

ABSTRACT
In this 12th annual installment of the Antibodies to Watch article series, we discuss key events in antibody therapeutics development that occurred in 2020 and forecast events that might occur in 2021. The coronavirus disease 2019 (COVID-19) pandemic posed an array of challenges and opportunities to the healthcare system in 2020, and it will continue to do so in 2021. Remarkably, by late November 2020, two anti-SARS-CoV antibody products, bamlanivimab and the casirivimab and imdevimab cocktail, were authorized for emergency use by the US Food and Drug Administration (FDA) and the repurposed antibodies levilimab and itolizumab had been registered for emergency use as treatments for COVID-19 in Russia and India, respectively. Despite the pandemic, 10 antibody therapeutics had been granted the first approval in the US or EU in 2020, as of November, and 2 more (tanezumab and margetuximab) may be granted approvals in December 2020.* In addition, prolgolimab and olokizumab had been granted first approvals in Russia and cetuximab saratolacan sodium was first approved in Japan. The number of approvals in 2021 may set a record, as marketing applications for 16 investigational antibody therapeutics are already undergoing regulatory review by either the FDA or the European Medicines Agency. Of these 16 mAbs, 11 are possible treatments for non-cancer indications and 5 are potential treatments for cancer. Based on the information publicly available as of November 2020, 44 antibody therapeutics are in late-stage clinical studies for non-cancer indications, including 6 for COVID-19, and marketing applications for at least 6 (leronlimab, tezepelumab, faricimab, ligelizumab, garetosmab, and fasinumab) are planned in 2021. In addition, 44 antibody therapeutics are in late-stage clinical studies for cancer indications. Of these 44, marketing application submissions for 13 may be submitted by the end of 2021. *Note added in proof on key events announced during December 1-21, 2020: margetuximab-cmkb and ansuvimab-zykl were approved by FDA on December 16 and 21, 2020, respectively; biologics license applications were submitted for ublituximab and amivantamab.
KEYWORDS: Antibody therapeutics, cancer, COVID-19, Food and Drug Administration, European Medicines Agency, immune-mediated disorders, Sars-CoV-2
Introduction
Each year since 2010, the ‘Antibodies to Watch’ articles have faithfully reported key events related to antibody therapeutics in late-stage development and regulatory review, and those that were recently approved.1–13 Events that occurred during 2020, however, were unprecedented due to coronavirus disease 2019 (COVID-19). During January – November 2020, over 60 million people worldwide were infected with the SARS-CoV-2 virus, and ~1.5 million people died of the disease. In terms of absolute numbers per country, the United States (US) had the highest number of infections, over 13 million, and the highest number of deaths, over 260 thousand.
It was clear early on that many new therapeutics and vaccines would be needed to manage the pandemic. The global response to this very substantial need has been truly extraordinary. Hundreds if not thousands of organizations in the biopharmaceutical, academic, government and nonprofit sectors pivoted from their usual activities, and started studying the virus and the disease, and developing therapeutics and vaccines. Remarkably, the programs initiated by the biopharmaceutical industry appear to have been accomplished in parallel with their ongoing activities involving late-stage clinical studies and regulatory review of antibody therapeutics. Regulatory authorities also managed the additional COVID-19-relatedworkload while maintaining timelines on existing marketing applications undergoing review.
‘Antibodies to watch in 2021ʹ includes sections dedicated to discussion of anti-SARS-CoV-2 antibodies and other antibody therapeutics that were repurposed as treatments for COVID-19. We focus on those in late-stage studies and the antibody therapeutics under review or authorized for COVID-19 in the US or other countries. The discussion and COVID-19 antibody therapeutics data are segregated from those related to the traditional topics (i.e., recently approved antibody products, antibodies in regulatory review, antibodies for which marketing applications may be submitted soon) to enable and facilitate comparisons with past and future installments of this article series. Data presented here were collected and analyzed during mid-August through mid-November 2020. Due to the large volume of literature for the molecules, we have reported primarily on publications, disclosures and events that occurred during 2020.
COVID-19 antibody therapeutics in late-stage studies
COVID-19 is characterized by cytokine storm-induced acute respiratory distress syndrome, moderate to severe pneumonia, tissue damage resulting from hyper-inflammation, and abnormal clotting. In particular, complement 5, interleukin (IL)-1, IL-6, interferon (IFN), and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been implicated in the pathology of the disease. As the pandemic began to spread in early 2020, over 60 antibody therapeutics already in clinical studies or marketed for other indications with similar pathology were repurposed as possible COVID-19 interventions. As of November 2020, late-stage clinical studies for numerous repurposed antibody therapeutics (Table 1) were recruiting patients, and emergency use authorizations (EUAs) had been requested or granted for 3, anti-IL-6 receptor levilimab, anti-CD6 itolizumab, and anti-C-C chemokine receptor type 5 (CCR5) leronlimab.
Table 1.
Monoclonal antibodies undergoing late-stage clinical studies or authorized for COVID-19*
| Primary sponsoring company | INN or code name | Molecular format | Target(s) | Most advanced phase for COVID-19 | Phase 2/3 or 3 clinical study conditions |
|---|---|---|---|---|---|
| Biocon, Equillium | Itolizumab# | Humanized IgG1 | CD6 | EUA in India | Hospitalized Patients With COVID-19 (NCT04605926 pending) |
| Biocad | Levilimab# | Human IgG1 | IL-6 R | EUA in Russia | Severe COVID-19 (NCT04397562) |
| AbCellera / Eli Lilly and Company | Bamlanivimab (LY-CoV555, LY3819253) | Human IgG1 | SARS-CoV-2 | EUA in US | Preventing SARS-CoV-2 Infection and COVID-19 (NCT04497987); Inpatients With COVID-19 (NCT04501978); Outpatients With COVID-19 (NCT04518410) |
| Regeneron Pharmaceuticals, Inc. | Casirivimab and imdevimab (REGN-COV2; REGN10933 + REGN10987) | Human IgG1 mAbs | SARS-CoV-2 | EUA in US | Ambulatory Adult Patients With COVID-19 (NCT04425629); Hospitalized Adult Patients With COVID-19 (NCT04426695); Preventing SARS-CoV-2 Infection in Household Contacts of Individuals Infected With SARS-CoV-2 (NCT04452318) |
| CytoDyn | Leronlimab | Humanized IgG4 | CCR5 | EUA requested in US | Severe or Critical COVID-19 (pivotal Phase 2 NCT04347239); Mild to moderate COVID-19 (pivotal Phase 2 NCT04343651) |
| InflaRx GmbH | Vilobelimab (IFX-1, CaCP29) | Chimeric IgG4 | C5 | Phase 2/3 | Severe COVID-19 Pneumonia (NCT04333420) |
| Alexion Pharmaceuticals | Ravulizumab-cwvz# | Humanized IgG2/4 | C5 | Phase 3 | Hospitalized adults with severe pneumonia or acute respiratory distress syndrome (NCT04369469) |
| Jiangsu Pacific Meinuoke Bio Pharmaceutical Co Ltd | Meplazumab | Humanized IgG2 | CD147 | Phase 2/3 pending | Hospitalized Adults With COVID-19 (NCT04586153) |
| Humanigen, Inc. | Lenzilumab | Human IgG1 | GM-CSF | Phase 3 | COVID-19 Pneumonia (NCT04351152) |
| Kiniksa Pharmaceuticals, Ltd. | Mavrilimumab | Human IgG4 | GM-CSFR | Phase 2/3 | COVID-19 Pneumonia and Hyper-inflammation (NCT04447469) |
| Swedish Orphan Biovitrum | Emapalumab# | Human IgG1 | IFN gamma |
Phase 2/3 | Hyper-inflammation and Respiratory Distress in Patients With SARS-CoV-2 Infection (NCT04324021) |
| R-Pharm JSC, Cromos Pharma | Olokizumab# | Humanized IgG4 | IL-6 | Phase 2/3 | Severe COVID-19 (NCT04380519, NCT04452474 pending) |
| Hoffmann-La Roche | Tocilizumab# | Humanized IgG1 | IL-6 R | Phase 3 | Hospitalized Patients With COVID-19 Pneumonia (NCT04372186, NCT04409262) |
| Sinocelltech Ltd. | SCTA01 | Humanized mAb | SARS-CoV-2 | Phase 2/3 pending | Hospitalized Patients With Severe COVID-19 (NCT04644185) |
| Vir Biotechnol./GlaxoSmithKline | VIR-7831/GSK4182136 | Human mAb | SARS-CoV-2 | Phase 2/3 | Early Treatment of COVID-19 in Outpatients (NCT04545060) |
| Celltrion | CT-P59 | Human mAb | SARS-CoV-2 | Phase 2/3 | Mild to Moderate COVID-19 (NCT04602000) |
| AstraZeneca | AZD7442 (AZD8895 + AZD1061) | Human mAbs | SARS-CoV-2 | Phase 3 | Pre-exposure Prophylaxis (NCT04625725); Post-exposure Prophylaxis (NCT04625972) |
*Data publicly available as of November 21, 2020.
#Product previously approved for a disease other than COVID-19 in at least one country.
Table notes: Table 1 includes only monoclonal antibodies evaluated in commercially sponsored, late-stage clinical studies that are listed on clinicaltrials.gov.
Abbreviations: COVID-19, coronavirus disease 2019; EUA, emergency use authorization; IFN, interferon; IL, interleukin; GM-CSF, granulocyte-macrophage colony-stimulating factor; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Also, in early 2020, over 100 organizations sought to discover new antibodies that bind the SARS-CoV-2 Spike protein, and thereby block viral entry into host cells by disrupting interactions of the Spike protein with the cellular angiotensin-converting enzyme 2 (ACE2) receptor. As of November 2020, details for over 120 anti-SARS-CoV-2 antibody therapeutics sponsored by commercial firms have been publicly disclosed (data available at www.antibodysociety.org/covid-19-biologics-tracker). In the remarkably short period of ~10 months, five advanced to late-stage clinical studies (Table 1), and the study results for two of these bamlanivimab (LY-CoV555) and casirivimab and imdevimab (REGN-COV2) were sufficiently positive that EUAs were granted by the US Food and Drug Administration (FDA). A Phase 2/3 study for another, SCTA01, is pending. All of these antibodies target the spike protein of the virus.
AZD7442 (AstraZeneca) is a combination of two monoclonal antibodies (mAbs) derived from convalescent patients with SARS-CoV-2 infection. Discovered at Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the mAbs were optimized by AstraZeneca with half-life extension (YTE modification; half-life 70–100 d) and reduced Fc receptor binding (L234F/L235E/P331S triple mutations). A Phase 1 study (NCT04507256) in healthy adults was started in August 2020, and Phase 3 trials are due to start in mid-November. The Phase 3 studies will evaluate AZD7442 in pre-exposure (PROVENT study, NCT04625725) and post-exposure (STORMCHASER study, NCT04625972) use, as well as outpatient treatment.
CT-P59 (Celltrion) is being evaluated in a placebo-controlled, Phase 2/3 study (NCT04602000) of outpatients with SARS-CoV-2 infection. The estimated enrollment is 1020 participants. This study started in late September 2020 but has a primary completion date in December 2020. Results for a Phase 1 study (NCT04593641) evaluating the safety, tolerability and virology of CT-P59 in patients with mild symptoms of SARS-CoV-2 infection were reported at the 2020 fall Conference of the Korean Society of Infectious Diseases on November 5, 2020.14 This study enrolled a total of 18 patients, 15 of which were randomized into three cohorts that received CT-P59 at 20 mg/kg, 40 mg/kg or 80 mg/kg, respectively. Three patients received matching placebo. Patients treated with CT-P59 experienced about 44% reduced mean clinical recovery time compared to the average for patients who received placebo. Celltrion expects to apply for an EUA, conditional on the results for the Phase 2/3 study of CT-P59.
VIR-7831 (also known as GSK4182136; Vir Biotechnology, GlaxoSmithKline) is being evaluated in a Phase 2/3 study (NCT04545060) as an early treatment of COVID-19 in patients who are at high risk of hospitalization. This study has an estimated enrollment of 1360 participants and a primary completion date in January 2021.
SCTA01 (Sinocelltech Ltd.) is being evaluated in a Phase 1 study (NCT04483375) of healthy subjects that started in July 2020, and a Phase 2/3 study is due to start in February 2021. The placebo-controlled Phase 2/3 trial (NCT04644185) will evaluate the efficacy and safety of SCTA01 in hospitalized patients with severe COVID-19.
Bamlanivimab (also known as LY-CoV555 and LY3819253; Eli Lilly and Company) is being evaluated for several types of patients (outpatient and hospitalized) and uses (prophylaxis and treatment). Results of an interim analysis of Phase 2 BLAZE-1 study (NCT04427501) involving outpatients with recently diagnosed mild or moderate COVID-19 were recently reported.15 Patients received intravenous (IV) administration of 700 mg, 2800 mg, or 7000 mg LY-CoV555 or placebo. The change from baseline to Day 11 in viral load was the primary outcome measure. These values were −0.20 (95% confidence interval [CI], −0.66 to 0.25; P = .38), −0.53 (95% CI, −0.98 to −0.08; P = .02), and 0.09 (95% CI, −0.37 to 0.55; P = .70) for the 700 mg, 2400 mg, and 7000 mg doses, respectively. However, the authors noted that the endpoint did not appear to be clinically meaningful because the viral load was substantially reduced at Day 11 for most patients. This outcome is consistent with the infection’s natural progression. The percentage of patients who were hospitalized on Day 29 was lower among those who received LY-CoV555 vs. placebo (1.6% vs. 6.3%, respectively), in particular for patients in high-risk subgroups (4.2% vs. 14.6%, respectively).
LY-CoV555 is being evaluated in Phase 3 BLAZE-2 trial (NCT04497987), which will assess the ability of the antibody to prevent SARS-CoV-2 infection and COVID-19 in residents and staff at nursing and assisted living facilities. It is also being evaluated in 2 Phase 2/3 platform studies (ACTIV-2 (NCT04518410) and ACTIV-3 (NCT04501978)) sponsored by the National Institute of Allergy and Infectious Diseases that will compare the effects of LY-CoV555 vs. placebo in outpatients with COVID-19 (ACTIV-2) or vs. remdesivir in hospitalized patients (ACTIV-3).
Casirivimab and imdevimab (also known as REGN-COV2 and REGN10933+ REGN10987; Regeneron) is being evaluated in multiple late-stage clinical studies. REGN-COV2 is a cocktail of two human antibodies derived from parallel efforts using both transgenic mice and B cells from recovered COVID-19 patients. Like LY-CoV555, REGN-COV2 is being evaluated in both outpatient and hospitalized patients, and for both prophylaxis and treatment. Results from the ongoing Phase 1/2/3 seamless trial (NCT04425629) evaluating high (8 g) and low (2.4 g) doses of REGN-COV2 in ambulatory adult patients with COVID-19 were announced in late October 2020.16 The study’s primary and key secondary endpoints, which assessed virologic endpoints based on viral load, seronegative status and dose group, and COVID-19-related medically attended visits in patients who had laboratory-confirmed COVID-19 at baseline, were met. No significant difference in virologic or clinical efficacy between the two doses was observed.
REGN-COV2 is also being evaluated in a seamless Phase 1/2/3 study (NCT04426695) of hospitalized COVID-19 patients. Prophylactic use of the cocktail is being evaluated in an ongoing Phase 3 study (NCT04452318), which will determine how well REGN-COV2 prevents SARS-CoV-2 infection in household contacts of individuals infected with SARS-CoV-2.
Antibody therapeutics under review or authorized for COVID-19 in the US
As of November 20, 2020, an EUA request for leronlimab for the treatment of COVID-19 had been submitted to the FDA, and both bamlanivimab and REGN-COV2 were authorized for emergency use by the FDA. Bamlanivimab and REGN-COV2 target the SARS-CoV-2 virus and thus reduce the viral load, while leronlimab targets CCR5 and is intended to treat symptoms of COVID-19.
Bamlanivimab (Eli Lilly and Company)
Lilly announced they had submitted an EUA request for bamlanivimab monotherapy in higher-risk patients on October 7, 2020, and on November 9, 2020, the FDA authorized its emergency use. The agency stated that “it is reasonable to believe that bamlanivimab may be effective for the treatment of mild to moderate COVID-19 in adults and pediatric patients with positive results of direct SARS-CoV-2 viral testing who are 12 y of age and older weighing at least 40 kg, and who are at high risk for progressing to severe COVID-19 and/or hospitalization, and that, when used under the conditions described in this authorization, the known and potential benefits of bamlanivimab when used to treat COVID-19 in such patients outweigh the known and potential risks of such product.”17
The EUA was based on review of the topline data from the planned interim analysis of BLAZE-1 (NCT04427501), which is ongoing. The EUA letter indicates that distribution of the authorized bamlanivimab (700 mg/20 mL vial) will be controlled by the United States Government for use consistent with the terms and conditions of the EUA, and that the EUA is effective only while circumstances exist justifying the authorization of emergency use during the COVID-19 pandemic. The recommended dose is 700 mg and the recommended concentration for infusion is 700 mg/200 mL, with a minimum infusion rate of 200 mL/h (i.e., 1-h total infusion time).
Lilly has an agreement with the US government to supply 300,000 vials of 700 mg doses of LY-CoV555 for 375 USD million.18 According to Lilly, up to 1 million doses may be available in Q4 2020. The price per vial has been set at 1,250 USD for wealthy countries, with one vial representing a full course of treatment. Tiered pricing for LY-Cov555 monotherapy will apply to middle- and low-income countries.19
Lilly is also pursuing development of a cocktail comprising LY-Cov555 and LYCoV016 (also known as JS016, LY3832479, CB6-LALA). LYCoV016 is a human anti-SARS-CoV-2 antibody with an Fc portion that was modified to include Leu234Ala and Leu235Ala (LALA) mutations, which disrupt effector functions.20 The two antibodies bind complementary regions of the SARS-CoV-2 spike protein. The company anticipates submission of an EUA request for combination therapy by the end of 2020 and may have data to support a biologics license application (BLA) submission for combination therapy as early as Q2 2021.
Casirivimab and imdevimab (Regeneron)
In early October 2020, Regeneron announced that they requested an EUA for casirivimab and imdevimab (REGN-COV2), and on November 20, 2020, the FDA authorized its emergency use.21 After reviewing the analysis of Phase 1 and 2 data from the ongoing Phase 1/2/3 NCT04425629 study, the agency concluded that “it is reasonable to believe that casirivimab and imdevimab, administered together, may be effective for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 y of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progressing to severe COVID-19 and/or hospitalization, and that, when used under the conditions described in this authorization, the known and potential benefits of casirivimab and imdevimab, administered together, outweigh the known and potential risks of such product.”
The EUA letter further states that distribution of REGN-COV2 will be directed by the US government, and its EUA will be effective until the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic is terminated or the EUA is revoked. The authorized dosage is 1,200 mg of casirivimab and 1,200 mg of imdevimab administered together as a single IV infusion over at least 60 min via pump or gravity as soon as possible after positive viral test for SARS-CoV-2 and within 10 d of symptom onset.
Regeneron was granted a 450 USD million contract to manufacture and supply REGN-COV2 by the US government, which has committed to making the doses available to Americans for free. The agreement covers a fixed number of bulk lots, as well as fill/finish and storage activities. Delivery of REGN-COV2 drug product started during the third quarter of 2020, and the company expects to have ~80,000 doses available by the end of November, ~200,000 total doses ready by the first week of January 2021, and ~300,000 total doses ready by the end of January 2021.
Leronlimab (Cytodyn Inc.)
Leronlimab is a humanized anti-CCR5 IgG4 antibody developed for a variety of indications, including HIV, stroke, graft-vs.-host disease (GvHD), triple-negative breast cancer (TNBC), as well as COVID-19. Among other roles, CCR5 modulates immune cell trafficking to sites of inflammation. Data from COVID-19 patients (n = 23) receiving 700 mg leronlimab on an open-label compassionate use basis have suggested that the drug may improve outcomes.22
CytoDyn completed a Phase 2 clinical trial (NCT04343651, CD10) that compared the efficacy and safety of leronlimab vs. placebo for mild to moderate COVID-19. Patients are being recruiting for a Phase 2b/3 trial (CD12, NCT04347239) to evaluate the efficacy and safety of leronlimab for patients with severe or critical COVID-19. In this adaptive-design multicenter study, patients are randomized to receive weekly doses of 700 mg leronlimab, or placebo, which are each administered via subcutaneous (SC) injection.
Cytodyn disclosed in an investment community conference call that they requested an EUA from FDA for leronlimab for mild to moderate COVID-19 based on data from the Phase 2 CD10 study. Top-level results of the study showed that, in patients with Total Clinical Symptom Scores of ≥4 at baseline (higher scores equate to poorer health state), at Day 3, more subjects treated with leronlimab reported improvement in total clinical symptom score compared to the placebo group (90% on leronlimab arm vs. 71% on placebo).23
Antibody therapeutics authorized for COVID-19 in Russia and India
As of November 2020, two repurposed antibodies, levilimab and itolizumab, had been registered for emergency use as treatments for COVID-19.
Levilimab (Ilsira), a human mAb targeting membrane-bound and soluble forms of IL-6 R, is registered in Russia for treatment of patients with severe COVID-19. Developed by Biocad, levilimab was originally developed for treatment of rheumatoid arthritis but had not yet been approved for this indication. Biocad initiated a Phase 3 study in COVID-19 patients in April 2020. The study included 204 participants who received a single SC administration of levilimab at a dose of 324 mg in combination with standard therapy. According to Biocad, the results of a clinical trial of the drug demonstrated that levilimab therapy can significantly reduce mortality among patients with COVID-19.24 Levilimab was registered in Russia on June 5, 2020, through an accelerated procedure established by the Government of the Russian Federation’s decree No. 441, which applies to emergency use of medical drugs for prophylaxis or treatment of disease caused by exposure to adverse chemical and biological conditions.
Itolizumab (Alzumab), a humanized IgG1 targeting CD6, was granted restricted emergency use in India for the treatment of cytokine release syndrome in COVID-19 patients with moderate to severe acute respiratory distress syndrome (ARDS). The EUA was issued by the Drugs Controller General of India on July 11, 2020. Developed by Biocon, itolizumab was previously approved in India for plaque psoriasis. Emergency use was granted based on a randomized, controlled, open-label study at four hospitals in India, enrolling a total of 30 hospitalized COVID-19 patients with moderate to severe ARDS. Twenty patients were randomized to receive itolizumab plus best supportive care, while 10 patients received best supportive care alone. The primary endpoint was reduction in mortality at 1 month.25
Antibody therapeutics granted a first approval in the US or EU in 2020
Companies within the biopharmaceutical industry achieved first marketing approvals for at least 6 antibody therapeutics, and as many as 13, each year starting in 2014 (Figure 1). In total, 61 products were approved in the past ~7 y (January 2014–November 2020). In contrast, only 34 products were approved during the previous 17 y (1997–2013). Despite the pandemic, 10 antibody therapeutics were granted the first approval in the US or European Union (EU) in 2020, as of November (Table 2). The products are teprotumumab-trbw (Tepezza®), eptinezumab-jjmr (VyeptiTM), isatuximab-irfc (Sarclisa®), sacituzumab govitecan-hziy (TrodelvyTM), inebilizumab-cdon (UpliznaTM), tafasitamab-cxix (Monjuvi®), belantamab mafodotin-blmf (Blenrep®), satralizumab-mwge (ENSPRYNGTM), atoltivimab/maftivimab/odesivimab-ebgn (InmazebTM), and naxitamab-gqgk (DANYELZA). It is important to note that EUAs for COVID-19 are not equivalent to approvals, but companies that receive EUAs may pursue marketing approvals for their products in this indication in the future.
Figure 1.
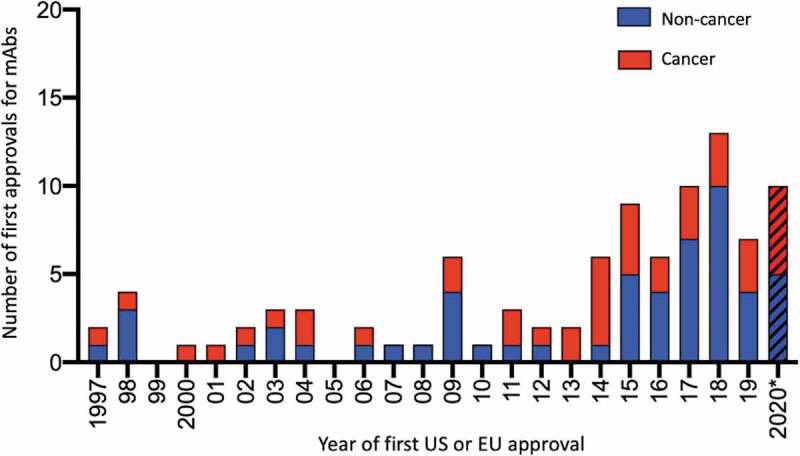
Number of antibody therapeutics first approved in the United States or European Union each year during 1997–2020*
*Data publicly available as of November 25, 2020. Red bars, first approvals for cancer. Blue bars, first approvals for non-cancer indications. Biosimilar antibody and Fc fusion protein products were excluded. A table of US and EU-approved antibody therapeutics is available at antibodysociety.org/resources/approved-antibodies/.
Table 2.
Antibody therapeutics granted first approvals in the European Union or the United States during 2020*
| International non- proprietary name | Brand name | Target; Format | Indication first approved | Date of first EU approval | Date of first US approval | FDA or EMA designations for first approved indication |
|---|---|---|---|---|---|---|
| Teprotumumab | Tepezza | IGF-1 R; Human IgG1 | Thyroid eye disease | NA | 21–01-2020 | BT, FT, US Orphan |
| Eptinezumab | Vyepti | CGRP; Humanized IgG1 | Migraine prevention | NA | 21–02-2020 | NA |
| Isatuximab | SARCLISA | CD38; Chimeric IgG1 | Multiple myeloma | 30–05-2020 | 02–03-2020 | EU/US Orphan |
| Sacituzumab govitecan | TRODELVY | TROP-2; Humanized IgG1 ADC | Triple-neg. breast cancer | NA | 22–04-2020 | BT, FT |
| Inebilizumab | Uplizna | CD19; Humanized IgG1 | Neuromyelitis optica and neuromyelitis optica spectrum disorders | NA | 11–06-2020 | BT, EU/US Orphan |
| Tafasitamab | MONJUVI | CD19; Humanized IgG1 | Diffuse large B-cell lymphoma | In review | 31–07-2020 | BT, FT, EU/US Orphan |
| Belantamab mafodotin | BLENREP | BCMA; Humanized IgG1 ADC | Multiple myeloma | 25–08-2020 | 05–08-2020 | BT, PRIME, EU/US Orphan |
| Satralizumab | ENSPRYNG | IL-6 R; Humanized IgG2 | Neuromyelitis optica spectrum disorders | In review, Accelerated assessment | 13–08-2020 | BT, EU/US Orphan |
| Atoltivimab, maftivimab, and odesivimab | Inmazeb | Ebola virus; Human IgG1 mixture (3 mAbs) | Treatment for Zaire ebolavirus (Ebola virus) infection | NA | 14–10-2020 | BT, EU/US Orphan |
| Naxitamab | Danyelza | GD2; Humanized IgG1 | Neuroblastoma | NA | 25–11-2020 | BT, Rare Pediatric Disease, EU/US Orphan |
*Data publicly available as of November 25, 2020.
Table notes: nonproprietary names for products approved in the US are atoltivimab/maftivimab/odesivimab-ebgn, belantamab mafodotin-blmf, eptinezumab-jjmr, inebilizumab-cdon, isatuximab-irfc, naxitamab-gqgk, sacituzumab govitecan-hziy, satralizumab-mwge, tafasitamab-cxix, teprotumumab-trbw.
Abbreviations: ADC, antibody-drug conjugate; BCMA, B cell maturation antigen; BT, Breakthrough Therapy; CGRP, calcitonin gene-related peptide; FT, Fast Track; IGF-1 R, insulin-like growth factor 1 receptor; IL, interleukin; NA, not applicable; PRIME, Priority Medicine; TROP-2, trophoblast cell-surface antigen 2.
Teprotumumab (Horizon Therapeutics Ireland DAC)
Teprotumumab (Tepezza®) is a human IgG1k antibody targeting insulin-like growth factor 1 receptor (IGF-1 R). It was originally developed in the mid-2000s by F. Hoffman-La Roche Ltd as a treatment for solid tumors.
On January 21, 2020, the FDA approved Tepezza (teprotumumab-trbw) for the treatment of adults with thyroid eye disease, which is associated with an outward bulging of the eye that can cause eye pain, double vision, light sensitivity or difficulty closing the eye. Teprotumumab was granted Fast Track, Breakthrough Therapy and Orphan Drug designations by the FDA for this indication. FDA’s approval was based on positive data from both Phase 2 (NCT01868997) and Phase 3 (OPTIC, NCT03298867) studies. The Phase 2 study evaluated the effects of teprotumumab or placebo when administered every 3 weeks (q3W) by IV infusion. A total of 88 participants were randomly assigned to the treatment phase in a 1:1 ratio to receive a starting dose of 10 mg/kg of teprotumumab or placebo. At Week 3, the teprotumumab dose was escalated to 20 mg/kg IV q3W. Following dose escalation, participants continued at this dose level for all subsequent infusions. Of the patients who were administered teprotumumab, 71% demonstrated a greater than 2 mm reduction in proptosis compared with 20% of those who received placebo at Week 24.26
In the randomized, placebo-controlled Phase 3 OPTIC study, participants received eight infusions of teprotumumab or placebo q3W for a total of 21 weeks. Teprotumumab 10 mg/kg was administered on Day 1 and teprotumumab 20 mg/kg was administered q3W for the remaining 7 infusions. Teprotumumab met the study’s primary endpoint, which was a responder rate of ≥2 mm reduction of proptosis in the study eye (without deterioration in the fellow eye) at Week 24. The study included a total of 83 patients, with 41 assigned to the teprotumumab arm and 42 to the placebo arm. A proptosis response was observed in 83% of patients receiving teprotumumab compared to 10% of patients receiving placebo at Week 24 (p < .001).27 All secondary endpoints in the study were also met.
Eptinezumab (Lundbeck Seattle BioPharmaceuticals, Inc.)
Eptinezumab (VyeptiTM) is a rabbit-derived, humanized, aglycosylated IgG1k antibody that targets calcitonin gene-related peptide. Heavy chain asparagine 297 was mutated to alanine to avoid FcγR and complement protein interactions.28 The mAb is produced in Pichia pastoris yeast cells by recombinant DNA technology. Development of eptinezumab was initiated by Alder BioPharmaceuticals, Inc., which was acquired by Lundbeck in October 2019.
On February 21, 2020, FDA approved eptinezumab-jjmr for the preventive treatment of migraine in adults. The recommended dosage is 100 mg as an IV infusion over approximately 30 min every 3 months; some patients may benefit from a dosage of 300 mg. The safety of Vyepti was evaluated in over 2000 patients with migraine who received at least one dose of the drug. The approval was supported by positive results from the PROMISE 1 (NCT02559895) and PROMISE 2 (NCT02974153) Phase 3 clinical trials, which investigated eptinezumab for episodic and chronic migraine prevention, respectively.1 In PROMISE-1, a total of 665 patients were randomized to receive placebo (n = 222), 100 mg Vyepti (n = 221), or 300 mg Vyepti (n = 222) every 3 months for 12 months. Mean migraine frequency at baseline was approximately 8.6 migraine days per month and was similar across treatment groups; mean change from baseline in monthly migraine days (MMD) with Vyepti compared with placebo months 1–3 was −3.9 d for 100 mg (p = .018), −4.3 d for 300 mg (p < .001), and −3.2 d for placebo. In PROMISE-2, a total of 1,072 patients were randomized to receive placebo (n = 366), 100 mg Vyepti (n = 356) or 300 mg Vyepti (n = 350) every 3 months for 6 months. Mean migraine frequency at baseline was approximately 16.1 migraine days per month and was similar across treatment groups. Mean change from baseline in MMD compared with placebo across Weeks 1 to 12 was −7.7 d for 100 mg (p < .0001), −8.2 d for 300 mg (p < .0001), and −5.6 d for placebo.29
H. Lundbeck A/S is evaluating IV administration of eptinezumab in a Phase 1 open-label, single-dose, pharmacokinetic study of children and adolescents with migraine that will be followed by an optional, multiple-dose, open-label extension period. Participants will receive up to four infusions of eptinezumab. The study started in August 2020. The estimated enrollment is 32 patients and the estimated primary completion date is March 2023.
Isatuximab (Sanofi)
Isatuximab (Sarclisa®, isatuximab-irfc) is a chimeric IgG1k that targets CD38 overexpressed on plasma cells in multiple myeloma (MM). Ixatuximab was granted Orphan Drug designations by the European Medicines Agency (EMA) and the FDA in 2014 and 2016, respectively. Isatuximab was first approved in the US on March 2, 2020, in combination with pomalidomide and dexamethasone for adult MM patients who have received at least two prior therapies including lenalidomide and a proteasome inhibitor.30 The European Commission granted a marketing approval on May 30, 2020, in the same indication. The recommended dose for isatuximab is 10 mg/kg IV every week for 4 weeks followed by every 2 weeks in combination with pomalidomide and dexamethasone until disease progression or unacceptable toxicity.
The approvals of Sarclisa® are based on data from the pivotal Phase 3 ICARIA-MM study (NCT02990338). This multicenter, multinational, randomized, open-label, two-arm trial enrolled 307 patients with relapsed and refractory MM who have received at least two prior therapies including lenalidomide and a proteasome inhibitor. Patients were randomized to receive either isatuximab with pomalidomide and dexamethasone (Isa-Pd) or pomalidomide and dexamethasone. The primary endpoint was progression-free survival (PFS). The study results showed an improvement of the median PFS, which was 11.53 months (95% CI: 8.94–13.9) in the Isa-Pd arm group vs. 6.47 months (95% CI: 4.47–8.28) for those who received only pomalidomide + dexamethasone.30 A subgroup analysis of the data from the ICARIA-MM study showed that Isa-Pd improved PFS, as well as overall response rate (ORR), and overall survival (OS) rates, in elderly patients.31 For patients ≥75 y, PFS was 11.40 months (Isa-Pd; n = 32) vs. 4.47 months (pomalidomide + dexamethasone only; n = 29) (hazard ratio (HR) 0.479; 95% CI, 0.242–0.946).
Sanofi has sponsored three other Phase 3 studies evaluating the safety and efficacy of isatuximab as a treatment for MM. In the IKEMA Phase 3 study (NCT03275285), isatuximab in combination with carfilzomib and dexamethasone was evaluated in 302 relapsed and/or refractory MM patients previously treated with 1 to 3 prior lines. Sanofi announced in May 2020 that the primary endpoint of PFS was met in this study. A risk reduction of 47% of PFS was observed in the isatuximab group compared to carfilzomib and dexamethasone alone.32 The Phase 3 IMROZ study (NCT03319667) is assessing the clinical benefit of isatuximab in combination with bortezomib (Velcade®), lenalidomide (Revlimid®) and dexamethasone versus bortezomib, lenalidomide and dexamethasone in 475 patients with newly diagnosed MM not eligible for transplant. The combination of isatuximab with lenalidomide and dexamethasone in high-risk smoldering MM is also being evaluated in a Phase 3 study (NCT04270409) with an estimated enrollment of 300 patients.
Sacituzumab govitecan (Immunomedics, Inc.)
Sacituzumab govitecan (IMMU-132, sacituzumab govitecan-hziy, TrodelvyTM) is an antibody-drug conjugate (ADC) comprising a humanized IgG1k antibody targeting TROP-2 fused to the active metabolite of irinotecan (SN-38).33 On April 22, 2020, FDA granted Trodelvy® an accelerated approval for adult patients with metastatic TNBC who received at least two therapies for metastatic disease.34 FDA had previously granted sacituzumab govitecan Breakthrough Therapy and Fast Track designations for metastatic TNBC, and the BLA received a Priority review.
FDA’s approval was based on findings from the pivotal, multicenter, single-arm clinical trial IMMU-132-01 (NCT01631552) that enrolled 108 previously treated patients with metastatic TNBC. Sacituzumab govitecan was administered at 10 mg/kg IV on Days 1 and 8 every 21 d. Tumor imaging was obtained every 8 weeks, and patients were treated until disease progression or intolerance to therapy. The primary efficacy endpoints were objective response rate using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 criteria and response duration. The ORR was 33.3% (95% CI: 24.6, 43.1) and the median response duration was 7.7 months (95% CI: 4.9, 10.8). Of the patients with a response to sacituzumab govitecan-hziy, 55.6% maintained their response for 6 or more months and 16.7% maintained their response for 12 or more months.34
The accelerated approval program allows FDA to approve drugs for serious conditions to fill an unmet medical need based on a surrogate endpoint, i.e., a result that is reasonably likely to predict a clinical benefit to patients, but additional clinical trials are required to confirm the clinical benefit. In July 2020, Immunomedics announced that the confirmatory Phase 3 ASCENT study (NCT02574455) of TrodelvyTM met its primary endpoint of PFS and key secondary endpoints in brain metastasis-negative patients with metastatic TNBC who have previously received at least two prior therapies for metastatic disease.35 While Immunomedics has indicated that they plan to submit a supplemental BLA to support full approval of TrodelvyTM in the US in the fourth quarter of 2020 and that they are on track to file for regulatory approval in the EU in the first half of 2021, in September 2020 Gilead Sciences, Inc. and Immunomedics announced that they entered into a definitive agreement pursuant to which Gilead will acquire Immunomedics.
Sacituzumab govitecan is also being evaluated as a treatment for hormonal receptor-positive, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer, lung cancer, and urinary bladder cancer. FDA has granted sacituzumab govitecan Fast Track designations for metastatic non-small cell lung cancer (NSCLC), metastatic SCLC, and metastatic urothelial cancer. The ADC has also been designated an Orphan Drug for SCLC by the FDA, and for pancreatic cancer by both FDA and EMA.
Inebilizumab (Viela Bio)
Inebilizumab-cdon (UpliznaTM) is a humanized anti-CD19 IgG1k antibody indicated as a treatment of neuromyelitis optica spectrum disorder (NMOSD). NMOSD is a rare autoimmune disorder of the central nervous system that primarily damages the optic nerve(s) and spinal cord, causing blindness, muscle weakness and paralysis. Most patients with this disorder have autoantibodies to aquaporin-4 (AQP4). Inebilizumab depletes CD19+ B cells and plasmablasts, which are responsible for the production of autoantibodies directed against AQP4. Inebilizumab was granted FDA’s Breakthrough Therapy designation for the treatment of NMOSD, as well as Orphan Drug designations by the FDA and the EMA.
On June 11, 2020, FDA approved inebilizumab-cdon for the treatment of NMOSD in adult patients who are anti-AQP4-antibody positive. The effectiveness of inebilizumab was demonstrated in the Phase 2/3 N-MOmentum trial (NCT02200770).36 This study enrolled 230 patients with and without the AQP4-IgG antibody who were randomized 3:1 to receive either 2 doses of 300 mg of inebilizumab as a monotherapy or placebo at Day 1 and Day 15, and then followed for a total of 28 weeks. The primary endpoint (time to first attack) and a majority of the secondary endpoints of the study were met. Of patients in the anti-AQP4 antibody positive group, 89% remained relapse-free during the post-treatment period compared to 58% of the patients taking placebo.37 There was no evidence of a benefit in patients who were anti-AQP4 antibody negative. Inebilizumab demonstrated a favorable safety and tolerability profile, with an adverse event rate similar to placebo. The recommended dosage of Uplizna includes an initial dose of 300 mg IV infusion followed 2 weeks later by a second 300 mg IV infusion, and then subsequent doses (starting 6 months from the first infusion) of single 300 mg IV infusion every 6 months.
Uplizna is the second antibody therapeutic to be approved in the US for NMOSD. FDA approved Soliris (eculizumab) injection for IV use for the treatment of NMOSD in adult patients who are anti-AQP4 antibody positive in June 2019. Soliris, which targets C5 was first approved by FDA in 2007 for paroxysmal nocturnal hemoglobinuria. A BLA for anti-IL-6 receptor satralizumab as a possible treatment for NMOSD was under review at FDA when Uplizna was approved for this indication.
Tafasitamab (MorphoSys AG)
Tafasitamab (Monjuvi®, tafasitamab-cxix), also known as MOR208 and XmAb5574, is a humanized anti-CD19 antibody with an engineered Fc domain that functions through apoptosis and Fc-dependent mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP).38 Tafasitamab was granted FDA’s Breakthrough Therapy and Fast Track designations, and Orphan Drug designations by both FDA and EMA for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL). Its BLA for this indication was granted Priority Review by FDA. A marketing authorization application (MAA) for tafasitamab for DLBCL is undergoing regulatory review by the EMA.
On July 31, 2020, FDA granted an accelerated approval for Monjuvi® in combination with lenalidomide for adult patients with relapsed or refractory DLBCL who are not eligible for autologous stem cell transplant. The approval was based on data from the single-arm Phase 2 L-MIND study (NCT02399085), which enrolled 81 patients. In this study, patients received tafasitamab at 12 mg/kg IV in combination with lenalidomide for a maximum of 12 28-d cycles, followed by tafasitamab as monotherapy. The primary endpoint was the best ORR and response duration. Of 80 patients who received tafasitamab plus lenalidomide, 60% had an objective response, 43% had a complete response and 18% had a partial response.39 The results of the study showed the best ORR in 71 DLBCL patients was 55%, with complete responses in 37% and partial responses in 18% of patients. Median response duration was 21.7 months.40
Belantamab mafodotin (GlaxoSmithKline)
Belantamab mafodotin (GSK2857916, belantamab mafodotin-blmf, Blenrep®) is a humanized, afucosylated IgG1k antibody targeting B cell maturation antigen (BCMA) conjugated to the cytotoxic agent maleimidocaproyl monomethyl auristatin (MMA) F. This ADC is produced using POTELLIGENT technology licensed from BioWa. FDA granted Orphan Drug and Breakthrough Therapy designations to belantamab mafodotin for MM, and its BLA for MM was granted a Priority review.
On August 5, 2020, Blenrep® was granted an accelerated approval by the FDA as monotherapy for the treatment of MM in adult patients who have received at least four prior therapies and whose disease is refractory to at least one proteasome inhibitor, one immunomodulatory agent, and an anti-CD38 mAb, and who have demonstrated disease progression on the last therapy.41 Belantamab mafodotin was evaluated in this indication in the open-label, multicenter Phase 2 DREAMM-2 (NCT03525678) study. Patients received IV administration of either belantamab mafodotin at 2.5 mg/kg or 3.4 mg/kg once every 3 weeks until disease progression or unacceptable toxicity. ORR was the primary endpoint of the study. Treatment with single-agent belantamab mafodotin, administered as 2.5 mg/kg doses every 3 weeks, resulted in an ORR of 31% (97.5% CI: 21%, 43%).42 The prescribing information for Blenrep® includes a boxed warning due to alterations in vision, including severe vision loss and corneal ulcer, and the drug is available only through a restricted distribution program.
Data from the DREAMM-2 Phase 2 study also served as the basis for the evaluation of belantamab mafodotin’s MAA, which was reviewed under EMA’s accelerated assessment program. Belantamab mafodotin was accepted in EMA’s PRIority MEdicines (PRIME) scheme, and it was designated as an Orphan medicinal product. The data reviewed by EMA included 13-month follow-up data, which showed an ORR of 32%, and median duration of response (DOR) and OS of 11 months and 13.7 months, respectively.43 The European Commission granted a conditional marketing authorization on August 25, 2020. Blenrep is indicated in the EU as monotherapy for the treatment of multiple myeloma in adult patients, who have received at least four prior therapies and whose disease is refractory to at least one proteasome inhibitor, one immunomodulatory agent, and an anti-CD38 mAb, and who have demonstrated disease progression on the last therapy.
Satralizumab (Genentech Inc.)
Satralizumab (ENSPRYNGTM) is a recombinant humanized IgG2k antibody targeting the receptor for IL-6, a pro-inflammatory cytokine implicated in the pathology of NMOSD. The antibody, which was invented by Chugai and licensed to Roche in 2016, is designed to have pH-dependent binding to soluble IL-6 receptor. The antibody thus has a longer duration compared to conventional antibodies because it can release bound IL-6 R in lysosomes and recycle via an FcRn-mediated salvage pathway. Satralizumab was granted Orphan Drug designations in the US, Europe and Japan. FDA granted a Breakthrough Therapy Designation for the treatment of NMOSD in December 2018. Prior to FDA’s approval, ENSPRYNG had been granted approvals in Canada, Japan and Switzerland. Applications are under review by other regulatory agencies, including in the EU and China.
On August 13, 2020, FDA approved satralizumab-mwge (ENSPRYNG) for the treatment of NMOSD in adult patients who are anti-AQP4 antibody positive. The efficacy of satralizumab for the treatment of NMOSD in adult patients was established in two studies. SAkuraStar (NCT02073279) was a randomized (2:1), placebo-controlled trial in 95 patients without concurrent immunosuppressive therapy (IST) in which 64 patients were anti-AQP4 antibody positive and 31 patients were anti-AQP4 antibody negative. SAkuraSky (NCT02028884) was a randomized (1:1), placebo-controlled trial in 76 adult patients with concurrent IST. Of these, 52 adult patients were anti-AQP4 antibody positive and 24 adult patients were anti-AQP4 antibody negative. In the SAkuraStar monotherapy study’s AQP4 antibody positive subgroup, 76.5% of satralizumab-treated patients were relapse-free at 96 weeks, compared to 41.1% with placebo.44 In the SAkuraSky study, which evaluated satralizumab when used concurrently with baseline IST, 91.1% of satralizumab-treated AQP4 antibody positive subgroup patients were relapse-free at 96 weeks, compared to 56.8% with placebo.45 Based on results of the clinical studies, the recommended loading dosage of ENSPRYNG for the first three administrations is 120 mg by SC injection at Weeks 0, 2, and 4, followed by a maintenance dosage of 120 mg every 4 weeks.
Atoltivimab, maftivimab, and odesivimab (Regeneron)
Atoltivimab, maftivimab, and odesivimab are IgG1 antibodies that bind glycoprotein on the surface of Ebola virus, thereby blocking attachment and entry of the virus into cells. Developed by Regeneron, a cocktail of these three antibodies was evaluated as a treatment for Ebola virus infection. FDA granted the atoltivimab, maftivimab, and odesivimab cocktail Breakthrough Therapy and Orphan Drug designations for this indication.
On October 14, 2020, FDA approved the triple antibody cocktail of atoltivimab, maftivimab, and odesivimab-ebgn (InmazebTM) for the treatment for Zaire ebolavirus (Ebola virus) infection in adult and pediatric patients.46 The effects of Inmazeb were evaluated in adult and pediatric patients with confirmed infections that occurred during an Ebola virus outbreak in the Democratic Republic of the Congo in 2018 and 2019.47 In the Pamoja Tulinde Maisha (PALM) study (NCT03719586), a 4-arm trial evaluating investigational therapies for Ebola virus infection initiated in November 2018, 154 patients received Inmazeb as a single IV infusion of 50 mg of each mAb. The primary efficacy endpoint was 28-d mortality. At this timepoint, 33% of those who received Inmazeb had died vs. 51% of those who received a control. Inmazeb was also made available in an expanded access program, which included an additional 228 patients who received Inmazeb. The PALM study was sponsored by the US National Institute of Allergy and Infectious Diseases, with collaborators from Institut National de Recherche Biomédicale (Democratic Republic of Congo); the Alliance for International Medical Action (Senegal); International Medical Corps, Los Angeles (US); Epicenter, Médecins sans Frontières (France); and the World Health Organization (Switzerland).
Naxitamab (Y-mAbs Therapeutics, Inc.)
Naxitamab (hu3F8, naxitamab-gqgk, Danyelza™), a humanized anti-GD2 IgG1k antibody, was developed by Memorial Sloan Kettering Cancer Center and licensed to Y-mAbs Therapeutics. FDA granted Breakthrough Therapy and Rare Pediatric Drug designations to naxitamab for the treatment of patients with neuroblastoma, and naxitamab was granted Orphan Drug designations in the EU and US for this indication.
On November 25, 2020, Y-mAbs announced that the FDA approved naxitamab-gqgk 40 mg/10 ml in combination with GM-CSF for the treatment of pediatric patients 1 y of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy. The approval for this indication was granted under FDA’s accelerated approval regulations based on the ORR and DOR.48
The BLA for naxitamab included efficacy and safety results from the Phase 1/2 study 12–230 (NCT01757626) and the ongoing pivotal Phase 2 study 201 (NCT03363373). In study 12–230, combination therapy of naxitamab with GM-CSF is being evaluated in patients with relapsed/refractory high-risk neuroblastoma. Data from several subsets of patients from Study 12–230 were presented during the International Society of Pediatric Oncology Annual Congress in 2019. In 28 patients with primary refractory high-risk neuroblastoma refractory to intensive induction therapy, the ORR was 78%, and the 2-y PFS rate was 50%. In the second subgroup of 35 patients with relapsed neuroblastoma resistant to salvage therapy, among 30 evaluable patients, the ORR was 37% and the 2-y PFS rate was 36%.49
The pivotal study 201 is investigating the efficacy and the safety of naxitamab plus GM-CSF as a treatment for high-risk neuroblastoma patients with primary refractory disease or incomplete response to salvage treatment in bone and/or bone marrow. The primary outcome measure of the study is ORR. Data from the study presented at the International Society of Pediatric Oncology Virtual Annual Congress in October 2020 indicated an ORR of 68% and rate of complete response of 59% for 22 patients.50 The estimated primary completion date of the study is November 2024.
Antibody therapeutics first approved outside the US or EU in 2020
Although antibody therapeutics are typically granted the first approval in the US or EU, this is not always the case. In 2020, prolgolimab and olokizumab were granted their first approval in Russia and cetuximab saratolacan sodium was first approved in Japan.
Prolgolimab (Biocad) approved in Russia
Prolgolimab (Forteca, BCD-100) is an anti-programmed cell death protein 1 (PD-1) human IgG1 (V-lambda-C-kappa) antibody with the Fc-silencing mutations leucine (L) to alanine (A) substitution at the position 234 and 235 (i.e., ‘LALA’ mutations). In April 2020, prolgolimab was granted the first market authorization by the Russian Ministry of Health for the treatment of unresectable or metastatic melanoma.51
Prolgolimab’s approval was based on positive clinical results from the open-label randomized Phase 2 MIRACULUM study (NCT03269565) in patients with advanced melanoma who received prolgolimab 1 mg/kg or 3 mg/kg once every 2 weeks until disease progression or intolerable toxicity. The primary endpoint, ORR, was met in both study arms. In the 1 mg/kg arm, 38% ORR was achieved, and the disease control rate (DCR) was 64%. In the 3 mg/kg arm, 29% ORR was achieved, and the DCR was 46%.52
Olokizumab (R-Pharm Group) approved in Russia
Olokizumab (Artlegia), an anti-IL-6 humanized IgG4k mAb, was first registered in Russia in May 2020 for the treatment of rheumatoid arthritis (RA). Olokizumab, formerly known as CDP-6038, was licensed by R-Pharm from UCB Pharma S.A in 2013.53
Olokizumab was evaluated in three Phase 3 studies evaluating two doses of olokizumab (64 mg SC every 2 or 4 weeks) in patients with RA. The primary outcome measures are the ACR20 response at Week 14. The Phase 3 NCT02760368 study (CREDO 1) evaluated the safety and effectiveness of olokizumab compared to placebo in RA patients who are already receiving, but not fully responding to, treatment with methotrexate. In study NCT02760407 (CREDO 2), the effectiveness and safety of olokizumab were being compared to placebo and adalimumab in RA patients who are taking methotrexate but have active disease. In study NCT02760433 (CREDO 3), the effects of olokizumab were compared to placebo in patients with RA who are already receiving but not fully responding to treatment with a tumor necrosis factor (TNF) inhibitor. Most patients who participated in these Phase 3 studies are continuing treatment in the long-term safety and efficacy study CREDO 4 (NCT03120949), which has a primary completion date in July 2021.
Cetuximab saratolacan sodium (Rakuten Medical) approved in Japan
Cetuximab saratolacan sodium (RM-1929, ASP-1929, Akalux® IV Infusion) was first approved in Japan in September 2020 for the treatment of unresectable locally advanced or recurrent head and neck cancer, along with the BioBlade® Laser System. Akalux® is composed of the anti-epidermal growth factor receptor (EGFR) IgG1k antibody cetuximab conjugated to the molecule IRDye® 700DX. When activated at the tumor site via the laser, the antibody-dye conjugate induces rapid destruction of the cancer cells.54
The Ministry of Health, Labor and Welfare granted cetuximab saratolacan a Sakigake Designation for head and neck cancers. The Sakigake Designation system facilitates the development of breakthrough therapies addressing unmet medical needs when companies initiate early development and seek initial product approval in Japan.55 FDA granted cetuximab saratolacan Fast Track designation for recurrent head and neck cancer squamous cell carcinoma.
Rakuten Medical does not yet have approvals in any other country and is currently running a randomized, double-arm, open-label, controlled Phase 3 trial (NCT03769506) of cetuximab saratolacan photoimmunotherapy versus physician’s choice standard of care for the treatment of locoregional, recurrent head and neck squamous cell carcinoma in patients who have failed or progressed on or after at least two lines of therapy, of which at least one line must be systemic therapy. The primary outcome measures are PFS and OS, and the estimated study completion date is December 15, 2020.
Antibody therapeutics undergoing first regulatory review in the US or EU
As of November 25, 2020, marketing applications for 16 investigational antibody therapeutics were undergoing regulatory review by either the FDA or EMA (Table 3). Of the 16 mAbs, 11 (tanezumab, narsoplimab, evinacumab, aducanumab, tralokinumab, teplizumab, inolimomab, ansuvimab, bimekizumab, anifrolumab, sutimlimab) are possible treatments for non-cancer indications and 5 (margetuximab, oportuzumab monatox, dostarlimab, balstilimab, loncastuximab tesirine) are potential treatments for cancer.
Table 3.
Investigational antibody therapeutics in regulatory review in the European Union or the United States*
| International non- proprietary name | Target; Format | Indication under review | Status in EU | Status in US; est. PDUFA date | FDA or EMA designations for indication under review |
|---|---|---|---|---|---|
| Margetuximab | HER2; Chimeric IgG1 | Breast cancer | NA | In review; Dec. 2020 | FT |
| Tanezumab | NGF; Humanized IgG2 | Osteoarthritis pain | In review | In review; Dec. 2020 | FT |
| Oportuzumab monatox | EpCAM; scFv immunotoxin | Bladder cancer | NA | Rolling BLA in review | FT |
| Dostarlimab | PD-1; Humanized IgG4 | Deficient mismatch repair endometrial cancer | In review | In review | NA |
| Narsoplimab | MASP-2; Human IgG4 | Hematopoietic stem cell transplant-associated thrombotic microangiopathy | NA | Rolling BLA in review | BT, EU/US Orphan |
| Evinacumab | Angiopoietin-like protein 3; Human IgG4 | Hypercholesterolemia | In review | In review; Feb 11, 2021 | BT, US Orphan |
| Aducanumab | Amyloid beta; Human IgG1 | Early Alzheimer’s disease | In review | Rolling BLA; Mar 7, 2021 | FT, PRIME |
| Tralokinumab | IL-13; Human IgG4 | Atopic dermatitis | In review | In review; Q1 2021 | NA |
| Loncastuximab tesirine | CD19; Humanized IgG1 ADC | Diffuse large B-cell lymphoma | NA | In review; May 21, 2021 | US Orphan |
| Teplizumab | CD3; Humanized IgG1 | Type 1 diabetes | NA | Rolling BLA in review | BT, PRIME |
| Inolimomab | CD25; Murine IgG1 | Acute graft-vs-host disease | NA | In review | EU/US Orphan |
| Ansuvimab | Ebola virus; Human IgG1 | Ebola virus infection | NA | In review | BT, US Orphan |
| Bimekizumab | IL-17A and IL-17 F; Humanized IgG1 | Psoriasis | In review | In review | NA |
| Balstilimab | PD-1; Human IgG4 | Cervical cancer | NA | Rolling BLA in review | FT |
| Anifrolumab | IFN a, b, ω receptor 1; Human IgG1 | Systemic lupus erythematosus | In review | In review; Sep 30, 2021 | FT |
| Sutimlimab | C1s; Humanized IgG4 | Cold agglutinin disease | NA | In review; Complete Response letter issued | BT, EU/US Orphan |
*Data publicly available as of November 15, 2020.
Abbreviations: BLA, biologics license application; BT, Breakthrough Therapy designation; C1s, complement component 1s; EMA, European Medicines Agency; EpCAM, epithelial cell adhesion molecule; FDA, Food and Drug Administration; FT, Fast Track designation; HER, human epidermal growth factor receptor; IL, interleukin; IFN, interferon; MASP-2, mannan-binding lectin-associated serine protease-2; MSI-H, microsatellite instability-high; NA, Not applicable; NGF, nerve growth factor; PD-1, programmed cell death protein 1; PDUFA, Prescription Drug User Fee Act.
Margetuximab (MacroGenics, Inc.)
Margetuximab (MGAH22) is a chimeric anti-HER2 IgG1k antibody derived from 4D5, the parent antibody of trastuzumab. Margetuximab and trastuzumab bind the same epitope of HER2 with similar high affinities, but the Fc region of margetuximab was optimized have higher affinity for both 158 V (high binding) and 158 F (low binding) alleles of the activating Fc receptor, CD16A, which may reduce population differences related to FcγR genotype. In January 2018, MacroGenics announced that FDA had granted Fast Track designation to margetuximab for the treatment of patients with metastatic or locally advanced HER2-positive breast cancer who have previously been treated with anti-HER2-targeted therapy. MacroGenics submitted a BLA for margetuximab for the treatment of patients with pre-treated metastatic HER2-positive breast cancer in combination with chemotherapy in December 2019. FDA’s first action on the application is anticipated by December 18, 2020.56
Margetuximab was investigated in the Phase 3 SOPHIA study (NCT02492711) comparing margetuximab plus chemotherapy versus trastuzumab plus chemotherapy in patients with HER2-positive metastatic breast cancer who have previously been treated with anti-HER2-targeted therapies. The primary endpoints include PFS and OS. As reported at the San Antonio Breast Cancer Symposium in December 2019, the median PFS was 5.8 months in the margetuximab arm compared with 4.9 months (95% CI, 4.14–5.45) in the trastuzumab arm (HR, 0.76; 95% CI: 0.59–0.98; P = .033).57 In patients with CD16A genotypes containing a 158 F allele, the median PFS was 6.9 months in the margetuximab arm vs. 5.1 months in the trastuzumab arm (HR, 0.68; 95% CI: 0.52–0.90; nominal P = .005). At that time, limited OS data were available, and the final pre-specified OS analysis was expected in the second half of 2020.58
Tanezumab (Pfizer, Eli Lilly and Company)
Tanezumab is an anti-nerve growth factor (NGF) humanized IgG2k antibody undergoing evaluation as a treatment of osteoarthritis (OA) pain and chronic lower back pain. FDA granted Fast Track designations to tanezumab for these indications. A BLA for tanezumab 2.5 mg administered to patients with chronic pain due to moderate-to-severe OA who have experienced inadequate pain relief with other analgesics was submitted to FDA, and a decision on the application is expected in December 2020. EMA is also reviewing a marketing application for tanezumab.
The marketing applications for tanezumab are supported by data from 39 Phase 1–3 clinical studies that included more than 18,000 patients. Of these, three were Phase 3 studies that evaluated SC administration of tanezumab in patients with moderate-to-severe OA. Results from the double-blind, randomized Phase 3 NCT02709486 study of the analgesic efficacy and safety of the SC administration of tanezumab 2.5 mg or 5 mg vs. placebo in patients with OA of the hip or knee were recently reported.59 In this study, 849 patients received tanezumab or matching placebo (tanezumab 2.5 mg n = 283, tanezumab 5 mg n = 284, placebo n = 282) every 8 weeks for 24 weeks (three doses). The three primary endpoints were (1) change from baseline to Week 24 in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Pain, (2) WOMAC Physical Function, and (3) Patient’s Global Assessment of OA (PGA-OA). Statistically significant improvements in WOMAC Pain and Physical Function were observed in the tanezumab 2.5 mg group, but not PGA-OA. For the tanezumab 5 mg group, statistically significant improvements were observed for all 3 endpoints, but rapidly progressive OA occurred more frequently in patients who received this dose of tanezumab (1.4% (4/283), 2.8% (8/284) and 0% (0/282) of patients in the tanezumab 2.5 mg, tanezumab 5 mg and placebo groups, respectively).
Ansuvimab (Ridgeback Biotherapeutics LP)
Ansuvimab (mAb114) is a human IgG1 mAb that targets the receptor-binding domain of Ebola virus. The antibody was isolated by National Institutes of Health (NIH) scientists and their collaborators from a human survivor of the 1995 Ebola outbreak in the Democratic Republic of the Congo. Ridgeback Biotherapeutics LP licensed the intellectual property related to mAb114 from NIH in 2018. Ansuvimab was granted US Orphan Drug and Breakthrough Therapy designations. A BLA for its use as a treatment for Ebola virus disease has been granted Priority Review by FDA.
Ansuvimab was included in a Phase 2/3 randomized, controlled trial (NCT03719586) that evaluated a total of 4 Ebola virus disease therapeutics.47 When initiated in November 2018, the study included 3 drugs, ansuvimab, ZMapp and remdesivir, but REGN-EB3 was added in January 2019. ZMapp, which served as the study control drug, and REGN-EB3 are both cocktails of three anti-Ebola virus antibodies, while remdesivir is a small molecule nucleotide analog RNA polymerase inhibitor. Patients in the ansuvimab arm of the study received 50 mg/kg administered IV on Day 1 as a single infusion, and those in the ZMapp arm received a dose of 50 mg/kg every third day beginning on Day 1 for a total of three doses. The study’s primary endpoint was death at 28 d. Patients (n = 681) were enrolled until August 9, 2019, when an interim analysis indicated ansuvimab and REGN-EB3 were superior in reducing the mortality of Ebola virus disease compared to ZMapp and remdesivir. Of 174 patients in the ansuvimab group, 61 (35.1%) died compared with 84 of 169 (49.7%) in the ZMapp group (P = .007). Mortality was additionally reduced (33.5%) in the REGN-EB3 group compared with the ZMapp subgroup (51.3%; subgroup comprises the patients in the ZMapp group who were enrolled on or after the time the REGN-EB3 group was added; P = .002).47
Evinacumab (Regeneron Pharmaceuticals, Inc.)
Evinacumab (REGN1500) is a human IgG4k antibody targeting angiopoietin-like 3 (ANGPTL3), which regulates the metabolism of plasma lipids, including low-density lipoprotein (LDL) cholesterol, high-density lipoprotein cholesterol, and triglycerides. The antibody was derived from Regeneron’s Velocimmune® technology platform and includes a stabilizing mutation in the hinge region to minimize half-antibody formation.
A BLA for evinacumab as an adjunct to other lipid-lowering therapies in patients with homozygous familial hypercholesterolemia (HoFH) is undergoing regulatory review in the US. FDA granted Breakthrough Therapy and Orphan Drug designations for evinacumab for HoFH. The BLA for evinacumab received FDA’s Priority Review, and the target action date for the FDA decision is February 11, 2021. Regeneron indicated that EMA recommended an accelerated assessment for evinacumab in June 2020.
Data from the Phase 3 ELIPSE trial (NCT03399786) were used as the basis of regulatory submissions. In this study, 65 patients were randomized to receive either IV administration of evinacumab 15 mg/kg every 4 weeks (n = 43) or placebo (n = 22), plus other lipid-lowering therapies. The primary endpoint was the percent change from baseline in the LDL cholesterol level at Week 24. At baseline, LDL cholesterol was 260 mg/dL in the evinacumab group and 247 mg/dL in the placebo group. The primary endpoint of the study was met. Relative to baseline, the LDL cholesterol level at Week 24 was reduced by 47.1% in the patients administered evinacumab but increased 1.9% in the placebo group.60
Aducanumab (Biogen Inc., Eisai, Co., Ltd.)
Aducanumab (BIIB037), an anti-amyloid beta (Aβ) IgG1k antibody, is undergoing regulatory review in the US as a possible treatment for early Alzheimer’s disease. Biogen licensed aducanumab from Neurimmune Holding AG in 2007 and has collaborated with Eisai on the global development and commercialization of aducanumab since 2017. FDA granted aducanumab Fast Track designation. Aducanumab was accepted into the EMA PRIME program, and EMA has accepted for review, following a standard timetable, an MAA for aducanumab for Alzheimer’s disease. In August 2020, Biogen and Eisai announced that FDA had accepted a BLA for aducanumab, and granted it a Priority Review.61 FDA’s first action on the application is expected by March 7, 2021.
The marketing applications include data from two placebo-controlled Phase 3 studies of patients with early Alzheimer’s disease, EMERGE (NCT02484547) and ENGAGE (NCT02477800), that were designed to evaluate the efficacy and safety of two dosing regimens of aducanumab. Both studies were discontinued in March 2019 because a pre-specified futility analysis predicted that the primary endpoints were unlikely to be met. However, further analysis of the EMERGE study data indicated that the change from baseline in the Clinical Dementia Rating–Sum of Boxes score at Week 78, which was the study’s primary endpoint, was significant in patients administered high-dose aducanumab (22% vs. placebo, P = .01). These patients also showed a consistent reduction of clinical decline as measured by the Mini-Mental State Examination (18% vs. placebo, P = .06), the Alzheimer’s disease Assessment Scale-Cognitive Subscale 13 Items (27% vs. placebo, P = .01), and the Alzheimer’s disease Cooperative Study-Activities of Daily Living Inventory Mild Cognitive Impairment Version (40% vs. placebo, P = .001). Amyloid plaque burden was reduced with low- and high-dose aducanumab compared to placebo at 26 and 78 weeks (P < .001).
Study data for aducanumab were evaluated by FDA’s Peripheral and Central Nervous System Drugs Advisory Committee. In an advisory meeting on November 6, 2020, most members of the committee voted that the study data did not support the drug’s efficacy. The four questions the members considered were (1) Does Study 302 (EMERGE), viewed independently and without regard for Study 301 (ENGAGE), provide strong evidence that supports the effectiveness of aducanumab for the treatment of Alzheimer’s disease? (votes: 1 yes, 8 no and 2 uncertain); (2) Does Study 103 (PRIME) provide supportive evidence of the effectiveness of aducanumab for the treatment of Alzheimer’s disease? (votes: 0 yes, 7 no and 4 uncertain); (3) Has the Applicant presented strong evidence of a pharmacodynamic effect of aducanumab on Alzheimer’s disease pathophysiology?” (votes: 5 yes, 0 no and 6 uncertain); and (4) In light of the understanding provided by the exploratory analyses of Study 301 and Study 302, along with the results of Study 103 and evidence of a pharmacodynamic effect on Alzheimer’s disease pathophysiology, it is reasonable to consider Study 302 as primary evidence of effectiveness of aducanumab for the treatment of Alzheimer’s disease? (votes: 0 yes, 10 no and 1 uncertain).62 FDA typically follows the advice of their advisory committees, but there have been exceptions in the past.
Tralokinumab (LEO Pharma Inc.)
Tralokinumab is a human IgG4λ antibody targeting IL-13, a pleiotropic T helper type 2 cytokine associated with atopic dermatitis and other inflammatory disorders. The antibody interferes with IL-13-mediated signaling by blocking its interactions with both IL-13 receptor α1 and IL-13 receptor α2.63 Originally identified by Cambridge Antibody Technology and developed by MedImmune as CAT-354, AstraZeneca sold the rights to tralokinumab in dermatology indications to LEO Pharma in 2016. Marketing applications for tralokinumab for atopic dermatitis are undergoing review in the US and EU. FDA’s first action on the BLA is expected in the second quarter of 2021.
The marketing applications are supported by data from three Phase 3 studies, ECZTRA 1 (NCT03131648), 2 (NCT03160885), and ECZTRA 3 (NCT03363854). The randomized, double-blind, placebo-controlled, multinational, 52-week ECZTRA 1 and ECZTRA 2 trials evaluated the safety and efficacy of tralokinumab (300 mg SC) as monotherapy in adults with moderate-to-severe atopic dermatitis who were candidates for systemic therapy. The ECZTRA 1 study included 802 patients and the ECZTRA 2 study included 794 patients. ECZTRA 3 was a double-blind, randomized, placebo-controlled, multinational 32-week study of 380 patients that evaluated the safety and efficacy of tralokinumab (300 mg SC) in combination with topical corticosteroid in adults with moderate-to-severe atopic dermatitis who were candidates for systemic therapy. Data from a Phase 2b study (NCT02347176) of 204 adults who received SC administration of 45, 150, or 300 mg tralokinumab, or placebo, every 2 weeks for 12 weeks with concomitant topical glucocorticoids64 supported the selection of the 300 mg dose for the Phase 3 studies.
The results of the three Phase 3 studies were recently published in the British Journal of Dermatology.65 The primary outcome measures for the studies included an Investigator’s Global Assessment (IGA) of 0 or 1 and 75% improvement in Eczema Area and Severity Index (EASI 75) at Week 16. At this time point in the ECZTRA 1 and ECZTRA 2 studies, 15.8% and 22.2% of patients who received tralokinumab achieved an IGA score of 0/1 vs. 7.1% and 10.9% of patients who received placebo, respectively. More patients who received tralokinumab also achieved EASI 75 compared to those who received placebo (25.0% vs. 12.7% and 33.2% vs. 11.4% in ECZTRA 1 and ECZTRA 2, respectively). In the ECZTRA 3 study, which included the use of topical corticosteroid, 38.9% of patients who received tralokinumab achieved IGA 0/1 vs. 26.2% of those who received placebo. EASI 75 was achieved in 56.0% of patients who received tralokinumab vs. 35.7% of those who received placebo.66
Narsoplimab (Omeros Corporation)
Narsoplimab (OMS721) is a human IgG4λ antibody that targets mannan-binding lectin-associated serine protease-2 (MASP-2), the effector enzyme of the lectin pathway of complement activation. An in vitro study has shown that inhibition of MASP2 by narsoplimab can reduce thrombotic microangiopathy (TMA) plasma-mediated human microvascular endothelial cell injury.67
Narsoplimab is undergoing review by FDA as a treatment for hematopoietic stem cell transplant (HSCT)-associated TMA, which is induced by factors associated with stem cell transplantation, such as conditioning regimens, immunosuppressant therapies, infection, and GvHD. FDA granted narsoplimab Breakthrough Therapy and Orphan Drug designations for HSCT-TMA, and narsoplimab was also granted Orphan Drug designation for HSCT in the EU. Omeros Corporation initiated submission of a rolling BLA for this indication in October 2019 and completed the submission in November 2020. The company also plans to submit an MAA to EMA.
The BLA includes results of the Phase 2 pivotal trial of narsoplimab in HSCT-TMA. In the 28-patient single-arm, open-label pivotal trial in adult HSCT-TMA patients, narsoplimab was administered IV once weekly for up to 8 weeks with an extended follow-up period. The complete response rate (CRR) was 61% (95% CI: 40.6% to 78.5%) in the full analysis set (FAS; patient receiving at least one dose of narsoplimab; p < .0001 compared to 15% efficacy threshold agreed with FDA). The CRR in the per-protocol (PP) population (patients receiving the protocol-specified narsoplimab treatment for at least 4 weeks) was 74% (95% CI: 51.6% to 89.8%; p < .0001 compared to the 15% threshold); 100-d survival was 68% in the FAS, 83% in the PP population and 94% in complete responders; and the median OS was 274 d in the FAS and 361 d in the PP population. OS could not be determined for the complete responders because more than half of the responders were alive at the last follow-up.68
The FDA has granted narsoplimab Breakthrough Therapy designation for IgA nephropathy, Fast Track designation for the treatment of patients with aHUS, and Orphan Drug designations for the prevention (inhibition) of complement-mediated thrombotic microangiopathies and for the treatment of IgA nephropathy. Narsoplimab was granted Orphan Drug designation for treatment of primary IgA nephropathy in the EU.
Teplizumab (Provention Bio, Inc.)
Teplizumab (PRV-031) is a humanized IgG1k that binds to an epitope of the CD3-epsilon chain expressed on mature T lymphocytes and thereby modulates the pathological immunologic responses underlying type 1 diabetes (T1D) and other autoimmune diseases. Teplizumab was granted FDA’s Breakthrough Therapy designation for the prevention or delay of clinical T1D, and EMA granted teplizumab PRIME designation for the prevention or delay of clinical T1D in individuals at risk of developing the disease. Provention Bio acquired worldwide development and commercialization rights to teplizumab from MacroGenics, Inc. in 2018.
A rolling BLA for teplizumab for the delay or prevention of clinical T1D in at-risk individuals, as indicated by the presence of two or more T1D-related autoantibodies, was started in April 2020 and completed by November 2020. The BLA includes data from the Phase 2 “At-Risk” study (NCT01030861), which evaluated whether administration of teplizumab can prevent or delay the development of T1D in high-risk autoantibody-positive non-diabetic relatives of patients with T1D. Participants received IV infusions of teplizumab given for 14 consecutive days (n = 44) or placebo (n = 32). Extended follow-up data showed that, compared to placebo, one course of teplizumab delayed insulin-dependence in presymptomatic T1D patients by a median of approximately 3 y.69 Participants in the “At-Risk” study who develop clinical T1D after the conclusion of that trial can enroll in an extension study (NCT04270942) and receive teplizumab treatment within 1 y of diagnosis of clinical T1D.
Teplizumab is also being evaluated in the Phase 3 PROTECT study (NCT03875729), which will determine whether teplizumab slows the loss of β cells and preserves β cell function in children and adolescents 8–17 y old who have been diagnosed with T1D in the previous 6 weeks. Estimated enrollment for the PROTECT study is 300 patients, and the estimated primary completion date is May 2022.
Inolimomab (ElsaLys Biotech SA)
Inolimomab (LEUKOTAC) is a murine IgG1k mAb that blocks the IL-2 receptor alpha chain (also known as CD25). The drug is undergoing regulatory review in the US as a possible treatment for steroid-refractory acute GvHD (SR-aGvHD) in grade II–IV adult patients.70 Inolimomab was granted Orphan Drug designation for GvHD in both the US and EU. ElsaLys Biotech acquired the development and commercialization rights of inolimomab from Jazz Pharmaceuticals in 2017, which had rights to the drug via its acquisition of EUSA Pharma. EUSA Pharma had acquired the rights for Leukotac from Biotest AG in 2003.
The BLA for inolimomab will be reviewed under FDA’s Real-Time Oncology Review pilot program, which allows the FDA to review key raw and derived datasets as well as results and analyses, and provide feedback before the applicant formally submits the complete application. This approach differs from rolling review, where only fully completed modules can be submitted, and not individual components of a module.
The submission is supported by data from a randomized, multicenter, controlled parallel-group Phase 3 study (INO-107 – EUDRACT 2007–005009-24) initiated in 2009 that compared inolimomab vs. anti-thymoglobulin (ATG) in patients with SR-aGVHD. The study included a total of 100 patients, with 49 patients in the inolimomab arm and 51 patients in the ATG arm. The primary endpoint, OS at 1 y without changing the therapy allocated at baseline, was not met, although 14 patients (28.5%) in the inolimomab arm survived 1 y vs. 11 patients (21.5%) in the ATG arm (adjusted HR, 0.722; P =.188) A long-term follow-up analysis of patient survival after 1 y showed additional clinical benefit was achieved by patients who received inolimomab (30% survival vs. 19.6% survival for the inolimomab vs. ATG arms, respectively).71
Bimekizumab (UCB)
Bimekizumab (UCB4940) is a humanized IgG1k antibody that selectively inhibits two (IL-17A and IL-17 F) of six structurally similar members of the IL-17 family of cytokines. These two pro-inflammatory cytokines share ~50% sequence identity and are expressed as homodimers and IL-17A/F heterodimers. Bimekizumab’s affinities for IL-17A and IL-17 F are 3.2 pM and 23 pM, respectively, and it can bind both the homodimers and heterodimers of these two cytokines.72 Marketing applications for bimekizumab as a treatment for psoriasis are undergoing regulatory review in the US and EU.
The clinical development program for bimekizumab in psoriasis included six Phase 3 studies, three of which included an active comparator arm, ustekinumab (Stellara®; BE VIVID study), adalimumab (Humira®; BE SURE study), or secukinumab (Cosentyx®; BE RADIANT study). Results for the Phase 3 BE VIVID study (NCT03370133) that compared bimekizumab to ustekinumab were recently presented as part of a virtual session for the American Academy of Dermatology 2020 Annual Meeting.73 The BE VIVID study enrolled 570 participants with chronic plaque psoriasis for at least 6 months prior to screening, and with an affected body surface area of at least 10% and Psoriasis Area and Severity Index (PASI) of at least 12 and IGA score ≥3 on a 5-point scale. The primary outcome measures were PASI90 response and IGA response at Week 16. Results at Week 16 and Week 52 were reported. At Week 16, 58.6% and 20.9% of bimekizumab-treated patients and ustekinumab-treated patients achieved PASI 100, respectively. PASI 90 rates (all comparisons p < .001) were bimekizumab: 85.0%; ustekinumab: 49.7%; and placebo: 4.8%. The IGA 0/1 rates were bimekizumab: 84.1%; ustekinumab: 53.4%; and placebo: 4.8%. At Week 52, 64.2% and 38% of bimekizumab-treated patients and ustekinumab-treated patients achieved PASI 100, respectively. More bimekizumab-treated patients also achieved IGA 0/1 and PASI 90 at Week 52 compared with ustekinumab-treated patients (77.9% vs. 60.7%, and 81.6% vs. 55.8%, respectively; p < .001).74
UCB has also reported positive results from the Phase 3 active-controlled BE SURE and BE RADIANT studies. BE SURE (NCT03412747) compared bimekizumab to the TNF inhibitor adalimumab in the treatment of adults with moderate-to-severe plaque psoriasis. BE SURE met its co-primary endpoints at Week 16, demonstrating the superiority of bimekizumab to adalimumab in achieving at least a 90% improvement in the PASI 90 and IGA response of clear or almost clear (IGA 0/1).75 BE RADIANT (NCT03536884) met its primary endpoint at Week 16 with statistical significance, demonstrating the superiority of bimekizumab over the IL-17A inhibitor secukinumab for complete skin clearance, as measured by a 100% improvement in the PASI 100.76
Oportuzumab monatox (Sesen Bio)
Oportuzumab monatox (Vicineum®, VB4-845) is an immunotoxin composed of a humanized single-chain antibody variable fragment directed against the epithelial cell adhesion molecule (EpCAM) fused to Pseudomonas aeruginosa exotoxin A. It is undergoing evaluation as a treatment of high-risk non-muscle invasive bladder cancer (NMIBC) that is unresponsive to treatment with bacillus Calmette-Guérin (BCG). In 2018, the FDA granted oportuzumab monatox Fast Track designation for the treatment of NMIBC.
In December 2019, Sesen Bio initiated a rolling BLA submission for oportuzumab monatox to the FDA based on data from the Phase 3 VISTA trial (NCT02449239). This open-label, multicenter, single-arm Phase 3 is evaluating the efficacy and tolerability of oportuzumab monatox for the treatment of high-risk, BCG-unresponsive NMIBC. The primary endpoints of the trial are the CRR and the DOR (defined as the time from complete response to treatment failure) in patients with carcinoma in situ (CIS) with or without papillary disease. Patients in the trial receive locally administered oportuzumab monatox twice a week for 6 weeks, followed by once-weekly treatment for another 6 weeks, then treatment every other week for up to 2 y. According to data available as of May 2019, the CRR at 3 months was 40% for CIS patients, and among patients who achieved a complete response at 3 months, 52% had a complete response for a total of 12 months or longer after starting therapy.77
Sesen Bio expects to complete the BLA submission in December 2020 and anticipates a potential approval in the US in 2021. They expect to submit an MAA for Vicineum to the EMA in early 2021 with a potential approval in the EU anticipated in early 2022. Sesen Bio licensed rights to the commercialization of oportuzumab monatox in Greater China (China, Hong-Kong, Macau and Taiwan) to Qilu Pharmaceutical. Sesen Bio retains full development and commercialization rights in the US and the rest of world excluding Greater China, where Qilu will be the marketing authorization holder.78
Dostarlimab (GlaxoSmithKline)
Dostarlimab (TS-042, GSK4057190A) is an anti-PD-1 humanized IgG4k antibody generated by Anaptysbio under partnership with Tesaro, which was acquired by GlaxoSmithKline in 2019. BLA and MAA submissions were accepted by the FDA and EMA, respectively, in the first quarter of 2020 for dostarlimab for the second-line treatment of advanced or recurrent deficient mismatch repair (dMMR) endometrial cancer.
Interim analyses of data for patients with MMR-deficient endometrial cancer with recurrent or advanced disease that progressed on a platinum doublet regimen enrolled in the Phase 1 GARNET study (NCT02715284) were reported at the European Society for Medical Oncology (ESMO) Virtual Congress in September 2020. Patients received 500 mg of dostarlimab every 3 weeks for the first 4 cycles, then 1,000 mg every 6 weeks until disease progression or discontinuation. The primary endpoints included confirmed ORR and DOR. The ORR was 44.7% in patients with dMMR disease and 13.4% in those with MMR-proficient (MMRp) disease. In the dMMR cohort (n = 103), 11 complete responses, and 35 partial responses were observed. Thirteen patients achieved stable disease, while 39 patients experienced disease progression. In the MMRp cohort (n = 142), 3 patients had CRs, 16 had PRs, 31 achieved stable disease, and 77 patients experienced progressive disease. At the time of data cutoff, with a median follow-up of 11.2 months, the median DOR had not been reached.79
Dostarlimab is also being evaluated as a treatment for various types of cancer in early-stage clinical studies, as well as two Phase 3 studies, RUBY and FIRST. The RUBY study (NCT03981796) is evaluating dostarlimab plus carboplatin-paclitaxel versus placebo plus carboplatin-paclitaxel in patients with recurrent or primary advanced endometrial cancer. The primary outcome measure is the PFS assessed by an investigator, and the primary completion date is October 2021. The FIRST study (NCT03602859) is a comparison of platinum-based therapy with dostarlimab and niraparib versus standard of care platinum-based therapy as first-line treatment of Stage III or IV non-mucinous epithelial ovarian cancer. The primary outcome measure is the PFS and the primary completion date is February 2023.
Balstilimab (Agenus Inc.)
Balstilimab (AGEN2034) is a human IgG4k antibody directed against PD-1, a negative regulator of immune activation expressed by T cells. Numerous antibodies that target PD-1 or its ligand PD-L1 (e.g., pembrolizumab, nivolumab, cemiplimab; atezolizumab, avelumab, durvalumab) are marketed as treatments for solid tumors as well as lymphoma. Agenus’ clinical studies of balstilimab have focused on its use, either as monotherapy or in combination with anti-CTLA4 zalifrelimab (AGEN1884), as a treatment for cervical cancer. FDA granted both balstilimab and the balstilimab/zalifrelimab combination Fast Track designations for the treatment of cervical cancer. As of September 2020, Agenus Inc. had initiated a rolling BLA submission for balstilimab as monotherapy for the treatment of recurrent/metastatic cervical cancer. Agenus controls worldwide rights to balstilimab, except for certain South American rights, which are controlled by Recepta Biopharma, and Greater China rights, which are exclusively licensed to Betta Pharmaceuticals.
Data from a randomized, blinded, non-comparative, two-arm Phase 2 study (NCT03894215) supported the BLA submission. This study evaluated the efficacy and safety of balstilimab as monotherapy or in combination with zalifrelimab for treatment of patients with advanced cervical cancer who relapsed or progressed after receiving first-line platinum-based chemotherapy for a maximum of 24 months, or until disease progression or unacceptable toxicity. The goal of the study is to evaluate the efficacy of each arm against its relevant historical controls. The primary outcome measure is the objective response rate to balstilimab administered with placebo (Treatment Arm 1 – monotherapy), or with zalifrelimab (Treatment Arm 2 – combination therapy). Data from the study were presented at the ESMO Virtual Congress held in September 2020.80 Of 160 patients included in the balstilimab monotherapy arm, the response rates in PD-L1-positive patients and all patients were 19% and 14%, respectively. Of 143 patients included in the balstilimab/zalifrelimab combination arm, the response rates in PD-L1-positive patients and all patients were 27% and 22%, respectively.
Loncastuximab tesirine (ADC Therapeutics SA)
Loncastuximab tesirine (ADCT-042) is an ADC composed of an anti-CD19 humanized IgG1k antibody conjugated via a linker to pyrrolobenzodiazepine (PBD)-dimer toxin that induces the killing of CD19-expressing malignant B cells. The FDA granted Orphan Drug designation to ADCT-402 for the treatment of diffuse large B-cell lymphoma (DLBCL) and mantle cell lymphoma. In September 2020, ADC Therapeutics SA announced the submission of a BLA to FDA for loncastuximab tesirine for the treatment of patients with relapsed or refractory DLBCL.81 FDA’s first action date is May 21, 2021.
The BLA submission was supported by data from the open-label, single-arm Phase 2 LOTIS 2 study (NCT03589469), which evaluated the safety and efficacy of loncastuximab tesirine for the treatment of patients with relapsed or refractory DLBCL following ≥2 lines of prior systemic therapy. A total of 145 patients received loncastuximab tesirine as an IV infusion over 30 min on Day 1 of each cycle (every 3 weeks) at a dose of 150 μg/kg for 2 cycles, then 75 μg/kg for subsequent cycles for up to 1 y or until disease progression, unacceptable toxicity, or other discontinuation criteria. The primary outcome measure is the ORR. Positive initial data from LOTIS 2 were presented during the virtual 25th Annual Congress of the European Hematology Association. The ORR was 48.3% (70/145 patients), the CRR was 24.1% (35/145 patients), and the median DOR was 10.25 months. The toxicity profile was manageable and no new safety concerns were identified.82
Anifrolumab (AstraZeneca)
Anifrolumab (MEDI-546) is an IFN alpha receptor 1 (IFNAR1)-specific human IgG1κ antibody. It binds to subunit 1 of IFNAR1, thereby blocking the action of different type I IFNs (IFN-α, IFN-β and IFN-ω). The heavy chain of the antibody incorporates three mutations, L234F, L235E, and P331S, to decrease effector functions.83 Marketing applications for anifrolumab for systemic lupus erythematosus (SLE) were submitted in the US and EU during the third quarter of 2020. Regulatory decisions are expected in the second half of 2021.84 FDA has granted anifrolumab Fast Track designation for SLE.
Data from the randomized, double-blind, placebo-controlled Phase 3 TULIP 2 study (NCT02446899), which included 180 SLE patients who received anifrolumab and 182 who received placebo, were recently reported.85 The primary endpoint was a response, defined by British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA), at Week 52. In the anifrolumab arm, 47.8% of patients had a BICLA response, with the percentage slightly higher (48.0%) for patients with a high IFN gene signature and lower (46.7%) among patients with a low IFN gene signature. In the placebo arm, the corresponding percentages were 31.5%, 30.7% and 35.5%, respectively. The difference between the two study arms in the percentage of patients with a BICLA response was statistically significant (95% CI, 6.3 to 26.3; P = .001).
Sutimlimab (Sanofi)
Sutimlimab is a humanized IgG4 antibody that targets and inhibits complement component 1s (C1s) in the complement system, thereby interfering with C1-activated hemolysis in cold agglutinin disease (CAD). FDA granted Sanofi a Breakthrough Therapy designation for sutimlimab in CAD, and FDA and EMA granted Orphan Drug designations for this indication. FDA granted priority review of the BLA for sutimlimab for the treatment of hemolysis in adult patients with CAD.
The BLA is based on results from part A of the open-label, single-arm pivotal Phase 3 CARDINAL study (NCT03347396) in patients with primary CAD. Of 24 patients who enrolled, 22 completed Part A of the study. The primary composite efficacy endpoint, defined as the proportion of patients who demonstrated an increase from baseline in Hgb level ≥2 g/dL or normalization of Hgb level ≥12 g/dL at the treatment assessment time point (mean value from Weeks 23, 25, and 26) and no blood transfusion from Week 5 through Week 26, was met. A total of 13 patients (54%) met the composite endpoint criteria, 62.5% (n = 15) of patients achieved a hemoglobin ≥12 g/dL or an increase of at least 2 g/dL, and 71% (n = 17) of patients remained transfusion-free after Week 5. Secondary endpoints were also met, indicating improvements in disease process, including improvements in hemoglobin, normalization of bilirubin, and improvements in Functional Assessment of Chronic Illness Therapy-Fatigue Score.86
On November 14, 2020, Sanofi announced that FDA issued a Complete Response Letter regarding sutimlimab’s BLA. No clinical or safety deficiencies were noted in the letter, but certain deficiencies were identified by FDA during a pre-license inspection of a third-party facility responsible for manufacturing. Sanofi is working to resolve the issues in a timely fashion.87 Prompt resubmission of the BLA may allow an approval to be granted in 2021.
Antibodies to watch in 2021: Non-cancer indications
Based on the information publicly available as of November 2020, 38 antibody therapeutics are in late-stage clinical studies for non-cancer indications other than COVID-19 (Table 4). Of these, BLA or MAA submissions for at least six (leronlimab, tezepelumab, faricimab, ligelizumab, garetosmab, and fasinumab) are planned in 2021.
Table 4.
Investigational monoclonal antibodies in late-stage clinical studies for non-cancer indications*
| Primary sponsoring company | INN or code name | Molecular format | Target(s) | Most advanced phase | Pivotal Phase 2, Phase 2/3 or 3 indications |
|---|---|---|---|---|---|
| Regeneron Pharmaceuticals, Inc. | Garetosmab (REGN2477) | Human IgG4 | Activin A | Phase 2 (pivotal); Phase 3 pending | Fibrodysplasia ossificans progressiva (NCT03188666; NCT04577820 (pending)) |
| Caelum Biosciences | CAEL-101, Ch mAb 11–1F4 | Chimeric IgG1 | Amyloid | Phase 3 | AL amyloidosis (NCT04512235, NCT04504825) |
| Hoffmann-La Roche | Gantenerumab | Human IgG1 | Amyloid beta | Phase 3 | Early or mild Alzheimer’s disease (NCT03443973, NCT03444870, NCT02051608, NCT01224106, NCT04339413, NCT04374253 (pending)) |
| Eisai Inc. | Lecanemab (BAN2401) | Humanized IgG1 | Amyloid β protofibrils | Phase 3 | Early Alzheimer’s disease (NCT03887455, NCT04468659) |
| Genentech | Etrolizumab | Humanized IgG1 | β7 subunit of α4-β7 and αE-β 7 integrins | Phase 3 | Crohn’s disease (NCT02394028, NCT02403323) |
| Novartis Pharmaceuticals Corp. | Ianalumab (VAY736) | Human IgG1 | BLyS/BAFF/TACI/BCMA receptor | Phase 2/3 | Autoimmune hepatitis (NCT03217422) |
| Chugai Pharmaceutical, Hoffmann-La Roche | Crovalimab (SKY59, RG6107, RO7112689) | Humanized IgG1 | C5 | Phase 3 | Paroxysmal nocturnal hemoglobinuria (NCT04434092, NCT04432584) |
| CytoDyn, Inc. | Leronlimab | Humanized IgG4 | CCR5 | Phase 2/3 | HIV infections (NCT03902522, NCT02859961) |
| SinoMab Pty Ltd | SM03 | Chimeric IgG1 | CD22 | Phase 3 | Rheumatoid arthritis (NCT04312815) |
| United Biopharma | UB-421, dB4C7 mAb | Humanized IgG1 | CD4 | Phase 3# | HIV infections (NCT03149211, NCT04406727 pending) |
| Regeneron Pharmaceuticals, Inc. | Pozelimab (REGN3918) | Human IgG4 | Complement 5 | Phase 3 | Paroxysmal nocturnal hemoglobinuria (NCT04162470); CD55-deficient protein-losing enteropathy (NCT04209634) |
| FibroGen | Pamrevlumab | Human IgG1 | Connective tissue growth factor | Phase 3 | Idiopathic pulmonary fibrosis (NCT03955146, NCT04419558); Duchenne muscular dystrophy (NCT04371666, NCT04632940 (pending)); Pancreatic cancer (NCT03941093); |
| Mapp Biopharmaceutical / LeafBio | Cosfroviximab, larcaviximab, porgaviximab (Zmapp) | Chimeric IgG1 mixture (3 mAbs) | Ebola virus | Phase 2/3 | Ebola virus infection (NCT03719586) |
| UCB Biopharma | Rozanolixizumab (UCB7665) | Humanized IgG4 | FcRn | Phase 3 | Myasthenia gravis (NCT03971422, NCT04124965); Primary immune thrombocytopenia (NCT04596995, NCT04224688, NCT04200456) |
| Momenta Pharmaceuticals | Nipocalimab (M281) | Human IgG1 | FcRn | Phase 2/3 | Warm autoimmune hemolytic anemia (NCT04119050) |
| GlaxoSmithKline | Otilimab (GSK3196165, MOR103) | Human IgG1 | GM-CSF | Phase 3 | Rheumatoid arthritis (NCT03980483, NCT03970837, NCT04134728, NCT04333147) |
| GC Pharma | Lenvervimab (GC1102) | Humanized IgG1 | Hepatitis B virus surface antigen | Phase 2/3 | Hepatitis B virus-associated liver transplant (NCT03519113) |
| Novartis Pharmaceuticals Corp. | Ligelizumab | Human IgG1 | IgE | Phase 3 | Chronic spontaneous urticaria (NCT03907878, NCT03580356, NCT04210843, NCT03580369) |
| Dermira, Inc. | Lebrikizumab | Humanized IgG4 | IL-13 | Phase 3 | Atopic dermatitis (NCT04146363, NCT04178967, NCT04392154, NCT04250350, NCT04250337) |
| Allergan | Brazikumab | Human IgG2 | IL-23 | Phase 3 | Crohn’s disease (NCT03961815, NCT03759288) |
| Eli Lilly & Co. | Mirikizumab | Humanized IgG4 | IL-23p19 | Phase 3 | Ulcerative colitis (NCT03519945, NCT03518086, NCT03524092, NCT04469062 (pending)): Psoriasis (NCT03482011, NCT03535194, NCT03556202); Crohn’s disease (NCT03926130, NCT04232553) |
| Maruho Co. Ltd. | Nemolizumab | Humanized IgG2 | IL-31 R alpha | Phase 3 | Atopic dermatitis (JapicCTI-173740, NCT03985943, NCT03989206, NCT03989349); Prurigo nodularis (NCT04204616, NCT04501666, NCT04501679) |
| Boehringer Ingelheim | Spesolimab (BI655130) | Humanized IgG1 | IL-36 R | Phase 3 | Ulcerative colitis (NCT03482635); pustular psoriasis (NCT03886246) |
| Vitaeris, Inc. | Clazakizumab | Humanized IgG1 | IL-6 | Phase 3 | Prevention of kidney transplant rejection (NCT03744910) |
| Takeda | Ontamalimab (SHP647) | Human IgG2 | Mucosal addressin cell adhesion molecule | Phase 3 | Ulcerative colitis (NCT03290781, NCT03259334); Crohn’s disease (NCT03627091, NCT03283085) |
| Regeneron Pharmaceuticals, Inc. | Fasinumab | Human IgG4 | Nerve growth factor | Phase 3 | Pain due to osteoarthritis of knee or hip (NCT03161093, NCT03245008, NCT02447276, NCT03304379, NCT02683239) and Low back pain (NCT03285646, NCT02620020) |
| Innovent Biologics (Suzhou) Co. Ltd. | Tafolecimab (IBI306) | Human IgG2 | PCSK9 | Phase 3 | Heterozygous familial hypercholesterolemia (NCT04179669); Homozygous Familial Hypercholesterolemia (NCT04031742) Non-familial Hypercholesterolemia (NCT04289285) |
| Synermore Biologics (Suzhou) Co., Ltd. | SYN023 | Humanized IgG1 mixture (2 mAbs) | Rabies virus | Phase 3 | |
| AstraZeneca | Nirsevimab (MEDI8897) | Human IgG1 | RSV | Phase 3 | Respiratory syncytial virus infections (NCT03979313, NCT03959488) |
| Aridis Pharmaceuticals, Inc. | Tosatoxumab (AR-301) | Human IgG1 | S. aureus alpha-toxin | Phase 3 | S. aureus ventilator-associated pneumonia (NCT03816956) |
| Allakos, Inc | Lirentelimab (AK002) | Humanized IgG1 | Siglec-8 | Phase 3 | Eosinophilic gastritis and/or eosinophilic duodenitis (NCT04322604); Eosinophilic Esophagitis (NCT04322708) |
| Alector, Inc. | AL001 | Human IgG1 | Sortilin | Phase 3 | Frontotemporal dementia due to heterozygous mutations in the progranulin gene (NCT04374136) |
| AstraZeneca | Tezepelumab | Human IgG2 | Thymic stromal lympho-poietin | Phase 3 | Severe uncontrolled asthma (NCT04048343, NCT03347279, NCT03968978, NCT03927157, NCT03706079, NCT03406078) |
| PhaseBio Pharmaceuticals Inc. | Bentracimab (PB2452) | Human Fab | Ticagrelor | Phase 3 | Reversal of the antiplatelet effects of ticagrelor (NCT04286438) |
| Pfizer | Marstacimab (PF-06741086) | Human IgG1 | Tissue factor pathway inhibitor | Phase 3 | Hemophilia A or B (NCT03938792) |
| Taisho Pharmaceutical Co., Ltd. | Ozoralizumab | Humanized bispecific nanobody | TNF, albumin | Phase 3 | Rheumatoid arthritis (JapicCTI-184031) |
| Kodiak Sciences Inc | KSI-301 | Antibody-biopolymer conjugate | VEGF | Phase 3 | Neovascular (wet) age-related macular degeneration (NCT04049266); Diabetic macular degeneration ((NCT04611152, NCT04603937) and macular edema due to retinal vein occlusion (NCT04592419) |
| Hoffmann-La Roche | Faricimab | Bispecific CrossMab | VEGF-A, Ang2 | Phase 3 | Diabetic macular edema (NCT03622593, NCT03622580, NCT04432831); Wet macular degeneration (NCT03823287, NCT03823300) |
*Data available as of November 1, 2020.
#Listed as Phase 3 in company pipeline.
Table notes: Companies may not pursue every indication for which a late-stage study is done. Pamrevlumab is included here because most late-stage studies are for non-cancer indications. ‘Pending’ category includes studies listed on clinicaltrials.gov that are not yet recruiting as of November 1, 2020.
Leronlimab (CytoDyn Inc.)
Leronlimab’s target, CCR5, has been implicated in many pathophysiological processes, such as human immunodeficiency virus (HIV)-1 entry into CD4 + T cells, promotion of tumor invasion and metastases, pathogenesis of nonalcoholic steatohepatitis, development of aGvHD, as well as inflammation. FDA has granted leronlimab a Fast Track designation as a combination therapy with highly active antiretroviral therapy for HIV-infected patients. CytoDyn Inc. submitted the non-clinical portion of a BLA for leronlimab (700 mg dose) as combination therapy with an antiretroviral regimen for HIV patients who are highly treatment-experienced, but the company announced in July 2020 that FDA refused to file the BLA due to the absence of information needed to complete a substantive review.88 CytoDyn intends to re-submit the BLA.
Tezepelumab (AstraZeneca/Amgen)
Tezepelumab (AMG157, MEDI-9929) is a human IgG2 λ antibody that targets thymic stromal lymphopoietin, a cytokine implicated in the pathogenesis of severe asthma and other immune-mediated disorders. FDA granted Breakthrough Therapy designation for tezepelumab in patients with severe asthma, without an eosinophilic phenotype, who are receiving inhaled corticosteroids/long-acting beta2-agonists with or without oral corticosteroids and additional asthma controllers. Amgen and AstraZeneca partnered on development of the drug in 2012. As of a July 2020 update, AstraZeneca anticipates a regulatory submission for tezepelumab for severe asthma in the first half of 2021.
The safety and efficacy of tezepelumab in patients with severe asthma are being evaluated in two Phase 3 studies with primary completion dates in September 2020, NAVIGATOR (NCT03347279) and SOURCE (NCT03406078). NAVIGATOR is a randomized, double-blind, placebo-controlled, parallel group study to evaluate the effects of tezepelumab in adults and adolescents with severe uncontrolled asthma. The primary outcome measure of the study is the annualized asthma exacerbation rate from baseline to Week 52. As announced in November 2020, this primary endpoint was met in the overall patient population, as well as in the subgroup of patients with baseline eosinophil counts less than 300 cells per microliter. SOURCE is a randomized, double-blind, placebo-controlled study to evaluate the effects of tezepelumab in reducing oral corticosteroid use in adults with oral corticosteroid (OCS)-dependent asthma. The primary outcome measure is the categorized percent reduction from baseline in the daily OCS dose while not losing asthma control at Week 48. Patients receive tezepelumab or placebo via SC injection in both studies.
Faricimab (F. Hoffmann-La Roche Ltd.)
Faricimab (RO6867461, RG7716) is a domain-exchanged bispecific antibody (CrossMab) targeting vascular endothelial growth factor-A (VEGF-A) and angiopoietin-2 (Ang-2). The antibody retains the format of a canonical human IgG1, but it is composed of two different heavy chains and two different light chains and has been Fc engineered to eliminate binding interactions with all Fcγ receptors, as well as FcRn.89 Hoffmann-La Roche plans to submit marketing application(s) for faricimab for diabetic macular edema (DME) and wet age-related macular degeneration (wAMD) in 2021. In the DME indication, faricimab is undergoing evaluation in two Phase 3 studies (YOSEMITE, RHINE) with primary completion dates in September 2020, while in the wAMD indication the antibody is being evaluated in 2 Phase 3 studies (TENAYA, LUCERNE) with primary completion dates in August 2021.
The randomized, double-masked, active comparator-controlled YOSEMITE (NCT03622580) and RHINE studies (NCT03622593) are comparing the effects of faricimab to those of aflibercept (Eylea®) in DME patients. The primary outcome measure is the average change from baseline in best-corrected visual acuity (BCVA) at 1 y. The YOSEMITE study has enrolled 940 patients and is not recruiting patients as of August 2020. The estimated enrollment of the RHINE study is 1070, and the study is enrolling patients by invitation as of August 2020.
TENAYA (NCT03823287) and LUCERNE (NCT03823300) are also randomized, double-masked, and active comparator-controlled studies of the effects of faricimab and aflibercept in wAMD patients. The primary outcome measure of both studies is the average change from baseline in BCVA at Week 48, and each study will enroll 640 patients. As of August 2020, the TENAYA study was active but not recruiting patients, and the LUCERNE study was enrolling patients by invitation.
Ligelizumab (Novartis Pharmaceuticals Corporation)
Ligelizumab (QGE031) is a humanized IgG1k antibody that binds IgE with high affinity (KD 17.8 pM). A recent study has shown that ligelizumab binds an epitope that only partially overlaps with that of the marketed anti-IgE antibody omalizumab (Xolair®), which has lower affinity to the target (KD 2659 pM). As a consequence of their molecular differences, the two antibodies have distinct inhibition profiles.90 In the US, Novartis Pharmaceuticals Corporation and Genentech work together to develop and co-promote Xolair; Novartis licensed ligelizumab from Tanox, which, as of 2007, was a subsidiary of Genentech. Novartis is planning to file regulatory submissions for ligelizumab for chronic spontaneous urticaria (CSU), which is an inflammatory condition also called hives, in the second half of 2021.
The safety and efficacy of ligelizumab in the treatment of CSU in adolescents and adults inadequately controlled with H1-antihistamines are being evaluated in two randomized, double-blind, active- and placebo-controlled Phase 3 studies, NCT03580369 and NCT03580356. Each study has 4 arms (2 dose levels of ligelizumab; 300 mg omalizumab; or placebo, with dosing once every 4 weeks), and each study has an estimated enrollment of 1050 patients. For both studies, the primary outcome measure is the absolute change from baseline in the Urticaria Activity Score 7 at Week 12. The estimated primary completion dates are January 28, 2021, and April 9, 2021, for NCT03580369 and NCT03580356, respectively.
Garetosmab (Regeneron Pharmaceuticals, Inc.)
Garetosmab (REGN2477) is a human IgG4k antibody that inhibits activin A, a member of the transforming growth factor-β family of growth and differentiation factors. This antibody is in development by Regeneron for fibrodysplasia ossificans progressiva (FOP), an ultra-rare genetic disorder characterized by the progressive replacement of soft tissue, such as muscles, tendons, and ligaments, by bone, a process known as heterotopic ossification (HO). The FDA granted Fast Track designation for garetosmab for the prevention of HO in patients with FOP, and garetosmab has been granted Orphan Drug designation in the US and EU for this indication. Regeneron plans regulatory submission(s) for garetosmab for FOP in 2021.
Regulatory submissions will include data from a Phase 1 study (NCT02870400) and the Phase 2 LUMINA-1 study (NCT03188666). The Phase 1 study was randomized, double-blind, and placebo-controlled, and assessed the safety, tolerability, pharmacokinetics, and pharmacodynamic effects of single ascending doses of IV and SC administered garetosmab in healthy women not of childbearing potential. Of 40 treated study participants, 24 received IV garetosmab (0.3 mg/kg, n = 6; 1 mg/kg, n = 6; 3 mg/kg, n = 6; 10 mg/kg, n = 6), 6 received SC garetosmab, and 10 received placebo (IV, n = 8; SC, n = 2). Study results indicated that the 10 mg/kg dose was well tolerated, and the target‐mediated pathway was saturated by this dose in Week 12. Based on these results, the 10 mg/kg dose was chosen for the LUMINA‐1 study.91
The randomized, placebo-controlled LUMINA-1 study assessed the safety, tolerability, pharmacokinetics, and effects on heterotopic bone formation of garetosmab in 44 adults (18–60 y of age) with FOP. Positron emission tomography (PET) imaging and computerized tomography (CT) scans were used to investigate the effect of garetosmab on change in HO in patients with FOP. The study included three periods: (1) a randomized, double-blind placebo-controlled treatment period (6 months); (2) an open-label treatment period during which placebo-treated patients cross over to garetosmab treatment (6 months); and (3) an open-label follow-up treatment. The primary analysis was recorded at Week 28. At this time, total lesion activity as measured by PET bone scans decreased 25% in the garetosmab arm compared to placebo (p = .07), with the number of new lesions decreasing by nearly 90%. The relative decrease in bone lesion volume as measured by CT showed similar results. Flare-ups reported by patients were reduced by 50% (nominal p = .03).92
A Phase 3 study (NCT04577820) to evaluate the efficacy and safety of garetosmab in Japanese adult patients with FOP is due to start in November 2020. This study has an estimated primary completion date of August 28, 2021.
Fasinumab (Regeneron Pharmaceuticals, Inc.)
Fasinumab (REGN475) is a human IgG1k antibody targeting NGF. In 2016, Regeneron entered into a worldwide development agreement with Teva Pharmaceutical Industries Ltd. to develop and commercialize fasinumab, excluding rights in Japan, Korea and nine other Asian countries, which were granted to Mitsubishi Tanabe Pharma Corporation in 2015. Regeneron anticipates regulatory submission(s) for fasinumab in OA pain in 2021.
Regeneron and Teva recently announced that two Phase 3 trials, FACT OA1 (NCT03161093) and FACT OA2 (NCT03304379), achieved the co-primary endpoints for fasinumab 1 mg monthly.93 FACT OA1 is a randomized, double-blind, multi-dose, placebo- and naproxen-controlled study to evaluate the efficacy and safety of two dose regimens of SC administered fasinumab in 3307 participants with pain due to OA of the knee or hip. FACT OA2 is a randomized, double-blind, multi-dose, placebo and non-steroidal anti-inflammatory drug (NSAID)-controlled (ZORVOLEX (diclofenac) and CELEBREX (celecoxib)) study to evaluate the efficacy and safety of fasinumab in 1650 patients with pain due to OA of the knee or hip.
In these studies, fasinumab demonstrated significant improvements in pain and physical function vs. placebo at Week 16 and Week 24, respectively. Fasinumab 1 mg monthly also showed nominally significant benefits in physical function in both trials and pain in FACT OA2, when compared to the maximum FDA-approved prescription doses of NSAIDs for OA. The alternate drug treatment arm of FACT OA1, fasinumab 1 mg every 2 months showed numerical benefit over placebo, but did not reach statistical significance. In the safety analysis, an increase in arthropathies was reported with fasinumab. These studies are active but not recruiting patients. Longer-term safety and efficacy data are expected to be reported in early 2021.
Antibodies to watch in 2021: Cancer indications
Based on the information publicly available as of November 2020, 44 antibody therapeutics are in late-stage clinical studies for cancer indications (Table 5). Of these, BLA or MAA submissions for at least 13 may be submitted by the end of 2021. In particular, application submissions for I-131 omburtamab, trastuzumab duocarmazine, tisotumab vedotin, amivantamab and ublituximab are anticipated by the end of 2020, and applications may be submitted in 2021 for sabatolimab, zalifrelimab, cusatuzumab, AK104, cosibelimab, mirvetuximab soravtansine, apamistamab-I-131, and KN046.
Table 5.
Investigational monoclonal antibodies in late-stage clinical studies for cancer indications*
| Primary sponsoring company | INN or code name | Molecular format | Target(s) | Most advanced phase | Pivotal Phase 2, Phase 2/3 or 3 indications |
|---|---|---|---|---|---|
| Y-mAbs Therapeutics, Inc. | I-131 Omburtamab | Murine IgG1 | B7H3 | Phase 3 | Neuroblastoma central nervous system/leptomeningeal metastases (NCT03275402) |
| OncoQuest Pharmaceuticals Inc. | Oregovomab | Murine IgG1 | CA125 (aka MUC16) | Phase 3 | Epithelial ovarian cancer (NCT04498117) |
| MacroGenics, Inc. | Flotetuzumab (MGD006) | Humanized scFv-scFv, bispecific | CD123, CD3 | Phase 2 (pivotal) | Acute myeloid leukemia (NCT02152956, NCT04582864 pending) |
| TG Therapeutics | Ublituximab | Chimeric IgG1 | CD20 | Phase 3 | Chronic lymphocytic leukemia (NCT02301156, NCT02612311); Non-Hodgkin’s lymphoma (Phase 2/3 NCT02793583); Multiple sclerosis (NCT03277261, NCT03277248, NCT04130997) |
| Hoffmann-La Roche | Mosunetuzumab | Humanized IgG1 bispecific | CD20, CD3 | Phase 1 (pivotal) | Follicular lymphoma (NCT04246086) |
| ADC Therapeutics Sarl | Camidanlumab tesirine | Human IgG1 ADC | CD25 (aka IL-2RA) | Phase 2 (pivotal) | Hodgkin lymphoma (NCT04052997) |
| Affimed N.V. | AFM13 | Human bispecific Tandem diabody (TandAb) | CD30, CD16A | Phase 2 (pivotal) | Peripheral T Cell lymphoma (NCT04101331) |
| I-Mab Biopharma, MorphoSys | Felzartamab (TJ202, MOR202) | Human IgG1 | CD38 | Phase 3 | Multiple myeloma (NCT03952091, pivotal Phase 2 NCT03860038) |
| Actinium Pharmaceuticals | Iodine (131I) apamistamab | Murine IgG1, radio-labeled | CD45 | Phase 3 | Ablation of bone marrow prior to hematopoietic cell transplantation in AML patients (NCT02665065) |
| Gilead Sciences | Magrolimab | Humanized IgG4 | CD47 | Phase 3 | Myelodysplastic syndromes (NCT04313881) |
| Sanofi | Tusamitamab ravtansine (SAR408701) | mAb, ADC | CEACAM5 | Phase 3 | Non-small cell lung cancer (NCT04154956) |
| Astellas Pharma Inc | Zolbetuximab | Chimeric IgG1 | Claudin-18.2 | Phase 3 | Gastric/gastro-esophageal junction adenocarcinoma (NCT03653507, NCT03504397) |
| AstraZeneca | Tremelimumab | Human IgG2 | CTLA-4 | Phase 3 | Small cell lung cancer (NCT03703297, NCT03043872); Urothelial cancer (NCT03682068, NCT02516241); Hepatocellular carcinoma (NCT03298451); Solid malignancies (NCT03084471); Squamous Cell carcinoma of the head and neck (NCT02369874, NCT02551159); Non-cell lung cancer (NCT02453282, NCT02542293, NCT03164616) |
| Agenus Inc. | Zalifrelimab (AGEN1884) | Human IgG1 | CTLA-4 | Phase 2 (pivotal) | Cervical cancer (NCT03894215, NCT03495882) |
| Innovent Biologics (Suzhou) Co. Ltd. | IBI310 | Human mAb | CTLA-4 | Phase 3 | Melanoma (NCT04277663) |
| Rakuten Aspyrian, Inc. | Cetuximab sarotalocan | Chimeric IgG1 conjugated to IR700; photoimmunotherapy | EGFR | Phase 3 | Head and neck squamous cell carcinomas (NCT03769506) |
| Janssen Research & Development, LLC | Amivantamab (JNJ-61186372) | Human IgG1, bispecific | EGFR, cMet | Phase 3 | Non-small cell lung cancer (NCT04487080, NCT04538664) |
| Five Prime Therapeutics, Zai Lab Limited | Bemarituzumab | Humanized IgG1 | FGFR2b | Phase 3 | Gastric/gastro-esophageal junction adenocarcinoma (NCT03694522) |
| Philogen SpA | Onfekafusp alfa, (L19IL2 + L19TNF combination) | scFv immunoconjugates | Fibronectin extra-domain B | Phase 3 | Melanoma (NCT03567889) |
| ImmunoGen | Mirvetuximab soravtansine | Humanized IgG1 ADC | Folate receptor alpha | Phase 3 | Ovarian cancer, primary peritoneal cancer or fallopian tube cancer (NCT02631876, NCT04209855, NCT04296890) |
| Immunocore Ltd | Tebentafusp | scFv bispecific immunoconjugate | gp100, CD3 | Phase 2 (pivotal) | Melanoma (NCT03070392) |
| Synthon Biopharmaceuticals BV | [vic-]trastuzumab duocarmazine | Humanized IgG1 ADC | HER2 | Phase 3 | Breast cancer (NCT03262935) |
| Bio-Thera Solutions | BAT8001 | Humanized IgG1 ADC | HER2 | Phase 3 | HER2+ Breast cancer (NCT04185649, CTR20180157) |
| RemeGen | Disitamab vedotin (RC48-ADC) | Humanized IgG1 ADC | HER2 | Phase 2 (pivotal), Phase 3 pending | Urothelial carcinoma (NCT03809013), Gastric cancer (NCT03556345), Breast cancer (NCT04400695 pending) |
| GlaxoSmithKline | Feladilimab (GSK3359609) | Humanized IgG4 | ICOS | Phase 3 | Head and neck squamous cell carcinoma (NCT04428333, NCT04128696) |
| Bristol-Myers Squibb | Relatlimab (BMS-986016) | Human IgG4 | LAG-3 | Phase 2/3 | Melanoma (NCT03470922) |
| AstraZeneca | Monalizumab | Humanized IgG4 | NKG2A | Phase 3 | Head and neck cancer (NCT04590963) |
| Shanghai Henlius Biotech | Serplulimab (HLX10) | Humanized IgG4 | PD-1 | Phase 3 | Esophageal squamous cell carcinoma (NCT03958890); Non-small cell lung cancer (NCT04033354, NCT03952403); Small cell lung cancer (NCT04063163); Gastric cancer (NCT04139135); Triple-neg. breast cancer (NCT04301739); Colorectal cancer (NCT04547166 pending); Hepatocellular carcinoma (NCT04465734 pending) |
| Taizhou Hanzhong Biomedical Co. Ltd. | HX008 | Humanized IgG4 | PD-1 | Phase 3 | Gastric cancer (NCT04486651) |
| MacroGenics, Inc. | Retifanlimab (INCMGA00012, MGA012) | Humanized IgG4 | PD-1 | Phase 3 | Gastric cancer/gastro-esophageal junction adenocarcinoma (NCT04082364); Non-small cell lung cancer (NCT04205812); Squamous Cell Carcinoma of the Anal Canal (NCT04472429) |
| Pfizer | Sasanlimab (PF-06801591) | Humanized IgG4 | PD-1 | Phase 3 | Non-muscle invasive bladder cancer (NCT04165317) |
| Akeso | Penpulimab (AK105) | Humanized IgG1 | PD-1 | Phase 3 | Non-small cell lung cancer (NCT03866993, NCT03866980); Hepatocellular carcinoma (NCT04344158 pending); Gastric and gastro-esophageal junction adenocarcinoma (NCT04385550 pending) |
| Genor Biopharma Co | Geptanolimab (CBT-501) | Humanized IgG4 | PD-1 | Phase 2 (pivotal) | Peripheral T cell lymphoma (NCT03502629) |
| Akesobio Australia Pty Ltd | AK104 | Humanized mAb-scFv, bispecific | PD-1, CTLA-4 | Phase 2 (pivotal) | Cervical cancer (NCT04380805) |
| MacroGenics, Inc. | Tebotelimab (MGD013) | Human IgG4 bispecific DART | PD-1, LAG-3 | Phase 2/3 | Gastric cancer/gastro-esophageal junction adenocarcinoma (NCT04082364) |
| Alphamab Oncology | Envafolimab (KN035) | VH-h-CH2-CH3 gamma1 chain dimer | PD-L1 | Phase 3 | Bile tract carcinoma (NCT03478488); dMMR or MSI-H solid tumors (pivotal Phase 2 NCT03667170); Pleomorphic sarcoma or myxofibrosarcoma (pivotal Phase 2 NCT04480502 not yet recruiting) |
| CBT Pharmaceuticals, Inc | TQB2450, CBT-502 | Humanized IgG1 | PD-L1 | Phase 3 | Head and neck cancer (NCT03855384); Small cell lung cancer (NCT04234607 pending); Renal cancer (NCT04523272); Triple-neg, breast cancer (NCT04405505); Non-small cell lung cancer (NCT04325763) |
| CStone Pharmaceuticals | Sugemalimab (CS1001) | Human IgG4 | PD-L1 | Phase 3 | Non-small cell lung cancer (NCT03789604, NCT03728556); Gastric/gastro-esophageal junction adenocarcinoma (NCT03802591); Esophageal squamous cell carcinoma (NCT04187352); Extranodal natural killer/T cell lymphoma (pivotal Phase 2 NCT03595657); classical Hodgkin lymphoma (pivotal Phase 2 NCT03505996) |
| Jiangsu HengRui Medicine Co., Ltd. | Adebrelimab (SHR-1316, HTI-1088) | Humanized IgG4 | PD-L1 | Phase 3 | Small cell lung cancer (NCT03711305 pending); Non-small cell lung cancer (NCT04316364 pending); Phase 3 studies ongoing in China |
| EMD Serono | Bintrafusp alfa (M7824) | Human IgG1 bispecific immunoconjugate | PD-L1, TGFβ | Phase 3 | Non-small cell lung cancer (NCT03631706); Biliary tract cancer (Phase 2/3 NCT04066491) |
| Alphamab (Australia) Co Pty Ltd. | KN046 | Humanized IgG1 bispecific | PD-L1, CTLA-4 | Phase 3 | Thymic carcinoma (pivotal Phase 2 NCT04469725 pending); Non-small cell lung cancer (NCT04474119) |
| Genentech | Tiragolumab (MTIG7192A, RO7092284) | Human IgG1 | TIGIT | Phase 3 | Small cell lung cancer (NCT04256421); Non-small cell lung cancer (NCT04294810, NCT04513925); Esophageal Cancer (NCT04540211); Esophageal squamous cell carcinoma (NCT04543617) |
| Novartis Pharmaceuticals Corp. | Sabatolimab (MBG453) | Humanized IgG4 | TIM-3 | Phase 3 | Myelodysplastic syndromes (NCT03946670, NCT04266301) |
| Genmab | Tisotumab vedotin | Human IgG1 ADC | Tissue factor | Phase 2 (pivotal) | Cervical cancer (NCT03438396) |
*Data publicly available as of November 1, 2020.
Table notes: Companies may not pursue every indication for which a late-stage study is done. Ublituximab is included here because the most advanced indication is CLL. ‘Pending’ category includes studies listed on clinicaltrials.gov that are not yet recruiting as of November 1, 2020.
I-131. Omburtamab (Y-mAbs Therapeutics, Inc.)
I-131 Omburtamab comprises the anti-B7H3 murine antibody 8H9 labeled with the radioisotope iodine 131. B7H3 is an immune checkpoint molecule expressed on various types of cancer cells. The antibody was created by Memorial Sloan Kettering Cancer Center and licensed to Y-mAbs Therapeutics, Inc. In June 2017, FDA granted Breakthrough Therapy designation to omburtamab, known as burtomab at that time, for treatment of pediatric patients with relapsed or refractory neuroblastoma with central nervous system (CNS) or leptomeningeal metastasis (LM). Omburtamab was also granted Orphan Drug and Rare Pediatric Disease designations from the FDA for the treatment of CNS/LM from neuroblastoma. Y-mAbs Therapeutics completed submission of a rolling BLA for I-131 omburtamab for the treatment of pediatric patients with CNS/LM from neuroblastoma in August 2020, but FDA refused to file the BLA due to deficiencies in the Chemistry, Manufacturing and Control (CMC) and Clinical modules. The company expects to resubmit the BLA by the end of 2020.94
The safety and efficacy of omburtamab were evaluated in the Phase 1 study 03–133 (NCT00089245) and the pivotal Phase 2/3 study 101 (NCT03275402). In the Phase 1 study, a dose escalation scheme was used to find the maximally tolerated dose of intrathecal 131I omburtamab, with patients entering in cohorts of 3 at each dose level from 10 mCi to 60 mCi and a cohort of 6 at each dose level from 70 mCi to 100 mCi. As reported in October 2019, 107 evaluable patients with CNS/leptomeningeal metastases from neuroblastoma received up to two doses of I-131 omburtamab, and study results showed that patients had a median survival of 50.8 months, with the final median survival not yet being reached.95
The Phase 2/3 study 101 is evaluating the effects of I-131 omburtamab in children up to 18 y with a neuroblastoma diagnosis and CNS/LM. Patients will receive up to two cycles of intracerebroventricular 131I-omburtamab, with one treatment cycle consisting of 2 131I-omburtamab doses, 2 mCi at Week 1 and 50 mCi at Week 2. The primary outcome measure is the OS rate at 3 y after the first treatment dose of 131I-omburtamab. Interim results for 17 patients enrolled in the 101 study were presented at the International Society of Pediatric Oncology Virtual Annual Congress held in October 2020. The 12-month OS was 87%, with a median follow-up of 26 weeks. The OS in a historic control group was 30%, as previously disclosed by the company.50 The 101 study’s estimated primary completion date is December 2020.
[vic]Trastuzumab duocarmazine (Byondis BV)
[vic]Trastuzumab duocarmazine (SYD985) is an ADC composed of trastuzumab, a humanized anti-HER2 IgG1k antibody, conjugated to the cleavable linker-drug valine-citrulline-seco-duocarmycin-hydroxybenzamide-azaindole. The unconjugated antibody trastuzumab (Herceptin®) was first approved in 1998 for metastatic HER2-positive breast cancer in patients who have received one or more chemotherapy regimens for their metastatic disease. Two ADCs comprising trastuzumab conjugated to different cytotoxic drugs, ado-trastuzumab emtansine (Kadcyla®) and fam-trastuzumab deruxtecan (Enhertu®), were first approved in the US for HER2-positive breast cancer in 2013 and 2019, respectively.
Byondis BV (formerly Synthon Biopharmaceuticals BV) is developing [vic]trastuzumab duocarmazine for metastatic breast cancer. In 2018, FDA granted Fast Track designation for [vic-]trastuzumab duocarmazine for patients diagnosed with HER2-positive metastatic breast cancer that has progressed during or after at least two HER2-targeting treatment regimens for locally advanced or metastatic disease, or progressed during or after Kadcyla® treatment. Byondis BV plans to submit a BLA for their ADC for HER2-positive breast cancer by the end of 2020.96
[vic]Trastuzumab duocarmazine is currently being investigated in the randomized, active-controlled TULIP Phase 3 study (NCT03262935) comparing the efficacy and safety of [vic-]trastuzumab duocarmazine to physician’s choice in patients with HER2-positive unresectable locally advanced or metastatic breast cancer. The active-control drugs are combinations of lapatinib, capecitabine, trastuzumab, vinorelbine, and eribulin. The study’s primary endpoint is PFS up to 2 y from the baseline, while OS and the objective response rate are secondary outcome measures. The estimated primary completion date of the TULIP study is July 2021, but Byondis expects results in the second half of 2020.96
Tisotumab vedotin (Genmab A/S, Seattle Genetics, Inc)
Tisotumab vedotin (HuMax-TF-ADC) is an ADC composed of a human anti-tissue factor IgG1k antibody conjugated to MMAE via a protease-cleavable linker. In August 2017, Genmab A/S and Seattle Genetics, Inc. announced that Seattle Genetics, Inc. had exercised its option to co-develop tisotumab vedotin. The companies may submit a BLA for an accelerated approval of tisotumab vedotin for cervical cancer by the end of 2020.
Regulatory submissions will include data from the pivotal single-arm Phase 2 innovaTV 204 study (NCT03438396) evaluating tisotumab vedotin as monotherapy in patients with previously treated recurrent and/or metastatic cervical cancer, which were reported at the ESMO Virtual Congress held in September 2020.97 Patients received tisotumab vedotin 2.0 mg/kg via IV every 3 weeks until progression or toxicity. The primary endpoint was confirmed objective response rate as assessed by independent central review in 101 patients treated with tisotumab vedotin. The objective response rate was 24% [95% CI: 15.9%-33.3%], including 7 patients (7%) with a complete response and 17 patients (17%) with a partial response. Secondary endpoints included DOR, time to response, PFS, and OS. The median DOR was 8.3 months (95% CI: 4.2, not reached) after a median follow-up of 10 months. The median time to response was 1.4 months (range, 1.1–5.1), while the median PFS and OS were 4.2 months (95% CI: 3.0, 4.4) and 12.1 months (95% CI: 9.6, 13.9), respectively.
Amivantamab (Janssen Research & Development, LLC)
Amivantamab (JNJ-61186372) is a human, low-fucose IgG1-based bispecific antibody targeting EGFR and mesenchymal epithelial transition factor (MET) that was created using Genmab’s DuoBody technology. Amivantamab has been shown to function through multiple mechanisms of action in preclinical models of NSCLC with EGFR exon 20 insertion driver mutations, which cause tumor cells to be insensitive to EGFR tyrosine kinase inhibitors.98 FDA granted Breakthrough Therapy designation to amivantamab for the treatment of patients with metastatic NSCLC with (EGFR) Exon 20 insertion mutations, whose disease has progressed on or after platinum-based chemotherapy. A BLA or MAA for amivantamab for NSCLC is included in Janssen’s new molecular entity filings planned during 2020.
Data from the CHRYSALIS Phase 1 study (NCT02609776) supported the Breakthrough Therapy designation. CHRYSALIS is a first-in-human, open-label, dose-escalation study evaluating the safety, pharmacokinetics, and efficacy of amivantamab as a monotherapy and in combination with lazertinib, an EGFR tyrosine kinase inhibitor, in adult patients with advanced NSCLC. The study has an estimated enrollment of 460 patients and is still enrolling patients as of November 2020. Part 1 of the study includes amivantamab monotherapy and combination dose escalations, while Part 2 includes amivantamab monotherapy and combination dose expansions. The study will determine the recommended Phase 2 doses for amivantamab monotherapy, the amivantamab/lazertinib combination, and amivantamab in combination with standard-of-care carboplatin and pemetrexed in 21-d treatment cycles for participants with advanced NSCLC. Primary outcome measures include ORR, DOR, and percentage of participants with clinical benefit.
Preliminary results from the CHRYSALIS study were presented at the 2020 American Society of Clinical Oncology (ASCO) Virtual meeting. The analysis included 50 enrolled patients with EGFR exon 20 insertion-mutated NSCLC who received the recommended Phase 2 dose of 1050 mg (1400 mg, patients ≥80 kg) amivantamab. The analysis showed an ORR of 36% (95% CI, 21–53) among the 39 response-evaluable patients, with a median follow-up of 4 months and 41% (95% CI, 24–61) in the 29 evaluable patients previously treated with platinum-based chemotherapy. The median DOR among 14 responders was 10 months. The median PFS was 8.3 months (95% CI, 3.0–14.8) for all response-evaluable patients and 8.6 months (95% CI, 3.7–14.8) for patients previously treated with platinum-based chemotherapy. The clinical benefit rate was 67% (95% CI, 50–81) for response-evaluable patients and 72% (95% CI, 53–87) for patients previously treated with platinum-based chemotherapy.99
Janssen is also investigating the efficacy and safety of amivantamab in combination with lazertinib versus osimertinib in locally advanced or metastatic NSCLC patients with EGFR mutations (Exon 19 deletions or Exon 21 L858R substitution) in the Phase 3 MARIPOSA study (NCT04487080), as well as amivantamab in combination with carboplatin-pemetrexed chemotherapy compared to carboplatin-pemetrexed in metastatic NSCLC with EGFR exon 20 insertions in the Phase 3 PAPILLON trial (NCT04538664).
Ublituximab (TG Therapeutics, Inc.)
Ublituximab (TG-1101) is a chimeric IgG1k antibody that targets a unique epitope on CD20, which is found on B cells and known to be involved in the pathogenesis of hematological malignancies such as chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL), as well as multiple sclerosis (MS). Initially developed by LFB Group but licensed to TG Therapeutics in 2012, ublituximab contains low fucose content in its Fc region, which enhances its ADCC and ADCP activity. FDA has granted Fast Track designation to the combination of ublituximab and umbralisib, which is an oral, once-daily, dual inhibitor of PI3K-delta and CK1-epsilon, for the treatment of adult patients with CLL. TG Therapeutics plans to submit a BLA to the FDA for ublituximab in combination therapy with umbralisib, for CLL in 2020 or in early 2021.100
The safety and efficacy of ublituximab in combination with umbralisib (TGR-1202) compared to obinutuzumab (GAZYVA®) plus chlorambucil in patients with previously untreated and relapsed or refractory CLL are being evaluated in Phase 3 UNITY-CLL study (NCT02612311). The primary endpoint is PFS and the secondary outcome is the ORR. In May 2020, TG therapeutics announced that, at a prespecified interim analysis, the trial met its primary endpoint of improved PFS (p < .0001) and will be stopped early.101
The 3-arm Phase 2/3 UNITY-NHL study (NCT02793583) is evaluating the effects of the combination of ublituximab plus umbralisib with or without bendamustine vs. umbralisib alone in patients with previously treated non-Hodgkin’s lymphoma, including diffuse large B-cell lymphoma, follicular, mantle cell lymphoma and marginal zone lymphoma. The primary and secondary endpoints are ORR and PFS, respectively. The estimated primary completion date is May 2021. Results from the UNITY-CLL Phase 3 trial evaluating the combination of umbralisib and ublituximab in patients with treatment naïve and relapsed/refractory chronic lymphocytic leukemia will be presented at the virtual 62nd American Society of Hematology annual meeting to be held in early December 2020.
Ublituximab in combination with ibrutinib vs. ibrutinib alone was evaluated in the open-label, multicenter, randomized Phase 3 GENUINE study (NCT02301156) in previously treated CLL patients with high-risk cytogenetic features, including deletion 17p, deletion 11q and/or TP53. Participants were randomized to receive either ublituximab 900 mg IV at Days 1, 8, and 15 followed by maintenance infusions plus ibrutinib at 420 mg oral daily dose or ibrutinib at fixed oral daily dose. The primary outcome measures were ORR and PFS. Results from the GENUINE Phase 3 were presented at the ASCO annual meeting in May 2020. Patients in the ublituximab plus ibrutinib study arm showed an improved ORR (93%) compared to those in the ibrutinib monotherapy arm (78%). At a median follow-up of 41.9 months, median PFS was not reached in the ublituximab plus ibrutinib arm and was 35.9 months in the ibrutinib monotherapy arm (HR 0.46).102,103
TG therapeutics is also evaluating ublituximab compared to teriflunomide as a treatment for relapsing MS in the Phase 3 ULTIMATE-1 (NCT03277261) and ULTIMATE-2 (NCT03277248) studies. For both studies, the primary outcome measure is annualized relapse rate and the estimated primary completion date is March 31, 2021. TG therapeutics expects to report topline results from these studies later in 2020 or early 2021.
Sabatolimab (Novartis Pharmaceuticals Corporation)
Sabatolimab (MBG453) is a humanized IgG4k antibody directed against T-cell immunoglobulin and mucin-domain domain-containing molecule-3 (TIM-3) developed by Novartis for the treatment of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). TIM-3 is expressed on malignant leukemia stem cells (LSC) and blast cells but not on normal hematopoietic stem cells, and it is expressed by multiple types of T cells, as well as macrophages and dendritic cells. In this context, the use of sabatolimab has a dual effect: (1) blockade of TIM-3 on immune cells may restore immune function, leading to enhanced effector T-cell responses, and (2) direct targeting of LSC.104 Novartis anticipates submission of marketing applications for sabatolimab for high-risk MDS in 2021.
The efficacy and safety of sabatolimab in combination with hypomethylating agents (azacitidine or decitabine) are currently being studied in a randomized, double-blind, placebo-controlled Phase 2 STIMULUS MDS-1 study (NCT03946670) of adult patients with high or very high-risk MDS not eligible for HSCT or intensive chemotherapy. The primary endpoints are the complete remission rate and PFS. Secondary endpoints include OS, leukemia-free survival, response rate, and duration of complete remission. The estimated primary completion date is February 2021.
The randomized, double-blind, placebo-controlled Phase 3 STIMULUS-MDS2 study (NCT04266301) initiated in June 2020 will also evaluate the clinical effects of sabatolimab in combination with azacitidine in adult subjects with intermediate, high or very high-risk MDS or chronic myelomonocytic leukemia-2 who have an indication for treatment with azacitidine in first-line setting and are not eligible for intensive chemotherapy or HSCT. Patients will be randomized to one of two study arms: (1) sabatolimab 800 mg + azacitidine 75 mg/m2; or (2) azacitidine 75 mg/m2 + placebo. The primary objective of this study is to compare OS in the two study arms, where OS is the time from randomization until death due to any cause.
Zalifrelimab (Agenus Inc.)
Zalifrelimab (AGEN1884) is a human anti-CTLA-4 IgG1k antibody discovered via collaborative efforts between Ludwig Cancer Research and 4-Antibody, which was acquired by Agenus Inc. in 2014. Zalifrelimab is undergoing evaluation in combination with anti-PD-1 balstilimab in several clinical studies of patients with cervical cancer. FDA recently granted Agenus Fast Track designation for this combination of antibody therapeutics for the treatment of patients with relapsed or refractory metastatic cervical cancer.
Agenus has reported positive results from a Phase 2 study (NCT03894215) evaluating the efficacy and safety of balstilimab in combination with zalifrelimab, as well as balstilimab as monotherapy, in patients with advanced metastatic cervical cancer. As described above in the discussion of balstilimab, data from this study supported a BLA submission for balstilimab as monotherapy for cervical cancer. Agenus plans to have a completed data packet supporting BLA submission of zalifrelimab in combination with balstilimab by the end of 2020.105
Cusatuzumab(Janssen Research & Development, LLC)
Cusatuzumab (JNJ-74494550, ARGX-110) is an afucosylated, humanized IgG1 lambda antibody that binds CD70, resulting in inhibition of CD70/CD27 signaling involved in the proliferation of malignant cells and Treg recruitment or activation within the tumor microenvironment.106 Cusatuzumab was generated using the SIMPLE antibody™ platform developed by ArgenX, which uses the active immunization of outbred llamas with a target antigen to deliver antibodies with variable regions that have 96–99% human sequence homology. These variable regions are then combined with human constant domains to generate human therapeutic antibodies. Cusatuzumab has also been modified using POTELLIGENT technology to enhance Fc-mediated cytotoxic mechanisms, such as ADCC, ADCP, and complement-dependent cytotoxicity, resulting in killing of CD70+ malignant cells, for the treatment of AML and MDS.
In December 2018, Argenx announced the closing of the exclusive, global correlation and license agreement for cusatuzumab with Cilag GmbH International, an affiliate of the Janssen Pharmaceutical Companies of Johnson & Johnson.107 Janssen’s July 2020 pipeline update indicated that they plan to submit a marketing application for cusatuzumab in AML during 2020–2023.108
Cusatuzumab is currently being evaluated in patients with hematological malignancies in the Phase 2 CULMINATE trial (NCT04023526) in combination with azacitidine (Vidaza®, Celgene) in patients with newly diagnosed AML and high-risk MDS who are unsuitable for chemotherapy. Participants will receive azacitidine 75 mg/m2 SC or IV on Day 1 through Day 7 and cusatuzumab 10 mg/kg IV on Day 3 and Day 17 of each 28-d cycle in Part 1. Part 1 findings will be reviewed by a data review committee, and when a dose of cusatuzumab is selected, randomization will stop and participants will be enrolled into an expansion cohort (Part 2) to evaluate efficacy. The primary endpoint is the percentage of participants with complete response. The estimated primary completion date for CULMINATE is December 29, 2020. The study has enrolled 103 patients, and is active but not recruiting. If the study is considered pivotal and study results are positive, Janssen may have an opportunity to submit a BLA for cusatuzumab for AML in 2021.
AK104 (Akeso Biopharma)
AK104 is a humanized tetravalent IgG1 bispecific antibody targeting PD-1 and CTLA-4 developed by Akeso Biopharma. The antibody was engineered to preferentially bind tumor-infiltrating lymphocytes over lymphocytes within peripheral tissue. FDA granted AK104 Fast Track designation as monotherapy for the treatment of patients with recurrent or metastatic squamous cervical cancer who have disease progression on or after platinum-based chemotherapy. Akeso Biopharma may submit marketing applications in China and the US as early as the second half of 2021 for AK104 in this indication.109,110
AK104 is under investigation in a pivotal, multicenter, open-label, single-arm Phase 2 study (NCT04380805) to evaluate the efficacy, safety, tolerability, pharmacokinetic properties and immunogenicity of AK104 monotherapy in adults with previously treated recurrent or metastatic cervical carcinoma. In total, 40 participants will receive AK104 IV monotherapy at a dose of 6 mg/kg on Day 1 and Day 15 of each 28-d treatment cycle. The primary endpoint is the ORR assessed by an Independent Radiological Review Committee. Secondary outcome measures include the ORR assessed by the investigator, the disease control rate, the DOR, and PFS. The estimated primary completion date is August 2021.
Cosibelimab (Checkpoint Therapeutics, Inc.)
Cosibelimab (CK-301) is a human IgG1λ antibody directed against the immune checkpoint inhibitor PD-L1. Blocking the interaction of PD-L1 with the PD-1 and B7.1 receptors removes the suppressive effects of PD-L1 and restores cytotoxic CD8+ T cell anti-tumor immune responses. As of November 2020, 3 anti-PD-L1 (atezolizumab, durvalumab, avelumab) have been granted marketing approvals in the US and EU. Like avelumab, cosibelimab has a functional Fc and is capable of inducing ADCC mechanisms. The Fc regions of both atezolizumab and durvalumab were engineered to eliminate FcγR binding and effector functions.111 Checkpoint Therapeutics licensed the exclusive worldwide rights to anti-PD-L1 antibodies from Dana-Farber Cancer Institute in March 2015. The company has indicated that they intend to submit marketing approval application submissions for cosibelimab in mid-2021,112 if positive results are observed in a pivotal Phase 1 study that includes a registration-enabling cohort of patients with metastatic cutaneous squamous cell carcinoma (mCSCC).
The single-arm, open-label, multicenter, dose-escalation Phase 1 trial (NCT03212404) is evaluating the safety, tolerability and efficacy of cosibelimab when administered IV as a single agent to subjects with selected recurrent or metastatic cancers, including lung neoplasms, NSCLC, small cell lung cancer, head and neck cancer, melanoma, Merkel cell carcinoma, renal cell carcinoma, urothelial carcinoma, endometrial cancer, NHL, Hodgkin lymphoma, and metastatic squamous cell carcinoma. Patients are treated with fixed doses (800 mg every 2 weeks; 1200 mg every 3 weeks) and then followed for up to 6 months of visits with survival follow-up for select cohorts. Following the dose-escalation part of the study, additional evaluable subjects may be included in order to further characterize safety and efficacy at selected doses or in specific patient sub-groups (dose expansion). The primary outcome measures include the confirmed objective response rate as per the RECIST Version 1.1 for the dose-expansion part of the study only in an average of 6 months. Recruitment is ongoing, and Checkpoint Therapeutics anticipates completing enrollment by the end of 2020. The estimated primary completion date is July 2021.
Interim results from the Phase 1 study were presented at the 2020 ESMO Virtual Congress. As of the interim analysis, 37 mCSCC patients were enrolled and evaluable for efficacy by investigator assessment with at least one post-baseline tumor assessment or discontinued treatment prior. For these patients, the objective response rate was 51.4% (95% CI: 34.4, 68.1) per RECIST 1.1, complete response was observed in 13.5% of patients, and 37.8% of patients achieved a partial response (2 pending confirmation at the next scan). The median DOR had not yet been reached, with 84.2% of responses ongoing. The longest response duration was 24 months (ongoing) at the time of analysis. Of the eligible responses, 91.7% had a duration of over 6 months.113
Mirvetuximab soravtansine (ImmunoGen, Inc.)
Mirvetuximab soravtansine (IMGN853) is an ADC composed of a humanized IgG1 antibody targeting folate receptor alpha (FRα), M9346A, conjugated to the maytansinoid drug DM4 via a cleavable disulfide linker. This ADC is owned and developed by ImmunoGen. The EMA and the FDA granted Orphan Drug designations to mirvetuximab soravtansine for patients with ovarian cancer in 2015. In June 2018, FDA also granted the ADC Fast track designation for the treatment of patients with medium to high FRα-positive platinum-resistant ovarian cancer who received at least one, but no more than three prior systemic treatment regimens, and for whom single-agent chemotherapy is appropriate as the next line of therapy.114
In 2019, ImmunoGen announced that the Phase 3 FORWARD I study (NCT02631876) did not meet the primary endpoint in either the entire treatment population or the pre-specified high FRα expression population. Promising efficacy signals were observed, however, in the pre-specified subset of patients with high FRα expression. After consulting with FDA, ImmunoGen initiated two new Phase 3 studies, SORAYA (NCT04296890), designed to allow accelerated approval of mirvetuximab soravtansine, and MIRASOL (NCT04209855), designed to allow full approval of the ADC. ImmunoGen anticipates the submission of a BLA for accelerated approval of mirvetuximab soravtansine for ovarian cancer during the second half of 2021.114
SORAYA is a single-arm trial designed to evaluate the efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant high-grade serous epithelial ovarian cancer, primary peritoneal, or fallopian tube cancer, whose tumors express a high-level of FRα. Enrolled patients receive mirvetuximab soravtansine at 6 mg/kg adjusted ideal body weight administered on Day 1 of every 3-week cycle. To be enrolled, patients must have received at least one but no more than three prior systemic lines of anticancer therapy, including at least one line of therapy containing bevacizumab, and single-agent therapy must be appropriate as the next line of treatment. The primary endpoint of this study is ORR by investigator assessment, and the key secondary endpoint is DOR. The estimated enrollment is 110 patients, and the estimated primary completion date is July 2021.
MIRASOL is a randomized, open-label Phase 3 study in which an estimated 430 patients will be randomized to either mirvetuximab soravtansine (6 mg/kg adjusted ideal body weight every 3 weeks) or Investigator’s Choice chemotherapy (paclitaxel, PEGylated liposomal doxorubicin, or topotecan). To be eligible, patients need to be diagnosed with platinum-resistant ovarian cancer expressing high levels of FRα using the PS2+ scoring method and have been treated with up to three prior regimens. The primary endpoint of this study is PFS by investigator assessment, and the key secondary endpoints include ORR and OS. The estimated primary completion date is June 2022.
Apamistamab-I-131 (Actinium Pharmaceuticals, Inc.)
Apamistamab-I-131 (Iomab-B) is an anti-CD45 IgG1k antibody (BC8) linked to the radioisotope iodine-131 developed by Actinium Pharmaceuticals, Inc. for targeted conditioning prior to an allogeneic bone marrow transplant (BMT) for patients with active, relapsed or refractory AML, who are age 55 or older. The antibody targets a pan-leukocytic antigen expressed on leukemia, lymphoma cells and normal immune cells. Preclinical data demonstrated that injection of Iomab-B effectively depletes greater than 90% of lymphocytes, including CD4+ and CD8+ effector T cells, CD19 + B cells, NK cells, as well as CD4+ Treg cells, and macrophages. Red blood cells, platelets, neutrophils, and bone marrow stem cells were preserved. Iomab-B binds preferentially in the lymph nodes, spleen, liver, and bone marrow, thereby avoiding the side effects of radiation on healthy tissues. Once attached to its target cells, radioactive energy from Iomab-B destroys cancer cells and ablates the bone marrow of patients. The FDA and EMA have granted Orphan Drug designations to Iomab-B as a conditioning treatment for patients with relapsed and refractory AML who will undergo HSCT. Actinium Pharmaceuticals plans to submit a BLA to the FDA in 2021 based on the results from the Phase 3 SIERRA clinical trial.
Iomab-B is currently being evaluated in the randomized, controlled, multi-center Phase 3 SIERRA trial (NCT02665065) to demonstrate the efficacy of Iomab-B prior to HSCT vs. conventional care in older patients with active, relapsed or refractory AML.115 The primary endpoint is the durable complete remission at 6 months and the secondary endpoint is OS at 1 y. In November 2020, Actinium Pharmaceuticals released interim results of the ongoing study. Of patients receiving the therapeutic dose of Iomab-B, 100% underwent BMT and successfully engrafted, whereas 17% of patients in the control arm underwent BMT. Of evaluable patients in the Iomab-B arm, the 100-d non-relapse transplant-related mortality rate was 5% (vs. 25% for the control arm). High doses of Iomab-B did not correlate with incidence of mucositis, febrile neutropenia, or sepsis. The estimated primary completion date of the SIERRA study is December 2021, but Actinium Pharmaceuticals plans to exercise an ad-hoc analysis that could generate topline data for the primary endpoint in late 2020 and early termination of the trial if positive.
KN046 (Alphamab Oncology)
KN046 is a humanized bispecific antibody that binds PD-L1 and CTLA4. It was derived from Charge Repulsion Induced Bispecific technology, which introduces point mutations to induce changes in charge and H-bond interactions, in addition to steric interactions, to favor heterodimerization over homodimerization of the Fc. FDA has granted Orphan Drug designations to KN046 for the treatment of thymic epithelial tumors and for the treatment of biliary tract cancer. Jiangsu Alphamab Biopharmaceuticals Co., Ltd, a wholly owned subsidiary of Alphamab Oncology, is sponsoring a Phase 3 study (NCT04474119) of KN046 in NSCLC patients, and several Phase 2 studies of KN046 in patients with other cancers.
Alphamab Oncology has announced that the Phase 2 clinical trial (NCT04469725) of KN046 to treat thymic carcinoma will support their plan to submit a New Drug Application for KN046 to China’s National Medical Products Administration and the US FDA in 2021.116 This single-arm, open-label, multi-center study will evaluate the efficacy, safety and tolerability of KN046 in thymic carcinoma patients who will be administered 5 mg/kg of the drug, every 2 weeks. The primary outcome measure is OS, and the estimated primary completion date is August 2022.
Notable set-backs
In addition to the advances documented here, notable setbacks also occurred in 2020. In particular, the biology of COVID-19 has proven to be highly complex, and the results of some clinical studies of three repurposed antibodies, anti-IL6R tocilizumab (Actemra®/RoActemra®) and sarilumab (Kevzara®), and anti-IL-1 beta canakinumab (ILARIS®), in COVID-19 patients have been disappointing. All three of these antibodies are marketed for other indications. In addition, primary endpoints were not met in late-stage studies of investigational agents spartalizumab in melanoma patients and etolizumab in ulcerative colitis patients.
Tocilizumab, sarilumab, and canakinumab in COVID-19
Tocilizumab is included as an intervention in over 45 clinical studies of patients with COVID-19 listed on clinicaltrials.gov. Although non-randomized studies have suggested some benefit, to date the results of randomized clinical trials of tocilizumab have not consistently shown clear evidence of efficacy.117 In July 2020, Roche announced that the Phase 3 COVACTA study (NCT04320615) of tocilizumab did not meet its primary endpoint of improved clinical status in hospitalized adult patients with severe COVID-19 associated pneumonia or key secondary endpoints, including the difference in patient mortality at Week 4.118 In September 2020, however, Roche announced that the Phase 3 EMPACTA study (NCT04372186) in hospitalized patients with COVID-19 pneumonia met its primary endpoint, which was the cumulative proportion of participants requiring mechanical ventilation by Day 28. In the EMPACTA study, patients who received tocilizumab plus standard of care were 44% less likely to progress to mechanical ventilation or death compared to patients who received placebo plus standard of care (log-rank p-value = 0.0348; HR [95% CI] = 0.56 [0.32, 0.97]).119 Another Phase 3 study (REMDACTA; NCT04409262) is evaluating the efficacy and safety of remdesivir plus tocilizumab compared with remdesivir plus placebo in hospitalized patients with severe COVID-19 pneumonia. The REMDACTA study’s primary endpoint is the time from randomization to hospital discharge in a time-frame of up to 28 d, and the estimated primary completion date is December 1, 2020.
Although some positive signals have been observed in the studies of tocilizumab, study endpoints were not met in two Phase 3 studies of sarilumab, which targets the same antigen. In July 2020, Sanofi and Regeneron announced that the US Phase 3 trial of sarilumab (400 mg) plus best supportive care in COVID-19 patients requiring mechanical ventilation did not meet its primary and key secondary endpoints compared to best supportive care alone. In September, Sanofi announced that the global Phase 3 trial investigating IV administered sarilumab (200 mg or 400 mg) in severely or critically ill patients hospitalized with COVID-19 did not meet its primary endpoint and key secondary endpoints when sarilumab was compared to placebo plus usual hospital care. Due to these results, Sanofi and Regeneron do not anticipate conducting further clinical studies for sarilumab in COVID-19.
In November 2020, Novartis announced that the US Phase 3 placebo-controlled CAN-COVID trial (NCT04362813) evaluating the efficacy and safety of canakinumab plus standard of care in hospitalized patients with COVID-19 pneumonia and cytokine release syndrome did not meet its primary endpoint of a greater chance of patient survival without the need for invasive mechanical ventilation, or its key secondary endpoint of reduced COVID-19 mortality, compared with standard of care only. The Phase 3 CanCovDia study (NCT04510493) of canakinumab in patients with COVID-19 and Type 2 diabetes, sponsored by the University Hospital, Basel, Switzerland, was still recruiting patients as of mid-November 2020.
Spartalizumab in advanced melanoma
Developed by Novartis, spartalizumab (PDR001) is a humanized IgG4 antibody that targets PD-1. The antibody has been or is being evaluated in over 50 clinical studies involving patients with various types of cancers. Novartis had planned to file for approval of spartalizumab in combination with Tafinlar® (dabrafenib) and Mekinist® (trametinib) for the treatment of unresectable or metastatic BRAF V600 mutation-positive cutaneous melanoma if results of the Phase 3 COMBI-i study (NCT02967692) were positive. On August 22, 2020, however, Novartis announced that the COMBI-i study did not meet its primary endpoint of investigator-assessed PFS.120 The company indicated that the development of spartalizumab will continue. As of November 2020, spartalizumab is undergoing evaluation in numerous Phase 2 studies that are recruiting cancer patients.
Etrolizumab in ulcerative colitis
Developed by Roche, etrolizumab is a humanized IgG1 antibody that targets the β7 subunit common to the integrins α4-β7 and αE-β7. Etrolizumab was undergoing evaluation in four Phase 3 studies in patients with ulcerative colitis, HIBISCUS I (NCT02163759) and II (NCT02171429), HICKORY (NCT02100696) and LAUREL (NCT02165215). HIBISCUS I and II investigated the efficacy and safety of etrolizumab in induction of remission, HICKORY evaluated etrolizumab’s efficacy at induction and maintenance of remission, and LAUREL evaluated etrolizumab’s efficacy at the maintenance of remission. Results of the studies were inconsistent.121 The HIBISCUS I study met the primary endpoint, percentage of participants with induction of remission with etrolizumab compared with placebo at Week 10 as determined by the Mayo Clinic Score, while the HIBISCUS II study did not. HIBISCUS I and II included patients without prior anti-TNF treatment. In the HICKORY study, which included patients with prior anti-TNF treatment, etrolizumab met the primary endpoint at induction but not at maintenance. In the LAUREL study, which included patients without prior anti-TNF treatment, etrolizumab did not meet its primary endpoint.
Late-stage clinical studies of etrolizumab as an induction and maintenance treatment in patients with moderately to severely active Crohn’s disease with and without prior anti-TNF treatment are ongoing. As of a July 2020 update, Roche is considering etrolizumab for Crohn’s disease as a possible regulatory submission in 2022.
Outlook for the future
As 2020 draws to a close, it is clear that controlling SARS-CoV-2 will be the world’s highest priority in 2021, and there is reason to hope that this goal will be accomplished. In November 2020, Pfizer, with their partner BioNTech, and Moderna announced that their COVID-19 vaccines were ~95% effective, and the companies will request EUAs from regulatory agencies. Pfizer has indicated that they plan to make 1.3 billion doses available by the end of 2021, while Moderna remains on track to manufacture 500 million to 1 billion doses in 2021. Other vaccines, including AZD1222, which was co-invented by the University of Oxford and its spin-out company, Vaccitech, will likely also be granted EUAs and distributed globally in 2021. Bamlanivimab and the casirivimab and imdevimab cocktail will be available, and it is likely that at least some of the many other anti-SARS-CoV-2 targeted therapeutics in preclinical and clinical development as of November 2020 (antibodysociety.org/covid-19-biologics-tracker) will also be authorized for emergency use in the future. The potential utility of these antibody products would be substantially broadened if they receive EUAs for prophylactic use in, for example, people who would respond poorly to vaccines, such as the immunocompromised and elderly, and those who are reluctant to undergo any vaccinations.
Looking beyond COVID-19, the number of approvals in 2021 is likely to reach double-digits, assuming companies remain on track with planned BLA submissions and regulatory agencies continue to meet deadlines, as they have done so well in 2020. The numbers of antibody therapeutics currently in early-stage clinical studies (i.e., Phase 1 or Phase 2) for indications other than COVID-19 are more than sufficient to replenish the Phase 3 pipeline, as molecules at that stage progress to regulatory review and approval (Figure 2) in 2021. Estimates as of November 2020 indicate that nearly 300 antibody therapeutics have advanced to Phase 2 studies and over 450 more are undergoing initial evaluations in Phase 1 studies. We look forward to documenting progress made with these and other ‘Antibodies to Watch’ in the next installment of this article series.
Figure 2.
Global commercial clinical pipeline of monoclonal antibodies*
*Data publicly available as of November 15, 2020. Red bars, most advanced clinical study is in cancer. Blue bars, most advanced clinical study is in non-cancer indications. Approved or authorized mAb products, investigational mAbs in regulatory review, and all biosimilar antibodies and Fc fusion proteins were excluded. Phase 1/2 studies were included with Phase 2; pivotal or registrational Phase 2 and Phase 2/3 studies were included with Phase 3.
Acknowledgments
The authors thank Vandna Prasad Rath and Andy Cook, Hanson Wade, for providing access to the Beacon Targeted Therapies database, and Rob Jones, Craic Computing LLC, for providing access to the Therapeutic Antibody Database.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Disclosure statement
HK is employed by a company that develops antibody therapeutics. JR is employed by The Antibody Society, a non-profit trade association funded by corporate sponsors that develop antibody therapeutics or provide services to companies that develop antibody therapeutics, and she is Editor-in-Chief of mAbs, a biomedical journal focused on topics relevant to antibody therapeutics development. Data in this publication were collected from publicly available sources.
Abbreviations
- Aβ
amyloid beta
- ACE2
angiotensin-converting enzyme 2
- AD
atopic dermatitis
- ADC
antibody-drug conjugate
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cellular phagocytosis
- aGvHD
acute graft-vs-host disease
- aHUS
atypical hemolytic uremic syndrome
- AML
acute myeloid leukemia
- Ang-2
angiopoietin-2
- ANGPTL3
angiopoietin-like protein 3
- AQP4
aquaporin-4
- ARDS
acute respiratory distress syndrome
- ASCO
American Society of Clinical Oncology
- ATG
anti-thymoglobulin
- BCG
bacillus Calmette-Guérin
- BCMA
B cell maturation antigen
- BCVA
best-corrected visual acuity
- BICLA
British Isles Lupus Assessment Group Based Composite Lupus Assessment
- BILAG
British Isles Lupus Assessment Group
- BMT
bone marrow transplant
- BLA
biologics license application
- CHMP
Committee for Medicinal Products for Human Use
- CAD
cold agglutinin disease
- CI
confidence interval
- CIS
carcinoma in situ
- CLL
chronic lymphocytic leukemia
- CMC
Chemistry, Manufacturing and Control
- CNS
central nervous system
- COVID-19
coronavirus disease 2019
- CRR
complete response rate
- CSU
chronic spontaneous urticaria
- CTLA-4
cytotoxic T lymphocyte antigen-4
- DCR
disease control rate
- DLBCL
diffuse large B-cell lymphoma
- dMMR
deficient mismatch repair
- DME
diabetic macular edema
- DM4
N2ʹ-deacetyl-N2ʹ-(4-mercapto-4-methyl-1-oxopentyl) maytansine
- DOR
duration of response
- EASI
eczema area and severity index
- EC
European Commission
- EGFR
epidermal growth factor receptor
- EMA
European Medicines Agency
- EpCAM
epithelial cell adhesion molecule
- ESMO
European Society for Medical Oncology
- EU
European Union
- EUAs
Emergency use authorization
- FAS
full analysis set
- FDA
US Food and Drug Administration
- Fc
fraction constant
- FOP
fibrodysplasia ossificans progressive
- FRα
folate receptor alpha
- GM-CSF
granulocyte-macrophage colony stimulating factor
- GvHD
graft-versus-host disease
- HER2
human epidermal growth factor receptor 2
- Hgb
hemoglobin
- HIV
human immunodeficiency virus
- HO
heterotopic ossification
- HoFH
homozygous familial hypercholesterolemia
- HSCT
hematopoietic stem cell transplant
- HR
hazard ratio
- IFN
interferon
- IFNAR1
interferon alpha receptor 1
- IgA
immunoglobulin A
- IgE
immunoglobulin E
- IGA
Investigator’s Global Assessment
- IGF-1R
insulin growth factor 1 receptor
- IL
interleukin
- IST
immunosuppressive therapy
- IV
intravenous
- LDL
low-density lipoprotein
- LM
leptomeningeal metastases
- LSC
leukemia stem cells
- MAA
marketing authorization application
- mAb
monoclonal antibody
- MASP-2
mannan-binding lectin-associated serine protease-2
- mCSCC
metastatic cutaneous squamous cell carcinoma
- MDS
myelodysplastic syndrome
- MET
mesenchymal epithelial transition factor
- MHLW
Ministry of Health, Labor and Welfare
- MM
multiple myeloma
- MMA
monomethyl auristatin
- MMD
monthly migraine days
- MMR
mismatch repair
- MMRp
MMR-proficient
- MS
multiple sclerosis
- MSI
microsatellite instability
- NDA
new drug application
- NGF
nerve growth factor
- NHL
non-Hodgkin’s lymphoma
- NIH
National Institutes of Health
- NMIBC
non-muscle invasive bladder cancer
- NMOSD
neuromyelitis optica spectrum disorder
- NSAIDs
nonsteroidal anti-inflammatory drugs
- NSCLC
non-small cell lung cancer
- OA
osteoarthritis
- OCS
oral corticosteroid
- ORR
overall response rate
- OS
overall survival
- PASI
Psoriasis Area and Severity Index
- PBD
pyrrolobenzodiazepine
- PD-1
programmed cell death protein 1
- PD-L1
programmed death ligand 1
- PDUFA
Prescription Drug User Fee Act
- PFS
progression free survival
- PGA
Patient’s Global Assessment
- PP
per-protocol
- PRIME
Priority Medicines
- Q3w
every 3 weeks
- RA
rheumatoid arthritis
- RECIST
Response Evaluation Criteria in Solid Tumors
- REMS
Risk Evaluation and Mitigation Strategy
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SC
subcutaneous
- SCLC
small cell lung cancer
- SD
stable disease
- SLE
systemic lupus erythematosus
- SR
steroid-refractory
- T1D
type 1 diabetes
- TNBC
triple-negative breast cancer
- TIM-3
T-cell immunoglobulin and mucin-domain domain-containing molecule-3
- TMAs
thrombotic microangiopathies
- TNF
tumor necrosis factor
- US
United States
- VEGF
human vascular endothelial growth factor
- wAMD
wet age-related macular degeneration
- WOMAC
Western Ontario and McMaster Universities Osteoarthritis Index
References
- 1.Kaplon H, Muralidharan M, Schneider Z, Reichert JM.. Antibodies to watch in 2020. MAbs. 2020;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9:167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reichert JM. Antibodies to watch in 2016. MAbs. 2016;8:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reichert JM. Antibodies to watch in 2015. MAbs. 2015;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reichert JM. Antibodies to watch in 2014: mid-year update. MAbs. 2014;6:799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichert JM. Antibodies to watch in 2014. MAbs. 2014;6:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichert JM. Antibodies to watch in 2013: mid-year update. MAbs. 2013;5:513–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichert JM. Which are the antibodies to watch in 2013? MAbs. 2013;5:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichert JM. Which are the antibodies to watch in 2012? MAbs. 2012;4:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichert JM. Antibody-based therapeutics to watch in 2011. MAbs. 2011;3:76–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichert JM. Antibodies to watch in 2010. MAbs. 2010;2:84–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Celltrion Healthcare . Celltrion presents efficacy and safety data for potential COVID-19 treatment candidate CT-P59 in patients with mild symptoms. November 5, 2020. press release. Accessed November6, 2020. www.celltrionhealthcare.com/en-us/board/newsdetail?modify_key=409&pagenumber=1&keyword=&keyword_type=
- 15.Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, Huhn G, Cardona J, Mocherla B, Stosor V, et al. BLAZE-1 Investigators. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med. 2020. October 28 . doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regeneron . Regeneron’s COVID-19 outpatient trial prospectively demonstrates that REGN-COV2 antibody cocktail significantly reduced virus levels and need for further medical attention. October 28, 2020. press release. Accessed November6, 2020. https://investor.regeneron.com/news-releases/news-release-details/regenerons-covid-19-outpatient-trial-prospectively-demonstrates
- 17.Food and Drug Administration. Bamlanivimab Emergency Use Authorization letter dated November 9, 2020. Accessed November10, 2020. www.fda.gov/media/143602/download
- 18.Eli Lilly and Company . Lilly announces agreement with U.S. government to supply 300,000 vials of investigational neutralizing antibody bamlanivimab (LY-CoV555) in an effort to fight COVID-19. October 28, 2020. press release. Accessed November6, 2020. https://investor.lilly.com/news-releases/news-release-details/lilly-announces-agreement-us-government-supply-300000-vials
- 19.Eli Lilly and Company . CEO Dave Ricks shares Lilly’s principles of COVID-19 antibody therapy pricing and access. October 28, 2020. press release. Accessed November6, 2020. https://www.lilly.com/news/stories/dave-ricks-covid19-antibody-therapy-pricing-access
- 20.Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S, et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–19. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- 21.Food and Drug Administration . Casirivimab and imdevimab Emergency Use Authorization letter dated November 21, 2020. Accessed November22, 2020. https://www.fda.gov/media/143891/download
- 22.Yang B, Fulcher JA, Ahn J, Berro M, Goodman-Meza D, Dhody K, Sacha JB, Naeim A, Yang OO. Clinical characteristics and outcomes of COVID-19 patients receiving compassionate use leronlimab. Clin Infect Dis. 2020. October 20;ciaa1583. doi: 10.1093/cid/ciaa1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cytodyn, Inc . CytoDyn announces clinically significant top-line results from its phase 2 trial in mild-to-moderate COVID-19 patients. August 11, 2020. press release. Accessed November6, 2020. https://www.cytodyn.com/investors/news-events/press-releases/detail/458/cytodyn-announces-clinically-significant-top-line-results.
- 24.Biocad . A drug against COVID-19 complications is registered in Russia. June 11, 2020. press release. Accessed October26, 2020. https://biocadglobal.com/post/Levilimab.
- 25.Biocon . Biocon presented insights into clinical study that enabled DCGI approval of itolizumab for COVID-19. July 13, 2020. press release. Accessed October26, 2020. www.biocon.com/biocon-presented-insights-into-clinical-study-that-enabled-dcgi-approval-of-itolizumab-for-covid19/.
- 26.Food and Drug Administration, Center for Drug Evaluation and Research . Clinical review, application number 761143Orig1s000. Accessed September22, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761143Orig1s000MedR.pdf
- 27.Douglas RS, Kahaly GJ, Patel A, Sile S, Thompson EHZ, Perdok R, Fleming JC, Fowler BT, Marcocci C, Marinò M, et al. Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med. 2020. January 23;382(4):341–52. doi: 10.1056/NEJMoa1910434. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Martinez LF, Raport CJ, Ojala EW, Dutzar B, Anderson K, Stewart E, Kovacevich B, Baker B, Billgren J, Scalley-Kim M, et al. Pharmacologic characterization of ALD403, a potent neutralizing humanized monoclonal antibody against the calcitonin gene-related peptide. J Pharmacol Exp Ther. 2020. July;374(1):93–103. doi: 10.1124/jpet.119.264671. [DOI] [PubMed] [Google Scholar]
- 29.Lipton RB, Goadsby PJ, Smith J, Schaeffler BA, Biondi DM, Hirman J, Pederson S, Allan B, Cady R. Efficacy and safety of eptinezumab in patients with chronic migraine: PROMISE-2. Neurology. 2020. March 31;94(13):e1365–e1377. doi: 10.1212/WNL.0000000000009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanofi . FDA approves Sarclisa® (isatuximab-irfc) for patients with relapsed refractory multiple myeloma. March 2, 2020. press release. Accessed September14, 2020. https://www.sanofi.com/en/media-room/press-releases/2020/2020-03-02-19-51-16.
- 31.Schjesvold FH, Richardson PG, Facon T, Alegre A, Spencer A, Jurczyszyn A, Sunami K, Frenzel L, Min CK, Guillonneau S, et al. Isatuximab plus pomalidomide and dexamethasone in elderly patients with relapsed/refractory multiple myeloma: ICARIA-MM subgroup analysis. Haematologica. 2020. June;25:253450. haematol.2020. doi: 10.3324/haematol.2020.253450. [DOI] [Google Scholar]
- 32.Sanofi . Sarclisa® (isatuximab) Phase 3 IKEMA trial meets primary endpoint early in patients with relapsed multiple myeloma. May 12, 2020. press release. Accessed September14, 2020. https://www.sanofi.com/en/media-room/press-releases/2020/2020-05-12-07-00-00.
- 33.Goldenberg DM, Sharkey RM. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs. 2019;11(6):987–95. Aug/Sep. doi: 10.1080/19420862.2019.1632115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.US Food and Drug Administration . FDA approves new therapy for triple negative breast cancer that has spread, not responded to other treatments. April 22, 2020. press release. Accessed September14, 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-new-therapy-triple-negative-breast-cancer-has-spread-not-responded-other-treatments.
- 35.Immunomedics, Inc . Immunomedics announces positive results from Phase 3 ASCENT study of TRODELVYTM. July 6, 2020. press release. Accessed September14, 2020. https://www.immunomedics.com/our-company/news-and-events/immunomedics-announces-positive-results-from-phase-3-ascent-study-of-trodelvytm/.
- 36.Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R, et al. N-MOmentum study investigators. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394(10206):1352–63. doi: 10.1016/S0140-6736(19)31817-3. [DOI] [PubMed] [Google Scholar]
- 37.Bio V. Viela Bio announces U.S. FDA approval of UPLIZNA™ (inebilizumab-cdon) for the treatment of neuromyelitis optica spectrum disorder (NMOSD). June 11, 2020. press release. Accessed September22, 2020. https://ir.vielabio.com/news-releases/news-release-details/viela-bio-announces-us-fda-approval-upliznatm-inebilizumab-cdon.
- 38.Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, Richards JO, Vostiar I, Joyce PF, Repp R, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res. 2008. October 1;68(19):8049–57. doi: 10.1158/0008-5472.CAN-08-2268. [DOI] [PubMed] [Google Scholar]
- 39.Salles G, Duell J, González Barca E, Tournilhac O, Jurczak W, Liberati AM, Nagy Z, Obr A, Gaidano G, André M, et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020. July;21(7):978–88. doi: 10.1016/S1470-2045(20)30225-4. [DOI] [PubMed] [Google Scholar]
- 40.MorphoSys AG FDA approves Monjuvi(R) (tafasitamab-cxix) in combination with lenalidomide for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). August 1, 2020. press release. Accessed September11, 2020. https://www.morphosys.com/media-investors/media-center/fda-approves-monjuvir-tafasitamab-cxix-in-combination-with-lenalidomide.
- 41.GlaxoSmithKline . FDA approves GSK’s BLENREP (belantamab mafodotin-blmf) for the treatment of patients with relapsed or refractory multiple myeloma. August 2, 2020. press release. Accessed September12, 2020. https://www.gsk.com/en-gb/media/press-releases/fda-approves-gsk-s-blenrep-belantamab-mafodotin-blmf-for-the-treatment-of-patients-with-relapsed-or-refractory-multiple-myeloma/.
- 42.Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, Abdallah AO, Callander N, Lendvai N, Sborov D, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207–21. doi: 10.1016/S1470-2045(19)30788-0. [DOI] [PubMed] [Google Scholar]
- 43.Helwick C Responses achieved with belantamab mafodotin in relapsed or refractory myeloma. The ASCO Post, July 10, 2020. Accessed November7, 2020. https://ascopost.com/issues/july-10-2020/responses-achieved-with-belantamab-mafodotin-in-relapsed-or-refractory-myeloma/.
- 44.Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, Yamamura T, Terada Y, Kawata Y, Wright P, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020. May;19(5):402–12. doi: 10.1016/S1474-4422(20)30078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, Patti F, Tsai CP, Saiz A, Yamazaki H, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381(22):2114–24. doi: 10.1056/NEJMoa1901747. [DOI] [PubMed] [Google Scholar]
- 46.U.S. Food and Drug Administration . FDA approves first treatment for Ebola virus. October 14, 2020. press release. Accessed October15, 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-ebola-virus.
- 47.Mulangu S, Dodd LE, Davey RT Jr, Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381(24):2293–303. doi: 10.1056/NEJMoa1910993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Y-mAbs Therapeutics, Inc . FDA approves Y-mAbs’ DANYELZA® (naxitamab-gqgk) for the treatment of neuroblastoma. November 25, 2020. press release. Accessed November26, 2020. https://ir.ymabs.com/news-releases/news-release-details/fda-approves-y-mabs-danyelzar-naxitamab-gqgk-treatment.
- 49.Y-mAbs Therapeutics, Inc . Y-mAbs announces naxitamab update. October 25, 2019. press release. Accessed September19, 2020. https://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-naxitamab-update
- 50.Y-mAbs Therapeutics, Inc . Y-mAbs announces update on naxitamab and omburtamab in neuroblastoma. October 16, 2020. press release. Accessed November7, 2020. https://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-update-naxitamab-and-omburtamab-neuroblastoma.
- 51.Biocad. New drug for melanoma treatment got approved. June 15 , 2020. press release. Accessed September20, 2020. https://biocadglobal.com/index.php?posts&post=52.
- 52.Tjulandin S, Fedyanin M, Demidov L, Moiseyenko V, Protsenko S, Odintsova S, Semiglazova TY, Zukov R, Lazarev S, Andreev A, et al. Final results of phase II trial (MIRACULUM) of the novel PD-1 inhibitor prolgolimab in patients with advanced melanoma. Annal Oncol. 2019. December;30(Supp 11). https://www.annalsofoncology.org/article/S0923-7534(20)34505-1/pdf [Google Scholar]
- 53.R-Pharm Group. R-Pharm has registered Artlegia (olokizumab). May 29 , 2020. press release. Accessed September20, 2020. http://r-pharm.com/en/press-center/news/467.
- 54.Kobayashi H, Furusawa A, Rosenberg A, Choyke PL. Near-infrared photoimmunotherapy of cancer: a new approach that kills cancer cells and enhances anti-cancer host immunity. Int Immunol. 2020. June;4:dxaa037. doi: 10.1093/intimm/dxaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagai S. Flexible and expedited regulatory review processes for innovative medicines and regenerative medical products in the US, the EU, and Japan. Int J Mol Sci. 2019;20(15):3801. doi: 10.3390/ijms20153801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macrogenics, Inc . MacroGenics provides update on FDA review of margetuximab for HER2-positive metastatic breast cancer. May 28, 2020. press release. Accessed September20, 2020. http://ir.macrogenics.com/news-releases/news-release-details/macrogenics-provides-update-fda-review-margetuximab-her2.
- 57.Rugo HS, Im S-A, Cardoso F, Cortes J, Curigliano G, Pegram MD, Musolino A, Bachelot T, Wright GS, De Laurentiis M, et al. Phase 3 SOPHIA study of margetuximab + chemotherapy vs trastuzumab + chemotherapy in patients with HER2+ metastatic breast cancer after prior anti-HER2 therapies: second interim overall survival analysis. Presented at: 2019 San Antonio Breast Cancer Symposium; December 10-14; San Antonio, TX. Abstract GS1-02. https://cancerres.aacrjournals.org/content/80/4_Supplement/GS1-02. [Google Scholar]
- 58.Macrogenics, Inc . MacroGenics presents results from the SOPHIA study of margetuximab in patients with HER2-positive metastatic breast cancer at the San Antonio Breast Cancer Symposium. San Antonio, TX. December 11, 2019. press release. Accessed September20, 2020. http://ir.macrogenics.com/news-releases/news-release-details/macrogenics-presents-results-sophia-study-margetuximab-patients. [Google Scholar]
- 59.Berenbaum F, Blanco FJ, Guermazi A, Miki K, Yamabe T, Viktrup L, Junor R, Carey W, Brown MT, West CR, et al. Subcutaneous tanezumab for osteoarthritis of the hip or knee: efficacy and safety results from a 24-week randomised phase III study with a 24-week follow-up period. Ann Rheum Dis. 2020;79(6):800–10. doi: 10.1136/annrheumdis-2019-216296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, Ali S, Banerjee P, Chan KC, Gipe DA, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383(8):711–20. doi: 10.1056/NEJMoa2004215. [DOI] [PubMed] [Google Scholar]
- 61.Biogen Inc., Eisai, Co., Ltd . FDA accepts Biogen’s aducanumab biologics license application for Alzheimer’s disease with priority review. August 7, 2020. press release. Accessed September8, 2020. https://investors.biogen.com/news-releases/news-release-details/fda-accepts-biogens-aducanumab-biologics-license-application.
- 62.Biogen . Update on FDA advisory committee’s meeting on aducanumab in Alzheimer’s disease. November 6, 2020. press release. Accessed November7, 2020. https://investors.biogen.com/news-releases/news-release-details/update-fda-advisory-committees-meeting-aducanumab-alzheimers.
- 63.Popovic B, Breed J, Rees DG, Gardener MJ, Vinall LM, Kemp B, Spooner J, Keen J, Minter R, Uddin F, et al. Structural characterisation reveals mechanism of IL-13-neutralising monoclonal antibody tralokinumab as inhibition of binding to IL-13Rα1 and IL-13Rα2. J Mol Biol. 2017. January 20;429(2):208–19. doi: 10.1016/j.jmb.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, Moate R, van der Merwe R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol. 2019;143(1):135–41. doi: 10.1016/j.jaci.2018.05.029. [DOI] [PubMed] [Google Scholar]
- 65.Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, Spelman L, Katoh N, Saeki H, Poulin Y, et al. Tralokinumab for moderate‐to‐severe atopic dermatitis: results from two 52‐week, randomized, double‐blind, multicentre, placebo‐controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2020. doi: 10.1111/bjd.19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silverberg JI, Toth D, Bieber T, Alexis AF, Elewski BE, Pink AE, Hijnen D, Jensen TN, Bang B, Olsen CK, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2020. doi: 10.1111/bjd.19573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 levels are elevated in thrombotic microangiopathies: association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin Exp Immunol. 2020. doi: 10.1111/cei.13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Omeros Corporation . Omeros reports final efficacy and safety data from the narsoplimab pivotal trial in HSCT-TMA. October 22, 2020. press release. Accessed November7, 2020. https://investor.omeros.com/news-releases/news-release-details/omeros-reports-final-efficacy-and-safety-data-narsoplimab.
- 69.Provention Bio, Inc . Provention Bio reports second quarter 2020 financial results and provides business update. August 6, 2020. press release. Accessed September8, 2020. http://investors.proventionbio.com/2020-08-06-Provention-Bio-Reports-Second-Quarter-2020-Financial-Results-and-Provides-Business-Update.
- 70.ElsaLys Biotech . ELSALYS BIOTECH announces submission of biologics license application to FDA for LEUKOTAC® (inolimomab) for the treatment of graft-versus-host disease in adult patients. July 23, 2020. press release. Accessed September9, 2020. https://www.elsalysbiotech.com/node/113.
- 71.Socié G, Milpied N, Yakoub-Agha I, Bay JO, Fürst S, Bilger K, Suarez F, Michallet M, Lewalle P, Liens D, et al. Long-term follow-up of a phase 3 clinical trial of inolimomab for the treatment of primary steroid refractory aGVHD. Blood Adv. 2019;3(2):184–86. doi: 10.1182/bloodadvances.2018028282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams R, Maroof A, Baker T, Lawson ADG, Oliver R, Paveley R, Rapecki S, Shaw S, Vajjah P, West S, et al. Bimekizumab, a novel humanized IgG1 antibody that neutralizes both IL-17A and IL-17F. Front Immunol. 2020;11:1894. doi: 10.3389/fimmu.2020.01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reich K, Papp KA, Blauvelt A, Langley R, Armstrong A, Warren RB, Gordon K, Merola JF, Madden C, Wang M, et al. Efficacy and safety of bimekizumab in patients with moderate-to-severe plaque psoriasis: results from BE VIVID, a 52-week Phase 3, randomized, double-blinded, ustekinumab- and placebo-controlled study. American Academy of Dermatology 2020 Annual Meeting (Virtual). https://aad.wistia.com/medias/xfapyslu8k. [Google Scholar]
- 74.UCB . First presentations of bimekizumab Phase 3 data demonstrate superior skin clearance over placebo and Stelara® at week 16 in adults with moderate-to-severe plaque psoriasis. June 12, 2020. press release. Accessed September22, 2020. https://www.ucb.com/stories-media/Press-Releases/article/First-Presentations-of-Bimekizumab-Phase-3-Data-Demonstrate-Superior-Skin-Clearance-Over-Placebo-and-Stelara-at-Week-16-in-Adults-with-Moderate-to-Severe-Plaque-Psoriasis.
- 75.UCB . Bimekizumab Phase 3 psoriasis study demonstrates superiority versus Humira[®]. December 6, 2019. press release. Accessed September22, 2020. https://www.ucb.com/stories-media/Press-Releases/article/Bimekizumab-Phase-3-Psoriasis-Study-Demonstrates-Superiority-Versus-Humira.
- 76.UCB . Bimekizumab superior to Cosentyx[®] in achieving complete psoriasis skin clearance. July 24, 2020. press release. Accessed September22, 2020. https://www.ucb.com/stories-media/Press-Releases/article/Bimekizumab-Superior-to-Cosentyx-in-Achieving-Complete-Psoriasis-Skin-Clearance.
- 77.Sesen Bio . Sesen Bio reports positive, preliminary data update from Phase 3 VISTA trial for high-risk non-muscle invasive bladder cancer. August 8, 2020. press release. Accessed September14, 2020. https://ir.sesenbio.com/news-releases/news-release-details/sesen-bio-reports-positive-preliminary-data-update-phase-3-vista.
- 78.Sesen Bio . Sesen Bio reports second quarter 2020 financial results and business update. August 10, 2020. press release. Accessed September14, 2020. https://ir.sesenbio.com/news-releases/news-release-details/sesen-bio-reports-second-quarter-2020-financial-results-and.
- 79.Oaknin A, Gilbert L, Tinker AV, Sabatier R, Boni V, O'Malley DM, Ghamande S, Duska L, Ghatage P, Guo W, et al. Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient (dMMR) or proficient (MMRp) endometrial cancer: results from GARNET. Presented at: 2020 ESMO Virtual Congress; September 19-21, 2020; Virtual. Abstract LBA36. Accessed November12, 2020. www.annalsofoncology.org/article/S0923-7534(20)42348-8/fulltext. [Google Scholar]
- 80.Agenus Inc . Agenus presents results from two large cervical cancer trials at ESMO. September 18, 2020. press release. Accessed September21, 2020. https://investor.agenusbio.com/news-releases/news-release-details/agenus-presents-results-two-large-cervical-cancer-trials-esmo.
- 81.ADC Therapeutics SA . ADC Therapeutics submits biologics license application to the U.S. Food and Drug Administration for loncastuximab tesirine for treatment of relapsed or refractory diffuse large B-cell lymphoma. September 21, 2020. press release. Accessed September22, 2020. https://ir.adctherapeutics.com/press-releases/press-release-details/2020/ADC-Therapeutics-Submits-Biologics-License-Application-to-the-U.S.-Food-and-Drug-Administration-for-Loncastuximab-Tesirine-for-Treatment-of-Relapsed-or-Refractory-Diffuse-Large-B-cell-Lymphoma/default.aspx.
- 82.Carlo-Stella C, Zinzani PL, Kahl B, Caimi PF, Solh M, Ardeshna K, Stathis A, Hamadani M, Ai W, Hess B, et al. Initial results of a Phase 2 study of loncastuximab tesirine, a novel pyrrolobenzodiazepine-based antibody-drug conjugate, in patients with relapsed or refractory diffuse large B-cell lymphoma. 25th Congress of the European Hematology Association (Virtual). May 28, 2020. presentation. Accessed September22, 2020. https://adctherapeutics.com/adctsite/wp-content/uploads/2020/09/Presentation_ADCT-402-201_Interim-data_EHA-2020.pdf. [Google Scholar]
- 83.Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018;5(1):e000261. doi: 10.1136/lupus-2018-000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.AstraZeneca . Year-to-date and Q3 2020 results. Accessed November11, 2020. https://www.astrazeneca.com/content/dam/az/PDF/2020/q3/Year-to-date_and_Q3_2020_results_Conferences_and_roadshows.pdf.
- 85.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, et al. TULIP-2 Trial Investigators. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382(3):211–21. doi: 10.1056/NEJMoa1912196. [DOI] [PubMed] [Google Scholar]
- 86.Sanofi . Positive results presented from pivotal Phase 3 trial of sutimlimab in people with cold agglutinin disease. December 10, 2019. press release. Accessed November14, 2020. https://www.sanofi.com/en/media-room/press-releases/2019/2019-12-10-13-30-00.
- 87.Sanofi . FDA issues Complete Response Letter for sutimlimab, an investigational treatment for hemolysis in adults with cold agglutinin disease. November 14, 2020. press release. Accessed November14, 2020. https://www.sanofi.com/en/media-room/press-releases/2020/2020-11-14-01-00-00.
- 88.CytoDyn Inc . Update on HIV-BLA-PDUFA. July 13, 2020. press release. Accessed November7, 2020. https://www.cytodyn.com/investors/news-events/press-releases/detail/449/update-on-hiv-bla-pdufa-fda-requested-more-information-to.
- 89.Regula JT, Lundh von Leithner P, Foxton R, Barathi VA, Cheung CM, Bo Tun SB, Wey YS, Iwata D, Dostalek M, Moelleken J, et al. Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med. 2016;8(11):1265–88. doi: 10.15252/emmm.201505889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gasser P, Tarchevskaya SS, Guntern P, Brigger D, Ruppli R, Zbären N, Kleinboelting S, Heusser C, Jardetzky TS, Eggel A. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat Commun. 2020;11(1):165. doi: 10.1038/s41467-019-13815-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vanhoutte F, Liang S, Ruddy M, Zhao A, Drewery T, Wang Y, DelGizzi R, Forleo-Neto E, Rajadhyaksha M, Herman G, et al. Pharmacokinetics and pharmacodynamics of garetosmab (anti-Activin A): results from a first-in-human Phase 1 study. J Clin Pharmacol. 2020. doi: 10.1002/jcph.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regeneron Pharmaceuticals, Inc . Regeneron announces encouraging garetosmab Phase 2 results in patients with ultra-rare debilitating bone disease. January 9, 2020. press release. Accessed September25, 2020. https://investor.regeneron.com/news-releases/news-release-details/regeneron-announces-encouraging-garetosmab-phase-2-results.
- 93.Regeneron Pharmaceuticals, Inc . Regeneron reports second quarter 2020 financial and operating results. August 5, 2020. press release. Accessed September25, 2020. https://investor.regeneron.com/news-releases/news-release-details/regeneron-reports-second-quarter-2020-financial-and-operating.
- 94.Y-mAbs Therapeutics, Inc . Y-mAbs provides regulatory update on omburtamab for the treatment of patients with neuroblastoma. October 5, 2020. press release. Accessed November7, 2020. https://ir.ymabs.com/news-releases/news-release-details/y-mabs-provides-regulatory-update-omburtamab-treatment-patients.
- 95.Y-mAbs Therapeutics, Inc . Y-mAbs announces positive omburtamab clinical data. October 29, 2019. press release. Accessed November 7, 2020. https://ir.ymabs.com/news-releases/news-release-details/y-mabs-announces-positive-omburtamab-clinical-data.
- 96.Byondis BV Synthon Biopharmaceuticals relaunches as Byondis. April 16, 2020. press release. Accessed September26, 2020. https://www.byondis.com/who-we-are/press-releases/synthon-biopharmaceuticals-rebranded-as-byondis.
- 97.Genmab and Seattle Genetics . Genmab and Seattle Genetics present data from tisotumab vedotin innovaTV 204 pivotal trial in recurrent or metastatic cervical cancer at ESMO Virtual Congress 2020. September 21, 2020. press release. Accessed September25, 2020. https://ir.genmab.com/news-releases/news-release-details/genmab-and-seattle-genetics-present-data-tisotumab-vedotin.
- 98.Yun J, Lee S-H, Kim S-Y, Jeong S-Y, Kim J-H, Pyo K-H, Park C-W, Heo SG, Yun MR, Lim S, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR–MET bispecific antibody, in diverse models of EGFR exon 20 insertion–driven NSCLC. Cancer Discovery 2020. doi: 10.1158/2159-8290.CD-20-0116. [DOI] [PubMed] [Google Scholar]
- 99.Park K, John T, Kim S-W, Lee JS, Shu CA, Kim D-W, Ramirez SV, Spira AI, Sabari JK, Han J-Y, et al. Amivantamab (JNJ-61186372), an anti-EGFR-MET bispecific antibody, in patients with EGFR exon 20 insertion (exon20ins)-mutated non-small cell lung cancer (NSCLC). J Clin Oncol. 2020;38(suppl; abstr 9512). doi: 10.1200/JCO.2020.38.15_suppl.9512. [DOI] [Google Scholar]
- 100.TG Therapeutics, Inc . TG Therapeutics provides business update and reports second quarter 2020 financial results. August 10, 2020. press release. Accessed September28, 2020. https://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-provides-business-update-and-reports-second-0.
- 101.TG Therapeutics, Inc . TG Therapeutics announces positive topline results from the UNITY-CLL Phase 3 study evaluating the combination of umbralisib and ublituximab (U2) for the treatment of patients with chronic lymphocytic leukemia. May 5, 2020. press release. Accessed September28, 2020. https://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-announces-positive-topline-results-unity-cll.
- 102.TG Therapeutics announces final results of the GENUINE Phase 3 study evaluating ublituximab plus ibrutinib in previously treated high-risk chronic lymphocytic leukemia at the 56th American Society of Clinical Oncology Annual Meeting (Virtual). May 29, 2020. press release. Accessed September28, 2020. https://ir.tgtherapeutics.com/news-releases/news-release-details/tg-therapeutics-announces-final-results-genuine-phase-3-study. [Google Scholar]
- 103.Sharman JP, Brander DM, Mato AR, Ghosh N, Schuster SJ, Kambhampati S, Burke JM, Lansigan F, Schreeder MT, Lunin SD, et al. Effect of adding ublituximab to ibrutinib on PFS, ORR, and MRD negativity in previously treated high-risk chronic lymphocytic leukemia: final results of the GENUINE phase III study. J Clin Oncol. 2020. May 20;38(15_suppl):8022–8022. doi: 10.1200/JCO.2020.38.15_suppl.8022. [DOI] [Google Scholar]
- 104.Novartis Pharmaceuticals Corporation . Reimagining AML & MDS: targeting immune and myeloid cells for dual impact. Accessed September28, 2020. https://www.virtualcongress.novartis.com/eha25/myeloid/aml-and-mds/.
- 105.Agenus Inc . Agenus announces ESMO oral presentation of two clinical trials of balstilimab alone and in combination with zalifrelimab in recurrent or metastatic cervical cancer. August 26, 2020. press release. Accessed October2, 2020. https://investor.agenusbio.com/news-releases/news-release-details/agenus-announces-esmo-oral-presentation-two-clinical-trials.
- 106.Silence K, Dreier T, Moshir M, Ulrichts P, Gabriels SME, Saunders M, Wajant H, Brouckaert P, Huyghe L, Van Hauwermeiren T, et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs. 2014;6(2):523–32. doi: 10.4161/mabs.27398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.argenx BV . argenx enters exclusive global collaboration and license agreement with Cilag GmbH International, an affiliate of Janssen, for cusatuzumab (ARGX-110). December 2, 2018. press release. Accessed October2, 2020. https://www.argenx.com/news/argenx-enters-exclusive-global-collaboration-and-license-agreement-cilag-gmbh-international.
- 108.Janssen Pharmaceutical Companies of Johnson & Johnson . Selected NME pharmaceutical pipeline as of July 16, 2020. Accessed October1, 2020. https://www.investor.jnj.com/pharmaceutical-pipeline.
- 109.Akeso Inc . 2020 Interim presentation, August 2020. Accessed September29, 2020. https://www.akesobio.com/media/1306/akeso-2020-interim-results-presentation.pdf.
- 110.China International Capital Corporation Limited. Akeso Inc . Equity Research, July 7, 2020. Accessed September29, 2020. http://pdf.dfcfw.com/pdf/H3_AP202007081390358126_1.pdf.
- 111.Chen X, Song X, Li K, Zhang T. FcγR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front. Immunol. 2019;10:292. doi: 10.3389/fimmu.2019.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Checkpoint Therapeutics, Inc . September 2020 corporate presentation. Accessed September27, 2020. https://s2.q4cdn.com/326900168/files/doc_presentations/2020/09/v2/Checkpoint-Presentation_CKPT_September-2020_2.pdf.
- 113.Checkpoint Therapeutics, Inc . Checkpoint Therapeutics announces positive interim results from registration-enabling trial of cosibelimab in metastatic cutaneous squamous cell carcinoma. September 17, 2020. press release. Accessed September27, 2020. https://ir.checkpointtx.com/press-releases/news-details/2020/Checkpoint-Therapeutics-Announces-Positive-Interim-Results-from-Registration-Enabling-Trial-of-Cosibelimab-in-Metastatic-Cutaneous-Squamous-Cell-Carcinoma/default.aspx
- 114.ImmunoGen, Inc . Form 10-K for the year ended December 31, 2019. Accessed September26, 2020. https://www.sec.gov/ix?doc=/Archives/edgar/data/855654/000155837020002399/imgn-20191231x10kd363eb.htm.
- 115.Actinium Pharmaceuticals, Inc . Actinium announces two oral presentations featuring data and findings from the Phase 3 SIERRA trial of Iomab-B at the 62nd American Society of Hematology Annual Meeting (Virtual). November 4, 2020. press release. Accessed November12, 2020. https://ir.actiniumpharma.com/press-releases/detail/372/actinium-announces-two-oral-presentations-featuring-data. [Google Scholar]
- 116.Alphamab Oncology . Alphamab Oncology initiates Phase II registration clinical trial (KN046-205) for bispecific antibody KN046 with a successful researchers’ conference. Shanghai, China. August 30, 2020. press release. Accessed October1, 2020. http://www.alphamabonc.com/en/html/news/2134.html [Google Scholar]
- 117.Parr JB. Time to reassess tocilizumab’s role in COVID-19 pneumonia. JAMA Intern Med. 2020. October 20. Published online. doi: 10.1001/jamainternmed.2020.6557. [DOI] [PubMed] [Google Scholar]
- 118.F Hoffman-La Roche Ltd . Roche provides an update on the phase III COVACTA trial of Actemra/RoActemra in hospitalised patients with severe COVID-19 associated pneumonia. July 29, 2020. press release. Accessed November11, 2020. www.roche.com/investors/updates/inv-update-2020-07-29.htm
- 119.F Hoffman-La Roche Ltd . Roche’s phase III EMPACTA study showed Actemra/RoActemra reduced the likelihood of needing mechanical ventilation in hospitalised patients with COVID-19 associated pneumonia. September 18, 2020. press release. Accessed November11. www.roche.com/investors/updates/inv-update-2020-09-18.htm.
- 120.Novartis . Novartis provides update on Phase III study evaluating investigational spartalizumab (PDR001) in combination with Tafinlar® + Mekinist® in advanced melanoma. August 22, 2020. press release. Accessed October26, 2020. www.novartis.com/news/media-releases/novartis-provides-update-phase-iii-study-evaluating-investigational-spartalizumab-pdr001-combination-tafinlar-mekinist-advanced-melanoma.
- 121.F. Hoffman-La Roche Ltd . Roche provides update on phase III studies of etrolizumab in people with moderately to severely active ulcerative colitis. August 10, 2020. press release. Accessed September24, 2020. https://www.roche.com/investors/updates/inv-update-2020-08-10.htm.