

Abstract
The inflammasome is a three-component (sensor, adaptor, and effector) filamentous signaling platform that shields from multiple pathogenic infections by stimulating the proteolytical maturation of proinflammatory cytokines and pyroptotic cell death. The signaling process initiates with the detection of endogenous and/or external danger signals by specific sensors, followed by the nucleation and polymerization from sensor to downstream adaptor and then to the effector, caspase-1. Aberrant activation of inflammasomes promotes autoinflammatory diseases, cancer, neurodegeneration, and cardiometabolic disorders. Therefore, an equitable level of regulation is required to maintain the equilibrium between inflammasome activation and inhibition. Recent advancement in the structural and mechanistic understanding of inflammasome assembly potentiates the emergence of novel therapeutics against inflammasome-regulated diseases. In this review, we have comprehensively discussed the recent and updated insights into the structure of inflammasome components, their activation, interaction, mechanism of regulation, and finally, the formation of densely packed filamentous inflammasome complex that exists as micron-sized punctum in the cells and mediates the immune responses.
Keywords: inflammation; ASC (apoptosis-associated speck-like protein containing a CARD), NLRP3; AIM2; NMR; protein structure; protein assembly
1. Introduction
Pathogen-associated molecular patterns (PAMPs) present in invading microbes and danger-associated molecular patterns (DAMPs) resulting from cellular insults are recognized by pathogen recognition receptors (PRRs). This recognition process results in the activation of a cytosolic supramolecular protein complex known as the inflammasome [1,2], which acts as a signaling platform and initiates an inflammatory response by triggering the production of proinflammatory cytokines (interleukin-1β (IL-1β) and interleukin-18 (IL-18)) [3]. Inflammasomes are divided into two categories, i.e., canonical inflammasomes, which activate caspase-1, and noncanonical inflammasomes that trigger activation of caspase-11 and caspase-4/5 in mouse and human, respectively [4,5,6]. The key components of canonical inflammasomes involve three classes of molecules, i.e., sensor, adaptor, and effector. These components tend to assemble via homotypic interactions between Death Domains (e.g., CARD-CARD (Caspase-activation and recruitment domain) and PYD-PYD (Pyrin domain)). In the presence of external stimuli or specific ligands, sensor proteins (ALRs (AIM2-like receptors) and NLRs (NOD-like receptors)) activate, oligomerize and nucleate the adaptor protein ASC (Apoptosis-associated speck-like protein containing a caspase-activation and recruitment domain (CARD)) through PYD-PYD interactions. ASC in turn recruits the effector protein procaspase-1 mediated by CARD-CARD homotypic recognition, thus serving as an activation platform of caspase-1.
Sensor proteins are divided into two classes: Absent in melanoma 2 (AIM2)-like receptors (ALRs) and NOD-like receptors (NLRs). AIM2 and IFI16 (Gamma-interferon-inducible protein) belong to the ALRs family and comprise an N-terminal PYD that interacts with ASC, and a C-terminal HIN (Hematopoietic, interferon-inducible, nuclear localization) domain for the recognition of double-stranded DNA (dsDNA) [7,8]. NLRs have tripartite domain organization and consist of: (1) N-terminal CARD or baculovirus inhibitor of apoptosis protein repeat (BIR) or PYD, which mediates homotypic protein–protein interactions for downstream signaling; (2) central nucleotide-binding domain (NBD) or NACHT domain, which elicits ATP-induced oligomerization; and (3) C-terminal manifold series of leucine-rich repeats (LRRs), which are responsible for ligand sensing and autoregulation [9,10,11,12]. The NBD domain belongs to the three subfamilies that include: NODs/NLRCs ((NLR family CARD domain-containing protein) NOD1–5), NLRPs/NALPs ((Nod-like receptor protein) NLRP1–14), and IPAF ((Ice protease activating factor) (NLRC4 and NAIP)). The human genome encodes for 23 NLRs; however, only a few NLR proteins such as NLRP1, NLRP3, NLRP6, NLRP7, NLRP9, NLRP12, and NLRC4 have been found to form inflammasomes and to activate caspase-1 [13,14,15,16,17,18]. These NLRs share structurally similar NACHT domains, but their activation is initiated by different stimuli [19,20]. A different type of inflammasome is formed by the sensor pyrin (also known as marenostrin or TRIM20), which contains a PYD domain, a B-box domain (zinc finger), a coiled-coil (CC) domain, and a B30.2/SPRY domain (absent in murine pyrin) [21]. Pyrin activates and assembles into the inflammasome complex in response to a bacterial infection that alters homeostasis and causes inactivation of RhoA GTPase [21]. Pyrin interacts with ASC through PYD-PYD homotypic interactions [21,22].
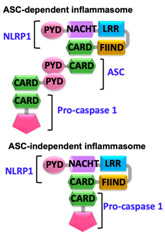
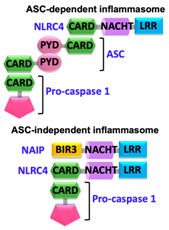
Upstream sensor proteins such as NLRP3 and AIM2 require the adaptor protein ASC for inflammasome assembly and activation, and thereby are known as ASC-dependent inflammasomes. Further homotypic interactions of ASCCARD with caspase-1CARD result in IL-1β and IL-18 release. These events facilitate the NF-κB (Nuclear factor kappa-light-chain-enhancer of activated B-cells), JNK (Jun N-terminal kinase), and p38 MAPK (Mitogen-activated protein kinase) signaling pathways that lead to high expression levels of cytokines and chemokines, and concomitant enlistment of immune cells to the site of pathogen invasion or tissue damage [23,24,25]. Other sensors with CARDs (e.g., NLRC4/NAIP and NLRP1) can directly activate caspase-1, and thus are representatives of ASC-independent inflammasomes [26,27]. However, reported studies on the function of the latter show that the presence of ASC enhances IL-1β secretion [28,29]. Details of activation signals, expression sites, and associated diseases of some canonical and non-canonical inflammasomes are summarized in Table 1.
Table 1.
Structural and functional details of canonical and non-canonical inflammasomes.
| Inflamma-some | Expression Site | Activation Signal | Diseases | Structure |
|---|---|---|---|---|
| Canonical Inflammasomes | ||||
| NLRP1 (NALP1, CARD7, CLR17.1, DEFCAP, VAMASI) | Adaptive immune cells and tissues, non- hematopoietic tissues | A/B toxin of Bacillus anthracis [31,32], Toxoplasma gondii infection [33], Muramyl dipeptide [28] | Vitiligo-associated multiple autoimmune disease [34], NLRP-1 associated autoinflammation with arthritis and dyskeratosis (NAIAD) [35], palmoplantar carcinoma and familial keratosis lichenoides chronica (FKLC) [36] |
|
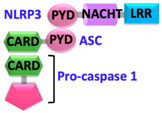
| NLRP3 (Cryopyrin, NALP3, CIAS1, CLR1.1, PYPAF1) | Monocytes, neutrophils, dendritic cells, lymphocytes, osteoblasts, and epithelial cells | Pathogen-associated molecular patterns (PAMPs) (bacteria and virus) [37,38,39,40], danger-associated molecular patterns (DAMPs) such as Monosodium urate [41], alum [42], silica [43], asbestos [43], calcium/potassium efflux [44], extracellular ATP [39], reactive oxygen species (ROS) [45] |
Cryopyrin-associated periodic fever syndrome (CAPS) [46], Muckle-Wells syndrome (MWS) [47], neonatal-onset multisystem inflammatory disease [48], Familial cold auto- inflammatory syndrome (FCAS) [49], Alzheimer’s disease [50] type II diabetes [51], cancer [52], chronic infantile neurological cutaneous and articular syndrome (CINCA, NOMID) [53] |
|
| NLRC4 (IPAF, CARD12, CLR2.1) | Macrophage and intestinal epithelial cells | Bacteria [54,55], Cytosolic flagellin [56] | Syndrome of enterocolitis and autoinflammation associated with mutation NLRC4 (SCAN4) [57], Macrophage activation syndrome (MAS) [58] |
|
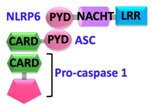
| NLRP6 | Cells of intestine and liver | Viral RNA [59], LTA of Gram-positive bacteria [60,61] | Colitis and colitis-induced tumorigenesis [62] |
|
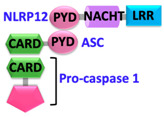
| NLRP12 (RNO, PYPAF7, Monarch-1) | Neutrophils, eosinophils, monocytes, macrophages, and dendritic cells | Bacterial components [15,63,64] | Familial cold auto- inflammatory syndrome 2 (FCAS2) [65] |
|
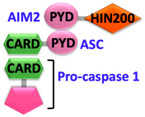
| AIM2 (PYHIN4) | Cytosol of hematopoietic cells | Bacterial and viral dsDNA [66,67,68,69] | Psoriasis [70], abdominal aortic aneurysm [71], systemic lupus erythematosus [72], prostate and colonic cancer [73] |
|
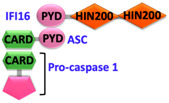
| IFI16 | Lymphocytes, monocytes, and epithelial cells | Viral and bacterial infections [74,75], Kaposi’s sarcoma-associated virus (KHSV) [76,77], HIV infections [78,79] | Systemic lupus erythematosus (SLE) [80] |
|
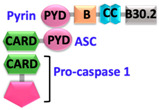
| Pyrin (Marenostrin, TRIM20) | Neutrophils, eosinophils, monocytes, dendritic cells and synovial fibroblast | Bacterial infection [81], RhoA-GTPase inactivation [82,83] | Familial Mediterranean fever (FMF) [84], pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) [85] |
|
| Non-Canonical Inflammasomes | ||||
| Human Caspase-4/5 | Macrophages, epithelial cells, and monocytes | LPS of Gram-negative bacteria [4], Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (oxPAPC) [86] | Inflammatory bowel disease and colorectal cancer [87] |

|
| Mouse Caspase-11 | Macrophages, epithelial cells, endothelial cells and neutrophils | LPS of Gram-negative bacteria [4], oxPAPC [5], Lipophosphoglycan (LPG) of Leishmania parasite [88], TIR-domain-containing adapter-inducing interferon β (TRIF) [89], secreted aspartyl proteinases [90] | Multiple sclerosis [91], Amyotrophic lateral sclerosis [92], Parkinson’s disease [93]. Inflammatory bowel diseases (IBDs) [94,95], Rheumatoid arthritis [86], Inflammatory respiratory diseases [96], Chronic obstructive pulmonary disease (COPD) [97] |
|
Non-canonical inflammasomes are not inflammasome particles per se as described above. Instead, caspase oligomerization and autoactivation are mediated by direct binding of the CARD domain of caspase-11 with cell wall components of bacteria such as intracellular lipid A and lipopolysaccharide (LPS) [5,26]. This binding induces caspase oligomerization and activation, with the subsequent cleavage of gasdermin D (GSDMD) and cell death by pyroptosis [30].
Dysregulation of inflammasome activation is responsible for several autoinflammatory diseases that are associated with high levels of IL-1β and/or IL-18 secretion (Table 1). Currently, significant progress has been made in our understanding of the activation, assembly and regulation of inflammasomes. However, the study of the molecular basis responsible for inflammasome assembly and its dissolution is still an emerging field. Comprehensive structural and biophysical studies will help in the identification of the factors influencing inflammasome formation and disassembly, as well as in the discovery of therapeutic targets aimed at designing novel anti-inflammatory drugs. This review describes structural and biophysical insights into the NLRP3 and AIM2 inflammasomes.
2. NLRP3
2.1. General Information on NLRP3
NLRP3 is expressed in myeloid cells, muscle cells, neurons, and endocrine cells [98]. In the resting state, it exists as an autoinhibited form that becomes activated upon stimulation and assembles into a large, micrometer-size cytosolic complex. In macrophages, NLRP3 becomes functional in a two-step process that includes priming and activation. In the priming process, PRRs such as Toll-like receptors (TLRs), or NODs and cytokines such as TNF-α trigger the activation of the transcription factor NF-κB, thus affecting the expression of the inflammasome components NLRP3, caspase-1, and pro-IL-1β [99,100]. Subsequently, NLRP3 undergoes post-translation modifications (PTMs) (ubiquitylation, phosphorylation, and sumoylation) [101,102,103], which stabilize the protein in a signal-competent auto-suppressed inactive state, later to be transformed into an activated state upon stimulation. In the activation step, NLRP3 inflammasome assembles into a mature multiprotein-complex particle composed of NLRP3, ASC, and procaspase-1, which is capable of activating IL-1β and IL-18.
2.2. Role of the NLRP3 Inflammasome in COVID-19
COVID-19 (Coronavirus disease 2019), caused by the SARS-Cov-2 (Severe acute respiratory syndrome coronavirus 2) virus, was declared a pandemic by the WHO (World health organization), and as of 16 December 2020, approx. 73.5 million cases with 1.64 million deaths have been reported worldwide [104,105,106]. The pandemic has been associated with severe social and economic consequences. An upsurge of IL-1β, IL-18, and LDH (Lactate dehydrogenase) has been reported in sera of COVID-19 patients, which hints the involvement of the inflammasome network [107,108,109]. Similarly, a recent study conducted on COVID-19 patients reveals the activation of the NLRP3 inflammasome [110]. In this study, microscopic analysis in combination with luminescent assays show the formation of NLRP3 and ASC puncta, caspase-1 activation, and IL-1β secretion in PBMCs (Peripheral blood mononuclear cells) of COVID-19 patients during the disease and in postmortem lung tissues [110]. In addition, the level of Casp1p20 and IL-18 in COVID-19 patients has been shown as an important marker for determining disease severity [110].
2.3. Structural Details of NLRP3
NLRP3 is composed of N-terminal PYD, central NACHT domain, and C-terminal LRR domain (Figure 1A). The structure of the PYD domain of human NLRP3 (NLRP3PYD) was determined by X-ray crystallography and solution-state NMR (Figure 1B,C) [111,112]. Both techniques reveal an overall similar architecture of the PYD domain with six helices (α1–α6) and five connecting loops, which is also analogous to the six-helix motif observed for the PYDs of NLRP1, NLRP4, NLRP7, NLRP10, and NLRP12 (Figure 1B) [111,112,113,114,115,116,117]. Among these, NLRP4 and NLRP10 show higher structural similarity with NLRP3 as compared to the other PYDs. However, pairwise structural alignments show that the orientation and length of the helices slightly differ in the NALP3PYD structure. The six helices adopt a canonical anti-parallel helical-bundle fold tightly packed by a central hydrophobic core made up of helices α1 (L10, A11, Y13, L14), α2 (F25, L29), α4 (L54, A55, M58), α5 (I74, F75), and α6 (A87). In addition, NLRP3PYD accommodates a second hydrophobic surface formed by F32, I39-P42, L57, and F61 residues [111], which stabilizes the shorter α3 helix by anchoring it to helix α2. Analysis of NLRP3PYD electrostatic surface shows a charge distribution that might be involved in interactions with ASCPYD or with other members of the Death Domain (DD) superfamily. In addition, structural homology and structure-based sequence alignment establish the presence of a conserved surface exposing a hydrophobic cluster (I39 and P40-P42 at α2–α3 loop and L57 and F61 at α4 helix) that could be responsible for inflammasome assembly and thus, caspase-1 activation [111]. The three-dimensional structures of NLRP3PYD and ASCPYD, as well as studies on their interactions, reveal that both electrostatic and hydrophobic interactions play important roles. In addition, there are several surfaces in the PYDs of the two proteins available for the interaction, which potentially enhance oligomerization. Conserved residues in NLRP3, C8 (α1 helix), and C108 (loop connecting PYD and NACHT) form a unique disulfide bond that might be involved in ROS (Reactive oxygen species) signaling and NLRP3 inflammasome activation [111]. Based on X-ray crystallographic studies, it has been speculated that the formation of the disulfide bond relieves the autoinhibitory state of NLRP3 upon activation by ROS. NLRP3PYD structures obtained from X-ray crystallography and solution-state NMR exhibit a high degree of resemblance (RMSD 1.66 Å); however, helices α2, α3, and α4 show slight differences between the crystal and solution structures (Figure 1C). Residue level calculation of B-factors obtained from X-ray crystallography suggests that NLRP3PYD is highly compact in agreement with the NMR studies [112]. In particular, helices α2, α3, α4, and α5, and the two hydrophobic cores of the NLRP3PYD are more rigid as reflected by the lower B-factor values, whereas the α2–α3 loop and C-terminus are more flexible.
Figure 1.

Structural details of NLRP3 and comparison with other pyrin domains (PYDs). (A) Schematic representation of NLRP3 domain organization; (B) 3D NMR solution-state structure of NLRP3PYD (PDB ID: 2NAQ) [112]; superposition of 3D solution structures of different PYDs with NLRP3PYD (blue, 2NAQ) which include NLRP1 (magenta, 1PN5); NLRP4, (ivory, 4EWI); NLRP7 (cyan, 2KM6); NLRP10 (orange, 2DO9); and NLRP12 (pink, 2L6A). Superimposed images are adapted from Figure 7A of [112]; (C) superposition of the crystallographic dimeric structure of NLRP3PYD (green, 3QF2 [111]) onto the monomeric NMR structure (blue); the dimeric interface is shown in the right panel. These superposed images are adapted from Figure 7B of [112].
Recently, cryo-EM (electron microscopy) studies revealed that NLRP3 folds into a characteristic earring-shaped conformation (Figure 2A) also observed for NLRC4, NOD2, and NAIP5 (NLR family of apoptosis inhibitory protein) [118]. The structure has a curved LRR domain (12 repeats) and a compact NACHT domain comprised of NBD, HD1 (helical domain 1), WHD (winged helical domain), and a variable HD2 (helical domain 2) module (Figure 1A) [118].
Figure 2.

Activation of NLRC4 and NLRP3. (A) Cryo-EM structure of leucine-rich repeats (LRR) and NACHT domains of NLRP3 in association with NEK7 (PDB ID: 6NPY) [118]. The ribbon diagram is generated with PyMOL molecular graphics software; (B) structural organization and activation of NLRC4. This image is adapted from Figure 2C of [137]; (C) modelling of NLRP3 active conformation and NLRP3 dimer formation in association with NEK7. This image is adapted from Figure 5A,B of [118].
2.4. Oligomerization of NLRP3PYD
The crystal structure of NLRP3PYD reveals the presence of a symmetric dimer, whereas NMR, MALS (Multiangle light scattering), and SEC (Size-exclusion chromatography) show the coexistence of monomeric with higher-order oligomeric forms of NLRP3PYD in solution. The existence of monomer or dimer depends on the concentrations of NLRP3PYD and salt, as well as pH [111,112]. The crystal structure of the NLRP3PYD (Figure 1C) dimer shows that residues E28 (α2), D29 (α2), and R41 (α3) in one protomer interact with the equivalent residues on the other protomer, which might result in repulsive interactions in the interface, thus suggesting that dimer structure is possibly an artifact from crystal packing [112]. The physiological relevance of the PYD dimer is still unclear. Some reports suggest that a PYD trimer, but not a dimer, can activate the inflammasome [119]; whereas others have found PYD dimer formation via a disulfide bond (e.g., NLRP12PYD) [120].
Sedimentation equilibrium and velocity results from analytical ultracentrifugation experiments and NMR studies indicate that NLRP3PYD forms concentration-dependent oligomers [112]. At low protein concentration and acidic pH (0.2 mM, 303 K), NLRP3PYD exists as a monomer that self-associates into higher-order oligomers at higher concentrations (0.6 mM, 303 K). The monomer–oligomer transition is drastically influenced by the presence of salt and temperature: Both the presence of salt and temperature increase favor oligomer formation even at low protein concentration (0.2 mM, 303 K, 100 mM NaCl). These results led to the conclusion that hydrophobic interactions play an important role in NLRP3PYD self-association.
2.5. Activation and Regulation of NLRP3
2.5.1. Post-Translational Modifications
Mutational studies uncovered that the LRR domain is not autoinhibitory because an LRR deleted variant demonstrates proinflammatory function similar to full-length (FL) NLRP3 in response to external stimuli [121]. These findings propose that the LRR domain is neither imperative for the assembly of active NLRP3 inflammasome nor crucial for the stabilization of the NLRP3 inactive state. It has been suggested that the importance of the LRR domain in NLRP3 activation could be related to: (1) Regulation by post-translational modifications due to the presence of ubiquitination and phosphorylation sites (K689 and Y861 in the human orthologue, and K687 and Y859 in the mouse orthologue) [121,122,123,124]; (2) the binding to NEK7, which is crucial for NLRP3 inflammasome activation [118]; and (3) the induction of TH2 cell (T helper type 2) responses [125]. TH2 cells facilitate adaptive immune responses against microorganisms and allergens by producing several interleukins (IL-4, IL-5, IL-9, IL-10, IL-13, and IL-25 (IL-17E)). The release of cytokines is further associated with antibody secretion, eosinophil/basophil activation, and various anti-inflammatory responses in order to provide phagocyte-independent protective responses. It is noteworthy that NLRP3 is also involved in the regulation of gene expression in TH2 cells in an inflammasome-independent manner. The transcription factor function of NLRP3 has been shown to be key for TH2 cell polarization. NLRP3 binds to DNA in TH2 cells at the Il4 promoter region and interacts with transcription factor IRF4 (Interferon regulatory factor) to regulate IL-4 synthesis. These results suggest that subcellular localization regulates NLRP3 functions by facilitating inflammasome assembly when expressed in the cytoplasm and promoting its transcriptional functions when localized in the nucleus.
Sequential phosphorylation–dephosphorylation events are also prerequisites for NLRP3 inflammasome activation. Several post-translational modification sites have been identified in human NLRP3 such as S5 (S3 in mouse), S198 (S194 in mouse), S295 (S291 in mouse), Y861 (Y859 in mouse), etc. [122,124,126,127]. Dephosphorylation by protein phosphatase 2 A (PP2A) at S5 located in the N-terminus PYD regulates the interaction of NLRP3 and ASC [122]. It was found that a phosphomimetic mutant of S5 that creates a negative charge completely attenuates NLRP3 activation by disturbing the interaction between NLRP3 and ASC [122]. Similar results were obtained from the phosphorylation of S295 by protein kinase A (PKA) and protein kinase D (PKD) [127,128]. In addition, dephosphorylation of Y861 by PTPN22 allows NLRP3 activation and subsequent IL-1β secretion [124]. The absence of PTPN22 in cells results in increased NLRP3 phosphorylation, which abolishes inflammasome assembly and reduces IL-1β secretion [124]. Another kinase, Jun N-terminal kinase1 (JNK1), has been shown to phosphorylate S198, providing a critical priming signal for NLRP3 self-association [126]. The phosphorylation/dephosphorylation interplay has been associated with the cryopyrin-associated periodic syndrome (CAPS), as it has been shown to be coupled to impaired phosphorylation at the S194 site in mouse [126]. Hence, the inhibition of NLRP3 phosphorylation/dephosphorylation processes is a potential pharmaceutical target for the treatment of NLRP3-associated diseases [126].
Ubiquitination and deubiquitination events are crucial for NLRP3 inflammasome regulation and involve a series of enzymatic steps mediated by ubiquitin-activating enzyme (E1)-ubiquitin-conjugating enzyme (E2)-ubiquitin ligase (E3), and deubiquitinating enzyme (DUBs), respectively [103,123]. The SCF (Skp-Cullin-F box) family member, FBXL2 (F-box L), is an anti-inflammatory molecule that binds at the W73 site and targets K689 for ubiquitin ligation and proteasomal degradation of NLRP3. In response to LPS exposure, an elevated level of FBXO3 (F-box O3), an E3 ligase, inhibits FBXL2 that in turn upregulates the NLRP3 expression level in cells and increases the release of proinflammatory cytokines. A small molecule, BC-1215, acts as an inhibitor for FBXO3 and reduces the secretion of matured IL-1β and IL-18 [123]. It has also been reported that neurotransmitter dopamine (DA) activates the ubiquitin E3 ligase, MARCH7 (membrane-associated ring finger (C3HC4) 7), that inhibits NLRP3 inflammasome activation via K48-linked polyubiquitination [129]. Similarly, another E3 ubiquitin ligase, Pellino2, assists in K63-linked ubiquitination and induces NLRP3 activation [130].
BRCC3 (BRCA1/BRCA2- containing complex, subunit 3), a murine deubiquitinating enzyme (human orthologue is BRCC36), is also involved in regulating NLRP3 activity. BRCC3, in combination with the protein ABRO1 (Abraxas brother 1), forms the cytosolic BRISC complex and deubiquitinates the LRR domain of NLRP3 by specifically cleaving the K63-linked polyubiquitin chain [103]. The ubiquitin isopeptidase inhibitor, G5 (NSC 144303), acts as an inhibitor for DUBs and inhibits deubiquitination and subsequent NLRP3 activation [103]. The protein NleA, secreted by enteropathogenic and enterohemorrhagic E. coli, inhibits deubiquitination of NLRP3 to limit inflammasome activation by hijacking the ubiquitin machinery [131]. A20, a ubiquitin modifying enzyme, acts as a negative regulator of NLRP3 [132,133]. Similarly, TRIM31 (tripartite motif 31) induces K48-linked ubiquitination and subsequent proteasomal degradation of NLRP3 [134]. Another E3 ubiquitin ligase, Shigella IpaH7.8, also activates NLRP3-inflammasome by targeting glomulin (GLMN), which is a member of Cullin ring ligase inhibitor [135].
2.5.2. NEK7 Mediated Activation of NLRP3
NIMA (Never In Mitosis gene A)-related kinase 7 or mitotic Ser/Thr kinase NEK7 regulates NLRP3 activation [118]. Cryo-EM structural analysis of the complex formed between NLRP3 without the PYD, the NEK7 C-lobe, and ADP bound to the NBD of NLRP3, reveals that the C-terminal lobe of NEK7 interacts with the NBD, HD2, and LRR regions of NLRP3 (Figure 2A). In addition, the dissociation constant of the complex between NLRP3 and NEK7 was determined to be, Kd 78.9 ± 38.5 nM. The binding between NLRP3 and NEK7 involves two interfaces: The first and second half of the NEK7 C-lobe interact with the LRR and NACHT domains (NBD and HD2), respectively. Residues Q129, R131, and R136 of NEK7 interact with the LRR domain, whereas residues D261, E265, and E266 interact with HD2, and D290, K293, and R294 interact with the NBD. Mutagenesis studies showed that both interfaces are required for NLRPE3-NEK7 complex formation. The substitution of G755 in LRR for the amino acids A or R leads to enhanced interaction between LRR and NEK7 [136]. In contrast, phosphorylation of Y859, located in the LRR domain, causes steric hindrance and charge repulsion, thus aborting the interaction with NEK7 [124].
A computational model of the oligomeric assembly of the NLRP3-NEK7 complex was generated to understand the NLRP3 activation mechanism using the structure of a full-length NLRC4 oligomer as a template [118]. It has been reported that the inactive form of NLRC4 undergoes a 90° rotation of the NBD-HD1 module with respect to the WHD-HD2-LRR module, which generates an active NLRC4 conformation (Figure 2B) [137,138]. The conformation of the NBD-HD1-WHD module of NLRP3 is similar to that of the inactive form of NLRC4; therefore, the active oligomeric structure of NLRP3 has been modeled from its inactive form by using the NLRC4 activation mechanism (Figure 2C) [118].
2.5.3. Role of Caspase-8 in Inflammasome Activation
Caspase-8 plays an important role in the regulation of inflammatory responses by direct cleavage of pro-inflammatory cytokines into their mature forms, and by activating the NLRP3 inflammasome [139,140]. Caspase-8 consists of a death effector domain (DED) at the N-terminus, and p18 and p10 catalytic subunits at the C-terminus. Cell studies showed that in the absence of caspase-1, pro-IL-1β processing is mediated by caspase-8 in response to LPS [141], and to a wide range of stimuli, such as activation of TLR4 in bone marrow-derived dendritic cells (BMDCs) [142] and Fas death receptor. Ligation to the bacterial and fungal C-type lectin receptor, dectin-1, triggers caspase-8 activation via CARD9-Bcl-10-MALT1 complex. Other stimuli include endoplasmic reticulum stress, chemotherapeutic agents, inhibition of c-FLIP (FLICE-like inhibitory protein), and histone deacetylases (HDAC) [143,144,145,146,147,148]. Activated caspase-8 cleaves pro-IL-1β at the same site (D117) utilized by caspase-1 [141,149].
Caspase-8 acts as positive and negative regulator of the NLRP3 inflammasome depending on specific cell types. In macrophages, caspase-8 together with FADD (Fas-associated protein with death domain) drives the priming and activation of the canonical and non-canonical inflammasomes [150]. It has been shown that in the absence of IAPs (inhibitor of apoptosis proteins), LPS or TNF-primed murine macrophages and dendritic cells show TLR-TRIF (TIR-domain-containing adapter-inducing interferon-β)-RIPK1 (Receptor-interacting protein kinase-1)-RIPK3-caspase-8 mediated activation of the NLRP3 inflammasome [142,151,152]. Similarly, human BlaER1 monocytes show TLR3 activation mediated by the TRIF-RIPK1-FADD-caspase-8 pathway [153]. In contrast, caspase-8 deficient murine BMDCs and macrophages, and human BlaER1 monocytes show RIPK1-RIPK3 dependent necroptotic activation of the NLRP3 inflammasome upon TLR4 ligation [152,154,155]. Ablation of IAPs and caspase-8 in TLR-primed macrophages and BMDCs leads to NLRP3 activation by RIPK3-MLK (Mixed lineage kinase domain-like pseudokinase), which suggests a negative role of caspase-8 in inflammasome activation [152,156]. In dendritic cells, it inhibits RIPK1-RIPK3-MLKL-mediated NLRP3 inflammasome activation [152,154]. Some studies indicate that caspase-8 is involved in pore formation in the plasma membrane by activating GSDMA or pannexin-1 (channel-forming protein) that in turn facilitates NLRP3 inflammasome activation via K+ efflux [157,158]. It has also been reported that caspase-8 is an integral part of the inflammasome particle, comprising MALT1, caspase-8, and ASC, and directly processes pro-IL-1β without caspase-1 involvement [159]. Caspase-8 interacts with ASCPYD via DED, which induces caspase-8 polymerization and subsequent activation of ASC [160].
3. AIM2
AIM2 is a cytosolic dsDNA sensor that is responsible for downstream signaling to the adaptor protein ASC in response to the presence of bacterial and viral DNA. AIM2 belongs to the PYHIN family (pyrin + HIN) and consists of an N-terminal PYD (1–87) and a C-terminal HIN domain (138–343) connected through a long linker [161,162] (Figure 3A). AIM2 interacts with ASC via PYD-PYD homotypic interaction and the HIN domain binds to dsDNA in a sequence-independent manner. In addition, AIM2 also heterodimerizes with other members of the PYHIN family such as p202, IFI16, and MNDA (Myeloid cell nuclear differentiation antigen) [69,163,164,165]. AIM2 interaction with ASC further activates procaspase-1 leading to pyroptosis, whereas the interaction with p202 inhibits AIM2-mediated inflammatory responses [68,166,167,168].
Figure 3.

Structural details of AIM2. (A) Schematic representation of AIM2 domain organization; (B) crystal structure of AIM2PYD with Maltose-binding protein (MBP) fusion tag (PDB ID: 3VD8) [161]; (C) crystal structure of AIM2HIN in complex with dsDNA (PDB ID: 3RN2) [162]. The ribbon diagrams are generated with PyMOL molecular graphics software.
3.1. AIM2PYD Domain Structure
The crystal structure of AIM2PYD reveals the six-helix bundle conformation characteristic of the DD superfamily [161] (Figure 3B). It shares structural homology with PYDs of NLRP3 and ASC with RMSD values of 1.8 Å and 1.6 Å, respectively [161]. However, AIM2PYD exhibits a short and highly dynamic α3 helix and long α6 helix with the most variable sequence among the known PYDs. Largely buried and highly conserved K26 residue located at α2 helix buttresses α3 helix via hydrogen bonding with L40 and A43 of the interconnecting loop, thereby stabilizing the α3 helix. The overall surface of AIM2PYD is populated by charged residues, thus resulting in distinct electrostatic charge distribution. These amino acids include acidic residues such as E7 and D15 in α1 helix; D19, E20, E21, and D23 in α2 helix; and basic residues such as K64, R67, and K71 in α5 helix; and K79, R80, K85, K87, K90, K93, and K97 in α6 helix. Among these, K64 and K85 are conserved in the PYHIN family, and E20 and E21 are specific to AIM2. Residues such as F27 and F28 create a solvent-exposed hydrophobic patch that shares similarities with the DED surface involved in homotypic interactions, suggesting that they may contribute to AIM2PYD self-association and AIM2-specific functions [161].
AIM2PYD tends to form large, insoluble oligomers in solution, and thus poses significant challenges for biophysical studies. To overcome oligomerization, AIM2PYD has been fused to an MBP (Maltose-binding protein) tag and a specific mutant, F27G, has been created to shift the monomer–oligomer equilibrium to the monomeric form [169]. The crystal structures of mouse AIM2PYD (mAIM2PYD), wild-type (WT) human AIM2PYD (hAIM2PYD), and hAIM2PYD F27G mutant very similar, as expected for an amino acid sequence identity of 56%. However, the conformational arrangement of helices α2 and α3 shows significant differences [169,170]. Helix α3 of mAIM2PYD is positioned adjacent to the N-terminus of helix α2, similarly to the reported conformation of these helices for hNLRP10PYD [169,170]. In contrast, helix α3 is positioned adjacent to the C-terminus of helix α2 in hAIM2PYD F27G mutant and shows intermediate orientation in the structure of WT hAIM2PYD. Differences in chain flexibility have also been observed; for example, the α2-α3 helical region is relatively ordered, with an average B-factor of 18.7 Å2 in hAIM2PYD F27G mutant and of ~120 Å2 in wild type AIM2PYD [169].
3.2. AIM2PYD Self-Association
Isolated AIM2PYD can self-associate and form filaments similar to ASCPYD. Homology modeling of the AIM2PYD filament using the cryo-EM structure of the ASCPYD filament as a template in combination with negative stating (ns)-EM data revealed that the AIM2PYD filament shows a three-fold symmetry arrangement of the PYD protomer structures [167,171]. EM analysis proposes that the AIM2PYD filament serves as a structural template for ASC polymerization [167]. It has been reported that mAIM2PYD maintains monomeric conformation at low pH (4.0) and low salt concentration (< 100 mM) due to repulsive electrostatic forces between positively charged molecules and by interfering with hydrophobic interactions, whereas high salt concentration promotes oligomerization through hydrophobic interaction of hydrophobic patches on the protein surface [170]. These results confirm that both electrostatic and hydrophobic interactions are necessary for AIM2PYD polymerization, which was also observed for the interaction between the PYDs of ASC and NLRP3 and for ASCPYD self-association [121,169,171]. Isolated AIM2HIN cannot form ordered macrostructures, thus pointing to a predominant role of Death Domains such as PYD in the formation of ordered polymers. In fact, AIM2PYD self-association has been shown to suffice for inducing the assembly and activation of the inflammasome [169].
Cryo-EM structural studies of GFP-AIM2PYD filament indicate that this truncated construct forms ~200 nm to ~1 μm long filaments with outer and inner diameters of ~90 Å and ~20 Å, respectively. In this filament structure, the GFP tag protrudes from a filament core formed by AIM2PYD [172]. Modeling studies propose that type I, II, and III interactions characteristic of Death Domains (PYDs and CARDs) play important roles in the helical organization of the AIM2PYD filament. In the type I interaction, residues of helices α1 and α4 (S3, K6, L10, L11, D31, and I46) located on the first protomer interact with residues on helices α2 and α3 (R24, F27, F28, and D31) located on the fourth protomer. In the type II interaction, residues of helix α4 (Q54 and N55) located on the first protomer interact with residues of helices α5 and α6 (N73, Y74, and L76) located on the sixth protomer. In type III interaction, residues of helix α3 (G38 and K39) of the first subunit interact with residues in helices α1 and α6 (D15, N16 and I17) of the third protomer. The type I interaction is mediated by hydrophobic contacts and dominates filament assembly. Thus, mutations of residues such as F27G/F27L (type Ib surface) and L10A/L11A (type Ia surface) inhibit AIM2PYD self-association and promote the monomeric form. Other structural studies have used the MBP fused to AIM2PYD to impede oligomerization. It has been found that residues L10 and L11 are located near the MBP in this construct, which agrees with the finding that these residues are involved in oligomerization via the type I interface [172].
3.3. AIM HIN Domain Structure
The HIN domain of AIM2, with ~200 amino acids, comprises two tandem OB (oligonucleotide/oligosaccharide binding) folds connected through a long linker (Figure 3C). Canonical OB folds contain five β-strands that fold into two sheets [162]. The proximal OB1 fold consists of β1–β5, among them β1, β4, and β5 split into two short strands (β and β’). Similarly, the distal OB2 fold (β1–β5) shows the splitting of β5 into two shorter strands. The linker connecting OB1 and OB2 is ~30 residues long and is folded into two alpha-helices. The two OB folds firmly interact with one another through conserved hydrophobic interactions. The HIN domains of AIM2, IFI16, and p202 are highly conserved and show an identical topological arrangement of OB folds [162,173].
3.4. AIM2 HIN:dsDNA Interaction
The crystal structure of the AIM2HIN domain in complex with dsDNA derived from the Vaccinia virus was determined using X-ray crystallography [162] (Figure 3C). This structure reveals that the highly positively charged surface of HIN interacts with the sugar-phosphate backbone of dsDNA mainly via electrostatic interactions. The N-terminus of the HIN domain is positioned far away from the DNA-binding surface, possibly facilitating the interaction between the N-terminal PYD of AIM2 with the adaptor protein ASC. The binding of the HIN domain to both the major and minor grooves of the DNA could explain AIM2-induced activation of the innate immune system in the presence of dsDNA, but not ssDNA [66,68,162]. Both OB folds and the connecting linker participate in DNA binding. Specifically, residues K160 (β1), K162 (β1), and K163 (β1–β1’ loop) of the OB1 fold, as well as residues L267 (β1), N287 (β2), K309 (β4), R311 (β4), K335 (β5), and I337 (β5), of the OB2 fold and linker residues such as R244 (α2), G247 (α2), and E248, T249, and K251 located at α2–α3 loop, participate in the binding between AIM2HIN domain and dsDNA mainly via hydrogen bonding, van der Waals interactions, and salt bridges. The crystal structure also shows the formation of bidentate hydrogen bonds between residue R311 and phosphate groups in the DNA backbone [162].
It has been reported that ~80 bp of dsDNA is the minimum size required for the induction of IL-1β by AIM2 activation [162]. Each HIN domain occupies four DNA base pairs, hence ~20 AIM2HIN domains wrap around the 80 bp of dsDNA with an observed axis tilt of 35°. Multiple sequence alignment suggests that most residues interacting with dsDNA are also conserved in IFI16HIN and mouse AIM2HIN domains. Site-directed mutagenesis studies indicate that mutations involving residues located on the interacting regions, such as the OB1-linker, the OB1-linker-OB2, and residue F165, lead to a diminished binding affinity of AIM2HIN to dsDNA. These results on AIM2HIN were corroborated by similar mutagenesis experiments conducted on full-length AIM2 (AIM2FL), which resulted in an impaired association of AIM2 with DNA and reduced IL-1β secretion [162]. Altogether, these studies suggest that an intact receptor binding surface is required for the association to dsDNA and the subsequent immune responses.
3.5. AIM2 PYD:HIN Interaction
Xiao et al. originally proposed that in the absence of a ligand, intramolecular interactions between the PYD and HIN domains in AIM2 retain the sensor in an autoinhibited state that prevents PYD-mediated oligomerization and suppresses HIN:DNA binding [162].
Docking analyses of crystal structures suggest that the negatively charged helix α2 of the PYD locates at the interface of the PYD:HIN interaction. In addition, ITC (Isothermal titration calorimetry) studies reveal that AIM2PYD interacts with AIM2HIN with a dissociation constant (Kd) of 23.5 µM. Furthermore, it has been shown that mutations of acidic residues located in helix α2 abolish the binding of the PYD and HIN domains [161]. These results confirm that the PYD-HIN interface is dominated by electrostatic interactions between negatively and positively charged residues in the PYD and HIN domains, respectively. The negatively charged surface of AIM2PYD that participates in the interaction with AIM2HIN is also involved in the binding to ASCPYD, hence ensuring downstream signaling to the adaptor ASC only after AIM2 is activated by dsDNA.
3.6. Importance of AIM2PYD in dsDNA Interaction and Oligomerization
The hypothesis of the autoinhibitory model was challenged by Sohn and colleagues [171]. Based on their studies, they proposed that AIM2PYD does not participate in AIM2 autoinhibition, instead, it actively helps in DNA binding and concomitant self-association. We mentioned above that the fusion of MBP to the N-terminus of AIM2FL interferes with PYD oligomerization. To interrogate whether the PYD has a role in dsDNA binding, fluorescence anisotropy experiments were conducted to compare the affinity of fluorescein amidite (FAM)-labeled 72-bp dsDNA with MBP-AIM2FL, MBP-AIM2HIN, untagged AIM2FL, and untagged AIM2HIN. The results show that MBP-AIM2FL binds 2-fold tighter to dsDNA than MBP-AIM2HIN, whereas untagged AIM2FL binds at least 20-fold more tightly than MBP-tagged AIM2 variants in presence of 160 mM KCl. Another important finding from the Sohn group is that the isolated HIN domain can oligomerize upon dsDNA binding and thus assists in filament formation. Salt concentration-dependent binding reveals that AIM2HIN oligomerizes on dsDNA in presence of 160 mM KCl, but fails to bind at 400 mM KCl. On the other hand, AIM2FL binds to dsDNA even at this high salt concentration, which hints the involvement of PYD in dsDNA binding. Furthermore, mutations of residues L10, L11, and F27 involved in AIM2PYD self-association and non-conservative mutations of AIM2PYD acidic residues D19, E20, E21, and D23 impede the binding of AIM2FL to dsDNA at 400 mM KCl, further supporting that the oligomerization of AIM2PYD plays an important role in dsDNA binding. Although the effects of these mutations in the 3D-fold of the PYD were not tested, it would be expected that these residues facilitate the transformation of AIM2 from the autoinhibited conformation to the activated form that can bind DNA even at high salt concentration. Therefore, the results obtained with these mutants contradict the inhibitory role of the AIM2PYD in DNA binding.
AIM2FL needs to bind to a larger dsDNA size (~12 bp) as compare to the HIN domain alone (~8 bp), confirming the relevance of oligomerization to potentiate dsDNA binding. Furthermore, binding of AIM2FL to dsDNA increases 1000-fold in the presence of 10-times longer DNA, indicating cooperativity between dsDNA size and AIM2 binding affinity. For the formation of AIM2FL-dsDNA complex, ~70 bp dsDNA and six molecules of AIM2FL are needed to cross the binding threshold (lag phase), and ~250–300 bp dsDNA and ~24 AIM2FL molecules are required for establishing an optimal oligomeric complex, as determined by fluorescence anisotropy competition binding assays using FAM-dsVACV72 (1.5 nM) and AIM2FL (70 nM) against various fragments of dsDNA at 400 mM KCl. The data were fit to competition binding equation; [171]. This observation is further confirmed by monitoring the increase of IL-1β secretion with increasing dsDNA size. Overall, these results propose that dsDNA size acts as a ‘molecular ruler’ to regulate AIM2 inflammasome assembly in a switch-like mechanism of PYD oligomerization and dsDNA binding.
In the presence of dsDNA excess, AIM2FL shows saturating or increased size-dependent FRET (Fluorescence resonance energy transfer) signals, but AIM2HIN displays decreased FRET signals, indicating that AIM2PYD is key for dsDNA binding and promotes oligomerization in presence of dsDNA excess [171]. Interestingly, ns-EM images illustrate that AIM2FL is able to self-oligomerize in the absence of dsDNA forming “Brussels sprout-like” filaments at high concentration (≥500 nM) [171]. Oligomerized AIM2PYD forms the filament core (~9 nm) and the HIN domains are observed at the periphery of this core, like Brussel sprouts. In contrast, filaments formed by AIM2FL bound to dsDNA are ~25 nm wide. The participation of the PYD in filament assembly is critical, as isolated AIM2HIN and MBP-AIM2FL do not show any ordered filament formation in absence of dsDNA, and isolated AIM2HIN displays random ‘beads on a string’-like cluster upon dsDNA addition. Moreover, mutagenesis studies show that both PYD and HIN domains are required for the oligomerization of AIM2FL in the presence or absence of dsDNA. FRET results reveal that the length of dsDNA regulates the assembly kinetics and lifetime of the dsDNA-AIM2 complex [174]. Modeling analysis based on cryo-EM and ns-EM observations propose that in the dsDNA-AIM2HIN filament complex of ~7.5 nm diameter, AIM2HIN is wrapped around the dsDNA core and each HIN molecule interacts with six adjacent HIN molecules [172]. Such an arrangement of AIM2HIN around the DNA core and long linker between both domains brings AIM2PYDs into close proximity where they form short helical filaments proposed to run parallel to the DNA and to act as a platform for ASCPYD filament nucleation.
Altogether, these results propose that in the absence of cytosolic dsDNA, AIM2 is expressed at a very low basal concentration level, and is therefore unable to oligomerize and induce downstream signaling via ASC. Pathogenic attack facilitates rapid oligomerization due to invasion of dsDNA in the cytosol, which hikes AIM2 local concentration [171]. The size of dsDNA acts as a molecular ruler and governs the AIM2 assembly.
3.7. Regulation of AIM2
3.7.1. Negative Regulators of AIM2 Inflammasome Activation
Regulation of inflammasome assembly is imperative for maintaining cellular homeostasis. The mouse protein p202 has been reported to sequester cytoplasmic dsDNA and inhibit AIM2 activation [69]. p202 consists of two HIN domains and lacks the PYD, rendering it unable to recruit ASC. The binding of p202 to DNA and AIM2 is proposed to attain a balance between pathological DNA-induced inflammation and physiological host defense. The crystal structure of mouse p202-dsDNA complex reveals that the p202HIN1 domain binds to DNA, whereas p202HIN2 interacts with AIM2 [173]. Full-length p202 (p202FL) forms a tetramer in cells as well as in vitro purified protein solutions. p202HIN2 first dimerizes in a parallel fashion using both OB folds (OB1-OB2 to OB1-OB2) and the formed dimers assemble into tetramers in a tail-to-tail orientation of the OB2 folds (OB2 to OB2) [166]. Although p202HIN2 lacks DNA binding capability, tetramer formation serves as a platform for p202HIN1 attachment to dsDNA, increasing the overall DNA binding affinity of p202FL as compared to AIM2. p202HIN1 shares structural similarity with mAIM2HIN and IFI16HIN2, but shows different charge distribution and opposite orientation of the dsDNA binding surface. Such a difference in surface electrostatic potential is responsible for the antagonist activity of p202 [166].
Unlike AIM2, the linker connecting the two OB folds of p202 does not participate in DNA binding. Positively charged residues located at the N-terminus and loop between β1–β2 of the OB1 fold engage with the DNA minor groove [166]. In the OB2 fold, residues located in the loop connecting β1–β2 and the loop between β4–β5 interact with the dsDNA major groove. Structure-based mutagenesis studies propose that among these, OB1 N-terminal residues K48, and K53, and OB2 residue R224 are crucial for HIN:DNA interactions. Most of these residues interact with the backbone of DNA; however, K53 side chain was found to make two hydrogen bonds with DNA bases.
p202HIN2 interacts with AIM2 through a short sequence motif (MFHATVAT) conserved in both proteins and buried in the core of the HIN domains [175,176]. The protein region, MFHATVAT, is required for p202 dimerization and subsequent interaction with AIM2 [175]. It has been reported that p202HIN2 does not block the DNA binding surface of AIM2; therefore, DNA binding affinity of AIM2 remains unaffected in the presence of p202HIN2 [166]. Computational docking studies showed that the binding of AIM2HIN domains with both ends of the p202HIN2 tetramer creates a spatial separation between AIM2PYDs, thus preventing ASC oligomerization [166]. Consequently, the knockdown of p202 increases the level of ASC and cross-linked ASC oligomers [166]. In this line, modeling studies propose that two adjacent mouse AIM2 (mAIM2) molecules bound to DNA are separated by less than 10 Å, thus generating AIM2 molecular crowding and favoring the interaction with ASCPYD and the subsequent activation of inflammasome assembly [173]. In contrast, p202 spans a larger dsDNA fragment and binds with higher affinity compared to AIM2. Therefore, when both p202 and AIM2 are present in equal amounts, the former competes with the latter for dsDNA binding and covers a larger surface area of dsDNA [173].
3.7.2. IFI16-β Mediated Regulation of AIM2 Inflammasome Activation
A novel human isoform of IFI16 designated as IFI16-β has been shown to selectively inhibit the formation and activation of AIM2 inflammasome assembly [177]. IFI16-β is ubiquitously expressed in various human cells and shows upsurge expression in leukocytes in case of viral infection. IFI16-β co-localizes with AIM2 in the cytoplasm and by sequestering cytoplasmic dsDNA, impedes its detection by AIM2. Analogously to p202, IFI16-β contains two HIN domains (HIN A and HIN B) and disrupts AIM2-ASC inflammasome activation by interacting with AIM2, competing with dsDNA binding as well as inhibiting AIM2 oligomerization [166,173,177]. Competition binding experiments suggest that IFI16-β binds with higher affinity to dsDNA than AIM2 because the IFI16-β-DNA complex shows a more prominent band in biotin-dsDNA pull-down assays as compared to AIM2 [177]. Altogether, dsDNA binding studies of p202 and IFI16-β indicate that proteins expressing two HIN domains bind to dsDNA more robustly than single HIN domain-containing proteins like AIM2 [166,173,177].
3.7.3. Post-Translational Modifications of AIM2
Very limited information is available on AIM2 post-translation modifications. However, it has been reported that TRIM11 (tripartite motif 11) acts as a negative regulator of the AIM2 inflammasome. TRIM11 binds AIM2 and undergoes poly-ubiquitination at K458, leading to the recruitment of autophagy cargo receptor p62, thus mediating the subsequent degradation of AIM2 [178,179]. In addition, studies conducted on mouse models of stroke and cultured primary microglia show elevated expression of HDAC3 (Histone deacetylases 3) linked to the regulation of the inflammatory process by activating the AIM2 inflammasome. RGFP966, a HDAC3 inhibitor, downregulates the AIM2 inflammasome by enhancing acetylation and inhibiting phosphorylation (at Y701 and S727) of STAT1 (Signal transducer and activator of transcription) in order to protect against ischemic brain injury [180].
4. ASC
ASC (PYCARD; PYD and CARD domain-containing or TMS; Target of Methylation-induced Silencing-1) is a ~24 kD bifunctional cytosolic adaptor protein that consists of an N-terminal PYD (1–89) and a C-terminal CARD (113–195) connected by a 23 residue-long linker (90–112) [181,182,183] (Figure 4A). ASC expresses in the nucleus of epithelial and immune cells, and in response to inflammatory stimuli, is redistributed to the cytoplasm where it assembles into a compact micrometer-size perinuclear structure referred to as ASC speck or ASC foci [181,182,184]. The ASC speck colocalizes with the sensor and the procaspase-1 by homophilic interactions mediated by the PYD and CARD domains, thus forming the inflammasome, which serves as the platform for caspase activation and pyroptotic cell death [168,185,186,187]. In addition to ASCFL, three other isoforms also exist: ASC-b, which also bears an N-terminal PYD and C-terminal CARD as with ASCFL, although connected by a short 3 amino acid-long linker; ASC-c, retaining the CARD, but only a partial PYD; and ASC-d, a 105-amino acid long polypeptide that only conserves residues 1–35 of the original ASCFL sequence [188]. These isoforms respond differently to inflammatory stimuli, exhibit irregularly shaped perinuclear aggregates and differential cellular expressions.
Figure 4.

Structural details of ASC. (A) Schematic representation of ASC domain organization; (B) 3D NMR solution-state structure of full-length ASC (PDB ID: 2KN6) [183]. The ribbon diagram is generated with PyMOL molecular graphics software.
ASCPYD interacts with the PYDs of NLRP3 and AIM2, whereas ASCCARD interacts with the CARDs of procaspase-1 and NLRC4 via homotypic interactions [20,189]. The 3D NMR-solution structure of ASC reveals that the PYD and CARD domains form rigid structures with RMDS of 0.78 ± 0.07 and 0.79 ± 0.08 Å, respectively. The two Death Domains do not interact with one another based on Nuclear Overhauser data (NOE) and are connected by a linker that shows the residual secondary structure and fast local motion on the picosecond time scale [183,190]. NMR-based secondary chemical shift analysis and NOE data indicate that the linker adopts low populated extended structures analogous to polyproline II-like conformation [183]. Rotational correlation times (τc) derived from NMR relaxation experiments for ASCFL and the individual domains suggest that both domains reorient at different rates, but feel the drag from each other due to the presence of the linker [183].
4.1. Structural Details of ASCPYD and Its Self-Association
ASCPYD adopts the classic six-helical bundle motif typical of the DD-fold showing a long loop between helices α2 and α3, a unique feature commonly found in PYDs [167,189] (Figure 4B). The electrostatic surface of ASCPYD is highly bipolar, with helices α1 and α4 containing mainly negatively charged residues, whereas helices α2, α3, and the connecting loop mostly accommodate positively charged residues [189]. Charge complementarity and the corresponding charge–charge interactions resulting from the bipolar distribution of the electrostatic surface are responsible for ASCPYD self-associations. Two oppositely charged surfaces of ASCPYD assemble back to back and self-associate with KD = 40–100 μM using the dominant type I interaction mode, resulting in a buried surface area of 880 Å2. In the type I interaction for ASCPYD self-association, helices α1 (E13), α4 (D51), the N-terminus of helix α5, and the α3-α4 loop (D48) of one surface interact with helices α2 (K21), α3 (R41), and the C-terminus of helix α5 of the opposite surface [112,167,189]. ASCPYD displays a structural difference compared to other PYDs such as NALP1PYD and NALP10PYD, as the latter shows considerable variation in the length of helices α1 and α6, and helix α3 is replaced by a disordered loop that may dictate their exclusive inflammatory functions [183].
Mutations of hydrophobic residues located in the PYD disrupt PYD-PYD filament formation, significantly increasing solubility at neutral pH while still retaining the monomeric folded conformation [190,191]. Similarly, NMR studies of ASC mutants in residues located in the type I and type III interfaces show complete or partially reduced ability of filament formation [167,190,192]. The L25A mutation in ASCPYD has been commonly used in structural and biophysics studies to avoid oligomerization. NMR-based chemical shift analysis showed that the L25A mutation causes structural perturbations around residues K24 and L45 located at the α2-α3 binding interface. Structural perturbation around K24 destabilizes the α3-helix, hence diminishing ASCPYD oligomerization by reducing the PYD binding ability. However, it is able to form dimers via the α1-α4 interface [112,190].
Cryo-EM studies revealed that ASCPYD subunits pack densely in a helical tube-like, three-fold symmetry structure with 53° right-handed rotation and 14.0 Å axial rise consisting of six molecules per turn with inner and outer diameter of ~20 Å and ~90 Å, respectively [167,193]. NMR and cryo-EM based structural analysis of ASCPYD and its comparison with other members of the Death Domain superfamily propose the involvement of all three interaction types in the stabilization of the PYD filament; i.e., intra-strand type I and inter-strand type II and III interactions [167,190]. These observations were further supported by mutagenesis experiments [182]. The type II interaction mode with a buried surface area of 524 Å2 involves contacts between helix α4 and the α4-α5 loop of one surface with the α5-α6 loop of the opposite surface. The type III interaction mode with a buried surface area of 360 Å2 involves contacts between helices α2 and α3 of one surface and the α1-α4 loop on another surface [167,182]. NMR and analytical centrifugation studies show that ASCPYD polymer formation is favored in the presence of salt. Cryo-EM and solid-state NMR experiments conducted on mouse ASCPYD with 71.8% sequence similarity to human ASCPYD reveal very similar polymer structures [182,194].
4.2. Structural Details of ASCCARD and Its Self-Association
CARDs adopt a conserved six-helix bundle fold and exclusively exhibit helix α1 divided into two small fragments, α1a and α1b, connected by a hinge [183,195,196]. Apaf-1 (Apoptotic protease activating factor), NOD1, ICEBERG, and RAIDD (RIP-associated ICH1/CED3-homologous protein with a death domain) are structurally homologous proteins, and structural comparison of their CARDs (Figure 5A) indicates differences in the length and orientation of the helices [183]. Among these, NOD1CARD exhibits an extended long helix composed of helices α5 and α6 due to their close proximity [183]. Electrostatic surface analysis of these CARDs shows polarized distribution of basic and acidic surfaces, which dictates specific protein–protein interactions Figure 5B [197]. NMR experiments reported the absence of a fragmented helix α1, variability in length and orientation of the helices and evenly distributed charge in the electrostatic surface of ASCCARD [183] (Figure 4B). ASCCARD self-oligomerizes with a dissociation constant of 50 μM [112,198]. This NMR study indicates that residues located in the turn preceding helix α1 and helices α2, α3, α5, and α6 are involved in the self-association of ASCCARD, and generate three contact regions involving; (1) the N-terminus of helix α1 and C-terminus of helix α6; (2) the C-terminus of helix α5 and the N-terminus of helix α6; and (3) helices α2 and α3 [190]. Negatively stained TEM images illustrate that ASCCARD assembles into two types/levels of filaments; ~3.4 ± 0.5 nm wide filaments that self-assemble into ~10 ± 0.5 nm wide bundles of filaments [190,198]. These NMR and TEM studies conclude that ASCCARD plays a key role in the structure and stabilization of ASC filaments [190].
Figure 5.

Structure comparison of caspase-activation and recruitment domains (CARDs) from different origins. (A) Superposition of Apaf-1-CARD (dark blue) [195] and iceberg (light blue) [199]. Superposition of RAIDD-CARD (pink) [200] and NOD-1-CARD (purple) [201]. Helix 1 (cylinder) in RAIDD-CARD, although not fragmented, is substantially bent and is shown as two cylinders; (B) electrostatic surface representation of CARDs in the same orientation as displayed in A. This figure is adapted from Figure S4 of [183].
In addition, cryo-EM studies reveal that ASCCARD can also polymerize into a helical tube-like filament with a diameter of ~8 nm and 3.6 subunits per turn, stabilized by type I, II, and III interactions [190,198,202,203]. The most predominant type I interaction involves charge–charge contacts between helix α2 (R119) in the surface of one protomer, and helices α1 and α4 (E130, D134 and R160) of the adjacent protomer. Hydrophobic residues such as W169 and Y187 participate in type II and III interactions. The type III interaction is also dominated by charge–charge interactions between R160 of helix α4, and D143 and E144 of helix α3 [202]. In the case of CARD polymerization, mutations involving residues that participate in type I (R119D, N128A/E130R, and D134K), type II (W169G, Y187A, Y187K), and type III interactions (D143K/E144K and R160E) completely abolish filament formation [202]. Analogously, mutations of E130, W131, and D134 by alanine impede the ability to oligomerize and thus ASC foci formation. These mutants form instead short and thin filaments as compared to WT ASCCARD. In addition, non-conservative mutations of negatively charged residues such as E130, D134, D191, and E193 by arginine completely impede filament formation [190].
4.3. ASC Filament Formation
TEM analysis of the dimensions of filaments and filament bundles formed by ASCFL and the individual domains, PYD and CARD, indicate that both domains form an integral part of the ASC filament, thus elevating the role of ASCCARD in filament formation, which has not been recognized in different studies of the truncated protein carrying only the PYD domain. In high-resolution TEM images, it is possible to discern the presence of stacked rings with an average diameter of 5 ± 0.6 nm, close to the dimensions of the experimentally-derived model of human ASCFL dimer of 6 nm [198]. In addition, single-molecule FRET experiments also reported that both domains in ASC form fibrils in which the CARD folds back onto the PYD domain [204]. These results altogether show that the ASC dimer serves as a building block for ASC oligomerization, and both PYD and CARD domains are crucial for filament assembly. These latter studies proposed that speck formation has two levels of compaction; firstly, type I interactions mediates homophilic PYD-PYD and CARD-CARD binding, and secondly, type II and III interactions organize ASC into larger assemblies [182]. Computational and FRET studies suggest that ASC speck formation is not simply unspecific aggregation, but instead self-association follows an organized scaffold [182]. To confirm the involvement of both PYD and CARD domains in ASC speck formation, mutagenesis studies have been conducted. Single-point mutations in ASC (human) of residues important for the specific domain interactions such as E13A, E19A, K21A, K26A, R41A, D48A, D51A, L68A, L73A, in the PYD region, and M159A and R160A in the CARD, disrupt filament as well as ASC speck formation when present in ASCFL [112,182,191,192,205,206]. Double mutations (K26A-R160A and L68A-R160A) generated in both CARD and PYD domains result in an inability to form filaments as well as ASC specks [182]. These data suggest that speck formation is due to the individual homophilic interactions mediated by the PYD and CARD. Altogether mutational, dynamics and structural studies of human ASC propose that speck formation is based upon two levels of compaction: One level involving a main type of homophilic interaction between PYD-PYD and CARD-CARD, and a second level organized by other interaction modes (e.g., type II and III interactions) [182].
These results match previous NMR studies on the 3D structure determination of ASC and its proposed model for polymerization indicating that the PYD-PYD and CARD-CARD domains are positioned in a confined space so that they do not cause steric interference with the binding interface of each other [183]. Dynamics resulting from a slightly structured linker lead to a back-to-back orientation of the two domains that increase the accessible space to facilitate the interaction of both PYD and CARD with the PYD of the sensor and the CARD of procaspase-1 [183,190].
4.4. Regulation of ASC
4.4.1. Regulation of ASC Mediated by ASC2
Humans encode for 10–13 kD single domain PYD-only proteins (POPs) such as POP1/ASC2, POP2 and POP3, and CARD-only proteins (COPs) such as Pseudo-ICE/COP, ICEBERG, INCA. ASC2 shares 63% sequence identity with ASCPYD [189,207,208,209], interferes with PYD-PYD interactions of inflammatory proteins, and has been shown to be crucial for modulating NF-κB and pro-caspase-1 regulation [208]. ASC2 binds to ASCPYD with KD = 4.08 ± 0.52 μM and serves as a negative regulator of ASC polymerization [209]. The L25A mutant of human ASCPYD is capable of interacting with ASC2 (KD= 3.81 ± 0.8 μM) via the α1–α4 interface, which indicates that L25A mutation does not affect ASCPYD-ASC2 interaction at least in one of the possible interacting interfaces [209]. Site-directed mutagenesis of residues located in helices α2 and α3 of human ASCPYD (K21, L25, K26, P40, and R41) disrupts ASCPYD self-association without disturbing the hydrophobic pocket, thus indicating that ASC2 binding site on ASCPYD is different from the site of self-association [191,209]. ASC2 displays a different orientation of helices α2 and α4 and has a disordered α3 helix. ASC2PYD and ASCPYD share similar 3D structures (RMSD = 1.5 Å) with comparable charge distributions across the surface [209]. NMR data indicate that the positively charged residues, K21 and R41, located on helices α2 and α3 of ASC2 interact via the type I interaction with the negatively charged residues D6, E13, D48, and D54 located on helices α1 and α4 of ASCPYD [209].
4.4.2. Post-Translational Modifications of ASC
Phosphorylation-dephosphorylation events are also important for ASC oligomerization and its activity. Tyrosine kinase-mediated phosphorylation of human ASC at Y60, Y137, and Y146 (Y144 in murine ASC) is necessary for inflammasome assembly and subsequent inflammatory response [210]. Similarly, the dephosphorylation of ASC tyrosine residues is also essential in the activation of the NLRP3 inflammasome. For example, it has been reported that the compound phenylarsine oxide (PAO), a tyrosine phosphatase (PTPase) inhibitor, suppresses ASC oligomerization and speck formation in LPS-primed human THP-1 cells by targeting the self-association nucleation step [210].
Differential ubiquitination of ASC by the K63 ubiquitin chain or linear ubiquitin plays an important role in ASC inflammasome activation. A ubiquitination enzyme complex LUBAC (linear ubiquitin chain assembly complex), which consists of HOIL-1, HOIP, and SHARPIN (Shank-associated RH domain-interacting protein) proteins, participates in linear ubiquitination of ASC via HOIL-1, HOIP E3 ligase activity [211,212] providing an activation signal. Another study reports that MAVS protein (mitochondrial antiviral signaling protein) recruits an E3 ligase, TRAF3 (TNF receptor-associated factor 3), that promotes ubiquitination of ASC at K174 position, which in turn increases ASC speck formation and secretion of IL-1β in response to viral infection [213]. In addition, it was found that a mitochondrial E3 ubiquitin ligase (Mul1 or MAPL or MULAN) abolishes inflammasome activation by K48-linked ubiquitination and subsequent proteasomal degradation of ASC [214].
5. Caspase-1
5.1. Structure and Activation of Caspase-1
Caspase-1 (ICE; interleukin 1β-converting enzyme) is an inflammatory initiator that belongs to the aspartate-specific cysteine protease family [215,216,217]. It is expressed as a 404 amino acid-long inactive monomeric form called procaspase-1 zymogen, which is converted into a catalytic active form by autoproteolysis upon proximity-induced association mediated by the macromolecular organization of the inflammasome or ASC pyroptosome [218,219,220,221]. Procaspase-1 consists of one prodomain (or propeptide) CARD (1–119) that interacts with upstream adaptor proteins and a catalytic domain consisting of subunits p20 (120–297) and p10 (317–404) [167,222,223]. Caspase-1 activation involves proteolytic removal of the N-terminal CARD and 19 residues of the interdomain linker (298–316) connecting the p20 and p10 subunits [218,224]. Although the catalytic residues C285 and H237 reside in the p20 subunit, both subunits are essential for the activity [225]. X-ray crystallographic studies revealed that a tetramer of two p20/p10 heterodimers is considered as the functional form [223,226]. In contrast, recent cellular studies have shown that caspase-1FL (p46) and transient species p33/p10 are dominant in the initial response to inflammasome assembly [219]. According to these studies, caspase-1FL (p46) is recruited to the inflammasome via CARD-CARD interaction and generates an active p46 dimer, which then is self-processed and enables the cleavage of the linker connecting the p20 and p10 subunits to generate p30/p10 active species. Subsequently, the separation of the CARD domain linker (CDL) from p33/p10 releases the unstable p20/p10 tetramer (catalytic domain) from the ASC-caspase-1 complex, leading to the formation of the caspase-1 active form, therefore triggering the inflammatory response [219]. Activated caspase-1 facilitates the maturation of pro-IL-1β and pro-IL-18 into their bioactive forms IL-1β and IL-18, respectively [227,228] (Figure 6). Cytokine IL-1β induces the proliferation, activation, and differentiation of immune cells and facilitates phagocytosis, degranulation, and oxidative burst activity [229,230]. IL-18 is an inducer of IFN-γ and is involved in the activation and differentiation of various T-cell populations [231,232]. In addition to cytokine production, caspase-1 also cleaves gasdermin D (GSDMD) into two subunits of approximately similar size: N- and C-terminal halves. The GSDMDNterm creates pores in the plasma membrane that are involved in cytokine secretion and facilitates cell-death by pyroptosis [224,233,234,235,236,237] (Figure 6). It has been shown that before CDL cleavage, dimerized caspase-1 retains its catalytic activity towards pro-IL-1β, pro-IL-18, and pro-gasdermin D, but once the CDL is detached caspase-1 protease activity deteriorates [219].
Figure 6.

Schematic representation of NLRP3 and AIM2 inflammasomes activation and assembly. Pathogen-associated molecular patterns such as LPS, crystalline/particulate ligands, K+ efflux, and ROS trigger the activation of NLRP3. TLR initiates the activation and nuclear translocation of NF-κB, which increases the synthesis of NLRP3 and IL-1β and IL-18 cytokines. AIM2 detects viral and bacterial dsDNA in the cytosol. Assembly of NLRP3 or AIM2 with ASC and procaspase-1 leads to the proximity-induced autoproteolytic maturation of caspase-1, functionalization of IL-1β and IL-18, and pyroptosis cell death mediated by the N-terminal fragment of gasdermin D. Inhibitors of NLRP3, ASC, caspase-1, and AIM2 are shown blue. The reference sources corresponding to the negative-staining electron micrographs are shown in square brackets in each image: ASCFL adapted from Figure 6 of [198], filamentous NLRP3PYD-NBD-ASCPYD binary complex adapted from Figure 2 of [167]; filament of AIM2FL from Figure 6 of [171], AIM2FL filament with 600 bp dsDNA adapted from Figure 3 (Copyright National Academy of Science) of [174], and His-GFP-caspase-1CARD/ASCFL/AIM2PYD ternary complex adapted from Figure 6 [167].
When separated from the rest of the protein, caspase-1CARD is able to polymerize into left-handed helical-tube macrostructures comprising four subunits per turn and with inner and outer diameters of ~10 Å and ~80 Å, respectively [231,238]. The formation of caspase-1CARD filament also involves the interaction between the three interfaces [231]. Caspase-1CARD filament shares helical symmetry with MAVSCARD filament and Myddosome DD complex. Fluorescence polarization results suggest that caspase-1CARD polymerization increases in the presence of ASCCARD or ASCFL [167]. Oligomerized ASCCARDs nucleate procaspase-1 through CARD-CARD interaction thus serves as a platform for polymerization, autocleavage, and caspase-1 activation [167,239,240,241].
5.2. Negative Regulation of Caspase-1 Activation
CARD-only proteins (COPs) inhibit inflammasome assembly and cytokine activation [242,243,244]. These inhibitors include COP-1 (Pseudo-ICE/CARD16), INCA (CARD17), and ICEBERG (CARD18), and share high sequence identity with caspase-1 CARD, i.e., 92%, 81%, and 53%, respectively [199,231,244,245]. COP-1 and ICEBERG can self-associate and form filaments, whereas INCA harbors monomer conformation [231,246]. In vitro and in vivo experiments show that ICEBERG is involved in the negative feedback of caspase-1 activation, and therefore suppresses IL-1β secretion [199]. ICEBERG has the ability to inhibit the interaction between RIP2 (Receptor-interacting protein 2, also known as or RIPK2 or RICK) and caspase-1CARD, and also dissociates the preformed RIP2:caspase-1 complex [199].
It was proposed that the negatively charged surface of ICEBERG exhibits competitive binding for the positively charged surface of caspase-1CARD with the upstream activator RIP2, which has a negatively charged patch [199]. Another study found that ICEBERG is not able to interact with RIP2, but COP1 can do so [231,246,247,248,249]. In vivo assays showed that both ICEBERG and COP1 hamper the binding of RIP2 to caspase-1 and reduce IL-1β expression by ~80% and 100%, respectively [246]. Two mutations, D27G and R45D, were created in caspase-1 in order to mimic the polypeptide sequence of INCA and ICEBERG [245]. These mutants were unable to activate NF-κB signaling due to loss of caspase-1 CARD-CARD interaction [245].
It has been reported as well that INCA inhibits caspase-1CARD polymerization at nanomolar concentration even in the presence of ASCCARD. The mechanism proposed for this inhibition involves the capping of the growing caspase-1CARD filament via CARD-CARD interaction, thus blocking the binding of upcoming caspase-1 molecules. This process would abrogate full polymerization of the caspase and subsequent autoactivation [231]. On the other hand, it was found that ICEBERG neither interacts with caspase-1CARD nor inhibits NLRP3 inflammasome activation, caspase-1 oligomerization and its activity [231]. These findings suggest that cellular and/or environmental factors may influence ICEBERG-mediated inflammasome inhibition. Therefore, comprehensive studies are required to unravel the structural and functional mechanisms governing these inhibitory processes [231].
6. Formation of Inflammasome Assembly
6.1. NLRP3-ASC Interaction
Electron microscopy results reveal that the localization of NLRP3PYD at one end of the ASCPYD filament indicates the nucleation of ASCPYD polymerization. However, the PYD-NBD (NLRP3ΔLRR or NLRP3PYD-NBD) fragment of NLRP3 is more efficient in triggering ASCPYD polymerization than the monomeric NLRP3PYD, suggesting that oligomerization by NLRP3 is crucial for ASCPYD polymerization. ASCPYD filament formation monitored by fluorescence polarization (FP) in the presence of different molar ratios of NLRP3PYD-NBD and ASCPYD reveal that one molecule of NLRP3ΔLRR can stimulate the polymerization of up to 1600 ASCPYD molecules [167]. NMR studies on the homotypic interaction between ASCPYD and NLRP3PYD reveal the involvement of two opposite surfaces: i.e., helices α1-α4 (R10, Y11, E13, D14, V18-L20, A47, D48, V50, D51, K84, and D88) and α5 helix (T4, G35, I37, F59, G61, E63, W66, A67, V70, W71, A74, E89, and K91). Further, based on NMR titration results and docking experiments, four different types of binding interfaces have been predicted for the ASCPYD-NLRP3PYD interaction that include ASC α1-α4/NLRP3 α1-α4, ASC α1-α4/NLRP3 α5, ASC α2-α3/NLRP3 α1-α4, and ASC α2-α3/NLRP3 α5 [112]. ASCPYD uses the same interfaces for self-association and binding to NLRP3PYD [112]. The interaction between ASCPYD and NLRP3PYD could result in the formation of hexameric ring structures stabilized by E15, K23, E64, and D82 residues of NLRP3PYD [112]. Similar ring architectures were also reported for other proteins assemblies such as the apoptosome and oligomeric forms of NLRP1 and NLRC4 with variable rotational symmetries [28,250,251]. Furthermore, based on NMR chemical shift perturbation results, amino acids L25, V30, and L45 were identified to mediate in ASCPYD-NLRP3PYD binding, thus highlighting the importance of hydrophobic interactions [112].
6.2. Interaction of ASC with Procaspase-1 and Formation of the NLRP3 Inflammasome
ASCCARD is an integral part of the ASC filament and participates in speck formation [183]. In addition, co-expression of ASCCARD with caspase-1 can also form foci similar to those formed by ASCFL [206]. Procaspase-1CARD has one negatively and one positively charged surface oriented in opposite sides of the domain, which facilitates the type I interaction that is prominent in the recruitment of procaspase-1CARD by ASCCARD. Mutational studies reported key residues responsible for the formation of foci: R10 (α1), D27 (α2), E41 and K42 (α3), R55 and D59 (α4) on caspase-1CARD, and R125, E130, D134, Y137, E144, R160, and D191 on ASCCARD [206,245,252]. The mutation of caspase-1CARD residues D27 and R55 completely interrupts ASC-caspase-1 signaling [206]. It is interesting to note that except for residues D143 and Y146, the rest of the mutants designed to perturb the type I interaction abolish foci formation. However, these ASC mutants can interact with caspase-1CARD and show oligomeric assemblies, albeit lacking the ability to propagate active signaling platforms, which results in concomitant loss of IL-1β secretion [206]. Mutations of residues D143 and Y146 do not destabilize the ASCCARD structure and allow foci formation, thus reflecting that a network of side-chain interactions might stabilize ASCCARD-ASCCARD binding even in the presence of mutations in the interface [196,206,252]. These results point out the crucial role of ASCCARD in foci formation and ASC-dependent inflammasome signaling [196,206,252].
The type III interaction (R45) is essential for caspase-1CARD auto-oligomerization and recruitment of RIP2 [245,252]. D27 (type I interaction) mutant of caspase-1CARD does not compromise auto-oligomerization; however, it fails to activate NF-κB signaling [206,245]. Similarly, caspase-1CARD R45 mutant can interact with ASC without affecting proteolytic activation, but fails to trigger NF-κB signaling [245]. These findings suggest the importance of two oppositely charged surfaces and the synergistic effect of R45 and D27 on RIP2-mediated activation of NF-κB signaling [206,245,252].
Immunoprecipitation and EM studies of the NLRP3 inflammasome triggered by monosodium urate (MSU) crystals in THP-1 cells indicate the formation of filamentous structures that cluster into ball-of-yarn-like particles upon overnight incubation [167]. Similarly, expression of eGFP-ASC in COS-1 cells against anti-ASC primary antibodies reveal the formation of a densely packed gigantic perinuclear punctum (~1–2 μm) in each cell [167]. Analogously to the AIM2-PYD/ASC/Caspase-1CARD assembly, a macro protein complex formed by NLRP3PYD/ASCFL/Caspase-1CARD also assembles into initial star-shaped structures [68,167,253]. Molecular modeling based on NMR data and supported by in-cell immunoprecipitation and in vitro reconstitution suggest that ASCCARD monomers could form 6–7 member ring structures via the type I interaction to which procaspase-1CARDs could stack, amplifying the disk-like structure while leaving the ASCCARDs accessible for interaction through type I, II, and III interactions [252]. The stacking of ASCFL onto this ring creates an additional ring of PYDs below the CARDs. Overall, this arrangement would help PYD:PYD and CARD:CARD self-association and its interaction with the NLRP3 and AIM2 sensors, and procaspase-1 [252]. The systematic formation of NLRP3-inflammasome has been illustrated in Figure 6.
6.3. Interaction of AIM2 with ASC and Formation of the AIM2 Inflammasome
Upstream cytosolic dsDNA sensor AIM2 induces the polymerization of downstream ASC via homotypic PYD-PYD interactions, which further activate procaspase-1 [167,238]. Filaments formed by AIM2PYD and ASCPYD exhibit similar helical symmetry, overall dimensions, and subunit organization [167,171,174].
EM results along with modeling studies show that the AIM2PYD filament nucleates the polymerization of the ASCPYD filament [174], resulting in the localization of the latter at the end of former [167]. Thus, although ASCPYD can self-polymerize, the presence of AIM2PYD enhances this process. Fluorescence polymerization experiments suggest that both AIM2PYD and the complex between AIM2FL and dsDNA participate in ASCPYD filament nucleation. Although it was found that the length of dsDNA regulates the self-association and propagation of AIM2 and ASCPYD polymerization, both of them can accelerate their assembly irrespectively of the presence or absence of dsDNA [174]. It has been suggested that AIM2PYD-ASCPYD assembly generates a three-stage continuous signal amplification in which AIM2PYD, ASCPYD, and AIM2PYD-ASCPYD complex filaments are all persistent. In order to initiate AIM2PYD-ASCPYD assembly, a critical concentration threshold is required. Both AIM2PYD and AIM2FL can induce ASCPYD polymerization in a concentration-dependent manner and such ability is augmented by 4-fold in the presence of dsDNA [174]. FRET assays reveal that pre-assembled ASCPYD polymers accelerate AIM2PYD polymerization as well as the assembly of AIM2FL on dsDNA, resulting in the regulation of the AIM2-ASCPYD assembly by positive feedback loops. The AIM2PYD/FL-ASCPYD complex filament is highly stable and once formed, biochemical data suggest that it cannot be disassembled [174].
6.4. AIM2 Ternary Complex: AIM2PYD:ASCFL:GFP-Casp1CARD
To reconstitute the ternary complex, His-MBP-AIM2PYD, His-MBP-ASCFL, and His-GFP-caspase-1CARD were mixed in a ratio of 1:1:3 and incubated with TEV protease to remove the MBP tags [167]. After purification of the ternary complex, AIM2 and ASC were subjected to immunogold labeling, whereas caspase-1CARD was labeled with Ni-NTA-nanogold conjugate. EM results reveal the formation of a star-shaped ternary complex in which AIM2PYD could nucleate the short filaments of ASCFL [167]. ASC subunits localize at the center of the complex-forming short filaments or rings, whereas the concomitant polymerization of AIMPYD and ASCPYD form long filaments. These results point to a critical role of ASCCARD in the control of ASC supramolecular assemblies, suggesting distinctive structural features of the latter compared to ASCPYD [198]. In the ternary complex, caspase-1CARD is observed along the arms of the stars, possibly polymerizing via interaction with ASCCARD [167]. The presence of the flexible linker connecting the PYD and CARD in ASC could facilitate the interaction with caspase-1CARD [183]. The formation of the AIM2 and NLRP3 inflammasomes has been illustrated in Figure 6.
7. Concluding Remarks and Perspective
Inflammasomes provide host defense from pathogens by means of IL-1β and IL-18 maturation and secretion. The complex and high-order oligomeric nature of inflammasome components pose significant challenges for protein expression by recombinant methods and for successful purification. Moreover, size and shape heterogeneity deter detailed structural investigation. However, recent advancements have provided new insights into the structure, function, activation, and regulation of NLRP3 and AIM2 inflammasomes. Structural techniques, including NMR, X-ray crystallography, and cryo-EM, in association with biophysical, biochemical, and cell-based studies, have revealed high-resolution structures that help to understand the molecular mechanisms of ligand/receptor-driven conformational changes, the release of auto-inhibition, oligomerization, and complex assembly, nucleation-induced polymerization of ASC and caspase-1, and further downstream signaling. PTMs play important roles in the control of inflammasome activation. Hence, PTM dysfunction leads to autoimmune diseases resulting from chronic inflammasome activation.
In spite of recent progress in inflammasome research, several fundamental questions still remain unanswered. For instance, the ligand-induced activation mechanism of NLRP3 and its contribution in pyroptosis, which in turn can cause serious injury to vital organs, are still unclear [254]. Little is known about the exact mechanism of phosphorylation and ubiquitin-mediated controlled activation of inflammasome components. AIM2 can detect damaged or mislocalized self-DNA, which is released into the cytosol due to the loss of nuclear envelope integrity resulting from the perturbation of cellular homeostasis [255,256]. However, detailed information on how AIM2 regulates the detection of self as well as foreign DNA is lagging. Similarly, NLRP3 is also involved in self-DNA sensing, but the exact mechanism of self-DNA induced activation is not known [257,258]. In addition to NLRP3 activation, NEK7 also regulates microtubule dynamic and spindle assembly during the cell cycle [259]. How this protein limits NLRP3 activity without affecting AIM2 activation during cell cycle progression is unknown. Potential roles of AIM2 in inflammasome-independent processes, such as neuronal morphology, anxiety, and memory of mice will uncover new functions of AIM2 [260]. Although remarkable progress has been made on the structure of inflammasome components and mechanism of ASC speck formation, more structural details are required to elucidate the whole ASC speck assembly consisting of receptor, the ASC adaptor, and caspase-1. Likewise, super-resolution microscopy studies at the cellular level are required to uncover the formation of endogenous specks and involvement of accessory proteins that contribute to speck size and organized shape in intact cells. In-depth investigation of these fundamental questions will open up new doors for the development of novel therapeutics, and faster and efficient anti-inflammatory therapies for the treatment of associated autoimmune and autoinflammatory diseases. Finally, the determination of specific structural components of SARS-CoV-2 involved in NLRP3 activation and decoding the subsequent downstream pathway that leads to cell death, will aid in the development of potential therapeutics for the treatment of COVID-19 in near future.
Acknowledgments
The authors acknowledge the support from the National Science Foundation Center grant; NSF-CREST Center for Cellular and Biomolecular Machines at the University of California, Merced (NSF-HRD-1547848), and the support from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R15AI146780 and R15AI146780-01S1 to EdA. “The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.”
Acronyms and Abbreviations
| ADP | Adenosine diphosphate |
| AIM2 | Absent in melanoma 2 |
| ALRs | (AIM2)-like receptors |
| Apaf-1 | Apoptotic protease activating factor |
| ASC | Apoptosis-associated speck-like protein containing a caspase-activation and recruitment domain (CARD) |
| Bcl | B-cell lymphoma |
| BIR | Baculovirus IAP (inhibitor of apoptosis protein) repeat |
| BMDCs | Bone marrow derived dendritic cells |
| BRCA1 | Breast cancer type-1 |
| BRCC3 | BRCA1/BRCA2- containing complex, subunit 3 |
| c-FLIP | FLICE-like inhibitory protein |
| CAPS | Cryopyrin-associated periodic syndrome |
| CARD | Caspase-activation and recruitment domain |
| CDL | CARD domain linker |
| CIAS1 | cold induced autoinflammatory syndrome 1 |
| CLR | C-type lectin receptor |
| COPs | CARD-only proteins |
| COS-1 | CV-1 in Origin with SV40 genes |
| COVID-19 | Coronavirus disease 2019 |
| DAMPs | Danger-associated molecular patterns |
| DD | Death domain |
| DEDs | Death-effector domain |
| DEFCAP | Death Effector Filament-forming Ced-4-likeApoptosis Protein |
| DUBs | Deubiquitinating enzyme |
| EM | Electron microscopy |
| FADD | Fas-associated protein with death domain |
| FBXL2 | F-box L |
| FBXO3 | F-box O3 |
| FLICE | Caspase-8/FADD-like IL-1β-converting enzyme |
| FP | Fluorescence polarization |
| FRET | Fluorescence resonance energy transfer |
| GFP | Green-fluorescent protein |
| GLMN | Glomulin |
| GSDMD | Gasdermin D |
| HD | helical domain |
| HDAC | Histone deacetylases |
| HIN | Hematopoietic, Interferon-inducible, Nuclear localization |
| IAPs | Inhibitor of apoptosis proteins |
| IC50 | Inhibitory concentration |
| ICE | Interleukin 1β-converting enzyme |
| ICE | Interleukin-1beta converting enzyme |
| IFI16 | Gamma-interferon-inducible protein |
| IFN | Interferon |
| IKK-γ | Inhibitor of nuclear factor kappa-B kinase subunit gamma |
| IL | Interleukin |
| INCA | Inhibitors of NFAT-calcineurin association |
| IPAF | Ice protease activating factor |
| IRF4 | Interferon regulatory factor |
| ITC | Isothermal titration calorimetry |
| JNK | Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| LRRs | Leucine- rich repeat |
| LTA | Lipoteichoic acid |
| LUBAC | linear ubiquitin chain assembly complex |
| MALS | Multiangle light scattering |
| MALT1 | Mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MAPL | Mitochondria-associated protein ligase |
| MARCH7 | Membrane associated ring finger (C3HC4) 7 |
| MAVS | Mitochondrial antiviral-signaling protein |
| MBP | Maltose-binding protein |
| MLKL | Mixed lineage kinase domain like pseudokinase |
| MNDA | Myeloid cell nuclear differentiation antigen |
| MSU | Monosodium urate |
| Mul1 | Mitochondrial E3 Ubiquitin Protein Ligase 1 |
| NAIP | NLR family of apoptosis inhibitory protein |
| NALP | NACHT, LRR and PYD domains-containing proteins |
| NBD | Nucleotide binding domain |
| NEK7 | NIMA-related kinase 7 |
| NEMO | NF-kappa-B essential modulator |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRC | NLR family CARD domain-containing protein |
| NLRP | Nod-like receptor protein |
| NLRP3 | NLR family pyrin domain containing 3 |
| NLRs | NOD-like receptors |
| NMR | Nuclear magnetic resonance |
| NOD | Nucleotide-binding oligomerization domain |
| OB | Oligonucleotide/oligosaccharide binding) |
| PAMPs | Pathogen-associated molecular patterns |
| PAO | Phenylarsine oxide |
| PKA | protein kinase A |
| PKD | protein kinase D |
| POPs | PYD-only proteins |
| PP2A | protein phosphatase 2 A |
| PRRs | Pathogen recognition receptors |
| PTM | Post-translation modifications |
| PTPase | Protein tyrosine phosphatase |
| PTPN22 | Protein tyrosine phosphatase nonreceptor type 22 |
| PYCARD | PYD and CARD domain-containing |
| PYD | Pyrin domain |
| PYHIN | Pyrin + HIN |
| PYPAF1 | PYRIN-containing APAF1-like protein 1 |
| RAIDD | RIP-associated ICH1/CED3-homologous protein with a death domain |
| RICK | RIP-like interacting CLARP kinase |
| RIP2 | Receptor-interacting protein 2 |
| RMSD | Root-mean-square deviation |
| RNO | Regulated by nitric oxide |
| ROS | Reactive oxygen species |
| SARS-Cov-2 | Severe acute respiratory syndrome coronavirus 2 |
| SCF | Skp-Cullin-F box |
| SEC | Size-exclusion chromatography |
| SHARPIN | Shank-associated RH domain-interacting protein |
| STAT1 | Signal transducer and activator of transcription |
| TEV | Tobacco etch virus. |
| TH2 | T-helper cell type 2 |
| TLR | Toll-like receptors |
| TMS | Target of Methylation-induced Silencing-1 |
| TNF | Tumor necrosis factor |
| TPA | 12-O-tetradecanoylphorbol-13-acetate |
| TRAF3 | TNF receptor associated factor 3 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| TRIM | Tripartite motif |
| WHD | winged helical domain |
| WHO | World health organization |
Funding
This research was funded by National Science Foundation Center grant; NSF-CREST Center for Cellular and Biomolecular Machines at the University of California, Merced (NSF-HRD-1547848), and the support from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R15AI146780 and R15AI146780-01S1 to EdA. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Akira S., Takeda K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Bauernfeind F., Hornung V. Of inflammasomes and pathogens–sensing of microbes by the inflammasome. EMBO Mol. Med. 2013;5:814–826. doi: 10.1002/emmm.201201771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 5.Zanoni I., Tan Y., Di Gioia M., Broggi A., Ruan J., Shi J., Donado C.A., Shao F., Wu H., Springstead J.R., et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kayagaki N., Wong M.T., Stowe I.B., Ramani S.R., Gonzalez L.C., Akashi-Takamura S., Miyake K., Zhang J., Lee W.P., Muszyński A. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 7.Rathinam V.A., Jiang Z., Waggoner S.N., Sharma S., Cole L.E., Waggoner L., Vanaja S.K., Monks B.G., Ganesan S., Latz E.J., et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010;11:395. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kerur N., Veettil M.V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., Chandran B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broz P., Dixit V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 10.Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Harton J.A., Linhoff M.W., Zhang J., Ting J.P. Cutting edge: CATERPILLER: A large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 2002;169:4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- 12.Inohara N., Nuñez G. The NOD: A signaling module that regulates apoptosis and host defense against pathogens. Oncogene. 2001;20:6473–6481. doi: 10.1038/sj.onc.1204787. [DOI] [PubMed] [Google Scholar]
- 13.Elinav E., Strowig T., Kau A.L., Henao-Mejia J., Thaiss C.A., Booth C.J., Peaper D.R., Bertin J., Eisenbarth S.C., Gordon J.I., et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khare S., Dorfleutner A., Bryan N.B., Yun C., Radian A.D., de Almeida L., Rojanasakul Y., Stehlik C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36:464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vladimer G.I., Weng D., Paquette S.W.M., Vanaja S.K., Rathinam V.A., Aune M.H., Conlon J.E., Burbage J.J., Proulx M.K., Liu Q., et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavarría-Smith J., Vance R.E. The NLRP 1 inflammasomes. Immunol. Rev. 2015;265:22–34. doi: 10.1111/imr.12283. [DOI] [PubMed] [Google Scholar]
- 17.Abderrazak A., Syrovets T., Couchie D., El Hadri K., Friguet B., Simmet T., Rouis M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi: 10.1016/j.redox.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer R., Rauch I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol. Aspects Med. 2020;76:100863. doi: 10.1016/j.mam.2020.100863. [DOI] [PubMed] [Google Scholar]
- 19.Davis B.K., Wen H., Ting J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schroder K., Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 21.Heilig R., Broz P. Function and mechanism of the pyrin inflammasome. Eur. J. Immunol. 2018;48:230–238. doi: 10.1002/eji.201746947. [DOI] [PubMed] [Google Scholar]
- 22.Pawlowski K., Pio F., Chu Z.L., Reed J.C., Godzik A. PAAD–a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem. Sci. 2001;26:85–87. doi: 10.1016/S0968-0004(00)01729-1. [DOI] [PubMed] [Google Scholar]
- 23.Liu T., Zhang L., Joo D., Sun S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017;2:1–9. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weston C.R., Davis R.J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Li D., Ren W., Jiang Z., Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol. Med. Rep. 2018;18:4399–4409. doi: 10.3892/mmr.2018.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones J.W., Kayagaki N., Broz P., Henry T., Newton K., O’Rourke K., Chan S., Dong J., Qu Y., Roose-Girma M., et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broz P., Newton K., Lamkanfi M., Mariathasan S., Dixit V.M., Monack D.M. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against. Salmonella Exp. Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faustin B., Lartigue L., Bruey J.M., Luciano F., Sergienko E., Bailly-Maitre B., Volkmann N., Hanein D., Rouiller I., Reed J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 29.Poyet J.L., Srinivasula S.M., Tnani M., Razmara M., Fernandes-Alnemri T., Alnemri E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001;276:28309–28313. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 30.Kayagaki N., Stowe I.B., Lee B.L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q.T., et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 31.Moayeri M., Sastalla I., Leppla S.H. infection, Anthrax and the inflammasome. Microbes Infect. 2012;14:392–400. doi: 10.1016/j.micinf.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyden E.D., Dietrich W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 33.Ewald S.E., Chavarria-Smith J., Boothroyd J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014;82:460–468. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin Y., Mailloux C.M., Gowan K., Riccardi S.L., LaBerge G., Bennett D.C., Fain P.R., Spritz R.A. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 2007;356:1216–1225. doi: 10.1056/NEJMoa061592. [DOI] [PubMed] [Google Scholar]
- 35.Grandemange S., Sanchez E., Louis-Plence P., Mau-Them F.T., Bessis D., Coubes C., Frouin E., Seyger M., Girard M., Puechberty J., et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis) Ann. Rheum. Dis. 2017;76:1191–1198. doi: 10.1136/annrheumdis-2016-210021. [DOI] [PubMed] [Google Scholar]
- 36.Zhong F.L., Mamaï O., Sborgi L., Boussofara L., Hopkins R., Robinson K., Szeverényi I., Takeichi T., Balaji R., Lau A., et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell. 2016;167:187–202. doi: 10.1016/j.cell.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Allen I.C., Scull M.A., Moore C.B., Holl E.K., McElvania-TeKippe E., Taxman D.J., Guthrie E.H., Pickles R.J., Ting J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poeck H., Bscheider M., Gross O., Finger K., Roth S., Rebsamen M., Hannesschläger N., Schlee M., Rothenfusser S., Barchet W., et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1β production. Nat. Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 39.Mariathasan S., Weiss D.S., Newton K., McBride J., O’Rourke K., Roose-Girma M., Lee W.P., Weinrauch Y., Monack D.M., Dixit V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 40.Duncan J.A., Gao X., Huang M.T.H., O’Connor B.P., Thomas C.E., Willingham S.B., Bergstralh D.T., Jarvis G.A., Sparling P.F., Ting J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009;182:6460–6469. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 42.Hornung V., Bauernfeind F., Halle A., Samstad E.O., Kono H., Rock K.L., Fitzgerald K.A., Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9:847. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B.T., Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B.L., Rajendiran T.M., Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tschopp J., Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 46.Keddie S., Parker T., Lachmann H.J., Ginsberg L. Cryopyrin-associated periodic fever syndrome and the nervous system. Curr. Treat. Options Neurol. 2018;20:43. doi: 10.1007/s11940-018-0526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawkins P.N., Lachmann H.J., Aganna E., McDermott M.F. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Off. J. Am. Coll. Rheumatol. 2004;50:607–612. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 48.Aróstegui J.I., Lopez Saldaña M.D., Pascal M., Clemente D., Aymerich M., Balaguer F., Goel A., Fournier del Castillo C., Rius J., Plaza S., et al. Rheumatism, A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: Novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 2010;62:1158–1166. doi: 10.1002/art.27342. [DOI] [PubMed] [Google Scholar]
- 49.Goldbach-Mansky R., Shroff S.D., Wilson M., Snyder C., Plehn S., Barham B., Pham T.H., Pucino F., Wesley R.A., Papadopoulos J.H. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Off. J. Am. Coll. Rheumatol. 2008;58:2432–2442. doi: 10.1002/art.23620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halle A., Hornung V., Petzold G.C., Stewart C.R., Monks B.G., Reinheckel T., Fitzgerald K.A., Latz E., Moore K.J., Golenbock D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008;9:857. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larsen C.M., Faulenbach M., Vaag A., Ehses J.A., Donath M.Y., Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–1668. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moossavi M., Parsamanesh N., Bahrami A., Atkin S.L., Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer. 2018;17:158. doi: 10.1186/s12943-018-0900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldbach-Mansky R., Dailey N.J., Canna S.W., Gelabert A., Jones J., Rubin B.I., Kim H.J., Brewer C., Zalewski C., Wiggs E.J., et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N. Engl. J. Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki T., Franchi L., Toma C., Ashida H., Ogawa M., Yoshikawa Y., Mimuro H., Inohara N., Sasakawa C., Nuñez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amer A., Franchi L., Kanneganti T.D., Body-Malapel M., Özören N., Brady G., Meshinchi S., Jagirdar R., Gewirtz A., Akira S., et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 2006;281:35217–35223. doi: 10.1074/jbc.M604933200. [DOI] [PubMed] [Google Scholar]
- 56.Franchi L., Amer A., Body-Malapel M., Kanneganti T.D., Özören N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A., et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 57.Romberg N., Al Moussawi K., Nelson-Williams C., Stiegler A.L., Loring E., Choi M., Overton J., Meffre E., Khokha M.K., Huttner A., et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014;46:1135–1139. doi: 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Canna S.W., de Jesus A.A., Gouni S., Brooks S.R., Marrero B., Liu Y., DiMattia M.A., Zaal K.J., Sanchez G.A.M., Kim H., et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014;46:1140–1146. doi: 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang P., Zhu S., Yang L., Cui S., Pan W., Jackson R., Zheng Y., Rongvaux A., Sun Q., Yang G. Nlrp6 regulates intestinal antiviral innate immunity. 2015, 350, 826–830. Science. 2015;350:826–830. doi: 10.1126/science.aab3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leng F., Yin H., Qin S., Zhang K., Guan Y., Fang R., Wang H., Li G., Jiang Z., Sun F., et al. NLRP6 self-assembles into a linear molecular platform following LPS binding and ATP stimulation. Sci. Rep. 2020;10:1–10. doi: 10.1038/s41598-019-57043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hara H., Seregin S.S., Yang D., Fukase K., Chamaillard M., Alnemri E.S., Inohara N., Chen G.Y., Núñez G. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates Gram-positive pathogen infection. Cell. 2018;175:1651–1664. doi: 10.1016/j.cell.2018.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen G.Y., Liu M., Wang F., Bertin J., Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaki M.H., Man S.M., Vogel P., Lamkanfi M., Kanneganti T.D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl. Acad. Sci. USA. 2014;111:385–390. doi: 10.1073/pnas.1317643111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanaja S.K., Rathinam V.A., Fitzgerald K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015;25:308–315. doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jeru I., Duquesnoy P., Fernandes-Alnemri T., Cochet E., Yu J., Lackmy-Port-Lis M., Grimprel E., Landman-Parker J., Hentgen V., Marlin S., et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA. 2008;105:1614–1619. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bürckstümmer T., Baumann C., Blüml S., Dixit E., Dürnberger G., Jahn H., Planyavsky M., Bilban M., Colinge J., Bennett K.L., et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009;10:266. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 67.Fernandes-Alnemri T., Yu J.W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roberts T.L., Idris A., Dunn J.A., Kelly G.M., Burnton C.M., Hodgson S., Hardy L.L., Garceau V., Sweet M.J., Ross I.L. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 70.de Koning H.D., Bergboer J.G., van den Bogaard E.H., van Vlijmen-Willems I.M., Rodijk-Olthuis D., Simon A., Zeeuwen P.L., Schalkwijk J. Strong induction of AIM 2 expression in human epidermis in acute and chronic inflammatory skin conditions. Exp. Dermatol. 2012;21:961–964. doi: 10.1111/exd.12037. [DOI] [PubMed] [Google Scholar]
- 71.Dihlmann S., Erhart P., Mehrabi A., Nickkholgh A., Lasitschka F., Böckler D., Hakimi M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol. Med. 2014;20:230–237. doi: 10.2119/molmed.2013.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choubey D., Panchanathan R. Absent in Melanoma 2 proteins in SLE. Clin. Immunol. 2017;176:42–48. doi: 10.1016/j.clim.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Man S.M., Karki R., Kanneganti T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016;46:269–280. doi: 10.1002/eji.201545839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diner B.A., Lum K.K., Toettcher J.E., Cristea I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio. 2016:7. doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson K.E., Chikoti L., Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 2013;87:5005–5018. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh V.V., Kerur N., Bottero V., Dutta S., Chakraborty S., Ansari M.A., Paudel N., Chikoti L., Chandran B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 2013;87:4417–4431. doi: 10.1128/JVI.03282-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roy A., Dutta D., Iqbal J., Pisano G., Gjyshi O., Ansari M.A., Kumar B., Chandran B. Nuclear innate immune DNA sensor IFI16 is degraded during lytic reactivation of Kaposi’s sarcoma-associated herpesvirus (KSHV): Role of IFI16 in maintenance of KSHV latency. J. Virol. 2016;90:8822–8841. doi: 10.1128/JVI.01003-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jakobsen M.R., Bak R.O., Andersen A., Berg R.K., Jensen S.B., Jin T., Laustsen A., Hansen K., Østergaard L., Fitzgerald K.A., et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA. 2013;110:E4571–E4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monroe K.M., Yang Z., Johnson J.R., Geng X., Doitsh G., Krogan N.J., Greene W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caneparo V., Landolfo S., Gariglio M., De Andrea M. The absent in melanoma 2-like receptor IFN-inducible protein 16 as an inflammasome regulator in systemic lupus erythematosus: The dark side of sensing microbes. Front. Immunol. 2018;9:1180. doi: 10.3389/fimmu.2018.01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Loeven N.A., Medici N.P., Bliska J.B. The pyrin inflammasome in host–microbe interactions. Curr. Opin. Microbiol. 2020;54:77–86. doi: 10.1016/j.mib.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park Y.H., Wood G., Kastner D.L., Chae J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 2016;17:914. doi: 10.1038/ni.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jamilloux Y., Magnotti F., Belot A., Henry T. The pyrin inflammasome: From sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018;76:fty020. doi: 10.1093/femspd/fty020. [DOI] [PubMed] [Google Scholar]
- 84.Belkhir R., Moulonguet-Doleris L., Hachulla E., Prinseau J., Baglin A., Hanslik T. Treatment of familial Mediterranean fever with anakinra. Ann. Intern. Med. 2007;146:825–826. doi: 10.7326/0003-4819-146-11-200706050-00023. [DOI] [PubMed] [Google Scholar]
- 85.Moghaddas F., Llamas R., De Nardo D., Martinez-Banaclocha H., Martinez-Garcia J.J., Mesa-del-Castillo P., Baker P.J., Gargallo V., Mensa-Vilaro A., Canna S., et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14–3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017;76:2085–2094. doi: 10.1136/annrheumdis-2017-211473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lacey C.A., Mitchell W.J., Dadelahi A.S., Skyberg J.A. Caspase-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect. Immun. 2018:86. doi: 10.1128/IAI.00361-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Flood B., Oficjalska K., Laukens D., Fay J., O’Grady A., Caiazza F., Heetun Z., Mills K., Sheahan K., Ryan E.J., et al. Altered expression of caspases-4 and-5 during inflammatory bowel disease and colorectal cancer: Diagnostic and therapeutic potential. Clin. Exp. Immunol. 2015;181:39–50. doi: 10.1111/cei.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Carvalho R.V., Andrade W.A., Lima-Junior D.S., Dilucca M., de Oliveira C.V., Wang K., Nogueira P.M., Rugani J.N., Soares R.P., Beverley S.M., et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 2019;26:429–437. doi: 10.1016/j.celrep.2018.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yi Y.S., Son Y.J., Ryou C., Sung G.H., Kim J.H., Cho J.Y. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014:270302. doi: 10.1155/2014/270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gabrielli E., Pericolini E., Luciano E., Sabbatini S., Roselletti E., Perito S., Kasper L., Hube B., Vecchiarelli A. Induction of caspase-11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect. Immun. 2015;83:1940–1948. doi: 10.1128/IAI.02895-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hisahara S., Yuan J., Momoi T., Okano H., Miura M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J. Exp. Med. 2001;193:111–122. doi: 10.1084/jem.193.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang S.J., Sanchez I., Jing N., Yuan J. Dissociation between neurodegeneration and caspase-11-mediated activation of caspase-1 and caspase-3 in a mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2003;23:5455–5460. doi: 10.1523/JNEUROSCI.23-13-05455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Furuya T., Hayakawa H., Yamada M., Yoshimi K., Hisahara S., Miura M., Mizuno Y., Mochizuki H. Caspase-11 mediates inflammatory dopaminergic cell death in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J. Neurosci. 2004;24:1865–1872. doi: 10.1523/JNEUROSCI.3309-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Demon D., Kuchmiy A., Fossoul A., Zhu Q., Kanneganti T.D., Lamkanfi M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014;7:1480–1491. doi: 10.1038/mi.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Williams T.M., Leeth R.A., Rothschild D.E., McDaniel D.K., Coutermarsh-Ott S.L., Simmons A.E., Kable K.H., Heid B., Allen I.C. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015;308:G139–G150. doi: 10.1152/ajpgi.00234.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheng K.T., Xiong S., Ye Z., Hong Z., Di A., Tsang K.M., Gao X., An S., Mittal M., Vogel S.M., et al. Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Investig. 2017;127:4124–4135. doi: 10.1172/JCI94495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eltom S., Belvisi M.G., Stevenson C.S., Maher S.A., Dubuis E., Fitzgerald K.A., Birrell M.A. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: An insight into the pathogenesis of COPD. PLoS ONE. 2014;9:e112829. doi: 10.1371/journal.pone.0112829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.O’Connor W., Harton J.A., Zhu X., Linhoff M.W., Ting J.P. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-κB suppressive properties. J. Immunol. 2003;171:6329–6333. doi: 10.4049/jimmunol.171.12.6329. [DOI] [PubMed] [Google Scholar]
- 99.Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B.G., Fitzgerald K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.He Q., You H., Li X.M., Liu T.H., Wang P., Wang B.E. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac. J. Cancer Prev. 2012;13:1365–1370. doi: 10.7314/APJCP.2012.13.4.1365. [DOI] [PubMed] [Google Scholar]
- 101.Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ghonime M.G., Shamaa O.R., Das S., Eldomany R.A., Fernandes-Alnemri T., Alnemri E.S., Gavrilin M.A., Wewers M.D. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 2014;192:3881–3888. doi: 10.4049/jimmunol.1301974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Py B.F., Kim M.-S., Vakifahmetoglu-Norberg H., Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 104.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 105.World Health Organization, Weekly Operational Update-14 December 2020. [(accessed on 14 December 2020)]; Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update.
- 106.Wu Z., McGoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 107.Chi Y., Ge Y., Wu B., Zhang W., Wu T., Wen T., Liu J., Guo X., Huang C., Jiao Y., et al. Serum cytokine and chemokine profile in relation to the severity of coronavirus disease 2019 in China. J. Infect. Dis. 2020;222:746–754. doi: 10.1093/infdis/jiaa363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Han Y., Zhang H., Mu S., Wei W., Jin C., Tong C., Song Z., Zha Y., Xue Y., Gu G. Lactate dehydrogenase, an independent risk factor of severe COVID-19 patients: A retrospective and observational study. Aging. 2020;12:11245. doi: 10.18632/aging.103372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rodrigues T.S., de Sá K.S., Ishimoto A.Y., Becerra A., Oliveira S., Almeida L., Gonçalves A.V., Perucello D.B., Andrade W.A., Castro R., et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020;218:e20201707. doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bae J.Y., Park H.H. Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 2011;286:39528–39536. doi: 10.1074/jbc.M111.278812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oroz J., Barrera-Vilarmau S., Alfonso C., Rivas G., de Alba E. ASC pyrin domain self-associates and binds NLRP3 protein using equivalent binding interfaces. J. Biol. Chem. 2016;291:19487–19501. doi: 10.1074/jbc.M116.741082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pinheiro A.S., Eibl C., Ekman-Vural Z., Schwarzenbacher R., Peti W. The NLRP12 pyrin domain: Structure, dynamics, and functional insights. J. Mol. Biol. 2011;413:790–803. doi: 10.1016/j.jmb.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hiller S., Kohl A., Fiorito F., Herrmann T., Wider G., Tschopp J., Grütter M.G., Wüthrich K. NMR structure of the apoptosis-and inflammation-related NALP1 pyrin domain. Structure. 2003;11:1199–1205. doi: 10.1016/j.str.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 115.Pinheiro A.S., Proell M., Eibl C., Page R., Schwarzenbacher R., Peti W. Three-dimensional Structure of the NLRP7 pyrin domain insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 2010;285:27402–27410. doi: 10.1074/jbc.M110.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eibl C., Grigoriu S., Hessenberger M., Wenger J., Puehringer S., Pinheiro A.S., Wagner R.N., Proell M., Reed J.C., Page R. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry. 2012;51:7330–7341. doi: 10.1021/bi3007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Su M.Y., Kuo C.I., Chang C.F., Chang C.I. Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS ONE. 2013;8:e67843. doi: 10.1371/journal.pone.0067843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sharif H., Wang L., Wang W.L., Magupalli V.G., Andreeva L., Qiao Q., Hauenstein A.V., Wu Z., Nunez G., Mao Y., et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570:338–343. doi: 10.1038/s41586-019-1295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sušjan P., Roškar S., Hafner-Bratkovič I. The mechanism of NLRP3 inflammasome initiation: Trimerization but not dimerization of the NLRP3 pyrin domain induces robust activation of IL-1β. Biochem. Biophys. Res. Commun. 2017;483:823–828. doi: 10.1016/j.bbrc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 120.Jin T., Huang M., Jiang J., Smith P., Xiao T.S. Crystal structure of human NLRP12 PYD domain and implication in homotypic interaction. PLoS ONE. 2018;13:e0190547. doi: 10.1371/journal.pone.0190547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hafner-Bratkovič I., Sušjan P., Lainšček D., Tapia-Abellán A., Cerović K., Kadunc L., Angosto-Bazarra D., Pelegrin P., Jerala R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018;9:1–18. doi: 10.1038/s41467-018-07573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stutz A., Kolbe C.C., Stahl R., Horvath G.L., Franklin B.S., van Ray O., Brinkschulte R., Geyer M., Meissner F., Latz E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017;214:1725–1736. doi: 10.1084/jem.20160933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Han S., Lear T.B., Jerome J.A., Rajbhandari S., Snavely C.A., Gulick D.L., Gibson K.F., Zou C., Chen B.B., Mallampalli R.K. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J. Biol. Chem. 2015;290:18124–18133. doi: 10.1074/jbc.M115.645549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Spalinger M.R., Kasper S., Gottier C., Lang S., Atrott K., Vavricka S.R., Scharl S., Gutte P.M., Grütter M.G., Beer H.D., et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Investig. 2016;126:1783–1800. doi: 10.1172/JCI83669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bruchard M., Rebé C., Derangère V., Togbé D., Ryffel B., Boidot R., Humblin E., Hamman A., Chalmin F., Berger H., et al. The receptor NLRP3 is a transcriptional regulator of TH 2 differentiation. Nat. Immunol. 2015;16:859–870. doi: 10.1038/ni.3202. [DOI] [PubMed] [Google Scholar]
- 126.Song N., Liu Z.S., Xue W., Bai Z.F., Wang Q.Y., Dai J., Liu X., Huang Y.J., Cai H., Zhan X.Y., et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell. 2017;68:185–197. doi: 10.1016/j.molcel.2017.08.017. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Z., Meszaros G., He W., Xu Y., de Fatima Magliarelli H., Mailly L., Mihlan M., Liu Y., Puig Gámez M., Goginashvili A., et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017;214:2671–2693. doi: 10.1084/jem.20162040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo C., Xie S., Chi Z., Zhang J., Liu Y., Zhang L., Zheng M., Zhang X., Xia D., Ke Y., et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 129.Yan Y., Jiang W., Liu L., Wang X., Ding C., Tian Z., Zhou R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73. doi: 10.1016/j.cell.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 130.Humphries F., Bergin R., Jackson R., Delagic N., Wang B., Yang S., Dubois A.V., Ingram R.J., Moynagh P.N. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 2018;9:1–17. doi: 10.1038/s41467-018-03669-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yen H., Sugimoto N., Tobe T. Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PLoS Pathog. 2015;11:e1005121. doi: 10.1371/journal.ppat.1005121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li M., Shi X., Qian T., Li J., Tian Z., Ni B., Hao F. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-κB and NLRP3 inflammasome activation in macrophages of mice. Int. J. Clin. Exp. Med. 2015;8:17430. [PMC free article] [PubMed] [Google Scholar]
- 133.Walle L.V., Van Opdenbosch N., Jacques P., Fossoul A., Verheugen E., Vogel P., Beyaert R., Elewaut D., Kanneganti T.D., van Loo G., et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Song H., Liu B., Huai W., Yu Z., Wang W., Zhao J., Han L., Jiang G., Zhang L., Gao C., et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016;7:1–11. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Suzuki S., Mimuro H., Kim M., Ogawa M., Ashida H., Toyotome T., Franchi L., Suzuki M., Sanada T., Suzuki T., et al. Shigella IpaH7. 8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl. Acad. Sci. USA. 2014;111:E4254–E4263. doi: 10.1073/pnas.1324021111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shi H., Wang Y., Li X., Zhan X., Tang M., Fina M., Su L., Pratt D., Bu C.H., Hildebrand S., et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hu Z., Zhou Q., Zhang C., Fan S., Cheng W., Zhao Y., Shao F., Wang H.W., Sui S.F., Chai J. Structural and biochemical basis for induced self-propagation of NLRC4. Science. 2015;350:399–404. doi: 10.1126/science.aac5489. [DOI] [PubMed] [Google Scholar]
- 138.Zhang L., Chen S., Ruan J., Wu J., Tong A.B., Yin Q., Li Y., David L., Lu A., Wang W.L., et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science. 2015;350:404–409. doi: 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Feltham R., Vince J.E., Lawlor K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017;6:e124. doi: 10.1038/cti.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee K.H., Kang T.B. The molecular links between cell death and inflammasome. Cells. 2019;8:1057. doi: 10.3390/cells8091057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maelfait J., Vercammen E., Janssens S., Schotte P., Haegman M., Magez S., Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J. Exp. Med. 2008;205:1967–1973. doi: 10.1084/jem.20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moriwaki K., Bertin J., Gough P.J., Chan F.K. A RIPK3–caspase 8 complex mediates atypical pro–IL-1β processing. J. Immunol. 2015;194:1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stammler D., Eigenbrod T., Menz S., Frick J.S., Sweet M.J., Shakespear M.R., Jantsch J., Siegert I., Wölfle S., Langer J.D., et al. Inhibition of Histone Deacetylases Permits Lipopolysaccharide-Mediated Secretion of Bioactive IL-1β via a Caspase-1–Independent Mechanism. J. Immunol. 2015;195:5421–5431. doi: 10.4049/jimmunol.1501195. [DOI] [PubMed] [Google Scholar]
- 144.Bossaller L., Chiang P.I., Schmidt-Lauber C., Ganesan S., Kaiser W.J., Rathinam V.A., Mocarski E.S., Subramanian D., Green D.R., Silverman N., et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012;189:5508–5512. doi: 10.4049/jimmunol.1202121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gringhuis S.I., Kaptein T.M., Wevers B.A., Theelen B., Van Der Vlist M., Boekhout T., Geijtenbeek T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012;13:246. doi: 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 146.Shenderov K., Riteau N., Yip R., Mayer-Barber K.D., Oland S., Hieny S., Fitzgerald P., Oberst A., Dillon C.P., Green D.R., et al. ER stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8-and TRIF-dependent pathway. J. Immunol. 2014;192:2029. doi: 10.4049/jimmunol.1302549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Antonopoulos C., El Sanadi C., Kaiser W.J., Mocarski E.S., Dubyak G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1β via caspase-8 in dendritic cells. J. Immunol. 2013;191:4789–4803. doi: 10.4049/jimmunol.1300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wu Y.H., Kuo W.C., Wu Y., Yang K., Chen S.T., Jiang S.T., Gordy C., He Y.W., Lai M.Z. Differentiation, Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 2014;21:451–461. doi: 10.1038/cdd.2013.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gurung P., Kanneganti T.D. Novel roles for caspase-8 in IL-1β and inflammasome regulation. Am. J. Pathol. 2015;185:17–25. doi: 10.1016/j.ajpath.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gurung P., Anand P.K., Malireddi R.S., Walle L.V., Van Opdenbosch N., Dillon C.P., Weinlich R., Green D.R., Lamkanfi M., Kanneganti T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vince J.E., Wong W.W.L., Gentle I., Lawlor K.E., Allam R., O’Reilly L., Mason K., Gross O., Ma S., Guarda G., et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36:215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 152.Lawlor K.E., Khan N., Mildenhall A., Gerlic M., Croker B.A., D’Cruz A.A., Hall C., Spall S.K., Anderton H., Masters S.L., et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015;6:1–19. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kang S., Fernandes-Alnemri T., Rogers C., Mayes L., Wang Y., Dillon C., Roback L., Kaiser W., Oberst A., Sagara J., et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015;6:1–15. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kang T.B., Yang S.H., Toth B., Kovalenko A., Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 155.Gaidt M.M., Ebert T.S., Chauhan D., Schmidt T., Schmid-Burgk J.L., Rapino F., Robertson A.A., Cooper M.A., Graf T., Hornung V. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 156.Conos S.A., Chen K.W., De Nardo D., Hara H., Whitehead L., Núñez G., Masters S.L., Murphy J.M., Schroder K., Vaux D.L., et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA. 2017;114:E961–E969. doi: 10.1073/pnas.1613305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Orning P., Weng D., Starheim K., Ratner D., Best Z., Lee B., Brooks A., Xia S., Wu H., Kelliher M.A., et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–1069. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen K.W., Demarco B., Heilig R., Shkarina K., Boettcher A., Farady C.J., Pelczar P., Broz P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP 3 inflammasome assembly. EMBO J. 2019;38:e101638. doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tummers B., Green D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017;277:76–89. doi: 10.1111/imr.12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vajjhala P.R., Lu A., Brown D.L., Pang S.W., Sagulenko V., Sester D.P., Cridland S.O., Hill J.M., Schroder K., Stow J.L., et al. The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J. Biol. Chem. 2015;290:29217–29230. doi: 10.1074/jbc.M115.687731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jin T., Perry A., Smith P., Jiang J., Xiao T.S. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 2013;288:13225–13235. doi: 10.1074/jbc.M113.468033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jin T., Perry A., Jiang J., Smith P., Curry J.A., Unterholzner L., Jiang Z., Horvath G., Rathinam V.A., Johnstone R.W., et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shaw N., Liu Z.J. Role of the HIN domain in regulation of innate immune responses. Cell. Biol. 2014;34:2–15. doi: 10.1128/MCB.00857-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Veeranki S., Duan X., Panchanathan R., Liu H., Choubey D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE. 2011;6:e27040. doi: 10.1371/journal.pone.0027040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cresswell K.S., Clarke C.J., Jackson J.T., Darcy P.K., Trapani J.A., Johnstone R.W. Biochemical and growth regulatory activities of the HIN-200 family member and putative tumor suppressor protein, AIM2. Biochem. Biophys. Res. Commun. 2005;326:417–424. doi: 10.1016/j.bbrc.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 166.Yin Q., Sester D.P., Tian Y., Hsiao Y.S., Lu A., Cridland J.A., Sagulenko V., Thygesen S.J., Choubey D., Hornung V., et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. 2013;4:327–339. doi: 10.1016/j.celrep.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lu A., Magupalli V.G., Ruan J., Yin Q., Atianand M.K., Vos M.R., Schröder G.F., Fitzgerald K.A., Wu H., Egelman E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sagulenko V., Thygesen S.J., Sester D.P., Idris A., Cridland J.A., Vajjhala P.R., Roberts T.L., Schroder K., Vince J.E., Hill J.M., et al. Differentiation, AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lu A., Kabaleeswaran V., Fu T., Magupalli V.G., Wu H. Crystal structure of the F27G AIM2 PYD mutant and similarities of its self-association to DED/DED interactions. J. Mol. Biol. 2014;426:1420–1427. doi: 10.1016/j.jmb.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hou X., Niu X. The NMR solution structure of AIM2 PYD domain from Mus musculus reveals a distinct α2–α3 helix conformation from its human homologues. Biochem. Biophys. Res. Commun. 2015;461:396–400. doi: 10.1016/j.bbrc.2015.04.046. [DOI] [PubMed] [Google Scholar]
- 171.Morrone S.R., Matyszewski M., Yu X., Delannoy M., Egelman E.H., Sohn J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015;6:1–13. doi: 10.1038/ncomms8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu A., Li Y., Yin Q., Ruan J., Yu X., Egelman E., Wu H. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015;1:1–14. doi: 10.1038/celldisc.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ru H., Ni X., Zhao L., Crowley C., Ding W., Hung L.W., Shaw N., Cheng G., Liu Z.J. Structural basis for termination of AIM2-mediated signaling by p202. Cell Res. 2013;23:855–858. doi: 10.1038/cr.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Matyszewski M., Morrone S.R., Sohn J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. USA. 2018;115:E1963–E1972. doi: 10.1073/pnas.1712860115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Choubey D., Walter S., Geng Y., Xin H. Cytoplasmic localization of the interferon-inducible protein that is encoded by the AIM2 (absent in melanoma) gene from the 200-gene family. FEBS Lett. 2000;474:38–42. doi: 10.1016/S0014-5793(00)01571-4. [DOI] [PubMed] [Google Scholar]
- 176.Koul D., Obeyesekere N.U., Gutterman J.U., Mills G.B., Choubey D. p202 self-associates through a sequence conserved among the members of the 200-family proteins. FEBS Lett. 1998;438:21–24. doi: 10.1016/S0014-5793(98)01263-0. [DOI] [PubMed] [Google Scholar]
- 177.Wang P.H., Ye Z.W., Deng J.J., Siu K.L., Gao W.W., Chaudhary V., Cheng Y., Fung S.Y., Yuen K.S., Ho T.H., et al. Inhibition of AIM 2 inflammasome activation by a novel transcript isoform of IFI 16. EMBO Rep. 2018;19:e45737. doi: 10.15252/embr.201845737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu T., Tang Q., Liu K., Xie W., Liu X., Wang H., Wang R.F., Cui J. TRIM11 suppresses AIM2 inflammasome by degrading AIM2 via p62-dependent selective autophagy. Cell Rep. 2016;16:1988–2002. doi: 10.1016/j.celrep.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 179.Yang J., Liu Z., Xiao T.S. Post-translational regulation of inflammasomes. Cel. Mol. Immunol. 2017;14:65–79. doi: 10.1038/cmi.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang M.J., Zhao Q.C., Xia M.X., Chen J., Chen Y.T., Cao X., Liu Y., Yuan Z.Q., Wang X.Y., Xu Y. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 2020;34:648–662. doi: 10.1096/fj.201900394RRR. [DOI] [PubMed] [Google Scholar]
- 181.Masumoto J., Taniguchi S.I., Ayukawa K., Sarvotham H., Kishino T., Niikawa N., Hidaka E., Katsuyama T., Higuchi T., Sagara J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999;274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 182.Sahillioglu A.C., Sumbul F., Ozoren N., Haliloglu T. Structural and dynamics aspects of ASC speck assembly. Structure. 2014;22:1722–1734. doi: 10.1016/j.str.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 183.de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC) J. Biol. Chem. 2009;284:32932–32941. doi: 10.1074/jbc.M109.024273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.McConnell B.B., Vertino P.M. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res. 2000;60:6243–6247. [PubMed] [Google Scholar]
- 185.Fernandes-Alnemri T., Wu J., Yu J., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E.S. Differentiation, The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ashkenazi A., Salvesen G. Regulated cell death: Signaling and mechanisms. Cell Dev. Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- 187.McIlwain D.R., Berger T., Mak T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013;5:a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bryan N.B., Dorfleutner A., Kramer S.J., Yun C., Rojanasakul Y., Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J. Inflamm. 2010;7:1–13. doi: 10.1186/1476-9255-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liepinsh E., Barbals R., Dahl E., Sharipo A., Staub E., Otting G. The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 2003;332:1155–1163. doi: 10.1016/j.jmb.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 190.de Alba E. Structure, interactions and self-assembly of ASC-dependent inflammasomes. Arch. Biochem. Biophys. 2019;670:15–31. doi: 10.1016/j.abb.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Moriya M., Taniguchi S., Wu P., Liepinsh E., Otting G., Sagara J. Role of charged and hydrophobic residues in the oligomerization of the PYRIN domain of ASC. Biochemistry. 2005;44:575–583. doi: 10.1021/bi048374i. [DOI] [PubMed] [Google Scholar]
- 192.Dick M.S., Sborgi L., Rühl S., Hiller S., Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016;7:1–13. doi: 10.1038/ncomms11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lu A., Wu H. Structural mechanisms of inflammasome assembly. FEBS J. 2015;282:435–444. doi: 10.1111/febs.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sborgi L., Ravotti F., Dandey V.P., Dick M.S., Mazur A., Reckel S., Chami M., Scherer S., Huber M., Böckmann A., et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA. 2015;112:13237–13242. doi: 10.1073/pnas.1507579112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhou P., Chou J., Olea R.S., Yuan J., Wagner G. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: A structural basis for specific adaptor/caspase interaction. Proc. Natl. Acad. Sci. USA. 1999;96:11265–11270. doi: 10.1073/pnas.96.20.11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Qin H., Srinivasula S.M., Wu G., Fernandes-Alnemri T., Alnemri E.S., Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 197.Park H.H., Lo Y.C., Lin S.C., Wang L., Yang J.K., Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007;25:561–586. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nambayan R.J.T., Sandin S.I., Quint D.A., Satyadi D.M., de Alba E. The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains, PYD and CARD. J. Biol. Chem. 2019;294:439–452. doi: 10.1074/jbc.RA118.004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Humke E.W., Shriver S.K., Starovasnik M.A., Fairbrother W.J., Dixit V.M. ICEBERG: A novel inhibitor of interleukin-1β generation. Cell. 2000;103:99–111. doi: 10.1016/S0092-8674(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 200.Chou J.J., Matsuo H., Duan H., Wagner G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell. 1998;94:171–180. doi: 10.1016/S0092-8674(00)81417-8. [DOI] [PubMed] [Google Scholar]
- 201.Manon F., Favier A., Núñez G., Simorre J.P., Cusack S. Solution structure of NOD1 CARD and mutational analysis of its interaction with the CARD of downstream kinase RICK. J. Mol. Biol. 2007;365:160–174. doi: 10.1016/j.jmb.2006.09.067. [DOI] [PubMed] [Google Scholar]
- 202.Li Y., Fu T.M., Lu A., Witt K., Ruan J., Shen C., Wu H. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. USA. 2018;115:10845–10852. doi: 10.1073/pnas.1810524115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matyszewski M., Zheng W., Lueck J., Antiochos B., Egelman E.H., Sohn J. Cryo-EM structure of the NLRC4CARD filament provides insights into how symmetric and asymmetric supramolecular structures drive inflammasome assembly. J. Biol. Chem. 2018;293:20240–20248. doi: 10.1074/jbc.RA118.006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gambin Y., Giles N., O’Carroll A., Polinkovsky M., Hunter D., Sierecki E. Single-molecule fluorescence reveals the oligomerization and folding steps driving the prion-like behavior of ASC. J. Mol. Biol. 2018;430:491–508. doi: 10.1016/j.jmb.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 205.Vajjhala P.R., Mirams R.E., Hill J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012;287:41732–41743. doi: 10.1074/jbc.M112.381228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Proell M., Gerlic M., Mace P.D., Reed J.C., Riedl S.J. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 2013;449:613–621. doi: 10.1042/BJ20121198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yang J.K., Wang L., Zheng L., Wan F., Ahmed M., Lenardo M.J., Wu H. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol. Cell. 2005;20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Stehlik C., Krajewska M., Welsh K., Krajewski S., Godzik A., Reed J.C. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappaB and pro-caspase-1 regulation. Biochem. J. 2003;373:101–113. doi: 10.1042/bj20030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Srimathi T., Robbins S.L., Dubas R.L., Chang H., Cheng H., Roder H., Park Y.C. Mapping of POP1-binding site on pyrin domain of ASC. J. Biol. Chem. 2008;283:15390–15398. doi: 10.1074/jbc.M801589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mambwe B., Neo K., Javanmard Khameneh H., Leong K.W.K., Vacca M., Muimo R., Mortellaro A. Tyrosine Dephosphorylation of ASC Modulates the Activation of the NLRP3 and AIM2 Inflammasomes. Front. Immunol. 2019;10:1556. doi: 10.3389/fimmu.2019.01556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Tokunaga F., Iwai K. Involvement of LUBAC-mediated linear polyubiquitination of NEMO in NF-kappaB activation. Tanpakushitsu Kakusan Koso Protein Nucleic Acid Enzym. 2009;54:635–642. [PubMed] [Google Scholar]
- 212.Rodgers M.A., Bowman J.W., Fujita H., Orazio N., Shi M., Liang Q., Amatya R., Kelly T.J., Iwai K., Ting J., et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 2014;211:1333–1347. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Guan K., Wei C., Zheng Z., Song T., Wu F., Zhang Y., Cao Y., Ma S., Chen W., Xu Q., et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J. Immunol. 2015;194:4880–4890. doi: 10.4049/jimmunol.1402851. [DOI] [PubMed] [Google Scholar]
- 214.Kim J.H., Lee Y., Suh G.Y., Lee Y.S. Mul1 suppresses Nlrp3 inflammasome activation through ubiquitination and degradation of ASC. BioRxiv. 2019:830380. doi: 10.1101/830380. [DOI] [Google Scholar]
- 215.Nuñez G., Benedict M.A., Hu Y., Inohara N. Caspases: The proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 216.Black R.A., Kronheim S.R., Merriam J.E., March C.J., Hopp T.P. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem. 1989;264:5323–5326. doi: 10.1016/S0021-9258(18)83546-3. [DOI] [PubMed] [Google Scholar]
- 217.Thornberry N.A., Bull H.G., Calaycay J.R., Chapman K.T., Howard A.D., Kostura M.J., Miller D.K., Molineaux S.M., Weidner J.R., Aunins J., et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 218.Elliott J.M., Rouge L., Wiesmann C., Scheer J.M. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J. Biol. Chem. 2009;284:6546–6553. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Boucher D., Monteleone M., Coll R.C., Chen K.W., Ross C.M., Teo J.L., Gomez G.A., Holley C.L., Bierschenk D., Stacey K.J., et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018;215:827–840. doi: 10.1084/jem.20172222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Romanowski M.J., Scheer J.M., O’Brien T., McDowell R.S. Crystal structures of a ligand-free and malonate-bound human caspase-1: Implications for the mechanism of substrate binding. Structure. 2004;12:1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 221.Yang X., Chang H.Y., Baltimore D. Autoproteolytic activation of pro-caspases by oligomerization. Mol. Cell. 1998;1:319–325. doi: 10.1016/S1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- 222.Walker N., Talanian R., Brady K., Dang L., Bump N., Ferenza C., Franklin S., Ghayur T., Hackett M., Hammill L., et al. Crystal structure of the cysteine protease interleukin-1β-converting enzyme: A (p20/p10) 2 homodimer. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 223.Wilson K.P., Black J.A.F., Thomson J.A., Kim E.E., Griffith J.P., Navia M.A., Murcko M.A., Chambers S.P., Aldape R.A., Raybuck S.A., et al. Structure and mechanism of interleukin-lβ converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 224.Li J., Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 225.Fuentes-Prior P., Salvesen G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Gu Y., Wu J., Faucheu C., Lalanne J., Diu A., Livingston D., Su M.S. Interleukin-1 beta converting enzyme requires oligomerization for activity of processed forms in vivo. EMBO J. 1995;14:1923–1931. doi: 10.1002/j.1460-2075.1995.tb07184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Martinon F., Burns K., Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 228.Dinarello C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 229.Li P., Allen H., Banerjee S., Franklin S., Herzog L., Johnston C., McDowell J., Paskind M., Rodman L., Salfeld J., et al. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 230.Turner M.D., Nedjai B., Hurst T., Pennington D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014;1843:2563–2582. doi: 10.1016/j.bbamcr.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 231.Lu A., Li Y., Schmidt F.I., Yin Q., Chen S., Fu T.M., Tong A.B., Ploegh H.L., Mao Y., Wu H. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 2016;23:416–425. doi: 10.1038/nsmb.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ghayur T., Banerjee S., Hugunin M., Butler D., Herzog L., Carter A., Quintal L., Sekut L., Talanian R., Paskind M. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature. 1997;386:619–623. doi: 10.1038/386619a0. [DOI] [PubMed] [Google Scholar]
- 233.Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D.C., Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 234.Ruan J., Xia S., Liu X., Lieberman J., Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62–67. doi: 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fink S.L., Cookson B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- 236.Shi J., Zhao Y., Wang K., Shi X., Wang Y., Huang H., Zhuang Y., Cai T., Wang F., Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 237.He W.T., Wan H., Hu L., Chen P., Wang X., Huang Z., Yang Z.H., Zhong C.Q., Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang B., Yin Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017;200:279–282. doi: 10.1016/j.jsb.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lin S.C., Lo Y.C., Wu H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wu B., Peisley A., Tetrault D., Li Z., Egelman E.H., Magor K.E., Walz T., Penczek P.A., Hur S. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell. 2014;55:511–523. doi: 10.1016/j.molcel.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yin Q., Fu T.M., Li J., Wu H. Structural biology of innate immunity. Annu. Rev. Immunol. 2015;33:393–416. doi: 10.1146/annurev-immunol-032414-112258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Le H.T., Harton J.A. Pyrin-and CARD-only proteins as regulators of NLR functions. Front. Immunol. 2013;4:275. doi: 10.3389/fimmu.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kersse K., Berghe T.V., Lamkanfi M., Vandenabeele P. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem. Soc. Trans. 2007;35:1508–1511. doi: 10.1042/BST0351508. [DOI] [PubMed] [Google Scholar]
- 244.Lamkanfi M., Denecker G., Kalai M., D’hondt K., Meeus A., Declercq W., Saelens X., Vandenabeele P. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1β generation. J. Biol. Chem. 2004;279:51729–51738. doi: 10.1074/jbc.M407891200. [DOI] [PubMed] [Google Scholar]
- 245.Kersse K., Lamkanfi M., Bertrand M.J., Berghe T.V., Vandenabeele P. Interaction patches of procaspase-1 caspase recruitment domains (CARDs) are differently involved in procaspase-1 activation and receptor-interacting protein 2 (RIP2)-dependent nuclear factor κB signaling. J. Biol. Chem. 2011;286:35874–35882. doi: 10.1074/jbc.M111.242321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Druilhe A., Srinivasula S., Razmara M., Ahmad M., Alnemri E. Differentiation, Regulation of IL-1β generation by Pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 2001;8:649–657. doi: 10.1038/sj.cdd.4400881. [DOI] [PubMed] [Google Scholar]
- 247.Dufner A., Pownall S., Mak T.W. Caspase recruitment domain protein 6 is a microtubule-interacting protein that positively modulates NF-κB activation. Proc. Natl. Acad. Sci. USA. 2006;103:988–993. doi: 10.1073/pnas.0510380103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kobayashi K., Inohara N., Hernandez L.D., Galán J.E., Núñez G., Janeway C.A., Medzhitov R., Flavell R.A. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- 249.Chin A.I., Dempsey P.W., Bruhn K., Miller J.F., Xu Y., Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- 250.Yuan S., Akey C.W. Apoptosome structure, assembly, and procaspase activation. Structure. 2013;21:501–515. doi: 10.1016/j.str.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Halff E.F., Diebolder C.A., Versteeg M., Schouten A., Brondijk T.H.C., Huizinga E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N-and C-terminal regions of flagellin. J. Biol. Chem. 2012;287:38460–38472. doi: 10.1074/jbc.M112.393512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Santiveri C.M., Oroz J., de Alba E. A ring-like model for ASC self-association via the CARD domain. Inflammasome. 2014;1:44–54. [Google Scholar]
- 253.Wu J., Fernandes-Alnemri T., Alnemri E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jiang D., Chen S., Sun R., Zhang X., Wang D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018;419:8–19. doi: 10.1016/j.canlet.2018.01.034. [DOI] [PubMed] [Google Scholar]
- 255.Di Micco A., Frera G., Lugrin J., Jamilloux Y., Hsu E.T., Tardivel A., De Gassart A., Zaffalon L., Bujisic B., Siegert S., et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl. Acad. Sci. USA. 2016;113:E4671–E4680. doi: 10.1073/pnas.1602419113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Schroder K., Muruve D.A., Tschopp J. Innate immunity: Cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 2009;19:R262–R265. doi: 10.1016/j.cub.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 257.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C., Englert J.A., Rabinovitch M., Cernadas M., Kim H.P., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yissachar N., Salem H., Tennenbaum T., Motro B. Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 2006;580:6489–6495. doi: 10.1016/j.febslet.2006.10.069. [DOI] [PubMed] [Google Scholar]
- 260.Wu P.J., Liu H.Y., Huang T.N., Hsueh Y.P. AIM 2 inflammasomes regulate neuronal morphology and influence anxiety and memory in mice. Scientific Report. 2016;6:32405. doi: 10.1038/srep32405. [DOI] [PMC free article] [PubMed] [Google Scholar]