


Abstract
The sequence of tRNAs is submitted to evolutionary constraints imposed by their multiple interactions with aminoacyl-tRNA synthetases, translation elongation factor Tu in complex with GTP (EF-Tu•GTP), and the ribosome, each being essential for accurate and effective decoding of messenger RNAs. In Staphylococcus aureus, an additional constraint is imposed by the participation of tRNAGly isoacceptors in the addition of a pentaglycine side chain to cell-wall peptidoglycan precursors by transferases FmhB, FemA and FemB. Three tRNAGly isoacceptors poorly interacting with EF-Tu•GTP and the ribosome were previously identified. Here, we show that these ‘non-proteogenic’ tRNAs are preferentially recognized by FmhB based on kinetic analyses and on synthesis of stable aminoacyl-tRNA analogues acting as inhibitors. Synthesis of chimeric tRNAs and of helices mimicking the tRNA acceptor arms revealed that this discrimination involves identity determinants exclusively present in the D and T stems and loops of non-proteogenic tRNAs, which belong to an evolutionary lineage only present in the staphylococci. EF-Tu•GTP competitively inhibited FmhB by sequestration of ‘proteogenic’ aminoacyl-tRNAs in vitro. Together, these results indicate that competition for the Gly-tRNAGly pool is restricted by both limited recognition of non-proteogenic tRNAs by EF-Tu•GTP and limited recognition of proteogenic tRNAs by FmhB.
INTRODUCTION
Aminoacyl-tRNAs are used as a source of activated amino acids for non-ribosomal synthesis of various biomolecules, including cell-wall peptidoglycan precursors (Figure 1A) (1), cyclodipeptides (2) and membrane lipids (3). In addition, Phe-tRNA and Leu-tRNA participate in protein aminoacylation, thereby triggering them for protein degradation by the proteasome (4). In Staphylococcus aureus, three aminoacyl-transferases of the Fem family (FmhB, FemA and FemB) use five aminoacyl-tRNAs to assemble a pentaglycine side chain onto peptidoglycan precursors (5–7). FmhB adds the first Gly whereas FemA and FemB each adds two residues. FmhB is an essential enzyme indicating that the complete absence of the peptidoglycan side chain is not compatible with synthesis of an osmoprotective peptidoglycan layer and bacterial growth (7). FemA and FemB are dispensable for growth in media of high osmolarity (8) and their absence is not compatible with expression of methicillin resistance mediated by the peptidoglycan transpeptidase PBP2a (9). In fact, the ‘Fem’ designation originates from early investigations based on random transposon mutagenesis that showed that the femA and femB genes encode factors essential for methicillin resistance (10). The essential role of the Fem transferases for growth or for methicillin resistance indicates that these enzymes are potential targets to develop drugs active on multidrug resistant S. aureus (11–13).
Figure 1.

Participation of tRNAs to peptidoglycan synthesis in Staphylococcus aureus. (A) Peptidoglycan assembly pathway. Aminoacyl-transferases FmhB, FemA and FemB assemble a pentaglycine side chain. The essential role of FmhB (for growth) and of FemA and FemB (for methicillin resistance) stems from the fact that the amino group at the extremity of the pentaglycine side chain is used for peptidoglycan cross-linking in the last step of peptidoglycan polymerization. (B) Alignment of S. aureus tRNAGly isoacceptors. P1 and P2, proteogenic isoacceptors; NP1, NP2 and NP3, non-proteogenic isoacceptors. Stars indicate the position of invariant nucleotides in the five sequences. Bases highlighted in blue and red are divergent within the proteogenic and non-proteogenic tRNA groups, respectively. The anticodon is highlighted in grey. (C) Cloverleaf representation of the five tRNAGly isoacceptors. Two arrows point to positions 8 and 19 corresponding to potential sites of posttranscriptional modification of non-proteogenic tRNA as discussed in the text (4-thiouridine and 5,6-dihydrouridine, respectively). The remaining arrow points to position 32 that determines the capacity of the unmodified UCC anticodon to decode all four tRNAGly. The UUC or UGC triplet in the T loop is boxed in red.
The participation of aminoacyl-tRNAs to non-ribosomal synthesis implies that the same aminoacyl-tRNAs are potentially used for protein and peptidoglycan synthesis. Alternatively, the sequence of tRNAGly isoacceptors may have divergently evolved so that particular aminoacyl-tRNAs are preferentially or exclusively used in either biosynthetic pathway. The chromosome of S. aureus harbors seven tRNAGly genes encoding isoacceptors with five distinct sequences (Figure 1B and C) (14,15). Previous analyses based on affinity chromatography showed that two of them, tRNAGly(GCC-1) and tRNAGly(UCC-1), display high affinity for the binary complex comprising the translation elongation factor Tu and GTP (EF-Tu•GTP) following acylation by the glycyl-tRNA synthetase (GlyRS), as expected for proteogenic aminoacyl-tRNAs (15). These tRNAs were designated P1 (GCC anticodon) and P2 (UCC-1) for proteogenic tRNAs. The remaining acylated isoacceptors (UCC-2, UCC-3 and UCC-4) eluted at low salt concentrations suggesting that they bind to EF-Tu•GTP with low affinity and could therefore preferentially participate in peptidoglycan synthesis. The tRNAGly(UCC-2), tRNAGly(UCC-3) and tRNAGly(UCC-4) were thus referred to as non-proteogenic isoacceptors NP1, NP2, and NP3, respectively. Analyses performed in the 60s and 70s on tRNAs purified from cultures of staphylococci resolved highly purified fractions containing non-proteogenic tRNAGly in S. aureus (16) and in closely related Staphylococcus epidermidis (17–20). The tRNAs present in these fractions displayed no binding affinity for ribosomes programmed with any of the four triplets specifying Gly and were unable to participate in in vitro protein synthesis (16–18). In addition, the non-proteogenic tRNA purified from S. epidermidis did not form a complex with EF-Tu•GTP (21), as also found for homologous tRNAs from S. aureus obtained by in vitro transcription in a more recent study (15). Two non-proteogenic tRNAs from S. epidermidis (tRNAGly Ia and Ib) were sequenced by biochemical methods (19) and were found to contain an unusually low content in modified bases (19,20), which was limited to one 4-thiouridine residue (position 8) and one 5,6-dihydrouridine residue (position 19) (Figure 1C). The absence of pseudouridine, which is ubiquitous in the T loop, was noteworthy. In agreement, the DNA sequence of the three non-proteogenic tRNAGly genes contains the TGC or TGT triplet in the T loop instead of the TTC triplet in proteogenic tRNAs (red box, Figure 1C) in which the second nucleotide corresponds to the position of the post-transcriptional modification of uridine to pseudouridine (14,15). The presence of an additional base-pairing in the anticodon stem of P1-GCC (seven instead of six in the three NP-UCCs) and the corresponding reduction in the number of bases in the anticodon loop (three in P1 instead of five in non-proteogenic tRNAGly) is also worth noting (22). The unusual features of the non-proteogenic tRNAGly were rationalized in terms of impaired interactions with EF-Tu and the ribosome [see (22) for discussion].
The interaction of the five S. aureus tRNAGly isoacceptors with components of the translation machinery has deserved much attention. In contrast, little is known on the recognition of the isoacceptors by the Fem transferases. There is a consensus for the potential participation of the five tRNAGly to peptidoglycan synthesis in S. aureus and S. epidermidis but the analyses were only qualitative (17,18). In this study, we determine whether FmhB preferentially uses specific tRNAGly isoacceptors for in vitro synthesis of peptidoglycan precursors. A positive answer to this first question prompted us to construct chimeric tRNAs in order to identify the tRNA regions that enable FmhB to discriminate between tRNAGly isoacceptors. Investigation of the mode of tRNA recognition by FmhB was also based on the synthesis of stable Gly-tRNAGly analogues with various nucleotide sequences that behaved as potent FmhB inhibitors. Since competitive binding of aminoacyl-tRNAs to FmhB and EF-Tu•GTP is expected to be the key factor determining the partition of members of the Gly-tRNAGly pool between peptidoglycan and protein synthesis, a second assay was developed to compare the relative affinity of these proteins for the aminoacylated tRNA isoacceptors. The observed weak interaction of non-proteogenic Gly-tRNAGly with EF-Tu•GTP, in combination with phylogenic analyses of the tRNAGly sequences from firmicutes, led to the conclusion that staphylococci produce a set of peculiar tRNAGly paralogues that preferentially participate in peptidoglycan synthesis, thereby preventing the potentially negative impact of a competitive use of the same aminoacyl-tRNAs in protein and peptidoglycan synthesis.
MATERIALS AND METHODS
Synthesis of FmhB substrates
In S. aureus, FmhB catalyzes the transfer of a glycyl residue from Gly-tRNAGly to the undecaprenyl-PP-MurNAc(-pentapeptide)-GlcNAc peptidoglycan precursor (lipid II; Figure 1A). To study the catalytic activity of FmhB in vitro, we developed synthetic routes for analogues of these two substrates. The tRNA analogues were obtained by in vitro transcription of DNA templates obtained by PCR (Supplementary Material, section 2, Supplementary Tables S1 and S2, and Supplementary Figure S1). The lipid II analogues were obtained by extraction of the UDP-MurNAc-pentapeptide nucleotide precursor (Figure 1A) from the cytoplasm of S. aureus followed by in vitro transfer of the phospho-MurNAc-pentapeptide moiety to commercial phospho-di-prenyl or phospho-hepta-prenyl lipids using the purified transferase, MraY. GlcNAc was added to the resulting lipid I by the purified MurG transferase (Supplementary Material, sections 3 and 4). Hepta-prenyl- and di-prenyl-containing lipid II analogues were purified by butanol/pyridinium-acetate extraction (23) and by reverse-phase HPLC (24), respectively.
Synthesis of FmhB inhibitors
Stable analogues of Gly-tRNAGly were obtained by organic synthesis of dinucleotides pdCpA-2′-azido and pdCpA-3′-azido (25,26) (Supplementary Material, section 5 and Supplementary Figure S3), CuI-catalyzed cycloaddition of commercially available 1,8-nonadiyne (Sigma-Aldrich), and ligation to tRNA analogues devoid of the terminal CA dinucleotide using T4 RNA ligase (26) (Supplementary Material, section 6).
Enzyme assays
Enzymes were produced in Escherichia coli and purified by nickel-affinity and size-exclusion chromatography (Supplementary Material, section 3, and Supplementary Figure S2). The catalytic efficacy of FmhB, FemA and FemB was tested in a coupled assay involving acylation of tRNA analogues by a glycyl-tRNA synthetase (GlyRS), which was used at a sufficiently high concentration (800 nM) to maintain full tRNA acylation during the entire reaction (24) (Supplementary Material, sections 7 and 8). Under the assay conditions the Gly-tRNA concentration ([S]) was negligible compared to Km so that turnover was equal to kcat × [S]/Km. Turnover was determined using several enzyme concentrations. Dividing turnover by [S] provided an estimate of the catalytic efficacy (kcat/Km) of FmhB for a particular tRNA, a relevant parameter for comparison of tRNAs with different sequences. Competitive inhibition of FmhB by stable Gly-tRNAGly analogues was assayed with a fixed and non-saturating concentration of Gly-tRNAGly containing the NP2 sequence and various concentrations of stable Gly-tRNAGly analogues. The inhibition parameter Ki was determined by fitting the Morrison equation to experimental data (27). Inhibition of FmhB following sequestration of Gly-tRNAGly by formation of a complex with EF-Tu•GTP was tested in the presence of EF-Ts and a solution of GTP that was pretreated with phosphoenolpyruvate kinase to remove any GDP (Supplementary Material, section 9, and Supplementary Table S3).
Detection of FmhB•tRNA complexes by size-exclusion chromatography
Isoacceptors P1 or NP2 were incubated with a four-fold molar excess of FmhB and formation of complexes was determined by size-exclusion chromatography (Supplementary Material, section 10).
Assessment of the purity of compounds synthesized in this study
Chromatographic analyses of dinucleotides and RNAs are reported in sections 11 and 12 of the Supplementary Material, respectively.
RESULTS
Glycylation of the tRNAGly isoacceptors by the GlyRS glycyl-tRNA synthetase from S. aureus
Our first objective was to identify the conditions required for efficacious glycylation of tRNAs obtained by in vitro transcription and to determine the concentration of Gly-tRNAGly. The five tRNAGly isoacceptors of S. aureus contain the necessary identity determinants for glycylation, namely G1-C72, C2-G70 and U73 in the acceptor stem and C35 and C36 in the anticodon (in bold in Figure 1B) (28). Accordingly, and as previously described (15), the five isoacceptors generated by in vitro transcription were aminoacylated by the S. aureus GlyRS. We did not detect any significant difference in the yield or kinetics of acylation of the five isoacceptors (Supplementary Figure S4 and data not shown). A concentration of GlyRS of 0.8 μM was used in further experiments to ensure full acylation of the isoacceptors in the GlyRS-FmhB coupled assay. Data presented in Supplementary Figure S4 also showed that increasing concentrations of GlyRS led to plateaus in the amounts of [14C]Gly-tRNAGly that provided an accurate quantification of the tRNAGly isoacceptors obtained by in vitro transcription.
Relative efficacy of FmhB for glycyl transfer from the five aminoacylated tRNAGly isoacceptors to peptidoglycan precursors
The GlyRS-FmhB coupled assay was used to investigate the impact of differences in the tRNAGly isoacceptor sequences on the catalytic activity of FmhB. Addition of [14C]Gly to the hepta- and di-prenyl analogues of lipid intermediate II (lipid II) was determined by thin-layer chromatography followed by scintillation counting of radioactive spots identified by autoradiography (Figures 2 and 3; Table 1).
Figure 2.

Impact of the tRNA sequence on the catalytic activity of FmhB in the coupled assay using the hepta-prenyl-containing lipid II analogue as substrate. (A and B) Representative autoradiographies of thin-layer chromatograms for kinetic analyses of FmhB with isoacceptors P1 and NP2, respectively. The arrow heads labeled with the letters a and b indicate the position of the radio-labeled product of the reaction ([14C]Gly-lipid II analogue) and of the [14C]Gly substrate, respectively. The arrow indicates the direction of migration. (C–G) Determination of FmhB activity with various tRNAs. For each isoacceptor, the top panel shows kinetics with three concentrations of FmhB using a fixed concentration of isoacceptor (0.2 μM for P1 and P2; 0.1 μM for NP1; 0.05 μM for NP2 and NP3). Preliminary investigations (data not shown) showed that these concentrations were rate limiting. The values of the velocity (V) of the formation of the radio-labeled product of the reactions ([14C]Gly-lipid II analogue) ± standard deviation were obtained by fitting a first order equation to the data obtained for each enzyme concentration. For each isoacceptor, the bottom panel shows the determination of the turnover (TO; V/FmhB concentration) ± standard deviation, which was obtained by fitting a first order equation to the data. Estimates of the efficacy of FmhB were obtained by dividing TO by the tRNA concentration.
Figure 3.

Impact of the tRNA sequence on the catalytic activity of FmhB in the coupled assay using the di-prenyl lipid II analogue as substrate. The coupled assay was performed with fixed concentrations of FmhB (20 nM), di-prenyl-containing lipid II analogue (60 μM), and tRNA isoacceptor (0.05 μM). Data are the mean ± standard deviation of three independent experiments for each tRNA. The rate of formation of the radio-labeled product of the reaction ([14C]Gly-lipid II analogue) and the standard error were determined by fitting a linear equation to the data. The rate was divided by the concentration of FmhB and that of the tRNA to provide the indicated estimate of the efficacy of FmhB for each isoacceptor.
Table 1.
FmhB efficacy for hepta- or di-prenyl- containing lipid II substrates
| FmhB activity (μM–1 min–1)a | |||
|---|---|---|---|
| tRNAGly sequence | Hepta-prenyl lipid II | Di-prenyl lipid II | Fold change |
| P1 | 51 ± 3 | 250 ± 9 | 4.9 ± 0.3 |
| P2 | 76 ± 6 | 570 ± 26 | 7.5 ± 0.7 |
| NP1 | 154 ± 13 | 1220 ± 50 | 7.9 ± 0.7 |
| NP2 | 140 ± 12 | 1130 ± 50 | 8.1 ± 0.8 |
| NP3 | 186 ± 8 | 1350 ± 50 | 7.3 ± 0.4 |
For the hepta-prenyl-containing lipid II, the relative efficacies of FmhB were determined with non-saturating concentrations of various isoacceptors (from 0.05 to 0.2 μM) and with three concentrations of FmhB (12.5, 25 and 50 nM) (Figure 2). The highest efficacy of FmhB was observed for the non-proteogenic isoacceptors (NP1, NP2 and NP3; 140–186 μM−1 min−1). The proteogenic isoacceptors were less effectively used by FmhB (P1 and P2; 51 ± 2 and 76 ± 3 μM−1 min−1).
The coupled assay used to generate the data presented in Figure 2 (above) requires the use of a detergent (Triton X-100; 1% final concentration in the assay) to solubilize the hepta-prenyl-containing lipid II analogue. To avoid any confounding factor linked to the use of a detergent and to facilitate substrate purification, we synthesized a soluble analogue of lipid intermediate II, which comprises two instead of seven prenyl units in the lipid moiety (Supplementary Figure S5). This water-soluble analogue was readily purified to homogeneity by reverse-phase HPLC and tested in the coupled assay with a non-saturating concentration (0.05 μM) of the isoacceptors and FmhB (20 nM) (Figure 3). FmhB displayed a higher efficacy with non-proteogenic than with proteogenic isoacceptors (1220 ± 50; 1130 ± 30; and 1350 ± 30 μM−1 min−1 for NP1, NP2 and NP3, respectively, versus 250 ± 10 and 570 ± 20 μM−1 min−1 for P1 and P2, respectively) (Figure 3). Replacement of the hepta-prenyl by the di-prenyl moiety in lipid II analogues improved the efficacy of FmhB to similar extents for all five isoacceptors (from 4.9- to 8.1-fold; Table 1). These results indicate, as might be expected, that the specificity of FmhB for the isoacceptors is not altered by modification of the number of prenyl units in the lipid II analogues used as substrates. The higher FmhB efficacy observed with the soluble lipid II analogue originates in part from a difference in the concentration of hepta- and di-prenyl-containing lipid II analogues used in the assay (30 μM versus 60 μM, respectively). In addition, the use of the soluble lipid II analogue might eliminate adverse effects of Triton X-100 on FmhB activity and on the accessibility of the substrate resulting from trapping of the hepta-prenyl-containing lipid II analogue in micelles.
Mapping FmhB specificity determinants in proteogenic and non-proteogenic tRNAs
Chimeric tRNAs harboring various portions of P1 and NP2 were constructed to identify regions of the isoacceptors that determine preferential usage of non-proteogenic tRNAs by FmhB (chimeras CHIM1, CHIM2, CHIM3, CHIM4 and CHIM5 in Figure 4A–C). Preliminary experiments showed that chimeras 1–5 were aminoacylated by GlyRS as effectively as P1 and NP2 (data not shown). These control experiments showed that differences in the efficacy of glycyl transfer from chimeric Gly-tRNAGly to the di-prenyl-containing lipid II analogue in the coupled assay could be attributed to differences in the activity of FmhB. As described above (Table 1), P1 is used 4.5-fold less effectively by FmhB than NP2 (250 ± 10 versus 1,130 ± 30 μM−1 min−1). We examined whether this difference could be specifically attributed to sequence polymorphisms restricted to the anticodon stem and loop or to the acceptor arm. Grafting the anticodon stem and loop of P1 onto NP2 (CHIM3) did not reduce (and in fact increased by 1.7-fold) the efficacy of the aminoacyl transfer reaction catalyzed by FmhB (1130 ± 30 versus 1920 ± 40 μM−1 min−1 for NP2 and CHIM3, respectively) (Figure 4D). Likewise, grafting the acceptor arm of P1 onto NP2 (CHIM1) did not reduce the efficacy of FmhB (1130 ± 30 versus 1430 ± 20 μM−1 min−1 for NP2 and CHIM1, respectively) (Figure 4D). A similar result was obtained for the graft of both the acceptor arm and anticodon stem and loop of P1 onto NP2 (1130 ± 30 versus 1410 ± 120 μM−1 min−1 for NP2 and CHIM5, respectively) (Figure 4C and F). These results suggest that neither the acceptor arm nor the anticodon stem and loop of NP2 contains any specificity determinant essential for effective aminoacyl transfer by FmhB. Conversely, these regions of P1 did not behave as anti-determinants in the context of the NP2 scaffold.
Figure 4.

FmhB specificity determinants in proteogenic and non-proteogenic tRNAs. (A–C) Cloverleaf representation of chimeric tRNAs containing P1 (red) and NP2 (blue) moieties. Divergent nucleotides in the acceptor stem and anticodon stem and loop are indicated by arrows. (D–F) Catalytic activity of FmhB with chimeric tRNAs: the coupled assay was performed with fixed concentrations of FmhB (20 nM), di-prenyl-containing lipid II analogue (60 μM), and tRNA (0.05 μM). Data are the mean ± standard deviation of three independent experiments for each tRNA. The rate of the reaction and the standard error were determined by fitting a linear equation to the data. The rate of the reaction was divided by the concentration of FmhB and of the tRNA to provide an estimate of the efficacy of FmhB for each isoacceptor.
The role of the anticodon stem and loop and of the acceptor arm of NP2 in the context of the P1 scaffold was analyzed by constructing additional chimeras (Figure 4B and E). Grafting the anticodon stem and loop of NP2 onto P1 (CHIM4) moderately increased (1.9-fold) the aminoacyl transfer efficacy of FmhB (250 ± 10 and 480 ± 20 μM−1 min−1 for P1 and CHIM4, respectively). In agreement with the results obtained above in the context of the NP2 scaffold (Figure 4A and D), these results indicate that the efficacy of the aminoacyl transfer by FmhB is not determined by sequence polymorphisms in the anticodon stem and loop moiety of the tRNAs. In contrast, grafting the acceptor arm of NP2 onto P1 (CHIM2, Figure 4E) unexpectedly increased the aminoacyl transfer efficacy by 3.3 fold (250 ± 10 and 820 ± 20 μM−1 min−1 for P1 and CHIM2, respectively). Thus, the acceptor arm of P1 might contain anti-determinants that function only in the context of the P1 scaffold.
In conclusion, results obtained with chimeras CHIM1–5 suggest that the efficacy of glycyl transfer catalyzed by FmhB is not determined by polymorphisms in the anticodon stem and loop of the tRNAs whereas data on the acceptor stem are inconclusive. The various parts of the tRNAs do not behave as independent entities and this obviously limits the power of analyses based on the construction of chimeras. This is particularly the case for the multiple interactions involving nucleotides in the T and D stems and loops. For this reason, we did not attempt the grafting approach for these tRNA moieties, which are the most likely candidates for the determination of the specificity of FmhB.
Detection of FmhB•tRNA complexes by size-exclusion chromatography
Binding of unacylated P1 and NP2 isoacceptors to FmhB was assayed by comparing the elution volume of the tRNA, FmhB, and a pre-incubated mixture of the two molecules (Figure 5 and Supplementary Figure S7 for the complete set of elution chromatograms recorded at 260 and 280 nm). A four-fold molar excess of FmhB was used to favor detection of shifts in the mobility of the tRNA at 260 nm. For P1, the chromatogram obtained for injection of a pre-incubated mixture of FmhB and the tRNA was similar to the addition of the chromatograms independently obtained for separate injections of FmhB and P1. Thus, there was no detectable interaction between FmhB and P1 in this assay (Figure 5A). In contrast, FmhB shifted NP2 elution toward a lower volume indicating formation of a high-molecular weight FmhB•tRNA complex (Figure 5B). These results indicate that FmhB displays a higher affinity for NP2 than for P1.
Figure 5.

Size-exclusion chromatography of FmhB, tRNAs, and mixtures of these two molecules. P1 (A) and NP2 (B) were used as representatives of proteogenic and non-proteogenic tRNAGly isoacceptors. The superimposed chromatograms (left panel) correspond to the injection (300 μl) of (a) FmhB alone (1200 pmol, ϵ280 nm = 55 800 M−1 cm−1), (b) tRNA alone (300 pmol, ϵ280 nm = 390 000 M−1 cm−1), or (c) a pre-incubated mixture of the same amounts of these two molecules. The superimposed chromatograms in the right panels correspond to the injection of the pre-incubated mixture of FmhB and the tRNA (chromatogram c in the left panels) and the addition of chromatograms (a) and (b) obtained by separate injections of FmhB and tRNA, respectively.
Impact of tRNA sequence variations on FmhB inhibition by stable Gly-tRNAGly analogues
Our next objective was to indirectly estimate the relative affinity of FmhB for the tRNAGly isoacceptors based on a competitive inhibition assay. For this purpose, we designed stable Gly-tRNAGly analogues that bind to FmhB but cannot be used as substrates by this enzyme. Semi-synthesis was used to replace the ester bond connecting Gly to the terminal ribose by a 1,4-triazole ring substituted by an alkyne side chain (Figure 6A). The 1,4-triazole linker was chosen for this replacement since the heterocycle is stable and behaves as a bio-isoster of ester and amide bonds (25). The nonadiyne molecule was chosen as it may mimic the Lys residue of the second substrate (lipid II) and contribute to stabilization of the inhibitor in the FmhB catalytic cavity. A prerequisite for the determination of the relative affinity of FmhB for Gly-tRNAGly analogues was the comparison of isomers containing the substituted triazole ring at the 2′ or 3′ position of the terminal ribose since both isomers are relevant in vivo. Indeed, GlyRSs are class II aminoacyl-tRNA synthetases known to acylate tRNAs at the 3′ position (29,30) (Figure 6B). The same isomer is used by the ribosome (31). However, spontaneous isomerization between the 2′ and 3′ positions occurs in solution at neutral pH with a rate and a thermodynamic equilibrium in the order of 5 s−1 and 1, respectively (32). Since this rapid equilibrium implies that the 3′ and 2′ isomers are both potential substrates of FmhB, we synthesized non-isomerizable Gly-tRNAGly analogues containing the substituted triazole at the 3′ or 2′ position (Figure 6D). Comparison of the 3′ and 2′ regioisomers containing the NP2 sequence revealed a 13-fold preference of FmhB for the 2′ regioisomer (Ki of 8.6 ± 2.0 versus 110 ± 6 nM) (Figure 6E). The 2′ isomers were therefore used to analyze the impact of variations in the sequence of the RNA moiety of Gly-tRNAGly analogues on the inhibition of FmhB.
Figure 6.

Competitive inhibition of FmhB by stable Gly-tRNAGly analogues. (A) Semi-synthesis of Gly-tRNAGly analogues. (B) Acylation of the 3′ position of tRNAGly by GlyRS and non-catalyzed isomerization. (C) Competitive binding of Gly-tRNAGly and stable analogues of Gly-tRNAGly to FmhB and inhibition of the transfer of Gly to the lipid II analogue in the coupled assay. (D) Stable and non-isomerizable Gly-tRNAGly analogues containing a stable 1,4-triazole ring substituted by an alkyne chain. Nucleotides appearing in red and blue fonts in P1 and NP2, respectively, highlight differences between the sequences of these isoacceptors. (E) Inhibition of FmhB by Gly-tRNAGly analogues. The Gly-tRNAGly analogues cannot be used as substrates by GlyRS or FmhB since the ester link connecting the hydroxyl of the tRNA to the carbonyl of Gly was replaced by a stable (uncleavable) 1,4-triazole linker. Preliminary control experiments showed that these stable Gly-tRNAGly analogues did not inhibit the acylation reaction catalyzed by the glycyl-tRNA synthetase (data not shown). The coupled assay was performed with a fixed concentration of NP2 (0.4 μM) and various concentrations of inhibitors containing the nucleotide sequence of P1 or NP2. Stable Gly-tRNAGly analogues acted as competitive inhibitors enabling determination of Ki values by fitting the Morrison equation to experimental data (27). (F) Stable Gly-tRNAGly analogues containing the acceptor arm of P1 or NP2.
Comparison of Gly-tRNAGly analogues containing the NP2 and P1 sequences revealed a 34-fold lower value of Ki for the former inhibitor (8.6 ± 2.0 versus 320 ± 10 nM) (Figure 6E). This observation indicates that FmhB displayed a 34-fold higher affinity for the inhibitor containing the NP2 sequence. This conclusion assumes a competitive inhibition mechanism (Figure 6C), which is highly likely considering the structure of the FmhB-related transferase FemX (33), which revealed a single binding site for the tRNA, and the structure of the substrate and the inhibitor, which only differs by the presence of an ester-linked glycyl group versus a triazole-linked alkyne chain.
Competitive inhibition was also tested with smaller Gly-tRNAGly analogues containing helices that only mimic the acceptor arm moiety of the tRNAs (Figure 6F). Similar Ki values were obtained for inhibitors based on the sequence of the P1 or NP2 acceptor arm (390 ± 80 versus 390 ± 60 nM, respectively, Figure 6E). This observation indicates that differences in the sequence of the P1 and NP2 acceptor arms did not modulate the affinity of FmhB for these inhibitors. In addition, the Ki values obtained with Gly-tRNAGly analogues containing the helices (390 ± 80 and 390 ± 60 nM) were similar to that obtained with the analogue containing the full tRNA sequence of P1 (320 ± 10 nM) but were significantly higher than that obtained with the analogue containing the full tRNA sequence of NP2 (8.6 ± 2.0 nM). This observation suggests that the affinity of FmhB for P1 is mainly determined by interactions involving the acceptor arm whereas NP2 forms additional contacts with the rest of the molecule.
Inhibition of FmhB by chimeric Gly-tRNAGly analogues
The inhibition assay depicted in Figure 6C was also performed with stable Gly-tRNAGly analogues containing the chimeric tRNA sequences that were tested above as substrates (Figure 4). Inhibition of FmhB was not affected by grafting the anticodon stem and loop of P1 onto NP2 (Ki of 5.4 ± 0.3 nM versus 8.6 ± 2.0 nM for CHIM3 and NP2, respectively) (Figure 7). Likewise, grafting the acceptor stem of P1 onto NP2 did not affect the value of Ki (7.6 ± 0.7 nM versus 8.6 ± 2.0 nM for CHIM1 and NP2, respectively). Grafting both elements of P1 onto NP2 (anticodon stem and loop plus acceptor stem) had a minor impact on the affinity (19 ± 2 nM versus 8.6 ± 2.0 nM for CHIM5 and NP2, respectively). Thus, the chimeras obtained by grafting the anticodon stem and loop or the acceptor stem of P1 onto NP2 retained the properties of NP2, indicating that these tRNA moieties did not harbor any specificity determinant. The same conclusion was reached from modifications introduced in the P1 scaffold. Indeed, grafting the anticodon stem and loop of NP2 onto P1 led to a minor increase (1.5-fold) in the affinity of FmhB for the inhibitors (Ki of 220 ± 40 versus 320 ± 10 nM for CHIM4 and P1, respectively). Grafting the acceptor arm of NP2 onto P1 did not increase the affinity of FmhB (in fact, a significant 3.1-fold decrease in affinity was observed; Ki of 1000 ± 160 versus 320 ± 10 nM for CHIM2 and P1, respectively).
Figure 7.

Competitive inhibition of FmhB by stable chimeric tRNA analogues. (A) Inhibition of FmhB by Gly-tRNAGly chimeric analogues containing a stable triazole ring. The color code highlights moieties of P1 (red) and NP2 (blue) in the sequence of chimeras CHIM1, CHIM2, CHIM3, CHIM4, and CHIM5. Chimeric analogues acted as competitive inhibitors enabling determination of Ki values by fitting the Morrison equation to experimental data. All inhibitors contained an alkyne-substituted 1,4-triazole ring linked to the 2′ position of the terminal nucleotide.
In conclusion, the results obtained by the analysis of stable Gly-tRNAGly analogues containing chimeric RNA moieties indicate that the higher affinity of FmhB for the inhibitor containing the NP2 sequence rather than the P1 sequence is not due to sequence polymorphism in the anticodon stem and loop or in the acceptor arm of the RNA moieties of these inhibitors. Thus, the difference in affinity should be determined by the remaining moieties of the tRNAs comprising the D and T stems and loops. Comparison of inhibitors containing the chimeric tRNAs or only the tRNA acceptor stems suggests that the D and T stems and loops of NP2 contain identity determinants that are absent from P1. Our results also indicate that introduction of the anticodon stem and loop and of the acceptor arm of P1 into the NP2 scaffold did not impair FmhB binding. Thus, the competitive binding assay did not reveal the presence of any antideterminant in the sequence of P1.
Comparison of the results obtained in the inhibition and activity assays
The same modifications were introduced in the tRNA moiety of stable Gly-tRNAGly analogues tested as inhibitors of FmhB (Figure 7) and in the tRNAs used to generate the Gly-tRNAGly substrates of FmhB in the coupled assay (Figure 4). Table 2 provides a comparison of the impact of these modifications on the affinity of FmhB for the inhibitors and on the enzymatic efficacy of FmhB. Variations in affinity and efficacy resulting from modifications of the RNA sequences were expected to be positively correlated since the tRNAs were used at non-saturating concentrations in the coupled assay. The expected correlation was observed for NP2, P1, and all chimeras except CHIM2 (Table 2). In the latter case, grafting the NP2 acceptor arm onto P1 had opposite effects on affinity (a 0.3-fold decrease) and on efficacy (a 3.3-fold increase). The basis for this discrepancy remains unknown. The amplitude of the difference observed for comparison of NP2 and P1 was greater for FmhB inhibition (a 34-fold difference) than for FmhB efficacy (a 4.5-fold difference) (Table 2). This observation is not surprising since the Ki value depends upon the ratio of the off to on constant (k-1/k1) whereas the Km Michaelis–Menten parameter additionally depends upon kcat (Km = k-1/(k1 + kcat). In conclusion, analyses of tRNA chimeras in two independent assays, coupled assay and inhibition by stable Gly-tRNAGly analogues, revealed that preferential use of NP2 by FmhB is due to the presence of specificity determinants located in the T and D stems and loops of the tRNA. The only discrepancy that was observed was a 3.3-fold improvement in FmhB efficacy for the introduction of the NP2 acceptor arm onto P1 in the coupled assay.
Table 2.
Impact of the tRNA sequence on the catalytic efficacy of FmhB and its affinity for inhibitors
| Amino acyl transfer catalyzed by FmhB | FmhB inhibition | |||
|---|---|---|---|---|
| tRNA sequence | Efficacy (μM–1 min–1) | Fold change relative to P1 | Affinity (nM) | Fold change relative to P1 |
| tRNAGly Isoacceptor | ||||
| P1 | 250 ± 9 | 1 | 320 ± 10 | 1 |
| NP2 | 1130 ± 30 | 4.5 ± 0.2 | 8.6 ± 2.0 | 37 ± 8 |
| Chimeras with the NP2 scaffold | ||||
| CHIM3 (NP2 with the P1 anticodon stem and loop) | 1920 ± 40 | 7.7 ± 0.3 | 5.4 ± 0.3 | 59 ± 4 |
| CHIM1 (NP2 with the P1 acceptor stem) | 1430 ± 20 | 5.7 ± 0.2 | 7.6 ± 0.7 | 42 ± 4 |
| CHIM5 (NP2 with P1 anticodon stem and loop and acceptor stem) | 1410 ± 120 | 5.6 ± 0.5 | 19 ± 2 | 17 ± 2 |
| Chimeras with the P1 scaffold | ||||
| CHIM4 (P1 with the NP2 anticodon stem and loop) | 480 ± 20 | 1.9 ± 0.1 | 220 ± 40 | 1.4 ± 0.2 |
| CHIM2 (P1 with the NP2 acceptor stem) | 820 ± 20 | 3.3 ± 0.2 | 1000 ± 160 | 0.32 ± 0.05 |
Errors for fold changes are calculated using the error propagation law.
Mapping identity determinants in the D and T stems and loops of P1 and NP2
Sequence polymorphisms in the D and T stems and loops of the P1 and NP2 isoacceptors were clustered in four regions (Supplementary Results section 17 and Figure S9). Construction of chimeric tRNAs indicated that each of these four regions may act as identity determinants in the transferase and inhibition assays (Supplementary Figures S10 and S11, respectively). These results suggest that preferential recognition of NP2 by FmhB involves several identity determinants that are scattered in the sequence of the D and T stems and loops.
Competitive binding of Gly-tRNAGly to FmhB and EF-Tu
In the cytoplasm of S. aureus, FmhB and EF-Tu•GTP potentially compete for the same pool of Gly-tRNAGly resulting from acylation of the P1, P2, NP1, NP2, and NP3 isoacceptors by GlyRS. In vitro, binding of Gly-tRNAGly to EF-Tu•GTP is expected to inhibit FmhB by substrate sequestration (Figure 8A). Thus, residual FmhB activity in the presence of increasing concentrations of EF-Tu•GTP was used to determine whether differences in the sequence of the isoacceptors affect competitive binding to EF-Tu•GTP and FmhB (Figure 8B and C). Glycyl transfer was inhibited by EF-Tu•GTP in a concentration-dependent manner for the five isoacceptors. Inhibition was higher for proteogenic (P1 and P2) than for non-proteogenic (NP1, NP2 and NP3) isoacceptors. Qualitatively, these results indicated that binding of Gly-tRNAGly to EF-Tu•GTP inhibited the glycyl transfer reaction catalyzed by FmhB in vitro. The low affinity of EF-Tu•GTP for Gly-tRNAGly containing the NP1, NP2, and NP3 sequences (13) largely prevented this inhibition. Thus, the low affinity of EF-Tu•GTP for Gly-tRNAGly-containing non-proteogenic tRNAGly could help to ensure an adequate supply of activated Gly for synthesis of peptidoglycan precursors by FmhB.
Figure 8.

Inhibition of FmhB by EF-Tu-mediated sequestration of Gly-tRNAGly. (A) Reaction scheme illustrating how binding of Gly-tRNAGly to EF-Tu•GTP leads to FmhB inhibition. The residual transferase activity of FmhB in the presence of increasing concentrations of EF-Tu•GTP was determined by quantification of [14C]Gly added onto the di-prenyl-lipid II analogue (Supplementary Table S3). (B) Autoradiography providing a representative example of the separation of the substrates and products of the reaction by thin-layer chromatography. The arrow indicates the direction of solvent migration. (C) Residual transferase activity of FmhB (%) was determined in the presence of increasing concentrations of EF-Tu•GTP. Inhibition was tested at a fixed concentration (0.1 μM) of proteogenic (P1 and P2) and non-proteogenic (NP1, NP2 and NP3) tRNAGly isoacceptors.
Relative efficacy of FemA and FemB for glycyl transfer from the P1 and NP2 Gly-tRNAGly isoacceptors to peptidoglycan precursor analogues
Synthesis of the complete pentaglycine side chain requires two aminoacyl-transferases, FemA and FemB, in addition to FmhB (Figure 1). FemA and FemB were purified and used in the coupled assay with P1 and NP2 as representatives of the proteogenic and non-proteogenic tRNAs, respectively. FemA and FemB displayed a 2.6 ± 0.2 and 3.4 ± 0.3-fold preference for the non-proteogenic tRNA, respectively (Supplementary Figure S6). FmhB displayed a 4.5 ± 0.2-fold preference for NP2 (Table 2). These results indicate that the non-proteogenic tRNA NP2 is preferentially used by the three aminoacyl-transferases involved in the synthesis of the pentaglycine side chain of the peptidoglycan precursors of S. aureus.
DISCUSSION
Although aminoacylated tRNAs offer a source of activated amino acids for the development of biosynthetic pathways, this opportunity seems to have been exploited or retained in a very limited number of cases in the evolution of the three kingdoms of life. For example, peptidoglycan synthesis in bacteria almost exclusively involves ATP-dependent activation of the carbonyl groups of amino acids as acyl-phosphates (1,34–36). Participation of tRNAs in the synthesis of peptidoglycan precursors is restricted to the side chain, a structure present in a limited number of species, mainly among the firmicutes and the actinobacteria (1,37–42). One possible limitation to the development of non-ribosomal tRNA-dependent biosynthetic pathways may stem from the potentially negative impact of the depletion of a specific aminoacyl-tRNA pool. Indeed, codon usage and the relative abundance of tRNAs have been adjusted to each other in complex ways during evolution to modulate the level of gene expression during translational elongation, to ensure the fidelity of translation, and to optimize protein folding as the latter is coupled to translation (see Rak et al. for a review) (43). The impact of variations in the relative abundance of tRNAs on these key cellular processes stems from the fact that aminoacyl-tRNAs compete to decode the same codon in the A site of the ribosome and that binding of aminoacyl-tRNAs to the A site is rate-limiting during elongation. In addition to bioinformatics analyses, experimental evidence has shown that modifications of the tRNA pools affect the fidelity of translation (44) and the rate of protein misfolding (45). Thus, the use of a large pool of aminoacyl-tRNAs for non-ribosomal synthesis is expected to be deleterious in the absence of a mechanism that maintains an adequate supply of aminoacyl-tRNAs for translation. Emergence of tRNA-dependent pathways may require the acquisition of mechanisms that ensure the harmonious partition of aminoacyl-tRNAs between the ribosomal and non-ribosomal synthesis pathways (13,46,47). From a quantitative standpoint, this requirement may be particularly important for the Fem pathway in S. aureus since this bacterium produces a thick peptidoglycan layer and Gly represents 5 out of the 10 amino acid residues present in the lipid II precursor (6,48–50) (Figure 1A). Quantitative estimates presented in the Supplementary Material (section 13, and supplementary Table S4) suggest that ca. 37% of the Gly-tRNAGly pool is directed toward protein synthesis and the remaining 63% toward peptidoglycan synthesis. The burden of the Fem pathway on the aminoacyl-tRNA pools is expected to be more limited in bacteria producing shorter side chains of mixed amino acid composition (39). The current study aimed to demonstrate the existence of mechanisms ensuring the harmonious partition of Gly-tRNAGly between translation and peptidoglycan synthesis and to identify tractable characteristics indicative of the coevolution of the ribosomal and non-ribosomal pathways.
It is first important to recall that the sequence of the tRNAs specifying Gly and the kinetics of their acylation indicate that all isoacceptors are acylated by the same glycyl-tRNA synthetase with similar efficacies (15) (Supplementary Figure S4). The T-box riboswitch that regulates transcription of the gene encoding GlyRS in S. aureus responds to both proteinogenic and non-proteinogenic isoacceptors indicating that the supply of tRNAGly to the glycyl transferase can be adjusted in response to accumulation of both types of unacylated tRNAs by the same mechanism (51). Thus, the tRNA acylation step does not seem to involve any evolutionary characteristics relevant to the partition of aminoacyl-tRNAs. In contrast, the tRNAGly isoacceptors clearly fall in two classes, proteogenic (P1 and P2) and non-proteogenic (NP1, NP2 and NP3), defined by their interaction with FmhB (this study), EF-Tu•GTP (15,21), and the ribosome (16,18). Here we show that non-proteogenic tRNAs are preferentially used as substrates by FmhB (Figures 2 and 3) and that this preference at least partially involves a difference in the relative affinity of FmhB for the two types of tRNAs, as directly established by the detection of FmhB•tRNA complexes by size-exclusion chromatography (Figure 5) and indirectly established by the competitive inhibition of FmhB by stable Gly-tRNAGly analogues (Figures 6 and 7). Thus, limited recognition of proteogenic tRNAs P1 and P2 by FmhB may contribute to ensure an adequate supply of aminoacyl-tRNAs for protein synthesis. Qualitatively, we also showed that EF-Tu•GTP inhibits FmhB in vitro by sequestering its substrate and that this inhibition is weaker for Gly-tRNAGly composed of non-proteogenic tRNAs (Figure 8), in agreement with the lower affinity of EF-Tu•GTP for these aminoacyl-tRNAs (15,52–55). These observations are relevant to the in vivo situation since the cytoplasmic concentration of EF-Tu is thought, at least in E. coli, to exceed that of aminoacylated tRNAs, which are present at a very low concentration in the free form due to the formation of high-affinity EF-Tu•GTP•aminoacyl-tRNA complexes (56,57). Together, these results indicate that the potential adverse impact of the competition between the two pathways for the Gly-tRNAGly pool is restricted by both limited recognition of non-proteogenic tRNAs by EF-Tu•GTP and limited recognition of proteogenic tRNAs by FmhB.
The basis for the lower affinity of EF-Tu•GTP for non-proteogenic than for proteogenic Gly-tRNAs can be deduced from the comparison of the sequences of the two types of tRNAs and the known sequence-specificity of EF-Tu. Recognition of both the tRNA body and the esterified amino acid contributes to the affinity of EF-Tu•GTP for aminoacyl-tRNAs (53). Variations in the thermodynamic contribution of these two interactions compensate for each other so that the affinity of EF-Tu•GTP for all proteogenic aminoacyl-tRNAs is similar, a feature thought to be of paramount importance for effective and accurate decoding of messenger RNAs by the ribosome (53). The variations in the affinity of EF-Tu•GTP for the tRNA body of aminoacyl-tRNAs are nearly exclusively determined by the first three base pairs of the T stem (54). The role of these six nucleotide residues in determining the relative binding affinity of EF-Tu•GTP for the tRNA body of aminoacyl-tRNAs is universally conserved in bacteria in spite of extensive sequence diversity in this portion of the T stem (55). The thermodynamic contribution of each of the three base pairs is additive enabling similar affinities to be reached with various nucleotide combinations (52). The contribution of the esterified amino acid to the binding affinity of EF-Tu•GTP for aminoacyl-tRNAs is one of the lowest for glycine, and consequently, this is compensated for by one of the highest affinities for the tRNAGly bodies (52,54). In S. aureus, the proteogenic P1 and P2 isoacceptors display, as expected, high-affinity base-pair combinations at the first three positions of the T stem (G49•U65, C50-G64 or U50-A64 and G51-C63, respectively, Figure 1C) (15,55). In contrast, the T stems of the NP1, NP2, and NP3 non-proteogenic tRNAs contain a less ‘tight’ base-pair combination (A49-U65, U50-A64 and A51-U63) with ΔG° values (–9.6 ± 0.3 and –9.7 ± 0.2 kcal mol−1, as estimated by two methods) significantly higher than the mean ΔG° value (-10.8 kcal mol−1) of bacterial Gly-tRNAs (a 7.9-fold increase in KD, from 8.8 to 69 nM) (55). This difference could account for the lower relative affinity of EF-Tu•GTP for the aminoacyl-tRNAs containing the NP1, NP2 or NP3 tRNA body rather than the P1 or P2 body reported by Giannouli et al. (15), based on a qualitative assay involving the determination of the salt concentration required to elute aminoacyl-tRNAs bound to EF-Tu•GTP immobilized on a solid support (15). This difference could also account for the difference in affinity between the two types of aminoacyl-tRNAs revealed by the competitive inhibition of FmhB by EF-Tu•GTP (Figure 8).
Since bias in codon usage affects the turnover of specific aminoacyl-tRNAs, the decoding characteristics of the isoacceptors should be considered in the analysis of the partition of Gly-tRNAGly between peptidoglycan and protein synthesis. As detailed in section 14 of the Supplementary Material, differences in the anticodons of proteogenic and non-proteogenic isoacceptors do not appear to play a role in tRNA partition.
Since partition of the isoacceptors between translation and peptidoglycan synthesis can be achieved by the presence of nucleotide polymorphisms that favor (determinants) or disfavor (anti-determinants) the interaction of isoacceptors with EF-Tu and FmhB, we determined whether functional differences could be traced to specific regions of the tRNAs. By using tRNA chimeras in the GlyRS-FmhB coupled assay (Figure 4) and stable Gly-tRNAGly analogues in the competitive binding assay (Figures 6 and 7), the relevant polymorphisms were found to be located in the T and D stems and loops of the tRNAs rather than in their anticodon stem and loop or acceptor arm. The use of inhibitors comprising only the tRNA acceptor arm led to the conclusion that the T and D stems and loops of NP2 harbor specificity determinants that may account for the higher affinity of FmhB for this non-proteogenic tRNA. The T and D stems and loops of NP2 may interact with the coiled-coil domain of FmhB as illustrated by the FemA:NP2 interaction model presented in Figure 9A. It is worth noting that the aminoacyl-transferase FemXWv from Weissella viridescens is devoid of the coiled-coil domain and recognizes Ala-tRNAAla by a distinct mechanism exclusively involving the acceptor arm (Figure 9B). This was established both by biochemical and structural approaches that led to the crystallization of FemXWv with a ‘bi-substrate’ comprising mimics of the peptidoglycan precursor and the tRNAAla acceptor arm (33). The current study indicates that deciphering the molecular mechanism of tRNAGly recognition by FmhB will require the challenging co-crystallization of FmhB with a molecule comprising the entire tRNAGly sequence since relevant interactions are scattered through several regions of the tRNAGly molecule.
Figure 9.
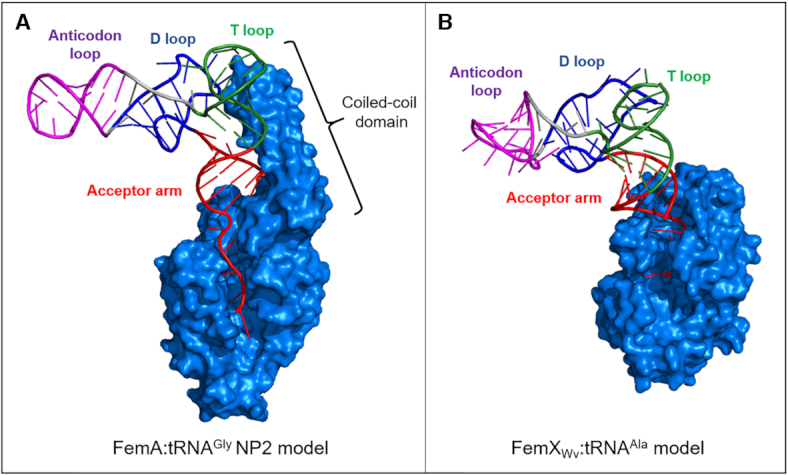
Models for tRNA recognition by aminoacyl-transferases of the Fem family. (A) FemA from S. aureus (58) in interaction with non-proteogenic isoacceptor NP2. (B) FemXWv from Weissella viridescens in interaction with tRNAAla (59). Construction of models for tRNA recognition by the Fem aminoacyl-transferases is described in section 15 of the Supplementary Material.
The evolutionary origin of the sequence polymorphisms in proteogenic and non-proteogenic tRNAs was investigated by constructing a phylogenic tree with the sequence of tRNAGly isoacceptors from a selected set of Gram-positive bacteria (Supplementary Material, section 16, Supplementary Table S5, and Figure S8). The non-proteogenic tRNAs NP1, NP2, and NP3, which are preferentially recognized by FmhB, form a distinct cluster, which is present in bacteria producing pentaglycine-containing peptidoglycan precursors and is notably absent from other bacteria. These observations suggest that the sequence of non-proteogenic tRNAs may have evolved to retain efficacious acylation by GlyRS and to minimize binding to EF-Tu•GTP (52,53,60), thus providing an adequate supply of aminoacyl-tRNAs for synthesis of peptidoglycan precursors by FmhB. In turn, FmhB may have evolved to preferentially use non-proteogenic isoacceptors, leaving the pool of proteogenic tRNAGly, which is similar to that of other firmicutes, available for protein synthesis.
The production of additional ‘non-proteogenic’ tRNAs preferentially participating in peptidoglycan synthesis has so far only been detected in S. aureus and S. epidermidis (13,55). This notion has been thoroughly explored in S. pneumoniae (61). In this bacterium, the MurM transferase is responsible for incorporation of the first residue of the Ala-Ala or Ser-Ala side chain of peptidoglycan precursors (62). S. pneumoniae is unusual among the firmicutes as the composition of the side chain is variable with a preponderance of L-Ala or L-Ser at the first position depending upon the presence of mosaic murM alleles generated by horizontal gene transfer (61,63). S. pneumoniae is also unusual as the peptidoglycan of certain clinical isolates mainly contains direct cross-links lacking the side chain, D-Ala4→L-Lys3 instead of D-Ala4→side chain-L-Lys3, due to the presence of murM alleles encoding weakly active transferases (62). In contrast, the sequence of the cross-links is generally uniformly conserved in members of the same species, a conservation that led to the historical proposal of using peptidoglycan structure for taxonomic purposes (37). In common with other firmicutes, the side chain is required for the expression of acquired β-lactam resistance in S. pneumoniae (penicillin resistance is clinically relevant in this case), as found for the pentaglycine side chain of methicillin-resistant S. aureus and the L-Ala-L-Ala side chain of cephalosporin-resistant E. faecalis (38). In clinical isolates of S. pneumoniae, variations in the relative abundance of tRNASer and tRNAAla were reported to correlate with incorporation of Ser and of Ala into the side chain of peptidoglycan precursors but this was not associated with any modification of the tRNA gene sequences (61). These results suggest that Fem transferases and EF-Tu•GTP compete for the same tRNA pool in most firmicutes, possibly with an adjustment of the tRNA concentration. However, a completely different type of interaction of Fem transferases with components of the translation machinery, i.e. editing of misacylated tRNAs, was detected in S. pneumoniae (64). In this bacterium, tRNAPhe contains an unusual U4•C69 mismatch that mimics the G3•U70 identity determinant of alanyl-tRNA synthetase (AlaRS), leading to synthesis of Ala-tRNAPhe and Ser-tRNAPhe. These misacylated tRNAs produced by AlaRS escape the editing activity of PheRS as the U4•C69 mismatch acts as an anti-determinant for deacylation of Ala-tRNAPhe by PheRS and this enzyme does not display any editing activity with tRNAPhe misacylated with Ser. Purified MurM was found to deacylate Ala-tRNAPhe and Ser-tRNAPhe as well as tRNAAla misacylated with Ser by AlaRS (64). It was thus proposed that MurM could contribute to the fidelity of translation by eliminating misacylated Ala-tRNAPhe, Ser-tRNAPhe and Ser-tRNAAla, and perhaps, productively direct the corresponding activated amino acids toward peptidoglycan synthesis. Although MurM is dispensable for growth in laboratory conditions, this enzyme may have a significant contribution in vivo, in addition to its role in penicillin resistance, since the harsh conditions that prevail during infection, in particular the presence of reactive oxygen species such as H2O2, are known to increase the rate of tRNA misacylation (43).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Zainab Edoo for proofreading the manuscript.
Contributor Information
Lauriane Rietmeyer, INSERM, Sorbonne Université, Université de Paris, Centre de Recherche des Cordeliers (CRC), F-75006 Paris, France.
Nicolas Fix-Boulier, INSERM, Sorbonne Université, Université de Paris, Centre de Recherche des Cordeliers (CRC), F-75006 Paris, France.
Chloé Le Fournis, INSERM, Sorbonne Université, Université de Paris, Centre de Recherche des Cordeliers (CRC), F-75006 Paris, France.
Laura Iannazzo, Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, Université de Paris, CNRS UMR 8601, Paris F-75006 France.
Camelia Kitoun, Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, Université de Paris, CNRS UMR 8601, Paris F-75006 France.
Delphine Patin, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France.
Dominique Mengin-Lecreulx, Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France.
Mélanie Ethève-Quelquejeu, Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, Université de Paris, CNRS UMR 8601, Paris F-75006 France.
Michel Arthur, INSERM, Sorbonne Université, Université de Paris, Centre de Recherche des Cordeliers (CRC), F-75006 Paris, France.
Matthieu Fonvielle, INSERM, Sorbonne Université, Université de Paris, Centre de Recherche des Cordeliers (CRC), F-75006 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale pour la Recherche [ANR-17-CE07-0041-02 (SyntRNA to M.E.Q. and M.A.)]; L.R. was funded by a Sorbonne Université doctoral contract. Funding for open access charge: ANR [ANR-17-CE07-0041-02].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mainardi J.L., Villet R., Bugg T.D., Mayer C., Arthur M.. Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram-positive bacteria. FEMS Microbiol. Rev. 2008; 32:386–408. [DOI] [PubMed] [Google Scholar]
- 2. Moutiez M., Belin P., Gondry M.. Aminoacyl-tRNA-utilizing enzymes in natural product biosynthesis. Chem. Rev. 2017; 117:5578–5618. [DOI] [PubMed] [Google Scholar]
- 3. Fields R.N., Roy H.. Deciphering the tRNA-dependent lipid aminoacylation systems in bacteria: novel components and structural advances. RNA Biol. 2018; 15:480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Watanabe K., Toh Y., Suto K., Shimizu Y., Oka N., Wada T., Tomita K.. Protein-based peptide-bond formation by aminoacyl-tRNA protein transferase. Nature. 2007; 449:867–871. [DOI] [PubMed] [Google Scholar]
- 5. Rohrer S., Berger-Bachi B.. FemABX peptidyl transferases: a link between branched-chain cell wall peptide formation and beta-lactam resistance in gram-positive cocci. Antimicrob. Agents Chemother. 2003; 47:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schneider T., Senn M.M., Berger-Bachi B., Tossi A., Sahl H.G., Wiedemann I.. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 2004; 53:675–685. [DOI] [PubMed] [Google Scholar]
- 7. Rohrer S., Ehlert K., Tschierske M., Labischinski H., Berger-Bachi B.. The essential Staphylococcus aureus gene fmhB is involved in the first step of peptidoglycan pentaglycine interpeptide formation. PNAS. 1999; 96:9351–9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Monteiro J.M., Covas G., Rausch D., Filipe S.R., Schneider T., Sahl H.G., Pinho M.G.. The pentaglycine bridges of Staphylococcus aureus peptidoglycan are essential for cell integrity. Sci. Rep. 2019; 9:5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hübscher J., Jansen A., Kotte O., Schäfer J., Majcherczyk P.A., Harris L.G., Bierbaum G., Heinemann M., Berger-Bächi B.. Living with an imperfect cell wall: compensation of femAB inactivation in Staphylococcus aureus. BMC Genomics. 2007; 8:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Lencastre H., Tomasz A.. Reassessment of the number of auxiliary genes essential for expression of high-level methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1994; 38:2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopp U., Roos M., Wecke J., Labischinski H.. Staphylococcal peptidoglycan interpeptide bridge biosynthesis: a novel antistaphylococcal target?. Microb. Drug Resist. 1996; 2:29–41. [DOI] [PubMed] [Google Scholar]
- 12. Hegde S.S., Shrader T.E.. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J. Biol. Chem. 2001; 276:6998–7003. [DOI] [PubMed] [Google Scholar]
- 13. Shepherd J., Ibba M.. Direction of aminoacylated transfer RNAs into antibiotic synthesis and peptidoglycan-mediated antibiotic resistance. FEBS Lett. 2013; 587:2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Green C.J., Vold B.S.. Staphylococcus aureus has clustered tRNA genes. J. Bacteriol. 1993; 175:5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giannouli S., Kyritsis A., Malissovas N., Becker H.D., Stathopoulos C.. On the role of an unusual tRNAGly isoacceptor in Staphylococcus aureus. Biochimie. 2009; 91:344–351. [DOI] [PubMed] [Google Scholar]
- 16. Bumsted R.M., Dahl J.L., Soll D., Strominger J.L.. Biosynthesis of the peptidoglycan of bacterial cell walls. X. Further study of the glycyl transfer ribonucleic acids active in peptidoglycan synthesis in Staphylococcus aureus. J. Biol. Chem. 1968; 243:779–782. [PubMed] [Google Scholar]
- 17. Petit J.F., Strominger J.L., Soll D. Biosynthesis of the peptidoglycan of bacterial cell walls. VII. Incorporation of serine and glycine into interpeptide bridges in Staphylococcus epidermidis. J. Biol. Chem. 1968; 243:757–767. [PubMed] [Google Scholar]
- 18. Stewart T.S., Roberts R.J., Strominger J.L.. Novel species of tRNA. Nature. 1971; 230:36–38. [DOI] [PubMed] [Google Scholar]
- 19. Roberts R.J. Staphylococcal transfer ribonucleic acids. II. Sequence analysis of isoaccepting glycine transfer ribonucleic acids IA and IB from Staphylococcus epidermidis Texas 26. J. Biol. Chem. 1974; 249:4787–4796. [PubMed] [Google Scholar]
- 20. Roberts R.J. Structures of two glycyl-tRNAs from Staphylococcus epidermidis. Nat.: New Biol. 1972; 237:44–45. [DOI] [PubMed] [Google Scholar]
- 21. Kawakami M., Tanada S., Takemura S.. Properties of alanyl-oligonucleotide, puromycin, and Staphylococcus epidermidis glycyl-tRNA in interaction with elongation factor Tu:GTP complex. FEBS Lett. 1975; 51:321–324. [DOI] [PubMed] [Google Scholar]
- 22. Chang A.T., Nikonowicz E.P.. Solution nuclear magnetic resonance analyses of the anticodon arms of proteinogenic and nonproteinogenic tRNAGly. Biochemistry. 2012; 51:3662–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hyland S.A., Anderson M.S.. A high-throughput solid-phase extraction assay capable of measuring diverse polyprenyl phosphate: sugar-1-phosphate transferases as exemplified by the WecA, MraY, and MurG proteins. Anal. Biochem. 2003; 317:156–165. [DOI] [PubMed] [Google Scholar]
- 24. Fonvielle M., Bouhss A., Hoareau C., Patin D., Mengin-Lecreulx D., Iannazzo L., Sakkas N., El Sagheer A., Brown T., Etheve-Quelquejeu M. et al.. Synthesis of lipid-carbohydrate-peptidyl-RNA conjugates to explore the limits imposed by the substrate specificity of cell wall enzymes on the acquisition of drug resistance. Chem. Eur. J. 2018; 24:14911–14915. [DOI] [PubMed] [Google Scholar]
- 25. Fonvielle M., Mellal D., Patin D., Lecerf M., Blanot D., Bouhss A., Santarem M., Mengin-Lecreulx D., Sollogoub M., Arthur M. et al.. Efficient access to peptidyl-RNA conjugates for picomolar inhibition of non-ribosomal FemX(Wv) aminoacyl transferase. Chem. Eur. J. 2013; 19:1357–1363. [DOI] [PubMed] [Google Scholar]
- 26. Chemama M., Fonvielle M., Villet R., Arthur M., Valery J.M., Etheve-Quelquejeu M.. Stable analogues of aminoacyl-tRNA for inhibition of an essential step of bacterial cell-wall synthesis. J. Am. Chem. Soc. 2007; 129:12642–12643. [DOI] [PubMed] [Google Scholar]
- 27. Copeland R.A. Evaluation of Enzyme Inhibitors in Drug Discovery. A Guide for Medicinal Chemists and Pharmacologists. Methods Biochem. Anal. 2005; 46:1–265. [PubMed] [Google Scholar]
- 28. Nameki N., Tamura K., Asahara H., Hasegawa T.. Recognition of tRNA(Gly) by three widely diverged glycyl-tRNA synthetases. J. Mol. Biol. 1997; 268:640–647. [DOI] [PubMed] [Google Scholar]
- 29. Eriani G., Delarue M., Poch O., Gangloff J., Moras D.. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990; 347:203–206. [DOI] [PubMed] [Google Scholar]
- 30. Niyomporn B., Dahl J.L., Strominger J.L.. Biosynthesis of the peptidoglycan of bacterial cell walls. IX. Purification and properties of glycyl transfer ribonucleic acid synthetase from Staphylococcus aureus. J. Biol. Chem. 1968; 243:773–778. [PubMed] [Google Scholar]
- 31. Nissen P., Hansen J., Ban N., Moore P.B., Steitz T.A.. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000; 289:920–930. [DOI] [PubMed] [Google Scholar]
- 32. Taiji M., Yokoyama S., Miyazawa T.. Transacylation rates of aminoacyladenosine moiety at the 3′-terminus of aminoacyl-transfer ribonucleic acid. Biochemistry. 1983; 22:3220–3225. [DOI] [PubMed] [Google Scholar]
- 33. Fonvielle M., Li de La, Sierra-Gallay I., El-Sagheer A.H., Lecerf M., Patin D., Mellal D., Mayer C., Blanot D., Gale N., Brown T. et al.. The structure of FemX(Wv) in complex with a peptidyl-RNA conjugate: mechanism of aminoacyl transfer from Ala-tRNA(Ala) to peptidoglycan precursors. Angew. Chem. Int. Ed. 2013; 52:7278–7281. [DOI] [PubMed] [Google Scholar]
- 34. Bouhss A., Mengin-Lecreulx D., Blanot D., van Heijenoort J., Parquet C.. Invariant amino acids in the Mur peptide synthetases of bacterial peptidoglycan synthesis and their modification by site-directed mutagenesis in the UDP-MurNAc:L-alanine ligase from Escherichia coli. Biochemistry. 1997; 36:11556–11563. [DOI] [PubMed] [Google Scholar]
- 35. Bellais S., Arthur M., Dubost L., Hugonnet J.E., Gutmann L., van Heijenoort J., Legrand R., Brouard J.P., Rice L., Mainardi J.L.. Aslfm, the D-aspartate ligase responsible for the addition of D-aspartic acid onto the peptidoglycan precursor of Enterococcus faecium. J. Biol. Chem. 2006; 281:11586–11594. [DOI] [PubMed] [Google Scholar]
- 36. Barreteau H., Kovac A., Boniface A., Sova M., Gobec S., Blanot D. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008; 32:168–207. [DOI] [PubMed] [Google Scholar]
- 37. Schleifer K.H., Kandler O.. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 1972; 36:407–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bouhss A., Josseaume N., Allanic D., Crouvoisier M., Gutmann L., Mainardi J.L., Mengin-Lecreulx D., van Heijenoort J., Arthur M.. Identification of the UDP-MurNAc-pentapeptide:L-alanine ligase for synthesis of branched peptidoglycan precursors in Enterococcus faecalis. J. Bacteriol. 2001; 183:5122–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dare K., Ibba M.. Roles of tRNA in cell wall biosynthesis. Wiley Interdiscip. Rev.: RNA. 2012; 3:247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong H.J., Hutchings M.I., Hill L.M., Buttner M.J.. The role of the novel Fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J. Biol. Chem. 2005; 280:13055–13061. [DOI] [PubMed] [Google Scholar]
- 41. Hong H.J., Hutchings M.I., Neu J.M., Wright G.D., Paget M.S., Buttner M.J.. Characterization of an inducible vancomycin resistance system in Streptomyces coelicolor reveals a novel gene (vanK) required for drug resistance. Mol. Microbiol. 2004; 52:1107–1121. [DOI] [PubMed] [Google Scholar]
- 42. Fiser A., Filipe S.R., Tomasz A.. Cell wall branches, penicillin resistance and the secrets of the MurM protein. Trends Microbiol. 2003; 11:547–553. [DOI] [PubMed] [Google Scholar]
- 43. Rak R., Dahan O., Pilpel Y.. Repertoires of tRNAs: the couplers of genomics and proteomics. Annu. Rev. Cell Dev. Biol. 2018; 34:239–264. [DOI] [PubMed] [Google Scholar]
- 44. Kramer E.B., Farabaugh P.J.. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yona A.H., Bloom-Ackermann Z., Frumkin I., Hanson-Smith V., Charpak-Amikam Y., Feng Q., Boeke J.D., Dahan O., Pilpel Y.. tRNA genes rapidly change in evolution to meet novel translational demands. Elife. 2013; 2:e01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shepherd J., Ibba M.. Bacterial transfer RNAs. FEMS Microbiol. Rev. 2015; 39:280–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blanquet S., Mechulam Y., Schmitt E.. The many routes of bacterial transfer RNAs after aminoacylation. Curr. Opin. Struct. Biol. 2000; 10:95–101. [DOI] [PubMed] [Google Scholar]
- 48. Matsuhashi M., Dietrich C.P., Strominger J.L.. Incorporation of glycine into the cell wall glycopeptide in Staphylococcus aureus: role of sRNA and lipid intermediates. PNAS. 1965; 54:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wyatt P.J. Cell wall thickness, size distribution, refractive index ratio and dry weight content of living bacteria (Staphylococcus aureus). Nature. 1970; 226:277–279. [DOI] [PubMed] [Google Scholar]
- 50. Vollmer W., Seligman S.J.. Architecture of peptidoglycan: more data and more models. Trends Microbiol. 2010; 18:59–66. [DOI] [PubMed] [Google Scholar]
- 51. Apostolidi M., Saad N.Y., Drainas D., Pournaras S., Becker H.D., Stathopoulos C.. A glyS T-box riboswitch with species-specific structural features responding to both proteinogenic and nonproteinogenic tRNAGly isoacceptors. RNA. 2015; 21:1790–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schrader J.M., Chapman S.J., Uhlenbeck O.C.. Understanding the sequence specificity of tRNA binding to elongation factor Tu using tRNA mutagenesis. J. Mol. Biol. 2009; 386:1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. LaRiviere F.J., Wolfson A.D., Uhlenbeck O.C.. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001; 294:165–168. [DOI] [PubMed] [Google Scholar]
- 54. Uhlenbeck O.C., Schrader J.M.. Evolutionary tuning impacts the design of bacterial tRNAs for the incorporation of unnatural amino acids by ribosomes. Curr. Opin. Chem. Biol. 2018; 46:138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schrader J.M., Uhlenbeck O.C.. Is the sequence-specific binding of aminoacyl-tRNAs by EF-Tu universal among bacteria?. Nucleic Acids Res. 2011; 39:9746–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Klumpp S., Scott M., Pedersen S., Hwa T.. Molecular crowding limits translation and cell growth. PNAS. 2013; 110:16754–16759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tubulekas I., Hughes D. Growth and translation elongation rate are sensitive to the concentration of EF-Tu. Mol. Microbiol. 1993; 8:761–770. [DOI] [PubMed] [Google Scholar]
- 58. Benson T.E., Prince D.B., Mutchler V.T., Curry K.A., Ho A.M., Sarver R.W., Hagadorn J.C., Choi G.H., Garlick R.L.. X-ray crystal structure of Staphylococcus aureus FemA. Structure. 2002; 10:1107–1115. [DOI] [PubMed] [Google Scholar]
- 59. Villet R., Fonvielle M., Busca P., Chemama M., Maillard A.P., Hugonnet J.E., Dubost L., Marie A., Josseaume N., Mesnage S. et al.. Idiosyncratic features in tRNAs participating in bacterial cell wall synthesis. Nucleic Acids Res. 2007; 35:6870–6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sanderson L.E., Uhlenbeck O.C.. The 51–63 base pair of tRNA confers specificity for binding by EF-Tu. RNA. 2007; 13:835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lloyd A.J., Gilbey A.M., Blewett A.M., De Pascale G., El Zoeiby A., Levesque R.C., Catherwood A.C., Tomasz A., Bugg T.D., Roper D.I. et al.. Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J. Biol. Chem. 2008; 283:6402–6417. [DOI] [PubMed] [Google Scholar]
- 62. Filipe S.R., Severina E., Tomasz A.. Functional analysis of Streptococcus pneumoniae MurM reveals the region responsible for its specificity in the synthesis of branched cell wall peptides. J. Biol. Chem. 2001; 276:39618–39628. [DOI] [PubMed] [Google Scholar]
- 63. Filipe S.R., Severina E., Tomasz A.. The murMN operon: a functional link between antibiotic resistance and antibiotic tolerance in Streptococcus pneumoniae. PNAS. 2002; 99:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shepherd J., Ibba M.. Lipid II-independent trans editing of mischarged tRNAs by the penicillin resistance factor MurM. J. Biol. Chem. 2013; 288:25915–25923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.