required. NMR spectroscopy has been proven to be suitable to probe the conformation state of GPCRs in solution. Here, by using chemical labeling with an isotope‐labeled methyl probe, we show that the activity and the conformation state of stabilized neurotensin receptor 1 variants obtained from directed evolution can be efficiently assayed in 2D NMR experiments. This strategy enables the quantification of the active and inactive conformation states and the derivation of an estimation of the basal as well as agonist‐induced activity of the receptor. Furthermore, this assay can be used as a readout when re‐introducing agonist‐dependent signaling into a highly stabilized, and thus rigidified, receptor by mutagenesis. This approach will be useful in cases where low production yields do not permit the addition of labeled compounds to the growth medium and where 1D NMR spectra of selectively 19F‐labeled receptors are not sufficient to resolve signal overlap for a more detailed analysis.
Keywords: dynamics, GPCRs, membrane, NMR spectroscopy, signaling
Active or inactive? We used NMR to probe the structural states of stabilized neurotensin receptor (NTR1) variants in complex with different ligands by chemical modification of surface‐exposed cysteine residues. This protocol was further used to design NTR1 variants with wild‐type‐like conformational switching properties that can be produced in bacterial hosts.
G protein‐coupled receptors (GPCRs) are important conformational switches in signal transduction across the cell membrane. The conformation state of a GPCR determines whether it is able to act as a guanine nucleotide exchange factor (GEF) on bound heterotrimeric G proteins. The analysis of the conformational transition between the inactive to the active state of a GPCR has been a subject of continuing efforts using structural methods. [1] Possible conformation states occurring along the activation process have been described recently. [2] There are two inactive states (S1 and S2) where the transmembrane helix 6 (TM6) is either tightly bound to the helical bundle or more mobile. Binding of the agonist increases the population of an intermediate state that is more mobile in the cytoplasmic part (S3 state), and this state becomes further opened and stabilized by binding to the G protein (S4 state). For high‐resolution structural investigations by crystallography or cryo‐EM, this cytoplasmic stabilization can also be achieved by binding to G protein fragments or a G protein mimetic. [3] NMR spectroscopy has been successfully used to probe ligand‐dependent conformational changes even without stabilizing binding partners, and it turned out to be a powerful tool for capturing slight changes in the populations of various states modulated by small molecule ligands and nanobody G protein mimetics. [4] Since many GPCRs are very unstable and cannot be produced in sufficient amounts for structural investigations, various approaches have been introduced to optimize their biophysical properties. In order to remedy stability issues, the use of a native lipid nanodisc environment [5] has been shown to increase thermodynamic as well as long‐term stability of a GPCR. [6] However, in order to resolve protein production issues, the GPCR itself needs to be modified. This has been achieved with systematic mutagenesis [7] or directed evolution. [8] Either of these approaches results in GPCRs that contain a varying number of mutations, raising concerns about the functionality of such stabilized GPCRs in general, or conversely, losing stability again. Thus, functional assays need to be conducted to probe the activity profile of each receptor variant. A prominent model system subjected to protein stabilization methods is the rat neurotensin receptor subtype 1 (rNTR1). While its successful production in Escherichia coli was described decades ago, [9] the overall yields and stability of the wild‐type receptor were still too low for X‐ray crystallographic structural studies. X‐ray structures of neurotensin‐bound rNTR1 could eventually be determined with stabilized receptor variants. [10] More recently, a structure of wild‐type human NTR1 in complex with a heterotrimeric G protein could be determined by cryo‐EM. [3a] Despite the apparent benefit of stabilized GPCRs for structural investigations it is essential to be able to assess their functionality in a reliable and quick manner. In particular, a more detailed estimation of the conformational equilibrium between the inactive and active state of a GPCR variant is highly beneficial to guide and motivate more detailed and time‐consuming structural and dynamical investigations with NMR spectroscopy.
Here, we used NMR spectroscopy to determine the ligand‐dependent conformational equilibria of stabilized rNTR1 variants, which is in very good agreement with biochemical GEF activity assay data. In order to record 2D 1H,13C correlation experiments to resolve NMR signals of the inactive and active states of the GPCR, we modified surface‐accessible cysteine residues in each rNTR1 variant with an 13C‐isotope‐labeled methyl tag. These probes turned out to be suitable to monitor changes in the conformational equilibrium of the receptor at the cytoplasmic G protein binding site induced by either an agonist or an antagonist ligand. After initial validation of the NMR results with functional assay data, we performed a systematic mutagenesis with a highly evolved rNTR1 variant and used our NMR setup to determine which mutation in the receptor is responsible for its markedly increased basal activity. Finally, we discuss structure‐activity relationships of the interplay between different mutations that might modulate the activity of the receptor in a cooperative manner. This experimental setup provides a fast and effective way to assay the conformation state of a GPCR in a ligand‐ or G protein‐dependent manner or for validation of stabilized receptors for subsequent NMR studies on structure and dynamics.
Directed evolution [11] has been employed to obtain optimized rNTR1 variants that can be produced in E. coli in high yields. Among others, two variants (named TM86 V and HTGH4) have been shown to be suitable for structure determination. [10b] However, the employed evolutionary stabilization procedure resulted in the stepwise accumulation of point mutations (11 in TM86 V, 26 in HTGH4, Figure 1a,b). Thus, we here probed the functionality of each receptor variant by G protein GTP exchange stimulation assays in comparison with wild‐type rNTR1 (Figure 1c). With wild‐type rNTR1, GTP exchange activity is very low if the receptor is present in the apo form or bound to the small molecule antagonist SR142948 (SR), [12] and highly elevated in complex with the native agonist peptide neurotensin (NT1), as expected for a functional GPCR with low basal activity (black bars). The rNTR1 variant TM86 V with fewer mutations, which has not been subjected to such a rigorous selection pressure as HTGH4, shows an increased basal activity but can still be stimulated in an agonist‐dependent manner (red bars). In order to ensure proper interaction of the GPCR with the G protein, a L1673.50R back mutation (BM) had to be introduced that reverts a leucine residue that has been introduced by directed evolution back to a highly‐conserved arginine residue (Figure 1a). The highly evolved rNTR1 receptor variant HTGH4‐BM shows a markedly increased basal activity, leading to a full GTP exchange signal in the chosen assay conditions even without bound agonist. Apparently, the additional mutations in HTGH4 lead to a striking increase in basal activity in the presence of BM, but only a slight one in its absence. Critical mutations are most likely inward‐facing positions where the amino acid side chains contribute to packing of the α‐helical bundle in the GPCR. A multiple sequence alignment of wild‐type rNTR1, TM86V and HTGH4 at critical regions in the protein reveals changes in charge, hydrophobic properties and chain length of amino acids by mutation (Figure 1d).
Figure 1.
Point mutations and G protein stimulation activity of stabilized rNTR1 variants. a) TM86V contains 11 point mutations. [13] b) In HTGH4, 15 further mutations are present from additional evolution for detergent stability. [14] c) G protein GTP exchange stimulation assay with wild‐type rNTR1 (black bars), TM86V‐BM (red bars), HTGH4‐BM (blue bars), and HTGH4 (green bars) in the apo form as well as bound to the peptide agonist NT1 or the small‐molecule antagonist SR. No data are available for TM86V‐BM in complex with SR, n.a.: not available. d) Selected regions of a multiple sequence alignment of rNTR1, TM86V and HTGH4 with important residues labeled.
Thus, we were interested in establishing a robust and reliable setup to probe not only macroscopic activity but also the conformation state of rNTR1 variants by solution‐state NMR spectroscopy in order to identify residues in HTGH4 that contribute to its characteristic and non‐selective activity profile. In order to introduce NMR‐active probes into rNTR1 variants, we utilized the selective chemical modification of surface‐exposed cysteine residues. HTGH4 and TM86 V contain 3 or 4 surface‐accessible cysteine residues, respectively, at positions 1723.55, 3326.59 (TM86 V only), 386 and 388 (both in helix 8; Figure 2a). In order to introduce a suitable probe close to the G protein binding interface and to be able to monitor the conformational change taking place upon GPCR activation, we introduced an additional cysteine residue at the cytoplasmic end of TM6 (V3006.27C). This position has been previously utilized to probe GPCR activation by fluorescence spectroscopy or 19F NMR.[ 4h , 15 ] The surface accessibility of the cysteines in HTGH4‐V300 C was confirmed by quantitative chemical modification yields with the alkylating agent S‐methyl‐13C‐methanethiosulfonate (MMTS), resulting in modified cysteine side chains that contain a disulfide‐bridged and 13C isotope‐labeled methyl tag, as detected by ESI‐mass spectrometry. The power of MMTS labeling for NMR has been demonstrated recently. [16] As shown in Figure 2b, we were able to detect the correct mass that is expected with 4 accessible cysteine residues in this rNTR1 variant. Similar results have been obtained with TM86V‐V300C containing five surface‐exposed cysteines. The thermal stability of both receptor variants is not affected by the chemical modification, giving rise to a cooperative thermal unfolding transition at 70 and 80 °C for TM86V and HTGH4, respectively (Figure 2c). These data also suggest that the cooperativity of thermal unfolding of TM86V is lower if in complex with the SR antagonist, presumably caused by the lower degree of evolution of this NTR1 variant as compared to HTGH4.
Figure 2.
Labeling of stabilized rNTR1 variants at accessible cysteine positions with 13C‐labeled MMTS. a) rNTR1 variants HTGH4 and TM86V contain four or five solvent‐accessible cysteine residues, respectively, that can be labeled. C332 next to the NT1 peptide binding site is only present in TM86V. b) ESI‐MS confirms the correct number of four attached −S−13CH3 labels in the case of HTGH4. c) CD‐detected thermal stability analysis of 13C‐MMTS‐labeled rNTR1 variants in complex with the agonist NT1 or the small‐molecule antagonist SR [12] in DDM micelles.
Next, we were interested to monitor the spectral features of the two rNTR1 variants in n‐dodecyl‐β‐d‐maltoside (DDM) micelles by 2D 13C,1H HMQC NMR experiments and probe the effect of the stabilizing mutation R1673.50L, which introduces a hydrophobic lock, [10b] but prevents G protein signaling. As shown in Figure 3, high‐quality spectra could be obtained with 13C‐MMTS‐labeled and otherwise natural abundance, i. e. non‐deuterated rNTR1 variants with the expected number of signals in each case. It is expected that additional deuteration of the receptor will further increase the NMR spectral quality. This is only possible in very few chases, though. Assignment of the NMR signals was achieved by mutagenesis of selected cysteine residues in the protein (Figure S1 in the Supporting Information), where 2D 13C,1H HMQC NMR spectra of HTGH4‐BM, HTGH4‐BM C386S and HTGH4‐BM V300C provided unambiguous information that could be transferred to TM86V‐BM. By comparing the spectra of HTGH4V300C and HTGH4‐BM V300C in the agonist or antagonist‐bound state we were able to obtain valuable insights on the observed change in GPCR activity (Figure 3a). With HTGH4V300C (containing L167), the spectra in each ligand‐bound state are almost identical, whereas the back mutation (L167R) leads to appearance of multiple conformation states and marked changes in the NMR peak intensity pattern between the agonist‐ and antagonist‐bound states. In this variant, we observe pronounced line broadening effects for position 172, indicating enhanced motions on the millisecond to microsecond timescale. In order to obtain insights into receptor stabilization by G protein binding, we added a peptide derived from the C‐terminal helix 5 of the G protein αi,1 subunit (Gαi,1; Figure S2). This resulted in additional line broadening at position 172 and a strong reduction of the intensity of the signals of the inactive states at position 300 (Figure 3a). Moreover, we observed the occurrence of two NMR signals for the active state, which most likely represents the S3 and the G protein‐bound S4 states. By plotting the observed intensity patterns along the indirect 13C dimension (Figure 3b), we could visualize and assign each observed peak to an active or inactive conformation state (S1 to S4 [2] ). The 2D NMR spectra of the TM86V‐BM V300C variant showed an additional NMR signal for Cys332, as well as multiple sub‐states that represent the inactive or active conformations of the receptor, respectively (Figure 3c).
Figure 3.
Probing the active state of rNTR1 variants by using 13C‐MMTS labeling and 2D NMR spectroscopy in DDM micelles. a) 2D 13C,1H HMQC spectra of 13C‐MMTS‐labeled rNTR1 variants in complex with an antagonist SR or agonist NT1 show marked differences. HTGH4, which harbors the ionic lock mutation L1673.50 does not adopt active states at the intracellular side with a bound agonist, whereas the back mutation (BM) L1673.50R leads to an active receptor, as indicated by the occurrence of a second signal for positions 300 and 172 that are located close to the G protein coupling interface. b) Slices of the spectra in (a) along the 13C dimension, as indicated by broken lines, can be used for quantification and the assignment of the involved structural states, as visualized in (e). c) 2D 13C,1H HMQC spectra of TM86 V‐BM bound to an antagonist SR or agonist NT1 and in complex with a peptide derived from the C‐terminal helix 5 of Gα. Asterisks mark methyl signals from the Gα peptide at natural abundance. d) Slices along the 13C dimension of the spectra in (c). For TM86V‐BM, an additional inactive state at ∼23.25 ppm 13C chemical shift as detected, consistent with a further inward rotation of TM6 at the intracellular side, was also observed in the crystal structures of the antagonist‐bound state. [18] e) Visualization of the structural states involved in GPCR activation, adapted from ref. [2]. f) Quantification of the peak intensities corresponding to the inactive or active species in all spectra shown in (a) and (c).
In addition, changes in signal intensity could be observed between the inactive and active states (Figure 3d), where in particular the intensity of the signal at position 172 was markedly weakened by line broadening in presence of an agonist, which is a clear indication for altered dynamics upon receptor activation. Furthermore, the addition of a peptide derived from the C‐terminal helix 5 of the G protein αi,1 subunit (Gαi,1) (Figure S2) to 13C‐MMTS‐labeled TM86V‐BM V300C resulted in chemical shift perturbations and changes in signal intensity where mostly the NMR signals corresponding to the active state were affected, further corroborating the assignment of the individual peaks to defined functional states (Figure 3c,d). A quantification of the inactive versus active states at the intracellular side in each of the shown receptor variants provides a detailed picture of their ligand‐dependent activity profile (Figure 3e,f). In the NMR spectra of active rNTR1 variants, we typically observe multiple peaks, similar to the observations of Kobilka and colleagues with the β2 adrenergic receptor, [2] which allowed them to define sub‐states occurring during receptor activation. These four states could be later confirmed using BLT2 receptor. [4e] Here, states S1 and S2 define the inactive conformation, S3 the intermediate, active conformation in the presence of agonist but without G protein, and S4 the active state bound to a G protein (Figure 3e). For a simplified analysis of the conformation states in rNTR1 variants we summed up the populations of the inactive (S1+S2) and the active states (S3+S4) for positions 1723.55 and 3006.27, respectively, that are both located in close proximity to the G protein binding site (Figure 3f). This analysis confirms the activity data derived from biochemical assays (Figure 1c). HTGH4, without the L167R back mutation, is always present in the inactive conformation as monitored at the intracellular side, even in complex with an agonist, consistent with the crystal structure of this complex. [10b] For HTGH4‐BM in complex with an antagonist the basal activity is increased compared to HTGH4, and bound to an agonist the relative population of the active state is increased to almost 60 %, which is further increased upon complex formation with a G protein. This increased population of the active state, even in the antagonist‐bound state, is consistent with the high basal activity of this receptor variant in the GTP exchange assay with a bound G protein (Figure 1c). To test whether antagonist‐bound HTGH4‐BM can still interact with a G protein, we added the Gα peptide to MMTS‐labeled receptor in complex with SR142948 and observed pronounced NMR spectral changes indicative of a pronounced stabilization of the active states (Figure S3). In contrast, TM86 V‐BM is almost inactive in complex with an antagonist, and the agonist‐dependent final population of the active state reaches only 50 %. Gα peptide binding generally leads to line broadening of the NMR signals originating from the methyl groups at positions 172 and 300, presumably caused by intermediate chemical exchange processes. Among those, position 172 appears to be more affected than position 300, in particular in the more active HTGH4‐BM variant.
After the initial NMR characterization of the active states of rNTR1 variants, which were in excellent agreement with the biochemical GTP exchange assay, we were wondering what mutations present in HTGH4‐BM might cause the apparent increase in basal activity that correlate with a strong decrease in the ligand‐dependent switching capability, present in a typical wild‐type GPCR. Thus, we designed a series of single‐point mutations in HTGH4‐BM that lead to back mutations to the wild‐type amino acid type that is also present in TM86 V. We selected five different positions (T1012.38R, D1242.61E, E1503.33D, A2605.61I, R2625.63N/R2635.64K) that are located at the G protein coupling site and at contact points between individual TMHs (Figure 4a). It has been shown that the negative charge of D1132.50, responsible for the Na+ sensitivity of agonist binding, is crucial for switching to the active conformation in NTR1 [17] as well as in the Adenosine A2A receptor. [4a] Mutation at this position consequently leads to an inactive receptor. We therefore included the D1132.50S mutation as a negative control that should not restore signaling. Simplified representations of the structural states that can occur during GPCR activation are shown in Figure 4b. The 1D NMR slices along the 13C dimension of an 2D 13C,1H HMQC experiment for these six variants of HTGH4‐BM V300 C show pronounced differences in the populations of the active and inactive states (Figure 4c). The 1D slices for HTGH4‐BM V300 C and TM86 V‐BM V300 C are also shown in the figure as a comparison. As expected, mutation of D113 to serine decreased the population of the active states (S3 and S4) of the receptor to an almost undetectable level in complex with both antagonist and agonist.
Figure 4.
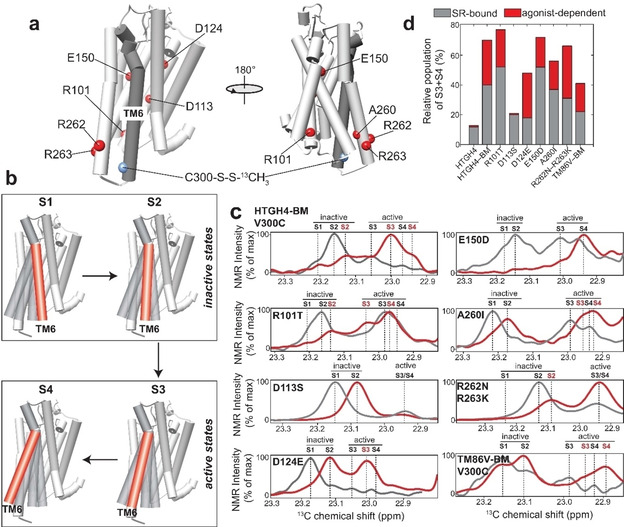
Modulation of ligand‐induced rNTR1 conformation states by mutagenesis. a) Single‐point mutations (to the wild‐type residue type) that have been introduced into the HTGH4‐BM V300C variant of rNTR1. b) Visualization of the structural states that could be monitored by NMR. c) Slices of 2D 13C,1H HMQC spectra of the MMTS resonance at position 300 of the rNTR1 variants shown in (a); red: bound to the agonist NT1, grey: bound to the antagonist SR142948. d) Relative populations of the active states (S3+S4 vs. total NMR signal) derived from the spectra in (c).
Interestingly, the receptor changes its conformation from the fully inactive S1 state, seen in complex with an antagonist, to the more dynamic S2 state when in complex with an agonist. In the S2 state, TM6 of the receptor is less tightly bound to the helical bundle of the GPCR (Figure 4b). However, due to the missing switch residue D113, the transition to the active state seems to be hindered, suggesting that a negative charge at the contact region between TM3, TM6 and TM7 (Figure 5a) is essential for a ligand‐induced transition to the active state. The back mutations at positions 101 and 262/263 caused a minor change of the conformational profile of the receptor, where 101 leads to a slight increase in the active states if bound to an antagonist and 262/263 to a decrease (Figure 4c). However, positions 124, 150 and 260 were found to be more crucial for rNTR1 activation. Mutation of Asp to Glu at position 124 resulted in a dramatic reduction in the population of the active states if bound to an antagonist. Upon activation by an agonist, mostly the states S2 and S3 were populated with an overall relative population of the active states of 45 %. The ligand‐dependent switching characteristics induced by this single‐point mutation in HTGH4‐BM is cleaner than the profile obtained with the less evolved TM86 V variant that also harbors a Glu residue at position 124. This behavior highlights the necessity to probe the conformation state of a receptor by rapid structural readouts. The location of residue 124 in TM2 in the structure of HTGH4 and TM86V suggests that the longer Glu side chain is able to form a salt bridge with Arg149 located in TM3, thus leading to a better interaction with the entire helical bundle of the GPCR (Figure 5b). An opposite tendency was observed with the E150D variant. In complex with an antagonist, all conformation states are present in the receptor to an almost equal extent, suggesting less stringent conformational switching. Bound to an agonist, this conformational equilibrium is completely shifted towards the fully active state S4 (Figure 4c).
Figure 5.
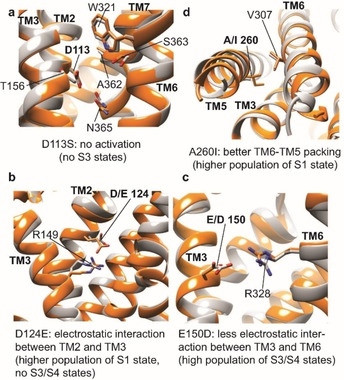
Structural basis of the modulation of rNTR1 activity by mutation with four single‐point mutations showing the most pronounced effects in the NMR assay. Except for the control D113S, all mutations are back mutations of HTGH4 (gray) to the residue present in TM86V (orange) or the wild‐type receptor in order to validate the influence of individual mutations on rNTR1 activation. a) D113S: removal of the highly conserved Asp1132.50 residue abolishes the agonist‐induced activation capability. b) D124E2.61: the slightly longer side chain of Glu as compared to Asp enhances electrostatic interactions with Arg1493.32 in TM3, presumably leading to a stabilized and compact helical bundle and consequently to a more populated inactive state with a lower basal activity. c) E150D: weakening of the electrostatic interaction between Asp1503.33 and Arg3286.55 in TM6 leads to a more populated active state of the receptor, predicted to have a markedly increased basal activity. d) A2605.61I: the longer Ile side chain provides better hydrophobic packing between TM5 and TM6, leading to an increase in the population in the inactive S1 state, which represents a conformation with a tightly associated TM6. Grey: HTGH4 (PDB ID: 4BWB), orange: TM86V (PDB ID: 4BUO). [10b]
When comparing the HTGH4 and TM86V structures (Figure 5c), it appears that the longer side chain of Glu1503.33 in HTGH4 forms a strong salt bridge with Arp3286.55, partially hindering agonist‐induced transition to the active state. This is the case in HTGH4 but not in TM86 V, where the wild‐type Asp150 is retained. A Glu‐mediated salt bridge at this position enhances the interaction between TM3 and TM6 and consequently hinders a conformational transition from the inactive to the active state. In contrast, the negative charge of the shorter Asp150 side chain, as well as the positive charge of Asp328, may play an important role in agonist‐induced receptor activation, in agreement with functional characterization of the D150A and R328M mutations in the wild‐type receptor. [18] This behavior highlights the complex interplay between various regions in the GPCR that affect its conformational landscape in a cooperative manner. Another important residue in TM86 V that has been mutated in HTGH4 is Ile260. In HTGH4, a smaller Ala side chain is present at this position, which most likely leads to a reduced interaction between TM5 and TM6 (Figure 5d), thus increasing the population of the active states of the receptor (in the presence of L167R). Back mutation of this residue in HTGH4 to Ile mainly affects the type and population of the inactive states. While predominantly the more mobile and active S2 state is present in HTGH4‐BM in complex with an antagonist, the A260I mutation enhances the interaction of TM6 with the helical bundle, leading to a markedly increased population of the inactive S1 state (Figure 4c), even though both active states S3 and S4 are still present. Upon activation by an agonist, a shift in the populations takes place to S2 and to the active S3 and S4 states. A summary of the investigated variants and their antagonist and agonist‐dependent populations of the active and inactive conformation states is shown in Figure 4d. In such a visualization, a wild‐type‐like GPCR should show a small grey and a large red section, indicating low population of the active states in complex with an antagonist and strong boost in activity induced by binding to an agonist. For both, HTGH4 and HTGH4‐BM V300 C containing the D113S mutation, an almost undetectable population of the active states and in addition no agonist‐dependent activation can be observed, confirming that these variants cannot undergo the conformational change at the intracellular side required for functional G protein binding, even in the presence of agonist, consistent with the nucleotide exchange results. HTGH4‐BM, as well as the R101T and the E150D variants show very high populations of the active conformation states in complex with the antagonist but can still be further activated by an agonist, suggesting G protein stimulation activity even in the antagonist‐bound form (Figure 1c). Variants A260I and R262N/R263K show a lower population of the active states with a bound antagonist and lower total activity in complex with the agonist. These features are similar to what has been measured with TM86 V, which carries fewer mutations. However, the most pronounced effect can be seen with the D124E variant of HTGH4‐BM, where the active states are barely populated in complex with an antagonist, as also seen for the inactive variants HTGH4 and HTGH4‐BM D113S, but the agonist‐induced increase in the active states is as high as with HTGH4‐BM. These characteristics bring HTGH4‐BM D124E closer to those of a wild‐type GPCR, which needs to display a low basal activity but high activation by an agonist, yet maintain stability beyond that of the wild‐type, which is a prerequisite for functional and structural studies (Figure 1c). Interestingly, this mutant also has a wider activation window than TM86 V. These results also emphasize the use of the presented NMR assay to probe the conformational landscape of a stabilized GPCR and apply this technology to rationally screen for gain‐of‐function variants, in the context of a stable variant, by mutagenesis.
In summary, we have presented a 2D NMR‐based approach to monitor the conformation states of a stabilized rNTR1 based on the chemical modification of intrinsic or engineered surface‐exposed cysteine residues with isotope‐labeled methyl tags. Due to the ability to record 2D 1H,13C HMQC experiments we could resolve signal overlap and capture the NMR signals of relevant conformation states in the receptor. Furthermore, using G protein stimulation profiles of two rNTR1 variants together with their 3D crystal structures, we were able to rationally design a set of point mutations and probe their effect on the population of the active or inactive conformation states at the intracellular side using the described method. This protocol facilitates the targeted functional rescue of stabilized GPCRs and can be used to rationally design receptor variants that can be produced in bacterial hosts in high yields and have functional signatures similar to the original wild‐type GPCR, representing a major bottleneck in NMR‐based structural biology of this highly important protein class.
Experimental Section
All experimental details on protein design, mutagenesis, protein production and purification, assays, as well as MMTS labeling and biophysical and NMR methods are described in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Ha 6105/3‐1) and the Helmholtz Society (VH‐NG‐1039; to F.H.). A.P. acknowledges support by the Schweizerische Nationalfonds grant 31003A_182334. We thank Mattia Deluigi and Christoph Klenk (University of Zürich) for critical reading of the manuscript. Open access funding enabled and organized by Projekt DEAL.
I. Goba, D. Goricanec, D. Schum, M. Hillenbrand, A. Plückthun, F. Hagn, ChemBioChem 2021, 22, 139.
References
- 1. Hilger D., Masureel M., Kobilka B. K., Nat. Struct. Mol. Biol. 2018, 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manglik A., Kim T. H., Masureel M., Altenbach C., Yang Z., Hilger D., Lerch M. T., Kobilka T. S., Thian F. S., Hubbell W. L., Prosser R. S., Kobilka B. K., Cell 2015, 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Kato H. E., Zhang Y., Hu H., Suomivuori C. M., Kadji F. M. N., Aoki J., Krishna Kumar K., Fonseca R., Hilger D., Huang W., Latorraca N. R., Inoue A., Dror R. O., Kobilka B. K., Skiniotis G., Nature 2019, 572, 80–85; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K., Nature 2011, 477, 549–555; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Carpenter B., Nehme R., Warne T., Leslie A. G., Tate C. G., Nature 2016, 536, 104–107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K., Nature 2011, 469, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Eddy M. T., Lee M. Y., Gao Z. G., White K. L., Didenko T., Horst R., Audet M., Stanczak P., McClary K. M., Han G. W., Jacobson K. A., Stevens R. C., Wüthrich K., Cell 2018, 172, 68–80 e12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Horst R., Liu J. J., Stevens R. C., Wüthrich K., Angew. Chem. Int. Ed. 2013, 52, 10762–10765; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10962–10965; [Google Scholar]
- 4c. Isogai S., Deupi X., Opitz C., Heydenreich F. M., Tsai C. J., Brueckner F., Schertler G. F., Veprintsev D. B., Grzesiek S., Nature 2016, 530, 237–241; [DOI] [PubMed] [Google Scholar]
- 4d. Bostock M. J., Solt A. S., Nietlispach D., Curr. Opin. Struct. Biol. 2019, 57, 145–156; [DOI] [PubMed] [Google Scholar]
- 4e. Casiraghi M., Damian M., Lescop E., Point E., Moncoq K., Morellet N., Levy D., Marie J., Guittet E., Banères J.-L., Catoire L. J., J. Am. Chem. Soc. 2016, 138, 11170–11175; [DOI] [PubMed] [Google Scholar]
- 4f. Ye L., Van Eps N., Zimmer M., Ernst O. P., Scott Prosser R., Nature 2016, 533, 265–268; [DOI] [PubMed] [Google Scholar]
- 4g. Solt A. S., Bostock M. J., Shrestha B., Kumar P., Warne T., Tate C. G., Nietlispach D., Nat. Commun. 2017, 8, 1795; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4h. Liu J. J., Horst R., Katritch V., Stevens R. C., Wüthrich K., Science 2012, 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G., J. Am. Chem. Soc. 2004, 126, 3477–3487; [DOI] [PubMed] [Google Scholar]
- 5b. Hagn F., Etzkorn M., Raschle T., Wagner G., J. Am. Chem. Soc. 2013, 135, 1919–1925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Hagn F., Nasr M. L., Wagner G., Nat. Protoc. 2018, 13, 79–98; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Klöpfer K., Hagn F., Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114–115, 271–283. [DOI] [PubMed] [Google Scholar]
- 6. Nasr M. L., Baptista D., Strauss M., Sun Z. J., Grigoriu S., Huser S., Plückthun A., Hagn F., Walz T., Hogle J. M., Wagner G., Nat. Methods 2017, 14, 49–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Magnani F., Shibata Y., Serrano-Vega M. J., Tate C. G., Proc. Natl. Acad. Sci. USA 2008, 105, 10744–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Sarkar C. A., Dodevski I., Kenig M., Dudli S., Mohr A., Hermans E., Plückthun A., Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Scott D. J., Plückthun A., J. Mol. Biol. 2013, 425, 662–677; [DOI] [PubMed] [Google Scholar]
- 8c. Schlinkmann K. M., Honegger A., Tureci E., Robison K. E., Lipovsek D., Plückthun A., Proc. Natl. Acad. Sci. USA 2012, 109, 9810–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tucker J., Grisshammer R., Biochem. J. 1996, 317, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. White J. F., Noinaj N., Shibata Y., Love J., Kloss B., Xu F., Gvozdenovic-Jeremic J., Shah P., Shiloach J., Tate C. G., Grisshammer R., Nature 2012, 490, 508–513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Egloff P., Hillenbrand M., Klenk C., Batyuk A., Heine P., Balada S., Schlinkmann K. M., Scott D. J., Schutz M., Plückthun A., Proc. Natl. Acad. Sci. USA 2014, 111, E655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarkar C. A., Dodevski I., Kenig M., Dudli S., Mohr A., Hermans E., Plückthun A., Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gully D., Labeeuw B., Boigegrain R., Oury-Donat F., Bachy A., Poncelet M., Steinberg R., Suaud-Chagny M. F., Santucci V., Vita N., Pecceu F., Labbe-Jullie C., Kitabgi P., Soubrie P., Le Fur G., Maffrand J. P., J. Pharmacol. Exp. Ther. 1997, 280, 802–812. [PubMed] [Google Scholar]
- 13. Schlinkmann K. M., Hillenbrand M., Rittner A., Kunz M., Strohner R., Plückthun A., J. Mol. Biol. 2012, 422, 414–428. [DOI] [PubMed] [Google Scholar]
- 14. Scott D. J., Kummer L., Egloff P., Bathgate R. A., Plückthun A., Biochim. Biophys. Acta 2014, 1838, 2817–2824. [DOI] [PubMed] [Google Scholar]
- 15. Dawaliby R., Trubbia C., Delporte C., Masureel M., Van Antwerpen P., Kobilka B. K., Govaerts C., Nat. Chem. Biol. 2016, 12, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Religa T. L., Ruschak A. M., Rosenzweig R., Kay L. E., J. Am. Chem. Soc. 2011, 133, 9063–9068. [DOI] [PubMed] [Google Scholar]
- 17. Krumm B. E., White J. F., Shah P., Grisshammer R., Nat. Commun. 2015, 6, 7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.M. Deluigi, A. Klipp, C. Klenk, L. Merklinger, S. A. Eberle, L. Morstein, P. Heine, P. R. E. Mittl, P. Ernst, T. M. Kamenecka, Y. He, S. Vacca, P. Egloff, A. Honegger, A. Plückthun, 2020, unpublished results.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary