


Abstract
Aptamers are single-stranded oligonucleotides that bind to a specific target with high affinity, and are widely applied in biomedical diagnostics and drug development. However, the use of aptamers has largely been limited to simple binders or inhibitors that interfere with the function of a target protein. Here, we show that an aptamer can also act as a positive allosteric modulator that enhances the activation of a receptor by stabilizing the binding of a ligand to that receptor. We developed an aptamer, named IR-A43, which binds to the insulin receptor, and confirmed that IR-A43 and insulin bind to the insulin receptor with mutual positive cooperativity. IR-A43 alone is inactive, but, in the presence of insulin, it potentiates autophosphorylation and downstream signaling of the insulin receptor. By using the species-specific activity of IR-A43 at the human insulin receptor, we demonstrate that residue Q272 in the cysteine-rich domain is directly involved in the insulin-enhancing activity of IR-A43. Therefore, we propose that the region containing residue Q272 is a hotspot that can be used to enhance insulin receptor activation. Moreover, our study implies that aptamers are promising reagents for the development of allosteric modulators that discriminate a specific conformation of a target receptor.
INTRODUCTION
Allosteric modulators bind to a receptor at a site distinct from the binding site of an orthosteric (endogenous) ligand and regulate receptor activation by changing the conformation of the receptor. They are classified according to their effect on the activity of orthosteric ligands and their capability to activate a receptor (1,2). Positive allosteric modulators (PAM), also known as allosteric enhancers, potentiate the affinity or efficacy of orthosteric ligands. Conversely, a substance that reduces the affinity or efficacy of an orthosteric ligand is called a negative allosteric modulator (NAM). Moreover, some PAMs and NAMs exhibit intrinsic agonism or antagonism at their target receptors, regardless of whether an orthosteric ligand is present. Allosteric modulators have several potential advantages as therapeutic agents compared with ligands binding to an orthosteric site, such as high specificity for receptor subtypes and reducing the risk of drug overdose (3,4). Therefore, studies of allosteric modulators are not only important to reveal mechanisms of receptor activation, but also to discover new drug candidates.
Aptamers are binding agents identified from in vitro selection using single-stranded oligonucleotide libraries (5,6). Aptamers bind to their targets with high affinity and specificity, similar to antibodies. However, unlike antibodies that primarily recognize the sequence of a protein as an epitope, aptamers interact with the topology of the surface structure of target proteins (7–9). Therefore, the specificity of aptamers tends to be very sensitive to small differences in the structure of a target (10–12). This characteristic makes aptamers ideally suited for the development of allosteric modulators, which are required to recognize ligand-induced conformational changes of a receptor. However, only a few aptamers that act as agonists and activate a receptor's function have so far been identified (13–18).
In this study, we developed a PAM aptamer, named IR-A43, that potentiates the activation of the insulin receptor by insulin. In the absence of insulin, IR-A43 has no effect on receptor activation, but it effectively enhances insulin binding to the insulin receptor. The enhancement of insulin binding is followed by increased autophosphorylation of tyrosine residues and enhanced downstream signaling of receptors. Furthermore, by utilizing the species-specific activity of IR-A43 at the human and mouse insulin receptor, we discover that residue Q272 in the cysteine-rich (CR) domain is a crucial hotspot for the insulin-enhancing activity of IR-A43. Based on the previously reported insulin receptor structures, we propose that IR-A43 acts as a ‘wedge’ to stabilize the insulin-induced active conformation of the receptor.
MATERIALS AND METHODS
Reagents and antibodies
Aptamers were synthesized by Aptamer Science, Inc. (Pohang, Korea). Bovine insulin, FITC-labeled insulin, dexamethasone, and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma-Aldrich. Recombinant proteins of the extracellular domain of the human insulin receptor and the insulin-like growth factor 1 receptor were purchased from R&D Systems. Adenosine triphosphate labeled on the gamma phosphate group with 32P was purchased from PerkinElmer. T4 polynucleotide kinase used for aptamer labeling was purchased from New England Biolabs. Anti-insulin receptor β-subunit (CT-3) antibody was purchased from Santa Cruz Biotechnology. Anti-phospho-tyrosine (4G10), anti-phospho-insulin receptor substrate-1 (Y612) human/(Y608) mouse, and anti-phospho-insulin receptor (Y1146) antibodies were purchased from Millipore. Anti-phospho-insulin receptor (Y960), anti-phospho-insulin receptor (Y1316), and anti-phospho-insulin receptor (Y1322) antibodies were purchased from Invitrogen. Anti-phospho-insulin receptor (pAb, Y1150/Y1151), anti-phospho-AKT (S473), anti-phospho-AKT (T308), anti-phospho-SHC (Y239/Y240) and anti-phospho-ERK1/2 (T202/Y204) antibodies were purchased from Cell Signaling Technology. IRdye 680LT-conjugated anti-rabbit and mouse antibodies were purchased from LI-COR. For western blotting, the anti-phospho-AKT (S473) antibody was used at a 1:2000 dilution, and other primary antibodies were used at a 1:1000 dilution. The secondary antibodies were used at a 1:20 000 dilution.
In vitro selection of IR aptamers
To identify insulin receptor-specific aptamers, we performed a SELEX process as previously described (18). Briefly, a modified single-stranded DNA (ssDNA) library with a 40 mer random region (N40) containing 5-[N-(1-naphthylmethyl)carboxamide]-2′-deoxyuridine (Nap-dU) in place of dT was prepared. The random regions were flanked by 20 mer constant regions for PCR with the following sequence: 5′-TATGAGTGACCGTCCGCCTG-N40-CAGCCACACCACCAGCCAAA-3′. Next, 100 pmol ssDNA library was incubated with 50 pmol his-tagged recombinant insulin receptor extracellular domain (His 28-Lys 944, R&D Systems) in selection buffer (40 mM HEPES (pH 7.5), 102 mM NaCl, 5 mM KCl, 5 mM MgCl2, and 0.05% Tween-20) at 37°C for 30 min. After ssDNA library binding, the insulin receptor proteins were immobilized by incubating with 20 μl Dynabeads TALON (Invitrogen) at 37°C for 15 min, and unbound ssDNAs were removed by washing five times with 100 μl selection buffer. ssDNAs were extracted from the insulin receptor proteins by adding 170 μl of 2 mM NaOH solution, and then neutralized by mixing 160 μl eluate with 40 μl of 8 mM HCl. The extracted ssDNAs were amplified using a 5′-OH terminal biotinylated reverse primer (IQ5 multicolor real-time PCR detection system, Bio-Rad). The amplified DNAs were captured on 25 μl Dynabeads MyOne streptavidin (Invitrogen) via the biotinylated antisense strand, and the sense strands were eluted by adding 180 μl of 20 mM NaOH at 37°C for 5 min. The eluted strand was discarded, and the beads were washed three times with selection buffer. To make the sense strand containing Nap-dU in place of dT, the beads were resuspended in 60 μl extension reaction mix (1× KOD DNA polymerase buffer; 500 pmol forward primer; 0.0625U KOD DNA polymerase; 0.5 mM each of dATP, dGTP, dCTP and Nap-modified dUTP) and incubated at 68°C for 60 min. After three washes with 180 μl selection buffer, the sense strands were eluted by adding 180 μl of 20 mM NaOH, and neutralized by mixing 175 μl eluate with 5 μl of 700 mM HCl and 5 μl of 180 mM HEPES. The eluted sense strands were used for the next round of selection. After eight rounds of SELEX, the enriched ssDNA pool was sequenced.
Aptamer binding assay
The binding affinities of the aptamer to the extracellular domains of the insulin receptor (His 28-Lys 944) and insulin-like growth factor 1 receptor (Gln 31Asn 932) were analyzed using a filter binding assay. First, the 5′-end of the aptamer was labeled with [α-32P]-ATP (the reaction mix contained 10x T4 polynucleotide kinase buffer 1 μl, 10 U/μl T4 polynucleotide kinase 0.25 μl, gamma-32P-ATP 3000 ci/mmol 0.25 μl, aptamer 1 pmol, made up to a volume of 10 μl with H2O and incubated at 37°C for 30 min). After labeling, unincorporated ATP was removed using size exclusion spin columns (MicroSpin G-50 columns, GE Healthcare). After heating at 95°C for 3 min, the mixture was slow-cooled to 37°C at 0.1°C/s in binding buffer (40 mM HEPES (pH 7.5), 120 mM NaCl, 5 mM KCl, 5 mM MgCl2 and 0.002% Tween-20) to reconstitute the aptamer structure. The aptamer was incubated with a purified recombinant insulin receptor or insulin-like growth factor 1 receptor at various concentrations for 30 min at 37°C. To pull-down the aptamer–protein complexes, the solution was incubated with 5.5 μl Zorbax silica beads (Agilent) for 1 min with shaking. The aptamer–protein complex bound to the beads was partitioned through nitrocellulose filter plates (Millipore) and washed in binding buffer to remove unbound aptamer. The amount of radiolabeled aptamer that interacted with the insulin receptor or insulin-like growth factor 1 receptor was detected by exposure to photographic film, and was quantified using Amersham Typhoon gel and blot imaging systems. The dissociation constant (Kd) of the aptamers was determined by fitting the binding data to a one-site saturation equation using the SigmaPlot program.
Cell culture and adipocyte differentiation
CHO-K1, 3T3-L1 and L6 myoblast cells were purchased from the American Type Culture Collection, and Rat-1 cells overexpressing the human insulin receptor (Rat-1/hIR) were kindly provided by Dr Nicholas J.G. Webster from the University of California, San Diego. Rat-1/hIR cells and 3T3-L1 pre-adipocytes were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM) with 10% (vol/vol) fetal bovine serum (FBS, Gibco); CHO-K1 cells were maintained in Ham's F-12K (Kaighn's) medium with 10% (v/v) FBS (Gibco); and L6 cells were maintained in low-glucose minimal essential medium (α-MEM) with 10% (vol/vol) FBS (Gibco) at 37°C under a humidified atmosphere containing 5% CO2. For adipocyte differentiation, 3T3-L1 pre-adipocytes were cultured for 2 days post-confluence. Differentiation was initiated by changing the medium to DMEM containing 1 μM dexamethasone, 500 nM IBMX, 850 nM insulin, and 10% FBS. After 2 days, the medium was replaced with DMEM containing 850 nM insulin and 10% FBS, and then incubated for 2 additional days. Finally, the medium was changed to DMEM containing only 10% FBS and incubated for 4–5 days until at least 90% of the cell population exhibited accumulation of lipid droplets.
Transfection
To overexpress wild-type or mutant insulin receptors in CHO-K1 or L6 cells, we performed transfections using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instruction. Briefly, 1 day prior to transfection, the cells were seeded in 12- or 24-well plates with Opti-MEM medium (Gibco) containing 5% FBS. For plasmid DNA transfection, 1 μg DNA and 2 μl P3000 reagent were added to 50 μl Opti-MEM, and 2 μl Lipofectamine 3000 was added to another 50 μl Opti-MEM. These mixtures were incubated at room temperature for 15 min and then added to cells (100 μl/well for 12-well plates and 50 μl/well for 24-well plates). After 24 h, the medium was replaced with DMEM or α-MEM containing 10% FBS. The cells were incubated for an additional 48 h before being used in experiments.
Sample preparation for western blotting
Cells for western blotting were prepared on 12-well plates. Before insulin or aptamer stimulation, the cells were incubated in medium without FBS for 3 h and then incubated in Krebs–Ringer HEPES buffer (25 mM HEPES (pH 7.4), 120 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.3 mM CaCl2 and 1.3 mM KH2PO4) for 1 h. For cell-based experiments, the aptamers and insulin were prepared in Krebs–Ringer HEPES buffer. All aptamer samples were heated for 5 min at 95°C and slowly cooled to room temperature to reconstitute the tertiary structure of the aptamer. After stimulation of the cells with insulin and/or aptamer for the described time, the cells were washed three times with 1 ml cold PBS. To prepare total cell lysate, harvested cells were lysed in lysis buffer (150 μl/well) (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 20 mM NaF, 10 mM β-glycerophosphate, 2 mM Na3VO4, 1 mM PMSF, 10% glycerol, 1% Triton-X and protease inhibitor cocktails). Soluble cell lysate was isolated by centrifugation at 14 000 rpm for 15 min at 4°C, and 120 μl supernatant was mixed with 30 μl of 5× Laemmli sample buffer. After SDS-PAGE, the proteins were transferred to nitrocellulose membranes and incubated in blocking buffer (PBS, 5% non-fat dried milk, and 0.1% NaN3) for 30 min at room temperature. The membrane was incubated with primary antibodies overnight at 4°C. After washing three times in TTBS buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween 20) for 10 min, the membranes were incubated with secondary antibodies for 1 h at room temperature. Membranes were washed three times with TTBS buffer for 10 min, and then blotting was performed using a LI-COR Odyssey infrared imaging system.
Flow cytometry
Rat-1 cells expressing the human insulin receptor (Rat-1/hIR) were grown in 100 mm dishes to 70% confluence. After cell detachment using PBS containing 5 mM EDTA, the cells were incubated with blocking buffer (PBS, 1% BSA and 0.1% NaN3) for 30 min at 4°C on a rotating shaker (10 rpm). Next, the cells were divided into equal aliquots (1 × 106 cells/sample). FITC-labeled insulin or FITC-labeled IR-A43 was diluted with blocking buffer and mixed with the cells in a 1:1 ratio (total 1 ml). Non-labeled ligands (insulin or IR-A43 without FITC) were also added at the same time as FITC-labeled ligands. The cells were incubated for 1 h at 4°C on a rotating shaker (10 rpm) to allow the binding reaction to reach equilibrium, and then the cells were washed twice with 1 ml cold PBS. After fixation with 1 ml PBS containing 4% paraformaldehyde for 30 min at room temperature, the binding of FITC-labeled insulin or FITC-labeled IR-A43 to the insulin receptor was measured by flow cytometry (BD Biosciences FACSCanto II).
2-Deoxy-d-glucose uptake
Cells used for glucose uptake assay were prepared on 24-well plates. The fully differentiated 3T3-L1 adipocytes were serum-starved for 3 h in DMEM without FBS and glucose-starved for 1 h in Krebs–Ringer HEPES buffer prior to insulin or aptamer stimulation. L6 cells were serum-starved for 1 h in α-MEM without FBS and glucose-starved for 1 h in Krebs–Ringer HEPES buffer prior to insulin or aptamer stimulation. After insulin or/and aptamer stimulation for 30 min (500 μl/well), the cells were incubated with 2-deoxy-[14C]-glucose (0.1 μCi/ml) for 10 min (500 μl/well) and washed three times in cold PBS containing 25 mM D-glucose (1 μml/well). The cells were lysed in 0.5 N NaOH and 1% SDS solution (500 μl/well), and 450 μl cell lysate was mixed with 4 ml of a liquid scintillation cocktail (Research Products International). Glucose uptake was measured using a liquid scintillation counter (Hidex 300 SL).
RESULTS
Insulin enhances the binding of the IR-A43 aptamer to the insulin receptor
We identified aptamers targeting the insulin receptor through SELEX (Systematic Evolution of Ligands by EXponential Enrichment) using the purified extracellular domain (His 28-Lys 944) of the insulin receptor. The single-strand DNA libraries used for the SELEX consist of a 40 mer random region and both sides of 20 mer constant regions. To enhance the affinity and specificity of protein–aptamer interactions, thymine nucleobases in the 40 mer random regions were replaced with 5-[N-(1-naphthylmethyl)carboxamide]-2′-deoxyuridine (Nap-dU) (19). Moreover, we did not use any special selection conditions to develop conformation-selective aptamers.
After the SELEX, 41 different aptamers containing Nap-dU modifications were obtained. To identify PAM aptamers, we compared the binding of these aptamers to the insulin receptor in the absence and presence of insulin. Only one of these aptamers, named IR-A43F, showed increased binding affinity for the extracellular domain of the insulin receptor in the presence of insulin. The dissociation constant (Kd) of IR-A43F alone was ∼70 nM, which significantly decreased to 2.85 nM in the presence of insulin. IR-A43F did not bind to the insulin-like growth factor 1 (IGF-1) receptor in the presence or absence of insulin, even though the IGF-1 receptor has a high degree of structural similarity with the insulin receptor (Figure 1A).
Figure 1.

Binding affinity and truncation of IR-A43F. (A) The affinity of IR-A43F for the insulin receptor (IR) and insulin-like growth factor type 1 receptor (IGF-1R) was measured using a filter binding assay in the absence or presence of 100 nM insulin. The dissociation constant (Kd) was determined by fitting the data to a one-site saturation model. Data are presented as mean ± standard deviation (SD) of two independent replicates. (B) Comparison of the sequences of IR-A43F (full length) and IR-A43 (truncated core sequence). ‘P’ indicates 5-[N-(1-naphthylmethyl)carboxamide]-2'-deoxyuridine (Nap-dU).
The full-length sequence of IR-A43 (IR-A43F) consists of 81 nucleotides containing two 20 mer constant regions at each terminal and a 41 mer variable region for target binding. To determine the core sequence of IR-A43F, we compared the affinity of IR-A43F truncation variants containing 3′ or 5′ sequential deletions. We identified a 31 nucleotide sequence that is essential for IR-A43F binding to the receptor (Figure 1B). There was no significant difference in the binding of the full-length (IR-A43F) aptamer and the core sequence (IR-A43) to the insulin receptor (data not shown). Thus, all subsequent experiments were performed using the truncated 31 mer core sequence (IR-A43).
Insulin and IR-A43 have mutual positive cooperativity
To confirm the effect of insulin on IR-A43 binding at holo-receptors, we measured the binding of FITC-labeled IR-A43 to the human insulin receptor overexpressed in Rat-1 cells (Rat-1/hIR) using flow cytometry. FITC-labeled IR-A43 (100 nM) alone caused a 20.9% shift in the peak fluorescent intensity, which indicates that IR-A43 normally binds to the insulin receptor in the absence of insulin. Consistent with results from the in vitro binding assay, insulin significantly increased the binding of FITC-labeled IR-A43 to the insulin receptor in a concentration-dependent manner (Figure 2A). Co-treatment of FITC-labeled IR-A43 with either 2 nM, 10 nM, or 50 nM insulin gradually increased the peak shift to 28.3%, 51.4% and 74.1%, respectively. This result suggests that IR-A43 binds more favorably to the insulin-bound active conformation of the receptor than the inactive conformation without insulin.
Figure 2.

Mutual positive cooperativity of insulin and IR-A43. Flow cytometry was used to measure the binding of insulin and IR-A43 to the holo-insulin receptor. Rat-1 cells expressing the human insulin receptor (Rat-1/hIR) were incubated with FITC-labeled insulin (FITC-Ins) or FITC-labeled IR-A43 (FITC-A43). (A) Cells incubated with FITC-IR-A43 (100 nM) were stimulated with either 2 nM insulin, 10 nM insulin, or 50 nM insulin for 1 h at 4°C. (B) Cells incubated with FITC-labeled insulin (100 nM) were stimulated with either 10 nM IR-A43, 50 nM IR-A43, or 250 nM IR-A43 for 1 h at 4°C.
Conversely, we measured the effect of IR-A43 on insulin binding to Rat-1/hIR cells using FITC-labeled insulin and observed that IR-A43 significantly increased the binding of insulin (Figure 2B). FITC-labeled insulin (100 nM) alone shifted the peak fluorescent intensity by 16.7%, and co-treatment with either 10, 50 or 250 nM IR-A43 increased the peak shift to 20.5%, 38.3% and 66.6%, respectively. Furthermore, we investigated insulin binding using a wide range of insulin concentrations (3.9 nM–4 μM) in the absence or presence of IR-A43, and showed that the presence of IR-A43 dramatically improved insulin binding (Supplementary Figure S1). Taken together, these results indicate that insulin and IR-A43 exhibit mutual positive cooperativity when binding to the insulin receptor.
IR-A43 allosterically enhances insulin signaling
Binding of insulin to the insulin receptor induces autophosphorylation of tyrosine residues in the intracellular domains, which initiates a signaling cascade of downstream proteins such as IRS (insulin receptor substrate), AKT (AKT8 virus oncogene cellular homolog), SHC (SH2-containing collagen-related proteins), and ERK (extracellular signal-regulated kinases) (20,21). Therefore, we evaluated whether the mutual positive cooperativity between insulin and IR-A43 can modulate insulin-induced receptor autophosphorylation and downstream signaling. Even though IR-A43 can bind to the insulin receptor in the absence of insulin, IR-A43 alone had no effect on the autophosphorylation of the insulin receptor in Rat-1/hIR cells. However, co-stimulation of the insulin receptor with insulin and IR-A43 amplified autophosphorylation of the insulin receptor. Moreover, the reversed sequence of IR-A43 (IR-A43-R) did not alter insulin-induced autophosphorylation of the insulin receptor, which indicates that the insulin-enhancing effect of IR-A43 is not caused by a non-specific interaction with oligonucleotides. In our previous study, we found that an allosteric agonist aptamer for the insulin receptor preferentially stimulates Y1150 phosphorylation in the kinase domain, and also has biased activity towards the IRS–AKT pathway (18). However, the phosphorylation of tyrosine residues was enhanced to a similar degree by IR-A43 without significant bias (Figure 3A), and the level of phosphorylation was dependent upon the concentration of IR-A43 (Figure 3B). The phosphorylation of downstream signaling proteins was also amplified when insulin and IR-A43 were bound to the insulin receptor, as was receptor autophosphorylation (Figure 3C).
Figure 3.
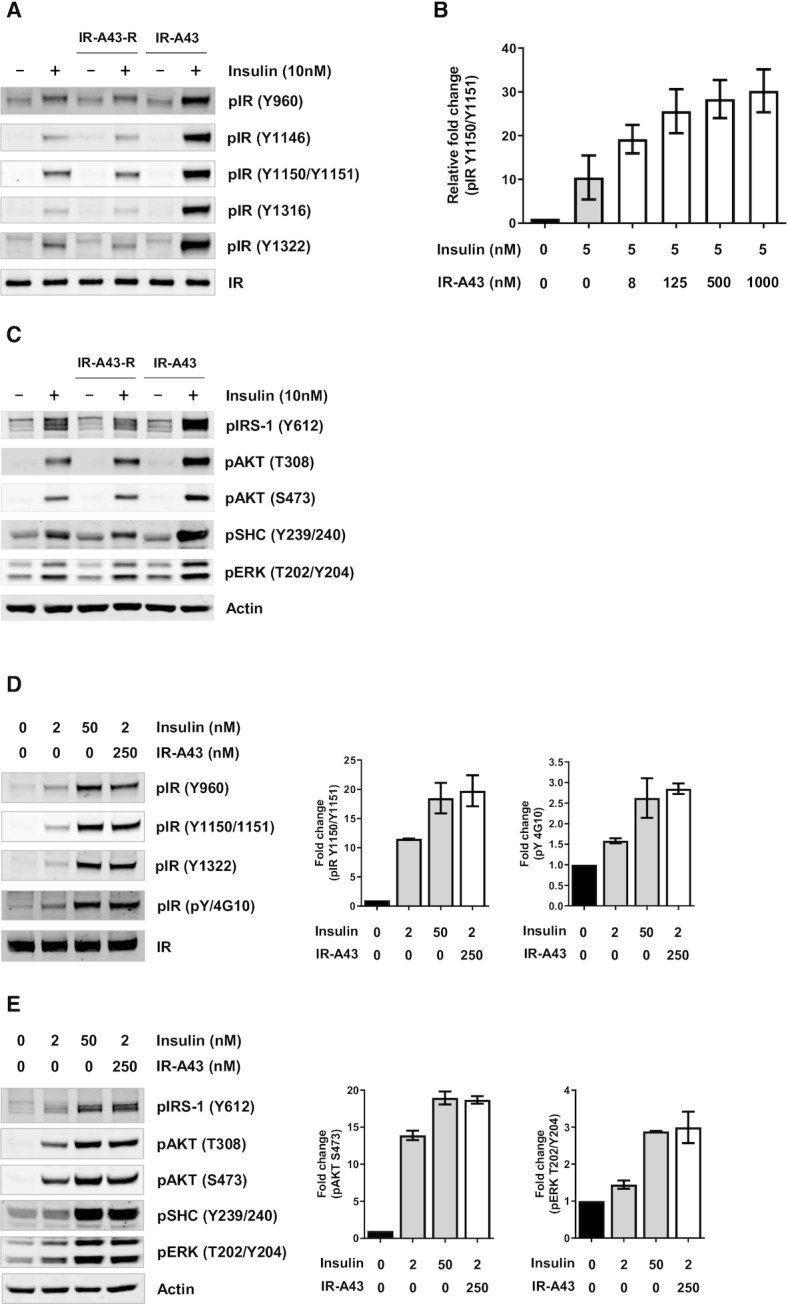
IR-A43 potentiates autophosphorylation of the insulin receptor and downstream insulin signaling. Rat-1 cells expressing the human insulin receptor (Rat-1/hIR) were co-stimulated with 10 nM insulin and 500 nM IR-A43 for 10 min. (A) Site-specific phosphorylation of tyrosine residues of the insulin receptor and (C) phosphorylation of molecules involved in downstream insulin signaling, such as pIRS-1, pAKT, pSHC and pERK, was detected by western blotting. (B) Rat-1/hIR cells were co-stimulated with 5 nM insulin and increasing concentrations of IR-A43 for 10 min; phosphorylation of the hIR (Y1150/1151) was detected by western blotting. IR-A43-R is an aptamer with the reversed sequence of IR-A43 (5′-CAGCGACPPACGGCPAPGACGCCPAPGTCCG-3′). (D, E) To quantitatively evaluate the insulin-enhancing activity of IR-A43, Rat-1/hIR cells were stimulated with a low concentration of insulin (2 nM) or a high concentration of insulin (50 nM), or were co-stimulated with a low concentration of insulin and IR-A43 (250 nM). All bar graphs are presented as mean ± standard deviation (SD) of two independent replicates.
To further evaluate the insulin-enhancing activity of IR-A43, we compared the effects of a low concentration of insulin enhanced by IR-A43 with the effects of a high concentration of insulin alone. When Rat-1/hIR cells were co-treated with 2 nM insulin and 250 nM IR-A43, the level of phosphorylation of the insulin receptor and downstream signaling molecules were similar to the level of phosphorylation produced by 50 nM insulin alone (Figure 3D, E). The mutual positive cooperativity between insulin and IR-A43 indicates that IR-A43 binds at a site that is distinct from the insulin binding site. Therefore, we conclude that IR-A43 is a PAM that amplifies the activity of insulin by stabilizing the binding of insulin to its receptor.
IR-A43 is specific for the human insulin receptor
Glucose uptake is a major function of the insulin receptor in peripheral tissues. Insulin signaling facilitates the translocation of GLUT4 (glucose transporter 4) to the plasma membrane in skeletal muscle and adipose tissue, which increases absorption of glucose from the blood into cells (22). To investigate the insulin-enhancing activity of IR-A43 on glucose uptake, we measured 2-deoxy-[14C]-glucose uptake in fully differentiated 3T3-L1 adipocytes. However, IR-A43 (500 nM) had no effect on glucose uptake induced by insulin (20 nM) (Figure 4A). This result was unexpected, given that IR-A43 (250 nM) increased the activity of insulin (2 nM) by ∼25-fold in Rat-1/hIR cells (Figure 3D).
Figure 4.
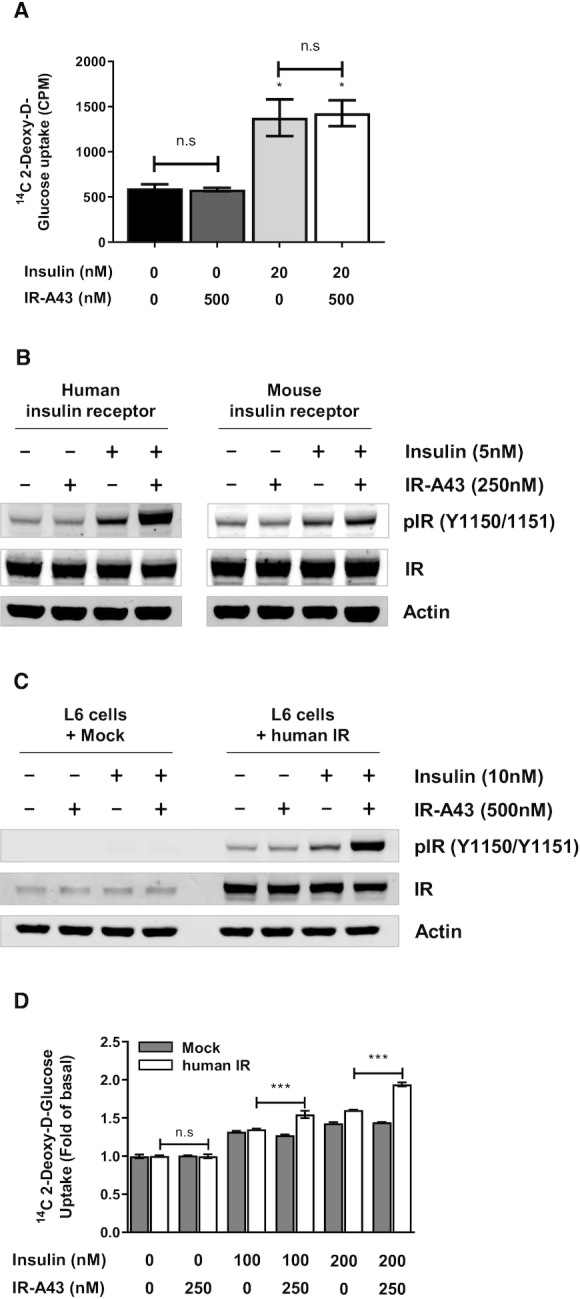
IR-A43 does not potentiate the activity of the mouse insulin receptor. (A) Fully differentiated 3T3-L1 adipocytes were used to measure insulin-stimulated glucose uptake. Cells were stimulated with 500 nM IR-A43, 20 nM insulin, or 20 nM insulin with 500 nM IR-A43 for 30 min. Data are presented as mean ± standard deviation (SD) of three biological replicates. (B) The human or mouse insulin receptor was transfected into CHO-K1 cells using Lipofectamine 3000 reagent. After 72 h, the cells were stimulated with 250 nM IR-A43, 5 nM insulin or 5 nM insulin with 250 nM IR-A43. (C) The human insulin receptor was transfected into L6 myoblast cells using Lipofectamine 3000 reagent. After 72 h, the cells were stimulated with 500 nM IR-A43, 10 nM insulin, or 10 nM insulin with 500 nM IR-A43. (D) L6 cells transfected with the human insulin receptor were used to measure insulin-stimulated glucose uptake. Cells were stimulated with 250 nM IR-A43, 100 nM insulin, 100 nM insulin with 250 nM IR-A43, 200 nM insulin, or 200 nM insulin with 250 nM IR-A43 for 30 min. Data are presented as mean ± standard deviation (SD) of three biological replicates. P-values were determined by one-way ANOVA followed by Tukey's multiple comparisons test (***P < 0.001 and ns, not significant).
3T3-L1 adipocytes used for the glucose uptake assay originates from mouse, whereas Rat-1/hIR cells used for the western blotting studies express the human insulin receptor. The amino acid sequences of the extracellular domain of human (hIR) and mouse (mIR) insulin receptors are highly homologous (95.9% residue identity), but not identical (Supplementary Figure S2). Thus, we hypothesized that the amino acid sequence of the IR-A43 binding site may not be present in the mIR, thus making IR-A43 specific for the hIR. To verify the species specificity of IR-A43, we transiently overexpressed hIR or mIR in CHO-K1 cells, and observed receptor autophosphorylation. Consistent with our expectation, IR-A43 did not enhance insulin-induced autophosphorylation of mIR (Figure 4B).
To confirm that IR-A43 can increase glucose uptake by enhancing hIR activation, we transfected hIRs into L6 myoblast cells. Consistent with results from Rat-1/hIR cells, IR-A43 normally enhanced insulin-induced autophosphorylation of hIRs in L6 cells (Figure 4C). Moreover, IR-A43 did not change insulin-induced glucose uptake in L6 cells not expressing hIRs, but insulin-induced glucose uptake increased in L6 cells overexpressing hIRs co-stimulated with IR-A43 (Figure 4D).
Residue Q272 is a hotspot for the insulin-enhancing activity of IR-A43
From a structural point of view, the species specificity of IR-A43 is useful to identify the binding site of IR-A43 and to understand the mechanism of the insulin-enhancing activity. There are 35 of mismatched amino acids between the ectodomains of hIR and mIR. We separated these amino acids into seven parts based on domains, and made hybrid hIRs containing each part of the mIR (Table 1, Figure 5A). IR-A43 enhanced insulin-induced autophosphorylation of each hybrid hIR receptor except for the hybrid hIR containing part 2 of the mIR (Figure 5B). To confirm that the amino acid sequence of part 2 of hIR is crucial for the activity of IR-A43, we made a hybrid mIR containing part 2 of the hIR, and showed that IR-A43 enhanced the activity of insulin in this hybrid (Figure 5C). These results demonstrate that at least one of the five residues located in part 2 (H264, K267, R271, Q272 and L300) of the hIR is directly involved in IR-A43 binding.
Table 1.
Summary of amino acids that differ between the ectodomains of human and mouse insulin receptors
| Residue Number | Human | Mouse | Domain | Part |
|---|---|---|---|---|
| 158 | I | V | CR | Part 1 |
| 210 | S | K | ||
| 218 | Q | E | ||
| 236 | R | Q | ||
| 264 | H | F | Part 2 | |
| 267 | K | R | ||
| 271 | R | K | ||
| 272 | Q | P | ||
| 300 | L | M | ||
| 313 | H | Q | L2 | Part 3 |
| 314 | L | I | ||
| 367 | Y | F | ||
| 386 | R | H | ||
| 477 | Y | F | FnIII-1 | Part 4 |
| 538 | L | Q | ||
| 547 | N | S | ||
| 631 | F | Y | FnIII-2α + ID-α | Part 5 |
| 665 | E | D | ||
| 676 | E | D | ||
| 680 | G | S | ||
| 726 | G | E | ID-β | Part 6 |
| 727 | D | E | ||
| 733 | V | A | ||
| 734 | A | T | ||
| 735 | V | T | ||
| 736 | P | L | ||
| 738 | V | L | ||
| 739 | A | P | ||
| 740 | A | D | ||
| 744 | T | V | ||
| 748 | S | I | ||
| 753 | P | Q | ||
| 790 | T | S | FnIII-2β + FnIII-3 | Part 7 |
| 792 | E | D | ||
| 886 | I | V |
Figure 5.

Residue Q272 in the CR domain is critically involved in the insulin-enhancing activity of IR-A43. (A) Schematic representation of the domain substitution between the human insulin receptor (hIR) and mouse insulin receptor (mIR) used to make hybrid hIRs containing portions of the mIR. (B) Phosphorylation of hybrid hIRs containing part 1, part 2, part 3, part 4, part 5, part 6, or part 7 of mIR. (C) Phosphorylation of a hybrid mIR containing part 2 of hIR. (D) Phosphorylation of a hIR mutant (R271K/Q272P) in which residue R271 was substituted to K271 and residue Q272 was substituted to P272. (E) Phosphorylation of hIR mutants in which R271 was substituted to K271 (R271K) or residue Q272 was substituted to P272 (Q272P). (F) Phosphorylation of a mIR mutant (P272Q) in which P272 was substituted to Q272. (G) The relative fold-change of insulin receptor phosphorylation of data presented in (F). Data are presented as mean ± standard deviation (SD) of two independent replicates. P-values were determined by one-way ANOVA followed by Tukey's multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, and ns, not significant). mIR or hIR DNAs were transfected into CHO-K1 cells using Lipofectamine 3000 reagent. Cells were stimulated with 5 nM insulin or 5 nM insulin with IR-A43 250 nM for 10 min.
To clarify which residues are directly associated with IR-A43 binding, we used three hIR mutants containing residues from part 2 of the mIR (H264F/K267R, R271K/Q272P, and L300M), and observed that the insulin-enhancing activity was impaired only in the hIR-R271K/Q272P mutant (Figure 5D, Supplementary Figure S3). Next, we evaluated the effect of R271K and Q272P mutations, and found that the Q272P mutation significantly impaired the insulin-enhancing activity of IR-A43 at the hIR (Figure 5E). Furthermore, the activity of IR-A43 was assessed in a mIR mutant (P272Q) in which residue P272 was replaced with residue Q272 of hIR. Consistent with the results of the hIR mutant (Q272P), this replacement recovered the insulin-enhancing activity of IR-A43 at the mIR (Figure 5F, G). Therefore, we conclude that residue Q272 located in the CR domain of hIR is directly involved in the binding of IR-A43 to the hIR, and the region containing Q272 is a hotspot for enhancing insulin activity by stabilizing insulin binding.
DISCUSSION
In this study, we developed the first PAM aptamer that potentiates the activation of a receptor by stabilizing the binding of a ligand. IR-A43 cannot activate the insulin receptor in the absence of insulin; rather the aptamer binds more favorably to the active, insulin-bound conformation of the receptor. Consequently, IR-A43 can amplify the activation of the insulin receptor by stabilizing the binding of insulin to its receptor. The mechanism of action of IR-A43 implies that the aptamer has high structural binding specificity to discriminate conformational changes between the active and inactive conformations of the target protein. Therefore, our study suggests that aptamers have the potential to be developed into allosteric modulators that recognize a specific conformation of a target protein.
The majority of aptamers developed to date are antagonists that simply disturb the functional interaction between a receptor and its ligand (23); only a few aptamers that act as agonists to activate receptor function have been reported. However, not all agonistic aptamers activate their target receptor by inducing a conformational change in their receptor upon binding. A monomer of these aptamers is devoid of agonistic activity when binding to a receptor. However, a bivalent form of the aptamers (dimeric aptamers) can initiate receptor activation by inducing receptor dimerization (13–16). To date, two aptamers have been reported to a receptor without the formation of an aptamer dimer, and only one of them functions as an allosteric agonist at its target receptor (17,18). Therefore, the insulin-enhancing activity of IR-A43, without inherent agonism, is a unique feature of this aptamer.
To understand the mechanism by which IR-A43 enhances the activity of insulin, the conformational change in the region containing residue Q272 should be elucidated. The structure of the inactive ectodomain of the insulin receptor was revealed by X-ray crystallography, which demonstrated that each α–β monomer is dimerized into a symmetric inverted ‘V’ shape (24,25). Recently, the active (insulin-bound) ectodomain structure was analyzed by single-particle cryo-electron microscopy (26). At a physiological insulin concentration, one insulin molecule binds to an insulin receptor, which induces the receptor into an asymmetric inverted ‘L’ shape by translocating the L1 domain towards the FnIII-1 (Fibronectin type III-1) domain of the opposite monomer (Figure 6A). The CR domain containing residue Q272 is located between two leucine-rich repeat domains (L1 and L2) (Figure 6B, C). The superimposition of the inactive and active structure of the L1–CR–L2 domains indicates that the L1–CR domains do not undergo substantial conformational changes upon insulin binding to the receptor (Figure 6D). After binding of insulin, the boundary between the L2 and CR domains (∼P303–P307) acts like a hinge, which brings the L1 and L2 domains close to each other (Figure 6D). In the inactive conformation, residue Q272 is located very close to part of the L2 domain (H313, L315 and E316) (Figure 6E). However, the insulin-induced conformational change leads to translocation of the L2 domain, which exposes residue Q272 on the outside of the structure (Figure 6F). Therefore, we suggest that insulin-mediated exposure of residue Q272 makes IR-A43 more favorably bind to the active conformation of the receptor than the inactive conformation without insulin. Moreover, when IR-A43 is bound to residue Q272, it may act as a ‘wedge’ to stabilize insulin binding by fixing the position of the L2 domain in the active conformation (Figure 6G). Our model is a plausible explanation for the mutual positive cooperativity that occurs when insulin and IR-A43 bind to the insulin receptor. To confirm this model, further studies of the structure of the insulin receptor and IR-A43 complex are required.
Figure 6.

Structural insight into the insulin-enhancing activity of IR-A43 based on insulin receptor structures in the Protein Data Bank (PDB). (A) Comparison between the inactive conformation (PDB ID: 4ZXB) and active conformations (PDB ID: 6HN4, 6HN5) of the ectodomain of the insulin receptor. The location of residue Q272 in L1–CR–L2 domains of (B) the inactive conformation (PDB ID: 4ZXB) and (C) active conformation (PDB ID: 6HN5). (D) Structural superimposition of L1–CR–L2 domains of the inactive conformation and the active conformation. A detailed view of the conformational change in the region close to residue Q272 between (E) the inactive conformation and (F) the active conformation of the insulin receptor. (G) A structural model for mutual positive cooperativity of insulin and IR-A43 at the insulin receptor.
According to our model, the insulin receptor is in the inactive state in the absence of insulin, which is an unfavorable condition for IR-A43 to bind to residue Q272. Therefore, it raises the question of how we were able to identify IR-A43 despite not using special selection conditions to expose residue Q272 on the outside of the structure. One plausible explanation is that the identification of IR-A43 may have resulted from structural heterogeneity of the recombinant extracellular domain of insulin receptor in the samples used for SELEX. To study the structure of the extracellular domain of the insulin receptor using X-ray crystallography, two antibodies that bind to the extracellular domain must be used for protein crystallization (24,25). Moreover, single-particle cryo-electron microscopy studies reveal that the extracellular domain shows a high degree of heterogeneity in the absence of insulin (26,27). Taken together, these results imply that the conformation of the extracellular domain without the transmembrane and intracellular domains is unstable and heterogeneous, which will make it difficult for the extracellular domain to constantly remain in the inactive conformation. Therefore, we assume that among heterogeneous structures in the SELEX samples, there were structures in which Q272 was exposed on the extracellular domain.
The monoclonal antibody XMetS is a PAM for the insulin receptor, similar to IR-A43 (28). XMetS increases the binding of insulin to the insulin receptor, and potentiates insulin-induced receptor autophosphorylation and downstream signaling. Unlike IR-A43, XMetS is not species-specific, and enhances the activation of both human and mouse insulin receptors. This finding indicates that the binding site of IR-A43 and its mechanism of action may differ from XMetS. The binding site of XMetS has not yet been reported, so it is difficult to directly compare the mechanisms of IR-A43 and XMetS.
Finding a way to effectively activate insulin signaling in peripheral tissues for the treatment of diabetes mellitus has been an active area of research for many years (29). Insulin therapy, in which exogenous insulin is administered to patients with diabetes, is the most effective way to decrease blood glucose levels but can also cause hypoglycemia due to the difficulty in controlling the concentration of injected insulin (30). One advantage of using PAMs as a drug is that they will only exert their effects in the presence of an endogenous ligand. In this way, PAMs can be used to precisely control receptor activity according to the concentration of endogenous ligands, which reduces the risk of adverse effects caused by drug overdose (4). Therefore, we anticipate that further investigations into PAMs of the insulin receptor, such as IR-A43, and hotspots on the insulin receptor associated with the insulin-enhancing activity of PAMs, will provide a solution to hypoglycemia caused by excessive insulin administration.
Supplementary Material
Contributor Information
Na-Oh Yunn, Department of Life Sciences, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Mangeun Park, School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Seongeun Park, Postech Biotech Center, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Jimin Lee, Department of Life Sciences, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Jeongeun Noh, Department of Life Sciences, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
Euisu Shin, Aptamer Sciences, Inc., Seongnam 13605, Republic of Korea.
Sung Ho Ryu, Department of Life Sciences, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea; School of Interdisciplinary Bioscience and Bioengineering, Pohang University of Science and Technology (POSTECH), Pohang 37673, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Global Research Laboratory (GRL) Program [NRF-2016K1A1A2912722]; Bio & Medical Technology Development Program [NRF-2017M3A9F6029753] through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT. Funding for open access charge: Grobal Research Laboratory (GRL) Program [NRF-2016K1A1A2912722]; Bio & Medical Technology Development Program [NRF-2017M3A9F6029753] through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT.
Conflict of interest statement. None declared.
REFERENCES
- 1. De Smet F., Christopoulos A., Carmeliet P.. Allosteric targeting of receptor tyrosine kinases. Nat. Biotechnol. 2014; 32:1113–1120. [DOI] [PubMed] [Google Scholar]
- 2. Changeux J.P., Christopoulos A.. Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell. 2016; 166:1084–1102. [DOI] [PubMed] [Google Scholar]
- 3. Nussinov R., Tsai C.J.. Allostery in disease and in drug discovery. Cell. 2013; 153:293–305. [DOI] [PubMed] [Google Scholar]
- 4. Wootten D., Christopoulos A., Sexton P.M.. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 2013; 12:630–644. [DOI] [PubMed] [Google Scholar]
- 5. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 6. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 7. Long S.B., Long M.B., White R.R., Sullenger B.A.. Crystal structure of an RNA aptamer bound to thrombin. RNA. 2008; 14:2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gelinas A.D., Davies D.R., Edwards T.E., Rohloff J.C., Carter J.D., Zhang C., Gupta S., Ishikawa Y., Hirota M., Nakaishi Y. et al.. Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J. Biol. Chem. 2014; 289:8720–8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jarvis T.C., Davies D.R., Hisaminato A., Resnicow D.I., Gupta S., Waugh S.M., Nagabukuro A., Wadatsu T., Hishigaki H., Gawande B. et al.. Non-helical DNA triplex forms a unique aptamer scaffold for high affinity recognition of nerve growth factor. Structure. 2015; 23:1293–1304. [DOI] [PubMed] [Google Scholar]
- 10. Kahsai A.W., Wisler J.W., Lee J., Ahn S., Cahill Iii T.J., Dennison S.M., Staus D.P., Thomsen A.R., Anasti K.M., Pani B. et al.. Conformationally selective RNA aptamers allosterically modulate the β2-adrenoceptor. Nat. Chem. Biol. 2016; 12:709–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexaki A., Hettiarachchi G.K., Athey J.C., Katneni U.K., Simhadri V., Hamasaki-Katagiri N., Nanavaty P., Lin B., Takeda K., Freedberg D. et al.. Effects of codon optimization on coagulation factor IX translation and structure: implications for protein and gene therapies. Sci. Rep. 2019; 9:15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zichel R., Chearwae W., Pandey G.S., Golding B., Sauna Z.E.. Aptamers as a sensitive tool to detect subtle modifications in therapeutic proteins. PLoS One. 2012; 7:e31948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramaswamy V., Monsalve A., Sautina L., Segal M.S., Dobson J., Allen J.B.. DNA aptamer assembly as a vascular endothelial growth factor receptor agonist. Nucleic Acid Ther. 2015; 25:227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soldevilla M.M., Villanueva H., Bendandi M., Inoges S., Lopez-Diaz de Cerio A., Pastor F.. 2-Fluoro-RNA oligonucleotide CD40 targeted aptamers for the control of B lymphoma and bone-marrow aplasia. Biomaterials. 2015; 67:274–285. [DOI] [PubMed] [Google Scholar]
- 15. Ueki R., Atsuta S., Ueki A., Hoshiyama J., Li J., Hayashi Y., Sando S.. DNA aptamer assemblies as fibroblast growth factor mimics and their application in stem cell culture. Chem. Commun. 2019; 55:2672–2675. [DOI] [PubMed] [Google Scholar]
- 16. Ueki R., Uchida S., Kanda N., Yamada N., Ueki A., Akiyama M., Toh K., Cabral H., Sando S.. A chemically unmodified agonistic DNA with growth factor functionality for in vivo therapeutic application. Science advances. 2020; 6:eaay2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang Y.Z., Hernandez F.J., Gu B., Stockdale K.R., Nanapaneni K., Scheetz T.E., Behlke M.A., Peek A.S., Bair T., Giangrande P.H. et al.. RNA aptamer-based functional ligands of the neurotrophin receptor, TrkB. Mol. Pharmacol. 2012; 82:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yunn N.O., Koh A., Han S., Lim J.H., Park S., Lee J., Kim E., Jang S.K., Berggren P.O., Ryu S.H.. Agonistic aptamer to the insulin receptor leads to biased signaling and functional selectivity through allosteric modulation. Nucleic Acids Res. 2015; 43:7688–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lollo B., Steele F., Gold L.. Beyond antibodies: new affinity reagents to unlock the proteome. Proteomics. 2014; 14:638–644. [DOI] [PubMed] [Google Scholar]
- 20. Youngren J.F. Regulation of insulin receptor function. Cell. Mol. Life Sci.: CMLS. 2007; 64:873–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haeusler R.A., McGraw T.E., Accili D.. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018; 19:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leto D., Saltiel A.R.. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012; 13:383–396. [DOI] [PubMed] [Google Scholar]
- 23. Zhou J., Rossi J.. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017; 16:181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKern N.M., Lawrence M.C., Streltsov V.A., Lou M.Z., Adams T.E., Lovrecz G.O., Elleman T.C., Richards K.M., Bentley J.D., Pilling P.A. et al.. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006; 443:218–221. [DOI] [PubMed] [Google Scholar]
- 25. Croll T.I., Smith B.J., Margetts M.B., Whittaker J., Weiss M.A., Ward C.W., Lawrence M.C.. Higher-Resolution structure of the human insulin receptor Ectodomain: Multi-Modal inclusion of the insert domain. Structure. 2016; 24:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weis F., Menting J.G., Margetts M.B., Chan S.J., Xu Y., Tennagels N., Wohlfart P., Langer T., Müller C.W., Dreyer M.K. et al.. The signalling conformation of the insulin receptor ectodomain. Nat. Commun. 2018; 9:4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scapin G., Dandey V.P., Zhang Z., Prosise W., Hruza A., Kelly T., Mayhood T., Strickland C., Potter C.S., Carragher B.. Structure of the insulin receptor-insulin complex by single-particle cryo-EM analysis. Nature. 2018; 556:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corbin J.A., Bhaskar V., Goldfine I.D., Bedinger D.H., Lau A., Michelson K., Gross L.M., Maddux B.A., Kuan H.F., Tran C. et al.. Improved glucose metabolism in vitro and in vivo by an allosteric monoclonal antibody that increases insulin receptor binding affinity. PLoS One. 2014; 9:e88684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boucher J., Kleinridders A., Kahn C.R.. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014; 6:a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCall A.L. Insulin therapy and hypoglycemia. Endocrinol. Metab. Clin. North Am. 2012; 41:57–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.