

Summary
Cellular factories engage numerous highly complex “molecular machines” to perform pivotal biological functions. 3D structural visualization is an effective way to understand the functional mechanisms of these biomacromolecules. The “resolution revolution” has established cryogenic electron microscopy (cryo-EM) as a preferred structural biology tool. In parallel with the advances in cryo-EM methodologies aiming at atomic resolution, several innovative approaches have started emerging where other techniques are sensibly integrated with cryo-EM to obtain additional insights into the biological processes. For example, combining the time-resolved technique with high-resolution cryo-EM enables discerning structures of short-lived intermediates in the functional pathway of a biomolecule. Likewise, integrating mass spectrometry (MS) techniques with cryo-EM allows deciphering structural organizations of large molecular assemblies. Here, we discuss how the data generated upon combining either time resolve or MS techniques with cryo-EM supplement structural elucidations with in-depth understanding of the function of cellular macromolecules when they participate in fundamental biological processes.
Subject areas: Biochemistry Methods, Cell Biology, Structural Biology
Graphical abstract
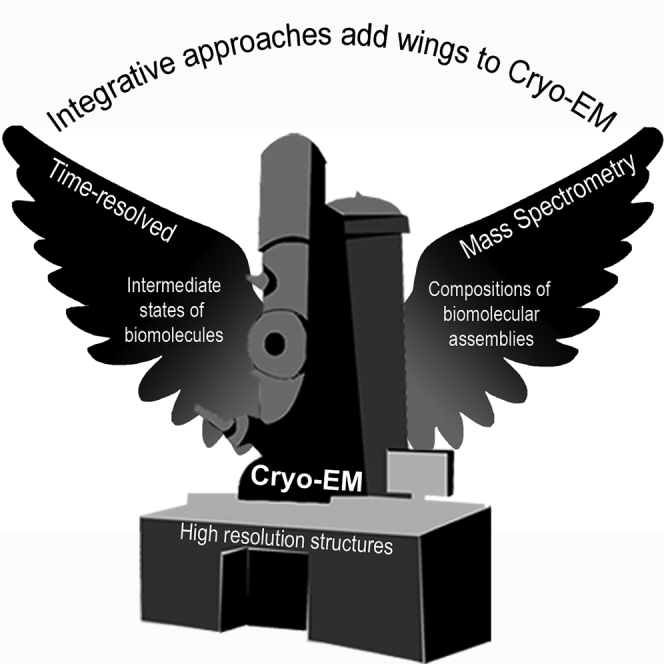
Highlights
-
•
The “resolution revolution” in cryo-EM field has greatly enhanced its application
-
•
Integrative approaches provide comprehensive understanding of biological processes
-
•
Time-resolved cryo-EM deciphers the intermediate structures in a functional pathway
-
•
Mass spectrometry aids in generating a detailed structural model of biomacromolecules
Biochemistry Methods; Cell Biology; Structural Biology
Introduction
Biological processes, often very complex in nature, are hard to be deciphered using any single technique. Structural visualizations of biological molecules provide significant insights into their function. Nonetheless, structural techniques with supplements from biophysical and biochemical experiments pave the way to depict a complex process with enhanced clarity (Schmidt and Urlaub, 2017). Pieces of information gathered from individual techniques when stitched together with structural data to model a functional biological system brings forth integrative structural biology.
Cryogenic electron microscopy (cryo-EM) in conjugation with 3D reconstruction techniques has emerged as a powerful method to study structures and dynamics of intrinsically flexible molecular assemblies. Previously, high-resolution structural data generated using NMR or X-ray crystallography were used to interpret limited resolution cryo-EM density maps at the molecular level. With time, cryo-EM has evolved and expanded into sub-domains, such as single particle analysis (SPA) (Van Heel et al., 2000; Orlova and Saibil, 2011; Vinothkumar and Henderson, 2016; Wu and Lander, 2020), cryogenic electron tomography (Oikonomou and Jensen, 2017; Beck and Baumeister, 2016), two-dimensional electron crystallography (Henderson and Unwin, 1975; Stahlberg et al., 2015), and, more recently, microcrystal electron diffraction (Nannenga and Gonen, 2019). Single-particle cryo-EM technique has advanced to a stage that enables structure determination of macromolecular complexes at nearly atomic resolution (Baker et al., 2010; Kuhlbrandt, 2014; Doerr, 2014; Cheng, 2018; Murata and Wolf, 2018). Technological advances have occurred in different fronts of the method, starting from sample preparation to image acquisition and image analysis (Akbar et al., 2020; Naydenova et al., 2019). High-resolution cryo-EM structures are manifestation of cumulative results of all these achievements.
In recent years, integration of different techniques is gradually taking up multi-dimensional shape in developing new methodological approaches to deal with complex biological systems. Cryo-EM, having received a great attention nowadays for determining 3D structures of intrinsically dynamic macromolecular assemblies (Lawson et al., 2020; Chen and He, 2020), mostly populates new integrative approaches (Figure 1). One such integrative structural biology technique is the correlative light electron microscopy method (CLEM), where the tomography approach of cryo-EM is combined with light microscopy (Koning et al., 2018). The significance of CLEM lies in the localization of a fluorescently tagged complex with the help of light microscopy followed by visualization of the same at higher resolution using cryo-EM (Schwartz et al., 2007; Sartori et al., 2007; Oikonomou and Jensen, 2017). Application of this integrative technique has captured crucial biological events in bacteria, as well as mammalian systems (Basler et al., 2012; Oikonomou and Jensen, 2017; Tuijtel et al., 2019) More recently, this approach has been further developed into cryo-CLEM where the fluorescence-based target detection is also carried out under cryo-conditions (Hampton et al., 2017). Another interesting integrative technique is an in situ approach which has refined our understanding of the complex cellular events (Von Appen et al., 2015; Guo et al., 2018). Bypassing the requirement of purification of macromolecules, cryo-electron tomography and sub-tomogram averaging in combination with focused ion beam (FIB) milling of the whole system has yielded significant structural insights in the cellular context (Schur, 2019).
Figure 1.

Synergistic approaches in structural biology to enrich information on functional mechanisms
In integrative structural biology approach, data generated from multiple techniques are combined to gain insights into a biological process. Structural biology tools like cryo-EM, X-ray crystallography, NMR, etc. elucidate 3D architecture of a macromolecular assembly, whereas biophysical techniques, like different wings of mass spectrometry and proteomics, illustrate the composition of and association within it. Integration of these diverse approaches leads to out-and-out understanding of a crucial biological pathway. For example, structure and function of the molecular machine ribosome has been studied with various techniques, and when all the results from these techniques are put together, the overall mechanism of protein synthesis (translation, shown in the center) has come to light.
To understand the molecular mechanism of a biological process, the short-lived, intermediate states of a biological pathway need to be structurally deciphered. A standalone technique has been developed recently, where a microfluidic device is used to prepare cryo-EM grids at specific time points of an on-going reaction in an automated and fast vitrification process (Maeots et al., 2020; Kontziampasis et al., 2019; Feng et al., 2017). This newly devised technique, popularly known as time-resolved cryo-EM (trCryo-EM), has started producing many significant and illustrative results of many complex biological processes (Dandey et al., 2020). Combination of biophysical methods like mass spectrometry (MS) with cryo-EM has also evolved as a promising integrative methodology (Schmidt and Urlaub, 2017).
A device integrating time resolved with cryo-EM has been designed (trCryo-EM) where a biochemical reaction is carried out directly on a microfluidic chip by controlled mixing of reactants. The reaction product is sprayed at different time points onto an EM grid which is rapidly plunge frozen to capture the various intermediates of the pathway (Dandey et al., 2020; Maeots et al., 2020). Several MS methodologies such as native MS, cross-linking MS (XL-MS), affinity purification MS (AP-MS), denaturing MS, and hydrogen-deuterium exchange MS (HDX-MS) have been developed which can be utilized to substantiate cryo-EM structural data. Several reviews have already well documented the details of technical aspects of integrative approaches in cryo-EM (Rout and Sali, 2019; Cerofolini et al., 2019; Srivastava et al., 2020). Here, we discuss additional knowledge gained by combining time-resolved and MS approaches with cryo-EM instead of focusing on technical details of the methods. We primarily focus on how trCryo-EM has aided in visualizing the sequential changes (within millisecond timescale) of various macromolecular assemblies in complex biological processes. Along with this, we also survey how MS results on molecular interactions or associations supplement cryo-EM structural data to study large bio-molecular assemblies.
Combining time-resolved approach with single-particle cryo-EM to capture intermediate functional states
Biomolecules, while participating in different fundamental cellular processes, rapidly interconvert among multiple processing states. The functionally relevant states have lower activation energy aiding the process under cellular thermal condition. In certain cases, protein factors, functioning as enzymes, reorganize different local minima in the energy landscape by assisting in lowering the energy barriers. The involvement of these factors modulates the timescale of the processes. Many structures of the factor-bound stable bio-molecular complexes have been solved till date. However, the crucial and interesting intermediates that exist in the microsecond to millisecond range are quite impossible to be captured by standard cryo-EM approaches due to the relatively slow, partially manual method of grid preparation (Frank, 2017; Stark and Chari, 2016; Passmore and Russo, 2016). The absence of structural information of the intermediates poses dilemma in tracing functional pathways of some biomolecules since multiple possibilities may exist. The trCryo-EM approach has the potential to resolve this issue. Traditionally, high-resolution cryo-EM structures depict intricate structural details of snapshot of a stable state of biological molecules. However, the complex molecular assemblies rapidly interconvert among multiple processing states in their functional pathway, and to gain full insight into a biological process, deciphering the structures of intermediate states is crucial.
The fleeting structural states of macromolecules generated during the functional pathway can be captured using trCryo-EM. A special microfluidic chip has been devised in which two reactants are mixed and at different time points samples from the reaction mixtures are sprayed on the grid which is then quickly plunged into liquid ethane (Figure 2). This automated mixing/spraying technique has the capability to capture reaction intermediates in the time range of 10 to 1000ms (Feng et al., 2017; Lu et al., 2009; Shaikh et al., 2014; Chen and Frank, 2016; Fu et al., 2016).
Figure 2.

Time-resolved cryo-EM allows visualizations of intermediate states of molecules in action
A spectacular time-resolved cryo-EM (trCryo-EM) study (Loveland et al., 2020) elucidating 33 states involved in tRNA proofreading process of the elongation step of protein synthesis is presented here. A schematic representation of the trCryo-EM technique is shown in the middle where different components (represented in red and yellow colored test tubes) are allowed to interact (mixing of components is shown in red and yellow color through a zigzag channel) and subsequently sprayed on the grids at different time points. Grids are plunged frozen and imaged by high-resolution cryo-TEM. The crests and troughs of the zigzag line are the time points where at each point the sample can be deposited onto the grid for image acquisition. Five representative structures of ribosomes (shown in different shades of blue) in the complex with EF-Tu are shown as for example (emd-21621, emd-21623, emd-21624, emd-21626, and emd-21629) revealing EF-Tu (shown in red) conformational changes during the process of tRNA delivery and subsequent release of EF-Tu upon GTP hydrolysis. The sequential intermediate structures trace the conformational changes from ribosome binding of EF-Tu in GTP-bound form (state I; GTP shown in green) to a state where EF-Tu is on the verge of leaving the ribosome upon GTP hydrolysis (state V).
The concept and need of trCryo-EM hatched long before designing of specific instrument. During early 2000, the maturation of the pro-capsid of herpes simplex virus into a capsid was investigated using cryo-EM. The complete process takes about 96 hr. The pro-capsids were isolated from a temperature-sensitive mutant, and samples were prepared at four different time points, i.e. 0, 24, 48, and 96 hr. The resulting density maps, in chronological order, portrayed an assembly sequence of capsid maturation which included significant switching events with relative rotation of the major capsid protein (Vp5) (Heymann et al., 2003).
Another example of trCryo-EM was to explore the process of DNA replication. Loading of two MCM (minichromosome maintenance protein complex) hexamer by the origin recognition complex (ORC) to form a head-to-head double hexamer (DH) around DNA is a prerequisite for bidirectional replication (Remus et al., 2009; Evrin et al., 2009). The mechanism of DH formation was unclear as single-molecule experiments and biochemical data pointed to contrasting hypothesis (Frigola et al., 2013; Coster and Diffley, 2017). Visualizing the intermediate states by preparing samples in a time course of 2, 6, and 20 min depicted that both the MCM hexamers are recruited by the same interaction between MCM and ORC C-terminal domains. The mechanism of coupled MCM loading was identified and thus put a possible end to the long-standing debate (Miller et al., 2019).
Translation, the protein biosynthesis process in any living cell, is the most exploited field in which trCryo-EM has grown and matured to its present status. Protein biosynthesis is accomplished via initiation, elongation, termination, and ribosome recycling steps on the ribosome, the key macromolecular machine for translation. Several translation factors control each of these steps in bacterial translation, namely, initiation (IF1, IF2, and IF3), elongation (EF-Tu, EF-G), release (RF1/RF2, RF3), and ribosome recycling (RRF along with EF-G and IF3) factors (Wilson and Nierhaus, 2003; Carter et al., 2001; Laursen et al., 2005). During the process, ribosome passes through several transient intermediate states which could not be structurally deciphered by a standard cryo-EM approach. The trCryo-EM technique enables visualization of these short-lived, physiologically important intermediates and thus allows tracing the sequence of events during the process.
In a trCryo-EM study of the initiation complexes (Kaledhonkar et al., 2019), five different short-lasting intermediates were captured at 20ms, 80ms, 200ms, and 600ms timescales. The intermediate structure captured within the first 20ms–80ms revealed that the formation of inter-subunit bridge B2a (forms between helix 69 of the 23S rRNA and helix 44 of the 16S rRNA) coincides with the time of the GTPase IF1 dissociation. Within 80ms and 200ms, IF2 bound to 70S initiation complex (IC) gets dissociated from a large fraction of population leading to the maturation of 70S IC to 70S elongation complex. This maturation event lasts till 600ms and beyond as it is largely dependent on the release of Pi from IF2 (even after IF2 hydrolyzes GTP) and the subsequent rapid liberation of GDP-IF2 from 70S IC. The structures obtained from this trCryo-EM study revealed the ratcheting movement of the ribosomal subunits induced by the IF2 dissociation in combination with a series of conformational alterations including (i) rearrangement of fMet-tRNAfMet from its P/I to P/P configuration, (ii) disentangling of the 3′ CCA-fMet tail; and (iii) placement of the fMet moiety into the peptidyl transferase center (Kaledhonkar et al., 2019). This visualization of the previously unperceived intermediates laid the foundation for better understanding of the initiation step.
Ribosome release factor (RRF) and elongation factor G (EF-G) in presence of GTP govern the ribosome recycling event (Fu et al., 2016). The sample imaged at 140ms revealed two classes, namely, PostTC.RRF140 (posttermination complex in presence of RRF) and PostTC.RRF.EF-G140 (posttermination complex in presence of RRF and EF-G). Juxtaposing these two structures highlighted the gyration of RRF domain II toward domain I occupying a place adjacent to the inter-subunit bridge B2a. The deacylated tRNA coheres to the 30S subunit even after termination of translation and later breaks off due to the competitive attachment of IF3. The transient states captured by trCryo-EM thus manifested the change in structural orientations and the sequence of events occurring during recycling (Fu et al., 2016).
Translocation of tRNAs is a part of the elongation step where a GTPase EF-G assists the shifting of the reading frame by one codon, making the A site free for the next incoming aminoacyl tRNA. The application of trCryo-EM has provided extensive information regarding the trajectory and dynamics of tRNA movement through the ribosome. The intermediate structures generated using trCryo-EM depicted the sequential break off and genesis of ribosome-tRNA contacts during the translocation process and also highlighted a spectrum of structural changes in the head and body region of the small subunit that are coupled to tRNA movement (Fischer et al., 2010).
High-resolution structures of the elongation factor Tu (EF-Tu)-bound 70S ribosome are available where the complexes were stalled either using non-hydrolyzable GTP analog or an antibiotic kirromycin (Voorhees et al., 2010; Fischer et al., 2015). Tentative functional pathway of the tRNA proofreading during protein biosynthesis has been proposed based on these structures. However, intricate details of EF-Tu conformational changes associated with this elongation step could be defined by deciphering high-resolution structures of 33 intermediate states using trCryo-EM in a recent study (Loveland et al., 2020). The trCryo-EM technique also has solved some previous disputes on bacterial translation. For instance, compact form of release factor crystal structure as compared to the “open” structures of its ribosome-bound state raised serious questions long back (Kjeldgaard, 2003). A recent trCryo-EM study resolved this long-standing issue by capturing a transient state of ribosome-bound release factor in the compact form (Fu et al., 2019). Evidently, resolving structures of the short-lasting intermediates using the time-resolved technique in combination with cryo-EM improves and fine-tunes our understanding of the mechanisms of fundamental cellular processes (Figure 2). Besides, the trCryo-EM approach holds the potential to disclose novel drug targets since larger the number of intermediate functional states captured, the greater would be the chance of getting a pool of drug targets.
Integrating mass spectrometry data with cryo-EM to elucidate organizations of multi-component biomolecules
Different sets of techniques can be intermingled to address crucial questions on specific biological processes. Mass spectrometry (MS) has been a widely used biophysical technique to understand various biological pathways. Different types of MS play their role in providing particular set of information (Rajabi et al., 2015; Lossl et al., 2016). In native MS, the mild experimental condition preserves the native state of the biomolecule (non-covalent interaction and natural folded state), thus giving us the glimpse of the whole complex (Mitra, 2019; Heck, 2008). Denaturing MS uses non-neutral pH and organic solvents, thus exposing the protonation sites (Schachner et al., 2016). The macromolecule is cleaved into smaller units giving rise to a normal spectral distribution. XL-MS uncovers the binding partners along with the residues involved in association (Liu et al., 2015). Using AP-MS, the co-purified interacting proteins can be analyzed (Bauer and Kuster, 2003). Solvent accessibility information obtained using HDX-MS helps to interpret protein structure and confirmation (Hamuro et al., 2003; Oganesyan et al., 2018). Handshaking of structural biology with MS techniques yields an application that has the potential to lead the way to describe the structural organization of different multimeric complexes (Figure 3).
Figure 3.

Integration of mass spectrometry data enhances understanding of biological pathways
A hybrid approach combining information from different types of mass spectrometry (MS) with high-resolution cryo-EM study has the potential to illuminate a biological process in more details. Results obtained from different types of MS experiments (posttranslational modifications of proteins [from native MS], composition of the sample [from denaturing MS], binding partners and residues involved [from XL-MS], characterization of the interacting partners [from AP-MS], and protein structure and confirmation [from HDX-MS]) are shown as droplets which together form a pool of information. Cutting-edge cryo-EM imaging and data processing techniques generate high-resolution 3D structures (shown in middle). Combining the information gathered from mass spectrometry with the cryo-EM structural analysis, for example, reveals the sequential steps of the ribosomal 50S subunit assembly (Davis et al., 2016) (depicted schematically at the bottom).
Nucleosome, the basic unit of chromatin, consists of roughly 146bp of DNA muffled around histone octamer (Luger et al., 1997). The dynamicity of chromatin plays an important role in various processes like replication, transcription, repair mechanisms (Venkatesh and Workman, 2015). Assembly of nucleosome into duplicated DNA is a prerequisite for replication. To fulfill this crucial step, a large number of histone proteins are synthesized and transported to the nucleus. Highly positively charged histones also contain strong hydrophobic patches making them prone to non-specific binding. The regulated transport of H3/H4 (histone 3/histone 4) dimer is assisted by importin4, the major nuclear importin that guides the transport of newly synthesized H3/H4 associated with Asf1a which is an essential histone chaperone. By using an integrative approach relying on XL-MS, X-ray crystallography, and negative stain electron microscopy , the structural and molecular interaction of the Importin4_H3/H4_Asf1a complex was unfolded highlighting the mechanism by which importin identifies H3/H4_Asf1a (Yoon et al., 2018).
XL-MS of Importin4_H3/H4_Asf1a complex pinpointed that the DNA binding surface of the histone interacted with importin (Yoon et al., 2018). It also highlighted that the C-terminal tail of importin4 recognizes the Nuclear Localization Signal (NLS) present on the N-terminal tail of H3 and not on H4. Limited proteolysis assay backed up this finding because the N-terminal of H3 was protected upon binding to importin4. The crystal structure of the C-terminal tail of importin4 bound to H3 N-terminal peptide uncovered the mechanism of identification and binding of importin4 and H3. Lysine 14 of H3 played a crucial role in attachment as its replacement to alanine reduced the binding affinity multiple folds. Structural models generated using data obtained from negative stain single-particle EM, X-ray crystallography, XL-MS, and small-angle X-ray scattering (SAXS) pictured the architecture of Importin4_H3/H4_Asf1a complex.
The nuclear transport of histone H1 requires the association (by liner histone H1.0) with two importins, i.e., importin 7 (Imp7) and importin β (Impβ). Cryo-EM structure of Imp7-Impβ-H1.0 trimeric complex revealed the cradle formed by the two importins to hold the linker histone. The globular domain of H1 was held together by Impβ, but the positively charged C-terminal tail was bound to and protected by both Imp7 and Impβ. XL-MS study showed that the H1 tail remains disordered and comes in close proximity with multiple sites of both importins. The C-terminal tail serves an important role in stabilizing the overall complex. C-terminal tail of Imp7 contains GGxxF and FxFG motifs which were essential for the complex formation and transport of histone. FxFG motifs of nucleoporins present on the nuclear side, in association with RanGTP, assist the disassembly of the trimeric complex to complete the transport of histone (Ivic et al., 2019). Integration of cryo-EM and XL-MS played a decisive role in this study. The sole utilization of cryo-EM would only provide the structure of the importin-histone complex where the dynamic regions are poorly resolved. The incorporation of XL-MS furnished the information on the interactions of lysine residues of H1.0 with acidic residues of Impβ and Imp7. The results also highlighted that a lysine of H1.0 cross-links various acidic residues of importin and similarly an acidic residue of importin cross-links different lysine residues of H1.0 indicating the absence of specific residue for H1.0 tail interaction. Thus, to supplement the poorly defined regions in cryo-EM structures, cross-linking coupled with MS plays a crucial role in providing the information on the amino acid proximity and interaction.
The collaboration of quantitative MS (qMS) and cryo-EM offered comprehensive understanding of ribosomal large subunit assembly by revealing parallel pathways in ribosome biogenesis process, one of the most crucial events in living cells, to ensure uninterrupted assembly of functional ribosome (Figure 3). A modified system lacking the gene rplQ encoding the ribosomal protein bL17 resulted in the decline of mature 70S particle and subsequent increase of the 30S subunit. Radiolabeled pulse experiment demonstrated that the LSUbL17dep (large subunit bL17 depleted) was competent for maturation upon addition of bL17. Even during bL17 starvation, the LSUbL17dep matured but at a very slow rate. Comparing the ribosomal protein profile of the LSU from cells grown under permissive and bL17-deprived conditions using qMS highlighted the variation in the protein profile. LSUbL17dep showed the presence of enriched assembly factors probably to help maturation. Chemical probing showed that the native rRNA secondary structure was conserved in the LSUbL17dep particles. Cryo-EM density maps of the obtained classes of LSUbL17dep pointed to the variation of the inter-subunit interface indicating the wide effect of bL17 depletion. Combining the information from the density maps with the data obtained from qMS and chemical probing using SHAPE-MaP (selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling) (Martin et al., 2019), the presence of parallel assembly pathways was concluded which allowed LSU maturation even in limitation of an initial protein (Davis et al., 2016).
These results imply that thoughtful integration of MS and cryo-EM can be an effective approach to unravel complex architectures and functional pathways of biological macromolecular assemblies.
Future directions and challenges
The studies discussed above evidence that integrative approaches in the cryo-EM field substantially enhance our understanding of complex biological systems. Both trCryo-EM and integrative MS approaches have been proved to be potentially effective in this respect.
Nowadays, trCryo-EM can elaborate biological processes in a timescale of milliseconds. However, many biological processes occur even in narrower timescales for which yet an advanced device to capture the ephemeral states of biological pathways is required. More efficient microfluidic devices to capture transitional states of biological macromolecules beyond millisecond timescales would be the new horizon (Maeots et al., 2020). A relatively old study in this direction showing tiny movements in nanosecond timescales (Fitzpatrick et al., 2013) may be explored further. Moreover, many biological reactions occur with more than two ingredients. The limitations of present-day microfluidic devices in trCryo-EM may be overcome by incorporating more than two channels for multi-dimensional reactions to be studied.
While trCryo-EM takes care of the homogeneous population of the sample, obtaining sufficiently thin layers of vitreous ice without affecting sample property is another challenge in focus for recent developments in optimal sample preparation techniques like spraying the sample onto the grid without any blotting step (Klebl et al., 2020). Getting random orientations of sample molecules on the grid is another hurdle for SPA where use of specialized grids depending on the sample properties may help. The property of the sample can be manipulated by biochemistry bench work and the property of the cryo-EM grids by technological advancement (Drulyte et al., 2018); there still remain future possibilities to biophysically tweak the orientation of the molecules, for example, by adjusting dipole moment of the molecular complex. Moving toward this direction, Tomasz Uchański and group used specially designed megabodies to circumvent the obstacle of orientation biasness. They resolved the structure of a relatively small membrane protein (GABAA-β3) having acute preferential orientation at a resolution of 2.5Å (Uchański et al., 2021).
Although usage of XL-MS to supplement and enhance the near atomic structures from cryo-EM is a wise synergistic approach, it can be even more fruitful if the resolution of cryo-EM structures reaches the atomic level. Taking a giant step forward to attain atomic resolution, cryo-EM has successfully deduced the structure of apoferritin with 1.25 Å (Yip et al., 2020) and 1.2 Å resolutions (Nakane et al., 2020). Nevertheless, it is needless to say, only the advancement in terms of obtaining higher resolution structures would not reveal the complete picture of the on-going processes inside cells.
An optimal strategy to visualize the biological molecules in liquid solution is probably the next step ahead. A recent enthusiastic approach to visualize molecules in liquid state holds promising opportunity to study complex biological processes in the most near-native condition. Development of specialized grids to deal with the liquid sample inside the microscope is a step ahead in this direction (Pu et al., 2020). Hopefully, combination of the time-resolved technique with liquid phase transmission electron microscopy (TEM) can explore yet unanswered questions in future. However, the problems associated with traditional TEM (e.g. radiation damage, electron-beam-induced motions of the molecules, etc.) are still pertinent issues associated with this approach.
Although till date various biological processes have been illuminated with these handshaking approaches, many pathways are still studded with mystery due to technological bottleneck of various methods. For example, an in situ approach using cryo-tomography and FIB milling in combination is mostly limited to thin samples, like the bacterial cells (application to mammalian cells is restricted). Improvement of the FIB milling method is required to access thicker cells and tissue samples. To be optimistic, development of innovative tools by synergistic utilization of existing technologies will also overcome the present-day difficulty to visualize the complexity in vivo.
Acknowledgments
We acknowledge financial supports from the Science and Engineering Research Board (SERB), Department of Science & Technology (India) sponsored projects (SB/SO/BB-0025/2014 and CRG/2019/001788), and CSIR-Indian Institute of Chemical Biology, Kolkata, India. A.B. and S.B. thank Department of Biotechnology (DBT) and Council of Scientific and Industrial Research (CSIR), India, respectively, for research fellowships. We thank our lab members Shirin Akbar, Krishnamoorthi Srinivasan, and Priya Baid for useful comments on the manuscript.
Author contributions
All authors searched the literature, contributed to data interpretation, preparation of the manuscript, and approved it for publication.
References
- Akbar S., Mozumder S., Sengupta J. Retrospect and prospect of single particle cryo-electron microscopy: the class of integral membrane proteins as an example. J. Chem. Inf. Model. 2020;60:2448–2457. doi: 10.1021/acs.jcim.9b01015. [DOI] [PubMed] [Google Scholar]
- Baker M.L., Zhang J., Ludtke S.J., Chiu W. Cryo-EM of macromolecular assemblies at near-atomic resolution. Nat. Protoc. 2010;5:1697–1708. doi: 10.1038/nprot.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M., Pilhofer M., Henderson G.P., Jensen G.J., Mekalanos J.J. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature. 2012;483:182–186. doi: 10.1038/nature10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A., Kuster B. Affinity purification-mass spectrometry. Powerful tools for the characterization of protein complexes. Eur. J. Biochem. 2003;270:570–578. doi: 10.1046/j.1432-1033.2003.03428.x. [DOI] [PubMed] [Google Scholar]
- Beck M., Baumeister W. Cryo-electron tomography: can it reveal the molecular sociology of cells in atomic detail? Trends Cell Biol. 2016;26:825–837. doi: 10.1016/j.tcb.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Carter A.P., Clemons W.M., Jr., Brodersen D.E., Morgan-Warren R.J., Hartsch T., Wimberly B.T., Ramakrishnan V. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science. 2001;291:498–501. doi: 10.1126/science.1057766. [DOI] [PubMed] [Google Scholar]
- Cerofolini L., Fragai M., Ravera E., Diebolder C.A., Renault L., Calderone V. Integrative approaches in structural biology: a more complete picture from the combination of individual techniques. Biomolecules. 2019;9:370. doi: 10.3390/biom9080370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Frank J. Two promising future developments of cryo-EM: capturing short-lived states and mapping a continuum of states of a macromolecule. Microscopy (Oxf) 2016;65:69–79. doi: 10.1093/jmicro/dfv344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., He J. Outlier profiles of atomic structures derived from X-ray crystallography and from cryo-electron microscopy. Molecules. 2020;25:1540. doi: 10.3390/molecules25071540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. Single-particle cryo-EM-How did it get here and where will it go. Science. 2018;361:876–880. doi: 10.1126/science.aat4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coster G., Diffley J.F.X. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science. 2017;357:314–318. doi: 10.1126/science.aan0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandey V.P., Budell W.C., Wei H., Bobe D., Maruthi K., Kopylov M., Eng E.T., Kahn P.A., Hinshaw J.E., Kundu N. Time-resolved cryo-EM using Spotiton. Nat. Methods. 2020;17:897–900. doi: 10.1038/s41592-020-0925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.H., Tan Y.Z., Carragher B., Potter C.S., Lyumkis D., Williamson J.R. Modular assembly of the bacterial large ribosomal subunit. Cell. 2016;167:1610–1622 e15. doi: 10.1016/j.cell.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr A. Single-particle electron cryomicroscopy. Nat. Methods. 2014;11:30. doi: 10.1038/nmeth.2779. [DOI] [PubMed] [Google Scholar]
- Drulyte I., Johnson R.M., Hesketh E.L., Hurdiss D.L., Scarff C.A., Porav S.A., Ranson N.A., Muench S.P., Thompson R.F. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallogr. D Struct. Biol. 2018;74:560–571. doi: 10.1107/S2059798318006496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C., Clarke P., Zech J., Lurz R., Sun J., Uhle S., Li H., Stillman B., Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. U S A. 2009;106:20240–20245. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X., Fu Z., Kaledhonkar S., Jia Y., Shah B., Jin A., Liu Z., Sun M., Chen B., Grassucci R.A. A fast and effective microfluidic spraying-plunging method for high-resolution single-particle cryo-EM. Structure. 2017;25:663–670 e3. doi: 10.1016/j.str.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N., Konevega A.L., Wintermeyer W., Rodnina M.V., Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- Fischer N., Neumann P., Konevega A.L., Bock L.V., Ficner R., Rodnina M.V., Stark H. Structure of the E. coli ribosome-EF-Tu complex at <3 A resolution by Cs-corrected cryo-EM. Nature. 2015;520:567–570. doi: 10.1038/nature14275. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A.W., Lorenz U.J., Vanacore G.M., Zewail A.H. 4D cryo-electron microscopy of proteins. J. Am. Chem. Soc. 2013;135:19123–19126. doi: 10.1021/ja4115055. [DOI] [PubMed] [Google Scholar]
- Frank J. Time-resolved cryo-electron microscopy: recent progress. J. Struct. Biol. 2017;200:303–306. doi: 10.1016/j.jsb.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J., Remus D., Mehanna A., Diffley J.F. ATPase-dependent quality control of DNA replication origin licensing. Nature. 2013;495:339–343. doi: 10.1038/nature11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Indrisiunaite G., Kaledhonkar S., Shah B., Sun M., Chen B., Grassucci R.A., Ehrenberg M., Frank J. The structural basis for release-factor activation during translation termination revealed by time-resolved cryogenic electron microscopy. Nat. Commun. 2019;10:2579. doi: 10.1038/s41467-019-10608-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Kaledhonkar S., Borg A., Sun M., Chen B., Grassucci R.A., Ehrenberg M., Frank J. Key intermediates in ribosome recycling visualized by time-resolved cryoelectron microscopy. Structure. 2016;24:2092–2101. doi: 10.1016/j.str.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q., Lehmer C., Martinez-Sanchez A., Rudack T., Beck F., Hartmann H., Perez-Berlanga M., Frottin F., Hipp M.S., Hartl F.U. In situ structure of neuronal C9orf72 poly-GA aggregates reveals proteasome recruitment. Cell. 2018;172:696–705 e12. doi: 10.1016/j.cell.2017.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton C.M., Strauss J.D., Ke Z., Dillard R.S., Hammonds J.E., Alonas E., Desai T.M., Marin M., Storms R.E., Leon F. Correlated fluorescence microscopy and cryo-electron tomography of virus-infected or transfected mammalian cells. Nat. Protoc. 2017;12:150–167. doi: 10.1038/nprot.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamuro Y., Coales S.J., Southern M.R., Nemeth-Cawley J.F., Stranz D.D., Griffin P.R. Rapid analysis of protein structure and dynamics by hydrogen/deuterium exchange mass spectrometry. J. Biomol. Tech. 2003;14:171–182. [PMC free article] [PubMed] [Google Scholar]
- Heck A.J. Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Henderson R., Unwin P.N. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- Heymann J.B., Cheng N., Newcomb W.W., Trus B.L., Brown J.C., Steven A.C. Dynamics of herpes simplex virus capsid maturation visualized by time-lapse cryo-electron microscopy. Nat. Struct. Biol. 2003;10:334–341. doi: 10.1038/nsb922. [DOI] [PubMed] [Google Scholar]
- Ivic N., Potocnjak M., Solis-Mezarino V., Herzog F., Bilokapic S., Halic M. Fuzzy interactions form and shape the histone transport complex. Mol. Cell. 2019;73:1191–1203 e6. doi: 10.1016/j.molcel.2019.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaledhonkar S., Fu Z., Caban K., Li W., Chen B., Sun M., Gonzalez R.L., Jr., Frank J. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature. 2019;570:400–404. doi: 10.1038/s41586-019-1249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldgaard M. The unfolding story of polypeptide release factors. Mol. Cell. 2003;11:8–10. doi: 10.1016/s1097-2765(03)00015-7. [DOI] [PubMed] [Google Scholar]
- Klebl D.P., Monteiro D.C.F., Kontziampasis D., Kopf F., Sobott F., White H.D., Trebbin M., Muench S.P. Sample deposition onto cryo-EM grids: from sprays to jets and back. Acta Crystallogr. D Struct. Biol. 2020;76:340–349. doi: 10.1107/S2059798320002958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning R.I., Koster A.J., Sharp T.H. Advances in cryo-electron tomography for biology and medicine. Ann. Anat. 2018;217:82–96. doi: 10.1016/j.aanat.2018.02.004. [DOI] [PubMed] [Google Scholar]
- Kontziampasis D., Klebl D.P., Iadanza M.G., Scarff C.A., Kopf F., Sobott F., Monteiro D.C.F., Trebbin M., Muench S.P., White H.D. A cryo-EM grid preparation device for time-resolved structural studies. IUCrJ. 2019;6:1024–1031. doi: 10.1107/S2052252519011345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrandt W. Biochemistry. The resolution revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Laursen B.S., Sorensen H.P., Mortensen K.K., Sperling-Petersen H.U. Initiation of protein synthesis in bacteria. Microbiol. Mol. Biol. Rev. 2005;69:101–123. doi: 10.1128/MMBR.69.1.101-123.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C.L., Berman H.M., Chiu W. Evolving data standards for cryo-EM structures. Struct. Dyn. 2020;7:014701. doi: 10.1063/1.5138589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Rijkers D.T., Post H., Heck A.J. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Lossl P., Van De Waterbeemd M., Heck A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016;35:2634–2657. doi: 10.15252/embj.201694818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland A.B., Demo G., Korostelev A.A. Cryo-EM of elongating ribosome with EF-Tu•GTP elucidates tRNA proofreading. Nature. 2020;584:640–645. doi: 10.1038/s41586-020-2447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Shaikh T.R., Barnard D., Meng X., Mohamed H., Yassin A., Mannella C.A., Agrawal R.K., Lu T.M., Wagenknecht T. Monolithic microfluidic mixing-spraying devices for time-resolved cryo-electron microscopy. J. Struct. Biol. 2009;168:388–395. doi: 10.1016/j.jsb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Maeots M.E., Lee B., Nans A., Jeong S.G., Esfahani M.M.N., Ding S., Smith D.J., Lee C.S., Lee S.S., Peter M., Enchev R.I. Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat. Commun. 2020;11:3465. doi: 10.1038/s41467-020-17230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Blankenship C., Rausch J.W., Sztuba-Solinska J. Using SHAPE-MaP to probe small molecule-RNA interactions. Methods. 2019;167:105–116. doi: 10.1016/j.ymeth.2019.04.009. [DOI] [PubMed] [Google Scholar]
- Miller T.C.R., Locke J., Greiwe J.F., Diffley J.F.X., Costa A. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature. 2019;575:704–710. doi: 10.1038/s41586-019-1768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra G. Application of native mass spectrometry in studying intrinsically disordered proteins: a special focus on neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2019;1867:140260. doi: 10.1016/j.bbapap.2019.07.013. [DOI] [PubMed] [Google Scholar]
- Murata K., Wolf M. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim. Biophys. Acta Gen. Subj. 2018;1862:324–334. doi: 10.1016/j.bbagen.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Nakane T., Kotecha A., Sente A., Mcmullan G., Masiulis S., Brown P.M.G.E., Grigoras I.T., Malinauskaite L., Malinauskas T., Miehling J. Single-particle cryo-EM at atomic resolution. Nature. 2020;587:152–156. doi: 10.1038/s41586-020-2829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nannenga B.L., Gonen T. The cryo-EM method microcrystal electron diffraction (MicroED) Nat. Methods. 2019;16:369–379. doi: 10.1038/s41592-019-0395-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenova K., Peet M.J., Russo C.J. Multifunctional graphene supports for electron cryomicroscopy. Proc. Natl. Acad. Sci. U S A. 2019;116:11718–11724. doi: 10.1073/pnas.1904766116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan I., Lento C., Wilson D.J. Contemporary hydrogen deuterium exchange mass spectrometry. Methods. 2018;144:27–42. doi: 10.1016/j.ymeth.2018.04.023. [DOI] [PubMed] [Google Scholar]
- Oikonomou C.M., Jensen G.J. Cellular electron cryotomography: toward structural biology in situ. Annu. Rev. Biochem. 2017;86:873–896. doi: 10.1146/annurev-biochem-061516-044741. [DOI] [PubMed] [Google Scholar]
- Orlova E.V., Saibil H.R. Structural analysis of macromolecular assemblies by electron microscopy. Chem. Rev. 2011;111:7710–7748. doi: 10.1021/cr100353t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore L.A., Russo C.J. Specimen preparation for high-resolution cryo-EM. Methods Enzymol. 2016;579:51–86. doi: 10.1016/bs.mie.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu S., Gong C., Robertson A.W. Liquid cell transmission electron microscopy and its applications. R. Soc. Open Sci. 2020;7:191204. doi: 10.1098/rsos.191204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi K., Ashcroft A.E., Radford S.E. Mass spectrometric methods to analyze the structural organization of macromolecular complexes. Methods. 2015;89:13–21. doi: 10.1016/j.ymeth.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Remus D., Beuron F., Tolun G., Griffith J.D., Morris E.P., Diffley J.F. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719–730. doi: 10.1016/j.cell.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout M.P., Sali A. Principles for integrative structural biology studies. Cell. 2019;177:1384–1403. doi: 10.1016/j.cell.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A., Gatz R., Beck F., Rigort A., Baumeister W., Plitzko J.M. Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. J. Struct. Biol. 2007;160:135–145. doi: 10.1016/j.jsb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Schachner L., Han G., Dillon M., Zhou J., Mccarty L., Ellerman D., Yin Y., Spiess C., Lill J.R., Carter P.J., Sandoval W. Characterization of chain pairing variants of bispecific IgG expressed in a single host cell by high-resolution native and denaturing mass spectrometry. Anal Chem. 2016;88:12122–12127. doi: 10.1021/acs.analchem.6b02866. [DOI] [PubMed] [Google Scholar]
- Schmidt C., Urlaub H. Combining cryo-electron microscopy (cryo-EM) and cross-linking mass spectrometry (CX-MS) for structural elucidation of large protein assemblies. Curr. Opin. Struct. Biol. 2017;46:157–168. doi: 10.1016/j.sbi.2017.10.005. [DOI] [PubMed] [Google Scholar]
- Schur F.K. Toward high-resolution in situ structural biology with cryo-electron tomography and subtomogram averaging. Curr. Opin. Struct. Biol. 2019;58:1–9. doi: 10.1016/j.sbi.2019.03.018. [DOI] [PubMed] [Google Scholar]
- Schwartz C.L., Sarbash V.I., Ataullakhanov F.I., Mcintosh J.R., Nicastro D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J. Microsc. 2007;227:98–109. doi: 10.1111/j.1365-2818.2007.01794.x. [DOI] [PubMed] [Google Scholar]
- Shaikh T.R., Yassin A.S., Lu Z., Barnard D., Meng X., Lu T.M., Wagenknecht T., Agrawal R.K. Initial bridges between two ribosomal subunits are formed within 9.4 milliseconds, as studied by time-resolved cryo-EM. Proc. Natl. Acad. Sci. U S A. 2014;111:9822–9827. doi: 10.1073/pnas.1406744111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Tiwari S.P., Miyashita O., Tama F. Integrative/hybrid modeling approaches for studying biomolecules. J. Mol. Biol. 2020;432:2846–2860. doi: 10.1016/j.jmb.2020.01.039. [DOI] [PubMed] [Google Scholar]
- Stahlberg H., Biyani N., Engel A. 3D reconstruction of two-dimensional crystals. Arch. Biochem. Biophys. 2015;581:68–77. doi: 10.1016/j.abb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Stark H., Chari A. Sample preparation of biological macromolecular assemblies for the determination of high-resolution structures by cryo-electron microscopy. Microscopy (Oxf) 2016;65:23–34. doi: 10.1093/jmicro/dfv367. [DOI] [PubMed] [Google Scholar]
- Tuijtel M.W., Koster A.J., Jakobs S., Faas F.G.A., Sharp T.H. Correlative cryo super-resolution light and electron microscopy on mammalian cells using fluorescent proteins. Sci. Rep. 2019;9:1369. doi: 10.1038/s41598-018-37728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchański T., Masiulis S., Fischer B., Kalichuk V., López-Sánchez U., Zarkadas E., Weckener M., Sente A., Ward P., Wohlkönig A. Megabodies expand the nanobody toolkit for protein structure determination by single-particle cryo-EM. Nature Methods. 2021;18:60–68. doi: 10.1038/s41592-020-01001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Heel M., Gowen B., Matadeen R., Orlova E.V., Finn R., Pape T., Cohen D., Stark H., Schmidt R., Schatz M., Patwardhan A. Single-particle electron cryo-microscopy: towards atomic resolution. Q. Rev. Biophys. 2000;33:307–369. doi: 10.1017/s0033583500003644. [DOI] [PubMed] [Google Scholar]
- Venkatesh S., Workman J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- Vinothkumar K.R., Henderson R. Single particle electron cryomicroscopy: trends, issues and future perspective. Q. Rev. Biophys. 2016;49:e13. doi: 10.1017/S0033583516000068. [DOI] [PubMed] [Google Scholar]
- Von Appen A., Kosinski J., Sparks L., Ori A., Diguilio A.L., Vollmer B., Mackmull M.T., Banterle N., Parca L., Kastritis P. In situ structural analysis of the human nuclear pore complex. Nature. 2015;526:140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees R.M., Schmeing T.M., Kelley A.C., Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330:835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D.N., Nierhaus K.H. The ribosome through the looking glass. Angew. Chem. Int. Ed. Engl. 2003;42:3464–3486. doi: 10.1002/anie.200200544. [DOI] [PubMed] [Google Scholar]
- Wu M., Lander G.C. Present and emerging methodologies in cryo-EM single-particle analysis. Biophys. J. 2020;119:1281–1289. doi: 10.1016/j.bpj.2020.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip K.M., Fischer N., Paknia E., Chari A., Stark H. bioRxiv; 2020. Breaking the Next Cryo-EM Resolution Barrier – Atomic Resolution Determination of Proteins! [DOI] [PubMed] [Google Scholar]
- Yoon J., Kim S.J., An S., Cho S., Leitner A., Jung T., Aebersold R., Hebert H., Cho U.S., Song J.J. Integrative structural investigation on the architecture of human Importin4_Histone H3/H4_Asf1a complex and its histone H3 tail binding. J. Mol. Biol. 2018;430:822–841. doi: 10.1016/j.jmb.2018.01.015. [DOI] [PubMed] [Google Scholar]