


Abstract
Quiescent cells reside in G0 phase, which is characterized by the absence of cell growth and proliferation. These cells remain viable and re-enter the cell cycle when prompted by appropriate signals. Using a budding yeast model of cellular quiescence, we investigated the program that initiated DNA replication when these G0 cells resumed growth. Quiescent cells contained very low levels of replication initiation factors, and their entry into S phase was delayed until these factors were re-synthesized. A longer S phase in these cells correlated with the activation of fewer origins of replication compared to G1 cells. The chromatin structure around inactive origins in G0 cells showed increased H3 occupancy and decreased nucleosome positioning compared to the same origins in G1 cells, inhibiting the origin binding of the Mcm4 subunit of the MCM licensing factor. Thus, quiescent yeast cells are under-licensed during their re-entry into S phase.
INTRODUCTION
Quiescent cells reside in G0 phase, a cell cycle stage that is characterized by the cessation of cell growth and proliferation (1–3). Quiescence is a conserved state that is common to all organisms and key to the long-term survival of stem cells (4–6). Budding yeast, Saccharomyces cerevisiae, has emerged as an excellent model system to study both the properties of quiescent cells and the factors that contribute to their development. Following the depletion of glucose from the culture medium at a point called diauxie, yeast cells enter stationary phase. During this period, cells differentiate into two distinct cell populations, one of which is comprised of non-quiescent cells that undergo apoptosis and necrosis, and a second that contains quiescent cells (7–9). Quiescent cells can be purified from stationary phase cultures by Percoll density gradient centrifugation and have been shown to exhibit the key features of G0 phase cells in mammalian cells (7,10,11).
Several studies have shown that quiescent yeast cells have distinct transcription and epigenomic profiles. During the development of quiescence following glucose depletion, transcription is repressed, ubiquitylated H2B is lost, and histone H3 and H4 acetylation levels are globally decreased (10,12–15). During this same period, histone methylation is retained on three H3 residues - lysine 4, lysine 36 and lysine 79 (10,16). As a consequence of this developmental program, purified quiescent cells contain a small pool of newly transcribed RNAs and a large cohort of stored RNAs that is central to their maintenance, and a chromatin landscape that is characterized by the presence of a specific set of modified histones (10,16–18). The molecular basis for transcriptional shut-off during the development of quiescent yeast cells has been attributed to several factors. First, at diauxie, the RPD3 histone deacetylase is targeted to many gene promoters and removes histone acetylation marks associated with the initiation of transcription (12). Second, the binding of condensin to nucleosome depleted regions of RNA polymerase II (RNAPII) regulated divergent gene promoters establishes domains of compacted chromatin that lead to global transcriptional repression (19). Third, the levels of the transcriptionally active forms of RNAPII are globally decreased shortly after diuaxie (10). Together, these factors reinforce the transition of the active transcriptional program in growing cells to the repressive program that characterizes quiescent cells.
In addition to the global repression of transcription, another key hallmark of quiescent cells is their ability to re-enter the cell cycle when prompted by appropriate stimuli. When glucose is added to quiescent yeast cells, these G0 cells enter a period in which transcription, translation and growth are restored (2,20,21). This period precedes the entry of quiescent cells into S phase, but it is not known if it is equivalent to the G1 phase of actively growing cells. The replication program used by proliferating yeast cells exiting from G1 into S phase is well defined, and is regulated by the ordered recruitment and activation of initiation factors at defined origins of replication (22,23). In contrast, little is known about the replication program employed by quiescent cells when they re-enter the cell cycle and whether it is the same or different from the program that characterizes the G1 to S phase transition. Using purified quiescent cells, we found that their entry into S phase was significantly delayed upon their return to growth medium. To investigate the basis for this delay, we examined a number of key factors known to be associated with the initiation of DNA replication in budding yeast. We found that these initiation factors, as well as the replicative DNA polymerases, were present at very low levels in quiescent cells, and that they were re-synthesized during the period of growth after glucose addition. However, once their protein levels were restored, the factors appeared to be recruited to active replication origins in the same temporal order as observed during the G1 to S phase transition. In addition to the delay in S phase entry, fewer origins of replication were activated in quiescent cells released into the cell cycle, which correlated with a longer S phase. The origins that were not activated in these cells were those that have been previously identified as late-firing and inefficient origins in cycling cells, and they showed reduced binding by the MCM origin licensing factor. The G0-inactive origins were also characterized by the presence of increased nucleosome occupancy and decreased nucleosome positioning compared to the nucleosomal profiles around origins in G1 cells. Together, the data indicate that the quiescent cell replication program is distinct from that of cycling cells, and regulated both by the levels of initiation factors and the chromatin context of replication origins in these G0 cells.
MATERIALS AND METHODS
Yeast strains and cell growth
The yeast strains listed in Supplemental Table S5 are all derived from W303. The strain used for BrdU profiling has an integrated BrdU-incorporating vector that expresses Herpes simplex virus thymidine kinase (HSV – TK) (24). Cells were grown in YPD medium (1% yeast extract, 2% peptone, 2% dextrose) at 30°C to a density of OD600 = 0.4 and arrested in G1 phase by the addition of α factor (5 μg/ml; United Biochemical Research) at 0, 45 and 90 min for a total incubation time of 135 min, or in G2/M phase by incubation in nocodazole (NOC; 20 μg/ml; Millipore-Sigma) for 2 h. G0 quiescent (Q) cells were isolated by Percoll gradient centrifugation 7 days after culture inoculation (7), and monitored for the presence of small unbudded cells. After isolation, the Q cells were washed with 50 mM Tris, pH 7.4 and released into YPD to initiate growth and re-entry into the cell cycle. Cells were released from G1 arrest by washing with 50 mM Tris, pH 7.4 before resuspension in YPD + 60 μg/ml pronase (Millipore-Sigma) and 5 mM CaCl2 as previously described (25).
Analysis of cell cycle progression
Cell cycle progression was monitored by both bud counts and flow cytometry. For bud counts, ∼0.15 OD600 units of G1 or G0 cells were harvested before and after release into YPD + pronase, CaCl2 (5 mM) and NOC (20 μg/ml) or YPD + NOC, respectively, and then fixed in ice-cold 70% ethanol. The fixed cells were washed once with 50 mM Tris, pH 7.4 and resuspended in 1 ml of 50 mM Tris. Cells were sonicated for 10 s (output 4 on a Branson 250 Sonifier), and 10 μl of cells were loaded onto a hemacytometer. The percentage of budded cells was determined by microscopic examination of 200 cells. For flow cytometry analysis, 0.4 OD600 units of G1 arrested or G0 Q cells were harvested before and after release into YPD medium and incubated with 0.1 M dithiothreitol (DTT) + 0.1% sodium azide at room temperature for 30 min (9). Cells were fixed in ice-cold 70% ethanol for 2 h or overnight, washed once with 50 mM Tris, pH 7.4, and resuspended in 50 mM Tris containing 1 mg/ml RNase A overnight at 37°C. After centrifugation, the cell pellet was suspended in 0.5 ml of pepsin solution (25 mg of pepsin in 5 ml of TE and 24 μl of concentrated HCl) and incubated for 1 h at 37°C. Following a second centrifugation, the cell pellet was resuspended in 0.5 ml of PBS (phosphate buffered saline, Millipore-Sigma) and sonicated for 10 s (Branson 250 Sonifier, output 4). Another 0.5 ml of PBS containing 0.2% NP-40 and 1 μM SYTOX-Green™ (Invitrogen™ Molecular Probes™) was added to stain nuclei for at least 2 h before loading samples onto a BD Biosciences FACS Calibur flow cytometer. BD CellQuest™ software was used for data acquisition (20 000 events) and analysis. Histograms were plotted, in which the y-axis represented the number of events and the x-axis represented the relative DNA content.
Western blot analysis
G1 arrested and G0 quiescent cells were released into YPD plus 20 μg/ml nocodozole and cells were collected over time. Cells were collected from a log phase culture and at various times during the development of quiescence, and seven days after culture inoculation quiescent cells were purified. Trichloroacetic acid (TCA) lysates were prepared from 6 OD600 units of cells and processed as previously described (26). Protein concentration was determined by the Bradford assay (Bio-Rad). Thirty micrograms of lysate were analyzed by SDS-PAGE (Criterion precast gels, Bio-Rad), followed by transfer to a PVDF membrane (Millipore-Sigma). Blots were incubated with primary antibodies overnight at 4°C, washed, and incubated for 2 h at room temperature with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (Millipore-Sigma). Antibodies and conditions are listed in Supplemental Table S5. Membrane-bound secondary antibodies were detected using Clarity ECL reagents (Bio-Rad), and blots were exposed to X-ray film. At least 2–3 independent time course experiments were performed, and the results of a representative experiment are shown.
Chromatin Immunoprecipitation
Twenty-four OD600 units of cells were collected and fixed with 1% formaldehyde for 15 min at room temperature. Fixation was stopped by the addition of glycine to a final concentration of 125 mM for 5 min and the cells were washed with 50 mM Tris, pH 7.4, followed by centrifugation. Cell pellets were resuspended in 0.6 ml of ice-cold FA lysis buffer (1% Triton X-100, 0.1% sodium deoxycholate, 50 mM HEPES–KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0) supplemented with 1× protease inhibitor cocktail (Millipore-Sigma) and 1 mM of phenylmethylsulfonyl fluoride (PMSF), and then vortexed at highest speed with an equal volume of acid-washed glass beads (Millipore-Sigma) for 25 min at 4°C. Cell lysates were sonicated using a Branson 250 Sonifier to generate DNA fragments of 200–500 bp. After centrifugation for 15 min at 17 900 × g, soluble chromatin was collected from the supernatant fraction and protein concentration was determined by a Bradford assay. Between 1 and 1.5 mg of chromatin was diluted in 1 ml of FA lysis buffer, and 20 μl was set aside as Input. Anti-Myc (Millipore-Sigma) or anti-HA (Biolegend) antibodies were added to the remaining chromatin solution and incubation was continued overnight at 4°C. Protein G agarose beads (Invitrogen) were added for 2 h at 4°C, and the beads were then sequentially washed with FA lysis buffer, wash buffer I (1% Triton X-100, 0.1% sodium deoxycholate, 50 mM HEPES–KOH, pH 7.5, 500 mM NaCl, 1 mM EDTA, pH 8.0), wash buffer II (0.5% sodium deoxycholate, 0.5% NP-40, 10 mM Tris, pH 8.0, 250 mM LiCl) and TE, pH 8.0. Immunocomplexes were eluted from beads by incubation with 0.2 ml of elution buffer (1% SDS in TE) for 20 min at 65°C. Immunoprecipitated and input samples were incubated with pronase solution (2 mg/ml pronase and 10 mM CaCl2) for 2 h at 42°C, followed by reverse crosslinking for 14–16 h at 65°C. DNA was purified with a Qiagen QIAquick PCR purification kit.
ChIP-qPCR
Following chromatin immunoprecipitation, IP and input DNAs were analyzed by quantitative real-time PCR on a StepOne™ Real-Time PCR machine (Applied Biosystems, ThermoFisher) operated by StepOne™ software (version 2.2.2). Each PCR reaction was prepared with 5 μl of DNA (diluted 1:10 for ChIP DNA and 1:50 for input DNA) in a 20 μl reaction mixture containing 0.75X Maxima SYBR Green/ROX qPCR Master Mix (ThermoFisher) and 0.15 μM of forward and reverse primers in nuclease-free water. Cycling conditions included a polymerase activation step at 95°C for 10 min, 40 cycles of amplification at 95°C for 15 s, and annealing/extension at 60°C for 1 min with melt curve analysis from 60 to 95°C in 0.3°C increments. All PCR reactions were performed in triplicate and then averaged. The relative quantity of DNA in each reaction was calculated by plotting Cq values determined by StepOne™ software against a standard curve generated from six, 10-fold serial dilutions of genomic DNA isolated by the same procedure used to prepare IP and input DNAs. All IP signals were first normalized to the corresponding input signal to adjust for DNA quantity and then normalized to the IP/input value at the ASI1 locus, which is ∼40 kb from the nearest origin of replication. The primers used for ChIP-qPCR are listed in Supplemental Table S7.
BrdU IP-seq
Sixty OD600 units of G1 arrested cells and 150 OD600 units of G0 Q cells were resuspended in 150 ml of YPD + pronase (60 μg/ml) + CaCl2 (5 mM) + 0.2M HU (Hydroxyurea, Millipore-Sigma) or 250 ml of YPD + 0.2 M HU, respectively. Immediately before and at various times after release of G1 and G0 cells, 35 ml of cell culture were transferred into a pre-warmed 100-ml flask and incubated with 800 μg/ml of BrdU (Sigma-Millipore) for overlapping periods of 20 min prior to harvesting. G1 released cells were collected at 25, 40, 55 and 70 min and G0 released cells were collected at 85, 100, 115, 130 and 160 min after BrdU pulse-labeling. Genomic DNA was isolated from each pulse-labelled sample by the ‘smash and grab’ protocol (27), and 0.2 mg/ml RNase A was added for 30 min at 37°C, followed by addition of 0.1 mg/ml of Proteinase K for 30 min at 50°C. DNA was purified with a Qiagen QIAquick PCR purification kit and its concentration was measured by an Invitrogen Qubit 2.0 Fluorometer. DNA was transferred to a 50 μl Covaris microTUBE and sheared to 200–250 bp using a Covaris M220 Focused-ultrasonicator (peak power: 75W; duty factor: 10%; cycles per burst: 200; treatment time: 400 s). One microgram of sheared DNA was end-repaired and ligated to Ion Torrent-compatible barcode adapters (Ion Xpress™ Barcode Adapters, Invitrogen) using an Ion Xpress Plus Fragment Library kit (Invitrogen). The adapter-ligated DNA was purified by Agencourt AMPure XP Reagent and DNA concentration was measured by Qubit. An equal concentration of each pulse-labeled G1 or G0 released DNA sample was pooled for immunoprecipitation (IP) and 20 ng of the mixture was set aside as input (IN) DNA (28). The pooled DNA was heated for 10 min at 95°C, followed by a snap cooling on ice. Immunoprecipitation was performed with a 1:250 dilution of anti-BrdU antibody (GE Healthcare, RPN202) for 2h at 4°C, followed by the chromatin immunoprecipitation procedure described above. The IP and Input DNA samples were amplified separately using an Ion Xpress Plus Fragment Library kit and sequenced with the Ion S5 System (ThermoFisher). The data represent the average of two independent experiments.
Mcm4 ChIP-seq
Six hundred OD600 units of G0 Q cells were isolated and released into YPD + 0.2 M HU for 90 and 105 min, and samples were collected. Ninety-six OD600 units of G1 arrested cells were also collected. Cells were fixed and processed for chromatin immunoprecipitation as described above, except the chromatin fragmentation step was performed in a 1-ml Covaris microTUBE and DNA was sheared to 200–250 bp using a Covaris M220 Focused-ultrasonicator (peak power: 75 W; duty factor: 10%; cycles per burst: 200; treatment time: 15 min for G1 samples; 25 min for G0 samples). Six microliters of anti-Myc antibodies were added to 1 mg of chromatin lysate and incubated overnight at 4°C, followed by incubation with 30 μl of Protein G Dynabeads (Invitrogen). Multiple immunoprecipitation reactions were carried out in order to obtain a total of ∼5 μg of immunoprecipitated DNA per sample. For each sample, 5 μg of DNA was extracted from lysates and processed in parallel as input DNA. IP and Input DNAs were processed for library preparation, including DNA end-repairing, barcoded adapter ligation and PCR amplification using Ion Xpress Plus Fragment Library kit (Invitrogen). The data represent the average of two independent experiments.
Data processing and analysis
All DNA sequencing reads were trimmed of barcode sequences using fastq-multx, with 5 bp removed to account for the barcode. For BrdU IP-seq analysis, the trimmed data were aligned to the Saccharomyces cerevisiae genome version sacCer3 (GCA_000146045.2) using Bowtie2 and then sorted and indexed using Samtools: http://samtools.sourceforge.net (29). BrdU IP-seq data analysis was performed as described previously (28) with the following modifications. For each labeling period, aligned sequences were filtered and sorted into 50-bp nonoverlapping bins using bamCompare from the deepTools package (30). Each IP bin was normalized after dividing by its corresponding Input bin to generate the IP/IN ratio, which was median-smoothed over a 2000 bp window across all time course experiments. Two biological replicates were averaged, and to facilitate comparisons between G1 and G0 experiments, the data were scaled to the maximum average G1 signal among the time course points, which was arbitrarily set as ‘1’. In parallel, BrdU enriched peaks were called by MACS2 (https://github.com/taoliu/MACS, version 2.1.3) using the following parameters: effective genome size of 12,100,000 and minimum FDR cutoff of 0.05. The exact command used is as follows: macs2 callpeak -t chip.bam -c input.bam -g 12100000 -n chipinput -q 0.05. Origin peaks were identified by their overlap with at least one ARS (autonomously replicating sequence; replication origin) across the samples during the time course experiment in both replicates. ARS co-ordinates were obtained from the oriDB database (31).
For Mcm4-Myc ChIP-seq analysis, the trimmed data were aligned to the S. cerevisiae genome version sacCer3 (GCA_000146045.2) using BWA (32). Aligned sequences for Mcm4 ChIP-seq samples from G1 arrested cells and from G0 cells released into YPD + HU for 90 and 105 min were filtered and binned into 50-bp nonoverlapping bins. The IP/IN ratio was median-smoothed over a 2000 bp window and normalization was performed as described above. Two biological replicates were averaged, and both the G1 and G0 data were scaled to the maximum average G1 signal, which was set as ‘1’. Mcm4-Myc enriched peaks were called by MACS2 using the same parameters described above. Origin peaks were identified by their overlap with at least one identified ARS. All additional computational analyses were conducted in R, v3.5.3.
To assess the correlation between nucleosome occupancy and positioning at origins in G0 cells, origins were classified as G0-active (BrdU+) and G0-inactive (BrdU-) based on BrdU IP-seq data. These origin classes were further classified as Mcm4-bound (Mcm4+) or -unbound (Mcm4-) based on Mcm4-Myc ChIP-seq data. Data on nucleosome occupancy in G1 and G0 cells were retrieved from (33) and (12), respectively. From these data, H3 ChIP signals 0.5 kb upstream of and downstream from the midpoint of the ACS (ARS consensus sequence; coordinates provided by Dr Toshio Tsukiyama, Fred Hutchinson Cancer Research Center) were Z-score normalized, and the average H3 ChIP signals (log2) were plotted for all origins in each sub-class. For nucleosome positioning analysis, MNase-seq data (representing 80% digestion to mononucleosomes) in log and G0 cells were retrieved from (12). The log2 MNase dyad counts were normalized to the genome average (excluding the rDNA locus) and then plotted for all origins in the sub-classes described above. The data were smoothed using the smooth spline function in the R package.
A two-tailed Welches t-test was used to determine if there was a statistically significant difference between the number of BrdU+ origins in G1 versus G0 cells, the replication timing (Trep) of G0-active vs G0-inactive origins, and the binding of Mcm4-Myc to G0-active or G0-inactive origins in G1 versus G0 cells.
For ChIP-qPCR experiments, the data are shown as the average with standard deviation from the results of at least three independent experiments.
RESULTS
Delayed entry into S phase occurs in G0 released cells
To compare the replication program used by quiescent cells upon their resumption of growth to the program that regulates the transition of G1 cells into S phase, we first monitored entry into S phase by bud emergence, which occurs coincidently with the initiation of DNA synthesis. We obtained G1 cells by arresting log phase cells with α-factor and isolated G0 quiescent cells from a 7-day stationary phase cell culture by Percoll density gradient centrifugation (7,11). Both G1 and G0 cells were then released into rich (YPD) medium containing nocodazole (NOC) to initiate growth of G0 cells and restrict analysis to a single cell cycle. Multiple experiments showed that bud emergence began in G1 cells ∼15–30 min after release but in G0 cells only 90 min after release, which represented an approximately ∼60–75 min delay in S phase entry (Figure 1). Additionally, G0 released cells had a longer S phase than G1 released cells. By measuring the length of time from the first appearance of buds to the plateau in budding, we found that G1 released cells completed S phase in ∼45–60 min, while G0 released cells took ∼90 min to progress through S phase (Figure 1). These results were supported by flow cytometry analysis of DNA content in G1 and G0 released cells, which also showed that G0 cells had a delayed entry into S phase as well as a lengthened S phase (Supplemental Figure S1). Interestingly, both the budding assay and flow cytometric analysis revealed that ∼20% of the quiescent cells released into YPD did not enter S phase, as has been previously reported in the W303 strain background (9). It is not known if these cells represent a fraction of senescent cells in the quiescent cell population or contamination of isolated quiescent cells with small non-quiescent cells that are unable to undergo DNA replication.
Figure 1.
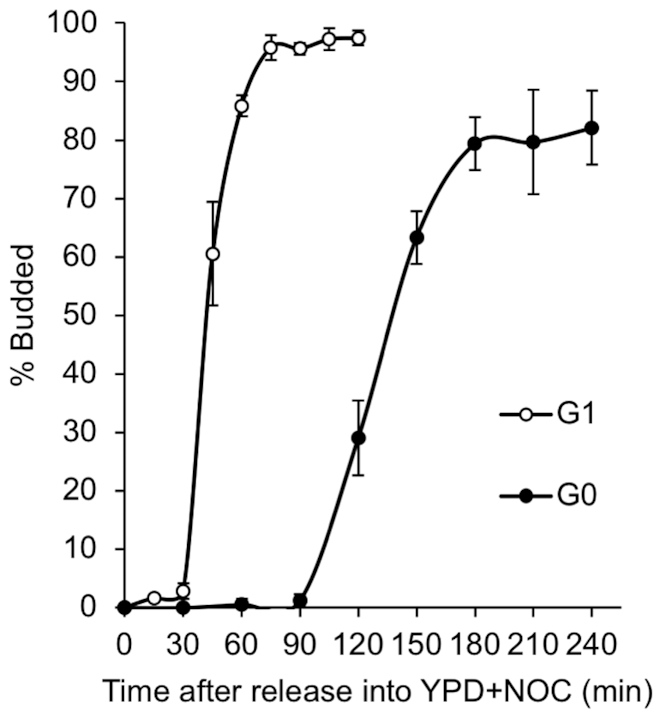
Entry of G0 released cells into S phase. G1 cells obtained by α-factor treatment and purified G0 quiescent cells were released into YPD containing 20 μg/ml of nocodazole (NOC) to block cells in G2/M following one round of DNA replication. Initiation of S phase was determined by counting the number of budded cells (100 cells) at various times after release. The results represent the average of three biological replicates with standard deviation.
Re-synthesis of replication factors occurs as G0 cells re-enter the cell cycle
The factors required to initiate DNA replication are generally present throughout the cell cycle in proliferating yeast cells (34). We wondered whether the delay in entry of quiescent cells into S phase might be a function of the level of replication initiation factors in these cells. During G1 phase, binding of the hexameric origin recognition complex (ORC) to origins co-ordinates the loading of the Mcm2–7 (MCM) helicase onto origin DNA to form the pre-replication complex (pre-RC), a key step in origin licensing (35). We first assessed the levels of several ORC proteins and the MCM helicase subunit, Mcm4, during the development of quiescent cells. Samples were collected from the starting culture during log phase, every 24 h during the development of stationary phase, and from quiescent cells isolated on Day 7. These time points encompassed the period of active growth and replication (Log), during the 1–2 rounds of slow DNA replication that occur after glucose exhaustion at diauxie (Days 1–3), and throughout the period when cells cease further growth and DNA replication (Days 3–7). Western blot analysis showed that the Orc1 and Orc2 proteins and Mcm4 were present at high levels in proliferating (Log) cells, decreased significantly after diauxie (Day 1), and were present at very low levels in purified quiescent cells (Q) (Figure 2A, B).
Figure 2.
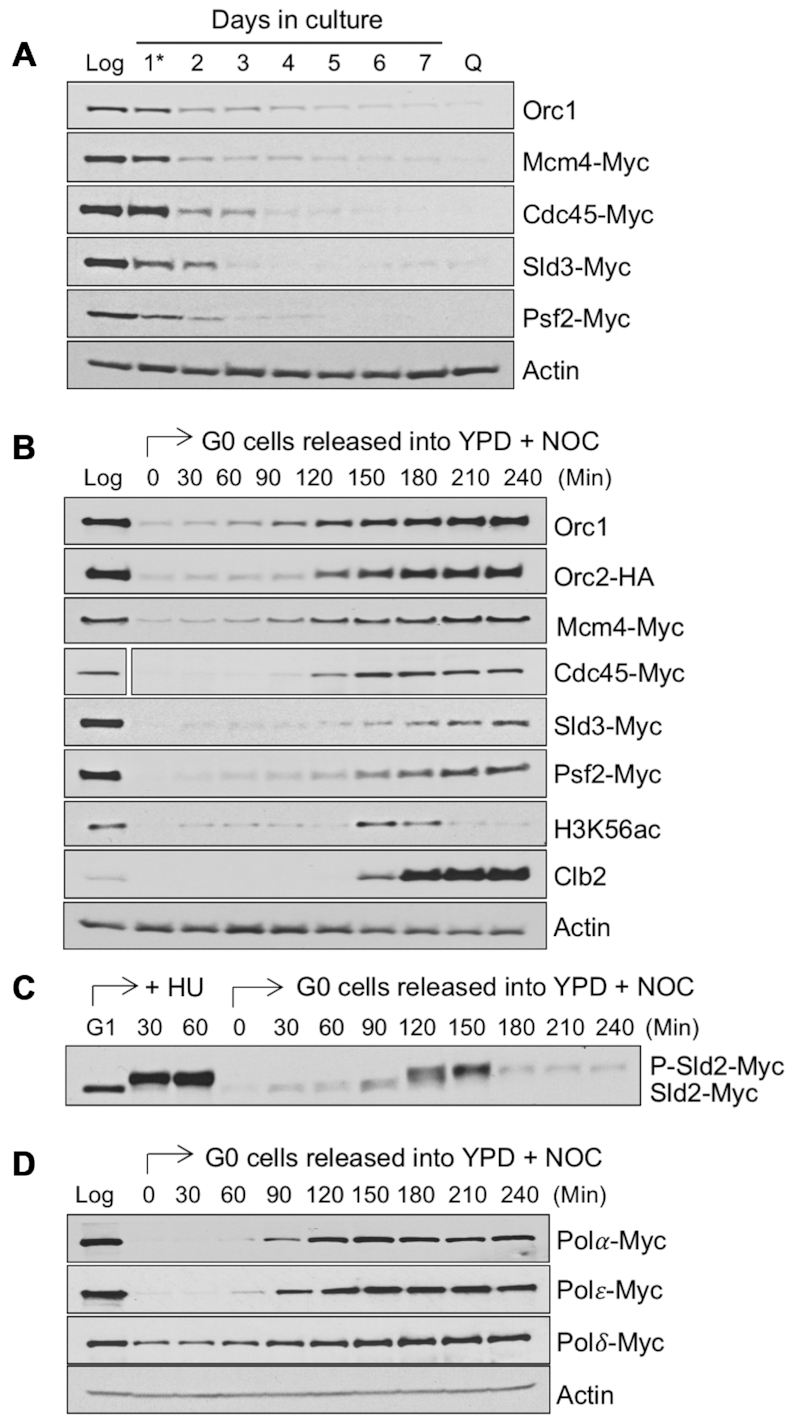
Levels of replication initiation factors during the development of quiescent cells and after release of G0 cells into the cell cycle. (A) Strains containing epitope tagged genes encoding various replication initiation factors were grown in YPD medium and allowed to develop into quiescence. Samples were removed during exponential growth (Log), upon glucose exhaustion at the diauxic shift (day 1*), and at one-day intervals thereafter. On day-7, purified quiescent (Q) cells were isolated. Western blot analysis was performed using anti-Myc antibodies or a native Orc1 antibody. Actin served as a loading control and a representative blot is shown. (B) G0 quiescent cells were isolated from strains containing epitope tagged replication initiation factors and released into YPD in the presence of 20 μg/ml of nocodazole (NOC). Samples were removed immediately before and at various times after release and western blot analysis was performed using antibodies against HA, Myc, or Orc1. The level of each protein in growing cells (Log) was also interrogated. Antibodies against H3K56ac and Clb2 were employed to identify S phase and G2/M phase, respectively. Actin served as a loading control and a representative blot is shown. The space gap after the Cdc45-Myc sample resulted from removal of irrelevant lanes from the blot. (C) CDK-dependent phosphorylation of Sld2 was examined by western blot analysis of lysates from cells released from G1 into hydroxyurea (HU) and from G0 into YPD + NOC using an anti-Myc antibody. (D) G0 Q cells were isolated from strains containing Myc tagged DNA polymerases (Pol α, Polϵ, Polδ) and released into YPD + NOC. The levels of these polymerases were measured by western blot analysis at various time after release using anti-Myc antibodies and compared to the levels in log phase cells. Actin served as a loading control and a representative blot is shown.
We next examined the levels of several additional factors that are required to initiate DNA synthesis. At the G1 to S phase transition, the concerted actions of the Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) activate MCM and induce bi-directional origin firing and DNA synthesis. First, DDK-dependent recruitment of Cdc45, Sld3 and Sld2 to licensed origins occurs, followed by CDK-dependent phosphorylation of Sld2. This, in turn, stimulates the recruitment of DNA polymerase epsilon (Polϵ) and the GINS complex to form a functional pre-initiation complex (pre-IC), which bi-directionally initiates DNA synthesis. Similar to what we observed for pre-RC factors during the development of stationary phase, the abundance of Cdc45, Sld3 and the GINS complex subunit, Psf2, declined dramatically after diauxie (Day 1), and the proteins were not present in purified quiescent cells (Figure 2A, B). Moreover, the replicative DNA polymerases, Polα, and Polϵ, were also not detectable in quiescent cells, and Polδ was present at lower levels in these cells than in proliferating cells (Figure 2D). Finally, transcriptome data indicated that the RNAs encoding replication factors and cell cycle regulators were also present at very low levels in purified quiescent cells (Supplemental Figure S2A, B). Together, the data suggest that the low levels of multiple replication factors in quiescent cells might account for the delay in initiating DNA synthesis when these cells are released into the cell cycle.
When quiescent cells are returned to glucose-rich medium, transcription and translation are stimulated (21,36). We therefore asked if replication factors were re-synthesized during this period of growth resumption and if their re-synthesis temporally correlated with the initiation of S phase. Quiescent cells were collected and released into YPD medium plus NOC, and samples were collected over time after release for western blot analysis of the same replication factors examined during the development of quiescence. Using H3 lysine 56 acetylation (H3K56ac) and Clb2 as markers for S phase and G2/M phase, respectively, we found that the levels of these replication factors increased over time after cells were released into YPD, accumulating just prior to or during S phase (Figure 2B–D; Supplemental Figure S3). Interestingly, as noted above, the lagging strand DNA polymerase, Polδ, was present at detectable levels in quiescent cells, and it was also present during early times after the release of these cells into the cell cycle (Figure 2D). This suggests that this DNA polymerase might play a special role during the first S phase that quiescent cells undergo. Together, the results support the view that the initiation of S phase in G0 cells released from quiescence depends in part on the restoration of growth in these cells, which promotes the re-synthesis of key replication factors.
We next used chromatin immunoprecipitation (ChIP) to examine the recruitment of the origin licensing and activation factors, Mcm4 and Cdc45, to a subset of well-characterized origins, three classified as early firing (ARS305, ARS306 and ARS607) and one classified as late firing (ARS501), in G1 arrested cells and in G0 cells before and after their release into YPD plus NOC. Mcm4 bound to both the early and late origins in G1 arrested cells, but it was present at low levels at these same origins in quiescent cells, in agreement with the reduced level of this protein in G0 cells (Supplemental Figure S4A, top panel). Mcm4 was eventually recruited to each of these origins in G0 cells 90 min after their release, a time that coincided with the increase in the level of the Mcm4 protein (Supplemental Figure S4A, bottom panel). In growing cells, Cdc45 associates with origins at the G1-S phase transition. Following the release of G1 arrested cells into hydroxyurea (HU) for 30 min, Cdc45 was associated with the three early origins but not the late origin, consistent with the inhibition of late origin activation by HU (Supplemental Figure S4B, top panel). In G0 released cells, Cdc45 did not bind to the early origins until its protein level had been re-established at ∼120 min after release, a time representing early S phase (Supplemental Figure S4B, bottom panel). Interestingly, Cdc45 was not recruited to the ARS501 late origin even at later times after release of G0 cells, suggesting that the utilization of some origins might be altered in these cells. The results suggest that key factors required for the initiation of DNA replication are recruited to origins in a similar temporal pattern when G1 and G0 cells are released into the cell cycle. However, in G0 cells the recruitment program is not initiated until these cells have resumed growth and re-synthesized replication factors.
Reduced genome-wide origin utilization during re-entry of G0 cells into the cell cycle
In addition to delayed entry into S phase, quiescent cells also exhibited a longer S phase once they had initiated DNA replication. We hypothesized that this lengthened S phase might be the consequence of a reduction in the number of replication origins activated in these cells. To test this hypothesis, we performed BrdU (5-bromo-2′-deoxyuridine) immunoprecipitation coupled with high-throughput DNA sequencing (BrdU IP-seq) to identify the genome-wide pattern of BrdU-labeled nascent DNA in both G1 and G0 cells. Each cell population was released into YPD in the presence of 0.2 M HU and pulse labeled with BrdU for overlapping intervals of 20 min before harvesting, using a modified BrdU IP-seq protocol (28) (Figure 3A). Examination of BrdU IP-seq signals across the genome revealed that in both G1 and G0 released cells BrdU peaks centered on well-defined origin sites; however, the number of peaks was markedly reduced in G0 released cells (Figure 3B, C; Supplemental Figure S5 for all chromosomes). For example, plots of BrdU incorporation along Chromosome XI in G1 released cells showed several distinct BrdU peaks representing early-firing, efficient origins (ARS1103, ARS1107.5, ARS1109. ARS1113.5, ARS1114, ARS1114.5, ARS1116) and several minor peaks representing late-firing, inefficient origins (ARS1104.5, ARS1106.3, ARS1106.7, ARS1107, ARS1112, ARS1120). In contrast, BrdU peaks were reduced at each of these origins in G0 released cells. In G1 cells, the major BrdU signals reached their peak ∼20–40 min after release, and at later times, the signals migrated divergently from these origins as DNA synthesis proceeded (Supplemental Figure S5). In G0 released cells, the peak of BrdU incorporation occurred later (∼95–115 min), and similar to G1 released cells, the BrdU signals also migrated bidirectionally from active origins (Supplemental Figure S5). Together, the data support the observation that the initiation of DNA replication was delayed during the re-entry of quiescent cells into the cell cycle, and that the extended S phase in these cells was likely related to the activation of fewer origins of replication.
Figure 3.

Genome-wide replication initiation during the re-entry of G0 cells into the cell cycle. (A) Schematic for BrdU pulse-labeling of G1 and G0 released cells. G1 cells were obtained by α-factor treatment and G0 cells were isolated from a day 7 stationary phase culture by Percoll gradient centrifugation. Both sets of cells were released into YPD containing 0.2 M hydroxyurea (HU) and pulse-labeled with 800 μg /ml of BrdU for overlapping intervals of 20 min before collection at the end of each pulse period. Pulse-labeled DNAs were pooled, immunoprecipitated with anti-BrdU antibody, and subjected to BrdU IP-seq analysis. (B) BrdU IP-seq was performed on two biological replicates and averaged data representing BrdU pulse labeling 20–40 min (G1) or 95–115 min (G0) after release are shown for chromosome XI. Arrowheads indicate MACS2-called peaks that align with known origins (ARS) in G1 (blue) and G0 (green) released cells. (C) The number of active (BrdU+) origins in G1 and G0 cells was calculated from BrdU IP-seq of samples pulse labelled with BrdU during 5–25 min and 20–40 min (G1) or 80–100 min and 95–115 min (G0) after release into YPD + HU. There is a statistically significant difference between the number of BrdU+ origins in G1 vs G0 released cells (P = 1.006e–7). (D) Data on origin replication timing (Trep) in proliferating cells were retrieved from Raghuraman et al. (38), and used to calculate the median Trep of G0-active (BrdU+) and G0-inactive (BrdU-) origins. The median, upper and lower quartiles, as well as upper and lower extreme data points, are indicated. The difference in Trep between G0-active and G0-inactive origins is statistically significant (P = 1.717e–9).
To identify the origins that were initiated during the entry of G1 and G0 cells into S phase, we aligned the BrdU peaks called by MACS2 with the 410 confirmed origins present in OriDB (31), and considered only those peaks that overlapped with known origin sequences for subsequent analysis. In G1 released cells, significant peaks of BrdU incorporation were detected at 237 origins (Figure 3C; Supplemental Table S1). In contrast, only 160 of these same origins were initiated in G0 released cells (Figure 3C; Supplemental Table S1), a 33% reduction in origin utilization. Importantly, even at later times after release, no additional BrdU-containing origin peaks were detected in G0 cells (Supplemental Figure S5). This was of interest because the DNA replication stress-induced phosphorylation of the Rad53 checkpoint effector was significantly impaired when G0 cells were released into HU (Supplemental Figure S6). We would therefore have expected that the constraints on late origin firing would have been lifted and have resulted in a larger number of origins incorporating BrdU (37). Together, the reduced number of BrdU positive peaks and weak S phase checkpoint response are consistent with G0 cells having fewer numbers of active origins when they re-entered the cell cycle.
Based on the BrdU IP-seq data, we characterized origins in G0 cells as either active (BrdU+) or inactive (BrdU-) (Supplemental Tables S1 and S2). To identify the features that distinguished G0-active from G0-inactive origins, we compared the replication timing (Trep) of these two classes of origins during the mitotic cell cycle using published data (38). This analysis revealed that the median Trep for the set of G0-active origins was 21.6 min, whereas the median Trep for G0-inactive origins was 27.4 min (Figure 3D; Supplemental Table S3). Thus, in cycling cells G0-active origins tend to be fired earlier than G0-inactive origins. Moreover, the G0-active origins were mostly associated with origins that have been classified as efficient, while the G0-inactive origins represented inefficient origins (39,40). Interestingly, 25% of the G0-inactive origins also represented origins that are regulated by Rpd3 (41), the HDAC that promotes the transition to transcriptional repression during the development of quiescent cells (Supplemental Figure S7). Taken together, the data suggest that the fewer number of origins activated after G0 cells re-enter the cell cycle represented a failure to initiate DNA replication at a class of late-firing and inefficient origins.
Reduced Mcm4 binding in G0 released cells affects origin utilization
A key early step in the initiation of DNA synthesis is the licensing of origins by the ORC-dependent binding of the MCM helicase. To assess the global licensing of origins by MCM during the entry of G0 quiescent cells into the cell cycle, we used ChIP-seq to identify the origin binding sites of Mcm4-Myc across the genomes of G0 and G1 cells. Samples were prepared for ChIP-seq from G1 arrested cells and at 0, 90 and 105 min after G0 cells were released into YPD + HU, with the latter two times chosen as they were close to when DNA replication was initiated. A number of Mcm4 peaks were associated with previously identified origins in G1 cells, while significantly fewer of these same origins were bound by Mcm4 in G0 cells even 105 min after their release (Figure 4A; Supplemental Figure S8). Out of the 410 confirmed origins listed in OriDB, 341 were bound by Mcm4 in G1 arrested cells and 234 were bound in G0 cells 90 and 105 min post-release (Figure 4B; Supplemental Table S4), representing an ∼33% reduction in the number of origins loaded with Mcm4. While the majority of origins bound by Mcm4 in G0 released cells were also bound by Mcm4 in G1 cells (228/234), six origins showed Mcm4 binding exclusively in G0 released cells (Figure 4B); however, while all of these origins were classified as active in G1 cells, only three were active in G0 released cells.
Figure 4.

Genome-wide association of Mcm4 with origins in G0 released cells. Cells containing Mcm4-Myc were arrested in G1 by treatment with α-factor and G0 Q cells were released into YPD + 0.2 M HU. Chromatin IP was performed with anti-Myc antibody from two biological replicates of G1 arrested cells and from G0 cells released into HU for 90 and 105 min. IP and IN samples were analyzed by ChIP-seq. (A) Mcm4 enrichment on chromosome XI in G1 arrested cells (blue) and G0 released cells (red). Arrowheads indicate MACS2-called peaks that correspond to known replication origins. (B) Number of origins bound by Mcm4 in G1 arrested or G0 released cells. The number of Mcm4-bound origins in G0 released cells was determined by counting MACS2-called origin peaks in the 90 and 105 min released samples. (C) Venn diagram showing Mcm4 binding at G0-active and G0-inactive origins in G1 arrested (left) or G0 released (right) cells. There is a statistically significant difference in the binding of Mcm4 to G0-inactive origins in G0 released cells compared to G1 cells (P = 0.0006989). The difference in the binding of Mcm4 to G0-active origins in G0 released versus G1 cells is not statistically significant (P = 0.1556).
To address the possibility that the reduced number of Mcm4-bound origins in G0 released cells was due to limiting MCM, we quantitated the levels of Mcm4 in quiescent cells after they were returned to the cell cycle, and compared these levels to the level of Mcm4 in G1 cells (Supplemental Figure S3). The data showed that by 90–120 min after G0 cells were returned to growth, Mcm4 reached levels slightly lower than those in proliferating cells. However, because MCM is present in significant excess relative to the number of origins, these levels were likely sufficient for its loading at all known origins (42,43). Importantly, at longer times after the re-entry of G0 cells into S phase, when Mcm4 levels were even higher, no additional origins were activated (compare Supplemental Figures S3 and S5). We next asked if Cdc6 and Cdt1 were limiting when quiescent cells re-entered the cell cycle, as these two factors play critical roles in the loading MCM at origins through their ORC association (43–45). In particular, we asked if the ratio of these factors to ORC was different between cells in G1 phase and in quiescent cells at several time points after their release into YPD. The results showed that while both loading factors were present at lower levels in quiescent cells compared to G1 cells, there was no difference seen in the Cdc6/Orc1 or Cdt1/Orc1 ratio between G1 cells and G0 released cells (Supplemental Figure S9). Thus, in G0 cells other factors likely contributed to the under-licensing of origins by MCM upon re-entry into the cell cycle.
Next, we investigated whether the decreased frequency of origin activation in G0 released cells was linked to reduced origin licensing by MCM by comparing the results of the BrdU IP-seq and Mcm4 ChIP-seq analyses (Figure 4C). For the 160 G0-active origins that were common to both G1 and G0 released cells, a similar number of origins had bound Mcm4 (G1: 141/160, 88%; G0: 130/160; 81%). However, for the 77 G0-inactive origins that were only active in G1 cells, the number of origins bound by Mcm4 was reduced from 77% (60/77) in G1 cells to 50% (39/77) in G0 released cells, a statistically significant difference. Overall, these data suggest that the fewer number of active origins in G0 released cells was a consequence of the reduced loading of MCM onto the class of G0 origins classified as inactive and inefficient.
Reduced origin utilization in G0 cells correlates with increased nucleosome occupancy and decreased nucleosome positioning
Chromatin structure, including nucleosome occupancy and positioning, has been implicated in the timing and efficiency of replication initiation (46–48). Given these links, we first analyzed nucleosome occupancy around the origins classified as either G0-active (BrdU+) or G0-inactive (BrdU-). The levels of nucleosome occupancy at these two classes of origins in G1 and G0 cells were retrieved from published H3 ChIP-seq datasets (12,33). The average H3 ChIP signals spanning 0.5 kb upstream and 0.5 kb downstream from the midpoint of the ACS (ARS consensus sequence) were then plotted (Figure 5A). In both G0 and G1 cells, the H3 occupancy profile around the two classes of origins revealed a typical nucleosome depleted region (NDR) that encompassed the ACS (46,47). However, the average H3 occupancy surrounding the NDR of both G0-active and G0-inactive origins was higher in G0 cells than in G1 cells (Figure 5A; Supplemental Figure S10). It has been reported that nucleosome density increases at transcription initiation sites during the transition of growing cells to quiescent cells, and that this chromatin structural change is intimately linked to the transcriptional shutoff that occurs during quiescent cell development (12,19). The present data support the view that nucleosome occupancy around replication origins also increases during the development of quiescent cells and may impair their utilization.
Figure 5.

Nucleosome occupancy at origins in G1 and G0 cells. (A) Average H3 ChIP signals (log2) around G0-active (left, BrdU+, n = 160) or G0-inactive (right, BrdU–, n = 77) in G1 cells (cyan) and G0 cells (red). Average H3 ChIP signals for each class of origins were aligned at the midpoint of the ACS (ARS consensus sequence). Data for H3 ChIP signals in G1 and G0 cells were retrieved from Rodriguez et al. (33) and McKnight et al. (12), respectively. (B) The difference in H3 ChIP signals (log2) between G1 and G0 cells around G0-active (left) or G0-inactive (right) origins.
To ask whether there might in fact be a difference in the pattern of nucleosome occupancy between G0-active and G0-inactive origins, we subtracted H3 occupancy at these origins in G1 cells from that in G0 cells, and plotted the difference across the G0-active and G0-inactive origins (Figure 5B). While both origin classes had increased H3 occupancy flanking the ACS in G0 cells compared to G1 cells, G0-inactive origins exhibited a greater increase in H3 occupancy in G0 cells than did G0-active origins, with the largest increase occurring immediately adjacent to the ACS. Together, the results suggest that G0-inactive origins tend to have increased nucleosome occupancy in G0 cells compared to G1 cells, and this, in turn, compromises the efficiency of origin firing when quiescent G0 cells re-enter the cell cycle. While G0-active origins also had higher nucleosome occupancy in G0 cells, we assume that this chromatin landscape is remodeled during the period of growth restoration to allow replication to be initiated.
In vitro and in vivo studies have shown that nucleosome density directly modulates the initiation of DNA replication at multiple steps, including origin licensing as well as origin activation (47–49). To investigate whether the reduced origin licensing observed in G0 released cells was linked to increased nucleosome occupancy, we compared the levels of H3 around the ACS of G0-active origins bound by Mcm4 (BrdU+ Mcm4+) and G0-inactive origins not bound by Mcm4 (BrdU– Mcm4–). H3 occupancy was higher at both classes of origins in G0 cells compared to G1 cells (Figure 6A, B). However, in G0 cells, H3 occupancy increased further at those origins that showed no Mcm4 binding and no BrdU incorporation (BrdU– Mcm4–) (Figure 6B). Interestingly, the pattern of H3 occupancy at this particular class of origins overlaps with the region where MCM binds to origins in G1 cells (33,48). These data suggest that the extent of nucleosome occupancy at a subset of origins plays an important role in preventing both origin licensing and activation when quiescent cells re-enter the cell cycle.
Figure 6.

Nucleosome occupancy in G0 cells influences both origin licensing and activation. (A) Average H3 ChIP signals (log2) in G1 (cyan) and G0 cells (red) around G0-active origins that are bound by Mcm4 (left, G0: BrdU+ Mcm4+, n = 110), and G0-inactive origins that are not bound by Mcm4 (right, G0: BrdU- Mcm4–, n = 22). Average H3 ChIP signals for each class of origins were aligned at the midpoint of the ACS. Data for H3 ChIP signals in G1 and in G0 cells were retrieved from Rodriguez et al. (33) and McKnight et al. (12), respectively. Mcm4 origin association with these classes of origins was determined as described in Figure 4 legend. (B) The difference in H3 ChIP signals (log2) between G1 and G0 cells around G0-active origins that are bound by Mcm4 (left) and G0-inactive origins that are not bound by Mcm4 (right).
In addition to the extent of nucleosome occupancy at origins, the positioning of nucleosomes around origins is also important for loading the Mcm2–7 helicase and the timing of origin activation (50). To determine whether there was a link between nucleosome positioning and origin activation in G0 cells, we retrieved nucleosome dyad signals from an MNase-seq data set generated in quiescent cells (12), and analyzed the signals across G0-active and G0-inactive origins (Figure 7A). The dyad signals at the ACS were very low for both classes of origins, consistent with the absence of well-positioned nucleosomes in this region. However, significant differences between G0-active and G0-inactive origins were seen in the adjacent upstream and downstream dyad signals. At G0-active origins, the dyad signals were strong and periodic, representing well-positioned nucleosomes (Figure 7A; BrdU+) (46,47). In contrast, at G0-inactive origins, the dyad signals were significantly weaker and lacked the precise periodicity seen around G0-active origins, indicating a decrease in nucleosome positioning (Figure 7A; BrdU–). Because around half of the G0-inactive origins were not bound by Mcm4, we also examined the nucleosome dyad signals for this class of origins (Figure 7B; BrdU– Mcm4–). Notably, these origins showed even more pronounced alterations in nucleosome positioning at both the NDR and origin-adjacent regions. In particular, the upstream –1 nucleosome was seen to encroach into the NDR. Together, the results suggest that the patterns of both nucleosome occupancy and positioning around inactive origins in G0 cells inhibit their licensing and firing when these cells are stimulated to re-enter the cell cycle.
Figure 7.
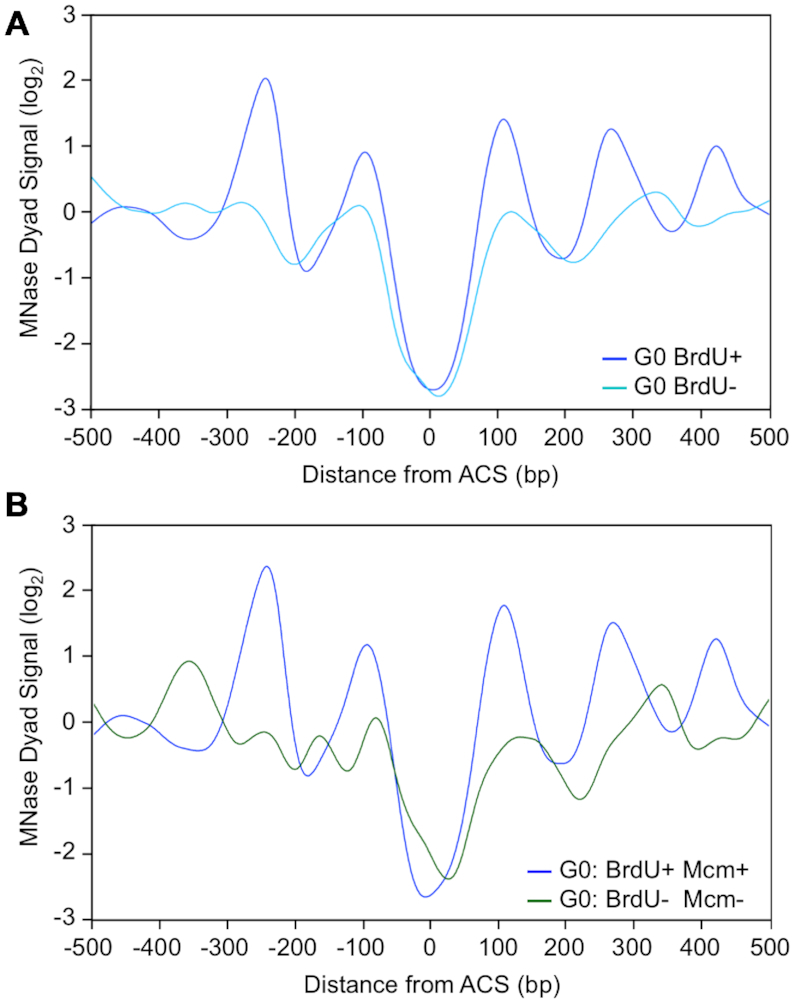
Nucleosome positioning at origins in G0 cells. (A) Nucleosome positioning around origins in G0 cells. Average MNase (micrococcal nuclease) dyad signals around G0-active (dark blue, G0: BrdU+, n = 160) and G0-inactive origins (light blue, G0: BrdU–, n = 77) in G0 cells. Data for MNase dyad signals in G0 cells (80% digestion to mononucleosomes) were retrieved from McKnight et al. (12). Data were aligned to the midpoint of the ACS. (B) Average MNase dyad signals around G0-active origins that are bound by Mcm4 (dark blue, G0: BrdU+ Mcm4+, n = 110), and G0-inactive origins that are not bound by Mcm4 (green, G0: BrdU– Mcm4–, n = 22) in G0 cells. Data for MNase dyad signals in G0 cells (80% digestion to mononucleosomes) were retrieved from McKnight et al. (12). Data were aligned to the midpoint of the ACS.
DISCUSSION
Little is known about the program that regulates the initiation of DNA replication during the re-entry of G0 phase quiescent cells into the cell cycle and how it compares to the program that regulates the initiation of DNA replication during the transition from G1 to S phase in growing cells. In this study, we used a yeast model of cellular quiescence to investigate this program in G0 cells during the first cell cycle after they were returned to growth. BrdU IP-seq analysis revealed that the initiation of DNA synthesis was delayed and fewer origins were activated when G0 cells entered the cell cycle compared to G1 cells, resulting in a longer S phase. G0 cells contained low levels of key replication initiation factors, and their delayed entry into S phase temporally correlated with a period of growth in which the re-synthesis of these factors occurred. Mcm4, a subunit of the MCM licensing factor, bound to a fewer number of replication origins during the re-entry of G0 cells into the cell cycle compared to G1 cells, and origins not associated with Mcm4 represented a class of late-firing, inefficient origins that failed to be activated. The inactive origins in G0 cells showed increased nucleosome occupancy and reduced nucleosome positioning compared to the same origins in G1 cells. Together, the data support the view that during cell cycle re-entry from quiescence, the chromatin context of a subset of origins in G0 cells contributes to origin under-licensing and reduced activation of DNA synthesis (Figure 8).
Figure 8.
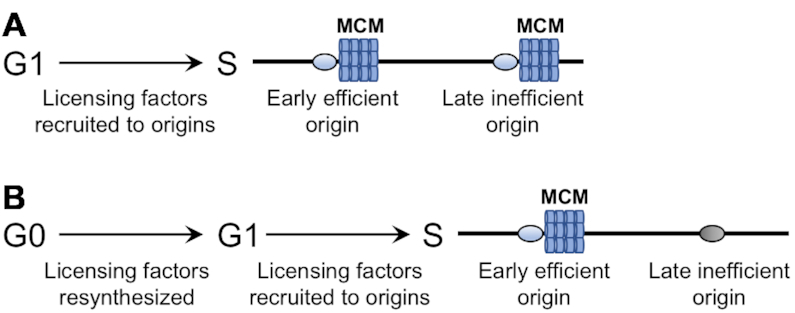
Origins are under-licensed during the re-entry of G0 cells into the cell cycle. (A) During G1 phase in proliferating cells, licensing factors are present and recruited to all origins during entry into S phase. Loading of the hexameric MCM helicase is a key step in origin licensing. (B) G0 cells have low levels of replication factors, including origin licensing factors. When G0 cells are returned to growth, these factors are re-synthesized and the cells enter G1. At S phase entry, MCM is preferentially loaded at early firing, efficient origins. The chromatin context of late firing, inefficient origins inhibits MCM loading, and thus G0 cells are under licensed when they re-enter the cell cycle.
In budding yeast, replication initiation factors are present in G1 phase cells and recruited to origins in a defined temporal order to first license and then activate DNA synthesis during the G1 to S phase transition (22,23). We found that many of these factors were present at very low levels in G0 cells, with their expression dropping precipitously during the development of quiescence, co-incident with the repression of transcription and translation that occurs during the same period (10,12,51). However, once quiescent cells were returned to growth, initiation factors were re-synthesized, and analysis of a key licensing factor subunit, Mcm4, and an activation factor, Cdc45, revealed that these proteins were recruited to a set of representative origins in a similar temporal order to that observed in G1 cells. This suggests that G0 cells first undergo a transition into a G1-like phase to render them competent to initiate DNA replication, and that a key determinant of this transition is the re-synthesis of proteins that execute the entry into S phase. This is similar to but distinct from the commitment point in G1 cells in which RNA and protein content increase and cells enter S phase when they reach a certain size. While the delayed entry of G0 cells into S phase temporally correlated with the transitional growth period in which replication proteins were re-synthesized, this may be just one of the factors contributing to the delay.
Temporal profiling of origin activation during S phase in mitotically dividing cells has defined two general classes of origins—those on average fired early and those fired late (52). BrdU IP-seq analysis revealed that fewer origins overall were activated when G0 cells entered S phase compared to G1 cells, and that the origins not fired in G0 cells primarily represented origins previously classified as late-firing and inefficient. Several mechanisms have been advanced to account for the temporal program of origin activation when G1 cells enter S phase. One mechanism proposes that origin firing is influenced by competition for the loading of rate-limiting factors onto origins (53,54). In support of this mechanism, several initiation factors, including Sld2, Sld3 and Cdc45, are present at low levels in yeast and preferentially bind to early-firing origins during the G1 to S phase transition (53,54). The levels of several of these proteins were even lower in G0 cells that had been returned to growth (Figure 2), and might account for the preferential activation of early origins when these cells entered S phase and the failure to activate late origins even after extended times. A second mechanism proposes that replication timing is regulated by the number of MCMs loaded at origins, with early origins having more MCMs loaded, leading to a greater probability that these origins will fire earlier in S phase (55). Mcm4 ChIP-seq data revealed that when G0 cells entered S phase, Mcm4 preferentially bound to active (BrdU+), early-firing origins over inactive (BrdU-), late-firing origins (Figure 4). As the maximum loading of Mcm4 onto origins in G0 released cells occurred prior to the restoration of Mcm4 levels to those observed in proliferating cells (Figure 2; Supplemental Figures S3 and S4A), MCM does not appear limiting for binding to origins in either these G0 cells or in G1 cells (56–58).
We propose that the local chromatin environment of origins in G0 cells plays an important role in identifying those origins that will be loaded by MCM for their eventual activation. Both nucleosome occupancy and positioning around replication origins have been linked to origin activation (33,48). Yeast origins are typically comprised of a nucleosome depleted region (NDR) containing a core DNA sequence (ACS) flanked by well-positioned nucleosomes, an environment that favors binding of ORC and MCM (46,47). A genome-wide analysis of nucleosome occupancy around origins in G1 cells revealed that there was a statistically significant correlation between low nucleosome occupancy and the early firing time of efficient origins (33). Moreover, MCM loading was highest at those origins having low nucleosome occupancy, consistent with a more open chromatin environment favoring pre-RC origin licensing (33,48). We found that while the average nucleosome occupancy at origins was higher in G0 cells compared to G1 cells, a larger increase in occupancy occurred at those origins classified as inactive (Figure 5). Additionally, nucleosome occupancy was also higher at inactive origins that did not bind Mcm4 upon the release of G0 cells into the cell cycle (Figure 6). Together, these results support the view that the more closed chromatin structure of a subset of origins in G0 cells prevented MCM loading and inhibited origin activation. Interestingly, origins that were activated upon release of G0 cells, most of which were bound by Mcm4, also had increased nucleosome occupancy in G0 cells compared to G1 cells (Figures 5 and 6). This apparent paradox between high nucleosome occupancy, efficient Mcm4 binding, and early origin firing time could be explained by assuming that nucleosomes were selectively depleted at these particular origins during the period of growth that preceded the entry of G0 cells into S phase. The identity of the factors facilitating nucleosome depletion and how they associate with particular origins are unknown, although one possibility is that the presence of MCM at G0-active origins could recruit chromatin remodelers such as SWI/SNF (49,59), FACT (60) or Asf1 (61) to open up chromatin around this class of origins. Alternatively, nucleosomes around these origins might be selectively remodeled by targeted histone acetylation, which occurs rapidly after quiescent cells are returned to growth and is correlated with origin activity (62).
Yeast origins are also characterized by the precise positioning of nucleosomes adjacent to the ACS element within the NDR (46,47). Nucleosome positioning is dependent on ORC binding to the origin, although, interestingly, this organization was not found to be related to origin efficiency or timing (47). The characteristic pattern of nucleosome positioning surrounding the ACS was present at active origins in G0 cells, while both nucleosome density and positioning on both sides of the ACS were altered at inactive origins (Figure 7A). Moreover, inactive origins that were not bound by Mcm4 after the release of G0 cells showed an even greater alteration in the pattern of nucleosome positioning, with encroachment of nucleosomes into the ACS region (Figure 7B). As ORC binding to origins is a prerequisite for Mcm4 recruitment, this suggests that G0-active origins were bound by ORC, while most G0-inactive origins had little or no ORC association. However, the levels of Orc1, Orc2, and Mcm4 were very low in G0 cells (Figure 2; Supplemental Figure S3), raising the question of how an ORC-dependent nucleosome positioning pattern could have been established at those origins destined to be activated when G0 cells enter the cell cycle. One possibility is that this pattern, as well as the level of nucleosome occupancy at origins, is set up during the period of DNA replication that takes place immediately after glucose deprivation as quiescent cells develop. A number of the G0 origins, particularly those classified as late-firing and inefficient, have been previously characterized as dependent on Rpd3 (41,63,64), which is globally recruited to yeast promoters to deacetylate histones and repress transcription during this same period (12). This suggests that Rpd3 may also establish a repressive chromatin architecture at these origins through deacetylation of histones on origin-adjacent nucleosomes as quiescent cells develop, thereby inhibiting their utilization (41,63,64). Thus, similar to the changes in chromatin structure at origins that occur between G2/M and G1 cells (33,48), dynamic changes in origin chromatin architecture may also occur during the formation of G0 cells. These changes, in addition to the concentration of replication initiation factors, may help to define the origins that will be active when these cells enter the cell cycle.
Other chromatin features have also been linked to origin selection and activation. As mentioned above, on a local level the presence of histone acetylation on nucleosomes surrounding replication origins has been proposed to create an open chromatin structure that advances replication timing (16,63,65). Although G0 cells contain very low levels of acetylated histones (10,12,16), acetylation is rapidly restored when these cells are returned to growth (16). While the global distribution of these newly acetylated histones is not known, one possibility is that acetylation occurs preferentially on nucleosomes flanking early firing origins, rendering these origins permissive for replication factor binding and activation. In addition to local chromatin structure, 3D chromatin structure has also been implicated in replication timing in both yeast (66) and mammalian cells (67). In G1 phase yeast cells, early firing origins are clustered by Fkh1/Fhk2 factors, programming these origins for activation (66). During the entry of yeast cells into quiescence, chromatin is also organized into large structural domains by the binding of condensin, leading to the global repression of transcription (19). Whether this organization also affects origin utilization when G0 quiescent cells are stimulated to enter the cell cycle remains unknown. Nonetheless, it is apparent that both local and global features of chromatin could have profound effects on origin definition, selection and timing in G0 cells.
Mammalian cells re-entering the cell cycle from quiescence are also under-licensed for the initiation of DNA synthesis, and this results from a compromised origin licensing checkpoint and slow MCM loading (68,69). While G0 yeast cells exiting quiescence also exhibit under-licensing and delayed replication initiation, an origin licensing checkpoint is not operative in budding yeast cells (70). Based on our results, we suggest that the contribution of slow MCM loading to under-licensing in mammalian cells may also be the consequence of origin chromatin context inhibiting MCM loading.
DATA AVAILABILITY
All ChIP-seq datasets have been be deposited to GEO, accession number GSE155831.
Link to a UCSC genome browser session displaying the uploaded sequence tracks:
http://genome.ucsc.edu/s/pohsuenlee/BrdU%20IP-seq%20%26%20Mcm4%20ChIP-seq.
Supplementary Material
ACKNOWLEDGEMENTS
The Analytical and Translational Genomics, Bioinformatics, and Flow Cytometry Shared Resources of the UNM Comprehensive Cancer Center provided excellent technical support. Toshi Tsukiyama is gratefully acknowledged for providing ARS coordinates, and Catherine Fox is thanked for a gift of ORC1 antibody. Eric Small is thanked for providing help with statistical analysis and helpful comments on the manuscript, and Olufunmilola M. Oyebamiji is acknowledged for her dedicated bioinformatics support.
Contributor Information
Po-Hsuen Lee, Department of Molecular Genetics and Microbiology University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA.
Mary Ann Osley, Department of Molecular Genetics and Microbiology University of New Mexico Health Sciences Center, Albuquerque, NM 87131, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health, National Institute of Aging [AG054494 to M.A.O.]; National Cancer Institute [P30 CA118100 to C.W.]. Funding for open access charge: National Institutes of Health, National Institute of Aging [AG054494 to M.A.O].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gray J.V., Petsko G.A., Johnston G.C., Ringe D., Singer R.A., Werner-Washburne M.. ‘Sleeping beauty’: quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004; 68:187–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orford K.W., Scadden D.T.. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008; 9:115–128. [DOI] [PubMed] [Google Scholar]
- 3. Cheung T.H., Rando T.A.. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013; 14:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dhawan J., Laxman S.. Decoding the stem cell quiescence cycle - lessons from yeast for regenerative biology. J. Cell Sci. 2015; 128:4467–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tümpel S., Rudolph K.L.. Quiescence: Good and Bad of Stem Cell Aging. Trends Cell Biol. 2019; 29:672–685. [DOI] [PubMed] [Google Scholar]
- 6. Valcourt J.R., Lemons J.M.S., Haley E.M., Kojima M., Demuren O.O., Coller H.A.. Staying alive: metabolic adaptations to quiescence. Cell Cycle. 2012; 11:1680–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Allen C., Büttner S., Aragon A.D., Thomas J.A., Meirelles O., Jaetao J.E., Benn D., Ruby S.W., Veenhuis M., Madeo F. et al.. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J. Cell Biol. 2006; 174:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li L., Lu Y., Qin L.-X., Bar-Joseph Z., Werner-Washburne M., Breeden L.L.. Budding yeast SSD1-V regulates transcript levels of many longevity genes and extends chronological life span in purified quiescent cells. Mol. Biol. Cell. 2009; 20:3851–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li L., Miles S., Melville Z., Prasad A., Bradley G., Breeden L.L.. Key events during the transition from rapid growth to quiescence in budding yeast require posttranscriptional regulators. Mol. Biol. Cell. 2013; 24:3697–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Young C.P., Hillyer C., Hokamp K., Fitzpatrick D.J., Konstantinov N.K., Welty J.S., Ness S.A., Werner-Washburne M., Fleming A.B., Osley M.A.. Distinct histone methylation and transcription profiles are established during the development of cellular quiescence in yeast. BMC Genomics. 2017; 18:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spain M.M., Swygert S.G., Tsukiyama T.. Preparation and analysis of Saccharomyces cerevisiae quiescent cells. Methods Mol. Biol. 2018; 1686:125–135. [DOI] [PubMed] [Google Scholar]
- 12. McKnight J.N., Boerma J.W., Breeden L.L., Tsukiyama T.. Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol. Cell. 2015; 59:732–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miles S., Li L., Davison J., Breeden L.L.. Xbp1 directs global repression of budding yeast transcription during the transition to quiescence and is important for the longevity and reversibility of the quiescent state. PLos Genet. 2013; 9:e1003854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galdieri L., Mehrotra S., Yu S., Vancura A.. Transcriptional regulation in yeast during diauxic shift and stationary phase. Omics J. Integr. Biol. 2010; 14:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miles S., Breeden L.. A common strategy for initiating the transition from proliferation to quiescence. Curr. Genet. 2016; 63:179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mews P., Zee B.M., Liu S., Donahue G., Garcia B.A., Berger S.L.. Histone methylation has dynamics distinct from those of histone acetylation in cell cycle reentry from quiescence. Mol. Cell. Biol. 2014; 34:3968–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aragon A., Quiñones G., Thomas E., Roy S., Werner-Washburne M.. Release of extraction-resistant mRNA in stationary phase Saccharomyces cerevisiae produces a massive increase in transcript abundance in response to stress. Genome Biol. 2006; 7:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aragon A.D., Rodriguez A.L., Meirelles O., Roy S., Davidson G.S., Tapia P.H., Allen C., Joe R., Benn D., Werner-Washburne M.. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures. Mol. Biol. Cell. 2008; 19:1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Swygert S.G., Kim S., Wu X., Fu T., Hsieh T.-H., Rando O.J., Eisenman R.N., Shendure J., McKnight J.N., Tsukiyama T.. Condensin-dependent chromatin compaction represses transcription globally during quiescence. Mol. Cell. 2019; 73:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Long L.J., Lee P.-H., Small E.M., Hillyer C., Guo Y., Osley M.A.. Regulation of UV damage repair in quiescent yeast cells. DNA Repair. 2020; 90:102861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martinez M.J., Roy S., Archuletta A.B., Wentzell P.D., Anna-Arriola S.S., Rodriguez A.L., Aragon A.D., Quiñones G.A., Allen C., Werner-Washburne M.. Genomic analysis of stationary-phase and exit in Saccharomyces cerevisiae: gene expression and identification of novel essential genes. Mol. Biol. Cell. 2004; 15:5295–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bell S.P., Labib K.. Chromosome duplication in Saccharomyces cerevisiae. Genetics. 2016; 203:1027–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parker M.W., Botchan M.R., Berger J.M.. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2017; 52:107–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viggiani C.J., Aparicio O.M.. New vectors for simplified construction of BrdU-incorporating strains of Saccharomyces cerevisiae. Yeast. 2006; 23:1045–1051. [DOI] [PubMed] [Google Scholar]
- 25. Trujillo K.M., Osley M.A.. A role for H2B ubiquitylation in DNA replication. Mol. Cell. 2012; 48:734–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wiest N.E., Houghtaling S., Sanchez J.C., Tomkinson A.E., Osley M.A.. The SWI/SNF ATP-dependent nucleosome remodeler promotes resection initiation at a DNA double-strand break in yeast. Nucleic Acids Res. 2017; 45:5887–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoffman C.S., Winston F.. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987; 57:267–272. [DOI] [PubMed] [Google Scholar]
- 28. Peace J.M., Villwock S.K., Zeytounian J.L., Gan Y., Aparicio O.M.. Quantitative BrdU immunoprecipitation method demonstrates that Fkh1 and Fkh2 are rate-limiting activators of replication origins that reprogram replication timing in G1 phase. Genome Res. 2016; 26:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Subgroup, 1000 Genome Project Data Processing . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nieduszynski C.A., Hiraga S., Ak P., Benham C.J., Donaldson A.D.. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007; 35:D40–D46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodriguez J., Lee L., Lynch B., Tsukiyama T.. Nucleosome occupancy as a novel chromatin parameter for replication origin functions. Genome Res. 2017; 27:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Enserink J.M., Kolodner R.D.. An overview of Cdk1-controlled targets and processes. Cell Div. 2010; 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blow J.J., Laskey R.A.. A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature. 1988; 332:546–548. [DOI] [PubMed] [Google Scholar]
- 36. Zhang J., Martinez-Gomez K., Heinzle E., Wahl S.A.. Metabolic switches from quiescence to growth in synchronized Saccharomyces cerevisiae. Metabolomics. 2019; 15:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimada K., Pasero P., Gasser S.M.. ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev. 2002; 16:3236–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raghuraman M.K., Winzeler E.A., Collingwood D., Hunt S., Wodicka L., Conway A., Lockhart D.J., Davis R.W., Brewer B.J., Fangman W.L.. Replication dynamics of the yeast genome. Science. 2001; 294:115–121. [DOI] [PubMed] [Google Scholar]
- 39. Goldar A., Marsolier-Kergoat M.-C., Hyrien O.. Universal temporal profile of replication origin activation in eukaryotes. PLoS One. 2009; 4:e5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rhind N., Yang S.C.-H., Bechhoefer J.. Reconciling stochastic origin firing with defined replication timing. Chromosome Res. 2010; 18:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Knott S.R.V., Viggiani C.J., Tavaré S., Aparicio O.M.. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev. 2009; 23:1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ibarra A., Schwob E., Méndez J.. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oehlmann M., Score A.J., Blow J.J.. The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J. Cell Biol. 2004; 165:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donovan S., Harwood J., Drury L.S., Diffley J.F.. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:5611–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fernández-Cid A., Riera A., Tognetti S., Herrera M.C., Samel S., Evrin C., Winkler C., Gardenal E., Uhle S., Speck C.. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol. Cell. 2013; 50:577–588. [DOI] [PubMed] [Google Scholar]
- 46. Berbenetz N.M., Nislow C., Brown G.W.. Diversity of eukaryotic DNA replication origins revealed by genome-wide analysis of chromatin structure. PLoS Genet. 2010; 6:e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eaton M.L., Galani K., Kang S., Bell S.P., MacAlpine D.M.. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010; 24:748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Belsky J.A., MacAlpine H.K., Lubelsky Y., Hartemink A.J., MacAlpine D.M.. Genome-wide chromatin footprinting reveals changes in replication origin architecture induced by pre-RC assembly. Genes Dev. 2015; 29:212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Azmi I.F., Watanabe S., Maloney M.F., Kang S., Belsky J.A., MacAlpine D.M., Peterson C.L., Bell S.P.. Nucleosomes influence multiple steps during replication initiation. eLife. 2017; 6:e22512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lipford J.R., Bell S.P.. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol. Cell. 2001; 7:21–30. [DOI] [PubMed] [Google Scholar]
- 51. Fuge E.K., Braun E.L., Werner-Washburne M.. Protein synthesis in long-term stationary-phase cultures of Saccharomyces cerevisiae. J. Bacteriol. 1994; 176:5802–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCarroll R.M., Fangman W.L.. Time of replication of yeast centromeres and telomeres. Cell. 1988; 54:505–513. [DOI] [PubMed] [Google Scholar]
- 53. Tanaka S., Nakato R., Katou Y., Shirahige K., Araki H.. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr. Biol. 2011; 21:2055–2063. [DOI] [PubMed] [Google Scholar]
- 54. Mantiero D., Mackenzie A., Donaldson A., Zegerman P.. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011; 30:4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Das S., Borrman T., Liu V., Yang S., Bechhoefer J., Rhind N.. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015; 25:1886–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harvey K.J., Newport J.. CpG methylation of DNA Restricts Prereplication Complex Assembly in Xenopus Egg Extracts. Mol. Cell. Biol. 2003; 23:6769–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mahbubani H.M., Chong J.P.J., Chevalier S., Thömmes P., Blow J.J.. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J. Cell Biol. 1997; 136:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Powell S.K., MacAlpine H.K., Prinz J.A., Li Y., Belsky J.A., MacAlpine D.M.. Dynamic loading and redistribution of the Mcm2-7 helicase complex through the cell cycle. EMBO J. 2015; 34:531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Flanagan J.F., Peterson C.L.. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999; 27:2022–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Foltman M., Evrin C., Piccoli G.D., Jones R.C., Edmondson R.D., Katou Y., Nakato R., Shirahige K., Labib K.. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Reports. 2013; 3:892–904. [DOI] [PubMed] [Google Scholar]
- 61. Groth A., Corpet A., Cook A.J.L., Roche D., Bartek J., Lukas J., Almouzni G.. Regulation of replication fork progression through histone supply and demand. Science. 2007; 318:1928–1931. [DOI] [PubMed] [Google Scholar]
- 62. Unnikrishnan A., Gafken P.R., Tsukiyama T.. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat. Struct. Mol. Biol. 2010; 17:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aparicio J.G., Viggiani C.J., Gibson D.G., Aparicio O.M.. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004; 24:4769–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Soriano I., Morafraile E.C., Vázquez E., Antequera F., Segurado M.. Different nucleosomal architectures at early and late replicating origins in Saccharomyces cerevisiae. BMC Genomics. 2014; 15:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cayrou C., Ballester B., Peiffer I., Fenouil R., Coulombe P., Andrau J.-C., Helden J., Méchali M.. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015; 25:1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Knott S.R.V., Peace J.M., Ostrow A.Z., Gan Y., Rex A.E., Viggiani C.J., Tavaré S., Aparicio O.M.. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell. 2012; 148:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yaffe E., Farkash-Amar S., Polten A., Yakhini Z., Tanay A., Simon I.. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010; 6:e1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Matson J.P., House A.M., Grant G.D., Wu H., Perez J., Cook J.G.. Intrinsic checkpoint deficiency during cell cycle re-entry from quiescence. J. Cell Biol. 2019; 218:2169–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carroll T.D., Newton I.P., Chen Y., Blow J.J., Näthke I.. Lgr5+ intestinal stem cells reside in an unlicensed G1 phase. J. Cell Biol. 2018; 217:1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hills S.A., Diffley J.F.X.. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014; 24:R435–R444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All ChIP-seq datasets have been be deposited to GEO, accession number GSE155831.
Link to a UCSC genome browser session displaying the uploaded sequence tracks:
http://genome.ucsc.edu/s/pohsuenlee/BrdU%20IP-seq%20%26%20Mcm4%20ChIP-seq.