

Abstract
Objective:
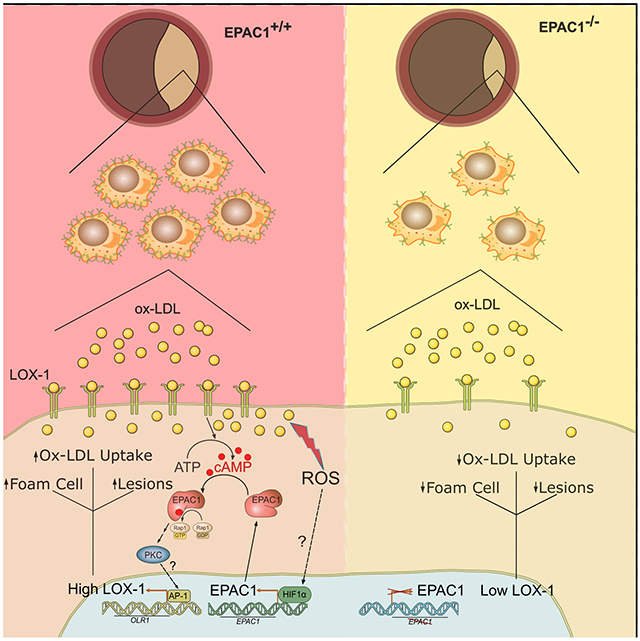
The cAMP second messenger system, a major stress-response pathway, play essential roles in normal cardiovascular functions and in pathogenesis of heart diseases. Herein, we test the hypothesis that the exchange protein directly activated by cAMP 1 (Epac1) acts as a major down-stream effector of cAMP signaling to promote atherogenesis and represents a novel therapeutic target.
Approach and Results:
To ascertain Epac1’s function in atherosclerosis development, a triple knockout mouse model (LTe) was generated by crossing Epac1−/− mice with atherosclerosis-prone LDb mice lacking both Ldlr and Apobec1. Deletion of Epac1 led to a significant reduction of atherosclerotic lesion formation as measured by post-mortem staining, accompanied by attenuated macrophage/foam cell infiltrations within atherosclerotic plaques as determined by immunofluorescence staining in LTe animals compared to LDb littermates. Primary bone-marrow derived macrophages (BMDM) were isolated from Epac1-null and wild type mice to investigate the role of Epac1 in lipid uptake and foam cell formation. Oxidized-LDL (ox-LDL) stimulation of BMDMs led to elevated intracellular cAMP and Epac1 levels, whereas an Epac-specific agonist, increased lipid accumulation in wild type, but not Epac1-null BMDMs. Mechanistically, Epac1 acts through PKC to upregulate oxidized low-density lipoprotein receptor 1 (LOX-1), a major scavenger receptor for ox-LDL uptake, exerting a feedforward mechanism with ox-LDL to increase lipid uptake and propel foam cell formation and atherogenesis.
Conclusions:
Our study demonstrates a fundamental role of cAMP/Epac1 signaling in vascular remodeling by promoting ox-LDL uptake and foam cell formation during atherosclerosis lesion development. Therefore, Epac1 represents a promising, unexplored therapeutic target for atherosclerosis.
Keywords: Atherosclerosis, atherogenesis, foam cells, cyclic nucleotide, LOX-1, Vascular Diseases, Cell Signaling/Signal Transduction, Vascular Biology, Coronary Artery Disease
Graphical Abstract
INTRODUCTION
Atherogenesis is a progressive inflammatory process of lipid-rich lesion growth that involves complex interactions among endothelial cells, macrophages, and vascular smooth muscle cells (VSMCs). Initial local shear stress and/or inflammation activates endothelial cells to recruit monocytes, which migrate into the sub-endothelial space differentiating into macrophages and subsequently “foam cells” by ingestion of native and modified/oxidized low density lipoproteins (ox-LDL). Foam cells loaded with cholesterol esters, together with T lymphocytes, form the fatty streak, a hallmark of atherosclerosis development. The presence of the fatty streak, a chronic inflammatory state, further activates VSMCs, manifested by phenotypic switching from contractile to synthetic properties, promoting their proliferation and migration from the media into the intima, where VSMCs readily deposit extracellular matrix proteins and take up modified lipoproteins. This feedforward process perpetuates and ultimately provokes the formation of atheromatous plaques in the inner lining of the arteries.1
The cAMP second messenger is a major stress-response signal found to perform important roles in cardiovascular functions. In vertebrates, with the exceptions of tissue-specific cyclic nucleotide regulated ion channels,2 the effects of cAMP are mainly transduced by two ubiquitously-expressed intracellular cAMP receptors, the classic protein kinase A (PKA) and the more recently discovered exchange protein directly activated by cAMP (Epac).3,4 Dependent upon relative abundance, tissue distribution and localization, as well as precise cellular environment, these two intracellular cAMP receptors may act independently, converge synergistically or oppose each other in regulating a specific cellular function.5,6 Two isoforms of Epac exist, Epac1 and Epac2, which are products of independent genes. While Epac1 is ubiquitously expressed in all tissues, Epac2 has a limited tissue distribution, detected predominantly in the brain, pancreas and adrenal gland.3,4 Although both isoforms are thought to act on the same immediate down-stream effectors, the Ras superfamily small GTPases Rap1 and Rap2, Epac1 and Epac2’s cellular functions are mostly non-redundant due to distinct tissue distribution and the ability to form discrete signalosomes at various cellular loci through interaction with specific cellular partners.7–10
Extensive studies, particularly recent in vivo analyses of Epac1 and Epac2 functions using genetic knockout mouse models and pharmacological probes, reveal that Epac proteins regulate a wide range of processes through coordinated interactions with a plethora of intracellular signaling molecules in a precise spatiotemporal fashion.7–10 Between the two isoforms, Epac1 is highly abundant in blood vessels.11 Recent studies from our laboratory, as well as others, in multiple independent vascular injury models demonstrate that the expression of Epac1 is upregulated in response to vascular injuries and that deletion of Epac1 suppresses neointimal formation, retinal neovascularization and neurodegeneration induced by vascular injuries.12–16 These results suggest that Epac1 plays an important role during vascular remodeling and supports involvement in the development of proliferative vascular diseases. In this study, we delineate pathophysiological vascular functions of Epac1 by defining a major role of Epac1 in the development of atherosclerosis through promotion of macrophage recruitment and accumulation of foam cells within atherosclerotic plaques, establishing Epac1 as a potential therapeutic target for an expansive repertoire of vascular proliferative diseases.
METHODS
Data Availability
Detailed methods are provided below and within the Extended Methods of the Supplemental Materials for all procedures carried out in this study. The authors declare that all supporting data are available within the article and the online supplementary files.
Study Design
The primary objectives of this project were to assess the function of Epac1 in the development of atherosclerosis and further investigate the underlying molecular mechanism. Building upon previous studies in restenosis animal models we initially hypothesized that inhibition of Epac1 would functionally protect against atherosclerosis development. We chose the LDb atherogenic-prone mouse model lacking both the Ldlr and Apobec1 genes for our model organism. While for our purposes the use of the LDb model requires the breeding of triple knockout mice (LTe; LDb × Epac1−/−) and is technically more challenging, this model is more clinically relevant as LDb mice develop atherosclerotic lesions spontaneously on standard laboratory diet and exhibit elevated plasma levels of apoB100-containing VLDL and LDL with reduced plasma levels of HDL closely mirroring human pathologies.17,18 To achieve a robust dataset, a minimum of 10 littermate pairs of age- and sex-matched animals were used for phenotypic characterization. A pre-defined endpoint of 8 months was initially set for all in vivo studies in accordance with previous knowledge of the model. Another endpoint at 11 months was added for female mice after observing sex dimorphism in lesion growth at 8 months. Phenotypic characterization of LTe and LDb mice at the whole-body and cellular levels led us to further hypothesize that Epac1 participates in foam cell formation through induction of scavenger receptors or reduction in lipid transport channels. For in vitro mechanistic studies, we elected to use primary BMDMs isolated from Epac1−/− and WT mice, over those derived from LTe and LDb mice, in order to demonstrate the primary observations were attributable to Epac1 signaling specifically and not confounded by the genetic deletion of other components in the intact animal. All methods described herein were conducted in adherence to the guidelines set forth by the AHA for experimental atherosclerosis studies.
Generation of Ldlr−/−/Apobec1−/−/Epac1−/− (LTe) Mice
C57BL/6NCrl Epac1−/− knockout mice were generated as described,19 while the C57BL/6J LDb (Ldlr−/−/Apobec1−/−) double knockout mice origin was previously documented.17 The LTe (Ldlr−/−/Apobec1−/−/Epac1−/−) triple knockout mice were generated by crossing Epac1−/− mice with LDb mice and confirmed by genotyping. Mice were kept in a semi-barrier facility on a 12/12 h light-dark cycle in virus-free facilities with free access to food and water and maintained on a standard laboratory diet (PicoLab Rodent Diet 20, LabDiet 5053*). Both male and female littermates were used for this study. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at Houston.
BMDM Isolation and Differentiation
In vivo experiments for profiling LDb and LTe animals’ disease development were conducted in both male and female animals, in adherence with guidelines described by the ATVB Council Statement for considering sex differences as a biological variable. In accordance with our initial in vivo data demonstrating more aggressive lesion development in male animals compared to females, the decision was made to focus on primary bone marrow derived from male mice for ex vivo studies. Since females also develop lesions in our animal model, cells derived from female marrow are anticipated to exhibit similar results ex vivo.
Four-month-old male mice were euthanized by isoflurane followed by cervical dislocation. Femur and tibiae were harvested and flushed with DPBS using a 25 gauge needle. Resulting cells were siphoned through a 40 μm nylon cell strainer, centrifuged at 500×g for 5 minutes at 4 °C, decanted, and resuspended in red blood cell lysing buffer Hybri-Max for 5 minutes RT. Cells were pelleted and washed twice, then resuspended in 5 mL of FBS to be counted and seeded at 5-6×106 cells per 10 cm uncoated, sterile Petri dish for 12-16 h in M0 media (DMEM High Glucose, 10% FBS, 1× Penicillin/Streptomycin, 1× Non-Essential Amino Acids, 20 ng/mL rm-MCSF) at 37 °C, 5% CO2. Floating and loosely adherent cells were transferred to an applicable cell culture-coated well for downstream experiments. After 24 h incubation, an equivalent volume of fresh M0 media was added to each dish and incubated for 72 h. Half of the media was replaced with fresh M0 media for an additional 48 h, this half-media exchange was repeated and cells incubated for 24 h before treatment. Plates were then serum starved for 2 h prior to treatment with either oxidized-LDL (ox-LDL) supplemented media (DMEM High Glucose, 2% FBS, 1× Penicillin/Streptomycin, 1× Non-Essential Amino Acids, ox-LDL [40 μg/mL, Kalen Biomedical]) for Mfoam cell differentiation or M0 media for treatment naïve macrophages (M0). . Cells were harvested for downstream functional and expression assays at 2 or 48 h following initiation of final differentiation.
Lipid Accumulation Assays in Bone-marrow Derived Macrophages
Differentiated Mfoam cells (48 h) were washed twice with cold DPBS, fixed in 10% neutral-buffered formalin for 30 min at 4 °C followed by three additional washes, and then equilibrated for 5 min in 78% methanol at room temperature. Cells were stained for 15 min with fresh 0.2% (w/v) Oil Red O (ORO) solution with constant agitation, prepared as described in the Supplemental Materials: Extended Methods section. Cells were de-stained for 1 min with 78% methanol then washed extensively with DPBS. Following ORO staining, cells were counterstained with Meyer’s hematoxylin and 10 random fields of view were captured for each well on a Nikon light microscope to attain an average visual representation of overall lipid staining. The ORO stain was eluted with 100% methanol for 10 min for quantitative measure of each well. Eluant was transferred to a 96-well plate where absorbance at 500 nm was measured by plate reader.
M0 BMDMs differentiated directly in 96-well clear bottom, black-walled plates (Greiner Bio-one, Germany) were serum starved for 3 h then treated with DiI-ox-LDL (10 μg/mL, Kalen Biomedical) for 2 h. Where described, additional treatment of cells with an Epac specific agonist, 8-(4-Chlorophenylthio)-2-O-methyladenosine-3′, 5′-cyclic monophosphate acetoxymethyl ester (007-AM, 5 μM), was conducted 10 min prior to treatment with ox-LDL. Very carefully, plates were washed thrice with DPBS following treatment to remove excess DiI-ox-LDL while retaining cell adherence. Fluorescence (ex/em: 525/580 nm) was measured by plate reader and cells fixed in 10% neutral-buffered formalin for 30 min at 4 °C followed by DAPI staining (200 ng/mL) for 20 min in the dark. DAPI signal was measured on the plate reader to normalize data for cell number. Cells were maintained in DPBS and fluorescent imaging was conducted using an A1R Nikon confocal microscope to visualize the DiI-ox-LDL uptake. A minimum 20 random fields of view for each associated genotype were captured through the automated pathfinding algorithms in NIS Elements software (Nikon) and fluorescence intensity for each image was measured.
Pharmacological Treatment of Cells
Human THP1 cells were sub-cultured in 10cm dishes in RPMI + 5% FBS at 37 °C, 5% CO2. Cells were seeded at approximately 0.7×106 cells per 12-well 24 h before treatment and serum starved for a minimum of 2 h prior to manipulation. Addition of inhibitors was conducted 30 min before stimulation including H89 (5 μM), GF109203X (3 μM), GO6976 (3 μM). Cells were then treated for 2 h with either ox-LDL (40 μg/mL), 007-AM (5 μM), forskolin (10 μM), or vehicle control (DMSO). Cells were promptly lysed, and mRNA harvested for downstream applications. BMDMs were treated with equivalent concentrations and times for cAMP/Epac1 agonist and PKC inhibitors as described for THP1 cells.
Statistical Analyses
Results are presented as mean ± standard error of the mean (SEM). Data was analyzed for normality and equal variance using the Shapiro-Wilk normality test and an F-test, respectively. For data exhibiting normal distribution, a Student t test was implemented to compare between two groups of equal variances, whereas a Welch t test was used in cases with unequal variance. In instances of non-normal distributions, a Mann-Whitney test was conducted to compare groups. Additionally, one-way ANOVA with a Bonferroni post hoc test were used to compare among groups of three or more with normal distributions. A p value less than 0.05 was considered as statistically significant.
RESULTS
Deletion of Epac1 protects LDb mice from developing atherosclerosis.
To ascertain the functional role of Epac1 in atherosclerosis development, we generated a triple knockout mice (LTe) by crossing Epac1−/− with atherosclerosis prone LDb mice lacking both the Ldlr and Apobec1 genes. LDb mice develop atherosclerotic lesions spontaneously on standard laboratory diet exhibiting elevated plasma levels of apoB100-containing VLDL and LDL and reduced plasma levels of HDL closely mirroring human pathologies.17,18 At eight months of age, male LDb mice developed significant atherosclerotic lesions, both in the aortic arch and thoracic aorta, determined by en face measurements (Fig. 1A&B) and aortic sinus lesion area (Fig. 1C&D). Concomitant with the formation of the atherosclerotic plaques, Epac1 expression was also observed to be elevated in LDb lesion areas compared to non-lesion areas (Fig. 1E&F). Deletion of Epac1 in male LTe mice resulted in a major reduction in atherosclerotic lesions in both arch and thoracic areas (Fig. 1A–D). In comparison, no significant difference was observed between female LTe and LDb mice, with minimal lesion development at 8 months of age (Fig. SI). However, significant lesions developed in female LDb mice after 11 months, where Epac1 deletion significantly reduced plaque development in female LTe at this time point (Fig. SI). Overall, our data suggests that the expression levels of Epac1 are increased in atherosclerotic lesions, and that deletion of Epac1 in atherosclerosis-prone LDb mice reduces atherogenesis in both male and female animals.
Figure 1. Epac1 deletion reduces atherosclerotic plaque formation.
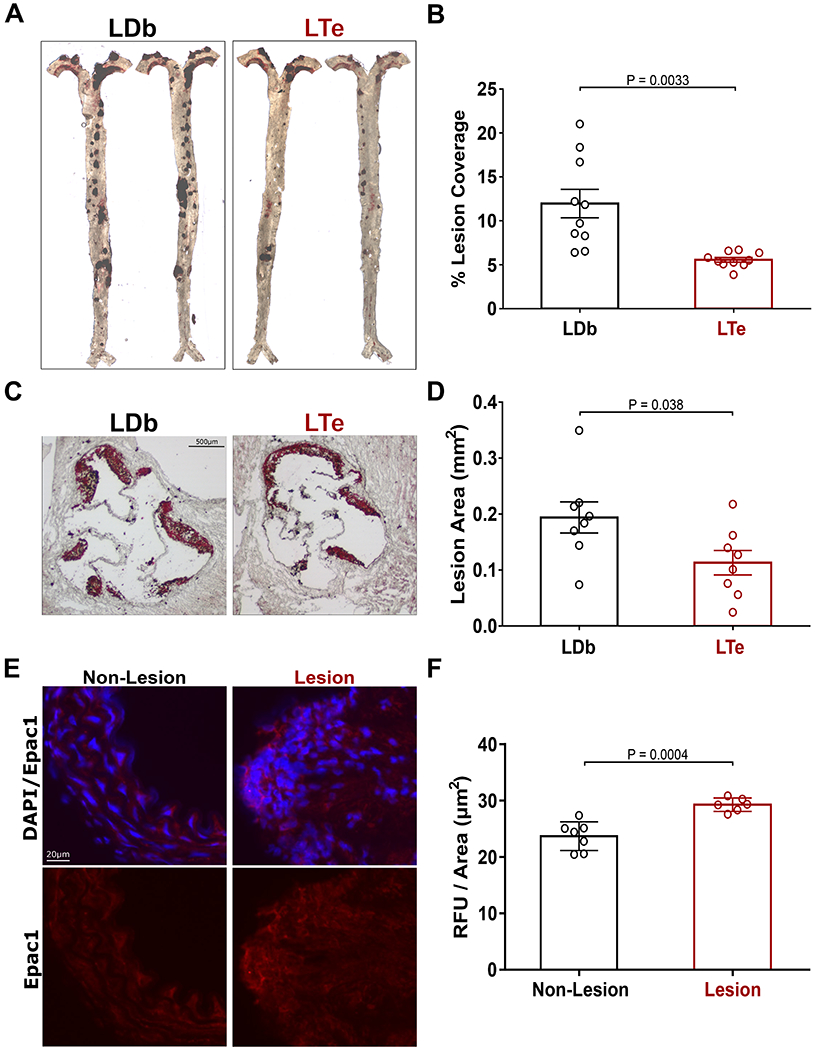
(A) Representative en face ORO stained images of aortic atherosclerotic plaques of male LDb and LTe mice at 8 months of age. (B) Lesion quantification of atherosclerotic plaques from the aortic arch to the iliac bifurcation (N = 10 independent animals for both genotypes). (C) Representative ORO stained images of aortic sinus cross-sections from male LDb and LTe mice at 8 months of age. (D) Quantification of aortic sinus lesion (N = 8 for both groups). (E) Representative images of Epac1 staining in aortic cross-sections. (F) Quantification of immunofluorescence signal per area for Epac1 in lesion and non-lesion areas (N = 7 and N = 6 for non-lesion and lesion, respectively). Data are shown as mean ± SEM.
Deficiency of Epac1 reduces macrophages/foam cells in atherosclerotic lesions.
To determine the potential causes of the apparent difference in atherosclerotic lesions between LTe and LDb mice, we examined plasma lipid profiles in both male and female mice. As shown in Figure SII, while the levels of total cholesterol, free cholesterol, high-density lipoprotein cholesterol (HDL-C), cholesteryl ester, phospholipid, triglyceride and non-esterified fatty acid (NEFA) in female mice were all lower than male mice, a significant change in lipid levels between LTe and LDb in both male and female mice was not detected. Similarly, no significant difference in populations of various immune cells was detected between LTe and LDb mice (Table SI), which is consistent with our prior finding showing Epac1 to be dispensable for immune cell development.20 Taken together, these results suggest that the protective effect of Epac1 deletion is unlikely due to changes in overall lipid profiles or immune functions at systemic levels, which led us to focus on local contributing factors within the lesions associated with vascular remodeling.
Since macrophage retention and foam cell accumulation within the vascular wall are critical steps for atherosclerosis development, we evaluated the levels of macrophage presence in atherosclerotic lesions by staining the aortic sinus with two macrophage markers, CD68 and MOMA2. As shown in Figure 2A, significant staining of both CD68 and MOMA2 were observed within the plaque lesions, but largely absent in non-lesion area. Importantly, the degree of macrophage infiltration reflected by the total CD68 and MOMA2 staining intensity and the density of the staining were significantly reduced in LTe mice as compared to the LDb controls (Fig. 2B). Closer examination revealed that the density of infiltrated cells in lesion areas was also significantly reduced in the LTe lesions (Fig. 2C&D), suggesting the apparent decrease in macrophage staining is due to reductions in foam cell number, not merely diminished cell size.
Figure 2. Deletion of Epac1 reduces macrophage/foam cell levels in aortic sinus valves of LTe mice.
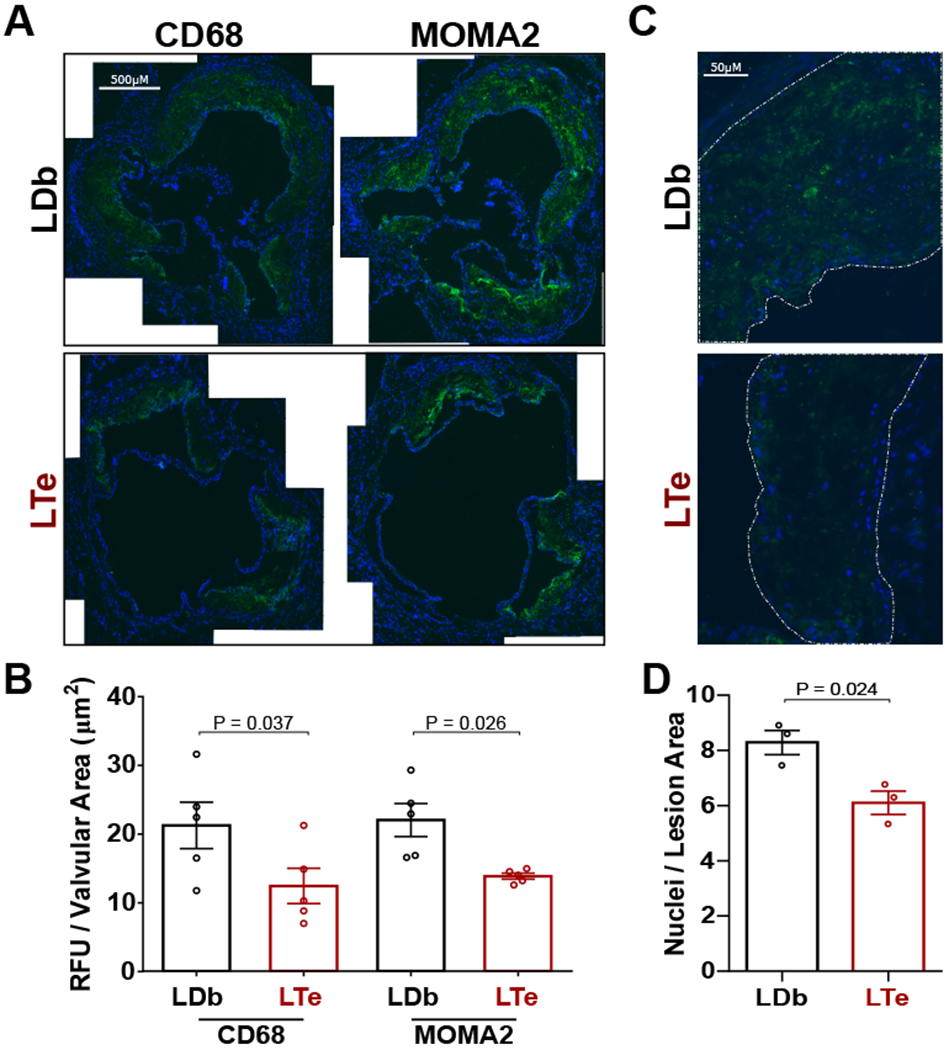
(A) Representative images of CD68 and MOMA2 immunofluorescence staining in aortic sinus cross-sections. (B) Quantification of immunofluorescence signal for CD68 and MOMA2 were processed at minimal in triplicate, sequential sections (N = 5 independent animals for LDb and LTe). (C) Representative images of the aortic sinus lesion cross-sections at increased magnification were used to measure infiltrating cell numbers in LDb and LTe tissue. (D) Particle analysis quantification of nuclei measured per area of lesion. A minimum of six independent fields of view were processed for each animal (N = 3). Data are presented as mean ± SEM.
cAMP/Epac1 signaling promotes uptake of ox-LDL and foam cell formation.
The significantly reduced macrophages/foam cells infiltration in plaque lesions of LTe mice suggests that Epac1 may be important for uptake of ox-LDL and foam cell formation. To ascertain this possible connection to Epac1 signaling, primary BMDM (M0) were isolated from WT and Epac1−/− mice and differentiated into foam cells (Mfoam) ex vivo by stimulating M0 with ox-LDL. Indeed, Mfoam cells derived from WT M0 had significantly more intracellular lipid accumulations revealed by ORO staining compared to Epac1−/− counterparts (Fig. 3A&B), suggesting a reduced capability of Epac1−/− macrophage to undergo ox-LDL-induced foam cell formation. Moreover, the expression levels of both Epac1 mRNA and protein were significantly upregulated in WT Mfoam over M0 (Fig. SIII), further supporting a potential functional role of Epac1 in foam cell formation. To determine if reduced foam cell formation was due to decreased ox-LDL accumulation by Epac1−/− macrophages, we further examined the dynamic of ox-LDL uptake in primary macrophages using fluorescently labeled ox-LDL, DiI-ox-LDL. As shown in Figure 3C&D, the ability of Epac1−/− macrophages to accrue DiI-ox-LDL after two hours was significantly reduced compared to WT macrophages. Moreover, activation of Epac in WT macrophage with an Epac-specific agonist, 007-AM, led to significant increases in DiI-ox-LDL uptake, while such 007-AM-dependent increases were absent in Epac1−/− macrophages (Fig. 3C&D). Taken together, these data suggest cAMP/Epac1 signaling promotes uptake of ox-LDL in macrophage and foam cell formation.
Figure 3. Epac1 promotes macrophage accumulation of ox-LDL.
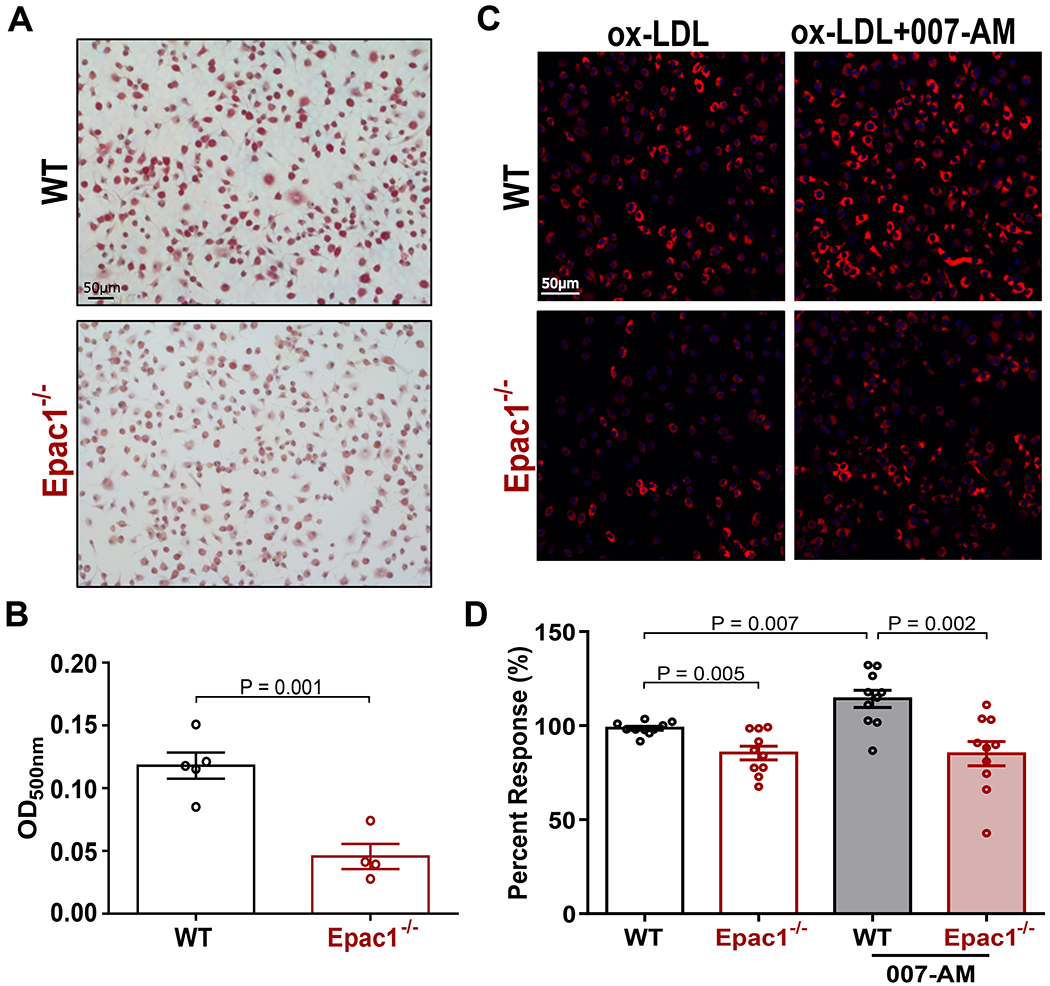
(A) Representative images of ORO stained WT and Epac1−/− foam cells (B) Quantification of ORO staining in A by methanol elution. Data are depicted as mean ± SEM (N = 5 or 4 for WT and Epac1−/−, respectively). (C) Representative fluorescence images from WT and Epac1−/− BMDMs treated with DiI-ox-LDL (10 μg/mL) for 2 h following serum starvation in the presence or absence of 007-AM (5 μM). (D) Quantification of DiI-ox-LDL fluorescence intensity in C based on automated capture of a minimum of 20 random fields of view for each genotype per well. Data are presented as mean percent response ± SEM compared to WT BMDM treated with DiI-ox-LDL alone (N = 10 BMDM cultures from independent animals for each genotype).
Loss of Epac1 obliterates ox-LDL-induced lectin-like ox-LDL receptor-1 (LOX-1) upregulation in macrophages.
Reduced lipid accrual in macrophages, as seen in our Epac1-null macrophages, suggests the intake or efflux of modified lipids in these cells may be altered. Therefore, we compared the expression levels of scavenger receptors as well as efflux transporters responsible for the uptake and export of ox-LDL in macrophages, respectively. Consistent with the literature, expression levels of lectin-like ox-LDL receptor-1 (LOX-1 or OLR-1) and cluster of differentiation 36 (CD36), major scavenger receptors important for ox-LDL uptake, were significantly enhanced in WT Mfoam over naïve M0 BMDM group (Fig. 4A&B). Interestingly, only the upregulation of LOX-1 in response to ox-LDL treatment was abolished by Epac1 deletion, while deletion of Epac1 had no significant effect on CD36 expression. Other scavenger receptors, such as macrophage scavenger receptor 1 (MSR1 or SR-A1), collectin subfamily member 12 (COLEC12 or SR-A4), scavenger receptor class B type 1 (SCARB1 or SR-B1) and CXC chemokine ligand 16 (CXCL16 or SR-G), were also investigated but demonstrated no difference between WT and Epac1-null genotypes (Fig. SV C). Turning to the efflux pathway, expression of ATP-binding cassette sub-family A member 1 (ABCA1) and ATP-binding cassette sub-family G member 1 (ABCG1), two principal transporters for lipid efflux associated with foam cell formation, was increased in WT Mfoam over naïve BMDM as expected, but deletion of Epac1 had no effect on this upregulation (Fig. 4C&D).
Figure 4. Oxidized LDL-mediated regulation of scavenger receptors and efflux transporters mRNA expression during foam cell formation.
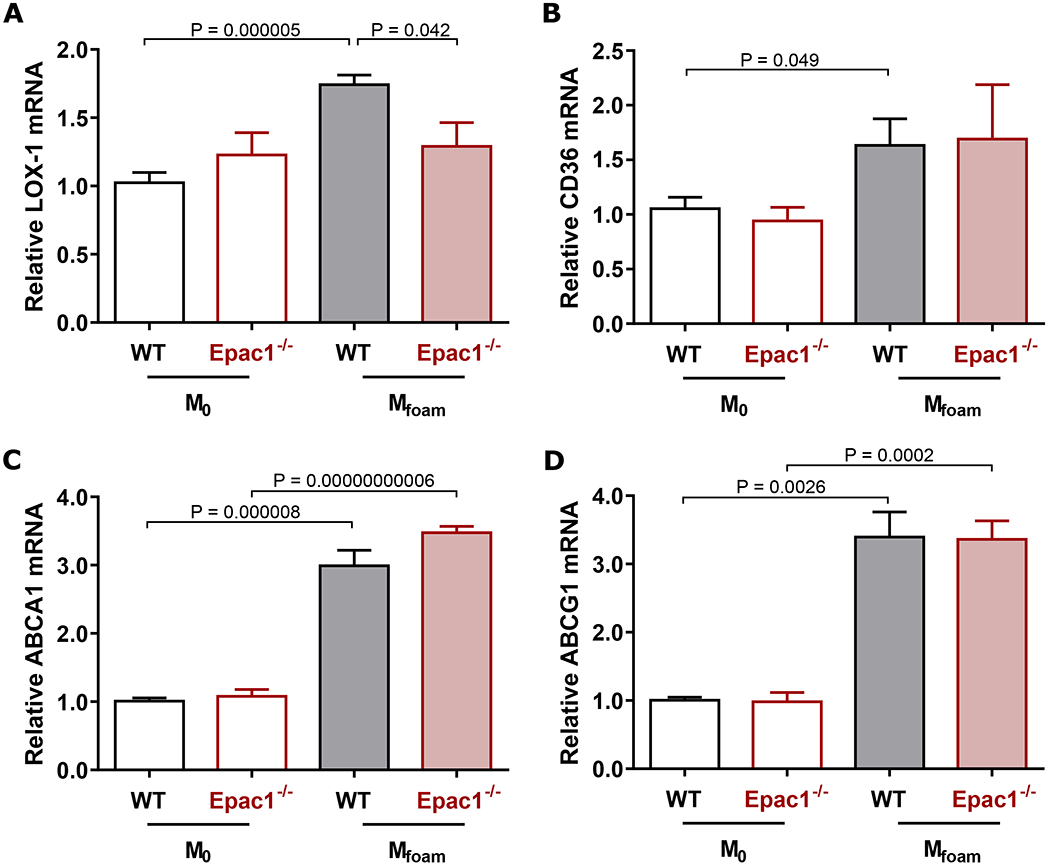
(A-D) Mfoam and M0 cells were prepared by incubating WT and Epac1−/− BMDMs with ox-LDL (40 μg/mL) or vehicle, respectively for 48 h. Expression levels of (A) LOX-1, (B) CD36, (C) ABCA1, and (D) ABCG1 were determined by qPCR. Each sample was run at minimum in triplicate for each independent animal (N = 5 or 4 for WT and Epac1−/− respectively). Data are presented as mean ± SEM.
To further support a role of Epac1 in LOX-1 regulation, we compared the protein expression levels of LOX-1 between WT and Epac1-null primary macrophages and observed an upregulation of ox-LDL-induced LOX-1 in WT macrophages following differentiation from M0 into Mfoam. This enhanced LOX-1 expression was impeded in Epac1−/− Mfoam cells (Fig. 5A&B). In fact, enhanced LOX-1 expression can be readily induced within two hours of ox-LDL treatment in WT, but not Epac1-null BMDMs (Fig. SV A&B). Consistent with the ex vivo observation in primary BMDMs, immunofluorescence staining for LOX1 in aortic arch sections revealed that although levels of LOX1 in the non-lesion areas were approximately equal between LDb and LTe groups, LOX-1 levels within LDb lesions were significantly elevated compared to the LTe counterparts (Figure 5C&D). These results support the notion that Epac1 is important for ox-LDL-mediated LOX-1 induction, which feeds forward to promote ox-LDL uptake and fuel atherogenesis in the intact animal.
Figure 5. Expression of LOX-1 protein is elevated in response to ox-LDL and within atherosclerotic lesions.
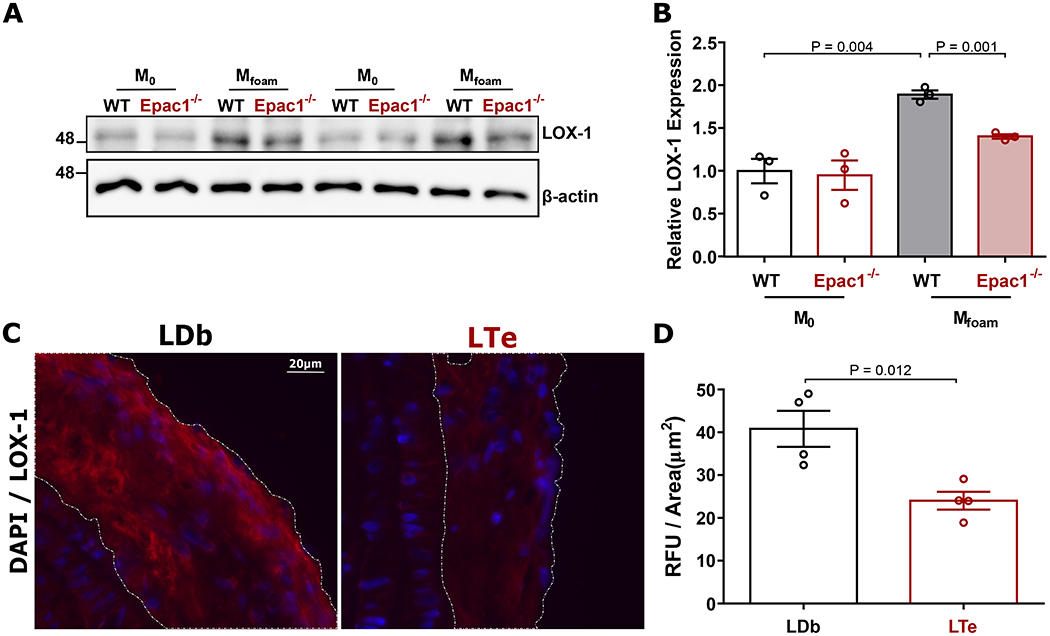
(A) Representative immunoblot of LOX-1 and β-actin expression in WT and Epac1−/− differentiated M0 and Mfoam BMDMs treated with vehicle or ox-LDL (40 μg/mL, 48 h), respectively. (B) Quantification of relative LOX-1 protein expression (N = 3 independent mice per genotype). (C) Representative images of LOX1 immunofluorescence staining in lesions from sagittal sections of the aortic arch. (D) Quantification of immunofluorescence signal per lesion area in LDb and LTe lesions, where a minimum of two independent fields of view were processed for each animal (N = 4 mice per genotype). Data are presented as mean ± SEM.
To strengthen the involvement of Epac1 in ox-LDL mediated LOX-1 upregulation during foam cell formation and exclude possible influence of secondary pathways, such as apoptosis and anti-inflammatory effects, which may also result in reduced foam cell formation, we investigated the effect of Epac1 deletion on apoptosis and inflammatory cytokine expression within the ex vivo macrophage model. As seen in Fig. SVI, ox-LDL treatment had no effect on cell survival or caspase 3 activation in either WT or Epac1-null BMDMs. Similarly, analysis of inflammatory cytokines generated in response to ox-LDL stimulation demonstrated a concurrent reduction of IL-1b in both WT and Epac1-null BMDMs. However, no difference was observed between the genotypes in the expression pattern of common inflammatory mediators IL-1b, IL-6, or TNF-α (Fig. SVII). These data suggest that Epac1 deletion mediated suppression of ox-LDL uptake/foam cell formation is unlikely due to increased apoptosis or anti-inflammatory effects.
Since VSMCs can also uptake ox-LDL and transdifferentiate into foam cells during atherogenesis,21,22 we tested if Epac1 may contribute to atherogenesis by playing a similar role in VSMCs in addition to macrophages. Unlike mouse primary BMDMs (Fig. SIII), VSMCs did not demonstrate an enhanced expression of Epac1 when stimulated with ox-LDL (Fig. SIV A) as did human THP1 macrophages (Fig. SIV A). Furthermore, both 007-AM and ox-LDL treatment were unable to increase LOX-1 expression in VSMCs (Fig. SIV B). Taken together, these results suggest that Epac1, promotes LOX-1 expression in response to ox-LDL stimulation in macrophages, but not in VSMCs.
Ox-LDL promotes LOX-1 transcriptional activation via increasing intracellular cAMP levels and upregulation of cellular Epac1 activity in macrophage.
Observing that Epac1 deletion is sufficient to attenuate ox-LDL stimulated LOX-1 expression, we sought to further elucidate whether ox-LDL was functioning directly through the cAMP/Epac1 pathway to activate Epac1 and subsequently modulate gene transcription of LOX-1. As an initial approach to determine the involvement of Epac1 activation following ox-LDL stimulation, we monitored the intracellular cAMP levels in response to ox-LDL stimulation and observe enhanced cAMP levels in isolated BMDMs (Fig. 6A), an observation that is consistent with previous studies showing that ox-LDL promotes cAMP production in endothelial cells and enhance monocyte binding.23–25 This observation prompted investigation into whether cAMP functions through Epac1 activation to promote LOX-1 transcriptional activation by monitoring LOX-1 expression in human THP1 cells and mouse BMDMs. As shown in Figure 6B, treatment of THP1 cells with forskolin, a potent stimulator that activates both Epac1 and PKA, resulted in an increased LOX-1 expression, which could not be blocked by the PKA inhibitor H89, suggesting a PKA-independent mechanism. Furthermore, stimulation of cells with an Epac-specific agonist, 007-AM, or ox-LDL mirrored the forskolin-induced LOX-1 expression in both human THP1 and mouse BMDMs, suggesting an Epac1-dependent, conserved regulatory mechanism across species (Fig. 6B&C and Fig. SV A). Importantly, the 007-AM induced LOX-1 expression was not observed in Epac1-null BMDMs (Fig. 6C) further supporting an Epac1-centric nature of the LOX-1 induction.
Figure 6. Epac1 modulates LOX-1 expression through PKC signaling pathway.
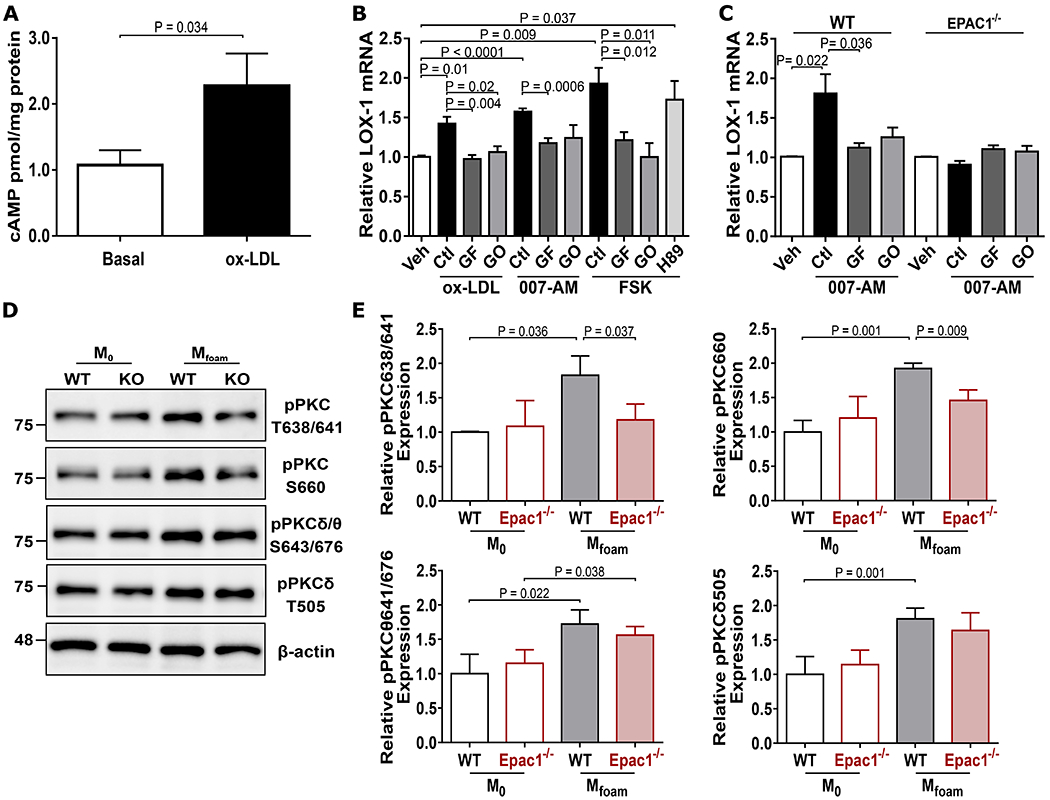
(A) Intracellular cAMP levels in BMDMs. Cells were serum starved for 2 h then stimulated with vehicle or ox-LDL (40 μg/mL) for 5 min. Lysates were subjected to ELISA measurement of cAMP. (B) Relative LOX-1 mRNA in THP1. Cells were pretreated with H89 (5 μM) or PKC inhibitors [GF109203X (GF, 3 μM) or G06976 (GO, 3 μM)] for 30 min, then stimulated with either ox-LDL (40 μg/mL), 007-AM (5 μM), or forskolin (10 μM) for 2 h. Gene expression of LOX-1 was monitored by qPCR (N = 5). (C) Relative LOX-1 mRNA in BMDMs. WT and Epac1−/− BMDMs were stimulated with 007-AM for 2 h in the presence or absence of pretreatment with PKC inhibitors, GF or GO. Gene expression of LOX-1 was determined by qPCR (N = 6). (D) Representative immunoblots of phosphor-PKC sites and β-actin expression in WT and Epac1−/− M0 and Mfoam BMDMs. (E) Quantification of relative protein expression from blots depicted in D (N = 3 independent mice per genotype). Data are presented as mean ± SEM.
Epac1 acts through PKC to promote LOX-1 transcriptional activation, ox-LDL uptake, and foam cell formation.
With converging actions of ox-LDL on the elevation of cAMP and Epac1-mediated increases in LOX-1 transcriptional activation, we next explored the cellular/molecular mechanism contributing to this response. Among known Epac1 down-stream effectors, we observed that treatment of WT BMDMs with ox-LDL significantly enhanced the phosphorylation of PKC at sites Thr638/641 in the autophosphorylation loop and Ser660 on the hydrophobic carboxy terminal while ox-LDL was unable to enhance PKC phosphorylation in Epac1−/− BMDMs (Fig. 6D&E). Moreover, the PKCδ/θ isoforms are unlikely involved since phosphorylation levels of key residues for these isoforms exhibit no discernible difference between WT or Epac1−/− BMDMs following ox-LDL stimulation. These results suggest the involvement of classical PKC isoforms in this process. To determine if Epac1 acts through PKC signaling to induce LOX-1 expression, we monitored the effect of PKC inhibition on Epac-mediated LOX-1 expression. As shown in Figure 6B, a pan-PKC inhibitor (GF109203X: α, β, δ, ε, ζ) was found to be most effective at attenuating ox-LDL and 007-AM-mediated LOX-1 gene induction in THP1 cells, while the PKCα/β inhibitor (GO6976) also demonstrated a trend to reduce the LOX-1 expression in macrophages under each condition. In FSK-treated THP1 cells, both inhibitors were also observed to reduce LOX-1 expression. These inhibitors were effective in suppressing 007-AM induced LOX-1 expression in WT BMDMs but had no effect on LOX-1 expression in Epac1−/− BMDMs, which were no-responsive to 007-AM stimulation (Fig. 6C). These data collectively suggest that activation of Epac1 can increase LOX-1 expression in human and mouse macrophages through a PKA-independent, PKC-dependent mechanism.
Finally, to connect this signaling mechanism back to the cellular phenotype we observed in the Epac1-null BMDMs and assess the functional consequence of PKC inhibition on ox-LDL engulfment, we treated WT and Epac1−/− BMDMs with ox-LDL for 48 hr in the presence or absence of the PKC inhibitor GF109203X. Supporting the LOX-1 expression data, inhibition of PKC in WT BMDMs effectively blocked ox-LDL update while GF109203X had no further effect on inhibiting ox-LDL intake in Epac1−/− BMDMs (Fig. 7A&B). Taken together, these data are evidence of a previously unidentified signaling pathway in which Epac1/PKC regulates downstream LOX-1 expression in macrophages to promote uptake of ox-LDL and foam cell formation during pathogenesis.
Figure 7. PKC inhibition recapitulates reduced ox-LDL accumulation observed in Epac1−/− BMDMs.
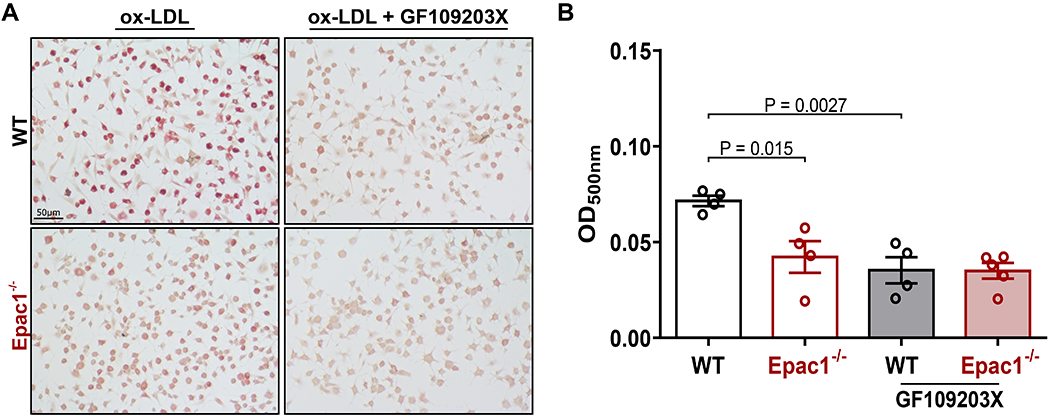
(A) Representative images of ORO stained WT and Epac1−/− BMDMs, pretreated with vehicle or PKC inhibitor GF109203X (3 μM) for 30 min, then stimulated with ox-LDL (40 μg/mL) for 48 h (N = 4 for each genotype). (B) Quantification of ORO stained BMDMs by methanol elution. Data are presented as mean ± SEM.
DISCUSSION
Our study demonstrates that deletion of Epac1 attenuates atherosclerosis in a human-like atherosclerosis mice model. At the cellular level, inhibition of Epac1 reduces ox-LDL accrual and foam cell formation, while Epac1 activation by an Epac-specific agonist enhances ox-LDL uptake in macrophages. Interestingly the expression levels of Epac1 are also upregulated by ox-LDL during the induction of foam cell formation, suggesting that ox-LDL uptake and Epac1 act as a feed-forward loop during atherogenesis. This ox-LDL induced Epac1 expression is likely mediated by the hypoxia-inducible factor 1α (HIF-1α) as the promoter region of Epac1 harbors a hypoxia responsive element (HRE) that can be modulated by hypoxia to facilitate Epac1 transcription, 26 while ox-LDL has been shown to enhance HIF-1α expression and activation in macrophages.27–29
Macrophage-derived foam cells play a crucial role in atherosclerotic plaque formation by inducing the expression of scavenger receptors to promote lipid uptake. Indeed, our study shows LOX-1, a major scavenger receptor for modified LDL, is upregulated in response to ox-LDL stimulation in WT macrophages. Although other scavenger receptors are also increased by the stimulation of ox-LDL as anticipated, including CD36 and MSR1, only LOX-1 upregulation is impaired in Epac1−/− macrophages suggesting an Epac1-dependent response. Upregulation of LOX-1 in developing atherosclerotic plaques is observed in patients.30 It has been reported that LOX-1 upregulation during stress-induced conditions accounts for greater than 40% of modified LDL intake.31,32 Further, in vivo studies with Apoe−/− mice overexpressing LOX-1 on high fat diet demonstrate increased modified lipid uptake and accelerated macrophage infiltration of arteries attributed to LOX-1.33,34 Together, these studies signify the importance of LOX-1 in atherogenesis. However, the molecular mechanisms governing regulation of LOX-1 expression during atherogenic development remain incomplete and underexplored. Therefore, our findings that cAMP/Epac1 signaling promotes LOX-1 transcriptional activation in macrophages in response to ox-LDL fill in a major gap.
To delineate an Epac-specific molecular mechanism for LOX-1 regulation, we have applied genetic and pharmacological approaches to probe Epac1 signaling in mouse and human-derived macrophages cells. Activation of Epac with 007-AM effectively induces LOX-1 expression, and inhibition of PKC blocks the 007-AM-mediated effect suggesting that PKC acts downstream of ox-LDL/Epac1 during LOX-1 transcriptional regulation. Indeed, ox-LDL stimulation in WT BMDMs, but not in Epac1−/− macrophages, activates classical PKC isoforms, such as PKCα/β. These results are consistent with observations that PKCα and PKCβ are activated by ox-LDL in human arterial endothelial cells.35,36 Furthermore, PKC inhibition blocks ox-LDL accumulation in WT macrophages, phenocopying the effects of Epac1 deletion. Crosstalk between Epac1 and PKC signaling has been well-established in various physiological contexts. For example, PLCε/PKCε function downstream of cAMP/Epac during mechanical hyperalgesia measurements of paw withdrawal in mice.37–39. Subsequent studies also implicate PKCα as a downstream effector of Epac1 in the development of inflammatory-mediated nociception.40,41 Epac1 also signals through PLCε/PKCε to inhibit annexin A2 surface translocation and plasminogen activation in endothelial cells.42 Similar signaling paradigms are observed in cardiomyocytes where Epac1 stimulation elevates CaMKII activity leading to RyR2 and PLN phosphorylation to enhance calcium dynamics and transients in a PKC-dependent manner.43,44 Epac-induced activation of PKC appears to occur via PLCε-mediated cleavage of phosphatidylinositol bisphosphate substrates dependent on the subcellular compartments where Epac and PLCε are localized. At the plasma membrane, cleavage of phosphatidylinositol-4, 5-bisphosphate (PIP2) into downstream mediators, DAG and inositol 1, 4, 5-triphosphate (IP3) can induce PKC activity and calcium release, while cleavage of phosphatidylinositol-4-bisphosphate (PI4P) leads to the formation of DAG and inositol 1, 4-bisphosphate (IP2) at the perinuclear Golgi.45,46 Moreover, the formation of biologically active signalosomes between Epac1, PLC, PKC, and protein kinase D, with A-kinase anchoring proteins further substantiates coordinated activation of PKC by Epac1.46 Our results demonstrate Epac1 as a major upstream regulator of PKC-mediated LOX-1 transcriptional regulation following ox-LDL stimulation in macrophage during atherogenesis.
In conclusion, our study reveals a novel function of Epac1 signaling as a major regulator of ox-LDL-mediated LOX-1 transcription activation in macrophages during atherogenesis. Mechanistically, Epac1 acts as a stress-response switch in response to ox-LDL stimulation to promote PKC activation and subsequent LOX-1 transcriptional activation, which, promotes a feed-forward mechanism of further ox-LDL intake propelling atherosclerotic development. These findings suggest Epac1 may be an attractive therapeutic target for atherosclerosis.
Supplementary Material
HIGHLIGHTS:
Deletion of Epac1 attenuates atherosclerotic lesion formation and macrophage/foam cell infiltrations within the atherosclerotic plaques in a human-like atherogenic mouse model.
Expression of Epac1 is elevated in atherosclerotic plaques, as well as in ex vivo differentiation of bone-marrow derived macrophages by ox-LDL stimulation.
Ablation of Epac1 expression is sufficient to reduce ox-LDL accrual in Epac1 null macrophages, while activation of Epac1 by Epac-specific agonist increases ox-LDL uptake in wild type macrophages.
Epac1 promotes ox-LDL uptake by upregulating LOX-1 scavenger receptor expression in a classical PKC-dependent, PKA-independent mechanism.
Chronic activation of Epac1 in response to oxidative stress such as modified LDL results in upregulation of LOX-1 in macrophages accelerating the ingestion of ox-LDL leading to a feed-forward mechanism of foam cell formation and atherogenesis, defining an unexplored, exploitable therapeutic target.
ACKNOWLEDGEMENTS:
We thank Dr. Wei Lin for helpful discussions.
SOURCE OF FUNDING:
This work was supported by grants from the National Institutes of Health R35GM122536.The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ABBREVIATIONS:
- 007-AM
8-(4-Chlorophenylthio)-2-O-methyladenosine-3′, 5′-cyclic monophosphate acetoxymethyl ester
- Apobec1
Apolipoprotein B mRNA editing enzyme catalytic subunit 1
- BMDM
Bone-marrow derived macrophages
- DiI
1, 1’-dioctadecyl- 3, 3, 3’, 3’-tetramethylindocarbocyanine perchlorate
- DPBS
sterile PBS without calcium or magnesium
- Epac
Exchange protein directly activated by cAMP
- Epac1
Epac isoform 1
- Epac2
Epac isoform 2
- FSK
Forskolin
- GF
GF109203X
- GO
GO6976
- HDL
High-density lipoproteins
- LDb
Genetically engineered mouse lacking Ldlr and Apobec1
- LDL
Low-density lipoproteins
- Ldlr
LDL receptor
- LOX-1
Oxidized low-density lipoprotein receptor 1
- LTe
Genetically engineered mouse lacking Ldlr, Apobec1 and Epac1
- M0
Naïve Macrophage
- Mfoam
Macrophage derived foam cell
- MOMA2
Monocyte and macrophage antibody 2
- ORO
Oil Red O
- Ox-LDL
Oxidized LDL
- PKA
Protein kinase A
- PKC
Protein kinase C
- VLDL
Very-low-density lipoproteins
- VSMC
Vascular smooth muscle cell
- WT
Wild type
Footnotes
CONFLICT OF INTEREST DISCLOSURES:
The authors declare no competing financial interests.
REFERENCES
- 1.Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. The Journal of cell biology. 2015;209(1):13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annual review of physiology. 2006;68:375–401. [DOI] [PubMed] [Google Scholar]
- 3.de Rooij J, Zwartkruis FJ, Verheijen MH, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. [DOI] [PubMed] [Google Scholar]
- 4.Kawasaki H, Springett GM, Mochizuki N, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282(5397):2275–2279. [DOI] [PubMed] [Google Scholar]
- 5.Mei F, Qiao J, Tsygankova O, Meinkoth J, Quilliam L, Cheng X. Differential signaling of cyclic AMP - Opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. Journal of Biological Chemistry. 2002;277(13):11497–11504. [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai). 2008;40(7):651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65(2):670–709. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee U, Cheng X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics. Gene. 2015;570(2):157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4). Gene. 2016;575(2 Pt 3):577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robichaux WG 3rd, Cheng X. Intracellular cAMP Sensor Epac: Physiology, Pathophysiology, and Therapeutics Development. Physiol Rev. 2018;98(2):919–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts OL, Dart C. cAMP signalling in the vasculature: the role of Epac (exchange protein directly activated by cAMP). Biochemical Society transactions. 2014;42(1):89–97. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Mei FC, Yang W, et al. Epac1 inhibition ameliorates pathological angiogenesis through coordinated activation of Notch and suppression of VEGF signaling. Sci Adv. 2020;6(1):eaay3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, Ha Y, Xia F, et al. Neuronal Epac1 mediates retinal neurodegeneration in mouse models of ocular hypertension. J Exp Med. 2020;217(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama U, Minamisawa S, Quan H, et al. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am J Physiol Heart Circ Physiol. 2008;295(4):H1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato Y, Yokoyama U, Yanai C, et al. Epac1 Deficiency Attenuated Vascular Smooth Muscle Cell Migration and Neointimal Formation. Arteriosclerosis, thrombosis, and vascular biology. 2015;35(12):2617–2625. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Robichaux WG, Wang Z, et al. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury. Sci Rep. 2016;6:36552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dutta R, Singh U, Li TB, Fornage M, Teng BB. Hepatic gene expression profiling reveals perturbed calcium signaling in a mouse model lacking both LDL receptor and Apobec1 genes. Atherosclerosis. 2003;169(1):51–62. [DOI] [PubMed] [Google Scholar]
- 18.Singh U, Zhong S, Xiong M, Li TB, Sniderman A, Teng BB. Increased plasma non-esterified fatty acids and platelet-activating factor acetylhydrolase are associated with susceptibility to atherosclerosis in mice. Clinical science (London, England : 1979). 2004;106(4):421–432. [DOI] [PubMed] [Google Scholar]
- 19.Yan J, Mei FC, Cheng H, et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol Cell Biol. 2013;33(5):918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almahariq M, Mei FC, Wang H, et al. Exchange protein directly activated by cAMP modulates regulatory T-cell-mediated immunosuppression. Biochem J. 2015;465(2):295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129(15):1551–1559. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Dubland JA, Allahverdian S, et al. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2019;39(5):876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parhami F, Fang ZT, Fogelman AM, Andalibi A, Territo MC, Berliner JA. Minimally modified low density lipoprotein-induced inflammatory responses in endothelial cells are mediated by cyclic adenosine monophosphate. J Clin Invest. 1993;92(1):471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parhami F, Fang ZT, Yang B, Fogelman AM, Berliner JA. Stimulation of Gs and inhibition of Gi protein functions by minimally oxidized LDL. Arteriosclerosis, thrombosis, and vascular biology. 1995;15(11):2019–2024. [DOI] [PubMed] [Google Scholar]
- 25.Cole AL, Subbanagounder G, Mukhopadhyay S, Berliner JA, Vora DK. Oxidized phospholipid-induced endothelial cell/monocyte interaction is mediated by a cAMP-dependent R-Ras/PI3-kinase pathway. Arteriosclerosis, thrombosis, and vascular biology. 2003;23(8):1384–1390. [DOI] [PubMed] [Google Scholar]
- 26.Lai TW, Lin SZ, Lee HT, et al. HIF-1alpha binding to the Epac1 promoter recruits hematopoietic stem cells to the ischemic brain following stroke. J Mol Cell Biol. 2012;4(3):184–187. [DOI] [PubMed] [Google Scholar]
- 27.Shatrov VA, Sumbayev VV, Zhou J, Brune B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101(12):4847–4849. [DOI] [PubMed] [Google Scholar]
- 28.Poitz DM, Augstein A, Weinert S, Braun-Dullaeus RC, Strasser RH, Schmeisser A. OxLDL and macrophage survival: essential and oxygen-independent involvement of the Hif-pathway. Basic Res Cardiol. 2011;106(5):761–772. [DOI] [PubMed] [Google Scholar]
- 29.Lee SJ, Thien Quach CH, Jung KH, et al. Oxidized low-density lipoprotein stimulates macrophage 18F-FDG uptake via hypoxia-inducible factor-1alpha activation through Nox2-dependent reactive oxygen species generation. J Nucl Med. 2014;55(10):1699–1705. [DOI] [PubMed] [Google Scholar]
- 30.Kataoka H, Kume N, Miyamoto S, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. 1999;99(24):3110–3117. [DOI] [PubMed] [Google Scholar]
- 31.Schaeffer DF, Riazy M, Parhar KS, et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. 2009;50(8):1676–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun H, Krauss RM, Chang JT, Teng BB. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion, and endothelial dysfunction. J Lipid Res. 2018;59(2):207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inoue K, Arai Y, Kurihara H, Kita T, Sawamura T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ Res. 2005;97(2):176–184. [DOI] [PubMed] [Google Scholar]
- 34.White SJ, Sala-Newby GB, Newby AC. Overexpression of scavenger receptor LOX-1 in endothelial cells promotes atherogenesis in the ApoE(−/−) mouse model. Cardiovasc Pathol. 2011;20(6):369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D, Liu L, Chen H, Sawamura T, Mehta JL. LOX-1, an oxidized LDL endothelial receptor, induces CD40/CD40L signaling in human coronary artery endothelial cells. Arteriosclerosis, thrombosis, and vascular biology. 2003;23(5):816–821. [DOI] [PubMed] [Google Scholar]
- 36.Li D, Liu L, Chen H, Sawamura T, Ranganathan S, Mehta JL. LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation. 2003;107(4):612–617. [DOI] [PubMed] [Google Scholar]
- 37.Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 2005;25(26):6119–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113(1-2):185–190. [DOI] [PubMed] [Google Scholar]
- 39.Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20(12):4680–4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu Y, Li G, Chen Y, Huang LY. Epac-protein kinase C alpha signaling in purinergic P2X3R-mediated hyperalgesia after inflammation. Pain. 2016;157(7):1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu Y, Li G, Huang LYM. Inflammation induces Epac-protein kinase C alpha and epsilon signaling in TRPV1-mediated hyperalgesia. Pain. 2018;159(11):2383–2393. [DOI] [PubMed] [Google Scholar]
- 42.Yang W, Mei FC, Cheng X. Epac1 regulates endothelial annexin A2 cell surface translocation and plasminogen activation. FASEB J. 2018;32(4):2212–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oestreich EA, Wang H, Malik S, et al. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in beta-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282(8):5488–5495. [DOI] [PubMed] [Google Scholar]
- 44.Oestreich EA, Malik S, Goonasekera SA, et al. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem. 2009;284(3):1514–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt M, Evellin S, Weernink PA, et al. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol. 2001;3(11):1020–1024. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Malik S, Pang J, et al. Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell. 2013;153(1):216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed methods are provided below and within the Extended Methods of the Supplemental Materials for all procedures carried out in this study. The authors declare that all supporting data are available within the article and the online supplementary files.