

ABSTRACT
Recently, by combining transcriptomics with functional splicing reporter assays we were able to identify GT > GC > TT as the three highest ranked dinucleotides of human 5ʹ splice sites (5’ss). Here, we have extended our investigations to the proteomic characterization of nuclear proteins that bind to canonical and noncanonical 5’ss. Surprisingly, we found that U1 snRNP binding to functional 5’ss sequences prevented components of the DNA damage response (DDR) from binding to the RNA, suggesting a close link between spliceosome arrangement and genome stability.
We demonstrate that all tested noncanonical 5’ss sequences are bona-fide targets of the U2-type spliceosome and are bound by U1 snRNP, including U1-C, in the presence of splicing enhancers. The quantity of precipitated U1-C protein was similar for all noncanonical 5’ss dinucleotides, so that the highly different 5’ss usage was likely due to a later step after early U1 snRNP binding.
In addition, we show that an internal GT at positions +5/+6 can be advantageous for splicing at position +1 of noncanonical splice sites. Likewise, and in agreement with previous observations, splicing inactive U1 snRNP binding sites could serve as splicing enhancers, which may also explain the higher abundance of U1 snRNPs compared to other U snRNPs. Finally, we observe that an arginine-serine (RS)-rich domain recruitment to stem loop I of the U1 snRNA is functionally sufficient to promote exon-definition and upstream 3’ss activation.
KEYWORDS: Noncanonical splicing, 5ʹ splice site, U1 snRNP, SR proteins, hnRNP proteins
Introduction
Alternative splicing enables the expression of tailored cellular proteomes from an identical repertoire of protein-coding genes [1]. Introns are in general removed from pre-mRNA by the major or U2-type spliceosome that consists of five uracil-rich small ribonucleoprotein particles (the U1, U2 and U4/U6.U5 tri-snRNP) and a rich repertoire of auxiliary proteins [2,3]. Spliceosomes recognize conserved sequence elements at the exon/intron boundaries, termed splice sites (ss) and the vast majority of the U2-type introns is flanked by GT-AG dinucleotides. However, at least approximately 0.9% of all human U2-type introns contain 5’ss with a GC dinucleotide at position +1/+2 [4]. In line with this, a recent study showed that GT > GC mutations not necessarily impair splicing [5]. Another rare class of introns (approximately 0.4%) is removed by the minor U12-type spliceosome [6–8]. Most of these introns are as well flanked by GT-AG dinucleotides, but some also exhibit AT-AC dinucleotides at the exon/intron borders. Finally, for physiological [9–11] and pathological 5’ss mutations [12–14] so-called noncanonical dinucleotides, i.e. other than the canonical GT or the non-canonical GC, were described.
Canonical 5’ss recognition was shown to be under combinatorial control of splicing regulatory elements (SREs) and the extent of sequence complementarity to the 5ʹ-end of the U1 snRNA [15]. SREs can act positively (as enhancer) or negatively (as silencer) on splice site use and are predominantly bound by members of the SR or hnRNP protein family [16]. SR proteins were shown to facilitate U1 snRNP binding through their RS domain forming physical contacts with the RS domain of the U1 snRNP specific protein U1-70k [17]. Yet, it remains largely undefined whether noncanonical 5’ss are recognized by components of the U2- or U12-type spliceosome. Accordingly, there is also lack of clarity about the principles underlying noncanonical 5’ss usage, although previous work by us and others rather implies that noncanonical 5’ss are substrates of the U2-type spliceosome. However, the importance of gaining a deeper understanding of canonical and noncanonical 5’ss usage becomes clear when looking at the continuously growing list of pathological 5’ss [13] and U1 snRNA mutations [18,19] that are associated with disease.
In this study, we show that noncanonical 5’ss are primarily targeted by the U1 snRNP of the U2-type spliceosome. We demonstrate that recognition of noncanonical 5’ss by the U1 snRNP relies on strong splicing enhancers in their neighbourhood and functional basepairing interactions with the 5ʹ-end of the U1 snRNA even more than their canonical counterparts. Finally, and in agreement with previous observations [20–22], we demonstrate that U1 snRNP can act as a splicing enhancer when located outside of the actual U1 snRNA binding location.
Material and methods
Oligonucleotides
Oligonucleotides were obtained from Metabion GmbH. Primers used for cloning, RT PCR analyses and RNA in vitro binding assays are listed in Table S1.
Plasmids
All SV-env/eGFP plasmids were cloned by substitution of the SacI/NdeI fragment with PCR products using appropriate forward primer and primer #640 as a reverse primer. They are either listed in Table S1 or have been described previously [23] . Also, proviral HIV plasmids were constructed as described previously [24]. U1 snRNA expression plasmids were constructed by insertion of a PCR product amplified with an appropriate forward primer and #3926 as a reverse primer, containing BglII and XhoI restriction sites into the template pUCBU1 (kindly provided by Alan M. Weiner) resulting in pUCBΔU1 [25]. Cloning of pUCBU1αD3 (+1G>C) was described elsewhere [24]. For cloning of pUCBU1 αD3 (+1G>C), SLII>MS2, the BclI/XhoI fragment of pUCBU1 αD3 (+1G>C) was replaced by a PCR product using primer pair #4391/#3926.
Cell culture and nucleic acid transfections
HEK 293 T cells were maintained in DMEM (Invitrogen) supplemented with 10% foetal calf serum (FCS) and 50 µg/ml of each penicillin and streptomycin (P/S) (Invitrogen). Plasmid transfections were performed in six-well plates with 2.5 × 105 HEK293 T cells using theTransIT®-LT1 reagent (Mirus Bio LLC) according to the manufacturer’s instructions.
RNA extraction and semi-quantitative RT-PCR
Total RNA samples were collected 48 h post-transfection. For RT-PCR analyses RNA was reversely transcribed using Superscript III (SSIII) Reverse Transcriptase (Invitrogen) and Oligo(dT) primer (Invitrogen). For semi-quantitative analyses of spliced mRNAs, cDNA was used in a PCR reaction with primer #3210 and #3211. To control for equal transfection efficiencies a separate PCR reaction was carried out with primer pair #1224/#1225 detecting coexpressed GH1-mRNA.All primer sequences used for semi-quantitative RT-PCR analyses are listed in Table S1. PCR products were separated on 8% non-denaturing polyacrylamide (PAA) gels and stained with ethidium bromide for visualization.
RNA in vitropulldown assays
For in vitro transcription of substrate RNAs, DNA templates were amplified from respective SV-env/eGFP plasmids with a forward primer containing a T7 promotor sequence and a single copy of an MS2 RNA binding site at the 5ʹ-end and a respective reverse primer (Table S1). RNA was synthesized using the RiboMaxTM large-scale RNA production system (P1300, Promega) according to the manufacturer’s instructions. Substrate RNAs were covalently linked to adipic acid dihydrazide-agarose beads as previously described [15,24,26]. Immobilized RNAs were incubated in 30% HeLa cell nuclear extract (Cilbiotech)/buffer D (20 mM HEPES-KOH [pH 7.9], 5% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM DTT) for 20 min at 30°C. Recombinant MS2 coat protein was added to nuclear extract dilutions to monitor for equal precipitation efficiencies. After washing, specifically bound fractions were eluted from the beads and subjected to immunoblotting. Membranes were probed with rat antibody against U1-C (4H12) from Sigma-Aldrich and rabbit antibody against MS2 (TC-7004) from Tetracore. For detection, we used horseradish peroxidase (HRP)-conjugated anti-rat antibody (A9037) and HRP-conjugated anti-rabbit antibody (A6154) from Sigma-Aldrich.
Mass spectrometry and mass spectrometric data analysis
Mass spectrometric analysis was essentially carried out as described earlier (Erkelenz et al. [24]). Briefly, protein samples (independent RNA pulldowns, n = 5 per group: CT, GA, GC, CT, TT, ΔGT, ΔEnhancer) were shorty separated (about 5 mm running distance) in an acrylamide gel and protein-containing band processed for mass spectrometric analysis by reduction, alkylation and tryptic digestion. Peptides were separated using C18 material on an Ultimate 3000 Rapid separation system (Thermo Fisher Scientific) using a 2 h gradient. Separated peptides were injected in an Orbitrap Elite hybrid mass spectrometer using an electrospray ionization nano-source interface. Precursor spectra were recorded in the orbitrap analyser and up to 20 precursor ions selected, fragmented using collision-induced dissociation and analysed in the linear ion trap part of the instrument.
Raw files were further processed with MaxQuant (version 1.5.0.30, Max Planck Institute for Biochemistry, Planegg, Germany) using standard parameters if not indicated otherwise for peptide and protein identification and quantification. Searches were carried out based on 20,187 homo sapiens entries downloaded on the 3 December 2014 from the SwissProt section of UniProtKB supplemented by a sequence of an MBP-MS2 fusion protein. The ‘match between runs’ function was enabled as well as label-free quantification. Protein and peptide identifications were accepted at a false discovery rate of 1% and only proteins identified with at least two different peptides and a minimum of three valid label-free quantification (LFQ) values in each analysed group further considered. Additional processing of quantitative mass spectrometric data was carried out using Perseus 1.6.6.0 (Max Planck Institute for Biochemistry, Planegg, Germany). For pairwise comparison of two sample groups, Student’s t-tests were calculated on log2 transformed LFQ intensities and cut-offs for significant differences determined based on the significance analysis of microarrays method [27] using an S0 of 0.1 and false discovery rate of 5%. One dimensional annotation enrichment analysis was carried out with Perseus [28] on the basis of the difference between the group-means of log2 transformed normalized (LFQ) intensities. Before analysis, identified protein groups were annotated with GOBP terms (Homo sapiens, build in from Perseus) as well as manually added annotations (RBM, hnRNP, SR, U1 snRNP, DNA damage, Supplemental Data 1). The cut-off for reporting significantly altered categories was a Benjamini–Hochberg corrected p-value < 0.05.
RNA-seq analysis
We studied 5′splice site usage in our human RNA-seq transcriptome data set of 54 human fibroblast samples originating from 27 individuals. The raw sequencing data had recently been made publicly accessible [29] and are deposited online (ArrayExpress, accession number: E-MTAB-4652). Reads were trimmed or discarded with Trimmomatic version 0.36 based on their base calling quality and their adapter content [30]. The extent of rRNA depletion was measured by mapping the reads to rRNA databases with the SortMeRNA algorithm version 2.1b [31]. For alignments and sequence analysis the human genome reference sequence GRCh38annotation data (release 99) was downloaded from ENSEMBL [32]and BioMart [33]. Alignments were calculated using the alternative two-pass mapping protocol of the STAR aligner (2.5.4b) [34]. Subsequent calculation of splice site localizations from gapped (exon junction) reads was carried out using CRAN package rbamtools [35]. Identified splice sites were then processed using Bioconductor package splice sites (R package version 1.8.3, https://bioconductor.org/packages/3.2/bioc/html/spliceSites.html) and summarized per genomic 5’ss position. FASTQ files and the alignments were prepared using custom BASH shell scripts in the environment of the High Performing Cluster of the Heinrich Heine University Düsseldorf. Computational support and infrastructure were provided by the ‘Centre for Information and Media Technology’ (ZIM) at the Heinrich Heine University Düsseldorf. 5’ss usage was normalized to the number of reads in the respective sample. To enable comparison, not only between samples, but between splice sites of differently expressed genes, the 5’ss usage was additionally normalized to the respective gene expression. Resulting Gene Normalized Reads (GNR) were calculated as the ratio in splice donor usage between a 5’ss and the most used 5’ss within the same gene. 5’ss usage information was then summarized from all samples leading to 298,197 5’ss with a GT dinucleotide at position +1/+2.269,186 5’ss were exclusively used with no competing secondary site within the 50 nt downstream region. The overall level of SRE-mediated splicing enhancement was estimated by calculating the average HEXplorer difference between the 50 nucleotides present up- and downstream of the 5’ss [36]. Affinity of the downstream sequences for U1 snRNA binding was calculated by assigning ‘Pseudo’-H-Bond scores (by insertion of a GT at position +1/+2) to every 11nt long sequence overlapping within the 50-nucleotide downstream sequences. ‘Pseudo’-H-Bond scores were summed up to form an integral measure for the general U1 snRNA affinity termed ”Pseudo-HBS integral”.
Results
Mutually exclusive binding of functional and non-functional splice site RNA substrates by U1 snRNP-associated proteins and components of the DNA damage response (DDR) machinery
To define the extended repertoire of U1 snRNP-associated proteins and globally screen for changes in the composition of ribonucleoprotein (RNP) complexes formed with either functional or non-functional splice site RNA substrates (Fig. 1A,B), we performed RNA pull down assays with nuclear extract followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. As expected, splice site RNA substrates with both, a strong splicing enhancer and consensus 5’ss sequence showed efficient binding to known core constituents of U1 snRNP, including all three U1-specific proteins U1-C (SNRNPC), U1-70K (SNRNP70) and U1-A (SNRPA), subunits of the heptameric Sm protein ring as well as more peripheral U1-associated proteins such as Luc7L (Fig. 1B and Supplemental Data 1). This finding was consistent with a high splicing efficiency of a respective splicing minigene (Fig. 1B) and the recruitment of U1 snRNP to the pre-mRNA. The specificity of the RNA pulldown was further supported by GO cluster analysis, demonstrating overrepresentation of GO terms associated with splicing (Fig. 1D and Supplemental Data 2). By contrast, binding of U1 snRNP proteins was completely abolished in the absence of a functional 5’ss sequence (Fig. 1B; “Enhancer-ΔGT“). Instead, proteins involved in DNA damage response (DDR) and DNA repair could be detected (Fig. 1B, D and Supplemental Data 1), indicating that complexation of nascent RNA by splicing complexes normally ousts these proteins from binding. We also compared RNP complexes formed with RNAs either lacking a strong enhancer or a functional 5’ss sequence (Fig. 1C and Supplemental Data 1). As expected, U1 snRNP recruitment strictly relied on the presence of a 5’ss sequence, while the enhancer appeared to be largely dispensable for precipitation of U1 snRNP proteins (Fig. 1C,E and Supplemental Data 1). Strikingly and quite unexpectedly, the splicing enhancer sequence was specifically bound by a considerable number of hnRNP proteins instead of SRSF7, suggesting that U1 snRNP binding is a prerequisite for SR protein stabilization on the pre-mRNA (Fig. 1C,E and Supplemental Data 1).
Figure 1.
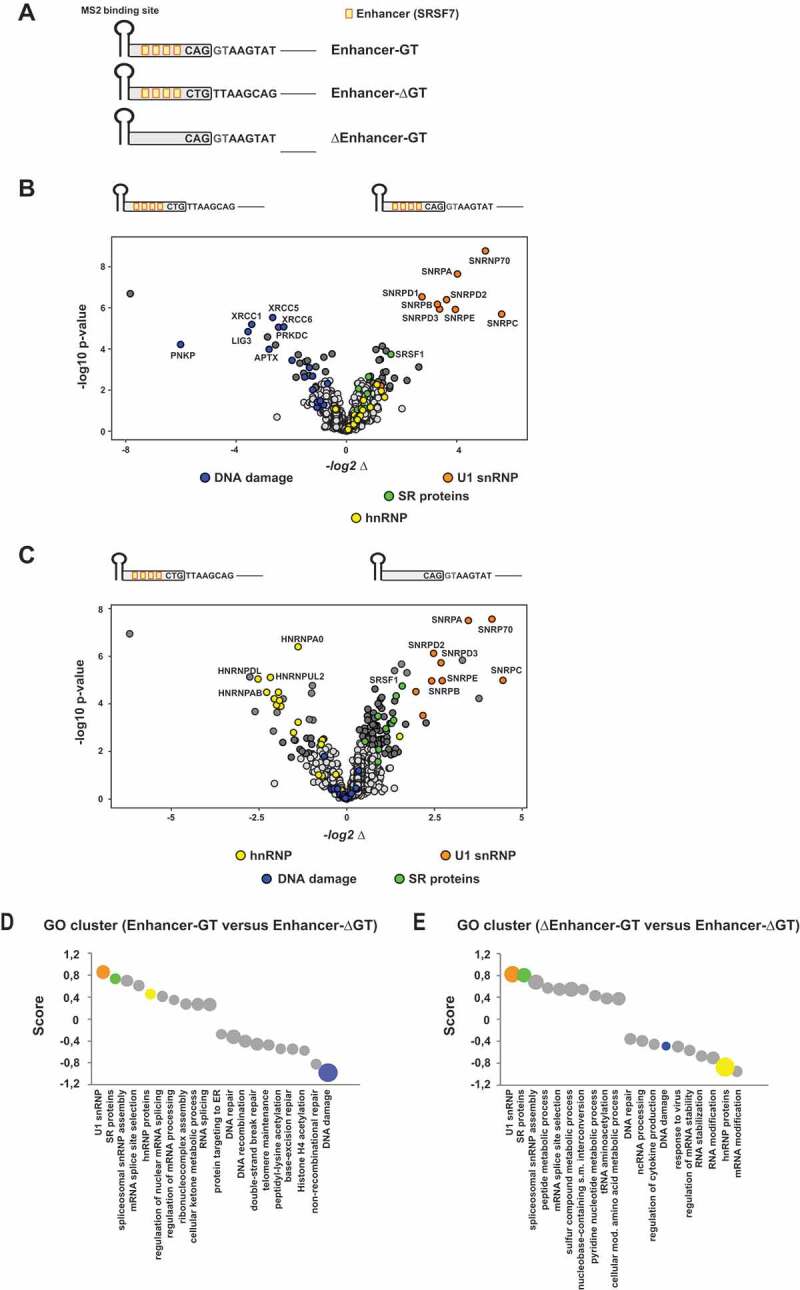
Profiling of the functional splice siteinteractome
(A) Schematic of the RNA substrates used for RNA in vitro pulldown assays. All in vitro transcribed RNAs contained an MS2 binding site at the 5ʹ-end that was recognized by recombinant MS2 coat protein (added to the nuclear extracts) and served as a precipitation control. RNAs were covalently coupled to agarose beads and bound fractions subsequently analysed by mass spectrometry (MS). Orange boxes indicate single splicing enhancer sequences that had been demonstrated before to efficiently enhance splice site activation [15,23,62]. (B, C) MS analysis identified the functional 5’ss (enhancer plus GT 5’ss) interactome (U1 snRNP proteins are highlighted in orange). Absence of the 5’ss sequence strongly reduced the levels of precipitated U1 snRNP proteins (B-C). In addition, the enhancer sequence revealed enriched binding for hnRNP proteins (C) (highlighted in green). Volcano Plots had been generated using InstantClue (www.instantclue.uni-koeln.de). (D, E) One dimensional Gene Ontology (GO) enrichment analysis was performed using Perseus. The top 10 non-redundant categories showing a shift to higher or lower abundances are presented. The size of the bubbles indicates the associated – log10 p-values. Ranges for – log10 p-values are 3.4 (smallest bubble) to 11.4 (largest Bubble) in E, 2–11.3 in F and 2.1–9.6 in G. The U1 snRNP complex annotation cluster is highlighted in orange, DNA repair cluster in blue and hnRNP cluster in yellow.
U1 snRNP recruitment generally mirrors 5’ss usage
Recently, we defined basic principles underlying the usage of noncanonical 5’ss [23]. We were able to rank them in the order of their intrinsic strengths GT > GC > TT > AT > GA > GG > CT. It is not fully understood yet why a TT dinucleotide at position −1/+1 compared favourably to all other tested dinucleotides except of a GC dinucleotide (Fig. 2A). Importantly, the splice site sequence CAG/TTAAGTAT in our experiments contained also a GT dinucleotide at −1/+1. This could theoretically entail alternative modes of U1 snRNA duplex formation by, e.g. bulging out a single nucleotide [37,38] or basepairing in a shifted register [39]. In fact, we could detect approximately equal proportions of −1 and +1 splicing for the TT sequence, indicating that competing basepairing registers dictate the splicing outcome. However, we hypothesized that the additional GT at positions −1 and +1 could also be responsible for further U1 snRNP stabilization by U1-C (SNRPC) and/or Luc7-like (Luc7L), stimulating splicing at position +1 [40]. Therefore, we aimed to analyse U1 snRNP complex binding and first, tested the levels of U1-C protein co-precipitating together with RNA substrates containing a single-nucleotide substitution at 5’ss positions +1 or +2 (Fig. 2B,C). Consistent with our ranking, the canonical GT site showed the highest levels of bound U1-C, directly followed by the GC site (Fig. 2B,C). However, contrary to our expectations the level of U1-C protein binding to TT sites was not elevated compared to the other tested noncanonical 5’ss such the CT dinucleotide with the lowest rank. Therefore, we carried on with a more unbiased proteomic approach and compared protein binding profiles of selected, noncanonical 5’ss (Fig. 2D–F). We found that the U1 snRNP binding profiles largely correlated with the relative 5’ss strength with the exception, that TT and CT sites recruited comparable levels of U1 snRNP proteins (Fig. 2F). These findings indicated that the selection and rejection of noncanonical splice sites might occur at a later step than U1 snRNP binding. Alternatively, TT splice sites could be recognized by spliceosomal factors other than the U1 snRNP such as U11-48K of the minor spliceosome [8]. Although we cannot unequivocally exclude this possibility, preceding experiments clearly demonstrated that TT splice site usage depended on the complementarity of the splice site sequence to the 5ʹ-end of the U1 snRNA [23].
Figure 2.
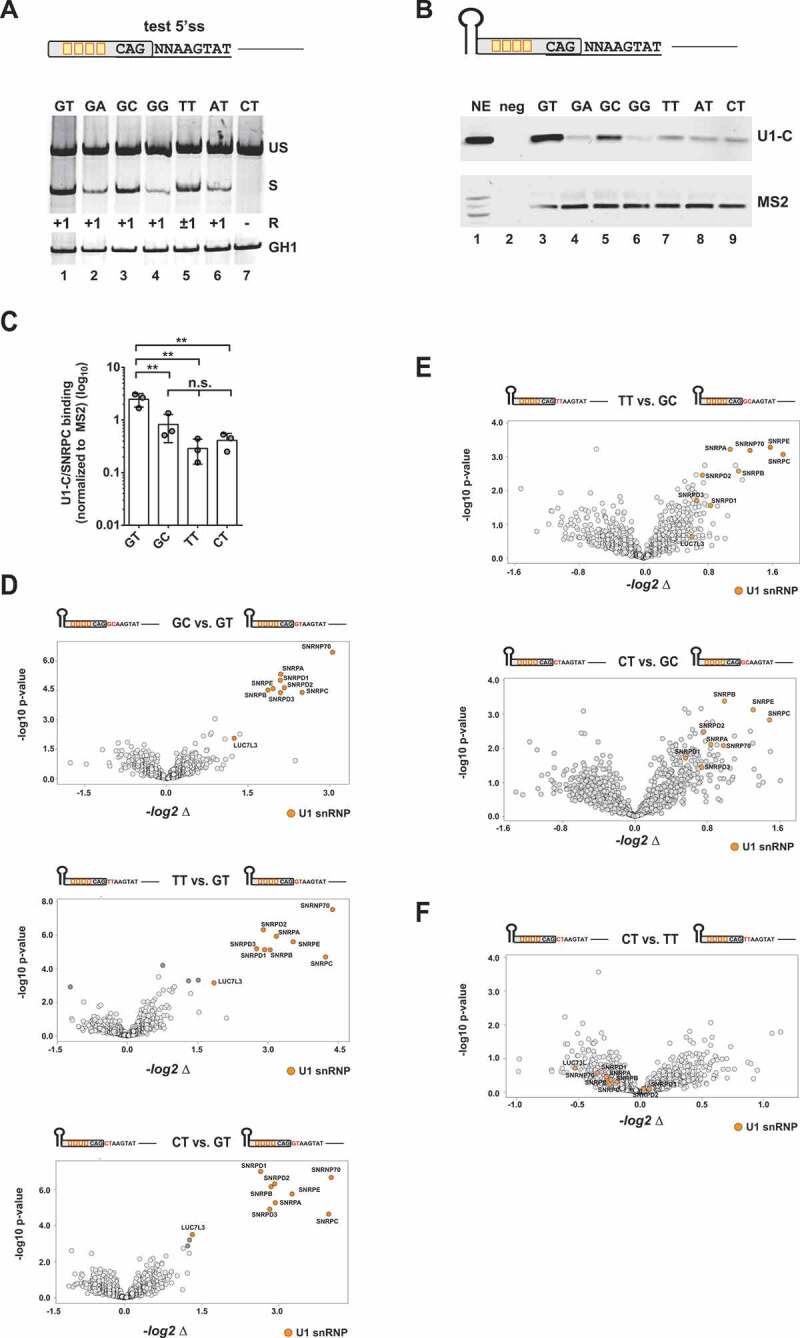
Binding of U1 snRNP-associated proteins to noncanonical splice sites
(A) Assessment of the usage of different noncanonical splice sites using an HIV-1-based SV-env/eGFP splicing reporter [23], which contains four enhancer binding sites upstream of the 5’ss test sequence CAGNNAAGTAT. HEK293T cells were transiently transfected with 1 µg of each: the respective SV-env/GFP splicing reporter and pXGH5 (expressing human growth hormone 1 [GH1] to monitor equal transfection efficiencies). RNA was isolated 30 h post-transfection and used for RT-PCR analysis as described in Material&Methods. Different splicing positions (R) obtained by sequencing of the RT-PCR products are indicated below the gel image. US: unspliced; S: spliced; +1: exclusive cleavage at position +1; −1/+1: cleavage at positions −1 and +1; +1/5: cleavage at positions +1 and +5. (B, C) RNA in vitro binding assays show U1-C/SNRPC binding to noncanonical 5’ss. As mentioned before, in vitro transcribed RNAs were equipped with an MS2 binding site at the 5ʹ-end bound by recombinant MS2 coat protein that had been added to the nuclear extracts (NE). RNAs were immobilized and analysed for binding of U1-C/SNRPC (U1 snRNP) and MS2 (precipitation control). Signal intensities were quantified by ImageJ and normalized to the mean band intensity of U1-C/SNRPC for each biological replicate (n = 3). Data show the mean value ± standard deviation (SD). ** p < 0.01 and n.s: not significant (one-way ANOVA). (D, F) Comparative MS analysis of the protein fractions eluted from the different in vitro RNA substrates largely agreed with a positive correlation between U1 snRNP protein precipitation efficiency and 5’ss usage. Vulcano Plots were created using InstantClue (www.instantclue.uni-koeln.de). U1 snRNP proteins are highlighted in orange. Significantly enriched proteins are highlighted in dark grey and dark orange (U1 snRNP). Dinucleotides are highlighted in red.
U1 snRNP binding to noncanonical 5’ss relies on enhancer-mediated U1 snRNA duplex stability
To address the question of whether U1 snRNP binding depends more on the support of splicing enhancers or base pairing between the U1 snRNA and the 5’ss sequence, we tested U1-C binding in presence and absence of splicing enhancers (Fig. 3A). Additionally, we compared enabled and disabled U1 snRNA binding by oligonucleotide-directed RNase H digestion of the U1 snRNA 5ʹ end. As before, we consciously decided for a minimal deviation from the consensus sequence and changed only a single nucleotide in one of the conserved GT positions at +1 or +2. Consistent with previous results from functional splicing assays [23], the tested noncanonical 5’ss sequences showed a higher enhancer dependency for U1-C binding (Fig. 3B, cf.upper panel ”GT”, lanes 1 and 3 with lower panel ‘TT’, lanes 1 and 3). In general, U1-C binding was decreased upon RNase H treatment (Fig. 3B, cf. lanes RNase H, – with +), suggesting that U1 snRNP recruitment to canonical as well as noncanonical splice sites relies on intact base pairing capacity of the endogenous U1 snRNA. Nonetheless, our results also clearly indicated that an efficient enhancer neighbourhood could partially overcome the need for basepairing interactions with the U1 snRNA (Fig. 3B, cf. lanes ‘both’ RNase H, – with +).
Figure 3.
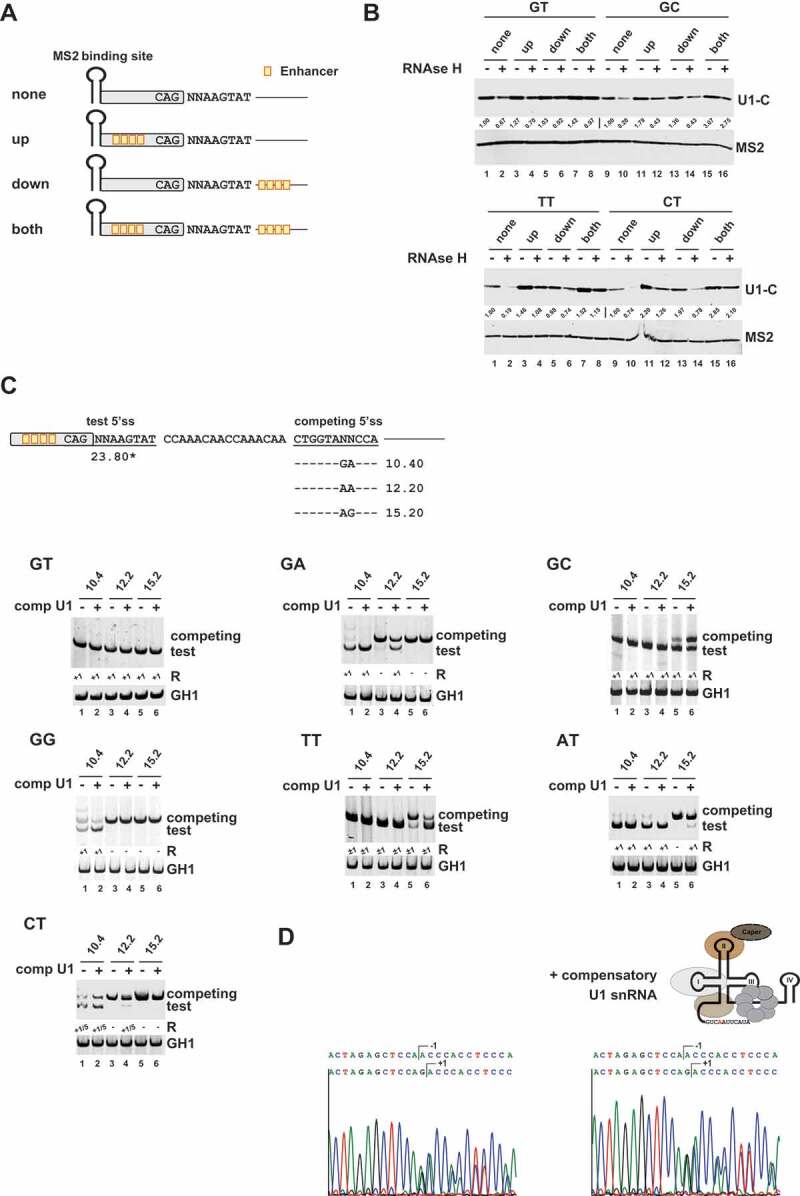
U1 snRNP binding to noncanonical splice sites relies on stable U1 RNA duplex formation
(A) Schematic of the RNA substrates used for RNA in vitro pulldown assays. Enhancer sites are indicated by orange boxes. (B) RNA in vitro pulldown assays showed that base pairing interactions with the U1 snRNA and splicing enhancers are major determinants for U1-C/SNRPC binding to noncanonical 5’ss sequences. HeLa cell nuclear were depleted (+) or mock-depleted (-) of U1 snRNA 5ʹ-end using short DNA oligonucleotides and RNase H. RNA pulldown assays were performed as described before. MS2 coat protein served as control. Signal intensities were quantified using the ImageJ software (https://fiji.sc/). The MS2 protein band intensities were used to normalize the U1-C/SNRPC signals. The normalized U1/C/SNRPC band intensities of the enhancer-less splice site substrates were set to 1 (‘none’, RNase H -) to calculate relative changes caused by RNase H treatment and/or presence of enhancer sequences in the upstream exon (‘up’) or downstream intron (‘down’). (C) Schematic of the HIV-1-based SV-env/eGFP splicing reporter containing the different pairs of competing splice sites (on top). Sequence variations (denoted by ‘NN’) and H-Bond scores (https://www2.hhu.de/rna/html/H-Bond_score.php) of the competing canonical splice sites are indicated below. The different splicing reporters were either coexpressed with a compensatory U1 snRNA (+) or not (-). RT-PCR analyses of spliced reporter mRNAs were performed as described before. Splicing positions (R) are indicated below.*: H-Bond score had been calculated by inserting a GT at position +1/+2; +1: cleavage at position +1; −1/+1: cleavage at positions −1 and +1; +1/5: cleavage at positions +1 and +5. (D) Sequencing results of TT splice site usage in the presence and absence of a compensatory U1 snRNA. Polyacrylamide (PAA) bands were isolated, reamplified with primer pair #3210/#3211 and sent to sequencing analysis using primer #3210.
The fact that U1 snRNP/noncanonical 5’ss interactions similarily required RNA duplex formation prompted us to determine whether noncanonical5’ss splicing can be activated by coexpression of a compensatory U1 snRNA. Therefore, we generated U1 snRNAs carrying single-nucleotide substitutions within their 5ʹ end that compensated for the mismatch at the central 5’ss dinucleotide positions +1 or +2. To investigate noncanonical5’ss usage in the presence or absence of these compensatory U1 snRNAs, we used a recently established competition splicing assay in which noncanonical5’ss are tested in competition to a set of canonical 5’ss of different strengths (Fig. 3C). Intriguingly, the coexpression of the modified U1 snRNA considerably strengthened the competitive ability of noncanonical 5’ss against their canonical counterpart. Accordingly, we already observed splice sites switching towards GA and CT in competition with the normally superior canonical splice site with an H-Bond Score (HBS) of 12.2 following coexpression of a compensatory U1 snRNA (Fig. 3C, cf. e.g. GA lanes 3 and 4, 12.2 – and +). Importantly, we efficiently redirected splicing towards the test TT splice site by U1 snRNA coexpression – even in presence of the relatively strong, competing 5’ss with an HBS of 15.2 (Fig. 3C, cf. TT lanes 5 and 6, 15.2 – and +). To our surprise, coexpression of an engineered U1 snRNA perfectly matching the GC site stimulated splicing at the competing 5’ss with an HBS of 15.2 (Fig. 3C, cf. GC lanes 5 and 6, 15.2 – and +). It is not clear yet whether this is the result of non-Watson-Crick G•U basepairing between the modified U1 snRNA and the competing 5’ss or stimulation of its usage by upstream U1 snRNA binding (see next section).
Noteworthy, sequencing of the spliced RT-PCR products revealed that coexpression of the compensatory U1 snRNAs did not change the superimposed splicing register. Despite increased splicing efficiencies, we still obtained approximately equal usage of the splicing register −1 and +1 for TT (Fig. 3D). Furthermore, for the CT sequence, coexpression of a compensatory U1 snRNA did not significantly change the ratio of splicing at positions +1 (~75%) and +5 (~25%) (Fig. S1). This supported the idea that initially positioning U1 snRNP did not necessarily define the later cleavage position. This is rather defined by U6 and/or U5 snRNA binding. Also, it argues for U1 snRNP-dependent TT splice site usage and against the idea that flexibility in the U1 basepairing registers was the main reason for the observed splicing at positions −1 and +1.
U1 snRNA binding sites can act as enhancers on nearby splice sites
Our studies indicated that a splicing inactive 5’ss with sufficiently high U1 snRNA complementarity should retain its capacity to interact with the U1 snRNP. In addition, we have previously shown that U1 binding sites that cannot be spliced by a G > C substitution at position +1 maintain their ability to enhance an upstream 3’ss, most likely through stimulation of cross-exon interactions [24]. Strikingly, the U1 snRNP is overrepresented within cells even though it is present in equal stoichiometry with the other U snRNP in spliceosomes, indicating additional functions outside of the spliceosome. Therefore, we addressed the question of whether splicing inactive U1 binding sites might have a general function in splicing regulation analogous to splicing regulatory elements (SREs) that serve as binding sites for SR or hnRNP proteins. Using a previously published large, human RNA-seq transcriptome data set of 54 human fibroblast samples taken from 27 different individuals [23,29], we specifically searched for exon junction reads from used 5’ss with a GT at position +1/+2. From a total set of identified 298,197 5’ss, 269,196 5’ss showed no evidence for the usage of a second 5’ss within their 50 nucleotide-sized downstream region and were used for further analyses. Cumulative support by SREs/the enhancing SRE landscape was defined as the average HEXplorer difference between the 50 nucleotides up- and downstream of a 5’ss [36]. The general capacity to serve as a U1 binding site was estimated by assigning ”Pseudo”-HBond scores (calculated using a fixed GT at position +1/+2) to every 11nt long sequence overlapping within the 50-nucleotides downstream of the used 5’ss that could be summed up to define a ‘Pseudo’-HBond score integral. Strikingly, SRE support and ‘Pseudo’-HBond score integral showed a significant negative correlation (r = −0.27, p < 0.01) (Fig. 4A,B). The higher the SRE support of a 5’ss, the lower the ‘Pseudo’-HBond score integral, indicating that low SRE support might be compensated by a higher affinity of the downstream sequence for the U1 snRNA.
Figure 4.
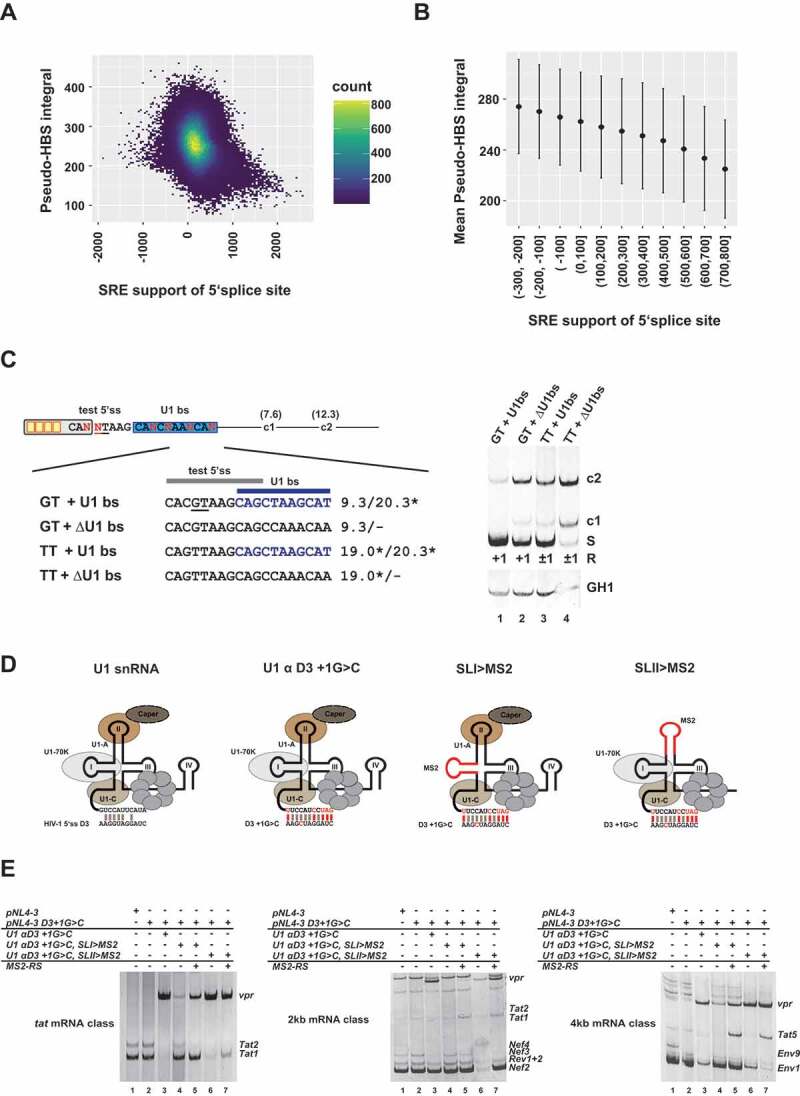
U1 snRNP binding act as splicing enhancer themselves
(A, B) Splicing regulatory element (SRE) support and Pseudo-HBS score integral of 50nt downstream sequence. (A) The density scatter plot illustrates each SRE support/‘Pseudo’-H-Bond score integral (Pseudo-HBS integral) pair grouped in small bins. Yellow colored bins indicate a high number of 5’ss with a specific SRE to Pseudo-HBS integral. (B) Mean Pseudo-HBS integral and standard deviation of SRE support groups. (C) U1 binding sites promote the activation of weak canonical and noncanonical 5’ss. Usage of a weak GT splice site (HBs: 9.30) and a noncanonical TT site (HBs: 19.0*) was tested in the presence or absence of a non-functional U1 binding site (U1 bs, HBs: 20.3*, highlighted in blue). Splicing positions (R) are indicated below.*: H-Bond score had been calculated by inserting a GT at position +1/+2; S: spliced; +1: cleavage at position +1; −1/+1: cleavage at positions −1 and +1; c1 and c2: cryptic splices sites in the downstream intron. (D) Schematic of the U1 snRNA mutants used in (E). (E) RT-PCR analysis show that RS domain binding to stem loop I of the U1 snRNA is important for exon-definition. HEK293T cells were transiently transfected with each of the proviral plasmids and U1 and/or MS2-RS fusion protein-expressing plasmids as indicated above the gel images. HIV-1 mRNA species are indicated to the right of the gel images according to the nomenclature published previously [63].
Our genome-wide search was in line with the concept that splicing inactive U1 binding sites might also acts as splicing enhancers. Indeed, previously it has been shown that tethering of U1 snRNAs to intronic locations can have a positive effect on splice site usage [20–22].
To examine whether U1 snRNA binding sites might positively act on the use of neighbouring 5’ss, we tested the activation of a weak canonical GT (HBS: 9.30) and a noncanonical TT 5’ss in the presence and absence of a downstream canonical GT U1 snRNA binding site (Fig. 4C).
Indeed, we found that the presence of the U1 binding site was linked to increased splicing of the upstream proximal canonical (GT) as well as the noncanonical (TT) 5’ss (Fig. 4C, cf. lanes 1 with 2 or 3 with 4).
Previously, we demonstrated that U1 snRNP binding to a splicing inactive, mutant CT 5’ss flanking HIV-1 exon 3 (D3 + 1 G > C) stimulates activation of the upstream 3’ss A2 used to form vpr-mRNAs by re-establishing exon-definition [24]. This was consistent with a new approach published at that time, which demonstrated that tethering of engineered U1 snRNAs (called Exon-Specific U1 snRNAs (ExspeU1)) to downstream intron positions can be used to correct exon skipping [20]. In the meantime, this technique had successfully been tested to rescue defective exon-definition in spinal muscular atrophy (SMA) [41–43] and several other diseases [44–46].
Since modified U1 snRNAs can be used to activate noncanonical 5’ss and enhance cross-exon interactions, we next wanted to define the molecular features of the U1 snRNA itself, which are critical for U1 snRNP-mediated splicing enhancement. For this purpose, we replaced the U1 snRNA stem loops I (SLI) and II (SLII) of a compensatory U1 snRNA that had been optimized for binding to mutant 5’ss D3 +1G>C (U1 αD3 +1G>C) by a single MS2 binding site (SLI > MS2 and SLII > MS2) (Fig. 4D) [24]. The U1 stem loop mutants were then coexpressed with the proviral mutant clone pNL4-3 D3 +1G>C and an expression plasmid coding for the bacteriophage MS2 coat protein or an MS2-RS domain fusion protein either to mimic U1-70K [17] or U1-A•CAPER [47] dependent RS domain binding to SL I or SL II (Fig. 4D). As it was seen before, a G-to-C substitution at position +1 of 5’ss D3 led to abrogation of exon 3 inclusion (Fig. 4E, cf. 4kb, lanes 1 and 2, Env9). However, vpr-mRNA – formed by activation of A2 – was efficiently induced by coexpression of U1 αD3 + 1 G > C (Fig. 4E, cf. e.g. 2kb, lanes 2 and 3) consistent with earlier findings that U1 binding re-establishes exon-definition [24]. Coexpressing U1 SLI > MS2 alone strongly reduced the shift towards 3’ss A2 as manifested by strongly reduced levels of vpr-mRNAs (Fig. 4E, cf. e.g. tat mRNA class cf. lanes 3 and 4). This indicated that U1-70K binding to SLI is necessary for upstream 3’ss A2 activation and exon-definition. Importantly, coexpression of U1 SLI > MS2 together with an MS2-RS fusion protein partially re-induced 3’ss A2 activation (Fig. 4E, cf. e.g. tat mRNA class lane 4 and 5). This substantiated the idea that U1-70K is required to stimulate exon-spanning interactions for A2 activation as it has been described before [43] and suggested that the U1-70K-dependent enhancement of exon-definition mostly relies on the RS domain. By contrast, and in agreement with Rogalska et al. [43], SLII appeared to be largely dispensable for activation of the upstream 3’ss A2 (Fig. 4E, cf. e.g. 4kb mRNA class lane 3 and 6), suggesting that U1-A was not critical for upstream exon-definition. Surprisingly, coexpression of the MS2-RS fusion protein also promoted activation of the downstream tat-specific 3’ss A3, regardless of whether the RS domain was tethered to stem loop I or II (Fig. 4E, cf. 4kb mRNA class lane 4 and 5 as well as 6 and 7). Taken together, our results showed that U1 snRNA binding sites can act as enhancer on proximal 5’ss as well as upstream 3’ss via the promotion of exon-defining interactions.
Noncanonical splice site usage is enhanced by proximal, canonical splice sites
Previous studies indicated that even significantly weaker canonical GT sequences can improve the use of noncanonical splice sites [23]. In addition, we could confirm here that an U1 snRNA binding site can enhance the recognition and usage of a neighbouring splice site (Fig. 4). To determine whether noncanonical splice site use might be driven by the presence of internal GT sites, we first analysed splicing of two comparably strong noncanonical GC sites (GC#1: 18.70* versus GC#2: 17.70*) but with unequally strong internal GTs at position +5/+6 (GC#1, GT: 9.30 versus GC#2, GT: 14.00) (Fig. 5A). It was found that the presence of a relatively strong GT at position +5/+6 considerably enhanced the splicing of the noncanonical GC site (Fig. 5A) without impacting the final splicing cleavage position (Fig. 5B). Both GC sites were also tested in competition. Although the selection of the enhancer-proximal splice site is normally preferred in a constellation of two competing, equally strong 5’ss ([15], Fig. 5C, lane 1), splicing was shifted to the downstream GC site when it possessed a stronger internal GT site than the upstream one (Fig. 5C, lane 3). This indicated that possession of a stronger internal GT is advantageous for the usage of noncanonical GC sites. By contrast, increasing the complementarities of the internal GTs to moderate intrinsic strength (GTinternal 1: HBS 11.9; GTinternal2: HBS 12.30) within a test TT splice site (Fig. 5D) was already sufficient to entirely abolish splicing at the TT in position +1/+2 (Fig. 5E). This suggested that TT sites, though benefiting from weak GT in their proximity, are outcompeted by canonical GT sites of sufficient strength.
Figure 5.
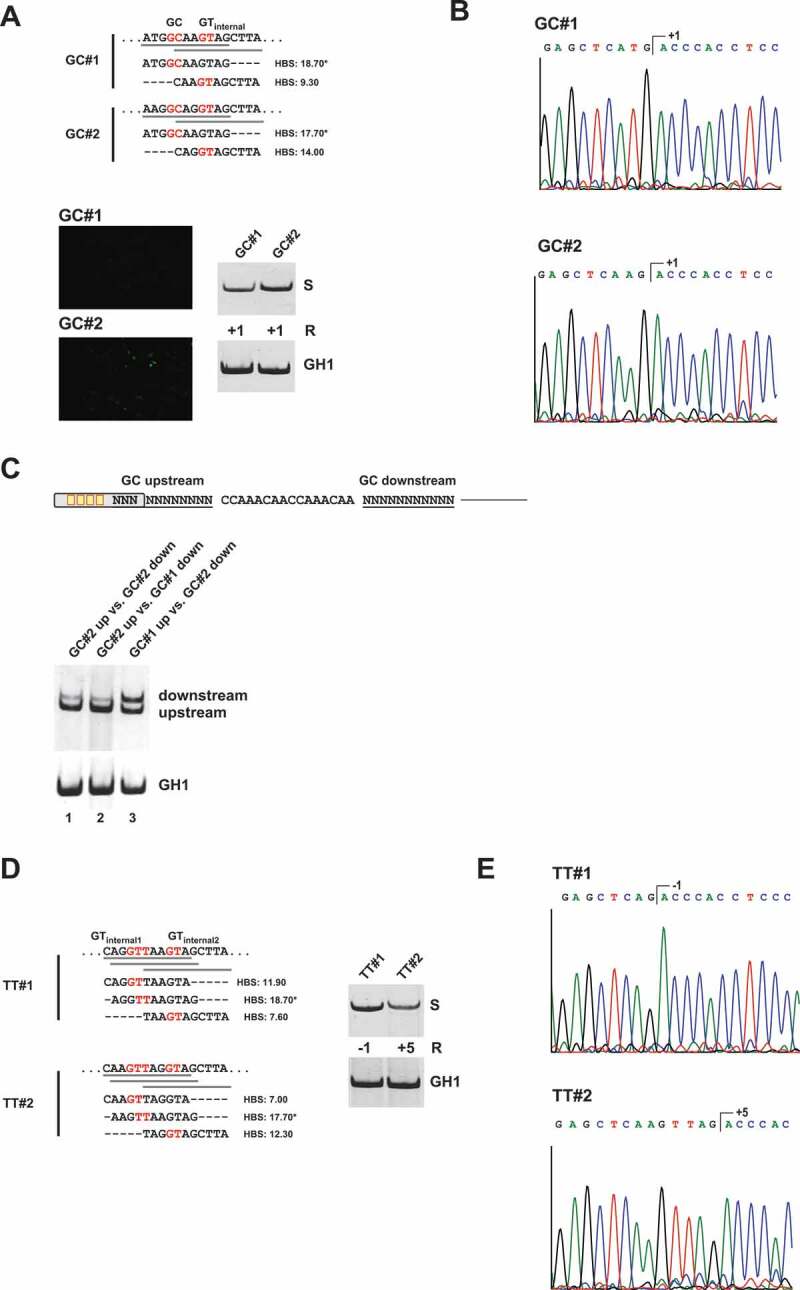
A noncanonical GC splice site benefits from presence of an internal GT site at position +5/+6
(A) Schematic of the HIV-1-based SV-env/eGFP splicing reporter containing different GC splice sites (on top). Sequence variations and H-Bond scores of the GC splice sites and the internal GT sequences at position +5/+6 are shown below. Splicing-dependent eGFP expression was monitored by fluorescence microscopy (see below). RT-PCR analyses of spliced reporter mRNAs were performed as described before. Splicing positions (R) are indicated below. (B) Sequencing results of GC splice site usage shown in (A). Polyacrylamide (PAA) bands were isolated, reamplified with primer pair #3210/#3211 and sent to sequencing analysis using primer #3210. (C) RT-PCR analysis of competing GC splice sites. (D, E) RT-PCR analysis and sequencing results of TT splice sites with variations of the internal GT sequence at position −1/+1 or +5/+6. *H-Bond score had been calculated by inserting a GT at position +1/+2; S: spliced; +1: cleavage at position +1; −1: cleavage at positions −1; +5: cleavage at position +5.
Discussion
Recognition of noncanonical splice sites by the major, U2-type spliceosome
Currently, there is only sparse knowledge about the recognition of noncanonical splice sites. Our previous experiments indicated predominant recognition of noncanonical 5’ss by the U1 snRNP and splicing by the major U2-type spliceosome, as the variation of the U1 snRNA complementarity led to correlative changes in splicing of a noncanonical test TT splice site [23]. In addition, coexpression of compensatory U1 snRNAs was shown to activate the splicing of noncanonical splice sites [12,48]. Our proteomic analysis of proteins interacting with noncanonical GC and TT splice sites as well as our U1-C/SNRPC binding assays also supports these observations. Furthermore, we demonstrated activation of all tested 5’ss, including AT, by compensatory U1 snRNAs. Our findings prompted us to conclude that noncanonical, but otherwise highly complementary 5’ss – when forming hybrid introns together with canonical AGs – might generally be recognized by the U2-type spliceosomes. This idea is further supported by previous studies demonstrating that U12-type introns require concerted recognition of both splice sites [14,49], whereas U2-type splice sites can be independently bound by U1 and U2 snRNP [50–52].
U1 binding sites act as splicing enhancer themselves
A large excess of the U1 snRNP over all other U snRNPs indicates that the U1 snRNP might have additional functions beyond its well-characterized role as an early, integral building block for spliceosome positioning. Interestingly, our and other experiments give reason to believe that one of these functions could play a role as a general enhancer of splice sites. Correspondingly, we found that the presence of U1 binding sites was connected to increased splicing of weak canonical and noncanonical splice sites (Fig. 6). In addition, our transcriptome-wide search indicated that regions of increased affinity for basepairing to U1 snRNA mimic conventional enhancer sequences that are bound by splicing regulatory SR and hnRNP proteins. In fact, previous studies could already demonstrate that targeting of so-called exon-specific U1 snRNAs (ExSpeU1) or engineered U1 snRNAs (eU1s) to intronic locations can be used to correct exon skipping caused by 5’ss, 3’ss or exonic mutations [20–22]. In particular, ExSpeU1s had been demonstrated to enhance splicing in numerous cases [20,41,45,53–59]with an already proven applicability to promote SMN expression in a mouse model of spinal muscular atrophy (SMA) [42]. Although the mechanism by which U1 snRNP binding can stimulate the recognition of nearby splice sites is not clear yet, previous work has suggested the simultaneous binding of multiple U1 snRNPs to RNA [60] and that additional U1 snRNP binding might affect splice site selection through stabilization of SR proteins [20,60]. Interestingly, we found that RS domain tethering (derived from SRSF1) to U1 snRNA stem loop I was sufficient to functionally replace U1-70K and its role in promoting exon-definition across the upstream exon, which was also consistent with findings from a preceding study probing the structural requirements of ExSpeU1s for splicing enhancement [43]. By contrast, stem loop II/U1-A binding turned out to be dispensable for U1 snRNP-mediated enhancement of upstream 3’ss again in accordance with functional splicing assays carried out with mutant ExSpeU1s [43]. These results suggest that physical interactions between RS domains from SR proteins and SR-related proteins associated with the U1 snRNP play a superior role for the activity of bound U1 snRNPs as enhancers of 3’ss activation. However, future experiments need to clarify whether SR protein stabilization is the main reason for U1 snRNP-driven activation of 3ʹ and 5’ss.
Figure 6.
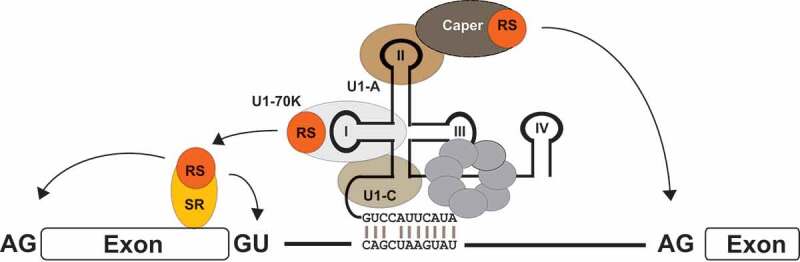
U1 snRNP binding sites serve as splicing enhancers through stabilization of SR protein binding
U1 snRNA binding site can functionally mimic SREs and enhance the usage of neighbouring 3’ss (presumably via RS-domain induced cross-exon interactions) as well as neighbouring 5’ss (by a so far undefined mechanism).
Use of modified, therapeutic U1 snRNAs for the activation of pathological, noncanonical splice sites
Modified U1 snRNAs are widely tested for their potential as a novel therapeutic drug to rescue splicing defects caused by pathological 5’ss mutations (e.g. [42,43,55,61]). However, our studies might disclose a potential shortcoming of modified U1 snRNA expression in therapy: while modified U1 snRNA coexpression can efficiently reactivate splicing, they cannot reset the correct cleavage position depending on the target 5’ss. Here, we found that the coexpression of a compensatory U1 snRNA could not change the mixed usage of the −1/+1 splicing register observed in the context of our test TT 5’ss. This was consistent with the idea that the selection of the cleavage position is determined at a step later than U1 snRNP binding, probably by the U6 and/or the U5 snRNA. Moreover, it argued against flexible U1/5’ss RNA duplex formation that occurs by basepairing with nonbulged or bulged sequence [38,39] as compensatory U1 coexpression increased splicing without changing the relative proportions of −1 and +1 usage.
A further risk could be caused by the apparent role of U1 snRNP as a splicing enhancer, which could lead to the accidental activation of cryptic, weak splice sites in the proximity [21]. These results suggest that modified U1 snRNAs, despite their undisputed therapeutic potential, still require very careful prior verification in individual cases.
Supplementary Material
Acknowledgments
We are grateful to Björn Wefers for excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) under Grant No. SCHA 909/4-1 (to H.S.).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [SCHA 909/4-1].
Disclosure statement
The authors report no conflict of interest.
Authors contributions
HS and SE conceived and designed all experiments and wrote the manuscript. SE performed the vast majority of all experiments. GP carried out the mass spectrometry (MS) analysis. JP conducted the computational, transcriptome-wide search for U1 binding sites.LM systematically compared SRE support for noncanonical TT 5’ss. All authors read and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Nilsen TW, Graveley BR.. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. 7280. doi: 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wahl MC, Will CL, Luhrmann R.. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. [DOI] [PubMed] [Google Scholar]
- [3].Wilkinson ME, Charenton C, Nagai K.. RNA splicing by the spliceosome. Annu Rev Biochem. 2020;89:359–388. [DOI] [PubMed] [Google Scholar]
- [4].Sheth N, Roca X, Hastings ML, et al. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006;34:3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lin JH, Tang XY, Boulling A, et al. First estimate of the scale of canonical 5ʹ splice site GT>GC variants capable of generating wild-type transcripts. Hum Mutat. 2019;40:1856–1873. [DOI] [PubMed] [Google Scholar]
- [6].Hall SL, Padgett RA. Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J Mol Biol. 1994;239:357–365. [DOI] [PubMed] [Google Scholar]
- [7].Hall SL, Padgett RA. Requirement of U12 snRNA for in vivo splicing of a minor class of eukaryotic nuclear pre-mRNA introns. Science. 1996;271:1716–1718. [DOI] [PubMed] [Google Scholar]
- [8].Turunen JJ, Niemela EH, Verma B, et al. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA. 2013;4:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brackenridge S, Wilkie AO, Screaton GR. Efficient use of a ‘dead-end’ GA 5ʹ splice site in the human fibroblast growth factor receptor genes. EMBO J. 2003;22:1620–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Parada GE, Munita R, Cerda CA, et al. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 2014;42:10564–10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Twigg SR, Burns HD, Oldridge M, et al. Conserved use of a non-canonical 5ʹ splice site (/GA) in alternative splicing by fibroblast growth factor receptors 1, 2 and 3. Hum Mol Genet. 1998;7:685–691. [DOI] [PubMed] [Google Scholar]
- [12].Hartmann L, Neveling K, Borkens S, et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet. 2010;87:480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Krawczak M, Thomas NS, Hundrieser B, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. [DOI] [PubMed] [Google Scholar]
- [14].Verma B, Akinyi MV, Norppa AJ, et al. Minor spliceosome and disease. Semin Cell Dev Biol. 2018;79:103–112. [DOI] [PubMed] [Google Scholar]
- [15].Erkelenz S, Mueller WF, Evans MS, et al. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA. 2013a;19:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cho S, Hoang A, Sinha R, et al. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shuai S, Suzuki H, Diaz-Navarro A, et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature. 2019;574:712–716. [DOI] [PubMed] [Google Scholar]
- [19].Suzuki H, Kumar SA, Shuai S, et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. 2019;574:707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fernandez Alanis E, Pinotti M, Dal Mas A, et al. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet. 2012;21:2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Singh NN, Del Rio-Malewski JB, Luo D, et al. Activation of a cryptic 5ʹ splice site reverses the impact of pathogenic splice site mutations in the spinal muscular atrophy gene. Nucleic Acids Res. 2017;45:12214–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Singh RN, Singh NN. A novel role of U1 snRNP: splice site selection from a distance. BBA Gene Regul Mech. 2019;1862:634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Erkelenz S, Theiss S, Kaisers W, et al. Ranking noncanonical 5ʹ splice site usage by genome-wide RNA-seq analysis and splicing reporter assays. Genome Res. 2018;28:1826–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Erkelenz S, Poschmann G, Theiss S, et al. Tra2-mediated recognition of HIV-1 5ʹ splice site D3 as a key factor in the processing of vpr mRNA. J Virol. 2013b;87:2721–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kammler S, Leurs C, Freund M, et al. The sequence complementarity between HIV-1 5ʹ splice site SD4 and U1 snRNA determines the steady-state level of an unstable env pre-mRNA. RNA. 2001;7:421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kammler S, Otte M, Hauber I, et al. The strength of the HIV-1 3ʹ splice sites affects Rev function. Retrovirology. 2006;3:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinf. 2012;13(Suppl 16):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaisers W, Boukamp P, Stark HJ, et al. Age, gender and UV-exposition related effects on gene expression in in vivo aged short term cultivated human dermal fibroblasts. PloS One. 2017;12:e0175657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. [DOI] [PubMed] [Google Scholar]
- [32].Cunningham F, Amode MR, Barrell D, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and bioconductor: a powerful link between biological databases and micro-array data analysis. Bioinformatics. 2005;21:3439–3440. [DOI] [PubMed] [Google Scholar]
- [34].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaisers W, Schaal H, Schwender H. rbamtools: an R interface to samtools enabling fast accumulative tabulation of splicing events over multiple RNA-seq samples. Bioinformatics. 2015;31:1663–1664. [DOI] [PubMed] [Google Scholar]
- [36].Erkelenz S, Theiss S, Otte M, et al. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014;42:10681–10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Roca X, Akerman M, Gaus H, et al. Wide-spread recognition of 5ʹ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012;26:1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tan J, Ho JX, Zhong Z, et al. Noncanonical registers and base pairs in human 5ʹ splice-site selection. Nucleic Acids Res. 2016;44:3908–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roca X, Krainer AR. Recognition of atypical 5ʹ splice sites by shifted base-pairing to U1 snRNA. Nat Struct Mol Biol. 2009;16:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Plaschka C, Lin PC, Charenton C, et al. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dal Mas A, Rogalska ME, Bussani E, et al. Improvement of SMN2 pre-mRNA processing mediated by exon-specific U1 small nuclear RNA. Am J Hum Genet. 2015b;96:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Donadon I, Bussani E, Riccardi F, et al. Rescue of spinal muscular atrophy mouse models with AAV9-exon-specific U1 snRNA. Nucleic Acids Res. 2019;47:7618–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rogalska ME, Tajnik M, Licastro D, et al. Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun. 2016;7:11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dal Mas A, Fortugno P, Donadon I, et al. Exon-specific U1s correct SPINK5 exon 11 skipping caused by a synonymous substitution that affects a bifunctional splicing regulatory element. Hum Mutat. 2015a;36:504–512. [DOI] [PubMed] [Google Scholar]
- [45].Donadon I, Pinotti M, Rajkowska K, et al. Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum Mol Genet. 2018;27:2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tajnik M, Rogalska ME, Bussani E, et al. Molecular basis and therapeutic strategies to rescue factor IX variants that affect splicing and protein function. PLoS Genet. 2016;12:e1006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shao W, Kim HS, Cao Y, et al. A U1-U2 snRNP interaction net-work during intron definition. Mol Cell Biol. 2012;32:470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yanaizu M, Sakai K, Tosaki Y, et al. Small nuclear RNA-mediated modulation of splicing reveals a therapeutic strategy for a TREM2 mutation and its post-transcriptional regulation. Sci Rep. 2018;8:6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Frilander MJ, Steitz JA. Initial recognition of U12-dependent introns requires both U11/5ʹ splice-site and U12/branchpoint interactions. Genes Dev. 1999;13:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Barabino SM, Blencowe BJ, Ryder U, et al. Targeted snRNP depletion reveals an additional role for mammalian U1 snRNP in spliceosome assembly. Cell. 1990;63:293–302. [DOI] [PubMed] [Google Scholar]
- [51].Michaud S, Reed R. A functional association between the 5ʹ and 3ʹ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes Dev. 1993;7:1008–1020. [DOI] [PubMed] [Google Scholar]
- [52].Tarn WY, Steitz JA. Modulation of 5ʹ splice site choice in pre-messenger RNA by two distinct steps. Proc Natl Acad Sci U S A. 1995;92:2504–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Balestra D, Barbon E, Scalet D, et al. Regulation of a strong F9 cryptic 5’ss by intrinsic elements and by combi-nation of tailored U1snRNAs with antisense oligonucleotides. Hum Mol Genet. 2015;24:4809–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Balestra D, Faella A, Margaritis P, et al. An engineered U1 small nuclear RNA rescues splicing-defective coagulation F7 gene expression in mice. J Thromb Haemost. 2014;12(2):177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Balestra D, Giorgio D, Bizzotto M, et al. Splicing mutations impairing CDKL5 expression and activity can be efficiently rescued by U1snRNA-based therapy. Int J Mol Sci. 2019;20(17):4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Balestra D, Scalet D, Ferrarese M, et al. A compensatory U1snRNA partially rescues FAH splicing and protein expression in a splicing-defective mouse model of tyrosinemia type I. Int J Mol Sci. 2020;21:2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Balestra D, Scalet D, Pagani F, et al. An exon-specific U1snRNA induces a robust factor IX activity in mice expressing multiple human FIX splicing mutants. Mol Ther Nucleic Acids. 2016;5:e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Scalet D, Balestra D, Rohban S, et al. Exploring splicing-switching molecules for seckel syndrome therapy. Biochim Biophys Acta Mol Basis Dis. 2017;1863:15–20. [DOI] [PubMed] [Google Scholar]
- [59].Scalet D, Sacchetto C, Bernardi F, et al. The somatic FAH C.1061C>A change counteracts the frequent FAH c.1062+5G>A mutation and permits U1snRNA-based splicing correction. J Hum Genet. 2018;63:683–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hodson MJ, Hudson AJ, Cherny D, et al. The transition in spliceosome assembly from complex E to complex A purges surplus U1 snRNPs from alternative splice sites. Nucleic Acids Res. 2012;40:6850–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Breuel S, Vorm M, Brauer AU, et al. Combining engineered U1 snRNA and antisense oligonucleotides to improve the treatment of a BBS1 splice site mutation. Mol Ther Nucleic Acids. 2019;18:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cavaloc Y, Bourgeois CF, Kister L, et al. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5:468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Purcell DF, Martin MA. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J Virol. 1993;67:6365–6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.