


Abstract
Whole-genome mapping technologies have been developed as a complementary tool to provide scaffolds for genome assembly and structural variation analysis (1,2). We recently introduced a novel DNA labeling strategy based on a CRISPR–Cas9 genome editing system, which can target any 20bp sequences. The labeling strategy is specifically useful in targeting repetitive sequences, and sequences not accessible to other labeling methods. In this report, we present customized mapping strategies that extend the applications of CRISPR–Cas9 DNA labeling. We first design a CRISPR–Cas9 labeling strategy to interrogate and differentiate the single allele differences in NGG protospacer adjacent motifs (PAM sequence). Combined with sequence motif labeling, we can pinpoint the single-base differences in highly conserved sequences. In the second strategy, we design mapping patterns across a genome by selecting sets of specific single-guide RNAs (sgRNAs) for labeling multiple loci of a genomic region or a whole genome. By developing and optimizing a single tube synthesis of multiple sgRNAs, we demonstrate the utility of CRISPR–Cas9 mapping with 162 sgRNAs targeting the 2Mb Haemophilus influenzae chromosome. These CRISPR–Cas9 mapping approaches could be particularly useful for applications in defining long-distance haplotypes and pinpointing the breakpoints in large structural variants in complex genomes and microbial mixtures.
INTRODUCTION
Restriction mapping has been applied in human genomics for physical mapping of genome fragments based on restriction enzyme cutting and was used extensively during the Human Genome Project to guide genome assembly (3–5). However, traditional restriction mapping is highly labor-intensive and requires large amounts of sample. More importantly, a traditional restriction map provides a ‘fingerprint’ of the genomic DNA, rather than an ordered sequence of restriction sites. A solution to the sequence assembly challenge that overcomes the drawbacks of traditional restriction mapping is optical mapping (6). The optical mapping method has been used to construct ordered restriction maps for whole genomes (7–9) and continues to be very useful in providing scaffolds for shotgun sequence assembly and validating sequence assemblies (10–12). More recently, a similar optical mapping technique has been introduced by combining sequence-specific labeling, along with consistent linearization of extremely long DNA molecules in nanochannel arrays (1). This provides an accurate, high-throughput, and robust whole-genome mapping technique, and optical mapping has been widely applied in assisting genome assembly, the detection and characterization of complex structural variants, and microbial comparative genomics (13–15).
The primary genome mapping strategy is based on measuring distances between short (6–8 bp) sequence motifs across the genome (16,17). However, the distribution of motifs is fixed for any given genome, which results in uneven spacing for different genomic regions. Often, there are no appropriate motifs within repetitive genomic regions, which results in large unmappable genomic intervals (14,18). Another challenge resides in detecting and typing specific structural variants for clinical diagnostic applications. Targeted variant-specific labeling is required to obtain accurate breakpoints, but this cannot be achieved by motif-mapping alone (19). In microbial comparative analyses, sequences with high similarities are often involved (20). Sequence motif mapping generally results in similar patterns in these regions. In principle, another important application of optical mapping could be long-range inference of haplotype structure, but use of motif-specific labels restricts analysis to polymorphisms that happen to impact genomic motifs.
Recently, we introduced a novel labeling strategy based on a CRISPR–Cas9 genome editing system using the nicking Cas9-D10A protein to address the above issues. This labeling strategy can target almost any 20 bp sequence (21). The method is especially powerful in targeting repetitive sequences or other sequences that rely on the distribution of restriction site motifs in the DNA. Since its introduction, the method has found many applications, including single-molecule telomere length measurements via fluorescent tagging of telomere repeats with guide RNA (gRNA) (22,23), global characterization of repeat-rich human subtelomere regions (24,25), mapping and identifying large-scale structural variants such as at acrocentric chromosomes (14), and identifying antibiotic resistance encoding plasmids present in bacterial isolates (26).
In this report, we present a couple of customized mapping strategies by CRISPR–Cas9 mediated DNA labeling. We demonstrate the overall effectiveness of the new mapping strategies using the bacterium Haemophilus influenzae strains as a model system, the standard lab strain Rd KW20 (RR722, NC_000907), and a marked derivative of clinical isolate 86-028NP (RR3131, NC_007416.2) (27,28). In the first strategy, we enable the CRISPR–Cas9 labeling to interrogate and differentiate the single allele difference in the NGG protospacer adjacent motif (PAM sequence). The same 20 base locus in two strains was either labeled or not depending on if an alternative allele other than G is present in PAM. Combining with sequence motif labeling, we can pinpoint the single-base differences in highly conserved sequences.
In the second strategy, we use a custom panel of sequence-specific sgRNAs to label multiple loci of a genomic region or a whole genome. Since nearly any 20mer sequence can be targeted (constrained only by the need for a 3′-NGG protospacer adjacent motif, or PAM) (21), one can design mapping patterns across a genome by selecting sets of specific single-guide RNAs (sgRNAs) for features of interest. To make this strategy viable and expand its utility requires: (a) reduced cost of sgRNA synthesis (a sgRNA still costs ∼$100 through commercial sources); (b) optimization of the total number of sgRNAs that can be combined into a single labeling reaction. By addressing the above issues, we first optimized and developed a sgRNA synthesis method based on T7 RNA transcription to generate a mixture of multiple sgRNAs in a single tube reaction (29), reducing synthesis costs by an order magnitude or greater. We then designed 162 sgRNAs targeting different chromosomal sites in the lab strain Rd and in vitro synthesized all of them in a single tube reaction. This mixture was then used to label two bacterial strains, followed by single-molecule optical mapping. Optical reads were successfully aligned to the custom-designed sgRNA map.
These CRISPR–Cas9 mapping approaches could be particularly powerful in defining long-distance haplotypes and pinpointing breakpoints of large structural variants in complex genomes (30,31), and they may enable microbial comparative analyses (17,32).
MATERIALS AND METHODS
High-molecular-weight DNA extraction
Two Haemophilus influenzae strains with complete genome sequences were used: the standard lab strain Rd KW20 (RR722, NC_000907) and a marked derivative of clinical isolate 86-028NP (RR3131, NC_007416.2, carrying novobiocin and nalidixic acid resistance alleles, NovR and NalR) (27,33,34). Bacterial culture followed standard protocols; cells were grown to stationary phase (OD600 nm = 1.2) in supplemented brain-heart infusion (10 μg/ml hemin 2 μg/ml NAD) shaking at 37°C, and then cells were harvested by centrifugation at 4000 rpm for 5 min before DNA extractions (35,36). Purification of ultra-high MW DNA fragments followed the Bionano Prep Cell Culture DNA Isolation Protocol. Briefly, cells were: (a) resuspended in cell buffer (∼5 × 109 CFU/ml); (b) embedded in 2% low-melt agarose (BioRad) plugs to minimize shearing forces; (c) lysed using Bionano cell lysis buffer supplemented with 167 μl Proteinase K (Qiagen) rocking overnight at 50°C; (d) RNase treatment by adding 50 μl of RNase A solution and incubating the plugs for 1 h at 37°C (Qiagen) and (e) washing in TE buffer with intermittent mixing. Finally, DNA was purified from low-melt agarose plugs by drop dialysis. Plugs were melted at 72°C, then incubated with 2 μl Agarase (Thermo Fisher Scientific) for 45 min. Melted plugs were dialyzed into TE buffer using 0.1 μm Millipore membrane filters for 45 min at a ratio of 15 ml buffer per ∼200 μl sample. DNA was allowed to homogenize overnight at room temperature before fluorometric quantification using the Qbit dsDNA BR kit (Thermo Fisher Scientific).
dsDNA synthesis
sgRNA oligos
sgRNAs were encoded on 55 nt DNA oligos with a 5′ T7 promoter sequence (5′-TTCTAATACGACTCACTATAG-3′), followed by the target 20mer sequence, complementary to the target gDNA sequence, and finally an overlap sequence (5′-GTTTTAGAGCTAGA-3′). Individually synthesized sgRNA oligos were then pooled into an equimolar mixture. sgRNA complementary oligo: An 80 nt long oligo was designed with the 3′ end complementary to the overlap sequence and remainder encoded the Cas9 binding sequence (5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′). All oligos were obtained from Integrated DNA Technology. The sgRNA oligo mix was hybridized to the sgRNA complementary oligo (at 10 μM each) in 1× NEBuffer2 (New England BioLabs, NEB) with 2 mM dNTPs at 90°C for 15 s followed by 43°C for 5 min. To complete dsDNA synthesis, the hybridization mixture was incubated at 37°C for 1hr with 5 U of Klenow Fragment 3′→5′ exo- (NEB). To degrade linear ssDNA remaining, the dsDNA was then treated with Exonuclease I in 1× Exonuclease I reaction buffer (NEB) for 1 h at 37°C. Finally, dsDNA was purified using QIAquick Nucleotide Removal Kit (Qiagen) and eluted in 30 ul elution buffer. Quality and concentration were assessed using agarose gel electrophoresis and the Synergy H1Hybrid Multi-Mode Reader (Bio Tek).
sgRNA synthesis
sgRNA was synthesized using HiScribe T7 High Yield RNA Synthesis Kit (NEB) following the Standard RNA Synthesis protocol. In summary, 1 μg dsDNA was incubated with 1× reaction buffer, 10 mM NTPs and T7 RNA polymerase enzyme mix at 37°C for 2 h followed by DNase I treatment at 37°C for 15 min to remove dsDNA from the reaction. sgRNA was then purified using RNA Clean & Concentrator Kits (Zymo Research). The concentration of the purified sgRNA was assessed using Synergy H1Hybrid Multi-Mode Reader (Bio Tek).
CRISPR–Cas9 labeling of chromosomal DNA
For DNA nicking using the 48 and 162 sgRNA mix (supplementary Tables S1 and S2),1.25 μM of the synthesized sgRNA was first incubated with 125 nM of Cas9 D10A (NEB) in 1× NEBuffer 3.1 (NEB) at 37°C for 15 min to form a sgRNA-Cas9 complex. 300 ng of the DNA sample was then added to the sgRNA–Cas9 complex mixture and incubated at 37°C for 60 min. For DNA nicking with both Cas9 and Nt.BspQI, 2.5 μM gRNA was first incubated with 63 nM of Cas9 D10A in 1X NEBuffer 3.1 at 37°C for 15min. After that, 300 ng of DNA and 5 U of Nt.BspQI (NEB) were added to the sample mixture and incubated at 37°C for 2 h. The nicked DNA samples were then labeled using 5 U Taq DNA Polymerase (NEB), 1× thermopol buffer (NEB), 266 nM free nucleotides mix (dATP, dCTP, dGTP (NEB) and Atto-532-dUTP (Jena Bioscience)) at 72°C for 60 min. The labeled sample was then treated with Proteinase K at 56°C for 30min and 1uM IrysPrep stop solution (BioNano Genomics) was added to the reaction.
DNA loading and imaging
Labeled DNA samples were stained and prepared for loading on an Irys Chip (BioNano Genomics) following manufacturer instructions. The sample was then linearized and imaged. The stained samples were loaded and imaged inside the nanochannels following the established protocol. Each Irys Chip contains two nanochannel devices, which can generate data from >60 Gb of long chromosomal DNA fragments (>150 kb). The image analysis was done using BioNano Genomics commercial software (IrysView 2.5) for segmenting and detecting DNA backbone YOYO-1 staining, similar to early optical mapping methods, and localizing the green labels by fitting the point-spread functions.
Data analysis
Single-molecule maps were de novo assembled and aligned to the reference as described in previous work (37). Briefly, the assembler is a custom implementation of the overlap-layout-consensus paradigm with a maximum likelihood model. An overlap graph was generated based on the pairwise comparison of all molecules as input. Redundant and spurious edges were removed. The assembler outputs the longest path in the graph and consensus maps were derived. Consensus maps are further refined by mapping single-molecule maps to the consensus maps and label positions are recalculated. Refined consensus maps are extended by mapping single molecules to the ends of the consensus and calculating label positions beyond the initial maps. After the merging of overlapping maps, a final set of consensus maps was output and used for subsequent analysis. RefAligner works similarly but compares molecules directly to an in silico nicked reference instead of first forming contigs. These maps were then opened in Irsyview visualization software from BioNano Genomics.
RESULTS AND DISCUSSION
Using CRISPR–Cas9 labeling to interrogate individual base, and tag specific genomic region of interest
The main strategy for long-range optical mapping is based on measuring the distances between the short sequence motifs recognized by nicking endonucleases (6–8 bp) on single long DNA molecules. The key information is the pattern of distances between motifs. Current labeling strategies can only detect single-base differences at polymorphisms that happen to coincide with nickase motifs, which has limited the potential applications of optical mapping. For example, the H. influenzae strains RR722 and RR3131 share a 100 kb region (819–916 kb of RR722, NC_000907, and 884–981 kb of RR3131, NC_007416) with 99% sequence similarity. The Nt.BspQI sequence motif maps for the two strains are almost identical for this region, except for one extra nick of the RR3131 genome, due to an adenine single-nucleotide difference from RR722, thus the nicking enzyme labels the RR3131’s allele but not RR722’s allele (Figure 1).
Figure 1.

Interrogation of individual bases with CRISPR–Cas9 labeling. Yellow lines indicate single molecules. The thick blue bars represent Nt.BSPQI reference map. The narrower blue bar represent consensus map of combined Nt.BSPQI CRISPR–Cas9 labeling. Red arrows and bases indicate the single base differences between the two strains. Additional details can be found in the Table 1.
We devised a strategy to use multiplexed CRISPR–Cas9 labeling to distinguish single-nucleotide variants affecting 3′-NGG PAM sites since the editing system has a strong requirement for the PAM immediately following the 20 bp recognition sequences. Genetic variation impacting PAM sites (i.e. if one of the G bases of a PAM in one genome is variant in another) is expected to strongly impact labeling, even if they share the 20 bp recognition sequence. Thus, we predicted that strong differential labeling at gRNA-guided PAM variants could reliably differentiate the single base difference between two genomes over long distances.
To demonstrate single-base resolution of multiplexed CRISPR–Cas9 labels at variation affecting PAM sites, we designed gRNAs targeting three distinct 20mer recognition sequences, but for each one of the two H. influenzae strains lacked a 3′-NGG PAM signal due to single nucleotide variation (Table 1). Labeling by both Nt.BspQI and CRISPR–Cas9 were performed in a single tube reaction, and the results of optical mapping are shown in Figure 1.
Table 1.
sgRNA target sequences used for single base differentiation in Figure 1
| Strains | Locations | Loci | Target sequence | gRNA sequence |
|---|---|---|---|---|
| RR722 | 819899 | 1 | AAAAATTGCTGCATCTTCTTTGA | AAAAATTGCTGCATCTTCTT |
| RR3131 | 885289 | 1 | AAAAATTGCTGCATCTTCTTTGG | |
| RR722 | 828196 | 2 | AACCATTCAAACGGCGATTGCGG | AACCATTCAAACGGCGATTG |
| RR3131 | 893590 | 2 | CACTATTCAAACGGCTATTGCTG | |
| RR722 | 903309 | 3 | AATATCCTTGCCTTGAGAGAACG | AATATCCTTGCCTTGAGAGA |
| RR3131 | 968698 | 3 | AATATCCTTGCCTTGAGAGAAGG |
The differing bases are shown in red for three locations.
Single-base variation away from either G in the PAM nearly eliminated the corresponding labeling. At ‘locus 1’ (NTHI0914-hypothetical protein of RR3131 and HI_0755-conserved hypothetical protein of RR722), the two strains share the same 20 bp recognition sequence (5′-AAAAATTGCTGCATCTTCTT-3′) as the gRNA, but RR3131 has a 3′-TGG PAM sequence, while RR722 has a TGA sequence instead. CRISPR–Cas9-mediated optical mapping clearly shows high-efficiency labeling at position 885289 in RR3131 (∼90% labeling), whereas RR722 molecules totally lacked labels (0%) at position 819899 (red arrow at ‘locus 1’ in Figure 1). Similarly, at ‘locus 3’ (NTHI0947-50S ribosomal protein L29 of RR3131), the labeling difference between two strains can only be explained by the presence of alternative alleles in the two strains, in which RR3131 becomes labeled at 98698 with a perfect AGG PAM sequence; RR722 is not labeled at the syntenic position because of an ACG variant non-PAM sequence. At ‘locus 2’ (ribB), the sgRNA matches RR722 at 828196 with a CGG PAM sequence, and correspondingly, over 90% of molecules spanning the position were labeled (red arrow at ‘locus 2’ in Figure 1). In RR3131, no labeling was seen at the best-matching genomic position (893590), but in addition to a non-PAM 3′-end (CTG), the first and third positions were also mismatched.
In summary, labeling efficiency was over 90% for gRNAs with an NGG PAM sequence, whereas almost none of the molecules were labeled if there is an alternative allele in the PAM sequences. This is in contrast to the variable labeling efficiencies seen for different mismatches from the 20 nt recognition sequences in the sgRNA experiments below. These results suggest that a customized optical mapping using gRNAs to target many of these polymorphisms (or ‘PAM SNPs’) could be an effective means to define long-distance haplotype structure in human genomes. It could also be applicable in other sample types, particularly mixed microbial specimens. The new DLE labeling strategy (6bp motif) from BioNano genomics provides 50% more labeling sites than Nt.BspqI labeling (7 bp motif) in the human genome, which may resolve some haplotype features. However, the density of 1 SNP per megabase in these motifs is still insufficient to construct whole-genome haplotypes, even given long average DNA read length of 300 kb.
We performed an in silico analysis of whole genomes from the 1000 genomes project (38,39) to determine the potential number and distribution of heterozygous PAM SNPs in the human genome, Out of 161 million NGG sites in hg38, on average, there are 220 000 heterozygous PAM SNPs in a single diploid human genome. In addition, there are on average 40 000 heterozygous indels (>4 bp) within potential CRISPR–Cas9 recognition sequences (20 bp + NGG); >2 bp heterozygous indels within the 20 bp gRNA recognition sequence preferentially target the matching allele. Together, the genomic density of these sites is ideal to generate long-distance haplotypes using CRISPR–Cas9 labeling of PAM sites with single molecules in these experiments longer than 100 kb.
Multiplexed sgRNA preparation in a single tube reaction
We adapted the previously described method to synthesize multiple sgRNAs in a single tube reaction (29). Figure 2 shows the synthesis scheme and workflow. The key difference between our approach and the available commercial kit (EnGen® sgRNA Synthesis Kit, S. pyogenes from NEB) is that we have a separate step to generate the dsDNA before the RNA transcription reaction. The mixture of multiple sgRNA oligos and the sgRNA complementary oligo was first mixed at a 1:1 ratio in reaction buffer. After Klenow exo- extension to generate dsDNA, the reaction was treated with Exonuclease I to remove extra ssDNA. The purity and size of dsDNA were further confirmed with gel electrophoresis before purification with PCR cleanup column. We typically get 5 μg dsDNA at 0.2 μg/μl concentration. After sgRNA synthesis using T7 RNA polymerase, the sample was treated with DNaseI to remove dsDNA and purified with an RNA cleanup column. We normally obtain 40μg sgRNA at 2 μg/μl concentration. This is enough to run ∼230 CRISPR–Cas9 labeling reactions with 300 ng target DNA sample each time. The purity and correct size of the dsDNA are critical to the synthesis of multiple sgRNAs. We successfully synthesized 162 sgRNAs in a single tube reaction.
Figure 2.

The workflow of sgRNA synthesis. The multiple oligos with a promoter sequence (red) and an overlap sequence (green) on either side of the target sequence are hybridized with a single complementary oligo that shares the overlap sequence.
Multiplexed sgRNA optical mapping
In the second customized mapping strategy, we customized the mapping patterns across a genome by selecting sets of specific single-guide RNAs (sgRNAs) for features of interest. This is particularly useful in designing different patterns to differentiate similar genomes or conserved sequences between strains or haplotypes. In designing the patterns, it is critical to avoid evenly distributed sgRNAs, because only long molecules across the entire pattern can be uniquely aligned. To test this, we first designed two custom optical mapping patterns using the different H. influenzae bacterial strains, lab strain Rd KW20 (RR722), and a marked derivative of clinical isolate 86-028NP (RR3131) as the model systems.
48 sgRNAs were designed to target a 300 kb region of RR722 (0–350 kb of NC_000907), which shares high sequence similarity with RR3131 strain (0–315 kb NC_007416). Each sgRNA was designed to have a single perfect match of 20 bases upstream of PAM NGGs based on the Rd reference genome (Supplementary Table S1). These 48 sgRNAs are evenly distributed across the 300 kb region of RR722 (RR722 reference map in Figure 3A). Dark lines on the blue bar indicate predicted sgRNA locations. Out of 48 sgRNAs, 33 sgRNAs also have a single perfect match of 20 bases upstream of a PAM NGG on the RR3131 strain. However, the predicted targeting locations of these 33 sgRNAs form an unevenly distributed mapping pattern (RR3131 reference map in Figure 3B), indicative of structural variation between the genomes.
Figure 3.

(A) Mapping results of RR722 molecules labeled with the 48 sgRNAs (Supplementary Table S1). The lines in the blue bar (designed reference map of RR722) represent the locations of the 48 sgRNAs on RR722. The yellow lines below the reference are labels with dark green dots representing where labels matched to the reference and light green dots representing labels not found in the reference. (B) Mapping results of RR3131 molecules labeled with the set of 48 sgRNAs (Supplementary Table S1). The lines in the blue bar (designed reference map of RR3131) represent the locations of the 48 sgRNAs on RR3131. The yellow lines below the reference are labels with dark green dots representing where labels matched to the reference map and light green dots representing labels not found in the reference map. The red arrows indicate the off-target labeling.
We then generated a single mixture of 48 sgRNAs, which was used to label and map targeted regions in both the RR722 and RR3131 genomes. The individual molecules are indicated as yellow lines that are aligned to blue references in Figure 3. The two data sets show similar characteristics with an average molecule length of 255 and 249 kb for RR722 and RR3131 respectively. But with the same amount of raw data, three times more molecules could be uniquely aligned to the RR3131 strain than the RR722 strain, even though RR3131 has fewer perfectly matched sgRNAs (Figure 3A and B, respectively). This is due to the fact that the shorter molecules will generate ambiguous alignments to the evenly distributed patterns. Longer molecules are needed to map across the whole evenly distributed reference, which results in fewer molecules aligned to RR722 sgRNA map. This clearly shows that an unevenly distributed mapping pattern could result in better mapping.
Main sources of off-target labeling
CRIPSR-Cas9 tagging is prone to off-target labeling. It is important to reduce off-target labeling as much as possible, especially when trying to use custom-target mapping to map sequences with high similarity. We aligned the 48 sgRNAs (20 base recognition sequence) against the RR3131 reference. Fifteen sgRNAs out of the above 48 sgRNAs that have imperfect matches to the RR3131 genome. Some of them result in off-target labeling in RR3131. In Figure 3B, many single molecules show off-target labels (light green dots) at six different locations, which are present in the RR722 genome, but not present in RR3131, therefore absent from the reference map.
Seven of these 15 sgRNAs show several partial matches (<8 bases) across the 300kb region, but without a PAM NGG next to the best match, which could not be labeled. These seven sgRNAs are designated as ‘N/A’ in Supplementary Table S1 and are unlikely to contribute to off-target labeling. Six of the remaining eight sgRNAs were matched the RR3131 reference at off-target loci with a PAM motif and a single mismatch in the 20 recognition sequences. These six were likely contributing to off-target labeling and designated as ‘off-target’ in Table 2. The final two sgRNAs of the 15 did not produce a label in RR3131 and are listed as ‘No label’. Of the two, the sgRNA at 219206 of RR722 (TTGTTTTACGATATAATACGNGG) also shows a single base mismatch on RR3371 strain, but did not result in off-target labeling. The sgRNA at 323878 of RR722 (TAATCAAGCATTAGATAGCTNGG) has several mismatches close to the 5′ end and also did not result in off-target labeling.
Table 2.
The off-target labeling of RR3131
| Strains | Locations | Labeling | Target sequence |
|---|---|---|---|
| RR722 | 21722 | GCTTTTTAGGATATCGTCCCNGG | |
| RR3131 | 21698 | off target | GCTTTTTAAGATATCGTCCCAGG |
| RR722 | 59529 | GCGGTATCCACCCCCACTGCNGG | |
| RR3131 | 60913 | off target | GCAGTATCCACCCCCACTGCAGG |
| RR722 | 86065 | GTTACATTACACACAAACTTNGG | |
| RR3131 | 86656 | off target | GTTACATTACACACAAATTTTGG |
| RR722 | 94393 | GGGGCGTAAATTCTTAACATNGG | |
| RR3131 | 151264 | off target | GGAGCGTAAATTCTTAACATTGG |
| RR722 | 253327 | CGAAGGGATAAATATTGCGANGG | |
| RR3131 | 316470 | off target | TGAAGGGATAAATATTGCGATGG |
| RR722 | 270963 | TAGCACTTAAAAGAGGAATGNGG | |
| RR3131 | 334078 | off target | TGGCACTTAAAAGAGGAATGGGG |
| RR722 | 219206 | TTGTTTTACGATATAATACGNGG | |
| RR3131 | 281336 | no label | TTGTTTTGCGATATAATACGAGG |
| RR722 | 296956 | TAATCAAGCATTAGATAGCTNGG | |
| RR3131 | 359914 | no label | GCGTAAAGCATTAGATAGCTTGG |
Two rows are shown for each of eight probes that did not have a perfect hit in the RR3131 genome. The second row is the designed probe named for its hit location on the RR722 genome. The upper row is the sequence found in the RR3131 strain, and named for its location. Bold indicates a PAM sequence motif (NGG). Red indicates a base that does not match the designed probe. The last two probes did not have a label seen consistently in the aligned data.
All six sgRNAs that caused high-frequency off-target labeling had a single mismatch to the target sequences of RR3131. Five of six had the single mismatch close to the 5′ end, distal from the PAM sequences, except the sgRNA at 86065 of RR722 (GTTACATTACACACAAACTTNGG) with the single mismatch at the 3rd base upstream of PAM. For example, the sgRNA at 21722 of RR722 (GCTTTTTAGGATATCGTCCCNGG) is designed to target the RR722 genome at coordinate 21722, but it also matches a synthetic position in RR3131 (at coordinate 21698) with a single mismatch (G/A) at the ninth base from the 5′ end. The off-target labeling of the RR3131 chromosome around 21698 was likely caused by this sgRNA. For the same reason, the sgRNA at 59529 of RR722 (GCGGTATCCACCCCCACTGCNGG) likely generated the off-target labeling on RR3131 around 60913 with a single mismatch at the third base. Notably, the off-target labeling on RR3131 is more efficient with sgRNA designed for RR722 at 59529 locus than the sgRNA of RR722 at 21722 locus, which may reflect that its mismatch is closer to the 5′ end.
Overall, these results are consistent with the observation that the last 8–10 seed bases of sgRNA upstream of the PAM are more important for reducing the off-target labeling (40–43), and that multiple mismatches also reduce off-target labeling.
Customized optical mapping of a whole bacterial genome
Based on our off-target labeling results and the reports that eight seeding bases immediately upstream of the PAM sequence (NGG) have higher discrimination (40,41), we optimized the design pipeline to select a set of sgRNAs spanning the full RR722 genome in a series of four stepwise filters: a) We first collected all possible sgRNAs with a single perfect match to the RR722 reference (all 20mers followed by a 3′ PAM NGG that occur only once in RR722); 40 870 such possible sgRNAs were available. (b) From those, we collected only the 8-base seeding sequences proximal to the PAM with single perfect hits to the reference. If an 8-base seed had multiple perfect hits to the reference, it was discarded since these had a high chance of contributing to off-target labeling. The remaining sgRNAs (15 339) all had a single perfect hit of 20 bases and a single perfect hit of the 8-base seeding sequences. (c) Since all eight base-seeding sequences have multiple hits with a single mismatch, we then applied a third filter to minimize the number of hits in the 8-base seeding sequences with single mismatches to RR722. This resulted in 1507 gRNAs with <5 singly mismatched hits in all 8-base seeding sequences. (d) From this dataset, we further tried to minimize off-target nicks by keeping the sgRNAs with one more mismatch in the first 12 bases from the 5′ end (415 remains). The sgRNA design flow chart is summarized in Figure 4. The final set of sgRNAs have only one perfect hit across the RR722 reference sequence in their 20-base recognition sequences and less than 5 hits with a mismatch in the 8-base PAM-proximal seeding sequence and another mismatch in 12 bases from the 5′ end respectively. After the four filters to minimize off-target labeling, a final manual adjustment was made to avoid evenly distributed mapping patterns. This resulted in a final set of 162 gRNAs (Supplementary Table S2) with an average density of 9 predicted labels per 100 kb on RR722. The labeling density is similar to Nt.BspQI labeling density used in commercial optical mapping kits (1).
Figure 4.
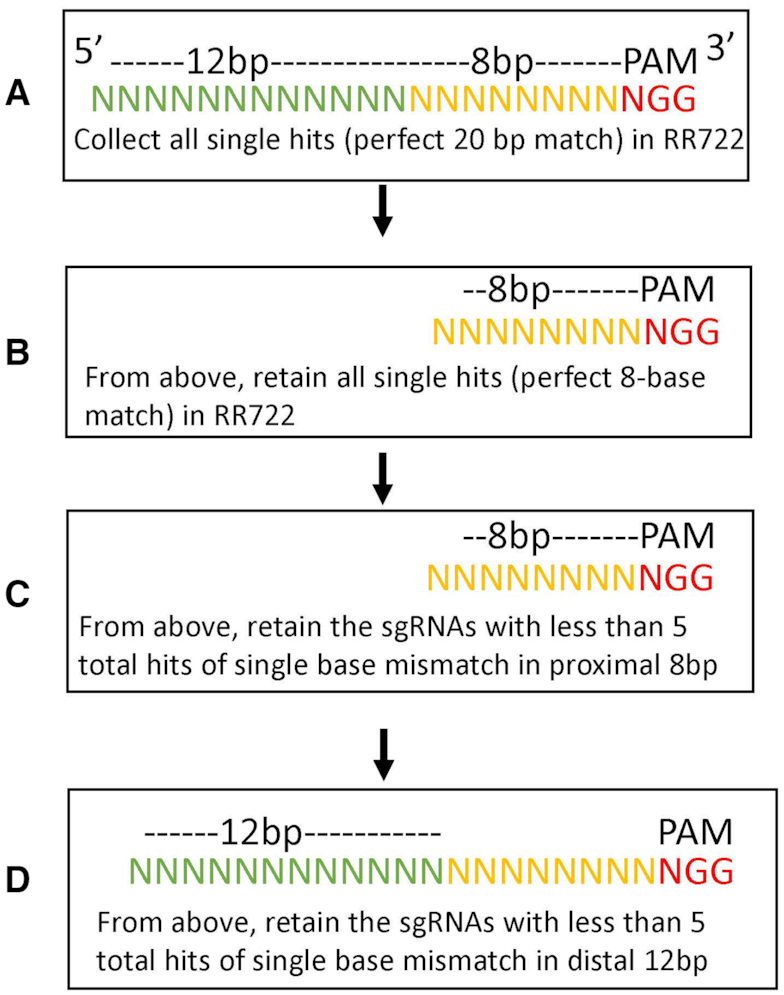
sgRNA design flow-chart.
This set of 162 sgRNAs was synthesized in a single-tube reaction and used to label RR722 chromosomal DNA. The resulting samples were run on the optical mapping setup described in the methods section. We collected total 0.5 Gb data with an average molecule length of 244 kb. Figure 5 shows a subset of single molecules (yellow lines) with good alignments to this custom-nicked reference (blue bars) with 100x overall coverage. As expected, no high-frequency off-target labels (>30%) were observed in this 162 set of sgRNAs. We then aligned the same set of 162 sgRNAs to the RR3131 reference sequence, only 90 of 162 perfect hits remained, and these form the RR3131 reference map shown in Figure 5B. When we aligned the labeled RR722 molecules to the RR3131 reference map, only 8 molecules aligned. These are shorter molecules around 100 kb that are aligned to two highly conserved regions, 884–981 kb of RR3131 (819–916 kb of RR722, NC_000907 and 884–981 kb of RR3131, NC_007416.02) and 1211–1254 kb of RR3131 (1177–1220 kb of RR722, NC_000907 and 1211–1254 kb of RR3131, NC_007416) respectively. If we apply the normal filter of molecules longer than 150 kb as shown in Figure 5A, none of the molecules aligns to RR3131 sgRNA map. This clearly demonstrated that the custom-designed sgRNAs can uniquely identify the genomic structure of the two strains.
Figure 5.

Mapping results of RR722 molecules labeled with the 162 sgRNAs (Supplementary Table S2). (A) The lines in the blue bar (designed reference map of RR722) represent the locations of the 162 sgRNAs on RR722. The yellow lines below the reference are labels with dark green dots representing where labels matched to the reference and light green dots representing labels not found in the reference. (B) Alignment results to RR3131.
CONCLUSION
Long-read sequencing technologies like Oxford nanopore and Pacific Biosciences can routinely reach an average of 10–30 kb read lengths and can provide assembled haplotype structures with N50 exceeding a Megabase and at single-base resolution (44). Recent advancements with DNA preparation and Oxford nanopore can have 50% of read lengths exceed 100 kb (45) and rare sequences can be more than a Megabase (45,46); in combination with Pacific Biosciences Sequel2 HiFi reads, this can generate fully contiguous human chromosome sequences (47), albeit still at a high cost. In general, obtaining accurate long sequence reads is dramatically more informative than only measuring spacings between short sequence motifs on long DNA fragments. Long-read sequencing will be the future. However, optical mapping provides a useful tool to infer extremely long-range haplotype information at a low cost, especially with the new ability to design custom labeling patterns. The average fragment length of optical reads can be ∼300 kb, and the long tail of extremely long fragments is correspondingly much larger (36). In turn, both mapping molecules to reference sequences and assembly-based inference can span much longer haplotype structures, especially in the regions containing long complex repetitive element arrays, and requiring less overall yield. This capability allows the optical mapping to detect large/complex SVs (>10 kb), which sequencing technologies would potentially miss (13). Optical mapping also provides an important independent validation tool for identifying misassemblies (47).
Here, we show for the first time that individual alleles can be differentiated at arbitrary loci by genome-scale optical mapping using CRISPR–Cas9 fluorescent labeling. Appropriately designed probe sets could provide an effective means to define long-distance haplotype structure in target regions of complex genomes like that of humans or to distinguish among strains and haplotypes in mixed microbial samples.
Traditionally, genome-scale optical mapping is based on measuring distances between short (6–8 bp) sequence motifs across the genome, which were labeled either via restriction enzyme cutting, or fluorescent tagging with nickase or methyltransferase. However, the distribution of motifs is fixed for any given genome. Here we also showed for the first time that one can customize the mapping patterns by designing a custom set of multiple sgRNAs to fluorescently tag any 20bp sequences with the CRISPR–cas9 genome editing system. This will greatly expand the applications of genome mapping in targeting specific features of interests, clinically relevant structural variants, repetitive regions, and other inaccessible regions by sequence motif labeling. Moreover, one added benefit is that our multiple sgRNAs provide more sequence information than motif mapping since they define multiple 20mers instead of the same 6–8mer. This will greatly increase the accuracy of pinpointing the breakpoints of structural variants and other specific genomic features. We have performed in silico mapping of repetitive elements such as ALU and SINE-1 in the human genome assembly, and we estimate that one sgRNA from ALU and one from LINE-1 will result in ∼90% coverage of the human genome. This coverage is similar to that of existing optical mapping schemes with Nt.Bspq1 and DLE labeling offered by Bionano Genomics, although understanding on- and off-target cutting by these gRNAs will be complicated. Thus, rational probe design might allow a small number of probes to interrogate long-range haplotype and repeat structures in complex genomes.
Since optical mapping does not rely on reading single base information, the cost of obtaining ultra-long optical reads is likely to remain low, although at the expense of full sequence information. Targeted enrichment of a single 0.3–1 Mb region for long-read sequencing remains challenging and costly. Our custom-designed optical maps require no target enrichment to define long-distance haplotype structure across target regions while maintaining a low cost at about $500 per diploid human genome. In targeted optical mapping, the cost can be further reduced by combining sets of sgRNAs designed to haplotype different regions. Careful design of sgRNA probes to target repetitive elements can further expand coverage and reduce probe costs. Thus, this expanded flexibility of genome-scale optical mapping shows it can continue to fill an important role for dissecting complex genomes and genomic variation, as long-read sequencing technologies continue to advance.
The custom-designed genomic labeling strategies described here could find wide applications for analyzing complex genomes like humans’, including determining long-range haplotype structure, higher precision breakpoint calling for complex structural variants, and improved resolution of complex repeat arrays. These strategies may also find applications in microbial comparative or community analyses since one can design gRNAs to identify characteristic markers on large genomic fragments of different microorganisms (e.g. pathogenic species) and virulence genes (e.g. antibiotic resistance genes and alleles) (15).
DATA AVAILABILITY
All additional data is available in the supplementary section.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Rosemary J. Redfield for bacterial strains.
Authors’ contributions: H.A., J.M., D.P., K.R. and D.V. generated data. H.A., J.M., D.P., K.R. J.C.M., D.V. and M.X. analyzed data. E.Y., J.C.M. and M.X. wrote the manuscript. M.X. designed and supervised the project. All authors have read and approved the manuscript.
Contributor Information
Heba Z Abid, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Eleanor Young, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Jennifer McCaffrey, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Kaitlin Raseley, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Dharma Varapula, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Hung-Yi Wang, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA.
Danielle Piazza, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA; Department of Microbiology and Immunology, College of Medicine, Drexel University, Philadelphia, PA, USA; Center for Genomic Sciences, Institute of Molecular Medicine and Infectious Disease, Drexel University, Philadelphia, PA, USA.
Joshua Mell, Department of Microbiology and Immunology, College of Medicine, Drexel University, Philadelphia, PA, USA; Center for Genomic Sciences, Institute of Molecular Medicine and Infectious Disease, Drexel University, Philadelphia, PA, USA.
Ming Xiao, School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, PA, USA; Center for Genomic Sciences, Institute of Molecular Medicine and Infectious Disease, Drexel University, Philadelphia, PA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health (NIH) [HG005946, in part]; the funders provided the financial support to the research but had no role in the design of the study, analysis, interpretations of data and in writing the manuscript. Funding for open access charge: US National Institutes of Health (NIH) [HG005946, in part].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lam E.T., Hastie A., Lin C., Ehrlich D., Das S.K., Austin M.D., Deshpande P., Cao H., Nagarajan N., Xiao M. et al.. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012; 30:771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Samad A., Huff E.J., Cai W.W., Schwartz D.C.. Optical mapping - a novel, single-molecule approach to genomic analysis. Genome Res. 1995; 5:1–4. [DOI] [PubMed] [Google Scholar]
- 3. Gillett W., Hanks L., Wong G.K.-S., Yu J., Lim R., Olson M.V.J.G.. Assembly of high-resolution restriction maps based on multiple complete digests of a redundant set of overlapping clones. Genomics. 1996; 33:389–408. [DOI] [PubMed] [Google Scholar]
- 4. Olson M.V. The human genome project. PNAS. 1993; 90:4338–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong G.K.-S., Yu J., Thayer E.C., Olson M.V.. Multiple-complete-digest restriction fragment mapping: generating sequence-ready maps for large-scale DNA sequencing. PNAS. 1997; 94:5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jing J., Reed J., Huang J., Hu X., Clarke V., Edington J., Housman D., Anantharaman T.S., Huff E.J., Mishra B.. Automated high resolution optical mapping using arrayed, fluid-fixed DNA molecules. PNAS. 1998; 95:8046–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Church D.M., Goodstadt L., Hillier L.W., Zody M.C., Goldstein S., She X., Bult C.J., Agarwala R., Cherry J.L., DiCuccio M.J.. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009; 7:e1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu C.-W., Schramm T.M., Zhou S., Schwartz D.C., Talaat A.M.. Optical mapping of the Mycobacterium avium subspecies paratuberculosis genome. BMC Genomics. 2009; 10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou S., Wei F., Nguyen J., Bechner M., Potamousis K., Goldstein S., Pape L., Mehan M.R., Churas C., Pasternak S.. A single molecule scaffold for the maize genome. PLos Genet. 2009; 5:e1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dong Y., Xie M., Jiang Y., Xiao N., Du X., Zhang W., Tosser-Klopp G., Wang J., Yang S., Liang J.. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013; 31:135–141. [DOI] [PubMed] [Google Scholar]
- 11. Latreille P., Norton S., Goldman B.S., Henkhaus J., Miller N., Barbazuk B., Bode H.B., Darby C., Du Z., Forst S.. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics. 2007; 8:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Teague B., Waterman M.S., Goldstein S., Potamousis K., Zhou S., Reslewic S., Sarkar D., Valouev A., Churas C., Kidd J.M.. High-resolution human genome structure by single-molecule analysis. PNAS. 2010; 107:10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L. et al.. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019; 10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levy-Sakin M., Pastor S., Mostovoy Y., Li L., Leung A.K.Y., McCaffrey J., Young E., Lam E.T., Hastie A.R., Wong K.H.Y. et al.. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019; 10:1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bogas D., Nyberg L., Pacheco R., Azevedo N.F., Beech J.P., Gomila M., Lalucat J., Manaia C.M., Nunes O.C., Tegenfeldt J.O. et al.. Applications of optical DNA mapping in microbiology. BioTechniques. 2017; 62:255–267. [DOI] [PubMed] [Google Scholar]
- 16. Lukinavicius G., Lapiene V., Stasevskij Z., Dalhoff C., Weinhold E., Klimasauskas S.. Targeted labeling of DNA by methyltransferase-directed transfer of activated groups (mTAG). J. Am. Chem. Soc. 2007; 129:2758–2759. [DOI] [PubMed] [Google Scholar]
- 17. Xiao M., Phong A., Ha C., Chan T.F., Cai D.M., Leung L., Wan E., Kistler A.L., DeRisi J.L., Selvin P.R. et al.. Rapid DNA mapping by fluorescent single molecule detection. Nucleic Acids Res. 2007; 35:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feuk L., Carson A.R., Scherer S.W.. Structural variation in the human genome. Nat. Rev. Genet. 2006; 7:85–97. [DOI] [PubMed] [Google Scholar]
- 19. Demaerel W., Mostovoy Y., Yilmaz F., Vervoort L., Pastor S., Hestand M.S., Swillen A., Vergaelen E., Geiger E.A., Coughlin C.R. et al.. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019; 29:1389–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tringe S.G., Von Mering C., Kobayashi A., Salamov A.A., Chen K., Chang H.W., Podar M., Short J.M., Mathur E.J., Detter J.C.. Comparative metagenomics of microbial communities. Science. 2005; 308:554–557. [DOI] [PubMed] [Google Scholar]
- 21. McCaffrey J., Sibert J., Zhang B., Zhang Y.G., Hu W.H., Riethman H., Xiao M.. CRISPR-CAS9 D10A nickase target-specific fluorescent labeling of double strand DNA for whole genome mapping and structural variation analysis. Nucleic Acids Res. 2016; 44:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCaffrey J., Young E., Lassahn K., Sibert J., Pastor S., Riethman H., Xiao M.. High-throughput single-molecule telomere characterization. Genome Res. 2017; 27:1904–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abid H.Z., McCaffrey J., Raseley K., Young E., Lassahn K., Varapula D., Riethman H., Xiao M.. Single-molecule analysis of subtelomeres and telomeres in Alternative Lengthening of Telomeres (ALT) cells. BMC Genomics. 2020; 21:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Young E., Abid H.Z., Kwok P.Y., Riethman H., Xiao M.. Comprehensive analysis of human subtelomeres by whole genome mapping. PLoS Genet. 2020; 16:e1008347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young E., Pastor S., Rajagopalan R., McCaffrey J., Sibert J., Mak A.C.Y., Kwok P.-Y., Riethman H., Xiao M.. High-throughput single-molecule mapping links subtelomeric variants and long-range haplotypes with specific telomeres. Nucleic Acids Res. 2017; 45:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muller V., Rajer F., Frykholm K., Nyberg L.K., Quaderi S., Fritzsche J., Kristiansson E., Ambjornsson T., Sandegren L., Westerlund F.. Direct identification of antibiotic resistance genes on single plasmid molecules using CRISPR/Cas9 in combination with optical DNA mapping. Sci. Rep. 2016; 6:37938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fleischmann R.D., Adams M.D., White O., Clayton R.A., Kirkness E.F., Kerlavage A.R., Bult C.J., Tomb J.-F., Dougherty B.A., Merrick J.M.. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995; 269:496–512. [DOI] [PubMed] [Google Scholar]
- 28. Hong W., Mason K., Jurcisek J., Novotny L., Bakaletz L.O., Swords W.E.. Phosphorylcholine decreases early inflammation and promotes the establishment of stable biofilm communities of nontypeable Haemophilus influenzae strain 86-028NP in a chinchilla model of otitis media. Infect. Immun. 2007; 75:958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gagnon J.A., Valen E., Thyme S.B., Huang P., Ahkmetova L., Pauli A., Montague T.G., Zimmerman S., Richter C., Schier A.F.. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. 2014; 9:e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. et al.. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007; 449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao M., Wan E., Chu C., Hsueh W.-C., Cao Y., Kwok P.-Y.. Direct determination of haplotypes from single DNA molecules. Nat. Methods. 2009; 6:199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagarajan N., Read T.D., Pop M.. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics. 2008; 24:1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mell J.C., Lee J.Y., Firme M., Sinha S., Redfield R.J.. Extensive cotransformation of natural variation into chromosomes of naturally competent Haemophilus influenzae. G3: Genes Genomes Genetics. 2014; 4:717–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mell J.C., Shumilina S., Hall I.M., Redfield R.J.. Transformation of natural genetic variation into Haemophilus influenzae genomes. PLoS Pathog. 2011; 7:e1002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poje G., Redfield R.J.. General methods for culturing Haemophilus influenzae. Methods Mol. Med. 2003; 71:51–56. [DOI] [PubMed] [Google Scholar]
- 36. Poje G., Redfield R.J.. Transformation of Haemophilus influenzae. Methods Mol. Med. 2003; 71:57–70. [DOI] [PubMed] [Google Scholar]
- 37. Mak A.C.Y., Lai Y.Y.Y., Lam E.T., Kwok T.-P., Leung A.K.Y., Poon A., Mostovoy Y., Hastie A.R., Stedman W., Anantharaman T. et al.. Genome-wide structural variation detection by genome mapping on nanochannel arrays. Genetics. 2016; 202:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McVean G.A., Altshuler D.M., Durbin R.M., Abecasis G.R., Bentley D.R., Chakravarti A., Clark A.G., Donnelly P., Eichler E.E., Flicek P. et al.. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Hsi-Yang Fritz M. et al.. An integrated map of structural variation in 2,504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anderson E.M., Haupt A., Schiel J.A., Chou E., Machado H.B., Strezoska Z., Lenger S., McClelland S., Birmingham A., Vermeulen A. et al.. Systematic analysis of CRISPR–Cas9 mismatch tolerance reveals low levels of off-target activity. J. Biotechnol. 2015; 211:56–65. [DOI] [PubMed] [Google Scholar]
- 41. Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S.. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014; 24:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L.H., Church G.M.. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013; 31:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y.L., Ge X.L., Yang F.Y., Zhang L.P., Zheng J.Y., Tan X.F., Jin Z.B., Qu J., Gu F.. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014; 4:5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q.. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020; 21:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jain M., Koren S., Miga K.H., Quick J., Rand A.C., Sasani T.A., Tyson J.R., Beggs A.D., Dilthey A.T., Fiddes I.T. et al.. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018; 36:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Payne A., Holmes N., Rakyan V., Loose M.. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics. 2018; 35:2193–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miga K.H., Koren S., Rhie A., Vollger M.R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G.A. et al.. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020; 585:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All additional data is available in the supplementary section.