

Abstract
Cyclotides are macrocyclic peptides with exceptionally stable structures and have been reported to penetrate cells, making them promising scaffolds for the delivery of inhibitory peptides to target intracellular proteins. However, their cellular uptake and cytosolic localization have been poorly understood until now, which has limited their therapeutic potential. In this study, the recently developed chloroalkane penetration assay was combined with established assays to characterize the cellular uptake and cytosolic delivery of the prototypic cyclotide, kalata B1. We show that kalata B1 enters the cytosol at low efficiency. A structure–activity study of residues in loop 6 showed that some modifications, such as increasing cationic residue content, did not affect delivery efficiency, whereas others, including introducing a single hydrophobic amino acid, did significantly improve cytosolic delivery. Our results provide a foundation for the further development of a structurally unique class of scaffolds for the delivery of therapeutic cargoes into cells.
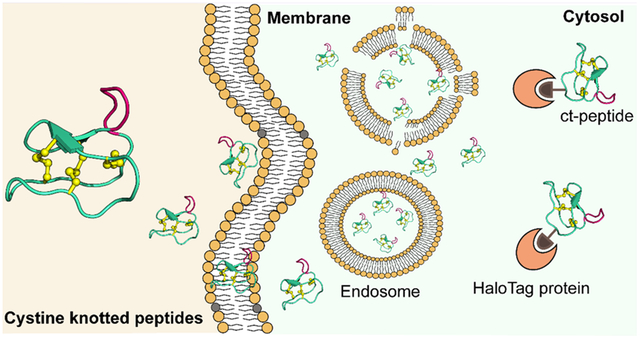
Graphical Abstract
INTRODUCTION
Peptides are promising modalities for drug design because they occupy a unique chemical space between small molecules and large biologics.1,2 Recent interest in their use as therapeutics has been driven by breakthroughs in bioactive peptide discovery and optimization, which have led to the development of many peptides that can modulate protein targets with high in vitro potency and selectivity.3–6 Despite these technological advances, targeting of intracellular protein–protein interactions remains a considerable challenge mainly because of the difficulty in delivery of drug leads into the cytosolic space.7,8
The discovery that the Tat peptide from the human immunodeficiency virus (HIV)-1 could shuttle exogenous cargo into the cytosol spurred the development of many other cell-penetrating peptides (CPPs).9–13 However, it is becoming clear that although these peptides and attached cargoes are taken up by the cell, most material ends up entrapped in endosomal compartments where they are physically separated from the majority of intracellular targets.14 The result is low effective concentrations of the cargo at the cytosolically localized protein target and unacceptably poor bioactivity, impeding further therapeutic development.
Variants of CPPs that have been conformationally constrained through cyclization have been reported to have increased rates of cellular uptake7 and facilitate delivery of large cargoes.15 For instance, a fluorescein-labeled cyclic Tat was shown to have higher total cellular uptake than the linear analogue.16 Other cyclic CPPs, FΦRRRRQ (where Φ is l-naphthylalanine) and [WR]4, are reported to enter cells and facilitate delivery into the cytosol.17–19 These studies tentatively support the benefit of cyclization for cellular uptake but raise the question of the mechanism by which cyclization might improve cellular uptake. One contributing factor might be that cyclization leads to improved proteolytic stability of the peptides, resulting in longer residence times in the extracellular environment and subsequently to higher amounts of intracellular localization over time.
MCoTI-II and kalata B1, as shown in Figure 1, are cyclic CPPs that have unique structural properties that underpin their potential for use in drug design.20,21 They belong to a large family of peptides called cyclotides that are distinguished by a head-to-tail cyclized backbone and a densely packed core of three disulfide bonds in a knotted arrangement (Figure 1A).22 These structural attributes engender them with exceptionally high resistance to thermal or enzymatic denaturation.23 Notably, they form a highly conserved scaffold within which bioactive peptide sequences can be introduced through a process known as molecular grafting.24 Over two dozen examples of grafted cyclotides have been reported to date.21,25 Most relevant to the current study are the examples in which insertion of an inhibitory peptide of HDM2/X into MCoTI-I activated the p53 tumor suppressor pathway in vitro and in vivo, and grafting of an antagonist against intracellular SET into MCoTI-II inhibited NF-κB-dependent gene expression.26,27 These examples nicely demonstrate the broader potential of cyclotides as cystine-knot scaffolds for drug design and intracellular delivery.
Figure 1.
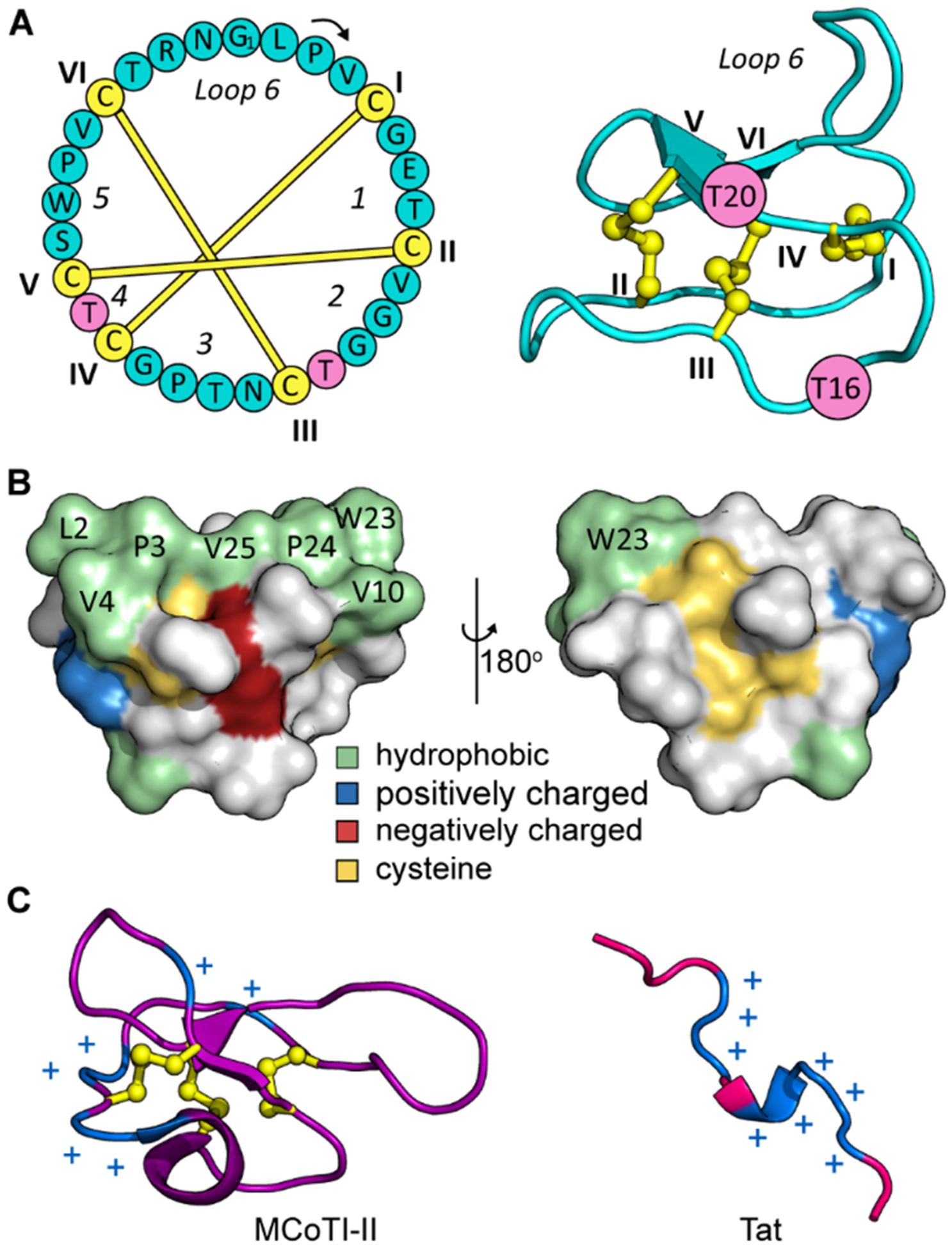
Sequence and 3D structure of the prototypical cyclotide, kalata B1 (kB1). (A) The amino acid sequence of kB1 (left panel) is illustrated as a cycle with its six cysteines highlighted in yellow and labeled using Roman numerals I to VI. Six sequence segments separated by cysteines are marked as loops 1–6. The knotted disulfide connectivity is indicated. The 3D structure of kB1 (PDB: 4TTM) is shown in the right panel with two threonine residues (T16 and T20) marked in pink. (B) Surface structure of kB1 with hydrophobic, positively and negatively charged residues colored in green, blue, and red, respectively; cysteines are colored yellow. (C) 3D structures of another cell-penetrating cyclotide MCoTI-II (left panel, PDB: 1IB9) and a cell-penetrating peptide Tat (right panel, PDB: 1TAC) with positively charged residues labeled with blue plus signs (+).
Kalata B1, although structurally similar to MCoTI-cyclotides, is significantly different chemically. MCoTI-I and -II are both highly positively charged, like the majority of CPPs investigated so far (Figure 1C), and they interact with the cell surface leading to uptake by endocytosis.28 By contrast, kalata B1 has a net neutral charge, comprising instead a high proportion of hydrophobic amino acids that cluster at the surface (Figure 1B). Its distinct amino acid composition compared to other CPPs is responsible for its unique mechanism of cell entry that involves stereospecific recognition of lipid headgroups, resulting in cellular uptake by both endocytic and direct translocation pathways.29,30 Furthermore, under some conditions, a lysine mutant of kalata B1 has been reported to have a total cellular uptake comparable to Tat, making it a promising scaffold for intracellular delivery.30 However, a better understanding of the cytosolic delivery of kalata B1 will be necessary to realize its full potential as a unique cell-penetrating scaffold.
Cytosolic localization has been traditionally difficult to quantify because, in assays commonly used to measure cell penetration, endosomal and cytosolic localization have been indistinguishable.31,32 The most common approach to measure cellular internalization is to track a fluorescently labeled peptide (e.g., labeled with Alexa Fluor 488) by flow cytometry and/or microscopy, but these approaches cannot accurately quantify the amount of peptide in the cytosolic space.31,33 Several groups have reported assays that can measure cytosolic localization without interference from endosomally trapped material.32,34 One prominent example of an assay that quantitates cytosolic delivery is the chloroalkane penetration assay (CAPA), recently developed by the Kritzer group.33,35 CAPA allows for quantitative measurement of the cytosolic delivery of peptides in a low-volume, high-throughput format.33
In this study, we aimed to provide a better understanding of the cell uptake and cytosolic delivery of kalata B1, the prototypical cyclic cystine knot scaffold. Previous studies have detected uptake of fluorescently labeled kalata B1 into cells by flow cytometry and microscopy but have not clearly demonstrated delivery into the cytosolic space. Here, we used CAPA as an independent assay to quantify cytosolic delivery. Flow cytometry and confocal microscopy were also performed to separately measure total cellular uptake, to deconvolute the effects of the required tags, and to allow cross-referencing to published results. We measured total cellular uptake and cytosolic delivery of many kalata B1 analogues with different tags, mutations, and loop grafts. We discovered that, although kalata B1 has been reported to penetrate cells, it enters the cytosol at low efficiency. Surprisingly, modification of kalata B1 by specific epitopes, including substitution with a single hydrophobic amino acid, resulted in significantly improved cytosolic delivery. The cytosolic localization of the most penetrant kalata B1 analogues was comparable to that of Tat. These data provide a basis for continued optimization of these unique scaffolds for therapeutic gain.
RESULTS
Peptide Synthesis and Labeling.
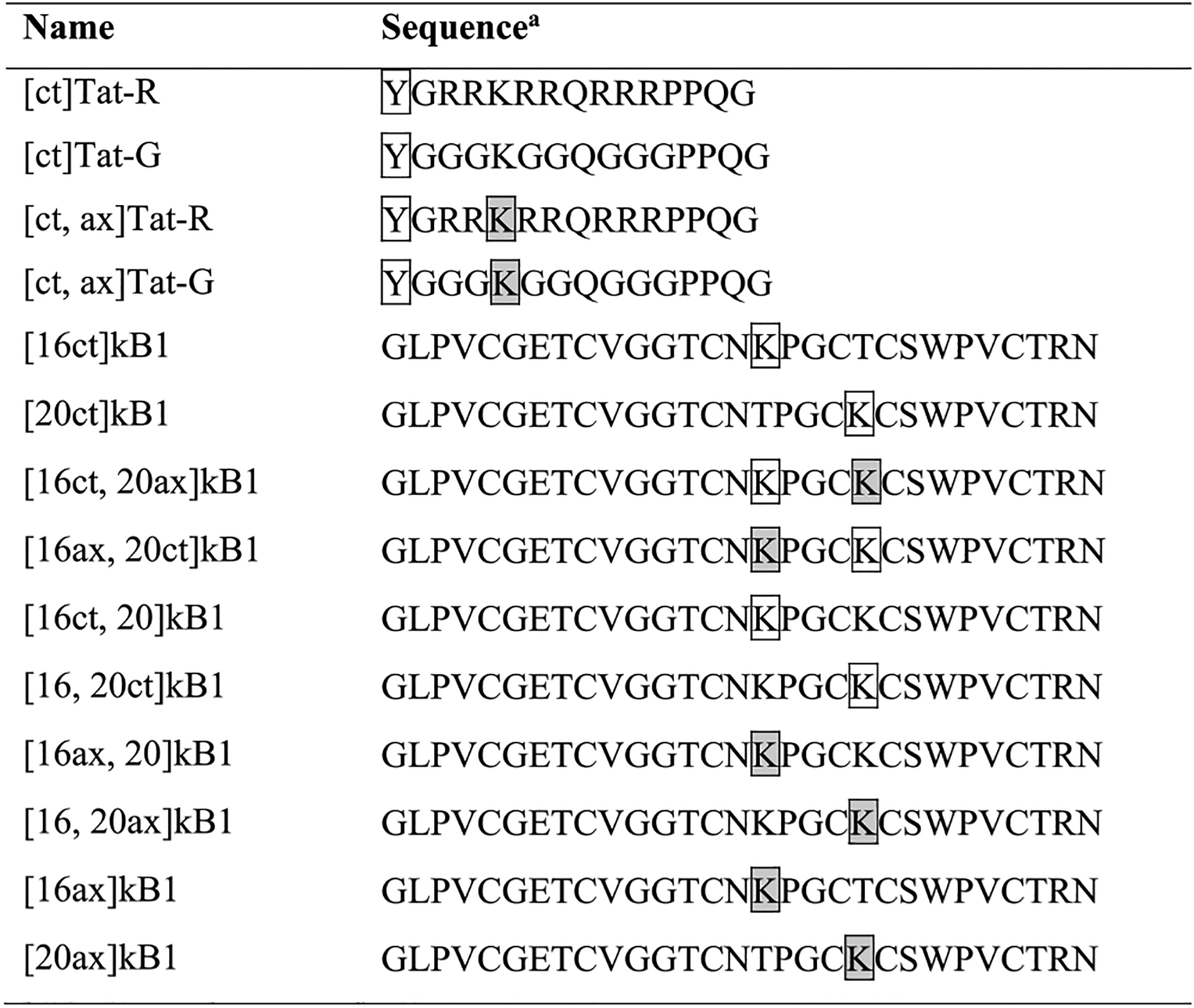
Peptides used in this study were synthesized using solid-phase peptide synthesis, with the sequences shown in Tables 1 and 2 and Supporting Information Table S1. Conjugation of Alexa Fluor 488 and chloroalkane tags was achieved by modification of peptide N-termini or side-chain lysine amino groups. As shown in Table 1, Tat-R was synthesized as a positive control, since Tat is known to exhibit high cellular uptake. Native Tat has two lysines, but for this study we used Tat-R, which has a single lysine residue to allow for defined attachment of tags.27,30,36 Another Tat variant, Tat-G, has glycine residues in place of arginines. Tat-G was used as a benchmark for low cellular uptake. Labeled variants of these peptides having a chloroalkane tag at their N-termini are referred to as [ct]Tat-R and [ct]Tat-G, respectively.
Table 1.
Names and Amino Acid Sequences of Tat and Kalata B1 (kB1) Peptides
|
Tat peptides are linear and have amidated C-termini; kB1 peptides are backbone cyclic and have three disulfide bonds as depicted in Figure 1. Y (boxed) denotes a tyrosine with a chloroalkane tag coupled to its N-terminus. K (boxed) denotes a lysine coupled to a chloroalkane tag, and K (boxed, shaded) denotes a lysine coupled to an Alexa Fluor 488 tag.
Table 2.
Names of Kalata B1 (kB1) Analogues and Their Loop 6 Amino Acid Sequences and Compositions
| Namea | Loop 6 Sequenceb | Hydrophobicc | Negatived | Positivee | Polarf |
|---|---|---|---|---|---|
| Native | |||||
| kB1 | TRNGLPV | 3 | 0 | 1 | 3 |
| Residue Mutants | |||||
| [Rmut, 16]kB1 | TRRGRPR | 1 | 0 | 4 | 2 |
| [Hmut, 16]kB1 | TRHGHPH | 1 | 0 | 4 | 2 |
| [Wmut, 16]kB1 | TRWGLPV | 4 | 0 | 1 | 2 |
| [Emut, 16]kB1 | TREGEPE | 1 | 3 | 1 | 2 |
| [Smut, 16]kB1 | TRSGSPS | 1 | 0 | 1 | 5 |
| [Gmut, 16]kB1 | TRGGGPG | 1 | 0 | 1 | 5 |
| Grafted Peptides | |||||
| [graft1, 16]kB1 | TRGFEWLDWEFGLPV | 8 | 3 | 1 | 3 |
| [graft2, 16]kB1 | TRGLDEETGEFGLPV | 5 | 4 | 1 | 5 |
| [graft3, 16]kB1 | TRGSFEGYDNGGLPV | 4 | 2 | 1 | 8 |
Chloroalkane (ct) and Alexa Fluor 488 (ax) tagged variants were used to study cellular uptake, but only unlabeled peptides are shown here for brevity. Tags were attached via a lysine at position 16.
Residue differences to the native peptide are highlighted with underlines. Refer to Figure 1 for the rest of the sequence of native kB1 and Table 1 for the rest of the sequence of the kB1 analogues.
Hydrophobic residues: V, P, L, W, F.
Negatively charged residues: D, E.
Positively charged residues: K, R, H.
Polar residues: S, T, Y, N, G.
To attach tags to kalata B1 (kB1), one or two lysine residues were introduced into its sequence to provide amino-group handles for conjugation. As shown in Figure 1A, two threonine residues (T16 and T20) were chosen as positions for lysine substitution because their lysine mutants have been well-characterized; here these variants are referred to as [16]kB1 and [20]kB1, respectively, instead of the [T16K]kB1 and [T20K]kB1 used previously.30,37 In this study, we will use “ct” to denote chloroalkane-tagged peptides and “ax” to denote Alexa Flour 488-tagged peptides (e.g., [16ax, 20ct]kB1 has an Alexa Fluor 488 conjugated to lysine at position 16 and a chloroalkane tag conjugated to lysine at position 20, Supporting Information Figure S3).
Cytosolic Delivery and Cellular Uptake of Kalata B1.
CAPA represents an independent assay to measure cytosolic delivery, and it complements assays previously used to characterize the total cellular uptake of kB1. The steps involved in CAPA are schematically illustrated in Figure 2A. CAPA differs from previous assays used to characterize kB1 (Figure 2B) because it measures only cytosolic delivery and uses a different tag for signal readout. In CAPA, after pulsing with a chloroalkane-tagged peptide (ct-peptide) and chasing with a chloroalkane-tagged fluorescent dye (ct-dye), flow cytometry is used to measure fluorescence intensity of the cell-associated ct-dye. This signal is inversely proportional to the amount of ct-peptide delivered to the cytosol during the pulse step. [16ct]kB1 and [20ct]kB1 were tested in CAPA up to concentrations of 30 μM, at which [20ct]kB1 began to exhibit toxicity, observed as changes in cell morphology. As shown in Figure 2C, [16ct]kB1 and [20ct]kB1 both showed reducing fluorescence intensity with increasing peptide concentration in the 3.75 to 15 μM range, indicating cytosolic localization. The amount of kB1 reaching the cytosol was more comparable to that of [ct]Tat-G than [ct]Tat-R, suggesting that cytosolic delivery of ct-tagged kB1 analogues was relatively low.
Figure 2.
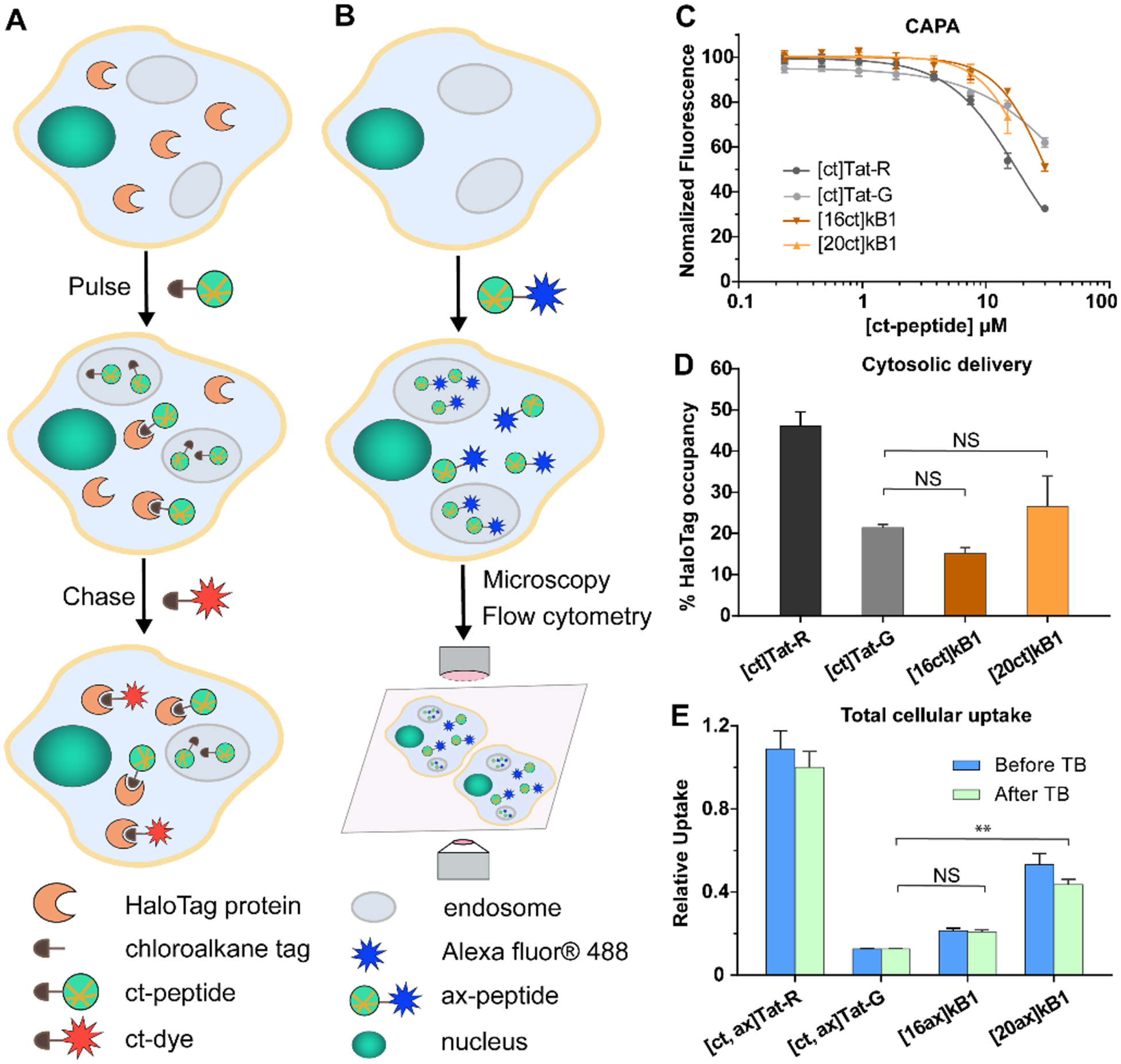
Cytosolic delivery and total cellular uptake of kB1. (A) A schematic illustration of the chloroalkane penetration assay (CAPA). Halo-GFP-Mito cells expressing the HaloTag protein are pulsed with chloroalkane-tagged peptide (ct-peptide), washed, and chased with chloroalkane-tagged dye (ct-dye). Then, the fluorescence intensity of the cell-associated ct-dye is measured using flow cytometry. CAPA results for ct-Tat analogues and ct-kB1 analogues are shown in panels B and C. (B) A schematic illustration of traditional fluorescence assays for measuring total cellular uptake, showing treatment of cells with Alexa Fluor 488-labeled peptide (ax-peptide) and detection by flow cytometry or microscopy. Compared to CAPA, these traditional assays cannot quantify cytosolic delivery. The flow cytometry assay, in particular, cannot distinguish between cytosolic delivery or endosomal entrapment. (C) Fluorescence intensity readout over a range of concentrations for ct-peptides. The data were normalized using a no-pulse control as 100% uptake and a no-pulse/no-chase control as 0% uptake. (D) Cytosolic delivery, as measured by HaloTag occupancy and normalized to no-dye (0% value) and no-molecule (100% value) controls, of ct-peptides at 8.75 μM. (E) Total cellular uptake of Alexa Fluor 488-tagged (ax) peptides at 4 μM. Mean fluorescence intensity of HeLa cells treated with ax-peptides before (blue) and after (green) the addition of trypan blue (TB) were measured by flow cytometry. Data are shown relative to the fluorescence signal of [ct, ax]Tat-R (positive control) after addition of TB. All experiments were done in triplicate, and the results are represented as mean ± SEM.
To contrast cytosolic penetration of kB1 analogs to their total cellular uptake and provide a cross-reference with previous studies, we measured total cellular uptake by quantifying the amounts of Alexa-tagged peptides using flow cytometry. We have previously used this approach to characterize cell penetration of kB1.20,30 In this assay, fluorescence intensity of the sample (after quenching extracellular fluorescence with the addition of trypan blue) is proportional to the amount of peptide taken up by cells. Like the CAPA results, kB1 peptides have low uptake relative to Tat-R (Figure 2E and Supporting Information Figure S4). The most noticeable difference was that the position of the lysine mutation and/or Alexa tag had an effect on cellular uptake measured by flow cytometry, which is consistent with a previous report;30 by contrast, the positional effect of the chloroalkane tag on cytosolic delivery as measured by CAPA was not statistically significant (Figure 2D). Overall, these data imply that kB1 analogues are taken up by cells and also enter the cytosol, when cells are treated at concentrations of 4 μM or higher for 4 h.
Effect of Chloroalkane and Fluorescent Tag.
In the next step, we further investigated the relationship between total cellular uptake (as measured by flow cytometry) and cytosolic delivery (as measured by CAPA), as well as the effect of the tags on these different assays. To do this, we prepared a kB1 analogue with lysine residues at positions T16 and T20 so that it could be conjugated to both chloroalkane and Alexa Fluor 488 tags. As shown in Table 1, [16ct, 20ax]kB1 has a chloroalkane tag attached at position 16 and an Alexa tag at position 20, whereas [16ax, 20ct]kB1 has the two tags switched in position. These doubly tagged peptides allowed us to use the same peptides in three different assays: CAPA, flow cytometry, and confocal fluorescence microscopy. Using these peptides, we found that the tags can affect cellular uptake and cytosolic delivery. For example, the CAPA results showed that doubly tagged peptides [16ct, 20ax]kB1 and [16ax, 20ct]kB1 have higher cytosolic delivery than singly tagged peptides [16ct, 20]kB1 and [16, 20ct]kB1, as shown in Figures 3A and B. Similarly, doubly tagged peptides have higher total cellular uptake as measured by quantitating the intensity of Alexa dye by flow cytometry, as shown in Figure 3C.
Figure 3.
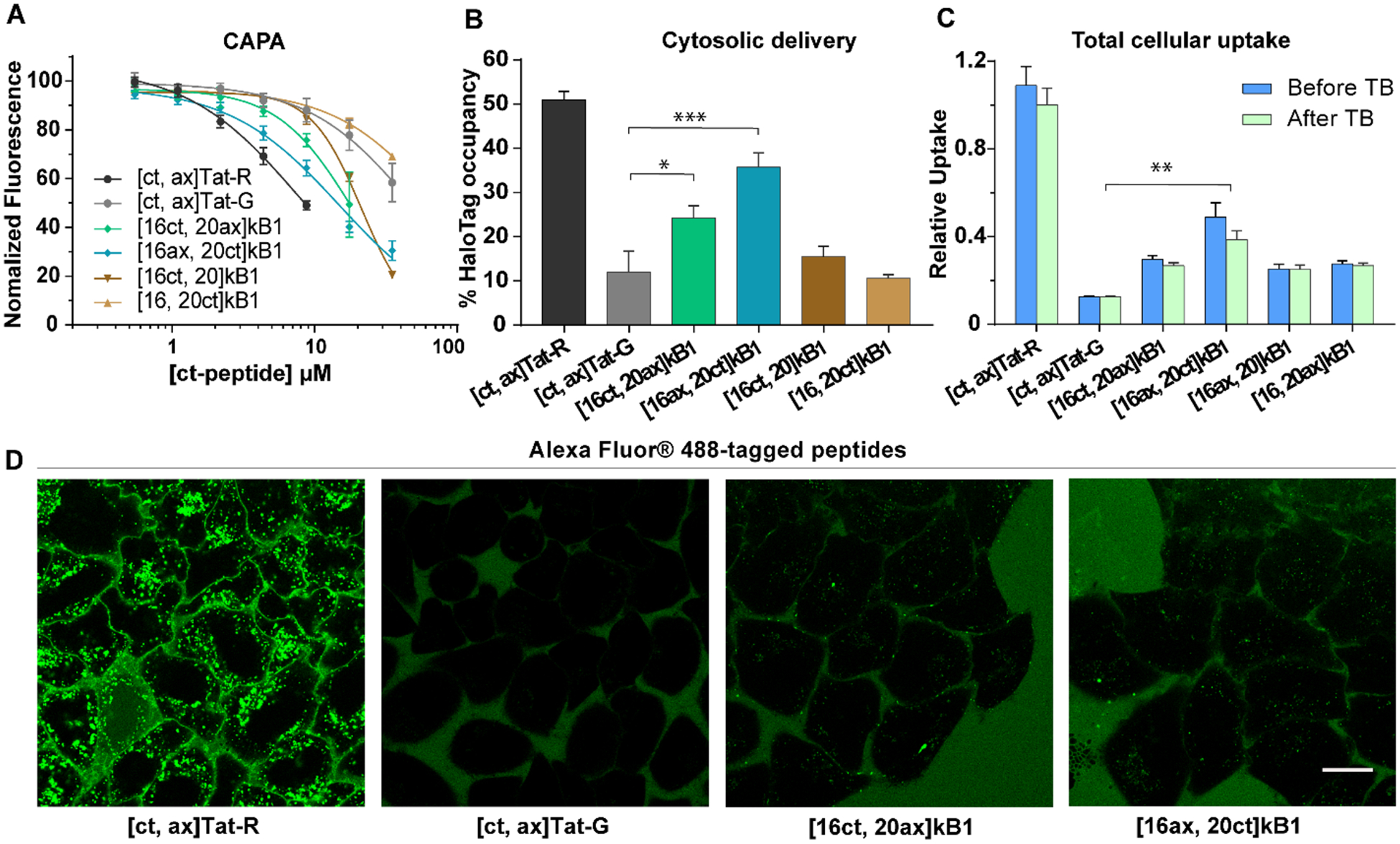
Cytosolic and total cellular uptake of doubly tagged peptides. (A) CAPA results of doubly tagged peptides over a range of concentrations. (B) The cytosolic delivery, as represented by HaloTag occupancy, of doubly tagged peptides at 8.75 μM. (C) Total cellular uptake of doubly tagged peptides at 4 μM before (blue) and after (green) the addition of TB. The mean fluorescence signal of HeLa cells treated with [ct, ax]Tat-R was used for normalization. All experiments were done in triplicate, and the results are shown as mean ± SEM. (D) Images of live cells captured using confocal microscopy of HeLa cells incubated with peptides with Alexa Fluor 488 and chloroalkane tags at 4 μM. Scale bar = 20 μm.
Together, the CAPA and Alexa-detecting flow cytometry results consistently showed that the doubly labeled kB1 peptides penetrate cells and enter the cytosol. To visualize subcellular localization, we used confocal microscopy to monitor the cellular fluorescence of [ct, ax]Tat-R, [ct, ax]Tat-G, [16ct, 20ax]kB1, and [16ax, 20ct]kB1 over time (Figure 3D and Supporting Information Figures S5, S6 and S7). Although we observed accumulation of peptides in cells between 0 and 4 h (Supporting Information Figures S5 and S6), confirming cellular uptake, we were unable to clearly identify cytosolic localization, even for Tat-R, which exhibited the highest cytosolic and cellular uptake for this set of peptides. Instead, all peptides accumulated primarily in punctate cellular structures (Figure 3D). These data suggest that the majority of peptide taken up by cells ends up entrapped inside endosomes, limiting their cytosolic concentration.
Design of Cyclotide Analogues to Investigate the Effect of Sequence on Cell Uptake.
Having shown that CAPA can provide independent and complementary information on cytosolic delivery for kB1, we were interested in applying it to understand structure–activity relationships. Many studies have shown that the sequence composition of peptides affects their cell internalization efficiency, with particular emphasis on the benefits of positive charge and hydrophobic and amphipathic compositions.38–40 Thus, as shown in Table 2, a series of analogues of kB1 (i.e., Rmut, Hmut, Wmut, Emut, Smut, and Gmut) were designed and synthesized to understand the influence of various properties on cytosolic delivery. Specifically, four types of mutations were introduced into loop 6, which is the most commonly modified loop of kB1 in peptide design and engineering studies. These mutations included positively charged (Arg, His), negatively charged (Glu), hydrophobic (Trp), and polar (Ser, Gly) amino acids. In addition, because kB1 has been widely used as a scaffold for stabilizing and delivering epitopes in molecular grafting,21 we were interested in how grafted epitopes affect cytosolic delivery. Therefore, three additional kB1 analogues having different epitopes (i.e., graft1, graft2, and graft3) of varying hydrophobic, charge, and polar amino acid compositions, as detailed in Table 2, were also synthesized.
Structural Characterization of Cyclotide Analogues.
Analysis of the chemical shifts of kB1 analogues was used to assess their tertiary folds. As shown in Figure 4A, the secondary Hα chemical shift pattern of the lysine mutant [16]kB1 was essentially identical to that of native kB1, indicating that [16]kB1 has native-like tertiary structure. All other analogues shown in Figure 4 have similar secondary Hα chemical shifts to the parent peptide over the region from loops 1 to 5, suggesting that the analogues were correctly folded and the introduction of mutations and epitopes did not significantly affect the overall structure of the scaffold. As expected, chemical shift differences were observed in the regions where changes were introduced, loop 6 and the region adjacent to it (part of loop 1).
Figure 4.
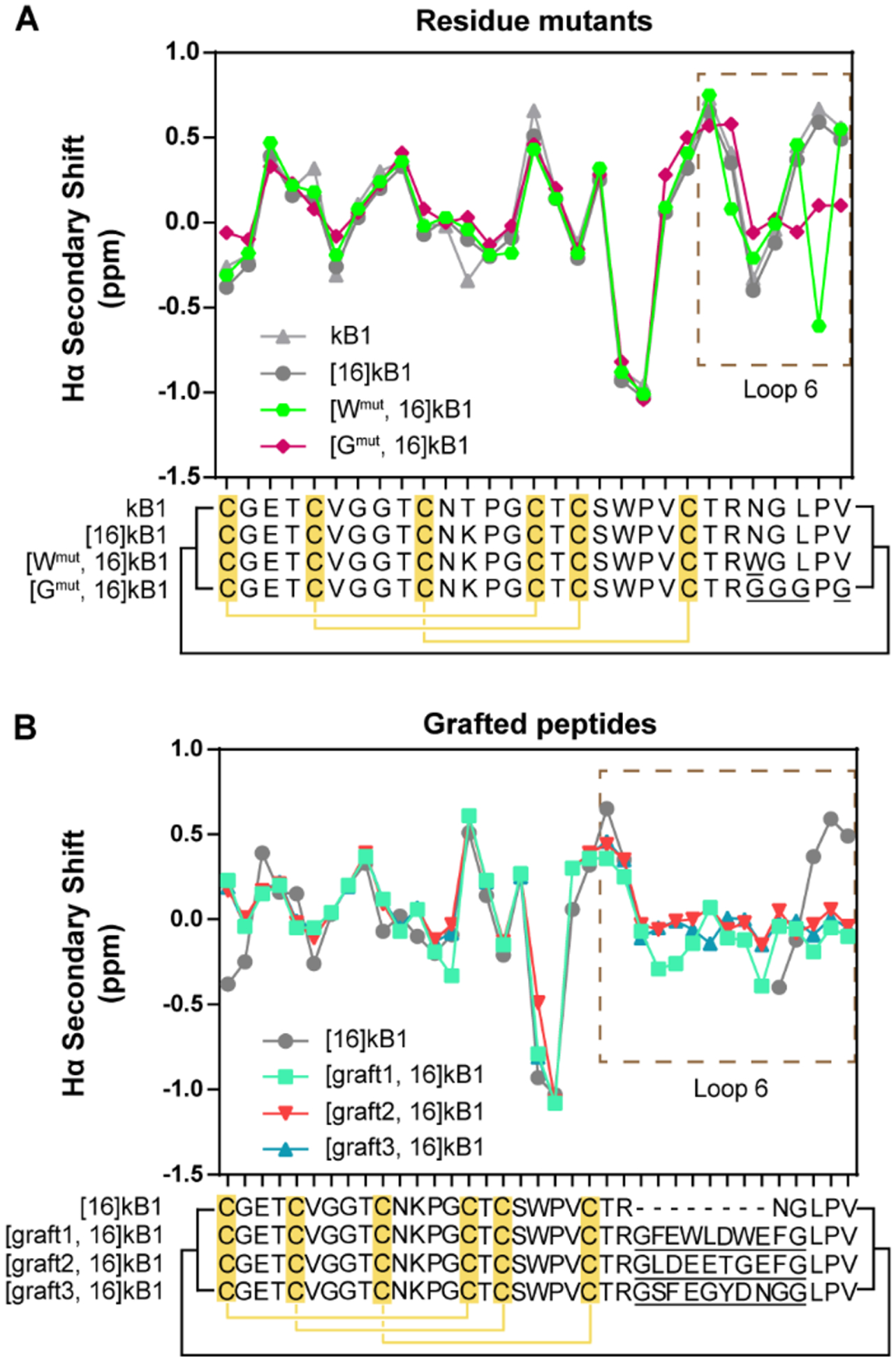
Structural characterization of cyclotide analogues using NMR. (A) Hα chemical shift analysis of kB1 analogues with mutations in loop 6 (residue mutants). (B) Hα chemical shift analysis of kB1 analogues with epitopes grafted into loop 6 (grafted peptides). Each point in the line graph presents the Hα secondary shift of an individual residue, and the sequence alignment of the peptides is shown below, with mutated residues in loop 6 underlined. The disulfide connectivity is indicated by yellow lines and the cyclic backbone by black lines. The region of kB1 with mutations and grafted epitopes (loop 6) is highlighted in a dashed line box.
Cytosolic Entry of Cyclotide Analogues.
Surprisingly, the CAPA results showed that nearly all mutations tested had no apparent influence on cytosolic delivery. The single exception was [Wmut, 16ct]kB1, which had a single asparagine to tryptophan substitution (Figure 5A). The cytosolic delivery of [Wmut, 16ct]kB1 was significantly higher than that of [16ct]kB1 and [ct]Tat-R, indicating that improvement in cytosolic delivery can be achieved by modifying loop 6.
Figure 5.
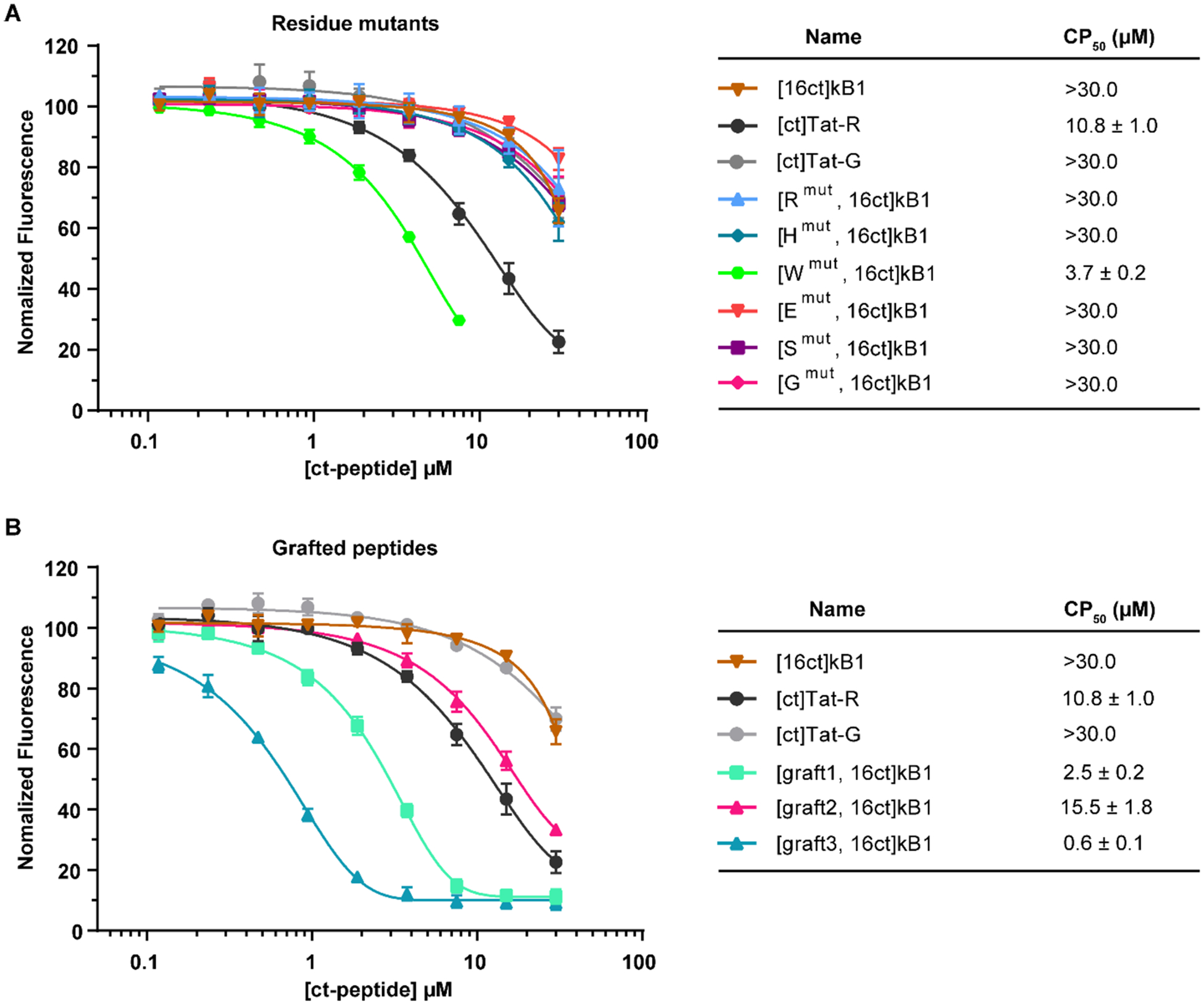
Cytosolic uptake of kB1 residue mutants and grafted peptides. Uptake was measured for chloroalkane tagged peptides using CAPA. (A) Results for kB1 analogues with mutations in loop 6. (B) Results for kB1 analogues with grafted epitopes in loop 6. [ct]Tat-R, [ct]Tat-G, and [16ct]kB1 were included as controls. CP50 values of peptides are shown in the right panel. The data were normalized using a no-pulse control as 100% signal and a no-pulse/no-chase control as 0% signal. All experiments were done in triplicate, and the results are represented as mean ± SEM.
The cytosolic delivery of the grafted peptides [graft1, 16ct]kB1, [graft2, 16ct]kB1, and [graft3, 16ct]kB1 were all higher than [16ct]kB1, as shown in Figure 5B. Of the three epitopes, graft1 and graft2 showed moderate improvements in cytosolic delivery compared to the native loop 6, while graft3 showed over 15-fold improvement in cytosolic delivery compared to the native loop 6. In fact, [graft3, 16ct]kB1 showed substantial cytosolic delivery at submicromolar concentrations, with a midpoint in the CAPA dose-dependence curve (CP50) of 0.6 μM. Overall, the cytosolic delivery of [Wmut, 16ct]kB1, [graft1, 16ct]kB1, and [graft3, 16ct]kB1 were each higher than [ct]Tat-R, which indicated that they could more efficiently enter the cytosolic space than this Tat analogue.
Total Cellular Uptake of Cyclotide Analogues.
To complement the CAPA results and further characterize the kB1 analogues, the peptides that exhibited improved cytosolic delivery, [Wmut, 16]kB1, [graft1, 16]kB1, [graft2, 16]kB1, and [graft3, 16]kB1, were labeled with Alexa Fluor 488 and their cell penetration assessed using flow cytometry and microscopy. [16]kB1, [Gmut, 16]kB1, Tat-R, and Tat-G were used as references for low and high uptake. While diffuse fluorescence is often taken as a qualitative sign of cytosolic delivery, it is impossible to exclude the signal from endosomally trapped material. Thus, confocal microscopy is not a quantitative measure of cytosolic delivery, only cellular uptake. In keeping with this conservative interpretation of confocal images, there were consistent trends across both flow cytometry and microscopy results. As shown in Figure 6 and Supporting Information Figure S8, results from both experiments showed that [ax]Tat-R had the highest cellular uptake and that [graft1, 16ax]kB1 and [Wmut, 16ax]kB1 had moderate cellular uptake. [graft2, 16ax]kB1, [graft3, 16ax]kB1, [Gmut, 16ax]kB1, [16ax]-kB1, and negative control [ax]Tat-G all showed low cellular uptake in both assays.
Figure 6.
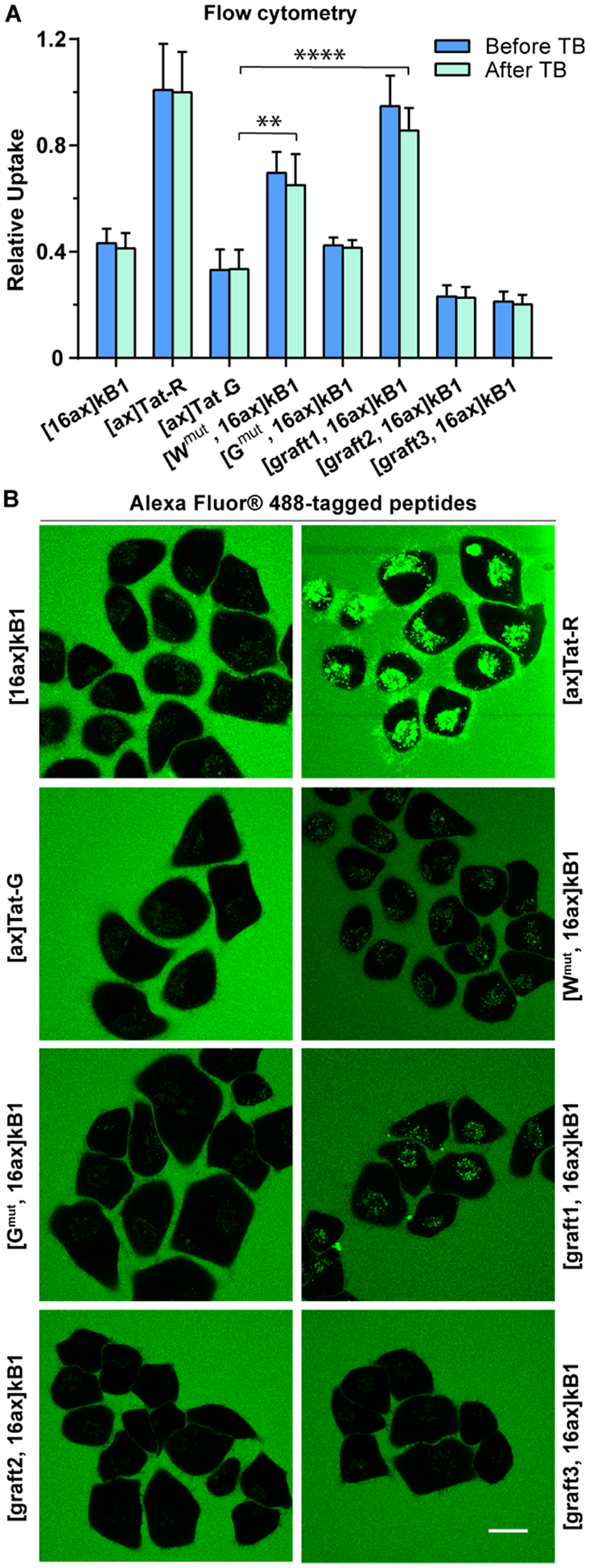
Total cellular uptake of kB1 residue mutants and grafted peptides. Uptake was measured for Alexa Fluor 488-tagged peptides at 4 μM using flow cytometry and microscopy. (A) Flow cytometry results before (blue) and after (green) the addition of TB. The mean fluorescence signal of cells treated with [ax]Tat-R after the addition of TB was used for normalization. All experiments were repeated in triplicate, and the results are represented as mean ± SEM. (B) Images of live cells captured using confocal microscopy of cells incubated with Tat-peptides and selected kB1 analogues. Images were acquired post addition of peptides, scale bar = 20 μm.
The results from direct flow cytometry and confocal fluorescence microscopy were largely consistent with the CAPA results (Figure 5). For example, [Wmut, 16]kB1 had higher cellular uptake compared to [16]kB1, [Gmut, 16]kB1, and Tat-G in flow cytometry and microscopy assays, reflecting the trend shown in the CAPA results. Similarly, the cellular uptake of [graft1, 16]kB1 was higher than that of [16]kB1 and [Gmut, 16]kB1, which is the same trend shown in the CAPA results. Both [Wmut, 16]kB1 and [graft1, 16]kB1 showed improved cell uptake and cytosolic delivery, suggesting that their epitopes were driving both endosomal uptake and endosomal escape.
However, for other analogues there were notable differences between the results from the Alexa-based assays compared to CAPA. For example, [graft3, 16ct]kB1 had high cytosolic delivery efficiency according to CAPA, but [graft3, 16ax]kB1 had low total cellular uptake in flow cytometry and microscopy assays. One explanation for these contrasting results is that the tags might alter uptake and/or endosomal escape. Additionally, CAPA results suggested that [Wmut, 16ct]kB1 and [graft1, 16ct]kB1 have higher cytosolic uptake than Tat-R, but the Alexa-based experiments suggested that corresponding kB1 analogues have lower total cellular uptake relative to Tat-R. This may also indicate different effects of the chloroalkane tag or Alexa dye, or it may indicate that a proportionally higher amount of Tat-R might be trapped in endosomes compared to either [Wmut, 16ct]kB1 or [graft1, 16ct]kB1. Overall, the flow cytometry and microscopy experiments provided a useful counterpoint to CAPA results and demonstrated that total cellular uptake and cytosolic delivery of kB1 can each be affected by modifying the composition of loop 6.
DISCUSSION AND CONCLUSIONS
Cyclotides have been widely used as peptide engineering scaffolds because of their exceptionally stable structures. Recently, new applications as intracellular delivery frameworks have been suggested due to their reported ability to internalize into cells.26,27 This study provides important new insights into both the total cellular uptake and the cytosolic penetration of the prototypic cyclotide, kB1. We also identified analogues with significantly improved cytosolic delivery.
In this work, we used traditional assays for the measurement of total cellular uptake as well as the recently developed CAPA to provide an independent measurement of cytosolic delivery. Each of these assays has strengths and limitations. A common caveat of these assays is that they all require the attachment of tags, which could potentially affect peptide uptake. We addressed this issue by studying different tags, singly tagged peptides labeled at different positions, and also doubly tagged peptides. Ideally, a tag-free assay would be used instead, but currently no such assay can be readily adapted to measuring many analogs in high-throughput format. Nevertheless, we found that kB1 can enter the cytosol but with lower efficiency than expected based on prior reports of its total cellular uptake.30 Comparison of the total cellular uptake with cytosolic delivery for kB1 and Tat-peptides showed that higher cellular uptake was generally associated with higher cytosolic delivery; however, this correlation was not always true. For example, Tat-R had higher total cellular uptake than the most cell-penetrant kB1 analogues, [Wmut, 16]kB1, [graft1, 16]kB1, and [graft3, 16]kB1, according to both flow cytometry and confocal microscopy measurements. However, Tat-R had lower cytosolic delivery according to CAPA. One explanation for these results is that Tat-R is taken up by cells more efficiently than kB1 analogues, but it then has a poorer ability to escape endosomes. This study emphasizes the importance of examining both total cellular uptake and cytosolic delivery in studies aiming to understand cell penetration.41
Cyclotides are structurally and chemically unique compared to other cell-penetrating peptides discovered thus far and might offer new opportunities to access underexplored pathways for intracellular delivery. Whereas cationic peptides penetrate cells by engaging in electrostatic interactions with the cell surface, entry of kB1 into cells is mediated by interactions between loops 1, 3, and 6 and phosphatidylethanolamine lipid head groups, and also between loops 2, 5, and 6 and hydrophobic components of the membrane.42,43 After binding the plasma membrane, kB1 appears to enter cells through endocytosis and/or membrane translocation pathways.30 It is possible that the kB1 analogues used in this study use similar pathways of cell entry because they exhibit native-like structures according to NMR and distribute into punctate patterns inside cells according to microscopy. However, their mechanisms could be subtly altered by the attachment of tags and dyes and/or by mutations in loop 6.
Analysis of the overall physicochemical properties of the kB1 analogues provides “structure–penetration relationships” to guide continued design of cyclotides with increased cytosolic delivery. Most strikingly, the kB1 analogues with the highest cytosolic delivery in CAPA and the highest total cellular uptake in our flow cytometry assay, [Wmut, 16]kB1 and [graft1, 16]kB1, both have increased overall hydrophobicity compared to wild-type kB1 (Supporting Information Figure S1). The additional hydrophobicity might add to the native hydrophobic interactions that help drive kB1 binding to cell surfaces. However, it was more difficult to derive clear conclusions on the importance of individual residues. We found that although overall hydrophobicity appears to be a determinant of both total uptake and cytosolic delivery, the number of hydrophobic residues did not correlate well with cytosolic delivery. For example, graft3 has the highest cytosolic delivery but also the least number of hydrophobic residues and the greatest number of polar residues among the three grafted peptides. Also, all three grafted peptides have higher cytosolic delivery despite the addition of multiple Glu/Asp residues in loop 6 compared to native kB1. However, negatively charged residues in loop 6 are also not correlated directly with improved cytosolic delivery because the single Glu mutant did not have improved delivery. While a previous study showing that an abundance of negative charge is correlated with poor cellular uptake for stapled peptides,44 the observation that loop 6 grafts with multiple negative charges result in kalata B1 analogues with substantial cytosolic penetration implies that cyclotides may be governed by more complex structure–penetration relationships.
Finally, we observed different but overlapping trends among kalata B1 analogues with respect to total cellular uptake (and entrapment in endosomes) versus cytosolic delivery. For example, graft1 promoted a high degree of total cellular uptake and improved cytosolic delivery, whereas graft3 promoted even better cytosolic delivery without a high degree of total cellular uptake. Effects of the tags notwithstanding, this result reinforces the importance of monitoring cytosolic delivery in peptide optimization projects because total cellular uptake can correlate with cytosolic delivery in some cases, but not in others.
Here, we report cyclotides with cytosolic delivery that is comparable to or better than a Tat peptide. Therefore, our results suggest that analogues of kB1 have potential as intracellular protein–protein interaction inhibitors. The most direct way to design inhibitors using kB1 is molecular grafting.21,45,46 As in other grafting studies, we demonstrated that analogues of kB1 having epitopes grafted into loop 6 could still adopt the native-like fold, retaining the features of kB1 that underpin its structural stability. Our study suggests that epitopes that increase hydrophobicity of kB1 are more likely to have improved cytosolic delivery and that cell-penetrant kB1 analogues can have multiple polar and negatively charged residues. These results provide valuable data for prioritizing epitopes for grafting studies targeting intracellular proteins. Another potential application of the peptides reported in this study is as vehicles for intracellular delivery. They could potentially be attached to small molecules, RNA, or other cargoes to enable intracellular delivery, mimicking the use of Tat in many applications. On the basis of the structure–penetration relationships shown here, we propose that further optimization of the kB1 scaffold will yield peptides with even higher cytosolic delivery.
Efficient approaches to transport biologics into the cytosol are highly sought after. Cyclotides are a unique class of cell-penetrating peptides that offer new solutions for the design of intracellular protein–protein interaction inhibitors and for delivery of biologics. This study provides new insights into the cellular uptake and cytosolic delivery of cyclotides, including alterations that improve delivery efficiency. This understanding has important implications for the fields of cell-penetrating peptides and peptide drug design, where both high delivery efficiency and well-formed conformations are necessary for achieving effective inhibition of intracellular protein targets.
METHODS
Peptide Synthesis and Purification.
Peptides were synthesized on 2-chlorotrityl chloride resin by Fmoc-based solid-phase peptide synthesis using an automated peptide synthesizer (Symphony, Gyros Protein Technologies).47 Linear peptides were cleaved from resin and deprotected in trifluoroacetic acid (TFA) with 2% (v/v) water and 2% (v/v) triisopropylsilane for 2.5 h while stirring. For cyclic peptides, their linear precursors were cleaved from resin by 10 repeated treatments of 1% (v/v) TFA in dichloromethane. The cleaved peptides were cyclized using the in-solution cyclization method as described previously.47 Briefly, linear peptide precursors were dissolved in dimethylformamide (DMF) with 5 mM 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate and 10 mM N,N-diisopropylethylamine (DIPEA) at a final peptide concentration of 2 mM and incubated for 6 h. Subsequently, deprotection was carried out in TFA containing 2% (v/v) triisopropylsilane and 2% (v/v) water by stirring for 2.5 h, and the crude peptides were precipitated with ice-cold diethyl ether, followed by extraction with 45% (v/v) acetonitrile/0.05% (v/v) TFA/water twice.
Linear and cyclic peptides were purified using preparative reversed-phase high-performance liquid chromatography (RP-HPLC) on a C18 column with a water/acetonitrile (0.05% v/v TFA) eluent system. Formation of disulfide bonds for all cyclotides was achieved by air oxidization in 0.1 M ammonium bicarbonate (pH 8.0), except for [Rmut, 16]kB1, which used a 0.1 M disodium phosphate/monosodium phosphate (pH 8.0) buffer instead. All oxidation buffers additionally contained 40% or 50% (v/v) 2-propanol and 2 mM reduced glutathione. Oxidation was performed overnight while stirring and stopped using TFA (2% v/v final concentration). Peptides were purified using RP-HPLC, their molecular weights measured by electrospray ionization mass spectrometry (ESI-MS) and purity analyzed by ultraperformance liquid chromatography (UPLC), as shown in Supporting Information Figures S1 and S2.
Design of Kalata B1 Grafted Analogues.
The grafted peptides [graft1]kB1, [graft2]kB1, and [graft3]kB1 were designed by replacing loop 6 of kB1 with three different foreign peptide epitopes. The epitopes were obtained by screening the PDB for structures of peptide fragments bound to proteins using an in-house script. They were selected based on their sequence length (3–15 amino acids) and amino acid composition to represent epitopes of varying hydrophobic, charged, and polar amino acid content. The introduced epitope of [graft1]kB1 is from a peptide that binds HDM2 (PDB ID: 2AXI), [graft 2]kB1 from a peptide that binds Keap1 (PDB ID: 6FMP), and [graft 3]kB1 from a peptide that binds Grb7 (PDB ID: 5EEL). A Gly residue was added to both ends of each epitope to increase solvent exposure of the epitope in the grafted peptide.
Chloroalkane Tag.
The chloroalkane carboxylic acid was made as previously described.33 To make chloroalkane N-hydroxysuccinimide, 22 mg of chloroalkane carboxylic acid, 50 mg of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide, and 100 mg of N-hydroxysuccinimide were added into 6 mL of 0.1 M 2-(N-morpholino)-ethanesulfonic acid (pH 5.8), 0.5 M sodium chloride, and 25% v/v acetonitrile, mixed and incubated for 15 min at RT. The mixture was diluted with 0.05% (v/v) TFA before isolation of the chloroalkane N-hydroxysuccinimide product using RP-HPLC on a C18 column with a water/acetonitrile (0.05% v/v TFA) eluent system.
Peptide Labeling.
To label peptides with chloroalkane, ~1 mg of peptide and 2.5 mg of chloroalkane N-hydroxysuccinimide were dissolved in 350 μL of DMF with 3.5 μL of DIPEA, mixed, and incubated at RT for 3 h. Ten microliters of TFA was added to stop the reaction, and the labeled peptides were purified using RP-HPLC after diluting with 0.05% (v/v) TFA. The purity and identities of labeled peptides were checked by UPLC and MALDI-TOF mass spectrometry Supporting Information Figures S1 and S2.
To label peptides with Alexa Fluor 488, ~1 mM of peptides and 1.5 mM Alexa Fluor 488 sulfodichlorophenol ester were dissolved in 98 μL of DMF with 2 μL of DIPEA, or in 100 μL of 0.1 M sodium bicarbonate (pH 8.0). After incubating at RT for >1 h, the sample was diluted with 0.05% (v/v) TFA before purification of the labeled peptide using RP-HPLC.
To make peptides labeled with both chloroalkane and Alexa Fluor 488, a two-step labeling procedure using orthogonally protected peptides was employed. Synthesis, oxidation, and purification of these peptides were carried out as described above. In the first step of the labeling procedure, chloroalkane was conjugated onto a primary lysine site using the method described above. During this reaction, the secondary lysine site intended for attachment to the Alexa dye was protected by an alloc-protecting group. Removal of the alloc group to allow for Alexa conjugation was performed in a solution of 2.5 mL of MeOH containing 1.5 mg of peptide, 0.45 mg of Pd(PPh3)4, and 2.4 mM K2CO3 overnight at RT.48 The reaction was diluted to 20 mL using 0.05% (v/v) TFA before purification using RP-HPLC.
Chloroalkane-TAMRA Synthesis.
As previously described,33 to make the chloroalkane-TAMRA (ct-TAMRA, ct-dye), 10 mg of 5/6-TAMRA-succinimidyl ester was dissolved in 200 μL of DMF, followed by the addition of 4.5 mg of the chloroalkane carboxylic acid and 32 μL of DIPEA into the solution. After stirring for 2 h at RT, the sample was diluted with 200 μL of water and purified by RP-HPLC.
Chloroalkane Penetration Assay.
CAPA was developed to measure cytosolic delivery of exogenously added molecules (Figure 2A).33 Briefly, the assay uses Halo-GFP-Mito cells, which stably express a fusion protein comprising HaloTag, GFP, and a mitochondria-targeting peptide. The fusion protein localizes on the outer surface of mitochondria, with the HaloTag component exposed in the cytosol.33,49 HaloTag protein rapidly catalyzes covalent bond formation between itself and a chloroalkane moiety, which is otherwise biorthogonal to cellular components.50,51 If a chloroalkane-tagged peptide (ct-peptide) can access the cytosol, it will covalently attach to the HaloTag, preventing reaction with a chloroalkane-tagged reporter dye (ct-dye) during a subsequent chase step. Fluorescence intensity measured using a flow cytometer provides a quantitative measure of the cytosolic localization of the ct-peptide, since the amount of ct-peptide that accessed the cytosol is inversely proportional to fluorescence intensity of the ct-dye.
Halo-GFP-Mito cells were cultured using Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 1% Pen/Strep, and 1 μg/mL puromycin and kept at 37 °C under 5% CO2. Cells were seeded in a 96-well plate with cell culture media approximately 16 h before the assay. Before performing the assay, the medium was replaced with 100 μL of fresh Opti-MEM. For the preparation of peptide samples, the concentration of peptides was measured using UV/vis spectroscopy at 280 nm (NanoDrop 2000c) and calculated extinction coefficients, and then peptide stocks in DMSO were serially diluted with 3% DMSO in water to maintain a consistent percentage of DMSO with the most concentrated peptide sample. Nine different concentrations were tested for each peptide, starting with a maximum peptide concentration of 30 μM, which was serially diluted 2-fold down to 0.117 μM. The cells in each well were treated with 25 μL of peptide solution, with the final concentration of DMSO at approximately 0.6% after addition into 96-well plates, and incubated at 37 °C with 5% CO2 for 4 h. After incubation, the medium was aspirated, and the cells were washed with fresh Opti-MEM. Then, the cells were chased using 5 μM ct-TAMRA (ct-dye) in Opti-MEM, except for control wells which received only Opti-MEM. After incubation for 15 min, the contents of the wells were aspirated and washed with fresh Opti-MEM for 30 min. Finally, cells were trypsinized and resuspended in PBS. The fluorescence intensity of live cells, gated as described using side scatter and green fluorescence intensity,33 was measured using a benchtop flow cytometer (Guava EasyCyte, EMD Millipore). On the basis of the percentage of live cells gated from the total input population, none of the peptides showed significant effects on cell viability, with the exception of [Wmut, 16ct]kB1, as shown in Supporting Information Figure S11. Concentrations causing apparent cell toxicity were excluded from further CAPA analysis. The data were normalized using a no-pulse control as 100% uptake and a no-pulse/no-chase control as 0% uptake, as described.33
Structural Characterization using Nuclear Magnetic Resonance (NMR).
NMR was used to characterize the structure of kalata B1 and its analogues. Briefly, peptides were dissolved in 90% H2O/10% D2O (v/v) with a final concentration of ~2 mg mL−1. All one- and two-dimensional TOCSY and NOESY spectra of peptides were acquired on a Bruker Avance-600 MHz spectrometer at 298 K, processed using Topspin 1.2 (Bruker), and analyzed using CCPNMR 2.2.2. The mixing time was 80 and 200 ms for TOCSY and NOESY spectra, respectively. 2,2-Dimethyl-2-silapentane-5-sulfonic acid (DSS) was used as an internal reference (0 ppm). Secondary Hα chemical shifts were calculated using previously reported random coil chemical shifts.52
Internalization Assay of Fluorescently Labeled Peptides Using Flow Cytometry.
The cellular uptake of Alexa Fluor 488-labeled peptides was measured using flow cytometry as described previously.36 In brief, HeLa cells were cultured using DMEM supplemented with 10% FBS and 1% Pen/Strep and kept at 37 °C under 5% CO2. Cells were seeded in a 24-well plate (1 × 105 cells/well) 24 h before the internalization assay. Stock solutions (10×) of Alexa Fluor 488 labeled peptides were prepared in 10% (v/v) DMSO and quantified using NanoDrop 2000c according to the extinction coefficient of Alexa Fluor 488 (71,000 M−1 cm−1 at 495 nm). Peptide solutions were further diluted in serum-free DMEM to the desired concentrations, i.e., 4 and 8 μM, and incubated with HeLa cells for 4 h at 37 °C. After 4 h of incubation, the peptide solutions were aspirated from each well, and the cells were rinsed with PBS, trypsinized, harvested by spinning down at 500g at 4 °C, and finally resuspended in ice-cold PBS. Fluorescence intensity of the cells, before and after the addition of trypan blue (160 μg/mL), was measured using a flow cytometry system (BD FACSCanto II) by analyzing 10,000 cells per sample, with excitation at 488 nm and emission at 530/30 nm.
Live Cell Imaging Using Confocal Microscopy.
To track the intracellular delivery of the Alexa Fluor 488-labeled peptides, confocal microscopy was used to monitor the localization of peptides inside cells. HeLa cells were seeded in an 8-well chamber slide (1 × 104 cells/well) 24 h prior to the assay. Peptide stock solutions were prepared and quantified as previously mentioned and diluted with serum-free DMEM (without phenol red) containing a Fluor 647 conjugate of wheat germ agglutinin (1:500, v/v) to a final concentration of 4 μM. Cells in the chambers were rinsed twice with serum-free DMEM (without phenol red) before incubation with peptide solutions individually. Time-lapse images were acquired using a Zeiss 880 LSM confocal equipped with a 63× 1.4 NA Plan Apochromat [OR 40× 1.3 NA Plan Apochromat] objective running Zeiss Zen Black software at various time points between 0 and 4 h. Alexa Fluor 488-labeled Tat and Tat-G were included as positive and negative controls. Bright field images indicated the peptides had no effect on cell morphology (Supporting Information Figure S9 and S10). During the experiment, cells were incubated at 37 °C with 5% CO2.
Cytotoxicity Assay.
The cytotoxicity of [20ct]kB1, [16ct]kB1, and [graft3, 16ct]kB1 against HeLa cells was evaluated using a resazurin-based method as described previously.36 Briefly, HeLa cells were cultured and seeded in a 96-well microplate (5000 cells/well) 16 h before the assay. The 100× DMSO stock solutions of ct-peptides were diluted with fresh DMEM to reach the desired starting concentration of 30 μM, followed by a 2-fold serial dilution with 1% DMSO in DMEM. Hela cells were treated with ct-peptides for 4 h at 37 °C before the addition of 0.005% resazurin solution. The fluorescence signal of resorufin (the reduced product of resazurin) was monitored at 584 nm using a microplate reader (Tecan M1000 Pro), and the percentage of cytotoxicity was calculated relative to cells treated with 0.1% Triton-X (100% inhibition). The experiments were performed in triplicate, and the results are shown in Supporting Information Figure 12.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Health and Medical Research Council (APP1107403), an Australian Research Council Australian Laureate Fellowship (FL150100146), and the USA National Institutes of Health (GM127585). H.Y. was supported by the China Scholarship Council. Microscopy was performed at the Australian Cancer Research Foundation (ACRF)/Institute for Molecular Bioscience Cancer Biology Imaging Facility, which was established with the support of the ACRF.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00297.
UPLC traces of peptides; MALDI-TOF mass spectra of peptides; flow cytometry measurements of peptide cellular uptake; confocal microscopy of peptides with live cells; sequence and chemical structure of tested peptides (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00297
Contributor Information
Kirsten Deprey, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States.
Nicholas D. Condon, Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia;.
Joshua A. Kritzer, Department of Chemistry, Tufts University, Medford, Massachusetts 02155, United States;.
REFERENCES
- (1).Morrison C (2018) Constrained peptides’ time to shine? Nat. Rev. Drug Discovery 17, 531–533. [DOI] [PubMed] [Google Scholar]
- (2).Craik DJ, Fairlie DP, Liras S, and Price D (2013) The future of peptide-based drugs. Chem. Biol. Drug Des 81, 136–147. [DOI] [PubMed] [Google Scholar]
- (3).Gavenonis J, Sheneman BA, Siegert TR, Eshelman MR, and Kritzer JA (2014) Comprehensive analysis of loops at protein-protein interfaces for macrocycle design. Nat. Chem. Biol 10, 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kale SS, Villequey C, Kong X-D, Zorzi A, Deyle K, and Heinis C (2018) Cyclization of peptides with two chemical bridges affords large scaffold diversities. Nat. Chem 10, 715–723. [DOI] [PubMed] [Google Scholar]
- (5).Nawatha M, Rogers JM, Bonn SM, Livneh I, Lemma B, Mali SM, Vamisetti GB, Sun H, Bercovich B, Huang Y, Ciechanover A, Fushman D, Suga H, and Brik A (2019) De novo macrocyclic peptides that specifically modulate Lys48-linked ubiquitin chains. Nat. Chem 11, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kintzing JR, and Cochran JR (2016) Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr. Opin. Chem. Biol 34, 143–150. [DOI] [PubMed] [Google Scholar]
- (7).Dougherty PG, Sahni A, and Pei D (2019) Understanding cell penetration of cyclic peptides. Chem. Rev 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Valeur E, Guéret SM, Adihou H, Gopalakrishnan R, Lemurell M, Waldmann H, Grossmann TN, and Plowright AT (2017) New modalities for challenging targets in drug discovery. Angew. Chem., Int. Ed 56, 10294–10323. [DOI] [PubMed] [Google Scholar]
- (9).Milletti F (2012) Cell-penetrating peptides: classes, origin, and current landscape. Drug Discovery Today 17, 850–860. [DOI] [PubMed] [Google Scholar]
- (10).Nakase I, Akita H, Kogure K, Gräslund A, Langel Ü, Harashima H, and Futaki S (2012) Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc. Chem. Res 45, 1132–1139. [DOI] [PubMed] [Google Scholar]
- (11).Hoyer J, and Neundorf I (2012) Peptide vectors for the nonviral delivery of nucleic acids. Acc. Chem. Res 45, 1048–1056. [DOI] [PubMed] [Google Scholar]
- (12).Patel KD, De Zoysa GH, Kanamala M, Patel K, Pilkington LI, Barker D, Reynisson J, Wu Z, and Sarojini V (2020) Novel cell-penetrating peptide conjugated proteasome inhibitors: Anticancer and antifungal investigations. J. Med. Chem 63, 334–348. [DOI] [PubMed] [Google Scholar]
- (13).Ramaker K, Henkel M, Krause T, Röckendorf N, and Frey A (2018) Cell penetrating peptides: a comparative transport analysis for 474 sequence motifs. Drug Delivery 25, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, and Pei D (2016) Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nischan N, Herce HD, Natale F, Bohlke N, Budisa N, Cardoso MC, and Hackenberger CPR (2015) Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem., Int. Ed 54, 1950–1953. [DOI] [PubMed] [Google Scholar]
- (16).Lattig-Tunnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, and Cardoso MC (2011) Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun 2, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lian W, Jiang B, Qian Z, and Pei D (2014) Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J. Am. Chem. Soc 136, 9830–9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mandal D, Nasrolahi Shirazi A, and Parang K (2011) Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem., Int. Ed 50, 9633–9637. [DOI] [PubMed] [Google Scholar]
- (19).Qian Z, Liu T, Liu Y-Y, Briesewitz R, Barrios AM, Jhiang SM, and Pei D (2013) Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cascales L, Henriques ST, Kerr MC, Huang Y-H, Sweet MJ, Daly NL, and Craik DJ (2011) Identification and characterization of a new family of cell-penetrating peptides: Cyclic cell-penetrating peptides. J. Biol. Chem 286, 36932–36943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang CK, and Craik DJ (2018) Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol 14, 417–427. [DOI] [PubMed] [Google Scholar]
- (22).Daly NL, Koltay A, Gustafson KR, Boyd MR, Casas-Finet JR, and Craik DJ (1999) Solution structure by NMR of circulin A: A macrocyclic knotted peptide having anti-HIV activity. J. Mol. Biol 285, 333–345. [DOI] [PubMed] [Google Scholar]
- (23).Colgrave ML, and Craik DJ (2004) Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 43, 5965–5975. [DOI] [PubMed] [Google Scholar]
- (24).Yin H, Craik DJ, and Wang CK (2019) Anchor residues guide form and function in grafted peptides. Angew. Chem., Int. Ed 58, 7652–7656. [DOI] [PubMed] [Google Scholar]
- (25).Poth AG, Chan LY, and Craik DJ (2013) Cyclotides as grafting frameworks for protein engineering and drug design applications. Biopolymers 100, 480–491. [DOI] [PubMed] [Google Scholar]
- (26).Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, Neamati N, Shekhtman A, and Camarero JA (2013) In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc 135, 11623–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).D’Souza C, Henriques ST, Wang CK, Cheneval O, Chan LY, Bokil NJ, Sweet MJ, and Craik DJ (2016) Using the MCoTI-II cyclotide scaffold to design a stable cyclic peptide antagonist of SET, a protein overexpressed in human cancer. Biochemistry 55, 396–405. [DOI] [PubMed] [Google Scholar]
- (28).Contreras J, Elnagar AYO, Hamm-Alvarez SF, and Camarero JA (2011) Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J. Controlled Release 155, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Henriques ST, Peacock H, Benfield AH, Wang CK, and Craik DJ (2019) Is the mirror image a true reflection? Intrinsic membrane chirality modulates peptide binding. J. Am. Chem. Soc 141, 20460–20469. [DOI] [PubMed] [Google Scholar]
- (30).Henriques ST, Huang YH, Chaousis S, Sani MA, Poth AG, Separovic F, and Craik DJ (2015) The prototypic cyclotide kalata B1 has a unique mechanism of entering cells. Chem. Biol 22, 1087–1097. [DOI] [PubMed] [Google Scholar]
- (31).Peraro L, and Kritzer JA (2018) Emerging methods and design principles for cell-penetrant peptides. Angew. Chem., Int. Ed 57, 11868–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Deprey K, Becker L, Kritzer J, and Plückthun A (2019) Trapped! A critical evaluation of methods for measuring total cellular uptake versus cytosolic localization. Bioconjugate Chem. 30, 1006–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Peraro L, Deprey KL, Moser MK, Zou Z, Ball HL, Levine B, and Kritzer JA (2018) Cell penetration profiling using the chloroalkane penetration assay. J. Am. Chem. Soc 140, 11360–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Méndez-Ardoy A, Lostalé-Seijo I, and Montenegro J (2019) Where in the cell is our cargo? Methods currently used to study intracellular cytosolic localisation. ChemBioChem 20, 488–498. [DOI] [PubMed] [Google Scholar]
- (35).Peraro L, Zou Z, Makwana KM, Cummings AE, Ball HL, Yu H, Lin Y-S, Levine B, and Kritzer JA (2017) Diversity-oriented stapling yields intrinsically cell-penetrant inducers of autophagy. J. Am. Chem. Soc 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Huang YH, Chaousis S, Cheneval O, Craik DJ, and Henriques ST (2015) Optimization of the cyclotide framework to improve cell penetration properties. Front. Pharmacol 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Huang Y-H, Colgrave ML, Clark RJ, Kotze AC, and Craik DJ (2010) Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J. Biol. Chem 285, 10797–10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ziegler A (2008) Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Delivery Rev 60, 580–597. [DOI] [PubMed] [Google Scholar]
- (39).Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, and Rothbard JB (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).D’Souza C, Henriques ST, Wang CK, and Craik DJ (2014) Structural parameters modulating the cellular uptake of disulfide-rich cyclic cell-penetrating peptides: MCoTI-II and SFTI-1. Eur. J. Med. Chem 88, 10–18. [DOI] [PubMed] [Google Scholar]
- (41).Shin M-K, Hyun Y-J, Lee JH, and Lim H-S (2018) Comparison of cell permeability of cyclic peptoids and linear peptoids. ACS Comb. Sci 20, 237–242. [DOI] [PubMed] [Google Scholar]
- (42).Henriques ST, Huang Y-H, Castanho MARB, Bagatolli LA, Sonza S, Tachedjian G, Daly NL, and Craik DJ (2012) Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem 287, 33629–33643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang CK, Wacklin HP, and Craik DJ (2012) Cyclotides insert into lipid bilayers to form membrane pores and destabilize the membrane through hydrophobic and phosphoethanolamine-specific interactions. J. Biol. Chem 287, 43884–43898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossmann TN, Yeh JTH, and Verdine GL (2015) Towards understanding cell penetration by stapled peptides. MedChemComm 6, 111–119. [Google Scholar]
- (45).Gunasekera S, Foley FM, Clark RJ, Sando L, Fabri LJ, Craik DJ, and Daly NL (2008) Engineering stabilized vascular endothelial growth factor-A antagonists: Synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides. J. Med. Chem 51, 7697–7704. [DOI] [PubMed] [Google Scholar]
- (46).Wong CTT, Rowlands DK, Wong C-H, Lo TWC, Nguyen GKT, Li H-Y, and Tam JP (2012) Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem., Int. Ed 51, 5620–5624. [DOI] [PubMed] [Google Scholar]
- (47).Cheneval O, Schroeder CI, Durek T, Walsh P, Huang YH, Liras S, Price DA, and Craik DJ (2014) Fmoc-based synthesis of disulfide-rich cyclic peptides. J. Org. Chem 79, 5538–5544. [DOI] [PubMed] [Google Scholar]
- (48).Vutukuri DR, Bharathi P, Yu Z, Rajasekaran K, Tran MH, and Thayumanavan S (2003) A mild deprotection strategy for allyl-protecting groups and its implications in sequence specific dendrimer synthesis. J. Org. Chem 68, 1146–1149. [DOI] [PubMed] [Google Scholar]
- (49).Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, and Chenoweth DM (2014) Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nat. Commun 5, 5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, and Wood KV (2008) HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- (51).Friedman Ohana R, Kirkland TA, Woodroofe CC, Levin S, Uyeda HT, Otto P, Hurst R, Robers MB, Zimmerman K, Encell LP, and Wood KV (2015) Deciphering the cellular targets of bioactive compounds using a chloroalkane capture tag. ACS Chem. Biol 10, 2316–2324. [DOI] [PubMed] [Google Scholar]
- (52).Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, and Sykes BD (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.