

Abstract
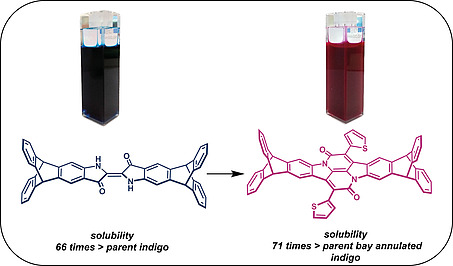
The synthesis of a highly soluble triptycene end‐capped indigo and its bay annulated derivative is reported. Both compounds have been studied by absorption and emission spectroscopy cyclic voltammetry, as well as theoretical calculations and compared to the parent indigo and bay annulated indigo. Besides a large improvement of solubility in organic solvents by the factor of approx. 70(!) the compounds also show a pronounced tendency to form crystals. Both properties, making these compounds promising electron acceptors for organic electronics.
Keywords: Indigo, Solubility, Triptycene, Dyes, Electron acceptors
Triptycene end‐capping is the solution to turn insoluble pigments into soluble dyes by a factor of 70, making them potential good electron‐acceptors for various organic electronics applications.
Indigo is one of the most widely used pigments in the textile industry.[ 1 ] Due to its distinct electronic properties, indigo and related indigoid compounds have recently gained interest for applications in organic electronics.[ 2 ] High planarity and hydrogen bonding leads to tight intermolecular packing, which explains the extremely poor solubility and high melting point (390–392 °C),[ 1 , 3 ] which is typical for an organic pigment. The poor solubility of indigo restricts its direct application e.g. as organic semiconductor.[ 2e ] To improve the solubility, different approaches have been used such as attaching bulky substituents or long alkyl, or alkoxy chains at the periphery of the benzene rings or at the nitrogen or carbonyl atoms of the indigo. For instance, a series of soluble 5,5' alkylated indigo derivatives were used for processing of optoelectronic devices.[ 4 ] Ngai et al. attached long alkoxy chains to the aromatic ring of the indigo unit for applications in n‐type organic field‐effect transistors.[ 5 ] Alkyl‐ or alkoxyphenyl‐substituted indigo derivatives were synthesized from the corresponding isatins and used to grow chromophoric liquid crystals.[ 6 ] tert‐Butoxy carbonyl units were attached to the nitrogen atoms of indigos to improve their solubility, allowing their solution processing into neat thin films for fabrication of organic solar cells.[ 7 ] Bo He and co‐workers reported the soluble bay annulated indigo as an electron accepting unit in semiconductor application.[ 8 ] Voss et al. presented a new group of 4,4,7,7‐tetraalkoxy‐5,5‐diaminoindigotins that are highly soluble in organic solvents.[ 9 ] Recently the Thamyongkit group reported indigo N‐arylimines and monoacyl‐substituted indigo which have shown good solubility in diesel (> 15 mg/mL) and other organic solvents.[ 10 ]
Despite the alkyl or alkoxy chains or bulky substituents helping to improve the solubility, the synthesis of soluble indigo derivatives without detrimental effects to the electrochemical properties of parent indigo is still demanding. It is worthwhile mentioning that in some cases it was even observed that the solubility of some long alkyl‐substituted indigos further decreased with increasing the length of alkyl chains, which was explained by the hydrophobic effect of alkyl chains in 1,2‐dichlorobenzene.[ 4 ] The attachment of long alkyl chains to larger aromatic compounds may increase solubilities but on the other hand, the tendency to form larger crystalline domains or even single crystals is hampered, which reduces the possibility for certain organic electronics applications.
In 2014, we observed that incorporating additional peripheral triptycene units to large triptycene based quinoxalinophenanthrophenazines (QPP) results in increased solubility when compared to, for example, alkylated analogues.[ 11 ] This was later explained by single crystal structure analysis, showing only very weak π–π interactions of the end‐capped triptycene units.[ 12 ] The solubility enhancement by triptycene end‐capping was further exploited for a pyrene‐fused pyrazaacene with eleven rectilinearly annulated rings, which was much more soluble than its analogues with four dodecyloxyphenyl chains.[ 13 ] In addition to its good solubility, the compound had a high tendency to form single‐crystals, which were analyzed by X‐ray diffraction. The concept is not restricted to linearly annelated π‐systems as has been further demonstrated with a triptycene end‐capped hexabenzovalene (HBO)[ 14 ] and diazadiebenzoanthracenes.[ 15 ]
Here, we report on the solubilizing effect of triptycene end‐capping to indigo which is a classical pigment of low solubility.
The synthesis of the triptycene end‐capped indigo starts with a Sandmeyer isatin synthesis based on 2‐amino triptycene 1 [ 16 ] by treatment with a mixture of chloral hydrate and sodium sulfate followed by addition of conc. HCl to give α‐isonitrosoacetanilide 2 in 95 % yield (Scheme 1).[ 17 ] The cyclization of α‐isonitrosoacetanilide 2 to isatin 3 was first attempted by using concentrated sulfuric acid.[ 18 ] No desired product could be isolated, possibly due to sulfonation at the peripheral phenylene rings of the triptycene core. Next, cyclization of compound 2 was tried using polyphosphoric acid (PPA) under different reaction conditions (Table S1) but all attempts were unsuccessful. Using BF3 ·Et2O shows the formation of isatin but the reaction did not proceed to completion, resulting in a complex mixture of products that needed HPLC for the purification.[ 19 ] When 2 was heated with methanesulfonic acid at 50 °C for 4 hours complete conversion was observed and isatin 3 was isolated as a deep red colored solid in 95 % yield after silica gel column chromatography. The successful conversion was determined by 1H NMR spectroscopy (Figure 1) and further confirmed by ESI‐MS, showing only one peak at m/z = 346.0838. To get triptycene end‐capped indigo 4, isatin 3 was finally heated at 100 °C with PCl5 in toluene for four hours and afterwards reduced in situ using thiophenol.[ 6 , 20 , 21 ] The crude product was purified using basic alumina column chromatography to give the desired product in a 57 % yield as a deep blue solid. Indigo 4 could be converted to bay‐annulated indigo 5 in 30 % yield, when refluxed in xylene with 2‐thiopheneacetyl chloride for 48 hours.[ 8 ] All compounds were fully characterized (1H, 13C NMR, MS, IR, UV/Vis, elemental analysis, for details, see Supporting Information).
Scheme 1.
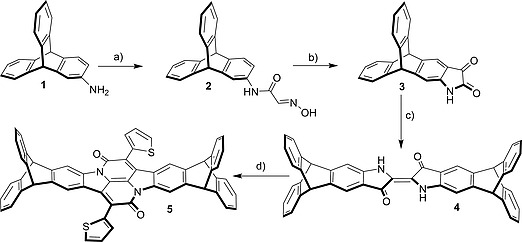
Synthesis of triptycene end‐capped indigo 4 and bay annulated indigo 5. a) Cl3CCH(OH)2, HONH2 ·HCl, HCl, 80 °C, 20 h, 95 %; b) MeSO3H, 50 °C, 4 h, 85 %; c) (i) PCl5, PhMe, 100 °C, 4 h; (ii) PhSH, 50 °C, 16 h, (57 % over two steps). d) 2‐Thiophene acetyl chloride, o‐xylene, 155 °C, 48 h, 30 %.
Figure 1.
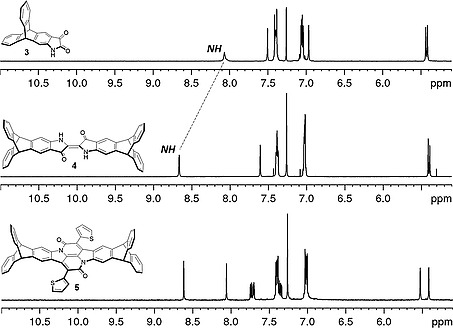
Comparison of 1H NMR (CDCl3) spectra of isatin 3, triptycene end capped indigo 4, and triptycene end capped bay annulated indigo 5.
Crystals of triptycene end‐capped indigo 4 suitable for single‐crystal X‐ray diffraction analysis were obtained by diffusion of n‐pentane into a solution of 4 in CDCl3 (Figure 2).[ 22 ] The compound crystallized in the triclinic space group P 1 with three molecules of chloroform enclathrated. The indigo unit of 4 is highly planar. The compound aggregates in a “step‐like” arrangement by intermolecular hydrogen bonding between the carbonyl oxygen and the amine hydrogen (d = 2.43 Å and 2.25 Å). Unlike in parent indigo (6), no π stacking interactions were observed between two molecules in the crystal structure,[ 3c ] which most likely improves the solubility of 4 (see discussion below). Triptycene end‐capped bay annulated indigo 5 was also characterized by single‐crystal X‐ray diffraction (Figure 2 c and d). Single crystals were grown by slowly cooling down a saturated solution of 5 in ethyl acetate.[ 22 ] The compound also crystallizes in the P 1 unit cell. Here, packing is dominated by π stacking of one of the peripheral triptycene phenylene rings with the central fused aromatic unit (shortest distance 3.27 Å) and some T‐shaped arranged π‐stacking of the thienyl moieties with the triptycene blades (d CH···Aryl = 2.80–2.85).
Figure 2.
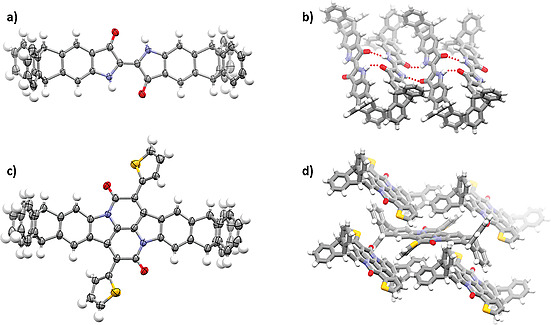
a) Single crystal structure of triptycene end‐capped indigo 4 (a and b) and triptycene end‐capped bay annulated indigo 5 (c and d). a) and c) molecular structures with ORTEP plot at 50 % probability level. Depicted is only one molecule of two different ones in the asymmetric unit. For a) and b) enclathrated chloroform has been omitted for clarity. c) Crystal packing of 4 emphasizing the “stair‐like” arrangement. Hydrogen bonds are displayed by red dashed lines. d) crystal packing of 5 by various types of π‐stacking.
The two new triptycene end‐capped dyes 4 and 5 were investigated by UV/Vis, cyclic voltammetry and DFT calculations and compared to their parent counterparts 6 and 7 (Figure 3).[ 8 ] Triptycene end‐capped indigo 4 shows three absorption maxima at λ 1 = 299 nm, λ 2 = 372 nm, λ 3 = 612 nm, comparable to parent indigo 6 (λ 1 = 286 nm and λ 2 = 606 nm, see Figure 4).[ 2 , 23 ] According to TD‐TDFT (see Figure S18, Supporting Information) the most red‐shifted adsorption corresponds to π–π* transitions (HOMO‐LUMO), λ 1 to HOMO‐1 to LUMO+1, and λ 2 to HOMO‐2 to LUMO transitions. The slight red‐shift of Δλ = +6 nm of the longest wavelength absorption for 4 compared to parent indigo 6 suggests only a small contribution of homoconjugation of the triptycene units to the indigo core.[ 24 ] Triptycene end‐capped bay annulated indigo 5 shows in dichloromethane absorption bands at λ 1 = 301 nm, λ 2 = 552 nm, λ 3 = 585 nm and again it is comparable to the parent compound 7 (λ 1 = 283 nm, λ 2 = 544 nm and λ 3 = 579 nm). The absorption band at 544 nm and 579 nm can be assigned to π–π* transition and intramolecular charge transfer.[ 8 ] All compounds have been investigated by cyclic voltammetry (Figure 5) in dichloromethane and show two quasi‐reversible reduction waves for 4 (E red,1 = –1.19 eV and E red,2 = –1.68 eV) as well as for 5 (E red,1 = –1.25 eV and E red,2 = –1.54 eV), which are comparable to the parent compounds 6 (E red,1 = –1.20 eV and E red,2 = –1.30 eV)[ 25 ] and 7 (E red,1 = –1.35 eV and E red,2 = –1.64 eV) (Figure 4, Table 1).[ 8 ] The stepwise reductions of all triptycene based compounds are reversible for at least 20 cycles, demonstrating the electrochemical stability of those (see Figures S20 and S29).
Figure 3.
Molecular structures of indigo 6 and bay‐annulated indigo 7.
Figure 4.
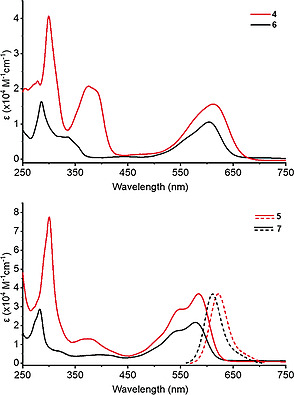
UV/Vis spectrum of a) triptycene end‐capped indigo 4 and parent indigo 6 in chloroform and b) UV/Vis and fluorescence spectrum of triptycene end‐capped bay annulated indigo 5 and parent bay annulated indigo 7 in dichloromethane.
Figure 5.
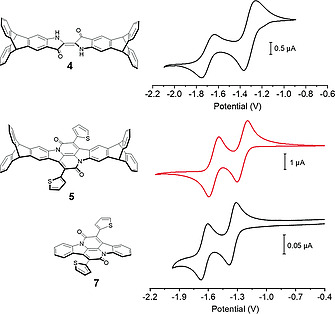
Cyclic voltammograms of 4, 5 and 7. DCM, nBu4NPF6 (0.10M) measured at room temperature with a Pt electrode and Fc/Fc+ as internal reference (scanning speed: 100 mV/s).
Table 1.
Summary of optoelectronic and electrochemical properties 4, 5, 6 and 7
| Compound[ a ] | λ abs [nm] | λ em(λ ex)[ b ] | Φ | Eg (opt) [ c ] | E red,1 [ d ] | E red,2 [ d ] | E LUMO,DFT [ e ] | E HOMO,DFT [ e ] | E g, DFT [ e ] |
|---|---|---|---|---|---|---|---|---|---|
| (log ε) | [nm] | [%] | [eV] | [V] | [V] | [eV] | [eV] | [eV] | |
| 4 | 612[ a ](4.20) | – | n.d.[ f ] | 1.9 | –1.19[ g ] | –1.68 | –3.0 | –5.4 | 2.4 |
| 5 | 585[ b ](4.59) | 619 (550) | 22.6 ± 0.6 | 1.9 | –1.25 | –1.54 | –3.1 | –5.5 | 2.4 |
| 6 [ 24 ] | 606 (4.02) | – | n.d.[ f ] | 1.9 | –1.20[ g ] | –1.30 | –3.0 | –5.5 | 2.5 |
| 7 | 579[ b ] (4.33) | 612 (545) | 58.5 ± 0.4 | 1.9 | –1.35 | –1.64 | –3.2 | –5.6 | 2.4 |
Measured in chloroform at RT.
Measured in DCM at RT, absorption maximum at the longest wavelength.
E g(opt) = 1242/λ onset.
Cyclic voltammogram measured in DCM with a Pt electrode and nBu4NPF6 as electrolyte. Scan speed: 100 mV/s, Fc/Fc+ was used an internal reference.
Obtained using DFT‐B3LYP/6‐311+G(d).
No significant emission detected.
E red,onset.
Frontier molecular orbitals were calculated by density functional theory [DFT B3LYP/6‐311+G(d)]. For 4 both the HOMO and LUMO orbitals are mainly distributed over the whole central planar indigo unit and only very small orbital coefficients are found at the lateral end‐capped benzene rings (Figure 6). Similar distribution has been found for bay annulated indigo 5. This explains similar absorption maxima and color for 4 and 5 in comparison to the parent indigos 6 and 7.
Figure 6.
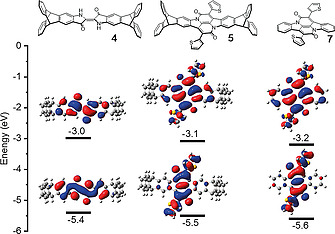
Energy level diagram (vs. vacuum) and frontier molecular orbitals of 4, 5 and 7 [calculated by DFT‐B3LYP/6‐311+G(d)].
As mentioned above, the objective of introducing end‐capped triptycene units to the pigments was to increase their solubility in organic solvents (Table 2). Triptycene end‐capped indigo 4 is with 10 mmol/L about 66 times more soluble in DCM than the parent indigo 6 itself (0.15 mmol/L). A similar observation was made for 5 and 7. Whereas 38 mmol/L of 5 were dissolved in DCM, only 0.53 mmol/L of the parent bay‐annulated indigo were brought into solution, which is a factor of 71(!) The more polar the solvent, the less significant is the difference between triptycene end‐capped dyes and the parent ones, due to the hydrophobic nature of the compounds. In CHCl3 4 is with 3.2 mmol/L still 10 times more soluble than 6 (0.31 mmol/L), which is approx. the same factor we observed previously for HBOs and QPPs.[ 14a ] In EtOAc the difference between triptycene end‐capped 4 and 6 reduces to the factor of 3 and in most polar methanol, no significant difference was determined. The bay‐annulated indigos 5 and 7 show similar trends. In EtOAc 5 is with 1.6 mmol/L about 8 times more soluble than 7 (0.19 mmol/L) and in methanol, again the factor is only 2.
Table 2.
Solubilities in mmol/L (at 25 °C) for triptycene end‐capped indigos 4 and 5 in comparison with the planar congeners 6 and 7
| Solvent | 4 | 6 | 5 | 7 |
|---|---|---|---|---|
| CH2Cl2 | 10 | 0.15 | 38 | 0.53 |
| EtOAc | 2.1 | 0.64 | 1.6 | 0.19 |
| CH3OH | 0.3 | 0.26 | 0.4 | 0.21 |
| CHCl3 | 3.2 | 0.31 | –[ a ] | 1.01 |
Decomposition.
To summarize, we expanded the triptycene end‐capping strategy to pigments of the indigo family, which are known to be very poorly soluble. By this strategy, solubility in dichloromethane was enhanced about 70(!) times without changing optoelectronic or electrochemical properties of the pigment core, which makes triptycene end‐capped indigo 4 and the bay annulated indigo congener 5 interesting electronic acceptors for organic electronic devices, such as for bulk‐heterojunction photovoltaics, where a solubility matching of acceptor and donor compounds can be crucial.[ 26 ]
Supporting information
Supporting Information
Acknowledgements
The authors like to thank the European Research Council ERC (consolidators grant CaTs n DOCs; grant agreement no. 725765) and Deutsche Forschungsgemeinschaft DFG (SFB 1249 “N‐heteropolycyclic compounds as functional materials” TP‐A04) for funding this project. We are further thankful to T. Cornelissen and M. Kemerink (CAM, Ruprecht‐Karls‐Universität Heidelberg) for preliminary studies and A. Rowse, for helpful discussions. Open access funding enabled and organized by Projekt DEAL.
References
- 1.a) Gordon P. F. and Gregory P., in: Organic chemistry in colour, Springer Science & Business Media, 2012; [Google Scholar]; b) Zollinger H., in: Color chemistry: syntheses, properties, and applications of organic dyes and pigments, 2003. [Google Scholar]
- 2.a) Irimia‐Vladu M., Głowacki E. D., Troshin P. A., Schwabegger G., Leonat L., Susarova D. K., Krystal O., Ullah M., Kanbur Y. and Bodea M. A., Adv. Mater, 2012, 24, 375–380; [DOI] [PubMed] [Google Scholar]; b) Głowacki E. D., Voss G. and Sariciftci N. S., Adv. Mater, 2013, 25, 6783–6800; [DOI] [PubMed] [Google Scholar]; c) Petermayer C. and Dube H., Acc. Chem. Res, 2018, 51, 1153–1163; [DOI] [PubMed] [Google Scholar]; d) Huang C.‐Y., Bonasera A., Hristov L., Garmshausen Y., Schmidt B. M., Jacquemin D. and Hecht S., J. Am. Chem. Soc, 2017, 139, 15205–15211; [DOI] [PubMed] [Google Scholar]; e) Kolaczkowski M. A. and Liu Y., Chem. Rec, 2019, 19, 1062–1077. [DOI] [PubMed] [Google Scholar]
- 3.a) Süsse P., Naturwissenschaften, 1980, 67, 453; [Google Scholar]; b) Süsse P., Steins M. and Kupcik V., Z. Kristallogr, 1988, 184, 269–273; [Google Scholar]; c) Kettner F., Hüter L., Schäfer J., Röder K., Purgahn U. and Krautscheid H., Acta Crystallogr., Sect. E, 2011, 67, o2867–o2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Watanabe M., Uemura N., Ida S., Hagiwara H., Goto K. and Ishihara T., Tetrahedron, 2016, 72, 4280–4287. [Google Scholar]
- 5. Ngai J. H., Leung L. M., So S. K. and Lee H. K., Org. Electron, 2016, 32, 258–266. [Google Scholar]
- 6.a) Porada J. H., Neudörfl J.‐M. and Blunk D., New J. Chem, 2015, 39, 8291–8301; [Google Scholar]; b) Porada J. H. and Blunk D., J. Mater. Chem, 2010, 20, 2956–2958. [Google Scholar]
- 7. Głowacki E., Voss G., Demirak K., Havlicek M., Sünger N., Okur A. and Monkowius U., Chem. Commun, 2013, 49, 6063–6065. [DOI] [PubMed] [Google Scholar]
- 8.a) He B., Pun A. B., Zherebetskyy D., Liu Y., Liu F., Klivansky L. M., McGough A. M., Zhang B. A., Lo K., Russell T. P., Wang L. and Liu Y., J. Am. Chem. Soc, 2014, 136, 15093–15101; [DOI] [PubMed] [Google Scholar]; b) Brebels J., Klider K. C. C. W. S., Kelchtermans M., Verstappen P., Van Landeghem M., Van Doorslaer S., Goovaerts E., Garcia J. R., Manca J., Lutsen L., Vanderzande D. and Maes W., Org. Electron, 2017, 50, 264–272. [Google Scholar]
- 9. Voss G., Gradzielski M., Heinze J., Reinke H. and Unverzagt C., Helv. Chim. Acta, 2003, 86, 1982–2004. [Google Scholar]
- 10. Modsiri S., Pongmaneerat P., Tawil S., Promarak V. and Thamyongkit P., ACS Omega, 2020, 5, 6039–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohl B., Rominger F. and Mastalerz M., Org. Lett, 2014, 16, 704–707. [DOI] [PubMed] [Google Scholar]
- 12. Kohl B., Rominger F. and Mastalerz M., Chem. Eur. J, 2015, 21, 17308–17313. [DOI] [PubMed] [Google Scholar]
- 13.a) Kohl B., Rominger F. and Mastalerz M., Angew. Chem. Int. Ed, 2015, 54, 6051–6056; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2015, 127, 6149; [Google Scholar]; b) Gao B., Wang M., Cheng Y., Wang L., Jing X. and Wang F., J. Am. Chem. Soc, 2008, 130, 8297–8306. [DOI] [PubMed] [Google Scholar]
- 14.a) Kohl B., Baumgärtner K., Rominger F. and Mastalerz M., Eur. J. Org. Chem, 2019, 2019, 4891–4896; [Google Scholar]; b) Baumgartner K., Chincha A. L. M., Dreuw A., Rominger F. and Mastalerz M., Angew. Chem. Int. Ed, 2016, 55, 15594–15598; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 15823; [Google Scholar]; c) Baumgärtner K., Rominger F. and Mastalerz M., Chem. Eur. J, 2018, 24, 8751–8755; [DOI] [PubMed] [Google Scholar]; d) Fresta E., Baumgärtner K., Cabanillas‐Gonzalez J., Mastalerz M. and Costa R. D., Nanoscale Horiz, 2020, 5, 473–480. [DOI] [PubMed] [Google Scholar]
- 15. Mastalerz M., Wang X., Kohl B., Rominger F. and Elbert S. M., Chem. Eur. J. 2020, DOI: 10.1002/chem.202002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chong J. H. and MacLachlan M. J., Inorg. Chem, 2006, 45, 1442–1444. [DOI] [PubMed] [Google Scholar]
- 17. Beaudoin D., Rominger F. and Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15599–15603; [DOI] [PubMed] [Google Scholar]; Angew. Chem, 2016, 128, 15828. [Google Scholar]
- 18. Sandmeyer T., Helv. Chim. Acta, 1919, 2, 234–242. [Google Scholar]
- 19. Loloiu G. and Maior O., Rev. Roum. Chim, 1997, 42, 67–69. [Google Scholar]
- 20.a) Baeyer A., Ber. Dtsch. Chem. Ges, 1878, 11, 1296–1297; [Google Scholar]; b) Baeyer A., Ber. Dtsch. Chem. Ges, 1879, 12, 456–461. [Google Scholar]
- 21. Katritzky A. R., Fan W. Q., Koziol A. E. and Palenik G. J., J. Heterocycl. Chem, 1989, 26, 821–828. [Google Scholar]
- 22. Deposition Numbers 2022650 (4) and CCDC No. 2023711 (5) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 23. Brode W. R., Pearson E. G. and Wyman G. M., J. Am. Chem. Soc, 1954, 76, 1034–1036. [Google Scholar]
- 24.a) Kawasumi K., Wu T., Zhu T., Chae H. S., Van Voorhis T., Baldo M. A. and Swager T. M., J. Am. Chem. Soc, 2015, 137, 11908–11911; [DOI] [PubMed] [Google Scholar]; b) Biegger P., Tverskoy O., Rominger F. and Bunz U. H., Chem. Eur. J, 2016, 22, 16315–16322; [DOI] [PubMed] [Google Scholar]; c) Menke E. H., Leibold D., Ullrich A. P., Vaynzof Y. and Mastalerz M., Org. Chem. Front, 2017, 4, 834–838. [Google Scholar]
- 25.a) Klimovich I. V., Leshanskaya L., Troyanov S., Anokhin D., Novikov D., Piryazev A., Ivanov D., Dremova N. and Troshin P., J. Mater. Chem. C, 2014, 2, 7621–7631; [Google Scholar]; b) Bond A. M., Marken F., Hill E., Compton R. G. and Hügel H., J. Chem. Soc., Perkin Trans. 2, 1997, 1735–1742; [Google Scholar]; c) Ju Z., Sun J. and Liu Y., Molecules, 2019, 24, 3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menke E. H., Leibold D., Lami V., Hofstetter Y. J., Mastalerz M. and Vaynzof Y., Org. Electron, 2017, 47, 211–219. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information