

Abstract
Objective
In a multicenter cohort of probable dementia with Lewy bodies (DLB), we tested the hypothesis that β-amyloid and tau biomarker positivity increases with age, which is modified by APOE genotype and sex, and that there are isolated and synergistic associations with the clinical phenotype.
Methods
We included 417 patients with DLB (age 45–93 years, 31% women). Positivity on β-amyloid (A+) and tau (T+) biomarkers was determined by CSF β-amyloid1-42 and phosphorylated tau in the European cohort and by Pittsburgh compound B and AV-1451 PET in the Mayo Clinic cohort. Patients were stratified into 4 groups: A−T−, A+T−, A−T+, and A+T+.
Results
A−T− was the largest group (39%), followed by A+T− (32%), A+T+ (15%), and A−T+ (13%). The percentage of A−T− decreased with age, and A+ and T+ increased with age in both women and men. A+ increased more in APOE ε4 carriers with age than in noncarriers. A+ was the main predictor of lower cognitive performance when considered together with T+. T+ was associated with a lower frequency of parkinsonism and probable REM sleep behavior disorder. There were no significant interactions between A+ and T+ in relation to the clinical phenotype.
Conclusions
Alzheimer disease pathologic changes are common in DLB and are associated with the clinical phenotype. β-Amyloid is associated with cognitive impairment, and tau pathology is associated with lower frequency of clinical features of DLB. These findings have important implications for diagnosis, prognosis, and disease monitoring, as well as for clinical trials targeting disease-specific proteins in DLB.
Classification of evidence
This study provides Class II evidence that in patients with probable DLB, β-amyloid is associated with lower cognitive performance and tau pathology is associated with lower frequency of clinical features of DLB.
β-Amyloid plaques and tau neurofibrillary tangles (NFT), the pathological hallmarks of Alzheimer disease (AD), are present in >50% of the patients with dementia with Lewy bodies (DLB) at autopsy.1 However, little is known about how these pathologies build up during life, contributing to disease progression and clinical phenotype in DLB.
β-Amyloid plaques and tau NFT contribute to clinical disease severity and accelerate disease progression,1–10 but contradictory findings also exist.3,5,7–9,11–16 A still unresolved and relevant question is whether the contribution of β-amyloid plaques and tau NFT to the clinical phenotype is isolated or synergistic. Some biomarker studies attempted to investigate their joint contribution using the ratio between β-amyloid and phosphorylated tau levels in the CSF.8,17–19 However, total tau CSF levels, a marker of unspecific neurodegeneration, were included in that ratio, hindering our understanding of the specific contributions of β-amyloid and tau biomarkers.
A promising approach is modeling the synergy between β-amyloid and tau biomarkers statistically, which has been challenged by the limited sample size of previous studies. We assembled a large-scale cohort by combining data from the European DLB Consortium (E-DLB)20 and Mayo Clinic16 that consisted of 417 patients with probable DLB. We hypothesized that the frequency of β-amyloid and tau biomarker positivity in patients with probable DLB would be higher with increasing age and in APOE ε4 carriers and that there would be sex differences. We also hypothesized that β-amyloid and tau biomarkers would have isolated and synergistic associations with the clinical phenotype in probable DLB.
Methods
Participants
The multicenter data were a combination of the E-DLB20 and the Mayo Clinic DLB cohort16 from the United States.
The E-DLB consortium is an effort to combine data from 40 clinics across Europe,21 including probable DLB, Parkinson disease with dementia, and AD. For the current study, we included patients with probable DLB from the 10 E-DLB centers that had data on CSF β-amyloid1-42 (Aβ42) and phosphorylated tau biomarkers available. The diagnostic procedure and clinical examinations are described elsewhere.21 Briefly, diagnosis was made according to the 2005 International Consensus Criteria for probable DLB22 and based on detailed history and clinical examinations, including physical, neurologic, and psychiatric examinations performed by a licensed neurologist. Patients with acute delirium, terminal illness, previous stroke, psychotic or bipolar disorder, or craniocerebral trauma or recently diagnosed with a major somatic illness were excluded.
The Mayo Clinic DLB cohort study is a prospective study from the Mayo Clinic sites located at Rochester, MN, and Jacksonville, FL. For the current study, we included consecutive cases assessed through the Mayo Clinic Alzheimer's Disease Research Center, which operates at both Mayo Clinic sites, between May 2015 and April 2019. We selected patients who had a PET scan with the 11C-Pittsburgh compound B (PiB) and 18F-AV-1451 tracers. All patients underwent a medical history review, informant interview, neurologic examination, and neuropsychological assessment.10 For the current study, patients with a diagnosis of probable DLB according to the 2005 International Consensus Criteria22 were included.
Centers from the E-DLB consortium and Mayo Clinic recorded whether patients fulfilled criteria for parkinsonism, visual hallucinations (VH), fluctuating cognition, and a clinical history of probable REM sleep behavior disorder (RBD).21 Presence/absence of clinical features was based on the 2005 International Consensus Criteria for probable DLB22 to allow harmonized diagnosis across all centers because many of the patients were assessed before the 2017 International Consensus Criteria. Polysomnogram confirmation was not required for RBD, although it was available for a proportion of the patients. In the current study, the Mini-Mental State Examination (MMSE) was selected as an outcome measure for global cognition. All these measures, i.e., parkinsonism, VH, fluctuating cognition, probable RBD, and MMSE, were used in our statistical models to investigate the clinical phenotype in probable DLB. In both the E-DLB centers and Mayo Clinic, DLB diagnosis was made by investigators blinded to CSF/PET biomarkers and APOE genotype.
Standard protocol approvals, registrations, and patient consents
Local ethics committee at each E-DLB center and the Mayo Clinic Institutional Review Board approved the study. Informed consent on participation was obtained from all of the patients or an appropriate surrogate according to the Declaration of Helsinki.
β-Amyloid and tau biomarkers
β-Amyloid (A) and tau NFT (T) were measured with CSF biomarkers in E-DLB centers and with PET imaging at Mayo Clinic. The procedures for collection and analysis of CSF and PET data are described elsewhere8,16,23 and briefly detailed in table 1. INNOTEST ELISAs (Fujirebio, Ghent, Belgium) were used for CSF phosphorylated tau in all samples. For CSF Aβ42, INNOTEST ELISAs were also used in all samples except for the 7 patients with DLB from the Stavanger center, for whom ELISA kits from Biosource Europe SA (Nivelles, Belgium) were used instead. All CSF analyses were performed locally following standard routines. CSF Aβ42 and phosphorylated tau values were classified as normal (−) or abnormal (+) with the use of well-established center-specific cut points as described previously.8,18 PET imaging at Mayo Clinic was performed on General Electric PET/CT scanners (General Electric Healthcare; Milwaukee, WI), operating in a 3-dimensional mode. The 11C-PiB scans consisted of four 5-minute dynamic frames acquired from 40 to 60 minutes after injection (596 MBq on average; range 292–729 MBq). For 18F-AV-1451 scans, an IV bolus injection of 370 MBq on average (range 333–407 MBq) was administered, followed by an 80-minute uptake period and a 20-minute scan. All PET images were visually inspected for technical quality. Details on PET data preprocessing are described fully in previous publications.16 Briefly, PET images were rigidly aligned to individuals' T1-weighted MRIs using SPM12 for the segmentation of gray and white matter.24 The global cortical PiB retention standardized uptake value ratio (SUVr) was obtained from the bilateral parietal, temporal, prefrontal, orbitofrontal, and anterior cingulate regions referenced to the cerebellum region.24 The AV-1451 SUVr was obtained from the bilateral amygdala, entorhinal cortex, fusiform, parahippocampal, and inferior temporal and middle temporal gyri referenced to the median value of the right and left cerebellar crus uptake.25 SUVr values were classified as normal (−) or abnormal (+) using a cut point of ≥1.48 for PiB and a cut point of ≥1.33 for AV-1451, as previously described.23
Table 1.
Methods for CSF biomarkers across E-DLB centers

Statistical analysis
Patients with probable DLB were classified as β-amyloid positive (A+) or negative (A−) and tau positive (T+) or negative (T−). By combining the A and T biomarkers, we could further classify patients with probable DLB into 4 groups as follows: negative β-amyloid and tau biomarkers (A−T−), positive β-amyloid and negative tau biomarkers (A+T−), negative β-amyloid and positive tau biomarkers (A−T+), and positive β-amyloid and tau biomarkers (A+T+). Following the current consensus, A+T− is interpreted as Alzheimer pathologic change and A+T+ as overlapping AD in patients with probable DLB.26
We first studied the demographic and clinical characteristics of the AT groups using means and SDs for continuous variables and counts and percentages for categorical variables. For each dependent variable (demographic and clinical characteristics), we analyzed differences related to AT group through independent mixed-effect linear or logistic regression models. We included AT group as a fixed-effect variable and center as a random variable, and we adjusted for age and sex as appropriate. The specific association of A and T with outcome variables and the interaction between them were investigated through separate models as described in the next paragraph. We assessed heterogeneity in the AT groups by center by comparing nested models: the above restricted model with AT groups and centers and a more general model with AT groups and AT groups within centers. The more general model allows each center to have different AT group effects. A significant p value would indicate significant heterogeneity in the AT groups by center (heterogeneity p values were not significant). We used the same approach to compare women and men and APOE ε4 noncarriers and carriers and to test for age associations. We assessed assumptions in the linear mixed models by examining normality and constant variance of the residuals, linearity of associations with continuous predictors, and influence of sites.27
To investigate isolated and synergistic contributions of β-amyloid and tau biomarkers to the clinical phenotype of probable DLB, we again studied demographic and clinical characteristics for A and for T groups using means and SDs for continuous variables and counts and percentages for categorical variables. For each dependent variable (demographic and clinical characteristics), we treated A and T in a factorial modeling approach by fitting A, T, and A × T interactions in the mixed-effect models while adjusting for age and sex as appropriate and including center as a random variable. We tested for heterogeneity using nested models. We estimated the predicted percentages of the AT groups by age using multinomial models with smooth curves. A value of p < 0.05 (2 tailed) was deemed significant in all these analyses.
Data availability
The E-DLB consortium and Mayo Clinic make data available to qualified researchers on reasonable request.
Results
Positivity of β-amyloid and tau biomarkers
From 1,208 patients with probable DLB included in the original E-DLB cohort21 and 71 patients with probable DLB in the Mayo Clinic cohort, 367 and 50 patients with probable DLB were eligible for the current study, respectively. Table 2 shows the demographic and clinical characteristics of the 4 AT groups. A−T− was the largest group, including 39% of the patients with probable DLB, followed by A+T−, which included 32% of the patients (figure 1A). T+ was less common and included <30% of the patients (A−T+ 13%, A+T+ 15%). This distribution was rather similar across centers (figure 1B). Complementary analyses showed that the distribution of AT groups in the PET center at the Mayo Clinic did not differ statistically from that in all CSF centers pooled together (p > 0.05). Figure 1C shows the AT groups with the deviation of each individual from cut points specific to center and biomarker modality (CSF or PET).
Table 2.
Demographic and clinical characteristics of the AT groups

Figure 1. Percentage of the AT groups (A) in the whole sample and (B) by center and (C) deviation from cut points.
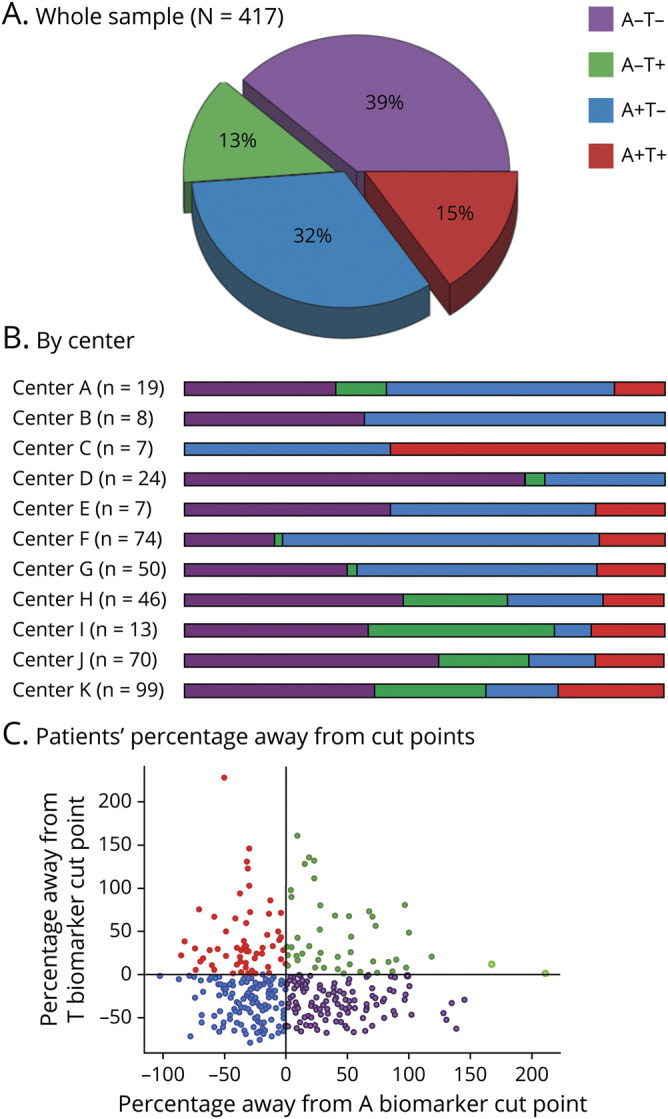
Center A = Fundació ACE Barcelona (Spain); Center B = University G d'Annunzio of Chieti-Pescara (Italy); Center C = Center for Age-Related Medicine, Stavanger University Hospital, Stavanger (Norway); Center D = Memory Clinic, Karolinska University Hospital, Huddinge (Sweden); Center E = University of Ljubljana (Slovenia); Center F = Clinical Memory Research Unit, Lund University (Sweden); Center G = Mayo Clinic at Rochester, MN (United States); Center H = Paracelsus-Elena-Klinik, Kassel (Germany); Center I = Motol University Hospital, Prague (Czech Republic); Center J = Neuropsychology Unit and Geriatric Day Hospital, University Hospital of Strasbourg, Strasbourg (France); Center K = VUMC Amsterdam (the Netherlands). (C) AT groups with the deviation of each individual from cut points (zero value), specific to center and biomarker modality (CSF or PET). A = β-amyloid; T = tau; + = abnormal values; − = normal values.
The distribution of AT groups was strongly modulated by age (p < 0.001). Figure 2A shows that the percentage of groups with an A+ biomarker doubled with older age (8% in A+T+ and 19% in A+T− at age 55 years vs 25% in A+T+ and 45% in A+T− at age 85 years). In contrast, the percentage of the A−T− group decreased to a third with older age (61% at age 55 years vs 20% at age 85 years). The percentage of the A−T+ group was relatively comparable across ages.
Figure 2. Predicted percentages of AT groups by age.

A = β-amyloid; T = tau; + = abnormal values; − = normal values.
Distribution of the AT groups also varied according to sex (p = 0.017) and APOE ε4 status (p < 0.001) (figure 3). A+T+ included a lower percentage of men (55%) than groups with an A− biomarker (A−T− and A−T+, both 75%) (table 2). APOE ε4 carriers were more common in groups with an A+ or T+ biomarker (A+T− 59%, A−T+ 50%, A+T+ 64%) than in the A−T− group (28%) (table 2). When plotting sex and APOE vs age, we observed that the percentage of A+T− and A+T+ seemed to increase in parallel with age in both men and women (figure 2B). In APOE ε4 carriers, we observed that the A+ groups achieved higher percentages at younger ages than in APOE ε4 noncarriers, which considerably reduced the percentage of A− groups over the age of 70 years in APOE ε4 carriers (figure 2C).
Figure 3. Percentage of AT groups by sex and APOE ε4 status.
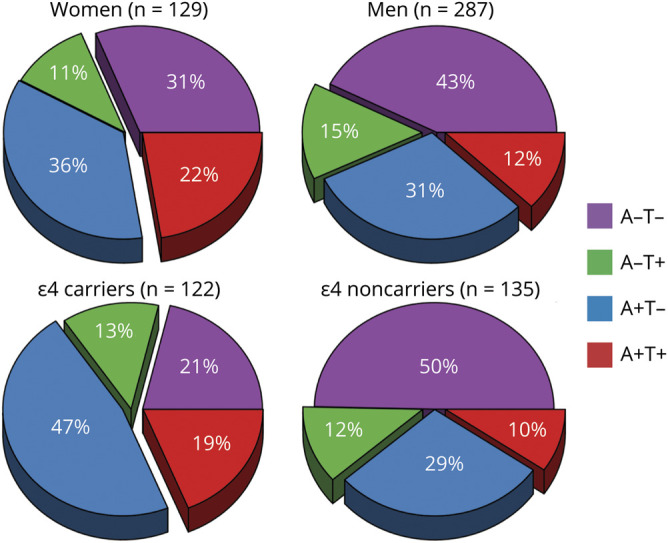
A = β-amyloid; T = tau; + = abnormal values; − = normal values.
Table 3 shows the demographic and clinical characteristics by sex and APOE. After controlling for age and center, women were older, more likely to have VH, and less likely to have parkinsonism (p < 0.05). After controlling for age, sex, and center, there were no differences in clinical characteristics between APOE ε4 carriers and noncarriers. None of the tests for heterogeneity across centers were statistically significant except for MMSE score by APOE ε4 carrier status (p = 0.02). After controlling for center, age had no association with APOE ε4 status, education, MMSE score, or clinical features of probable DLB.
Table 3.
Sex- and APOE-related differences

Isolated and synergistic contributions of β-amyloid and tau biomarkers
Table 4 shows the isolated association of A+ and T+ biomarkers with outcome variables. These data, together with those reported for the 4 AT groups in table 2, show that groups with 1 or 2 positive biomarkers were older (p < 0.001) and had lower MMSE scores (p < 0.05) compared with the A−T− group. The differences in MMSE score were independent of age and were associated with both the A+ and T+ biomarker positivity, and no significant interaction between A+ and T+ was found (p = 0.67). Furthermore, only A+ remained statistically significant (p < 0.001) when included together with T+ (p = 0.094) in the same model. This pattern could be due partially to increased likelihood of T+ in A+ patients, that is, due to multicollinearity in A+ and T+. APOE ε4 carriers were more common in A+ than in A− patients with probable DLB (61% vs 34%, p < 0.001). In addition, patients with an A+ biomarker were less likely to be male (p < 0.05).
Table 4.
Isolated contributions of β-amyloid and tau biomarkers

Regarding the clinical features, patients with a T+ biomarker were less likely to have parkinsonism (p = 0.007) or RBD (p = 0.006) and tended to have fewer cognitive fluctuations (p = 0.053) (table 4). A+ did not have any significant association with the clinical features. Furthermore, no significant interaction between A+ and T+ was found in relation to demographic and clinical variables, nor did we find any heterogeneity by center.
Discussion
There is currently a significant interest in the biological interplay between α-synuclein, β-amyloid plaques, and tau NFT, as well as in the contribution of these proteins to the clinical phenotype in DLB.28,29 The 3 pathologies often coexist at autopsy, which suggests a synergistic contribution of α-synuclein, β-amyloid plaques, and tau NFT to the pathogenesis in DLB.28,29 This synergistic contribution is also supported by animal and cell research.28,29 However, how β-amyloid plaques and tau NFT build up during life, contributing to disease progression and clinical phenotype in DLB, cannot be addressed in postmortem or animal and cell studies. We tackled this question in vivo using tau and β-amyloid biomarkers in the largest probable DLB cohort with tau and β-amyloid biomarkers to date. More specifically, we investigated the frequency of β-amyloid and tau biomarkers across a wide age span using cross-sectional data and tested for the association of β-amyloid and tau biomarkers with MMSE score and clinical features of probable DLB.
We found that A−T− was the largest group, but its percentage decreased substantially from ≈60% at age 55 years to ≈20% at age 85 years. In turn, A+T− and A+T+ groups were more common by age 85 years. This finding converges with postmortem studies showing that the presence of AD pathology increases with age in Lewy body disease.30 Our study extends the finding of previous post-mortem studies by demonstrating the frequency of β-amyloid and tau biomarkers and several influencing factors in patients with probable DLB during life across a wide age range. In this context, we interpret A+T− as an Alzheimer pathologic change and A+T+ as AD,26 that is, AD likely concomitant to Lewy body disease pathology in this cohort.
The current study further improves our understanding of the association between β-amyloid and tau biomarkers in probable DLB during life. In particular, the role of NFT tau pathology in DLB pathogenesis and whether NFT tau and β-amyloid pathologies interact in their contribution to the clinical phenotype are unclear in the previous literature. First, while a correlation between β-amyloid and tau pathology has been reported in patients with DLB in both postmortem1,10,31 and PET studies,16 that correlation is not consistently demonstrated in studies across the Lewy body disease spectrum.5,9 Second, AV-1451 uptake on PET imaging differs in probable DLB from AD in regional distribution.16 Third, tau biomarkers can be abnormal in the absence of β-amyloid pathology in patients with probable DLB.6,9,16 In our study, the proportion of patients with isolated tau biomarker positivity (A−T+, 13%) is comparable to the proportion of patients with both β-amyloid and tau biomarker positivity (A+T+, 15%). Because the percentage of A−T+ remained essentially stable with increasing age but the percentage of A+T+ seemed to increase, paralleling the slope of isolated β-amyloid pathology (A+T−), our data suggest that tau pathology may be independent of the increasing β-amyloid pathology with age in ≈15% of patients with probable DLB (i.e., A−T+). Therefore, in a proportion of patients with probable DLB, tau pathology alone may be a distinct form of copathology accruing in synergy with α-synuclein pathology, as previously noted in postmortem data and cell models.29 In contrast, our data show that in another 15% of patients with probable DLB tau pathology occurs along with β-amyloid pathology (i.e., A+T+; AD) and perhaps downstream to β-amyloid pathology with tau biomarkers becoming positive at older ages in A+ patients with probable DLB. In our study, we used cut points for tau biomarkers. While this is a popular approach that aligns with the ATN biomarker scheme32 and favors clinical interpretation and pooling of data across centers, we did not investigate the full range of tau pathology. Investigating the full range of tau pathology may be important to better understand the extent of tau pathology in individuals with minimal accumulation of β-amyloid pathology who may be at the borderline for A− and T+. Previous studies showed that the magnitude of tau biomarkers in probable DLB is much less than that observed in AD.16
The positivity of β-amyloid biomarkers was more common in APOE ε4 carriers, especially at older ages. We show that the frequency of A+ reaches higher levels at younger ages in APOE ε4 carriers than noncarriers. Whether this increase in A+ percentage in APOE ε4 carriers is a direct influence of APOE ε4 on β-amyloid pathology or is indirectly through an influence on Lewy body pathology needs to be elucidated further because postmortem data suggest that APOE ε4 is also associated with a greater severity of Lewy body pathology independently of β-amyloid and tau pathology.33
A+T+ was more common in women than men compared with the A−T− and A−T+ groups. The basis for this sex difference in the frequency of AD pathology in probable DLB needs further investigation, but this finding may have important implications for the individualized interventions targeting specific proteins in patients with probable DLB.
Regarding the association of β-amyloid and tau pathology with clinical features of probable DLB, the literature is scarce and contradictory. A postmortem study showed that the presence of tau NFT is associated with a lower percentage of VH but not of parkinsonism.34 Another postmortem study showed that the presence of tau NFT but not β-amyloid plaques is associated with a lower percentage of clinical features of DLB.35 PET studies suggest that β-amyloid and tau biomarkers in isolation do not seem to be associated with VH, cognitive fluctuations, parkinsonism, or probable RBD.4,5,14–16 When combined with total tau CSF levels, an abnormal β-amyloid biomarker does not seem to be associated with parkinsonism but is associated with a lower percentage of VH.11,17 In addition, a recent study from the E-DLB Consortium showed that abnormal CSF phosphorylated tau levels were associated with a lower percentage of parkinsonism.19 In the current study, we found that A+ was the main predictor of lower cognitive performance and that T+ was associated with the clinical features: T+ was associated with a lower percentage of parkinsonism and probable RBD and tended to be associated with a lower percentage of cognitive fluctuations. The association of β-amyloid and tau biomarkers with MMSE is a frequent finding in probable DLB,2,4,6–9 although no association has also been reported.3,5,7–9,11–13,15,16 Given the contradictory results in the previous literature, by investigating both CSF and PET biomarkers across a wide age range in a large cohort, our current study may help to clarify the associations of β-amyloid and tau biomarkers with the clinical phenotype in probable DLB in vivo.
The synergistic contribution of β-amyloid and tau pathology to the clinical phenotype of probable DLB has been addressed by investigating the ratio between β-amyloid and phosphorylated tau CSF levels.8,17–19 However, total tau CSF levels, a marker of nonspecific neurodegeneration, are often included in that ratio, which makes it impossible to determine whether the observed association is related to β-amyloid and tau pathology or to neurodegeneration. For instance, β-amyloid and phosphorylated tau CSF levels do not seem to increase mortality risk in isolation,13 whereas total tau CSF levels were reported to increase mortality risk.13 Hence, β-amyloid CSF levels predict mortality risk when combined with total tau CSF levels.11 To overcome this, we applied linear mixed models to test for the statistical interaction between β-amyloid and tau biomarkers on the clinical phenotype. We did not find a significant interaction between β-amyloid and tau biomarkers in relation to global cognitive performance (MMSE score), VH, cognitive fluctuations, parkinsonism, or probable RBD. Therefore, our findings suggest that although β-amyloid plaques and tau NFT often coexist in probable DLB, they have different contributions to the clinical phenotype.
Together, our results demonstrated that β-amyloid and tau biomarkers do not interact with each other when it comes to explaining the presence of clinical features or cognitive performance. β-Amyloid pathology was the main predictor of lower cognitive performance, and only tau pathology was associated with the clinical features, with T+ patients with probable DLB showing a lower likelihood of parkinsonism and probable RBD. Hence, with regard to the notion that β-amyloid and tau pathology may contribute to the timing of dementia onset relative to parkinsonism in Lewy body diseases,28 our cross-sectional analyses suggest that this timing may be partially explained by β-amyloid leading to earlier dementia onset and by tau pathology decreasing (or delaying the onset of) the signs of parkinsonism. How the interaction among α-synuclein, β-amyloid, and tau pathology contributes to the timing of clinical features of DLB is a relevant question that can be elucidated only with the availability of in vivo α-synuclein biomarkers.
Multicenter studies have the added value of increased statistical power and ability to generalize the findings. However, multicenter studies may also introduce methodologic variation in data collection across centers. In this study, several strategies were conducted to minimize variation and inconsistencies across centers. First, clinical methods were standard and comparable across centers, aligning with international guidelines and diagnostic criteria. All centers conducted detailed history and clinical examinations by licensed neurologists. Although we do not have an estimation of intercenter reliability for CSF biomarkers, CSF methods were largely comparable across centers, as shown in table 1. Second, well-established center-specific cut points were used to determine biomarker abnormality. Figure 1C shows that variation in cut points is expected to have a minimal impact on individuals' classification into AT groups. Third, all our statistical models accounted for potential residual heterogeneity in our data by including the center as a random variable. All of these strategies may have reduced the heterogeneity of the data across centers because our heterogeneity analyses were nonsignificant except for the association of APOE ε4 genotype with MMSE score. We determined abnormality in A and T biomarkers by using cut points derived from AD research. However, the levels of clinically relevant β-amyloid and tau pathology and corresponding cut points in DLB may be distinct from those in AD. Still, the literature to validate cut points for A and T biomarkers in DLB is very limited. We recently proposed an autopsy-validated cut point for PiB PET in DLB in the Mayo Clinic cohort,36 but such a validation at present is not available for the E-DLB centers or for T biomarkers. The academic centers included in our study enrolled participants through specialized memory clinics and movement disorder clinics, so the reported frequencies of abnormal A and T biomarkers may not be entirely representative of the patients with DLB in the general population. Combining PET and CSF data may also be a limitation. The concordance between A biomarkers measured in CSF and PET is good in AD,37,38 but little is known about their concordance in DLB. The literature on T biomarkers is very scarce, but there is an association between T biomarkers measured in CSF and PET in AD,39 and a recent study expanded on the concordance between these 2 biomarker modalities.40 Because there is no previous literature on the concordance between CSF and PET biomarkers in patients with DLB, we investigated Alzheimer’s Disease Neuroimaging Initiative cases with a pathologic diagnosis of Lewy body disease.41 We observed a perfect concordance between A biomarkers measured in CSF and PET (n = 8). This finding is reassuring for combining PET and CSF data for A positivity in DLB. PET biomarkers of T, however, were not available in this Alzheimer’s Disease Neuroimaging Initiative subsample. Another reassuring finding is that the distribution of AT groups in the PET center did not differ statistically significantly from CSF centers pooled together, and the heterogeneity analyses did not reveal any deviation of the PET center. Nonetheless, the concordance between PET and CSF biomarkers of A and T is a prospect for the future and will help us understand the combinability of biomarker modalities in DLB. Another limitation is that global cognition was measured with the MMSE due to availability in all the centers. Although, cross-sectional data suggested that the Montreal Cognitive Assessment is more sensitive than the MMSE in detecting cognitive impairment in patients with probable DLB,42 our longitudinal data using the E-DLB cohort showed that both the MMSE and Montreal Cognitive Assessment are comparable in measuring cognitive decline over time.43 Future studies should investigate the potential synergistic contribution of β-amyloid and tau biomarkers to specific cognitive domains. Due to the multicenter nature of this study, we focused on core clinical features of DLB, but investigating supportive clinical features such as sensitivity to antipsychotics is warranted in future studies. Finally, large cohorts of patients with probable DLB including longitudinal biomarker data are nonexistent at present.44 We estimated percentages of β-amyloid and tau biomarkers from cross-sectional data across age, but initiatives at the global scale need to be conducted in the future to substantiate our findings with large longitudinal biomarker datasets.
This study provides data for the occurrence of β-amyloid and tau pathology in probable DLB during life from a multinational cohort, as well as their impact on the clinical phenotype. Our data from multiple sites across Europe and the United States indicate that β-amyloid pathology is common in probable DLB, increases with age, and contributes to global cognitive impairment. Tau pathology is relatively less common, and when present, it seems to influence Lewy body disease clinical phenotype with a lower frequency of clinical features of probable DLB. β-Amyloid co-occurs with tau pathology in 15% of patients with probable DLB. Hence, 15% of patients with probable DLB fulfill the biological definition of AD,26 most likely with mixed Lewy body disease pathology. These data may help to advance our ability to distinguish the clinical phenotype associated with concomitant AD pathology in patients with probable DLB,29 with potential implications for diagnosis, prognosis, and disease monitoring, as well as for clinical trials targeting disease-specific proteins in DLB.
Acknowledgment
The authors thank AVID Radiopharmaceuticals, Inc, for provision of the AV-1451 precursor, chemistry production advice and oversight, and Food and Drug Administration regulatory cross-filing permission and documentation needed for this work. They particularly thank the patients and their family members for participating in this research.
Glossary
- Aβ42
β-amyloid1-42
- AD
Alzheimer disease
- DLB
dementia with Lewy bodies
- E-DLB
European DLB Consortium
- MMSE
Mini-Mental State Examination
- NFG
neurofibrillary tangles
- PiB
Pittsburgh compound B
- RBD
REM sleep behavior disorder
- SUVr
standardized uptake value ratio
- VH
visual hallucinations
Appendix. Authors
Footnotes
Study funding
This study was supported by the NIH (U01- NS100620, P50-AG016574, U01-AG006786, R37-AG011378, R01-AG041851, R01-AG040042, C06-RR018898 and R01-NS080820), the Foundation Dr. Corinne Schuler, the Mangurian Foundation for Lewy Body Research, the Elsie and Marvin Dekelboum Family Foundation, the Little Family Foundation, the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program, the Western Norway Regional Health Authority, Karolinska Institutet travel grants, and the Foundation for Geriatric Diseases at Karolinska Institutet.
Disclosure
D. Ferreira, S.A. Przybelski, T.G. Lesnick, A.W. Lemstra, E. Londos, F. Blanc, Z. Nedelska, and C.G. Schwarz report no disclosures relevant to the manuscript. J. Graff-Radford receives research support from the NIH. M.L. Senjem, J.A. Fields, D.S. Knopman, and R. Savica report no disclosures relevant to the manuscript. T.J. Ferman receives funding from the Mangurian Foundation for Lewy Body Research and the NIH. N.R. Graff-Radford reports no disclosures relevant to the manuscript. V.J. Lowe serves as a consultant for Bayer Schering Pharma, Philips Molecular Imaging, Piramal Imaging, and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (National Institute on Aging, National Cancer Institute), and the MN Partnership for Biotechnology and Medical Genomics. C.R. Jack has consulted for Lily and serves on an independent data monitoring board for Roche and as a speaker for Eisai, but he receives no personal compensation from any commercial entity. He receives research support from the NIH and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic. R.C. Petersen, B. Mollenhauer, S. Garcia-Ptacek, C. Abdelnour, J. Hort, L. Bonanni, K. Oppedal, and M.G. Kramberger report no disclosures relevant to the manuscript. B.F. Boeve has served as an investigator for clinical trials sponsored by Biogen and Alector. He receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge Medicine, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from the NIH, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, and the Little Family Foundation. D. Aarsland has received research support and/or honoraria from AstraZeneca, H. Lundbeck, Novartis Pharmaceuticals, and GE Health and served as paid consultant for H. Lundbeck, Eisai, and Evonik. E. Westman reports no disclosures relevant to the manuscript. K. Kantarci serves on the data safety monitoring board for Takeda Global Research and Development Center, Inc and receives research support from Avid Radiopharmaceuticals and Eli Lilly and funding from the NIH and Alzheimer's Drug Discovery Foundation. Go to Neurology.org/N for full disclosures.
References
- 1.Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 2017;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoonenboom NS, Reesink FE, Verwey NA, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology 2012;78:47–54. [DOI] [PubMed] [Google Scholar]
- 3.Sarro L, Senjem ML, Lundt ES, et al. Amyloid-β deposition and regional grey matter atrophy rates in dementia with Lewy bodies. Brain 2016;139:2740–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomperts SN, Locascio JJ, Marquie M, et al. Brain amyloid and cognition in Lewy body diseases. Mov Disord 2012;27:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SH, Cho H, Choi JY, et al. Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord 2018;33:262–272. [DOI] [PubMed] [Google Scholar]
- 6.Bousiges O, Cretin B, Lavaux T, et al. Diagnostic value of cerebrospinal fluid biomarkers (phospho-tau181, total-tau, Aβ42, and Aβ40) in prodromal stage of Alzheimer's disease and dementia with Lewy bodies. J Alzheimers Dis 2016;51:1069–1083. [DOI] [PubMed] [Google Scholar]
- 7.Reesink FE, Lemstra AW, van Dijk KD, et al. CSF α-synuclein does not discriminate dementia with Lewy bodies from Alzheimer's disease. J Alzheimers Dis 2010;22:87–95. [DOI] [PubMed] [Google Scholar]
- 8.Abdelnour C, van Steenoven I, Londos E, et al. Alzheimer's disease cerebrospinal fluid biomarkers predict cognitive decline in Lewy body dementia patients. Mov Disord 2016;31:1203–1208. [DOI] [PubMed] [Google Scholar]
- 9.Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 2016;73:1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferman TJ, Aoki N, Crook JE, et al. The limbic and neocortical contribution of α-synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimers Dement 2018;14:330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemstra AW, de Beer MH, Teunissen CE, et al. Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2017;88:113–118. [DOI] [PubMed] [Google Scholar]
- 12.Parnetti L, Tiraboschi P, Lanari A, et al. Cerebrospinal fluid biomarkers in Parkinson's disease with dementia and dementia with Lewy bodies. Biol Psychiatry 2008;64:850–855. [DOI] [PubMed] [Google Scholar]
- 13.Boström F, Hansson O, Blennow K, et al. Cerebrospinal fluid total tau is associated with shorter survival in dementia with Lewy bodies. Dement Geriatr Cogn Disord 2009;28:314–319. [DOI] [PubMed] [Google Scholar]
- 14.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging 2012;33:2091–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graff-radford J, Murray ME, Lowe VJ, et al. Dementia with Lewy bodies: basis of cingulate island sign. Neurology 2014;83:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kantarci K, Lowe VJ, Boeve BF, et al. AV-1451 tau and β-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017;81:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Zande JJ, Steenwijk MD, ten Kate M, Wattjes MP, Scheltens P, Lemstra AW. Gray matter atrophy in dementia with Lewy bodies with and without concomitant Alzheimer's disease pathology. Neurobiol Aging 2018;71:171–178. [DOI] [PubMed] [Google Scholar]
- 18.van Steenoven I, Aarsland D, Weintraub D, et al. Cerebrospinal fluid Alzheimer's disease biomarkers across the spectrum of Lewy body diseases: results from a large multicenter cohort. J Alzheimers Dis 2016;54:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Censo R, Abdelnour C, Blanc F, et al. CSF tau proteins correlate with an atypical clinical presentation in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2020;91:109–110. [DOI] [PubMed] [Google Scholar]
- 20.Oppedal K, Ferreira D, Cavallin L, et al. A signature pattern of cortical atrophy in dementia with Lewy bodies: a study on 333 patients from the European DLB Consortium. Alzheimers Dement 2019;15:400–409. [DOI] [PubMed] [Google Scholar]
- 21.Kramberger MG, Auestad B, Garcia-Ptacek S, et al. Long-term cognitive decline in dementia with Lewy bodies in a large multicenter, international cohort. J Alzheimers Dis 2017;57:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 23.Jack CR, Wiste HJ, Botha H, et al. . The bivariate distribution of amyloid-β and tau: relationship with established neurocognitive clinical syndromes. Brain 2019;142:3230–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CRJ, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowe VJ, Kemp BJ, Jack CR, et al. . Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med 2009;50:878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fox J, Weisberg S. An R Companion to Applied Regression, 3rd ed. Thousand Oaks: Sage; 2019. Available at: socialsciences.mcmaster.ca/jfox/Books/Companion/. Accessed December 17, 2019. [Google Scholar]
- 28.Jellinger KA. Dementia with Lewy bodies and Parkinson's disease-dementia: current concepts and controversies. J Neural Transm (Vienna) 2018;125:615–650. [DOI] [PubMed] [Google Scholar]
- 29.Irwin DJ, Hurtig HI. The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimers Dis Parkinsonism 2018;8: 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol 2010;119:421–433. [DOI] [PubMed] [Google Scholar]
- 31.Walker L, McAleese KE, Thomas AJ, et al. Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 2015;129:729–748. [DOI] [PubMed] [Google Scholar]
- 32.Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dickson DW, Heckman MG, Murray ME, et al. APOE e4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 2018;91:e1182–e1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merdes AR, Hansen LA, Jeste DV, et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 2003;60:1586–1590. [DOI] [PubMed] [Google Scholar]
- 35.Tiraboschi P, Attems J, Thomas A, et al. Clinicians' ability to diagnose dementia with Lewy bodies is not affected by β-amyloid load. Neurology 2015;84:496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kantarci K, Lowe VJ, Chen Q, et al. β-Amyloid PET and neuropathology in dementia with Lewy bodies. Neurology 2020;94:e282–e291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 2018;14:1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doecke JD, Rembach A, Villemagne VL, et al. Concordance between cerebrospinal fluid biomarkers with Alzheimer's disease pathology between three independent assay platforms. J Alzheimers Dis 2018;61:169–183. [DOI] [PubMed] [Google Scholar]
- 39.Leuzy A, Chiotis K, Lemoine L, et al. Tau PET imaging in neurodegenerative tauopathies: still a challenge. Mol Psychiatry 2019;24:1112–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer PF, Binette AP, Gonneaud J, Breitner JCS, Villeneuve S. Characterization of Alzheimer disease biomarker discrepancies using cerebrospinal fluid phosphorylated tau and AV1451 positron emission tomography. JAMA Neurol 2019;77:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toledo JB, Cairns NJ, Da X, et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol Commun 2013;1:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang CS, Pai MC, Chen PL, Hou NT, Chien PF, Huang YC. Montreal Cognitive Assessment and Mini-Mental State Examination performance in patients with mild-to-moderate dementia with Lewy bodies, Alzheimer's disease, and normal participants in Taiwan. Int Psychogeriatr 2013;25:1839–1848. [DOI] [PubMed] [Google Scholar]
- 43.Biundo R, Weis L, Bostantjopoulou S, et al. MMSE and MoCA in Parkinson's disease and dementia with Lewy bodies: a multicenter 1-year follow-up study. J Neural Transm (Vienna) 2016;123:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oppedal K, Borda MG, Ferreira D, Westman E, Aarsland D. European DLB consortium: diagnostic and prognostic biomarkers in dementia with Lewy bodies, a multicenter international initiative. Neurodegener Dis Manag 2019;9:247–250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The E-DLB consortium and Mayo Clinic make data available to qualified researchers on reasonable request.