

Abstract
The ubiquitous fungal pathogen Aspergillus fumigatus is the primary cause of opportunistic mold infections in humans. Aspergilli disseminate via asexual conidia passively traveling through air currents to germinate within a broad range of environs, wherever suitable nutrients are found. Though the average human inhales hundreds of conidia daily, A. fumigatus invasive infections primarily affect the immunocompromised. At-risk individuals can develop often fatal invasive disease for which therapeutic options are limited. Regrettably, the global insurgence of isolates resistant to the triazoles, the frontline antifungal class used in medicine and agriculture to control A. fumigatus, is complicating the treatment of patients. Triazole antifungal resistance in A. fumigatus has become recognized as a global, yet poorly comprehended, problem. Due to a multitude of factors, the magnitude of resistant infections and their contribution to treatment outcomes are likely underestimated. Current studies suggest that human drug-resistant infections can be either environmentally acquired or de novo host selected during patient therapy. While much concerning development of resistance is yet unknown, recent investigations have revealed assorted underlying mechanisms enabling triazole resistance within individual clinical and environmental isolates. This review will provide an overview of triazole resistance as it is currently understood, as well as highlight some of the prominent biological mechanisms associated with clinical and environmental resistance to triazoles in A. fumigatus.
Keywords: Aspergillus, antifungal, triazole, resistance, ergosterol
Introduction
Among species of human filamentous fungal pathogens, one of the most pestilent is Aspergillus fumigatus, a saprophytic fungus extant in much of the world (Anastasi, Varese, and Marchisio 2005; Nierman et al. 2005; Denning 1998). This hyaline septate mold is ubiquitous, found in a plethora of natural climates and ecosystems as well as urban settings. While every human is estimated to inhale hundreds of conidia daily of this opportunistic pathogen, invasive aspergilloses, (IA,) primarily affect humans with a compromised immune system. A. fumigatus represents a major cause of filamentous fungal infections among susceptible patient populations as well as affecting many domesticated animals including dogs, wild animals such as birds, and even insects (Seyedmousavi et al. 2015; Beernaert et al. 2009; Verweij, Chowdhary, et al. 2016; Bowyer, Bromley, and Denning 2020; Valdes et al. 2018). Infection culminates in a spectra of disease severity ranging from allergic conditions to invasive and disseminated aspergillosis (Groll and Walsh 2001; van der Linden et al. 2015). Overall mortality varies depending on the severity and type of underlying infection and on patient-specific factors such as severity of immunosuppression. Regardless of treatment, invasive disease poses a universally high mortality rate ranging from 30% to 90% for most patient types even with current antifungal therapies (Rybak, Fortwendel, and Rogers 2019; Denning et al. 2011; Verweij, Chowdhary, et al. 2016; Tekaia and Latge 2005; Falcone et al. 2011). Aspergillosis can be chronic, requiring continual and prolonged antifungal treatment lasting years in duration. In the recent past, invasive aspergilloses have seen a dramatic rise in incidence in multiple areas of the world. This epidemiological pattern of increasing incidence is largely due to prolonged survival of immunocompromised and immunosuppressed individuals, but is also linked to other shifting host-specific variables, changing environmental conditions, and to pathogenic adaptation to selective pressures (Brissaud et al. 2012; Groll et al. 1996; Groll and Walsh 2001; Lass-Florl and Cuenca-Estrella 2017; Verweij, Chowdhary, et al. 2016).
Aspergillus fumigatus is a saprophytic mold which primarily acts as a decomposer of organic matter (Latge 1999; Dagenais and Keller 2009). Aspergilli primarily reproduce asexually by producing many tiny structures known as conidia which are designed for dispersal through air (Levetin 2004). When some force such as weather, construction, landscaping, agricultural activity, animal activity or wind disturbs the mold’s environment, conidia are dislodged. These structures can travel in contaminated material, air, and/or water and will germinate to grow wherever conditions and nutrients permit. Consequently, A. fumigatus is commonly found growing in both outdoor spaces and human structures. The fungus can be found subsisting on a wide range of ecological spaces such as within farmlands, greenhouses and in aquatic habitats including on surfaces including leaves, leaf litter, rotting wood, in soils and compost (Paulussen et al. 2017; Basenko et al. 2018; Ren et al. 2017; Zhang et al. 2017). Moreover, the species exists within indoor locales circulating in air as buoyant spores or in bioaerosols, on various surfaces, and within virtually any material or substance with adequate organic residues (Paulussen et al. 2017). A. fumigatus is even found growing within concrete and flooring material, especially following water damage (Andersen et al. 2011). For example, one source suggests that between 10% and 50% of buildings within North America and Europe exhibit levels of moisture suitable for fungal growth and between 15% and 40% of homes harbor mold within the structure (Andersen et al. 2011).
Conidia of A. fumigatus can be two to three micrometers in diameter, small enough to become lodged in lung alveoli. Both animals and humans commonly inhale conidia from A. fumigatus. The fungal spores are usually cleared by innate immune mechanisms. However, conidia that withstand host defenses can germinate, grow, and subsist by destroying surrounding tissue for nutrients (Dagenais and Keller 2009). The pathogen can spread and disseminate to further sites via hyphal growth or angioinvasion and even conidiate inside a host within an aspergilloma (Eggimann et al. 2006; Dagenais and Keller 2009). While allergic bronchopulmonary aspergillosis (ABPA) and aspergillomas can occur, infections in healthy individuals are rare (Latge 1999).
One of the most common causes of invasive fungal infections in immunocompromised humans, A. fumigatus poses a threat to populations with predisposing conditions including solid-organ and stem cell transplant recipients, individuals undergoing certain chemotherapies, steroid use, those with acquired or inherited immunodeficiencies, severe liver disease, and with conditions affecting the lungs such as asthma, COPD, cystic fibrosis, bronchiectasis, sarcoidosis, respiratory infections such as influenza and very likely the COVID-19 coronavirus (Koehler et al. 2017; Lopez-Medrano et al. 2016; Steinbach et al. 2012; Wiederhold and Verweij 2020; Kosmidis and Denning 2015; Falcone et al. 2011; Resendiz Sharpe et al. 2018). In such individuals, infections typically begin in the lung following inhalation of fungal conidia but may then disseminate through tissue and bone to other locales. While the general inoculum required to cause disease and the incubation period for development of aspergillosis are not defined, infection likely depends on various aspects including the size of the fungal exposure, virulence of the strain, and host-specific factors (CDC 2019; Dagenais and Keller 2009; Paulussen et al. 2017). Excessive contact with conidia, for example, can lead to invasive disease for even immunocompetent humans (Kosmidis and Denning 2015). Moreover, while the link between fungal exposures and development of allergy such as ABPA, allergic Aspergillus sinusitis (AAS,) or Aspergillus-induced asthma (AIA) are clear, host specific factors such as genetics appear to determine susceptibility to allergic disease more than any external factor (Andersen et al. 2011; Shah and Panjabi 2016).
With developments and advances in modern medical strategies came the extended survival of immunocompromised populations and concurrent increases in certain diseases such as IA (Latge 1999). Multiple studies have attempted to approximate the global impact of aspergilloses and to discern any appreciable changes in incidence over time. Surveys have recently estimated the current burden of aspergilloses worldwide, approximating 4.8 million cases of ABPA, 3 million cases of chronic pulmonary aspergillosis (CPA,) and between 250,000 and 400,000 cases of IA annually (Brown et al. 2012; Bongomin et al. 2017; Bongomin et al. 2020; CDC 2019). IA occurs in up to 14% of heart transplant recipients, up to 7% of bone marrow transplant recipients, and up to 10% of hematological malignancy and lung transplant patients (Eggimann et al. 2006).
Unfortunately, within the United States, aspergillosis is not considered a reportable infection. Certain areas such as Seattle, Washington, where a children’s hospital has had fourteen cases and lost seven patients since 2001 to nosocomial aspergillosis linked to contaminated air circulation systems, are moving to require reporting of aspergillosis to promote better responses to the disease (Staff 2019). However, the CDC does not require medical personnel to recount cases of confirmed or suspected disease in human patients, (though the CDC did prompt passive surveillance specifically for triazole resistant clinical isolates of A. fumigatus beginning in 2011) (Pham et al. 2014). Consequently, accurate numbers of active or past cases in the U.S. are difficult to procure and often depend on data from retrospective studies. However, reasonable approximations for the weight of these infections on public health have been proposed based on available literature.
One of the first active evaluations in the U.S. to detect the burden of invasive fungal infections assessed data within the San Francisco Bay area from 1992 to 1993 and predicted the incidence of IA as 12.4 cases per million people annually. This study found aspergillosis to be the fourth most common invasive fungal infection (IFI) at that time. However, this study was limited to 45 hospital laboratories in a specific region of California (Rees et al. 1998). According to one study which evaluated information on a national hospital database, hospitalizations associated with IA rose by about 3% annually in the U.S. between the years 2000 and 2013, (approximately 169,110 IA-related hospitalizations total.) Using current census data, the same study estimated the rate of IA related hospitalizations to have increased in that time frame from about 32.8 cases per million people to 46 cases per million (Vallabhaneni et al. 2017). In a study utilizing available data from 2001 to 2006 to estimate the burden of IFI in the U.S., IA was the most common IFI detected (Kontoyiannis et al. 2010). Of the 16,200 hematopoietic stem cell transplant (HSCT) patients within the 23 participating centers of the study, 425 proven or probable cases of IA were found, with 187 confirmed as A. fumigatus specifically. The median time from transplant to onset of invasive aspergillosis infection was 99 days. For the 15,820 HSCT patients for which follow-up data was available, the total 12-month cumulative incidence (CI) of IA was 1.6%, though the incidence tended to show an increase over time. The 1-year mortality in this study among patients with IA was reported as 25.4% (Kontoyiannis et al. 2010). Unfortunately, details concerning patient treatment were unavailable for many of the cases and this study only evaluated HSCT patients. Another study of data from participating healthcare centers within Idaho and Utah from 2006 to 2016 found IA to be the third most common IFI, detecting 301 cases total, (40 confirmed specifically as A. fumigatus,) and estimating a mean annual incidence of 2.4 cases per 100,000 patients (Webb et al. 2018). This study reported a crude 1-year mortality for IA patients of 48.8% (Webb et al. 2018). The occurrence of IA among the healthcare centers remained stable throughout the period of study. However, this study only includes information within southern Idaho and Utah (Webb et al. 2018). The overall consensus in the literature is that incidence of IA both within the U.S. and across the globe has increased over the past thirty years, although approximations of the burden of aspergilloses on public health may vary depending on regional and local variables (Pfaller et al. 2018; CDC 2019; Hokken et al. 2019).
The list of approved therapies to combat aspergilloses include three specific classes of antifungals comprised of the polyenes, the echinocandins, and the triazoles. Primary therapy for IA begins with the triazoles voriconazole and isavuconazole, followed by the polyene amphotericin B (lipid formulations) and the echinocandin caspofungin. For patients who show no clinical response to primary treatment, salvage therapies also include other triazoles as well as amphotericin B, caspofungin, or combination therapy with multiple antifungals (Patterson et al. 2016). Antifungal prophylaxis, such as with the triazole posaconazole, is commonly used for at-risk individuals to preclude fungal infection. Moreover, surgery to remove infected tissue is sometimes required, such as for pulmonary aspergilloma (Walsh et al. 2008; Bellete et al. 2010; Patterson et al. 2016). Treatment approach depends largely on the type of infection and patient-specific variables. For example, patients with ABPA may require treatment with anti-inflammatory corticosteroids to moderate lung function in addition to triazole antifungal doses sufficient to clear infection (Walsh et al. 2008; Denning, Pleuvry, and Cole 2013).
The triazole class of antifungals were introduced for medical use in the late 1990’s and are currently utilized for therapy in human infections as well as certain animal species including birds (Beernaert et al. 2009; Hokken et al. 2019). This class of antimicrobial can be given orally with relatively low patient toxicity and effective activity against many fungal pathogens (Denning et al. 1989; Oakley, Moore, and Denning 1998; Heeres, Backx, and Van Cutsem 1984; George, Miniter, and Andriole 1996; Denning, Radford, et al. 1997; Verweij, Chowdhary, et al. 2016). Triazoles chiefly act to inhibit a key enzyme of ergosterol biosynthesis in fungi known as lanosterol 14α-demethylase, (Erg11 in many fungal species or the Cyp51 enzymes in Aspergillus) (Basenko et al. 2018; Arnaud et al. 2010). These compounds possess affinity for the substrate binding site of the fungal enzymes, thus blocking access for the natural substrate and inhibiting the synthesis of ergosterol, the key membrane sterol of fungi.
Unfortunately, the continued utility of the triazoles against Aspergillus spp. infections is now threatened by the global insurgence of triazole resistance. Studies indicate that triazole resistant invasive infection is positively associated with treatment failure and with patient mortality ranging from 30% to approaching 90% for some patient types (van der Linden et al. 2015; Lestrade et al. 2019; Falcone et al. 2011). Clinical cases of documented and confirmed A. fumigatus infections resistant to triazoles have originated from outdoor locales and within medical centers in various countries across the globe (Denning 1998; Groll and Walsh 2001; Chowdhary et al. 2015; Snelders et al. 2008; Snelders et al. 2009). While susceptibility of a strain to an azole antifungal is difficult to determine in-vivo, the common method by which antifungal activity is measured in-vitro involves the broth microdilution method, though methods such as disk-diffusion are also useful. Much of the literature employs minimum inhibitory concentration (MIC) values obtained through this method to approximate the susceptibility of an isolate to a given compound. Two standardized protocols exist whereby MIC values may be obtained; the European Committee on Antibiotic Susceptibility Testing (EUCAST) method, and a protocol provided by the Clinical and Laboratory Standards Institute (CLSI) (Alastruey-Izquierdo et al. 2015). While the CLSI has not yet set clinical breakpoints (CBP) of MIC values to mold-active medical triazoles for filamentous fungi, epidemiological cutoff values (ECV) based on the CLSI and EUCAST methods suggest that strains of A. fumigatus possessing an MIC for voriconazole or itraconazole above 1.0 μg/mL, isavuconazole above 2 μg/mL, or posaconazole above 0.25 μg/mL may be non-wild-type and should be considered for resistance (Alastruey-Izquierdo et al. 2015).
Investigations have attempted to uncover the prevalence of azole resistance among clinical and environmental isolates and estimate local or global incidence of resistant IA cases. A global surveillance study of 497 A. fumigatus isolates collected from 2008 to 2009 detected a total of 29 isolates with heightened triazole MIC values (Lockhart et al. 2011). A study of 1,972 isolates of Aspergillus from across the globe identified eight isolates of A. fumigatus with an MIC to itraconazole exceeding 8ug/mL (Pfaller et al. 2018). Another group evaluated isolates submitted to the Mycology Reference Centre Manchester from 1992 to 2007 and then again between 2008 and 2009. Of the 519 isolates submitted to the center between 1992 and 2007, a total of 34 isolates were deemed resistant to at least one triazole, (possessing an MIC greater than 2μg/mL to itraconazole or voriconazole or >0.5μg/mL to posaconazole.) Interestingly, most of the patient isolates possessed cyp51A gene mutations, including TR34/L98H, G54(E,V, or R,), G138, M220(K or T,) and G448S, which are associated with triazole resistance in A. fumigatus and which will be discussed further in this review (Howard et al. 2009). At least 5 of the patient’s deaths in this earlier study were blamed on progression of aspergillosis despite treatment with triazoles and, for some, efforts with alternative therapy. The frequency of resistance was only 1% from 1999 to 2004, but then rose to 8% from 2004–2007. The latter study found that resistance among isolates was 14% in 2008 and jumped to 20% in 2009 (Howard et al. 2009; Bueid et al. 2010). Another work published in 2008 reported similarly prominent cyp51A mutation occurrences among resistant isolates from the Netherlands, however in this investigation, the majority of isolates possessed only the TR34/L98H mutation rather than a variety of mechanisms (Snelders et al. 2008). Again in the Netherlands, a retrospective investigation into the prevalence of triazole resistance within patients in a university hospital ICU suffering from IA from 2011 to 2013 determined that of the 38 patients whose patient cultures grew A. fumigatus, 10 patients’ isolates were deemed azole resistant. The 90-day mortality among the 28 patients with azole susceptible isolates was 82% (23 out of 28,) while mortality for the 10 patients with resistant disease was 100% (van Paassen et al. 2016). Isolates from the 10 patients with resistant IA harbored the cyp51A TR34/L98H and TR46/Y121F/T289A mutations. Microsatellite typing indicated the isolates were not likely related (van Paassen et al. 2016). In another survey for resistant A. fumigatus isolates within 16 participating hospitals and medical centers in the Netherlands published in 2016, the rate of resistance among tested isolates ranged from 5–10% overall, but was especially elevated within certain patient subsets where rates reached 30% (Lestrade et al. 2016). Resistant strains of A. fumigatus have now been identified within six of the seven continents (Dos Reis et al. 2019; Resendiz Sharpe et al. 2018).
Though the first A. fumigatus isolates found with resistance to triazoles originated in California in the late 1980’s, some studies indicate that triazole resistance is less prevalent within the U.S. than in other regions of the world such as in Europe and the Netherlands (Walker et al. 2018). While the U.S. has fewer investigations than in European countries where active surveillance for triazole resistance has occurred regularly, some data is available for prevalence of resistant A. fumigatus. In a study of transplant recipients within the United States, occurrence of resistance within patient-isolated Aspergillus reached 7% (Baddley et al. 2009). Another study screened 1,026 clinical isolates of Aspergillus collected from 22 states in the U.S. from October of 2011 to October of 2013 for resistance to itraconazole (Pham et al. 2014). Of the 51 isolates which produced an MIC above 1ug/mL to the drug, one isolate with an MIC above 32ug/mL possessed an M220 mutation, (which, again, will be discussed later in this review.)
The true scope of triazole resistance in this pathogen and its effect on public health are likely underestimated due to the profound lack of regular screening for resistance by laboratories and centers both within the United States and around the world (Krishnan Natesan et al. 2012). Moreover, the frequency of negative patient cultures for this pathogen, despite confident diagnosis of aspergillosis supported by serological diagnostics, renders detection of clinical resistance problematic (Denning et al. 2011). For example, systematic analysis of patient health records and matching autopsy records of adult patients admitted to ICU from 1966 to 2011 within the U.S. revealed that aspergillosis was one of the four most common potentially lethal patient misdiagnoses which likely resulted in patient death (Winters et al. 2012; Webb et al. 2018). To conclusively diagnose aspergillosis typically demands a positive culture from a normally sterile sample location as well as histological evidence for a patient Aspergillus infection, (though radiological scans, PCR-based assays, and blood galactomannan or beta-D-glucan assays can also aid diagnosis.) Consequently, infections with either resistant or wild-type Aspergillus strains are often misdiagnosed or otherwise undetected, making accurate assumptions of the true national and global impact of this pathogen difficult.
The origins, drivers, and effects of clinically and environmentally resistant isolates have been a subject of intense research since the first report of clinically resistant isolates correlated with patient treatment failure in 1997 (Denning, Venkateswarlu, et al. 1997). Unfortunately, due to the similarity in essential chemical structure and antifungal biomolecular activity, even agricultural fungicides can select for resistance to medical triazole drugs (Snelders et al. 2009; Schoustra et al. 2019; Faria-Ramos et al. 2014; Zhang et al. 2017). Indeed, this environmentally selected resistance is blamed for more clinically resistant patient infections than de novo resistance acquisition during extended medical triazole therapy (van der Linden et al. 2013). Regardless of the route of acquisition, mechanisms underpinning triazole resistance in A. fumigatus often come at no pathogenic fitness cost (Buil et al. 2019). Regrettably, much concerning the specific biomechanical causes, (whether genetically based or epigenetic,) clinical versus environmental derivation, and overall burden of triazole resistant A. fumigatus to public health within the United States and globally remains undetermined. This brief review aims to provide a succinct description of the selection of resistance in this species as well as to highlight some of the known environmental and clinical resistance mechanisms, especially those contributing to patient treatment failures and associated with increased mortality among infected immunocompromised patients.
Section One: The Rise of Resistance
The first instances of well-documented triazole-resistant A. fumigatus associated with treatment failure occurred in California during 1989–1990 and the results were published in 1997 (Denning, Venkateswarlu, et al. 1997). This study identified itraconazole resistant isolates from two separate patients with invasive aspergilloses undergoing triazole therapy. The isolates each exhibited an in vitro broth micro-dilution MIC value to itraconazole exceeding 16 μg/mL. In vitro resistance was confirmed to translate in vivo through experiments utilizing murine neutropenic models of infection (Denning, Venkateswarlu, et al. 1997). Differences in the phenotypes and sterol profiles of the resistant patient isolates pointed to the existence of at least two entirely separate stratagems of triazole drug resistance. One likely involved the increased expression, altered drug binding affinity, and / or enhanced activity of the fungal 14α- demethylase to counteract the drug’s action whereas the other likely involved reduced intracellular accumulation of drug potentially as a result of increased membrane transporter activity. Observations of this initial report have held true to the current view that multiple mechanistic strategies putatively underlie triazole resistance in A. fumigatus (Denning, Venkateswarlu, et al. 1997).
It is now understood that there are likely two general routes for selection of resistance in A. fumigatus: de novo selection within the host during therapy with triazoles or through selection in the environment (Verweij, Chowdhary, et al. 2016; Snelders et al. 2009; Buil et al. 2019; Verweij et al. 2013; Camps, van der Linden, et al. 2012; Hagiwara et al. 2014). Environmental saprobic fungi such as A. fumigatus are typically both phenotypically and genotypically plastic, a trait that allows for continual acclimation to ever-changing and generally adverse conditions (Groll and Walsh 2001). In turn, stressful stimuli within the environment often cause enduring adaptive responses and can select for beneficial and often heritable genetic or epigenetic changes within such organisms (Buil et al. 2019; Sniegowski and Gerrish 2010; Yona, Frumkin, and Pilpel 2015). Fungi can survive stress and outcompete other microbes within their environment through intracellular changes enabled by genetic alterations or via responses such as activation of efflux mechanisms or stress response pathways. Unfortunately, when demethylase-inhibiting fungicides are introduced into natural settings, as during agricultural work, fungi within these locales become exposed to the compounds and can readily adapt to the selective antifungal pressures (Groll and Walsh 2001; Chowdhary et al. 2015; Snelders et al. 2009). Though A. fumigatus is not a phytopathogen, the species acts as an important decomposer of organic matter, thriving wherever suitable nutrients and conditions are found including within outdoor environments and human-inhabited settings (Verweij, Chowdhary, et al. 2016). Multiple agriculturally employed azole fungicides are chemically similar and each possess the same primary mechanism of action against the fungal ergosterol biosynthetic pathway (Verweij, Snelders, et al. 2009; Stensvold, Jørgensen, and Arendrup 2012; Snelders et al. 2012; Zhang et al. 2017). Once introduced into water or soil, triazole fungicides can persist for at least several months (Ribas et al. 2016). Consequently, A. fumigatus present in such settings may become strained by these agricultural fungicides and counter with adaptive stress responses (Buil et al. 2019; Alvarez-Moreno et al. 2019). For example, three separate laboratory-based studies have reported the induction of target gene mutations similar to those found in clinical isolates via exposure to agricultural triazole compounds including tebuconazole, metconazole, bromuconazole, difenoconazole, propiconazole, hexaconazole and epoxiconazole. These compounds have been in use for agricultural applications and have been shown to select for strains possessing heightened MIC to the medical drugs voriconazole, itraconazole, posaconazole, or multi- or pan-azole resistance (Zhang et al. 2017; Snelders et al. 2012; Ren et al. 2017).
Exposures to resistant strains derived from the environment are blamed for a preponderance of the causal mechanisms identified within patient isolates (van der Linden et al. 2015; Chowdhary et al. 2015; Chowdhary et al. 2013; Verweij, Snelders, et al. 2009; van der Linden et al. 2009; Verweij, Chowdhary, et al. 2016; Verweij et al. 2013; van der Linden et al. 2013). However, the remainder of resistant clinical infections seem to result primarily due to prolonged or successive triazole treatments selecting for resistant fungi within the host (Howard et al. 2009; Chryssanthou 1997; Camps, Dutilh, et al. 2012; Verweij et al. 2002; Howard et al. 2013). Extended phases of antifungal treatments even spanning several months can provide sufficient time for the development of resistant strains within the human host (Camps, Dutilh, et al. 2012; Denning et al. 2003). This is especially pertinent given that patients have been known to suffer from chronic aspergilloses requiring therapy exceeding 12 years in duration (Denning et al. 2003). A de novo resistant infection will likely no longer respond to treatment with the original triazole used and may additionally exhibit pan-azole resistance (van der Linden et al. 2015; Howard et al. 2009; Camps, Dutilh, et al. 2012; Verweij et al. 2002). Consequently, development of resistant infection during therapy can contribute to treatment failures and increased mortality (Dannaoui et al. 2001; Camps, Dutilh, et al. 2012).
It is, however, often difficult to prove the origins of a resistant strain and the occurrence of later infection with a resistant strain from the patient’s environment cannot always be dismissed (Dannaoui et al. 1999). Analyses of the genomes of cultured strains are often utilized to propose the origins of a resistant infection (Dannaoui et al. 1999; Camps, Dutilh, et al. 2012). The presence of genetically diverse isolates possessing the same resistance mechanism in azole-naïve patients can imply isolates were selected for resistance within the environment (Wiederhold et al. 2016; Snelders et al. 2009; Walsh et al. 2008; Verweij et al. 2002). However, whole-genome sequencing experiments indicate A. fumigatus can undergo rather drastic genomic changes during invasive growth within the host niche, bringing such assumptions into question (Hagiwara et al. 2014). To provide clarity on isolate lineage, microsatellite typing has been employed to indicate whether serially collected isolates from a patient infection are “isogenic,” having identical or very similar genotypes. As in the work by Hagiwara et. al., though related strains accumulated many separate mutations detectable through sequencing, they retained identical microsatellite results (Hagiwara et al. 2014). When genetic relationships are confirmed utilizing this method, a series of isogenic patient isolates which become resistant during extended patient treatment may suggest de novo resistance (Camps, Dutilh, et al. 2012). Regardless of the mode of resistance acquisition, infection with an azole-resistant strain of A. fumigatus is associated with an escalated cost of hospitalization and care, treatment failures, and increased mortality (Zilberberg et al. 2018; van der Linden et al. 2009; Lestrade et al. 2019; van der Linden et al. 2013). Resistant invasive Aspergillus infection elevates the observed mortality rate from about 50% for an azole-susceptible strain to approximately 90% for a resistant infection (van der Linden et al. 2011; Howard et al. 2009; Lestrade et al. 2019). Some of the most common resistance-associated mechanisms within both environmental and de-novo originated strains include triazole target or non-target alterations and are summarized in Table 1.
Table 1:
List of mechanisms associated with clinical triazole resistance in isolates of A. fumigatus.
| Gene Name | Systematic Name Description |
Alteration | Reference |
|---|---|---|---|
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
TR34/L98H |
Mellado, E., et al., 2007 Snelders, E., et al., 2008 Gsaller, F., et al., 2016 |
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
TR46/Y121F/T289A |
Chowdhary, A., et al., 2014 Snelders, E., et al., 2015 |
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
G54 | Diaz-Guerra, T.M., et al., 2003 |
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
G138 |
Manavathu, E.K., et al., 2000 Xiao, L., et al., 2004 |
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
M220 |
Mellado, E., et al., 2004 Howard, S.J., et al., 2013 |
| cyp51A | Afu4g06890 14-alpha sterol demethylase |
G448S |
Krishnan Natesan, S., et al., 2012 Bellete, B., et al., 2010 |
| hapE | Afu2g14720 Sequence-specific CCAAT DNA binding transcription factor |
P88L | Camps, S.M., et al., 2012 |
| hmg1 | Afu2g03700 Hydroxymethylglutaryl-CoA (HMG-CoA) reductase |
S269F | Hagiwara, D., et al., 2018 |
| hmg1 | Afu2g03700 Hydroxymethylglutaryl-CoA (HMG-CoA) reductase |
F262 deletion | Rybak, J.M., et al., 2019 |
| hmg1 | Afu2g03700 Hydroxymethylglutaryl-CoA (HMG-CoA) reductase |
S305P | Rybak, J.M., et al., 2019 |
| hmg1 | Afu2g03700 Hydroxymethylglutaryl-CoA (HMG-CoA) reductase |
I412S | Rybak, J.M., et al., 2019 |
| cdr1B/ abcC | Afu1g14330 Putative ABC transporter |
Overexpression |
Fraczek, M.G., et al., 2013 Hagiwara, D., et al., 2016 |
| atrF | Afu6g04360 Putative ABC transporter |
Overexpression | Slaven, J.W., et al., 2002 |
| mdr1 | Afu5g06070 ABC multidrug transporter |
Overexpression |
Fraczek, M.G., et al., 2013 Nascimento, A.M., et al., 2003 Rajendran, R., et al., 2011 |
| mdr2 | Afu4g10000 ABC multidrug transporter |
Overexpression |
Fraczek, M.G., et al., 2013 Nascimento, A.M., et al., 2003 Rajendran, R., et al., 2011 |
| Mdr3 | Afu3g03500 Putative multidrug resistance protein |
Overexpression | Nascimento, A.M., et al., 2003 |
| Mdr4 | Afu1g12690 ABC multidrug transporter |
Overexpression | Nascimento, A.M., et al., 2003 |
Section Two: Target gene-dependent resistance
Mutations affecting the target enzymes of triazoles, the fungal sterol 14α-demethylases, represent the most commonly identified mechanisms associated with triazole resistance in A. fumigatus (Snelders et al. 2010; Denning et al. 2011; van der Linden et al. 2013). A. fumigatus possesses two Cyp51 sterol 14α demethylase enzymes; Cyp51A and Cyp51B. Studies predict that Cyp51B has better binding affinity with triazole compounds. Aberrations have been identified within both genes encoding 14α demethylases of A. fumigatus isolates. However, only alterations affecting the cyp51A sterol-demethylase gene and its protein product have thus far been convincingly associated with clinically relevant triazole resistance in this pathogen (Rybak, Fortwendel, and Rogers 2019; Ferreira et al. 2005). Target enzyme lesions have been found within both environmental and clinical isolates and have been recovered both from patients who have had extended contact with triazoles, as well as from infections of azole-naïve individuals (Wiederhold et al. 2016; Howard et al. 2006; Verweij, Chowdhary, et al. 2016). Sterol 14α-demethylase mutations have been identified in patient isolates and implicated in treatment failures among clinical patients in multiple countries (Wiederhold et al. 2016; Bueid et al. 2010; Howard et al. 2006; Chowdhary et al. 2015; van der Linden et al. 2013).
Depending on the type of mutation and its location in the genetic sequence, the resulting strain can exhibit a range of resistant phenotypes including a spectrum of triazole susceptibilities as well as single or pan-azole resistant qualities (Wiederhold et al. 2016; Howard et al. 2009; Chowdhary et al. 2015; van der Linden et al. 2013). Several principal stratagems exist whereby alterations in the coding sequence or upstream regulatory sequence of cyp51A enable resistance. The most commonly found Cyp51A mutations associated with triazole resistance occur at the gene sequence codons G54, L98, Y121 and T289, G138, and M220, with nonsynonymous mutations at position 98, 121 and 289 typically accompanied by a tandem repeat within the upstream genetic regulatory promoter region (Denning et al. 2011; Wiederhold et al. 2016). This category includes the mutation combinations TR34/L98H and TR46/Y121F, and TR46/T289A, which exist within patient derived and environmental isolates of A. fumigatus from multiple continents (Chowdhary et al. 2015; Verweij, Mellado, and Melchers 2007; Mellado et al. 2007; Snelders et al. 2008; Camps, Rijs, et al. 2012; Pham et al. 2014; Verweij, Howard, et al. 2009; Bader et al. 2013; Snelders et al. 2010; Wiederhold et al. 2016; Chowdhary et al. 2014; Snelders et al. 2009; Snelders et al. 2015; Stensvold, Jørgensen, and Arendrup 2012). Several of these will be discussed in more detail.
TR34/L98H is named such because of a tandem repeat of a length spanning 34 base pairs within the upstream promoter region of cyp51A and is also characterized by a substitution of a leucine at the 98th amino acid position to a histidine in the resultant protein (Gsaller et al. 2016b, 2016a; Bader et al. 2013; Snelders et al. 2010; Verweij, Mellado, and Melchers 2007; Abdolrasouli et al. 2015; Lockhart et al. 2011; Mellado et al. 2007; Rybak et al. 2019; Fuhren et al. 2015). This mechanism enhances transcription of the cyp51A gene, as well as altering the resultant protein enzyme’s structure [Figure 1B] (Mellado et al. 2007). In A. fumigatus, the residue at position 98 lies near the first recognition site for the enzyme’s substrate within the Cyp51A enzyme (Mellado et al. 2007; Snelders et al. 2011). Isolates possessing this mechanism tend to show reduced susceptibility to multiple medical triazoles including voriconazole, posaconazole, and itraconazole as well as the experimental azole compound ravuconazole and to multiple agricultural and industrial azole fungicides (Mellado et al. 2007; Mann et al. 2003; Snelders et al. 2008; Snelders et al. 2011). Cyp51A TR34/L98H has been well established as a functional cause of increased triazole resistance within human infections and is often associated with patient treatment failures and increased mortality (Pham et al. 2014). Strains with a TR34/L98H mechanism also retain virulence in a mouse model of invasive aspergillosis and possess no distinct defects in growth in comparison to wild-type isolates (Mavridou et al. 2013; Wang et al. 2018). The mechanism has been suggested to be the dominant causal contributor to azole resistance among both environmental and clinical isolates of A. fumigatus and exists in samples from across the globe including the U.K., China, the Netherlands, Belgium, Austria, Norway, Denmark, Spain, France, Germany, Iran, India, and the U.S. (Gsaller et al. 2016b, 2016a; Bader et al. 2013; Snelders et al. 2010; Verweij, Mellado, and Melchers 2007; Abdolrasouli et al. 2015; Lockhart et al. 2011; Mellado et al. 2007; Chowdhary et al. 2012; Snelders et al. 2009; Chowdhary et al. 2015; Wiederhold et al. 2016; Fuhren et al. 2015; Pham et al. 2014). Among clinically resistant infections, TR34/L98H appears to be the result of both de novo resistance in azole-naïve patients and from environmental resistance involving strains selected in nature prior to human infection, the latter situation being more likely (Gsaller et al. 2016b, 2016a; Verweij, Mellado, and Melchers 2007; Verweij et al. 2002; Mellado et al. 2007; Snelders et al. 2008). Studies have illustrated the potential for this mechanism to arise in response to antifungal pressures. For example, duplication of the 34bp tandem repeat can be induced in an isolate possessing a TR34/L98H mutation following exposure to demethylase-inhibiting agricultural fungicides in a laboratory setting (Snelders et al. 2012).
Figure 1. Illustration of genetic circuitry enabling enhanced cyp51A expression in isolates possessing one of two representative azole resistance mechanisms.
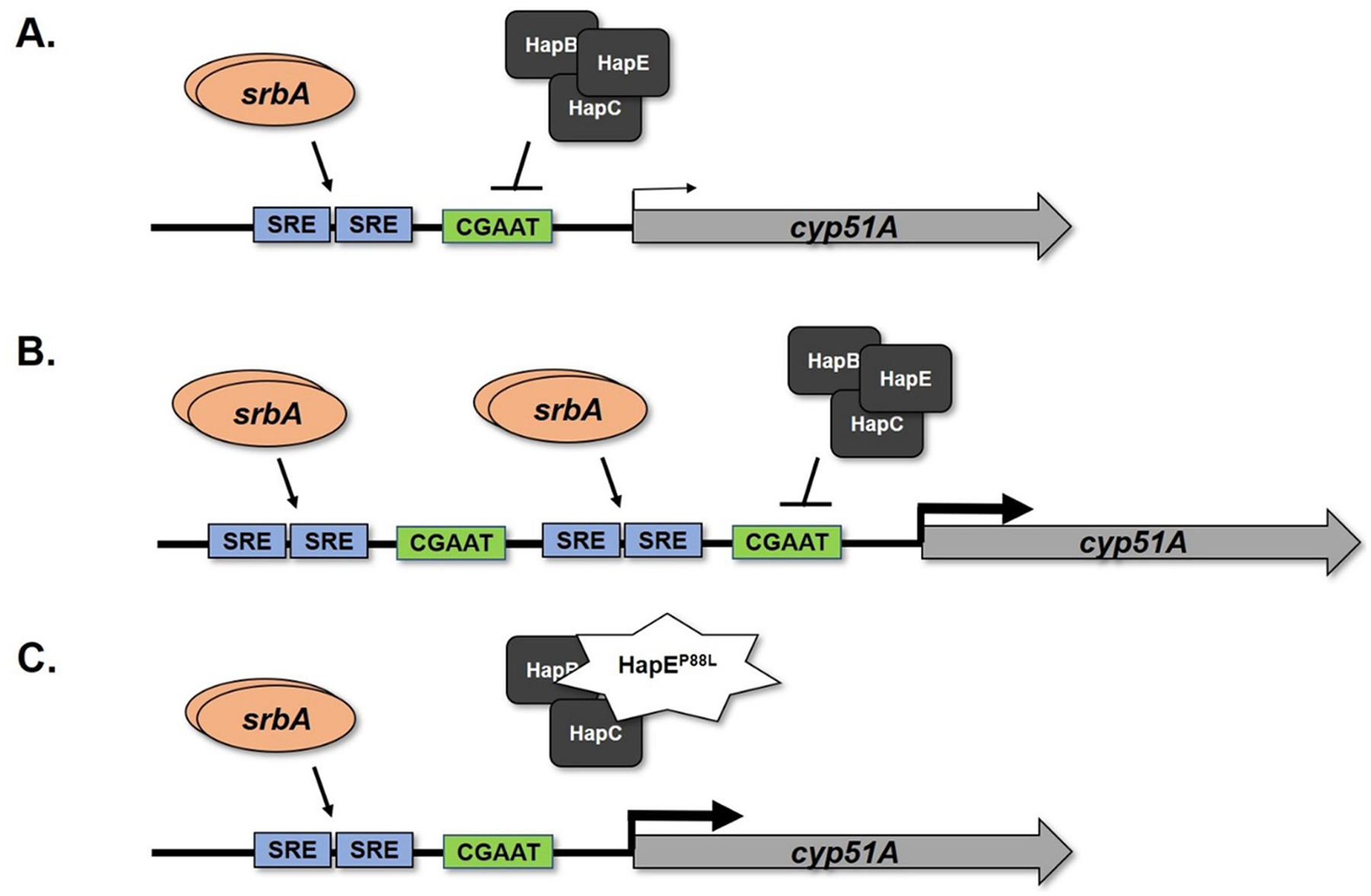
(A) The wild type configuration of the cyp51A promoter is shown. Under normal regulation, proper cyp51A expression levels are orchestrated by at least two transcriptional regulators. The activator, SrbA, binds to upstream sterol regulatory elements (SRE) to induce cyp51A expression, whereas the multi-subunit CCAAT-Binding Complex (CBC) binds to a conserved CGAAT motif downstream of the SREs to inhibit cyp51A expression. (B) Tandem repeat mutations of the cyp51A promoter wherein the SREs are duplicated, as occurs in isolates bearing the TR34 and TR46 mutations, lead to increased binding of SrbA and activation of gene expression. Although the CBC binding motif is also duplicated in these mutations, physical blockage of additional binding of the CBC complex occurs by the increased presence of the SrbA activator. (C) An illustration of a target gene-independent clinical triazole resistance mechanism involving a single mutation within the gene encoding a member of the heterotrimeric HAP complex, HapE. This mutation reduces binding of the entire CBC to its regulatory binding site in the cyp51A promoter. Note that, as in (B), reduced binding of the CBC complex results in loss of repression and consequently increased gene expression. Both representative mechanisms ultimately enable increased transcription of cyp51A mRNA, thus increasing the level of Cyp51A protein produced within fungi and enabling the biosynthesis of ergosterol.
While introduction of only the tandem repeat into a previously susceptible strain of A. fumigatus increases mRNA transcription in laboratory mutant strains, the introduction of both lesions simultaneously is required to recreate the phenotype of the clinically resistant strains (Mellado et al. 2007). Studies reveal that the 34bp repeat in the promoter region is bound by several important transcription factors which are known to affect the transcription of genes related to sterol biosynthesis as well as other pathways (Gsaller et al. 2016a, 2016b). One such transcriptional regulator, SrbA, binds to the repeated sequences in the cyp51A promoter in this mutant (Gsaller et al. 2016b). SrbA acts as a sterol regulatory element binding protein or “SREBP.” Increased SrbA binding to the cyp51A promoter results in increased expression, thus likely exacerbating triazole resistance observed within these mutant strains (Gsaller et al. 2016b). Moreover, SrbA competes with the transcriptional repressor CCAAT-DNA binding complex (CBC) for DNA binding on the cyp51A promoter. Increased binding of SrbA blocks binding of the repressor resulting in de-repression of cyp51A transcription (Gsaller et al. 2016b).
The TR46/Y121F/T289A resistance mechanism also involves a tandem repeat of bases within the regulatory region as well as nonsynonymous mutations within the cyp51A gene (Wiederhold et al. 2016; Chowdhary et al. 2014; Snelders et al. 2015; van der Linden et al. 2013; Fuhren et al. 2015). The TR46/Y121F/T289A mechanism was initially found in the Netherlands within multiple voriconazole-resistant isolates of fifteen individual patients from six individual geographically distant hospitals, the earliest of which dated to December of 2009 (van der Linden et al. 2013). These patients suffered a high incidence of treatment failures and mortality through the study, especially among those given voriconazole. The same study sampled ten separate locations and recovered isolates possessing the same mechanism at six of the ten environmental locales (van der Linden et al. 2013). This resistance mechanism can spontaneously arise in response to antifungal pressure, as a study has reported generation of TR46/Y121F/T289A and G448S mutation combinations upon exposure of A. fumigatus strains to agricultural triazoles (Ren et al. 2017). The G448S mutation will be discussed in more detail later in this review. Another study found that introduction of the 46-basepair repeat and the Y121F mutation together without the T289A substitution into a previously susceptible strain appears sufficient to recapitulate the multi-azole resistant nature of clinically obtained resistant isolates with the TR46/Y121F/T289A mutation pattern (Snelders et al. 2015). Isolates bearing this mutation typically have elevated MIC values for voriconazole and also attenuation of itraconazole and posaconazole susceptibility (van der Linden et al. 2013). The residue Y121 supposedly hydrogen bonds with the heme group within Cyp51A according to homology models, supporting the assumption of this residue as the more important site altering antifungal susceptibility when mutated [Figure 2A] (Snelders et al. 2015). In contrast, genetic introduction of either TR46 and the T289A mutation or the T289A mutation alone had little effect on susceptibility, though it may alter the enzyme’s association with voriconazole (Snelders et al. 2015).
Figure 2. Protein model illustrating impact of cyp51A mutations on binding of the frontline anti-Aspergillus triazole, voriconazole.
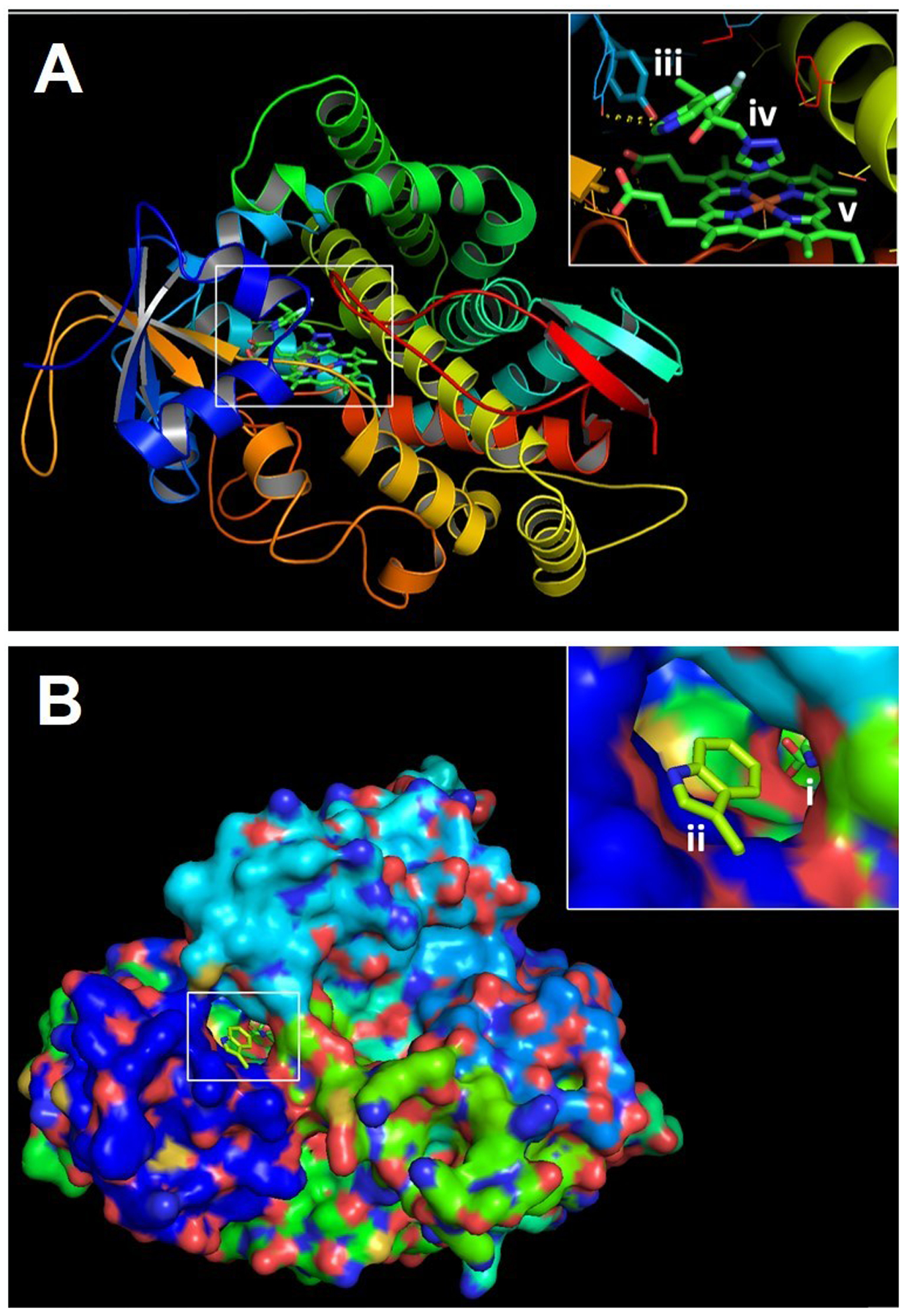
PyMOL 2 was used to create three dimensional models of the A. fumigatus Cyp51A protein using the previously resolved crystal structure of the A. fumigatus Cyp51B as a scaffold. A) Three-dimensional ribbon model of Cyp51A. Inset depicts the Y121 residue (iii) which stabilizes the interaction between voriconazole (iv) and heme (v). B) Three-dimensional model of the surface of the Cyp51A protein. Inset depicts the ligand access channel and entrance to the Cyp51A catalytic site which is occupied by both heme and voriconazole (i). Also depicted is the G54W amino acid substitution (ii) predicted to interfere with the binding of the long lipophilic sidechains of itraconazole and posaconazole.
Mutations in the sequence for A. fumigatus Cyp51A at position G54 are also associated with cross-resistance to multiple triazole compounds including posaconazole and itraconazole (Chowdhary et al. 2015; Diaz-Guerra et al. 2003; Verweij, Howard, et al. 2009; Camps, van der Linden, et al. 2012; Mann et al. 2003; Nascimento et al. 2003; Bader et al. 2013; Mellado et al. 2007; Snelders et al. 2010; Pfaller et al. 2008; Rodriguez-Tudela et al. 2008; Krishnan Natesan et al. 2012; Denning et al. 2011). In the predicted tertiary structure of Cyp51A, the glycine residue at position 54 appears to interact directly with triazole drugs and is situated within the substrate access channel [Figure 2B] (Xiao et al. 2004; Snelders et al. 2010; Rybak, Fortwendel, and Rogers 2019; Diaz-Guerra et al. 2003). Multiple amino acid substitutions at glycine 54 are known including G54E, G54W and G54R (Mellado et al. 2007; Diaz-Guerra et al. 2003; Xiao et al. 2004; Tashiro and Izumikawa 2016). Replacement of the glycine residue at position 54 with an arginine (R) or a glutamate (E) in a previously azole-susceptible strain has been shown to increase MIC values to posaconazole by 30-fold while replacement with a tryptophan residue (W) produces a 250-fold MIC increase in laboratory mutants. These mutations also greatly increase MIC values to itraconazole but typically have no effect on in vitro voriconazole susceptibility (Xiao et al. 2004; Mann et al. 2003; Nascimento et al. 2003; Manavathu et al. 2000; Pfaller et al. 2008; Diaz-Guerra et al. 2003; Tashiro and Izumikawa 2016).
Another known mutation occurs at position G138 of the enzyme (Snelders et al. 2010; Manavathu et al. 2000; Xiao et al. 2004; Howard et al. 2006; Denning et al. 2011). The cyp51A G138C mutation was identified in a patient with chronic cavitary aspergillosis undergoing extended triazole therapy whose isolates simultaneously developed resistance to voriconazole and itraconazole (Howard et al. 2006). Isolates containing this amino acid substitution tend to be resistant to voriconazole but not to posaconazole according to both in vivo and in vitro measures of susceptibility or response to treatment (Xiao et al. 2004; Manavathu et al. 2000; Rybak, Fortwendel, and Rogers 2019). The residue at 138 is also situated within the ligand access channel near the heme cofactor of Cyp51A (Rybak, Fortwendel, and Rogers 2019). A G138R substitution likely disrupts the heme cofactor environment in the active site, disrupting the binding of voriconazole to the enzyme. The side-chain interactions of posaconazole may accommodate better association with Cyp51A and overcome the altered active site conformation (Xiao et al. 2004; Rybak, Fortwendel, and Rogers 2019). The G138 mutation has been spontaneously generated through exposure of A. fumigatus to triazoles in a laboratory setting, hinting at the potential for triazole antifungal stress to select for this mechanism. One such study induced the formation of a G138L mutation by exposing strains to itraconazole (Snelders et al. 2012).
Furthermore, one common lesion associated with azole resistance occurs at position M220 of the Cyp51A protein sequence (Mellado et al. 2004; Chen et al. 2005; Snelders et al. 2010; Mellado et al. 2007; Pfaller et al. 2008; Diaz-Guerra et al. 2003; Rodriguez-Tudela et al. 2008; Denning et al. 2011). Amino acid substitutions replacing the Methionine at residue 220 of the gene for Cyp51A have also been associated with an itraconazole resistant phenotype in vivo and elevated MIC values to itraconazole, voriconazole, and posaconazole in vitro (Mellado et al. 2004; Chen et al. 2005; Snelders et al. 2010; Mellado et al. 2007; Pfaller et al. 2008; Diaz-Guerra et al. 2003; Rodriguez-Tudela et al. 2008). This amino acid substitution has been detected within cultures recovered from diseased patients as well as through ultra-sensitive, real-time PCR assays for A. fumigatus cyp51A within patient samples which were not culturable (Denning et al. 2011; Howard et al. 2013; Howard et al. 2009). One such group of isolates were collected from a patient with chronic pulmonary aspergillosis (CPA) undergoing extended itraconazole treatment who developed a resistant infection. Upon the patient’s death, researchers found that the individual’s left lung had nearly entirely been destroyed; reduced to fibrotic tissue and interconnected aspergillomas (Howard et al. 2013). Individual strains cultured from separate aspergillomas displayed cyp51A mutations M220K or M220T and exhibited elevated MIC to itraconazole as well as to either voriconazole or posaconazole (Howard et al. 2013).
Finally, isolates possessing a G448S mutation have been associated with resistance to voriconazole and itraconazole (Krishnan Natesan et al. 2012; Bellete et al. 2010; Fuhren et al. 2015; Howard et al. 2009) and these mutants tend to remain susceptible to posaconazole (Xiao et al. 2004; Manavathu et al. 2000; Pfaller et al. 2008; Mellado et al. 2004; Howard et al. 2009; Krishnan Natesan et al. 2012). The residue at position G448 is also located near the heme cofactor in Cyp51A (Xiao et al. 2004). The mutation G448S has been established as a causal contributor to triazole resistance within the context of patient infections as well as an animal model of invasive pulmonary aspergillosis (Howard et al. 2009; Verweij, Howard, et al. 2009; Krishnan Natesan et al. 2012; Xiao et al. 2004; Manavathu et al. 2000). One individual developed a voriconazole resistant infection bearing the cyp51A G448S mutation after a year of treatment with voriconazole (Bellete et al. 2010). Though the patient was switched to lipid formulation amphotericin B and caspofungin therapy following the susceptibility testing results, in this case the patient ultimately died shortly after this modification of antifungal therapy. Sequencing of isolates recovered during the patient’s extended care revealed that though the strains were identical according to microsatellite genotyping, the earlier collected strains which were recovered during treatment were susceptible to itraconazole and voriconazole while the later isolates possessed resistance to itraconazole, voriconazole, or both (Bellete et al. 2010). Among the four patient isolates and a reference strain, no other mutations existed in the cyp51A sequence in either the sense or antisense directions (Bellete et al. 2010). Laboratory mutant strains ectopically expressing cyp51A containing a G448S mutation introduced into a previously susceptible strain also recapitulate voriconazole resistance in vitro (Hagiwara et al. 2018).
Section Three: Target gene mutation-independent resistance
Often, sequence analysis of isolates with heightened MIC values unveils multiple strains which possess no Cyp51A-related lesions (Arendrup et al. 2010; Wiederhold et al. 2016; Fraczek et al. 2013; Verweij, Chowdhary, et al. 2016; Buil et al. 2019). Indeed, a poorly understood shift from target enzyme-based mechanisms to alternative resistance mechanisms appears to be occurring in certain areas of the world (Fraczek et al. 2013). Common non-target gene mechanisms currently recognized include genetic and epigenetic occurrences such as alterations to transcription factor components, alteration of the sterol biosynthetic pathway, overexpression of specific drug efflux pumps or other means by which intracellular accumulation of triazole is reduced in the fungal cells, and enhanced adaptability due to biochemical or genomic plasticity (Gsaller et al. 2016a, 2016b; Camps, Dutilh, et al. 2012; Bellete et al. 2010). Selected genetic lesions which are not within the sequences or promoter regions for 14α demethylases have been well studied and convincingly associated with drug resistance and treatment failures in patients (Nascimento et al. 2003; Camps, Dutilh, et al. 2012; Fraczek et al. 2013; Ferreira et al. 2005). While a comprehensive list of all known or suggested azole resistance mechanisms is beyond the scope of this review, a selection of such methods will be discussed.
One such resistance mechanism involves a single point mutation in the protein coding sequence for a subunit of the fungal CCAAT-binding transcription factor complex, CBC, known as HapE in A. fumigatus. The CBC acts as a transcriptional repressor affecting the expression of genes such as cyp51A (Gsaller et al. 2016b; Camps, Dutilh, et al. 2012). The hapE P88L mutation was initially discovered within the sequences of multiple isolates obtained from a single patient with invasive pulmonary aspergillosis given extended triazole therapy (Camps, Dutilh, et al. 2012). In the case of this study, the patient ultimately failed therapy and succumbed to the invasive pulmonary infection. This outcome was very likely due to the resistant nature of the infection (Camps, Dutilh, et al. 2012). Microsatellite typing and sequencing analyses confirmed the isogenic nature of the early susceptible patient isolates of A. fumigatus and the later resistant samples. Analyses also revealed no changes within the gene sequences for Cyp51A and Cyp51B nor their promoter regions and only the hapE P88L mutation was able to recapitulate the invulnerable phenotype of the isolates. The P88L mechanism of resistance was reasonably verified as the causal mutation conferring a pan-azole resistant phenotype in the patient isolates through homologous gene replacement and sexual crossing studies with subsequent analyses of susceptibility and growth (Camps, Dutilh, et al. 2012). The repercussions of the P88L mutation involve loss of CBC activity in the mutant strain which results in de-repression thus allowing a higher level of cyp51A transcription within the pathogen, enabling resistance to triazole treatment [Figure 1C] (Gsaller et al. 2016b; Camps, Dutilh, et al. 2012).
A recent finding showing that altered sterol biosynthesis pathway activity underpins resistance in A. fumigatus clinical isolates came from work with the pathway rate-limiting HMG-CoA reductase, hmg1 (Buil et al. 2019; Rybak et al. 2019; Hagiwara et al. 2018). One study which screened clinical isolates collected from multiple patients and from multiple medical centers in Japan for novel resistance mechanisms identified hmg1 mutations among azole-resistant isolates (Hagiwara et al. 2018). The mutation S269F was present in each isolate which exhibited elevated triazole MIC collected from one patient with chronic pulmonary aspergillosis. The residue at S269 of hmg1 resides within the beginning of the enzyme’s predicted sterol-sensing domain (Hagiwara et al. 2018). However, several of the patient’s isolates from this study also bore a G448S mutation in cyp51A and all resistant isolates from the individual also possessed an A350T mutation in erg6, a major sterol methyltransferase (Hagiwara et al. 2018). This study further showed that ectopic expression of mutant hmg1 and erg6 alleles caused no shift in triazole MIC, suggesting that these mutations are not drivers of resistance (Hagiwara et al. 2018). However, another study positively linked mutations residing in the predicted sterol-sensing domain of hmg1 within clinically resistant isolates to triazole resistance (Rybak et al. 2019). In this work, one of three mutations identified as unique to triazole resistant isolates or a wild-type hmg1 allele were directly swapped with the native hmg1 genomic locus of a previously susceptible laboratory strain (Rybak et al. 2019; Al Abdallah, Ge, and Fortwendel 2017). The resulting mutants with either an S305P or I412S mutation or an in-frame deletion of F262 showed increased MIC at least 4-fold to voriconazole, itraconazole, posaconazole, and isavuconazole, whereas the control wild-type allele matched the susceptibility of the parental strain. Furthermore, genetic knock-in of a wild-type hmg1 sequence into the backgrounds of the hmg1-mutant resistant clinical isolates abrogated resistance (Rybak et al. 2019). These findings suggested that the sterol-sensing function of Hmg1 is an essential element for mediation of pathway activity and triazole susceptibilities in A. fumigatus. Further supporting this, sterol profiling confirmed an altered accumulation of precursors to ergosterol in strains carrying hmg1 mutations, though the expression of A. fumigatus sterol demethylases was not abnormal (Rybak et al. 2019).
Apart from transcriptional or enzyme activity-based de-regulation of sterol biosynthesis, increased triazole export is another commonly identified mechanism of resistance. Multiple putative drug efflux transporters are transcriptionally upregulated in response to itraconazole treatment in A. fumigatus, implying a role in stress responses in wild-type strains (Fraczek et al. 2013). Drug efflux pump overexpression has been well-established in yeast species as a triazole resistance mechanism, but their contribution to resistance in molds has been somewhat less studied. However, multiple reports have suggested that constitutive overexpression of specific efflux pumps may be a mechanism of triazole resistance in clinical isolates of Aspergillus (Denning, Venkateswarlu, et al. 1997; Slaven et al. 2002; Nascimento et al. 2003; Rajendran et al. 2011; Fraczek et al. 2013; Nierman et al. 2005; Ferreira et al. 2005). The A. fumigatus genome contains genes encoding efflux transporters of the multidrug resistance (MDR) variety including major facilitator superfamily (MFS) and ATP-binding cassette (ABC) superfamily, few of which are currently associated with clinical or environmental resistance to azole compounds (Fraczek et al. 2013; Paul, Diekema, and Moye-Rowley 2013; Ferreira et al. 2005).
One study of resistant clinical isolates obtained both from patients who had prior triazole exposure and from azole-naïve individuals revealed clinically resistant strains overexpressing the gene cdr1B and which possessed increased MIC values to itraconazole, posaconazole, and/ or voriconazole (Fraczek et al. 2013). The A. fumigatus efflux pump-encoding gene cdr1B, also known as abcB, abcC, or abcG1, encodes an ATP-binding cassette (ABC) transporter (Fraczek et al. 2013; Rybak et al. 2019). Deletion of cdr1B in susceptible and itraconazole resistant isolates results in reduced itraconazole MIC values relative to controls, showing that loss of cdr1B increases susceptibility to the triazole (Fraczek et al. 2013; Hagiwara et al. 2016). Another study confirmed a genetic deletion of cdr1B within a triazole resistant background containing the TR34/L98H mutation resulted in a significant decrease of resistance (Paul, Diekema, and Moye-Rowley 2013). Likewise, constitutive overexpression of the ABC transporter atrF also has been implicated in resistance (Slaven et al. 2002; Fraczek et al. 2013; Nascimento et al. 2003; Wang et al. 2018).
Several putative multidrug resistance (MDR) transporters in A. fumigatus have also been suggested to contribute to triazole resistance (Nascimento et al. 2003; Rajendran et al. 2011). The putative A. fumigatus efflux pumps Mdr1 and Mdr2, coded by the genes mdr1 and mdr2, may underlie resistance (Tobin, Peery, and Skatrud 1997; Nascimento et al. 2003). In a study of 20 putative efflux transporters in A. fumigatus, levels of mRNA transcript for mdr1 in the wild-type strain Af293 were found to be increased 14-fold over basal expression following four hours exposure to 1 mg/L itraconazole (Fraczek et al. 2013). Furthermore, gene deletion of mdr1 in a wild-type strain of A. fumigatus results in increased susceptibility to itraconazole according to MIC results, indicating that this transporter is at least important in response to triazole-induced stress (Fraczek et al. 2013). Aberrations in the transcriptional levels of additional putative MDR efflux pumps in A. fumigatus have also been reported and have been suggested to play a role in resistance (Nascimento et al. 2003; Wang et al. 2018). A transcriptional analysis of a collection of isolates with high itraconazole MIC values identified strains possessing increased expression of a gene with homology to a major facilitator (MFS) transporter mdr3 as well as to a gene with homology to an ABC transporter, mdr4 (Nascimento et al. 2003). These strains displayed either constitutive expression of these transporters or heightened induction upon triazole exposure. However, genetic evidence confirming their roles in increased triazole MICs is currently lacking.
The same biochemical or genetic plasticity which enables fungi to adapt to stressors ranging from climate changes and nutrient availability to internalized toxins from competing microbiota can also allow for adaptation to antifungals (Verweij, Chowdhary, et al. 2016; Covo 2020; Hokken et al. 2019; Buil et al. 2019). Loss of genetic stability in fungal species such as Candida albicans and A. fumigatus has been associated with lessened susceptibility to triazole antifungals (Buscaino 2019; Selmecki et al. 2009). While frequent genomic changes can often result in inviable or weakened isolates under normal conditions, a moderate level of genetic instability can allow for selection of isolates inherently resistant to an adverse condition (Verweij, Zhang, et al. 2016; Dos Reis et al. 2018; Buscaino 2019). Genetic instability can result in ploidy changes, chromosomal rearrangement, and mutations in the genetic code. Mutations or disruption of genes encoding elements of DNA damage and cell cycle checkpoint pathways have been found to affect genomic stability and susceptibility to triazoles. For example, deletion of the genes encoding the AtmA and AtrA kinases in Aspergillus fumigatus, which are involved in the response to DNA damage and activation of DNA repair and cell cycle checkpoint pathways (Dos Reis et al. 2018). Moreover, loss of either gene atmA or atrA enhances adaptability to triazole stress to higher levels than wild-type, indicating that alterations in DNA damage or cell checkpoint players may affect evolvability and acquisition of resistant phenotypes (Dos Reis et al. 2018). The same group also reported that loss of the gene encoding DNA mismatch repair (MMR) protein MshA in A. fumigatus results in genomic instability that promoted enhanced evolvability upon exposure to posaconazole to resistant level MIC. Moreover, researchers detected the presence of mshA genetic variants among clinical and environmental isolates (Dos Reis et al. 2019). This study mirrors studies of C. albicans strains lacking the related MMR protein MSH2 which similarly resulted in genomic instability and frequent occurrence of triazole resistant mutant strains, as well as a report that half of sequenced C. glabrata strains possess mutations in MSH2 which may contribute to resistance acquisition (Legrand et al. 2007; Healey et al. 2016). Moreover, study of Cryptococcus neoformans isolates which mutated at a high rate revealed strains bearing MSH2 mutations (Boyce et al. 2017). Although mutations that alter genetic stability have not been yet described as drivers of resistance in clinical isolates, these examples validate the possibility that genetic instability from aberrations in pathways such as DNA repair may enable acquisition of triazole resistance within A. fumigatus.
In addition to the genetic means outlined in this review, other mechanisms have been put forth as strategies by which A. fumigatus may undermine and survive the effects of triazole exposures. These include mitochondrial dysfunction, the formation of biofilms, epigenetic changes, RNA interference, alterations in intracellular networks, and impermeability to drug due to cell wall changes (Matthaiou et al. 2018; Bowyer, Bromley, and Denning 2020; Li et al. 2020; Hokken et al. 2019; Bromley et al. 2016; Valdes et al. 2018; Abdolrasouli et al. 2015). Many other stratagems likely exist which have yet to be detected or linked to resistance in environmental or clinical strains of A. fumigatus (Buil et al. 2019). While a discussion of every potential contributor to triazole antifungal resistance is beyond the scope of this review, such additional pathogen attributes may well impact the health of aspergillosis patients or alter the makeup of ecological niches and fragile microbiomes.
Conclusions
Fungal pathogens have become a considerable threat to human health as well as to animals and ecosystems globally. The rise of antifungal resistance only exacerbates this situation. The impact that A. fumigatus triazole resistance has on environmental microbiomes and patient treatment outcomes is only recently being appreciated. It is clear from the data collected thus far that drug resistance in A. fumigatus is associated with multiple distinct adaptive strategies. Often, the aberrant induction of gene transcription enables a strain to withstand drug activity by overproducing the drug’s target enzyme or drug efflux mechanisms (Nascimento et al. 2003). Overexpression can also occur concomitantly with ORF mutations in the A. fumigatus gene encoding the triazole target, Cyp51A, to alter the binding affinity of triazoles to the enzyme. Such mutations can occur without detected changes in enzyme expression to confer triazole resistance. Moreover, resistance has also been associated with the overexpression of drug efflux transporters and various additional stratagems, including alteration of sterol biosynthesis in the mevalonate pathway, have been suggested to enable resistance to triazole compounds in this opportunistic pathogen. It is likely that we are only scratching the surface of the myriad mechanisms that filamentous fungi can utilize to subvert the activity of triazole compounds.
Although triazole resistance in A. fumigatus appears to be selected both during prolonged exposure to azoles within a host and in the environment by agricultural triazoles, the incidence of each route and the potential for common mechanisms acquired through each is still unclear. Increased surveillance for resistant A. fumigatus isolates in agricultural settings, compost, indoor ventilation, and patient samples will be of high value in order to appreciate and respond to this threat to public health (Verweij, Mellado, and Melchers 2007; Zilberberg et al. 2018; Chowdhary et al. 2012). Adequate methods of surveillance and detection of pathogenic Aspergillus species, especially drug-resistant variants, are needed to predict the onset of allergic symptoms, nosocomial infections, and to guide treatment decisions in patient centers densely concentrated with at-risk individuals (Streifel et al. 1983; Rose 1972; Patterson et al. 2016). Unfortunately, studies on the fiscal and mortal burdens of Aspergillus diseases within the United States are few in number and often outdated (Zilberberg et al. 2018; Dasbach, Davies, and Teutsch 2000; Fraser et al. 1979; Steinbach et al. 2012). Improved surveillance to determine the prevalence of resistant strains, epidemiology, patient outcomes with resistant infections, contemporary effective treatment stratagems to combat invasive aspergillosis, and the occurrence of triazole resistance within both environmental samples and clinical patient isolates of A. fumigatus is essential (Steinbach et al. 2012; Neofytos et al. 2010; Patterson et al. 2016). This will enable effective study of the distribution and impact of resistant pathogens on ecosystems and human health (Fisher et al. 2012). Better understanding of the process of resistance development within A. fumigatus is also needed (Verweij, Chowdhary, et al. 2016). Probes into the process whereby strains become resistant may reveal stratagems to halt the rise of resistance in this opportunistic pathogen. Moreover, illuminating triazole-specific intracellular stress responses could identify novel targets and inform strategies to address resistant infections in novel stand alone or combination therapies (Snelders et al. 2008).
Originality-Significance Statement:
This work addresses the understudied but significant issue of triazole antifungal resistance in the human and animal pathogen Aspergillus fumigatus. Though resistance within this species represents a threat to ecology and public health, much information concerning its origination and causes is limited, frustrating comprehension and progress in this area. This work concisely summarizes the process of triazole antifungal resistance progression within both patients and environmental settings as it is currently understood, as well as discussing multiple reported biological mechanisms associated with clinical and environmental triazole resistance in Aspergillus fumigatus.
Acknowledgements
This work was supported by (R21AI142509 and R21AI139388) to JRF and by (R01AI143197) to PDR and JRF. All authors declare no conflict of interest.
References:
- Abdolrasouli A, Rhodes J, Beale MA, Hagen F, Rogers TR, Chowdhary A, Meis JF, Armstrong-James D, and Fisher MC. 2015. ‘Genomic Context of Azole Resistance Mutations in Aspergillus fumigatus Determined Using Whole-Genome Sequencing’, mBio, 6: e00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Abdallah Q, Ge W, and Fortwendel JR. 2017. ‘A Simple and Universal System for Gene Manipulation in Aspergillus fumigatus: In Vitro-Assembled Cas9-Guide RNA Ribonucleoproteins Coupled with Microhomology Repair Templates’, mSphere, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Moreno C, Lavergne RA, Hagen F, Morio F, Meis JF, and Le Pape P. 2019. ‘Fungicide-driven alterations in azole-resistant Aspergillus fumigatus are related to vegetable crops in Colombia, South America’, Mycologia, 111: 217–24. [DOI] [PubMed] [Google Scholar]
- Anastasi A, Varese GC, and Marchisio VF. 2005. ‘Isolation and identification of fungal communities in compost and vermicompost’, Mycologia, 97: 33–44. [DOI] [PubMed] [Google Scholar]
- Andersen B, Frisvad JC, Sondergaard I, Rasmussen IS, and Larsen LS. 2011. ‘Associations between fungal species and water-damaged building materials’, Appl Environ Microbiol, 77: 4180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendrup MC, Mavridou E, Mortensen KL, Snelders E, Frimodt-Moller N, Khan H, Melchers WJ, and Verweij PE. 2010. ‘Development of azole resistance in Aspergillus fumigatus during azole therapy associated with change in virulence’, PLoS One, 5: e10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud MB, Chibucos MC, Costanzo MC, Crabtree J, Inglis DO, Lotia A, Orvis J, Shah P, Skrzypek MS, Binkley G, Miyasato SR, Wortman JR, and Sherlock G. 2010. ‘The Aspergillus Genome Database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community’, Nucleic Acids Res, 38: D420–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddley JW, Marr KA, Andes DR, Walsh TJ, Kauffman CA, Kontoyiannis DP, Ito JI, Balajee SA, Pappas PG, and Moser SA. 2009. ‘Patterns of susceptibility of Aspergillus isolates recovered from patients enrolled in the Transplant-Associated Infection Surveillance Network’, J Clin Microbiol, 47: 3271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader O, Weig M, Reichard U, Lugert R, Kuhns M, Christner M, Held J, Peter S, Schumacher U, Buchheidt D, Tintelnot K, Gross U, and Partners MykoLabNet D. 2013. ‘cyp51A-Based mechanisms of Aspergillus fumigatus azole drug resistance present in clinical samples from Germany’, Antimicrob Agents Chemother, 57: 3513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basenko EY, Pulman JA, Shanmugasundram A, Harb OS, Crouch K, Starns D, Warrenfeltz S, Aurrecoechea C, Stoeckert CJ Jr., Kissinger JC, Roos DS, and Hertz-Fowler C. 2018. ‘FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes’, J Fungi (Basel), 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beernaert LA, Pasmans F, Van Waeyenberghe L, Dorrestein GM, Verstappen F, Vercammen F, Haesebrouck F, and Martel A. 2009. ‘Avian Aspergillus fumigatus strains resistant to both itraconazole and voriconazole’, Antimicrob Agents Chemother, 53: 2199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellete B, Raberin H, Morel J, Flori P, Hafid J, and Manhsung RT. 2010. ‘Acquired resistance to voriconazole and itraconazole in a patient with pulmonary aspergilloma’, Med Mycol, 48: 197–200. [DOI] [PubMed] [Google Scholar]
- Bongomin F, Asio LG, Baluku JB, Kwizera R, and Denning DW. 2020. ‘Chronic Pulmonary Aspergillosis: Notes for a Clinician in a Resource-Limited Setting Where There Is No Mycologist’, J Fungi (Basel), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongomin F, Gago S, Oladele RO, and Denning DW. 2017. ‘Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision’, J Fungi (Basel), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer P, Bromley MJ, and Denning DW. 2020. ‘Linking calcium signaling and mitochondrial function in fungal drug resistance’, Proc Natl Acad Sci U S A, 117: 1254–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce KJ, Wang Y, Verma S, Shakya VPS, Xue C, and Idnurm A. 2017. ‘Mismatch Repair of DNA Replication Errors Contributes to Microevolution in the Pathogenic Fungus Cryptococcus neoformans’, mBio, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissaud O, Guichoux J, Harambat J, Tandonnet O, and Zaoutis T. 2012. ‘Invasive fungal disease in PICU: epidemiology and risk factors’, Ann Intensive Care, 2: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromley M, Johns A, Davies E, Fraczek M, Mabey Gilsenan J, Kurbatova N, Keays M, Kapushesky M, Gut M, Gut I, Denning DW, and Bowyer P. 2016. ‘Mitochondrial Complex I Is a Global Regulator of Secondary Metabolism, Virulence and Azole Sensitivity in Fungi’, PLoS One, 11: e0158724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC. 2012. ‘Hidden killers: human fungal infections’, Sci Transl Med, 4: 165rv13. [DOI] [PubMed] [Google Scholar]
- Bueid A, Howard SJ, Moore CB, Richardson MD, Harrison E, Bowyer P, and Denning DW. 2010. ‘Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009’, J Antimicrob Chemother, 65: 2116–8. [DOI] [PubMed] [Google Scholar]
- Buil JB, Hare RK, Zwaan BJ, Arendrup MC, Melchers WJG, and Verweij PE. 2019. ‘The fading boundaries between patient and environmental routes of triazole resistance selection in Aspergillus fumigatus’, PLoS Pathog, 15: e1007858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscaino A 2019. ‘Chromatin-Mediated Regulation of Genome Plasticity in Human Fungal Pathogens’, Genes (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps SM, Dutilh BE, Arendrup MC, Rijs AJ, Snelders E, Huynen MA, Verweij PE, and Melchers WJ. 2012. ‘Discovery of a HapE mutation that causes azole resistance in Aspergillus fumigatus through whole genome sequencing and sexual crossing’, PLoS One, 7: e50034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps SM, Rijs AJ, Klaassen CH, Meis JF, O’Gorman CM, Dyer PS, Melchers WJ, and Verweij PE. 2012. ‘Molecular epidemiology of Aspergillus fumigatus isolates harboring the TR34/L98H azole resistance mechanism’, J Clin Microbiol, 50: 2674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps SM, van der Linden JW, Li Y, Kuijper EJ, van Dissel JT, Verweij PE, and Melchers WJ. 2012. ‘Rapid induction of multiple resistance mechanisms in Aspergillus fumigatus during azole therapy: a case study and review of the literature’, Antimicrob Agents Chemother, 56: 10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. 2019. ‘Aspergillosis Statistics’, Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), Division of Foodborne, Waterborne, and Environmental Diseases (DFWED), Accessed 8/24/2020 https://www.cdc.gov/fungal/diseases/aspergillosis/statistics.html. [Google Scholar]
- Chen J, Li H, Li R, Bu D, and Wan Z. 2005. ‘Mutations in the cyp51A gene and susceptibility to itraconazole in Aspergillus fumigatus serially isolated from a patient with lung aspergilloma’, J Antimicrob Chemother, 55: 31–7. [DOI] [PubMed] [Google Scholar]
- Chowdhary A, Kathuria S, Randhawa HS, Gaur SN, Klaassen CH, and Meis JF. 2012. ‘Isolation of multiple-triazole-resistant Aspergillus fumigatus strains carrying the TR/L98H mutations in the cyp51A gene in India’, J Antimicrob Chemother, 67: 362–6. [DOI] [PubMed] [Google Scholar]
- Chowdhary A, Kathuria S, Xu J, and Meis JF. 2013. ‘Emergence of azole-resistant aspergillus fumigatus strains due to agricultural azole use creates an increasing threat to human health’, PLoS Pathog, 9: e1003633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhary A, Sharma C, Kathuria S, Hagen F, and Meis JF. 2014. ‘Azole-resistant Aspergillus fumigatus with the environmental TR46/Y121F/T289A mutation in India’, J Antimicrob Chemother, 69: 555–7. [DOI] [PubMed] [Google Scholar]
- Chowdhary Anuradha, Sharma Cheshta, Kathuria Shallu, Hagen Ferry, and Meis Jacques F.. 2015. ‘Prevalence and mechanism of triazole resistance in Aspergillus fumigatus in a referral chest hospital in Delhi, India and an update of the situation in Asia’, Frontiers in Microbiology, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chryssanthou E 1997. ‘In vitro susceptibility of respiratory isolates of Aspergillus species to itraconazole and amphotericin B. acquired resistance to itraconazole’, Scand J Infect Dis, 29: 509–12. [DOI] [PubMed] [Google Scholar]
- Covo S 2020. ‘Genomic Instability in Fungal Plant Pathogens’, Genes (Basel), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagenais TR, and Keller NP. 2009. ‘Pathogenesis of Aspergillus fumigatus in Invasive Aspergillosis’, Clin Microbiol Rev, 22: 447–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannaoui E, Borel E, Monier MF, Piens MA, Picot S, and Persat F. 2001. ‘Acquired itraconazole resistance in Aspergillus fumigatus’, J Antimicrob Chemother, 47: 333–40. [DOI] [PubMed] [Google Scholar]
- Dannaoui E, Persat F, Monier MF, Borel E, Piens MA, and Picot S. 1999. ‘In-vitro susceptibility of Aspergillus spp. isolates to amphotericin B and itraconazole’, Journal of Antimicrobial Chemotherapy, 44: 553–55. [DOI] [PubMed] [Google Scholar]
- Dasbach EJ, Davies GM, and Teutsch SM. 2000. ‘Burden of aspergillosis-related hospitalizations in the United States’, Clin Infect Dis, 31: 1524–8. [DOI] [PubMed] [Google Scholar]
- Denning DW 1998. ‘Invasive aspergillosis’, Clin Infect Dis, 26: 781–803; quiz 04–5. [DOI] [PubMed] [Google Scholar]
- Denning DW, Park S, Lass-Florl C, Fraczek MG, Kirwan M, Gore R, Smith J, Bueid A, Moore CB, Bowyer P, and Perlin DS. 2011. ‘High-frequency triazole resistance found In nonculturable Aspergillus fumigatus from lungs of patients with chronic fungal disease’, Clin Infect Dis, 52: 1123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning DW, Pleuvry A, and Cole DC. 2013. ‘Global burden of allergic bronchopulmonary aspergillosis with asthma and its complication chronic pulmonary aspergillosis in adults’, Med Mycol, 51: 361–70. [DOI] [PubMed] [Google Scholar]
- Denning DW, Radford SA, Oakley KL, Hall L, Johnson EM, and Warnock DW. 1997. ‘Correlation between in-vitro susceptibility testing to itraconazole and in-vivo outcome of Aspergillus fumigatus infection’, J Antimicrob Chemother, 40: 401–14. [DOI] [PubMed] [Google Scholar]
- Denning DW, Riniotis K, Dobrashian R, and Sambatakou H. 2003. ‘Chronic cavitary and fibrosing pulmonary and pleural aspergillosis: case series, proposed nomenclature change, and review’, Clin Infect Dis, 37 Suppl 3: S265–80. [DOI] [PubMed] [Google Scholar]
- Denning DW, Tucker RM, Hanson LH, and Stevens DA. 1989. ‘Treatment of invasive aspergillosis with itraconazole’, Am J Med, 86: 791–800. [DOI] [PubMed] [Google Scholar]
- Denning DW, Venkateswarlu K, Oakley KL, Anderson MJ, Manning NJ, Stevens DA, Warnock DW, and Kelly SL. 1997. ‘Itraconazole resistance in Aspergillus fumigatus’, Antimicrob Agents Chemother, 41: 1364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Guerra TM, Mellado E, Cuenca-Estrella M, and Rodriguez-Tudela JL. 2003. ‘A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus’, Antimicrob Agents Chemother, 47: 1120–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Reis TF, Silva LP, de Castro PA, Almeida de Lima PB, do Carmo RA, Marini MM, da Silveira JF, Ferreira BH, Rodrigues F, Malavazi I, and Goldman GH. 2018. ‘The Influence of Genetic Stability on Aspergillus fumigatus Virulence and Azole Resistance’, G3 (Bethesda), 8: 265–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Reis TF, Silva LP, de Castro PA, do Carmo RA, Marini MM, da Silveira JF, Ferreira BH, Rodrigues F, Lind AL, Rokas A, and Goldman GH. 2019. ‘The Aspergillus fumigatus Mismatch Repair MSH2 Homolog Is Important for Virulence and Azole Resistance’, mSphere, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggimann P, Chevrolet JC, Starobinski M, Majno P, Totsch M, Chapuis B, and Pittet D. 2006. ‘Primary invasive aspergillosis of the digestive tract: report of two cases and review of the literature’, Infection, 34: 333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcone M, Massetti AP, Russo A, Vullo V, and Venditti M. 2011. ‘Invasive aspergillosis in patients with liver disease’, Med Mycol, 49: 406–13. [DOI] [PubMed] [Google Scholar]
- Faria-Ramos I, Farinha S, Neves-Maia J, Tavares PR, Miranda IM, Estevinho LM, Pina-Vaz C, and Rodrigues AG. 2014. ‘Development of cross-resistance by Aspergillus fumigatus to clinical azoles following exposure to prochloraz, an agricultural azole’, BMC Microbiol, 14: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ME, Colombo AL, Paulsen I, Ren Q, Wortman J, Huang J, Goldman MH, and Goldman GH. 2005. ‘The ergosterol biosynthesis pathway, transporter genes, and azole resistance in Aspergillus fumigatus’, Med Mycol, 43 Suppl 1: S313–9. [DOI] [PubMed] [Google Scholar]
- Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, and Gurr SJ. 2012. ‘Emerging fungal threats to animal, plant and ecosystem health’, Nature, 484: 186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, and Bowyer P. 2013. ‘The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus’, J Antimicrob Chemother, 68: 1486–96. [DOI] [PubMed] [Google Scholar]
- Fraser DW, Ward JI, Ajello L, and Plikaytis BD. 1979. ‘Aspergillosis and other systemic mycoses. The growing problem’, JAMA, 242: 1631–5. [PubMed] [Google Scholar]
- Fuhren J, Voskuil WS, Boel CH, Haas PJ, Hagen F, Meis JF, and Kusters JG. 2015. ‘High prevalence of azole resistance in Aspergillus fumigatus isolates from high-risk patients’, J Antimicrob Chemother, 70: 2894–8. [DOI] [PubMed] [Google Scholar]
- George D, Miniter P, and Andriole VT. 1996. ‘Efficacy of UK-109496, a new azole antifungal agent, in an experimental model of invasive aspergillosis’, Antimicrob Agents Chemother, 40: 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll AH, Shah PM, Mentzel C, Schneider M, Just-Nuebling G, and Huebner K. 1996. ‘Trends in the postmortem epidemiology of invasive fungal infections at a university hospital’, J Infect, 33: 23–32. [DOI] [PubMed] [Google Scholar]
- Groll AH, and Walsh TJ. 2001. ‘Uncommon opportunistic fungi: new nosocomial threats’, Clin Microbiol Infect, 7 Suppl 2: 8–24. [DOI] [PubMed] [Google Scholar]
- Gsaller F, Hortschansky P, Furukawa T, Carr PD, Rash B, Capilla J, Muller C, Bracher F, Bowyer P, Haas H, Brakhage AA, and Bromley MJ. 2016a. ‘Correction: Sterol Biosynthesis and Azole Tolerance Is Governed by the Opposing Actions of SrbA and the CCAAT Binding Complex’, PLoS Pathog, 12: e1006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsaller F, Hortschansky P, Furukawa T, Carr PD, Rash B, Capilla J, Muller C, Bracher F, Bowyer P, Haas H, Brakhage AA, and Bromley MJ. 2016b. ‘Sterol Biosynthesis and Azole Tolerance Is Governed by the Opposing Actions of SrbA and the CCAAT Binding Complex’, PLoS Pathog, 12: e1005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D, Arai T, Takahashi H, Kusuya Y, Watanabe A, and Kamei K. 2018. ‘Non-cyp51A Azole-Resistant Aspergillus fumigatus Isolates with Mutation in HMG-CoA Reductase’, Emerg Infect Dis, 24: 1889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D, Takahashi H, Watanabe A, Takahashi-Nakaguchi A, Kawamoto S, Kamei K, and Gonoi T. 2014. ‘Whole-genome comparison of Aspergillus fumigatus strains serially isolated from patients with aspergillosis’, J Clin Microbiol, 52: 4202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara D, Watanabe A, Kamei K, and Goldman GH. 2016. ‘Epidemiological and Genomic Landscape of Azole Resistance Mechanisms in Aspergillus Fungi’, Front Microbiol, 7: 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey KR, Jimenez Ortigosa C, Shor E, and Perlin DS. 2016. ‘Genetic Drivers of Multidrug Resistance in Candida glabrata’, Front Microbiol, 7: 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeres J, Backx LJ, and Van Cutsem J. 1984. ‘Antimycotic azoles. 7. Synthesis and antifungal properties of a series of novel triazol-3-ones’, J Med Chem, 27: 894–900. [DOI] [PubMed] [Google Scholar]
- Hokken MWJ, Zwaan BJ, Melchers WJG, and Verweij PE. 2019. ‘Facilitators of adaptation and antifungal resistance mechanisms in clinically relevant fungi’, Fungal Genet Biol, 132: 103254. [DOI] [PubMed] [Google Scholar]
- Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, and Denning DW. 2009. ‘Frequency and evolution of Azole resistance in Aspergillus fumigatus associated with treatment failure’, Emerg Infect Dis, 15: 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard SJ, Pasqualotto AC, Anderson MJ, Leatherbarrow H, Albarrag AM, Harrison E, Gregson L, Bowyer P, and Denning DW. 2013. ‘Major variations in Aspergillus fumigatus arising within aspergillomas in chronic pulmonary aspergillosis’, Mycoses, 56: 434–41. [DOI] [PubMed] [Google Scholar]
- Howard SJ, Webster I, Moore CB, Gardiner RE, Park S, Perlin DS, and Denning DW. 2006. ‘Multi-azole resistance in Aspergillus fumigatus’, Int J Antimicrob Agents, 28: 450–3. [DOI] [PubMed] [Google Scholar]
- Koehler P, Hamprecht A, Bader O, Bekeredjian-Ding I, Buchheidt D, Doelken G, Elias J, Haase G, Hahn-Ast C, Karthaus M, Kekule A, Keller P, Kiehl M, Krause SW, Kramer C, Neumann S, Rohde H, La Rosee P, Ruhnke M, Schafhausen P, Schalk E, Schulz K, Schwartz S, Silling G, Staib P, Ullmann A, Vergoulidou M, Weber T, Cornely OA, and Vehreschild MJ. 2017. ‘Epidemiology of invasive aspergillosis and azole resistance in patients with acute leukaemia: the SEPIA Study’, Int J Antimicrob Agents, 49: 218–23. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, Ito J, Andes DR, Baddley JW, Brown JM, Brumble LM, Freifeld AG, Hadley S, Herwaldt LA, Kauffman CA, Knapp K, Lyon GM, Morrison VA, Papanicolaou G, Patterson TF, Perl TM, Schuster MG, Walker R, Wannemuehler KA, Wingard JR, Chiller TM, and Pappas PG. 2010. ‘Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: overview of the Transplant-Associated Infection Surveillance Network (TRANSNET) Database’, Clin Infect Dis, 50: 1091–100. [DOI] [PubMed] [Google Scholar]
- Kosmidis C, and Denning DW. 2015. ‘The clinical spectrum of pulmonary aspergillosis’, Thorax, 70: 270–7. [DOI] [PubMed] [Google Scholar]
- Krishnan Natesan S, Wu W, Cutright JL, and Chandrasekar PH. 2012. ‘In vitro-in vivo correlation of voriconazole resistance due to G448S mutation (cyp51A gene) in Aspergillus fumigatus’, Diagn Microbiol Infect Dis, 74: 272–7. [DOI] [PubMed] [Google Scholar]
- Lass-Florl C, and Cuenca-Estrella M. 2017. ‘Changes in the epidemiological landscape of invasive mould infections and disease’, J Antimicrob Chemother, 72: i5–i11. [DOI] [PubMed] [Google Scholar]
- Latge JP 1999. ‘Aspergillus fumigatus and aspergillosis’, Clin Microbiol Rev, 12: 310–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M, Chan CL, Jauert PA, and Kirkpatrick DT. 2007. ‘Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans’, Eukaryot Cell, 6: 2194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestrade PP, Bentvelsen RG, Schauwvlieghe Afad, Schalekamp S, van der Velden Wjfm, Kuiper EJ, van Paassen J, van der Hoven B, van der Lee HA, Melchers WJG, de Haan AF, van der Hoeven HL, Rijnders BJA, van der Beek MT, and Verweij PE. 2019. ‘Voriconazole Resistance and Mortality in Invasive Aspergillosis: A Multicenter Retrospective Cohort Study’, Clin Infect Dis, 68: 1463–71. [DOI] [PubMed] [Google Scholar]
- Lestrade PP, Meis JF, Arends JP, van der Beek MT, de Brauwer E, van Dijk K, de Greeff SC, Haas PJ, Hodiamont CJ, Kuijper EJ, Leenstra T, Muller AE, Oude Lashof AM, Rijnders BJ, Roelofsen E, Rozemeijer W, Tersmette M, Terveer EM, Verduin CM, Wolfhagen MJ, Melchers WJ, and Verweij PE. 2016. ‘Diagnosis and management of aspergillosis in the Netherlands: a national survey’, Mycoses, 59: 101–7. [DOI] [PubMed] [Google Scholar]
- Levetin E 2004. ‘An atlas of fungal spores’, J Allergy Clin Immunol, 113: 366–8. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang Y, Zhang C, Wang H, Wei X, Chen P, and Lu L. 2020. ‘Mitochondrial dysfunctions trigger the calcium signaling-dependent fungal multidrug resistance’, Proc Natl Acad Sci U S A, 117: 1711–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SR, Frade JP, Etienne KA, Pfaller MA, Diekema DJ, and Balajee SA. 2011. ‘Azole resistance in Aspergillus fumigatus isolates from the ARTEMIS global surveillance study is primarily due to the TR/L98H mutation in the cyp51A gene’, Antimicrob Agents Chemother, 55: 4465–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Medrano F, Fernandez-Ruiz M, Silva JT, Carver PL, van Delden C, Merino E, Perez-Saez MJ, Montero M, Coussement J, de Abreu Mazzolin M, Cervera C, Santos L, Sabe N, Scemla A, Cordero E, Cruzado-Vega L, Martin-Moreno PL, Len O, Rudas E, de Leon AP, Arriola M, Lauzurica R, David M, Gonzalez-Rico C, Henriquez-Palop F, Fortun J, Nucci M, Manuel O, Pano-Pardo JR, Montejo M, Munoz P, Sanchez-Sobrino B, Mazuecos A, Pascual J, Horcajada JP, Lecompte T, Moreno A, Carratala J, Blanes M, Hernandez D, Farinas MC, Andres A, Aguado JM, the Group for the Study of Infection in Transplant Recipients of the Spanish Society of Clinical Microbiology Spanish Network for Research in Infectious Diseases, the Study Group for Infections in Compromised Hosts of the European Society of Clinical Microbiology Infectious Diseases, Diseases Infectious, and Study the Swiss Transplant Cohort. 2016. ‘Clinical Presentation and Determinants of Mortality of Invasive Pulmonary Aspergillosis in Kidney Transplant Recipients: A Multinational Cohort Study’, Am J Transplant, 16: 3220–34. [DOI] [PubMed] [Google Scholar]
- Manavathu EK, Cutright JL, Loebenberg D, and Chandrasekar PH. 2000. ‘A comparative study of the in vitro susceptibilities of clinical and laboratory-selected resistant isolates of Aspergillus spp. to amphotericin B, itraconazole, voriconazole and posaconazole (SCH 56592)’, J Antimicrob Chemother, 46: 229–34. [DOI] [PubMed] [Google Scholar]
- Mann PA, Parmegiani RM, Wei SQ, Mendrick CA, Li X, Loebenberg D, DiDomenico B, Hare RS, Walker SS, and McNicholas PM. 2003. ‘Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14alpha-demethylase’, Antimicrob Agents Chemother, 47: 577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaiou EI, Sass G, Stevens DA, and Hsu JL. 2018. ‘Iron: an essential nutrient for Aspergillus fumigatus and a fulcrum for pathogenesis’, Curr Opin Infect Dis, 31: 506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavridou E, Meletiadis J, Jancura P, Abbas S, Arendrup MC, Melchers WJ, Heskes T, Mouton JW, and Verweij PE. 2013. ‘Composite survival index to compare virulence changes in azole-resistant Aspergillus fumigatus clinical isolates’, PLoS One, 8: e72280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Cuenca-Estrella M, and Rodriguez-Tudela JL. 2004. ‘Substitutions at methionine 220 in the 14alpha-sterol demethylase (Cyp51A) of Aspergillus fumigatus are responsible for resistance in vitro to azole antifungal drugs’, Antimicrob Agents Chemother, 48: 2747–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Melchers WJ, Verweij PE, Cuenca-Estrella M, and Rodriguez-Tudela JL. 2007. ‘A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations’, Antimicrob Agents Chemother, 51: 1897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento AM, Goldman GH, Park S, Marras SA, Delmas G, Oza U, Lolans K, Dudley MN, Mann PA, and Perlin DS. 2003. ‘Multiple resistance mechanisms among Aspergillus fumigatus mutants with high-level resistance to itraconazole’, Antimicrob Agents Chemother, 47: 1719–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neofytos D, Fishman JA, Horn D, Anaissie E, Chang CH, Olyaei A, Pfaller M, Steinbach WJ, Webster KM, and Marr KA. 2010. ‘Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients’, Transplant Infectious Disease, 12: 220–29. [DOI] [PubMed] [Google Scholar]
- Nierman WC, May G, Kim HS, Anderson MJ, Chen D, and Denning DW. 2005. ‘What the Aspergillus genomes have told us’, Med Mycol, 43 Suppl 1: S3–5. [DOI] [PubMed] [Google Scholar]
- Oakley KL, Moore CB, and Denning DW. 1998. ‘In-vitro activity of voriconazole against Aspergillus spp. and comparison with itraconazole and amphotericin B’, J Antimicrob Chemother, 42: 91–4. [DOI] [PubMed] [Google Scholar]
- Patterson TF, Thompson GR 3rd, Denning DW, Fishman JA, Hadley S, Herbrecht R, Kontoyiannis DP, Marr KA, Morrison VA, Nguyen MH, Segal BH, Steinbach WJ, Stevens DA, Walsh TJ, Wingard JR, Young JA, and Bennett JE. 2016. ‘Practice Guidelines for the Diagnosis and Management of Aspergillosis: 2016 Update by the Infectious Diseases Society of America’, Clin Infect Dis, 63: e1–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Diekema D, and Moye-Rowley WS. 2013. ‘Contributions of Aspergillus fumigatus ATP-binding cassette transporter proteins to drug resistance and virulence’, Eukaryot Cell, 12: 1619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen C, Hallsworth JE, Alvarez-Perez S, Nierman WC, Hamill PG, Blain D, Rediers H, and Lievens B. 2017. ‘Ecology of aspergillosis: insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species’, Microb Biotechnol, 10: 296–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Messer SA, Boyken L, Rice C, Tendolkar S, Hollis RJ, and Diekema DJ. 2008. ‘In vitro survey of triazole cross-resistance among more than 700 clinical isolates of Aspergillus species’, J Clin Microbiol, 46: 2568–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Rhomberg PR, Wiederhold NP, Gibas C, Sanders C, Fan H, Mele J, Kovanda LL, and Castanheira M. 2018. ‘In Vitro Activity of Isavuconazole against Opportunistic Fungal Pathogens from Two Mycology Reference Laboratories’, Antimicrob Agents Chemother, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham CD, Reiss E, Hagen F, Meis JF, and Lockhart SR. 2014. ‘Passive surveillance for azole-resistant Aspergillus fumigatus, United States, 2011–2013’, Emerg Infect Dis, 20: 1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran R, Mowat E, McCulloch E, Lappin DF, Jones B, Lang S, Majithiya JB, Warn P, Williams C, and Ramage G. 2011. ‘Azole resistance of Aspergillus fumigatus biofilms is partly associated with efflux pump activity’, Antimicrob Agents Chemother, 55: 2092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees JR, Pinner RW, Hajjeh RA, Brandt ME, and Reingold AL. 1998. ‘The epidemiological features of invasive mycotic infections in the San Francisco Bay area, 1992–1993: results of population-based laboratory active surveillance’, Clin Infect Dis, 27: 1138–47. [PubMed] [Google Scholar]
- Ren J, Jin X, Zhang Q, Zheng Y, Lin D, and Yu Y. 2017. ‘Fungicides induced triazole-resistance in Aspergillus fumigatus associated with mutations of TR46/Y121F/T289A and its appearance in agricultural fields’, J Hazard Mater, 326: 54–60. [DOI] [PubMed] [Google Scholar]
- Resendiz Sharpe A, Lagrou K, Meis JF, Chowdhary A, Lockhart SR, Verweij PE, and Isham Ecmm Aspergillus Resistance Surveillance working group. 2018. ‘Triazole resistance surveillance in Aspergillus fumigatus’, Med Mycol, 56: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas E Ribas AD, Spolti P, Del Ponte EM, Donato KZ, Schrekker H, and Fuentefria AM. 2016. ‘Is the emergence of fungal resistance to medical triazoles related to their use in the agroecosystems? A mini review’, Braz J Microbiol, 47: 793–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Tudela JL, Alcazar-Fuoli L, Mellado E, Alastruey-Izquierdo A, Monzon A, and Cuenca-Estrella M. 2008. ‘Epidemiological cutoffs and cross-resistance to azole drugs in Aspergillus fumigatus’, Antimicrobial Agents and Chemotherapy, 52: 2468–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose HD 1972. ‘Mechanical control of hospital ventilation and Aspergillus infections’, Am Rev Respir Dis, 105: 306–7. [DOI] [PubMed] [Google Scholar]
- Rybak JM, Fortwendel JR, and Rogers PD. 2019. ‘Emerging threat of triazole-resistant Aspergillus fumigatus’, J Antimicrob Chemother, 74: 835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak JM, Ge W, Wiederhold NP, Parker JE, Kelly SL, Rogers PD, and Fortwendel JR. 2019. ‘Mutations in hmg1, Challenging the Paradigm of Clinical Triazole Resistance in Aspergillus fumigatus’, MBio, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoustra SE, Debets AJM, Ajmm Rijs J. Zhang, Snelders E, Leendertse PC, Melchers WJG, Rietveld AG, Zwaan BJ, and Verweij PE. 2019. ‘Environmental Hotspots for Azole Resistance Selection of Aspergillus fumigatus, the Netherlands’, Emerg Infect Dis, 25: 1347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, and Berman J. 2009. ‘Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance’, PLoS Genet, 5: e1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedmousavi S, Guillot J, Arne P, de Hoog GS, Mouton JW, Melchers WJ, and Verweij PE. 2015. ‘Aspergillus and aspergilloses in wild and domestic animals: a global health concern with parallels to human disease’, Med Mycol, 53: 765–97. [DOI] [PubMed] [Google Scholar]
- Shah A, and Panjabi C. 2016. ‘Allergic Bronchopulmonary Aspergillosis: A Perplexing Clinical Entity’, Allergy Asthma Immunol Res, 8: 282–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaven John W., Anderson Michael J., Sanglard Dominique, Dixon Graham K., Bille Jacques, Roberts Ian S., and Denning David W.. 2002. ‘Increased expression of a novel Aspergillus fumigatus ABC transporter gene, atrF, in the presence of itraconazole in an itraconazole resistant clinical isolate’, Fungal Genetics and Biology, 36: 199–206. [DOI] [PubMed] [Google Scholar]
- Snelders E, Camps SM, Karawajczyk A, Rijs AJ, Zoll J, Verweij PE, and Melchers WJ. 2015. ‘Genotype-phenotype complexity of the TR46/Y121F/T289A cyp51A azole resistance mechanism in Aspergillus fumigatus’, Fungal Genet Biol, 82: 129–35. [DOI] [PubMed] [Google Scholar]
- Snelders E, Camps SM, Karawajczyk A, Schaftenaar G, Kema GH, van der Lee HA, Klaassen CH, Melchers WJ, and Verweij PE. 2012. ‘Triazole fungicides can induce cross-resistance to medical triazoles in Aspergillus fumigatus’, PLoS One, 7: e31801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelders E, Huis In ‘t Veld RA, Rijs AJ, Kema GH, Melchers WJ, and Verweij PE. 2009. ‘Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles’, Appl Environ Microbiol, 75: 4053–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelders E, Karawajczyk A, Schaftenaar G, Verweij PE, and Melchers WJ. 2010. ‘Azole resistance profile of amino acid changes in Aspergillus fumigatus CYP51A based on protein homology modeling’, Antimicrob Agents Chemother, 54: 2425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelders E, Karawajczyk A, Verhoeven RJ, Venselaar H, Schaftenaar G, Verweij PE, and Melchers WJ. 2011. ‘The structure-function relationship of the Aspergillus fumigatuscyp51A L98H conversion by site-directed mutagenesis: the mechanism of L98H azole resistance’, Fungal Genet Biol, 48: 1062–70. [DOI] [PubMed] [Google Scholar]
- Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, and Verweij PE. 2008. ‘Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism’, PLoS Med, 5: e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, and Gerrish PJ. 2010. ‘Beneficial mutations and the dynamics of adaptation in asexual populations’, Philos Trans R Soc Lond B Biol Sci, 365: 1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff KING 5. 2019. ‘King County to make Aspergillus mold infections reportable to public health’. https://www.king5.com/article/news/health/seattle-childrens-aspergillus-mold-reporting/281-6ae6b740-e479-4334-a9e3-78c80f3494f1. [Google Scholar]
- Steinbach WJ, Marr KA, Anaissie EJ, Azie N, Quan SP, Meier-Kriesche HU, Apewokin S, and Horn DL. 2012. ‘Clinical epidemiology of 960 patients with invasive aspergillosis from the PATH Alliance registry’, J Infect, 65: 453–64. [DOI] [PubMed] [Google Scholar]
- Stensvold, Christen Rune, Lise Nistrup Jørgensen, and Maiken Cavling Arendrup. 2012. ‘Azole-Resistant Invasive Aspergillosis: Relationship to Agriculture’, Current Fungal Infection Reports, 6: 178–91. [Google Scholar]
- Streifel AJ, Lauer JL, Vesley D, Juni B, and Rhame FS. 1983. ‘Aspergillus fumigatus and other thermotolerant fungi generated by hospital building demolition’, Appl Environ Microbiol, 46: 375–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro M, and Izumikawa K. 2016. ‘The Current Status of Drug-resistant Aspergillus’, Med Mycol J, 57: J103–12. [DOI] [PubMed] [Google Scholar]
- Tekaia F, and Latge JP. 2005. ‘Aspergillus fumigatus: saprophyte or pathogen?’, Curr Opin Microbiol, 8: 385–92. [DOI] [PubMed] [Google Scholar]
- Tobin MB, Peery RB, and Skatrud PL. 1997. ‘Genes encoding multiple drug resistance-like proteins in Aspergillus fumigatus and Aspergillus flavus’, Gene, 200: 11–23. [DOI] [PubMed] [Google Scholar]
- Valdes ID, van den Berg J, Haagsman A, Escobar N, Meis JF, Hagen F, Haas PJ, Houbraken J, Wosten HAB, and de Cock H. 2018. ‘Comparative genotyping and phenotyping of Aspergillus fumigatus isolates from humans, dogs and the environment’, BMC Microbiol, 18: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhaneni S, Benedict K, Derado G, and Mody RK. 2017. ‘Trends in Hospitalizations Related to Invasive Aspergillosis and Mucormycosis in the United States, 2000–2013’, Open Forum Infect Dis, 4: ofw268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden JW, Arendrup MC, Warris A, Lagrou K, Pelloux H, Hauser PM, Chryssanthou E, Mellado E, Kidd SE, Tortorano AM, Dannaoui E, Gaustad P, Baddley JW, Uekotter A, Lass-Florl C, Klimko N, Moore CB, Denning DW, Pasqualotto AC, Kibbler C, Arikan-Akdagli S, Andes D, Meletiadis J, Naumiuk L, Nucci M, Melchers WJ, and Verweij PE. 2015. ‘Prospective multicenter international surveillance of azole resistance in Aspergillus fumigatus’, Emerg Infect Dis, 21: 1041–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden JW, Camps SM, Kampinga GA, Arends JP, Debets-Ossenkopp YJ, Haas PJ, Rijnders BJ, Kuijper EJ, van Tiel FH, Varga J, Karawajczyk A, Zoll J, Melchers WJ, and Verweij PE. 2013. ‘Aspergillosis due to voriconazole highly resistant Aspergillus fumigatus and recovery of genetically related resistant isolates from domiciles’, Clin Infect Dis, 57: 513–20. [DOI] [PubMed] [Google Scholar]
- van der Linden JW, Snelders E, Kampinga GA, Rijnders BJ, Mattsson E, Debets-Ossenkopp YJ, Kuijper EJ, Van Tiel FH, Melchers WJ, and Verweij PE. 2011. ‘Clinical implications of azole resistance in Aspergillus fumigatus, The Netherlands, 2007–2009’, Emerg Infect Dis, 17: 1846–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden JW, Warris A, van der Meer JW, Bresters D, Melchers WJ, and Verweij PE. 2009. ‘[Azole-resistant invasive aspergillosis]’, Ned Tijdschr Geneeskd, 153: A765. [PubMed] [Google Scholar]
- van Paassen J, Russcher A, In ‘t Veld-van Wingerden AW, Verweij PE, and Kuijper EJ. 2016. ‘Emerging aspergillosis by azole-resistant Aspergillus fumigatus at an intensive care unit in the Netherlands, 2010 to 2013’, Euro Surveill, 21. [DOI] [PubMed] [Google Scholar]
- Verweij PE, Chowdhary A, Melchers WJ, and Meis JF. 2016. ‘Azole Resistance in Aspergillus fumigatus: Can We Retain the Clinical Use of Mold-Active Antifungal Azoles?’, Clin Infect Dis, 62: 362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij PE, Howard SJ, Melchers WJ, and Denning DW. 2009. ‘Azole-resistance in Aspergillus: proposed nomenclature and breakpoints’, Drug Resist Updat, 12: 141–7. [DOI] [PubMed] [Google Scholar]
- Verweij PE, Kema GH, Zwaan B, and Melchers WJ. 2013. ‘Triazole fungicides and the selection of resistance to medical triazoles in the opportunistic mould Aspergillus fumigatus’, Pest Manag Sci, 69: 165–70. [DOI] [PubMed] [Google Scholar]
- Verweij PE, Mellado E, and Melchers WJ. 2007. ‘Multiple-triazole-resistant aspergillosis’, N Engl J Med, 356: 1481–3. [DOI] [PubMed] [Google Scholar]
- Verweij PE, Snelders E, Kema GH, Mellado E, and Melchers WJ. 2009. ‘Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use?’, Lancet Infect Dis, 9: 789–95. [DOI] [PubMed] [Google Scholar]
- Verweij PE, Te Dorsthorst DT, Rijs AJ, De Vries-Hospers HG, and Meis JF. 2002. ‘Nationwide survey of in vitro activities of itraconazole and voriconazole against clinical Aspergillus fumigatus isolates cultured between 1945 and 1998’, J Clin Microbiol, 40: 2648–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij PE, Zhang J, Debets AJM, Meis JF, van de Veerdonk FL, Schoustra SE, Zwaan BJ, and Melchers WJG. 2016. ‘In-host adaptation and acquired triazole resistance in Aspergillus fumigatus: a dilemma for clinical management’, Lancet Infect Dis, 16: e251–e60. [DOI] [PubMed] [Google Scholar]
- Walker TA, Lockhart SR, Beekmann SE, Polgreen PM, Santibanez S, Mody RK, Beer KD, Chiller TM, and Jackson BR. 2018. ‘Recognition of Azole-Resistant Aspergillosis by Physicians Specializing in Infectious Diseases, United States’, Emerg Infect Dis, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh TJ, Anaissie EJ, Denning DW, Herbrecht R, Kontoyiannis DP, Marr KA, Morrison VA, Segal BH, Steinbach WJ, Stevens DA, van Burik JA, Wingard JR, Patterson TF, and America Infectious Diseases Society of. 2008. ‘Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America’, Clin Infect Dis, 46: 327–60. [DOI] [PubMed] [Google Scholar]
- Wang HC, Huang JC, Lin YH, Chen YH, Hsieh MI, Choi PC, Lo HJ, Liu WL, Hsu CS, Shih HI, Wu CJ, and Chen YC. 2018. ‘Prevalence, mechanisms and genetic relatedness of the human pathogenic fungus Aspergillus fumigatus exhibiting resistance to medical azoles in the environment of Taiwan’, Environ Microbiol, 20: 270–80. [DOI] [PubMed] [Google Scholar]
- Webb BJ, Ferraro JP, Rea S, Kaufusi S, Goodman BE, and Spalding J. 2018. ‘Epidemiology and Clinical Features of Invasive Fungal Infection in a US Health Care Network’, Open Forum Infect Dis, 5: ofy187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold NP, Gil VG, Gutierrez F, Lindner JR, Albataineh MT, McCarthy DI, Sanders C, Fan H, Fothergill AW, and Sutton DA. 2016. ‘First Detection of TR34 L98H and TR46 Y121F T289A Cyp51 Mutations in Aspergillus fumigatus Isolates in the United States’, J Clin Microbiol, 54: 168–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold NP, and Verweij PE. 2020. ‘Aspergillus fumigatus and pan-azole resistance: who should be concerned?’, Curr Opin Infect Dis, 33: 290–97. [DOI] [PubMed] [Google Scholar]
- Winters B, Custer J, Galvagno SM Jr., Colantuoni E, Kapoor SG, Lee H, Goode V, Robinson K, Nakhasi A, Pronovost P, and Newman-Toker D. 2012. ‘Diagnostic errors in the intensive care unit: a systematic review of autopsy studies’, BMJ Qual Saf, 21: 894–902. [DOI] [PubMed] [Google Scholar]
- Xiao L, Madison V, Chau AS, Loebenberg D, Palermo RE, and McNicholas PM. 2004. ‘Three-dimensional models of wild-type and mutated forms of cytochrome P450 14alpha-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding’, Antimicrob Agents Chemother, 48: 568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona AH, Frumkin I, and Pilpel Y. 2015. ‘A relay race on the evolutionary adaptation spectrum’, Cell, 163: 549–59. [DOI] [PubMed] [Google Scholar]
- Zhang J, van den Heuvel J, Debets AJM, Verweij PE, Melchers WJG, Zwaan BJ, and Schoustra SE. 2017. ‘Evolution of cross-resistance to medical triazoles in Aspergillus fumigatus through selection pressure of environmental fungicides’, Proc Biol Sci, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg MD, Nathanson BH, Harrington R, Spalding JR, and Shorr AF. 2018. ‘Epidemiology and Outcomes of Hospitalizations With Invasive Aspergillosis in the United States, 2009–2013’, Clin Infect Dis, 67: 727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]