

Untreated HIV-1-infected individuals who durably control HIV-1 replication below detection thresholds of commercial viral load assays (here termed “elite controllers”, ECs) may represent the closest possible approximation to a natural cure of HIV-1 infection1. Previous studies have linked elite HIV-1 control to specific variations of the human HLA class I gene locus2, and to the presence of highly-functional cellular immune responses3 with stronger abilities to kill virally-infected cells3, target mutationally-constrained epitopes4 and limit viral escape5. Although the persistence of small, replication-competent proviral reservoirs has been documented in ECs6,7, the characteristics and possible distinguishing features of reservoir cells in this specific group of individuals remain poorly defined.
To address this question, we applied FLIP-Seq (Full-Length Individual Proviral Sequencing)8 to profile the proviral reservoir landscape at single-genome resolution to a large cohort of ECs who maintained undetectable HIV-1 plasma viral loads for a median of 9 (range: 1–24) years based on commercially-available PCR assays. A reference cohort of HIV-1-infected individuals treated with suppressive antiretroviral therapy (ART) for a median of 9 (range: 2–19) years was recruited for comparative purposes (Extended Data Table 1). Collectively, our analysis of a large number of individual HIV-1 proviral genomes (n=1,385 from 64 ECs and n=2,388 from 41 ART-treated individuals) demonstrated that the median number of proviral amplification products (intact and defective) per person was significantly lower in ECs relative to ART-treated individuals (Fig. 1a). Frequencies of near-full length, genome-intact proviral sequences (IPs) lacking defined lethal sequence defects were also markedly reduced in ECs, although their quantitative spectrum varied considerably (Fig. 1b). Of note, IPs made up a significantly larger proportion of all proviral sequences in ECs at both the cohort level (Fig. 1c) and the per study participant level (Fig. 1d), compared to ART-treated individuals; in four ECs, IPs accounted for 100% of detected proviral species. Intra-individual proviral sequence diversity, determined by pair-wise comparisons of all IPs within a given study participant, was smaller in ECs (Fig. 1e, Extended Data Fig. 1a). Interestingly, within IPs from ECs, optimal cytotoxic T lymphocyte (CTL) epitope sequences restricted by autologous HLA class I isotypes displayed more limited evidence of mutational escape (Fig. 1f, Extended Data Fig. 1c–f). These data suggest that IPs from ECs were seeded early in the disease process and persisted long-term.
Figure 1: Proviral reservoir landscape in HIV-1 ECs.
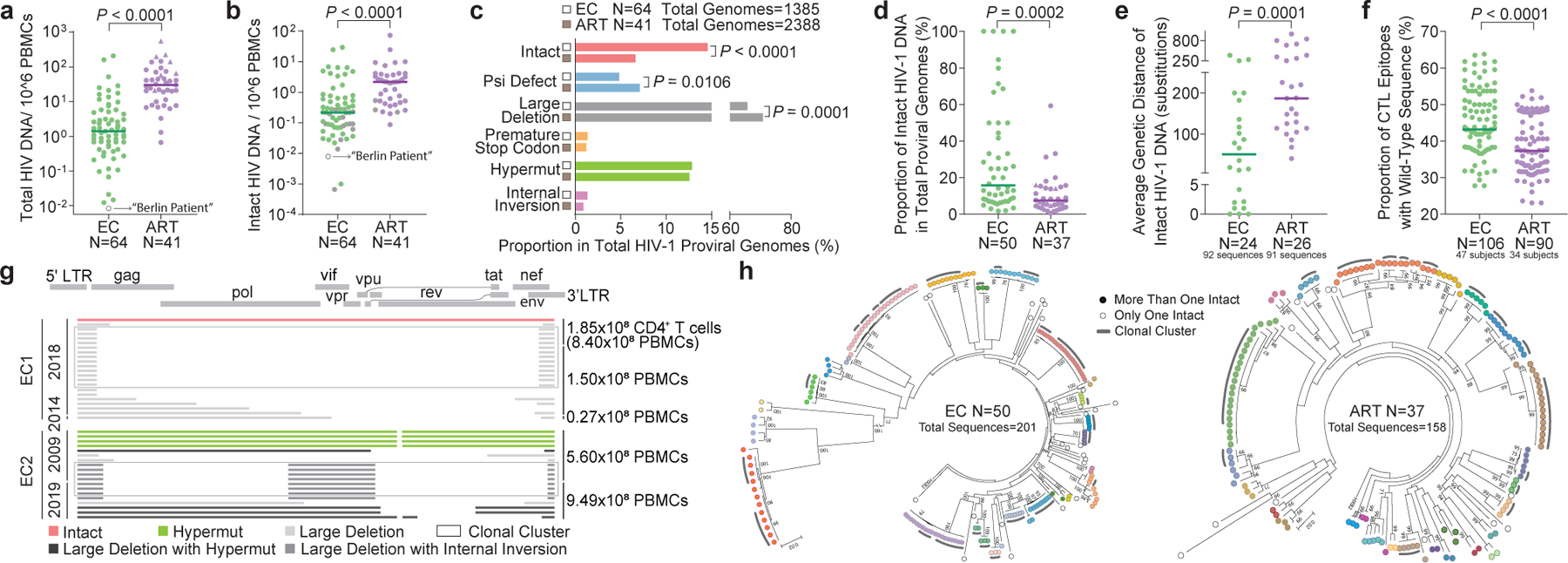
(a-b): Relative frequencies of total (a) and near full-length intact (b) HIV-1 DNA sequences in ECs and ART-treated individuals (ART). Grey symbols: Limit of detection (expressed as 1 copy/total number of analyzed cells without target identification). Circles: Proviral sequences obtained from unfractionated PBMC; triangles: proviral sequences retrieved from isolated CD4+ T-cells and normalized to PBMC. (c): Proportions of proviral sequences that are genome-intact or display defined structural defects among all proviral genomes. (d): Proportion of IPs among all proviral genomes from each study participant. Only individuals with at least one detected IP are shown. (e): Average genetic distance between distinct IPs obtained from each study participant. Participants with at least two detectable IPs are included. (f): Proportion of optimal CTL epitopes (restricted by autologous HLA class I isotypes) with wild-type clade B consensus sequences. Each dot represents one IP. Clonal sequences are counted once. (g): Diagrams reflecting all proviral HIV-1 sequences isolated from EC1 and EC2. Left vertical axis: Dates of sample collection; right vertical axis: Numbers of cells analyzed. (h): Circular maximum-likelihood phylogenetic trees for all IPs from ECs and ART-treated individuals. Dots with the same colors indicate IPs detected in the same individuals. Clonal sequences, defined by complete sequence identity, are indicated by grey arches. Bootstrap analysis with 1000 replicates was performed to assign confidence to tree nodes; bootstrap support values >70% are shown in the trees. Two-tailed Mann Whitney U tests were used for panels a-b, d-f; False Discovery Rate (FDR)-adjusted two-tailed Fisher’s exact tests were used for panel c.
For a deeper analysis of the proviral reservoir structure, we initially focused on two ECs in whom no IPs were observed in our initial analysis. In EC1, an individual who had maintained drug-free HIV-1 control for a recorded time of 12 years with only one documented viremia of 56 HIV-1 RNA copies/ml in 23 viral load tests spanning this period (Extended Data Fig. 2), escalating the number of analyzed PBMC to the limit of available cells yielded a single IP from a total of 1.02 billion PBMC; 21 defective proviruses, many of which belonged to a sequence-identical cluster, were also observed (Fig. 1g). In EC2, in whom only a single documented episode of 93 HIV-1 RNA copies/ml was noted in 39 viral load tests spanning >24 years of follow-up without ART exposure (Extended Data Fig. 2), we failed to detect even a single IP from more than 1.5 billion PBMC, while 19 defective proviral species, including near full-length sequences with lethal hypermutations, were observed, clearly documenting that this individual had been infected with HIV-1 in the past (Fig. 1g). Members of a sequence-identical cluster of defective proviral sequences with large deletions were noted in samples collected in 2009 and in 2019 from EC2, demonstrating a profound durability of a clonal population of cells harboring this sequence.
Moreover, a subsequent quantitative viral outgrowth assay (qVOA) with 340 million resting CD4+ T-cells isolated from approximately 1 billion PBMC (collected in 2019), and an additional qVOA involving 41 million total CD4+ T-cells isolated from 158.5 million PBMC (collected in 2009) did not retrieve a single replication-competent viral species. The recently-developed intact proviral DNA assay (IPDA) did not reveal evidence of IPs in 14 million resting CD4+ T-cells, while confirming the presence of defective HIV-1 DNA sequences (Extended Data Fig. 1b). In addition, an analysis of 7.72 million gut cells collected by colonoscopy from the rectum (2.08 million CD45+ mononuclear cells and 2.30 million CD45− cells) and terminal ileum (1.99 million CD45+ mononuclear cells and 1.35 million CD45− cells) by FLIP-Seq did not reveal any intact or defective proviruses in EC2.
To the authors’ knowledge, the absence of IPs in such extremely large numbers of analyzed cells has only been documented in the “Berlin Patient” who underwent an allogeneic hematopoietic stem cell transplantation from a donor who was homozygous for CCR5Δ329, which resulted in what is widely considered a sterilizing cure of HIV-1 infection. Indeed, in our hands, an analysis of 113 million available PBMC from the “Berlin Patient” (collected in 2017 and 2018) retrieved not a single intact or defective proviral sequence using FLIP-Seq (Fig. 1a–b). Although the logic of scientific discovery10 will never allow us to confirm that EC2 has achieved a sterilizing cure of HIV-1 infection through natural immune-mediated mechanisms, it is noteworthy that we have failed to falsify this hypothesis, despite analyzing massive amounts of cells with a range of complementary, highly-sensitive detection techniques.
We next performed a phylogenetic analysis of all IPs obtained from 50 ECs and 37 ART-treated individuals. In both groups, we readily observed large clusters of sequences that were completely identical over entire analyzed viral genomes (Fig. 1h), strongly suggesting that they originate from clonally-expanded HIV-1-infected cells that pass on identical copies of IPs during cell divisions. The proportions of these clonally-expanded IPs were significantly higher in ECs compared to ART-treated individuals (Extended Data Fig. 1g–h). A number of these sequences were also retrieved from qVOA, documenting that these IPs are indeed fully replication-competent (Fig. 2–3).
Figure 2: Increased frequency of IPs integrated in centromeric satellite DNA in ECs.
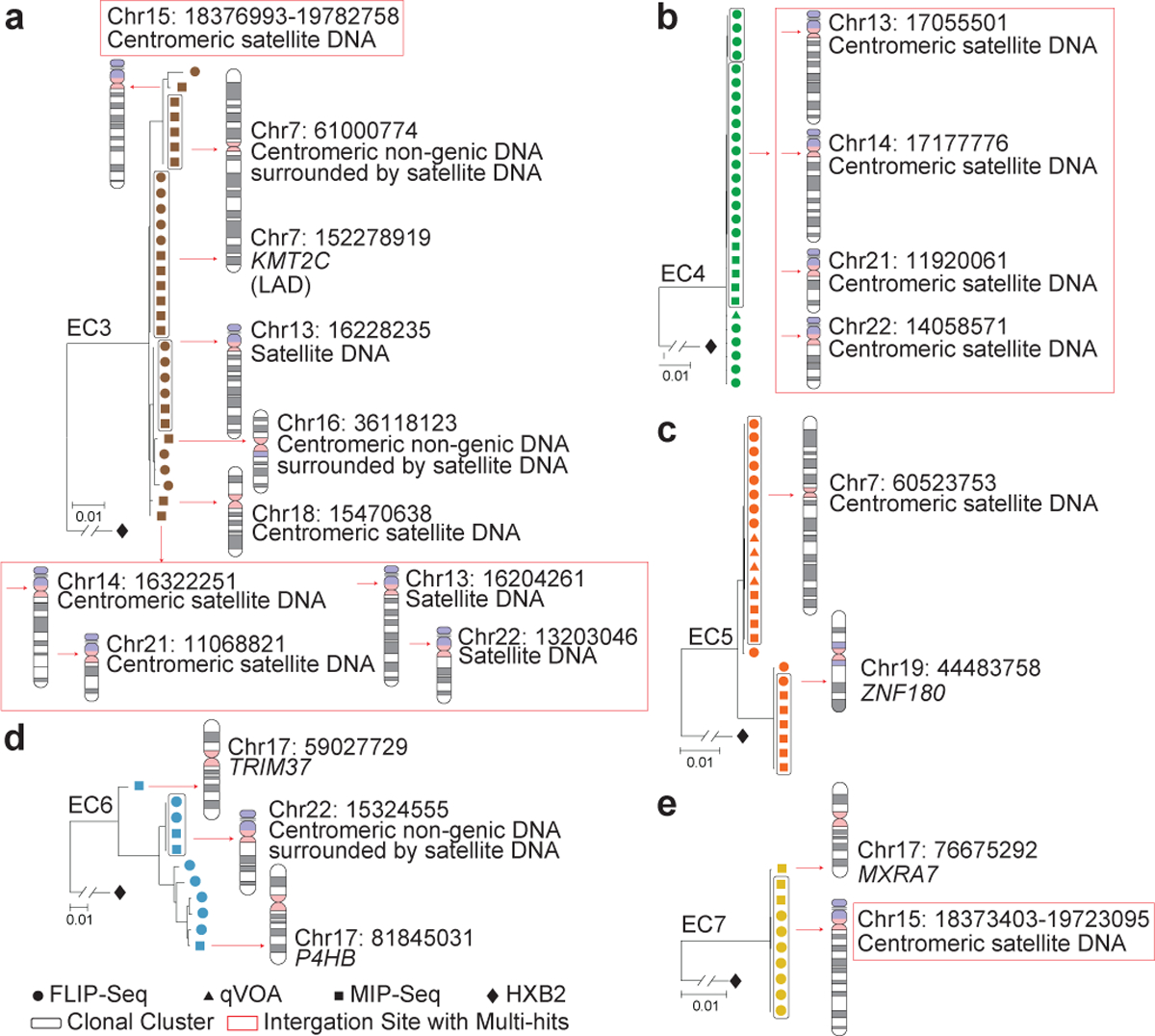
(a-e): Data indicate linear maximum-likelihood phylogenetic trees for IPs from five ECs. Coordinates and relative positioning of IS are depicted; genes harboring IS are italicized. Clonal IPs, defined by identical proviral sequences and identical corresponding IS, are highlighted in curved black boxes. Red boxes reflect multi-hit IS that cannot be definitively mapped to one particular genomic location due to positioning in repetitive centromeric satellite DNA present in multiple regions of the human genome. LAD, lamina associated domain.
Figure 3: Preferential location of IPs from ECs in genes encoding for KRAB-ZNF proteins.
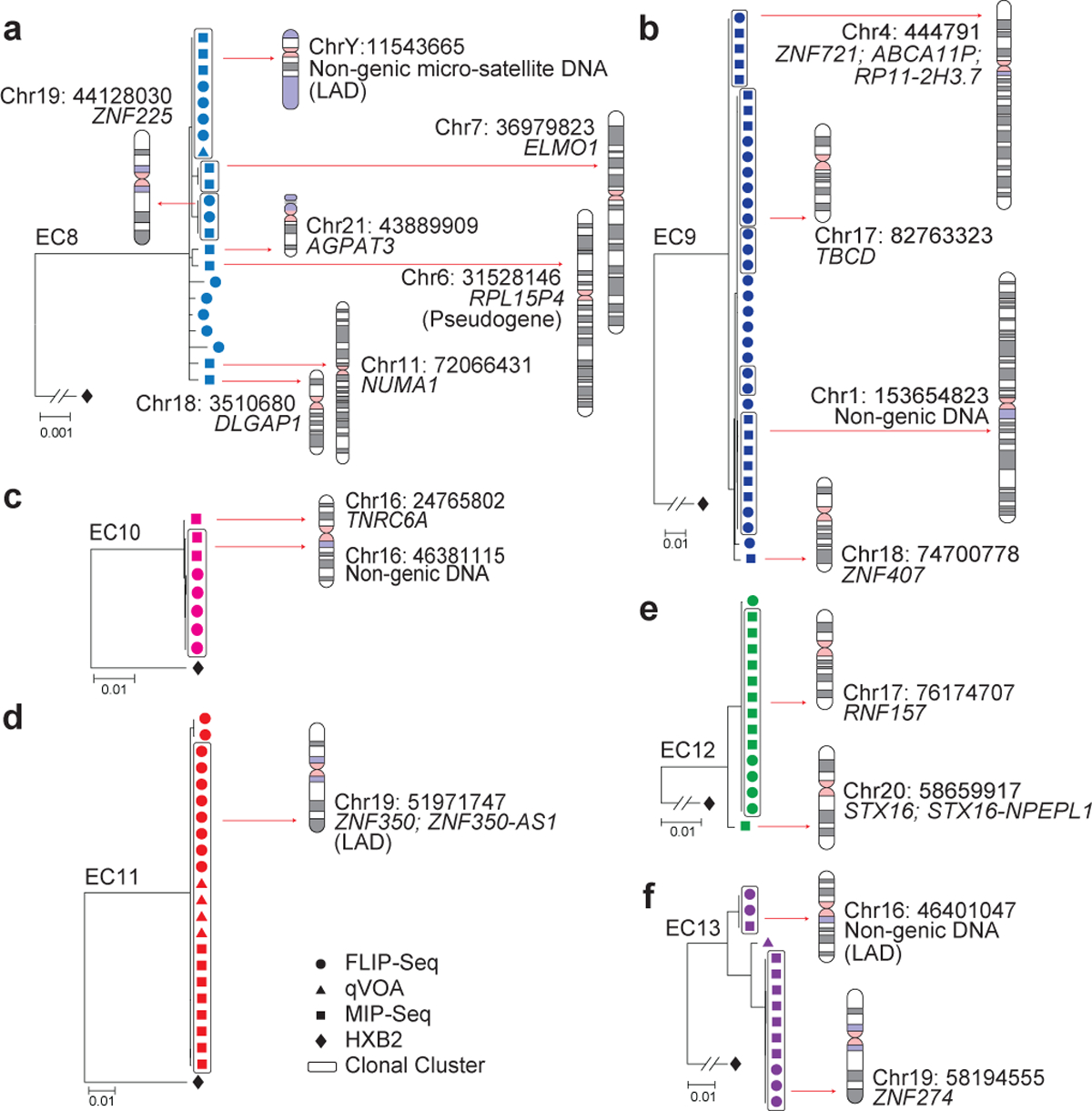
(a-f): Linear maximum-likelihood phylogenetic trees demonstrate IPs from indicated study participants. Coordinates and relative positioning of IS are depicted. Other pertinent information is as defined in the legend to Figure 2.
For a detailed analysis of the viral reservoir landscape in ECs, we focused on eleven ECs (EC3–13) in whom large clusters of identical IPs were detected and from whom sufficient numbers of cells were available. In these ECs, we frequently observed oligoclonal, and sometimes almost monoclonal compositions of the entire intact proviral reservoir landscape (Figures 2–3, Extended Data Fig. 3). Notably, such a narrowly-focused viral reservoir configuration consisting of rather few distinct IPs but displaying relatively large expansions of identical IP clones is compatible with very low, if any, levels of ongoing viral replication in these ECs. This viral reservoir structure is atypical relative to the more diverse spectrum of IPs previously described in long-term ART-treated individuals8,11. Instead, the viral reservoir landscape in EC3–13 is more reminiscent of the oligoclonal viral reservoir structure of IPs typically observed in chronic HTLV-1 infection, a retroviral disease characterized by deep proviral latency that limits active viral transcription and replication, such that viral propagation occurs almost exclusively by mitotic spread during clonal proliferation of infected T cells12. Based on these considerations, we hypothesized that IPs from ECs maintain a state of deep, long-lasting latency, possibly due to chromosomal integration into genomic regions not permissive to active viral transcription.
To investigate chromosomal positions of IPs, we used MIP-Seq (Matched Integration site and Proviral Sequencing)13 to analyze integration sites (IS) in conjunction with corresponding proviral sequences. Briefly, proviral DNA was diluted to single genome levels, subjected to phi29-catalyzed whole-genome amplification, and subsequently exposed to FLIP-Seq8 and IS analysis using “integration site loop amplification”14 or ligation-mediated PCR15. These experiments, performed in the eleven ECs (EC3–13), identified a total of 92 IS corresponding to IPs, of which 33 were associated with unique chromosomal locations (Supplementary Table 1). These IS of IPs were preferentially located in chromosomes 7, 17 and 19, and to a lesser extent in chromosomes 16 and 18 (Fig. 4a, Extended Data Fig. 5a). Consistent with previous studies13 in which a total of 100 pairs of IPs and corresponding IS (n=73 IPs with unique IS) were analyzed in long-term ART-treated individuals, proviral species that displayed complete sequence identity shared the same IS, confirming their clonal origin. Notably, upstream HIV-1 long terminal repeat regions, which are not included in typical FLIP-Seq assays8,11 but were specifically amplified in these individuals, also displayed complete sequence identity within analyzed proviral clones (Extended Data Fig. 4).
Figure 4: Distinct genomic and epigenetic features of IS of IPs from ECs.
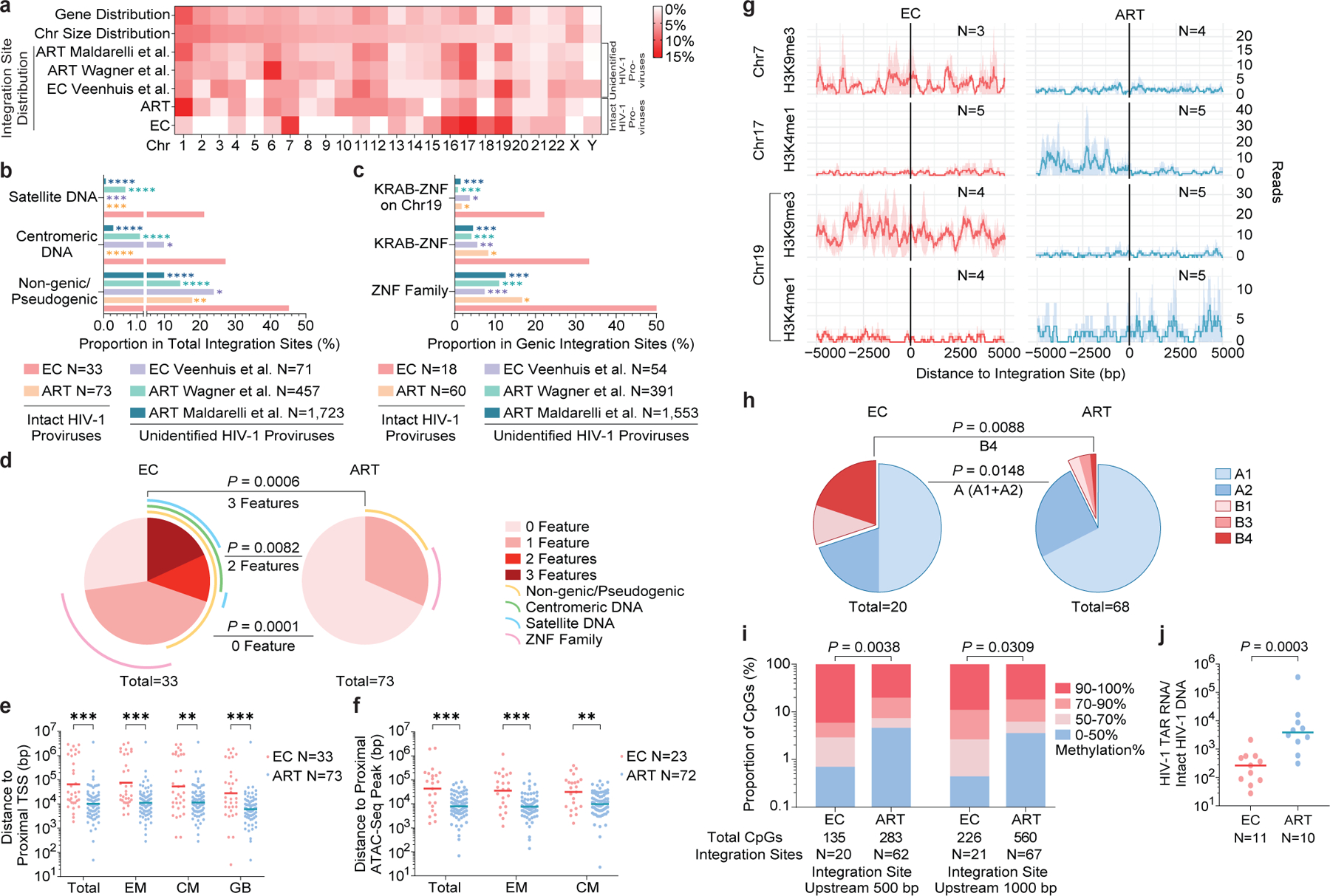
(a): Relative proportion of proviral IS of IPs in each chromosome. Contributions of each chromosome to total number of genes (first row) and to total size of human genome (second row) are included as references. (b-c): Proportion of IPs located in indicated genomic regions. (a-c): Data from IPs in ART-treated individuals13 (ART) and from unselected (intact and defective) proviral sequences in ECs7 and in ART-treated individuals14,16 are shown as references. (d): SPICE diagrams demonstrating proportions of IPs with indicated IS features in ECs and ART-treated individuals. (e-f): Chromosomal distance between IS of IPs and the most proximal TSS in autologous total, EM or CM CD4+ T-cells or from Genome Browser (GB) (e), or to the most proximal ATAC-Seq peaks (f) in autologous total, EM and CM CD4+ T-cells. Horizontal lines reflect the geometric mean. (g): Numbers of DNA sequencing reads associated with activating (H3K4me1) or repressive (H3K9me3) histone protein modifications in proximity to IS from ECs and long-term ART-treated individuals13; median and confidence intervals (one standard deviation) of ChIP-Seq data from primary memory CD4+ T-cells included in the ROADMAP repository25 are shown. (h): Proportions of IPs located in structural compartment A and B (and associated sub-compartments), as determined by Hi-C-Seq data28. IS in regions not covered in ref.28 were excluded. (i): Numbers of cytosine residues with indicated levels of methylation (derived from CD4+ T-cells in the iMethyl database30) in proximity (500 or 1000 bp upstream of the 5’ LTR host-viral junction) to IS from ECs and ART-treated individuals. (j): Frequencies of HIV-1 RNA transcripts in PBMC from ECs and ART-treated individuals, normalized to the corresponding number of IPs determined by FLIP-Seq. (a-i): Clonal sequences were only counted once. (f-i): Sequences in genomic regions included in the ENCODE blacklist27 were excluded. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05; (b/c/d/h): two-sided Fisher’s exact tests; (e/f/j): two-sided Mann Whitney U tests; (i): two-tailed Chi-square test; (b/c/e/f/i): FDR-adjusted p-values; (d/h/j): nominal p-values. All comparisons were made between ECs and reference groups.
Interestingly, IS analysis revealed that a significantly larger proportion of IPs from ECs were located in non-genic/pseudogenic regions, relative to IPs from long-term ART-treated individuals analyzed using the same approach13 (45% vs. 17.8% of distinct IPs, respectively, p=0.0051; 40.2% vs. 13% of all IPs, respectively, p<0.0001), and in comparison to prior studies analyzing HIV-1 IS in ART-treated individuals14,16 without distinguishing intact from defective proviruses (Fig 4b, Extended Data Fig. 5b). A closer investigation revealed that the non-genic IS of IPs from ECs were frequently positioned in or surrounded by centromeric satellite or microsatellite DNA (EC3–7, Fig. 2a–e), non-coding regions of the human genome that consist of dense heterochromatin “gene deserts”17 that are typically disfavored for HIV-1 integration18. Localization of proviral sequences in such centromeric satellite DNA has been associated with deep viral latency in functional viral reactivation studies19,20 and was exquisitely rare21 or entirely undetectable in prior investigations involving ART-treated individuals13. In our study, the integration of IPs into centromeric satellite or microsatellite DNA was observed for a total of 8 unique IPs (24% of distinct IPs, 20.7% of all IPs) and occurred at least once in five (EC3–5, EC7–8; Fig. 2a–c, e; 3a) of the 11 ECs analyzed. Additionally, three IS of IPs were located in centromeric non-genic DNA surrounded by satellite DNA (EC3, EC6; Fig. 2a, d). Notably, as many as six different IS of IPs were located in or surrounded by centromeric satellite DNA in EC3 (Fig. 2a). In addition to this highly disproportionate overrepresentation of centromeric satellite DNA among IS of IPs from ECs, EC10 and EC13 harbored integrations of clonal IPs in a large non-genic region in proximity to non-centromeric micro-satellite DNA on chromosome 16 (Fig. 3c, f). Thus, in total, 39.4% of all 33 distinct IPs (32.6% of all 92 IPs) from ECs were located within or in proximity to satellite or microsatellite DNA.
Corresponding to the disproportionate enrichment of non-genic IS in ECs, we noted that the number of genic IS associated with IPs was significantly diminished in ECs, relative to ART-treated individuals13. These genic IS were almost exclusively located in introns of genes that, in comparison to long-term ART-treated individuals, showed weaker transcriptional activity (Extended Data Fig. 7a) and displayed an opposite orientation relative to the harboring host gene in approximately 60% of all sites analyzed (Extended Data Fig. 7b–c). Genes encoding for members of the Zinc-Finger protein (ZNF) family, in particular for Krueppel-associated box domain-containing ZNF (KRAB-ZNF)22, accounted for 33% of all 18 genes harboring distinct IPs in ECs (corresponding to 49% of all 55 genic integration events of IPs), a notable enrichment relative to ART-treated individuals (Fig. 4c, Extended Data Fig. 5c). Of note, clonal IPs were frequently integrated into KRAB-ZNF genes located in defined regions of chromosome 19 (Ref.23), which are extensively occupied by the heterochromatin proteins CBX1 and SUV39H124 and display highly distinct chromatin features, with profound enrichment for repressive chromatin marks that extensively cover the bodies of ZNF genes, but selectively spare the corresponding host transcriptional start sites (TSS)24. Interestingly, a prior computational, genome-wide analysis of chromatin states based on a combinatorial evaluation of multiple different chromatin marks in their respective spatial context revealed that repetitive satellite DNA and ZNF genes share a common, highly distinct chromatin state (referred to as “ZNF genes & repeats”)25. When combined, IPs located either in satellite DNA or in ZNF genes represented >45% of all 33 independent IPs and >60% of all 92 IPs in ECs, proportions that were significantly increased relative to ART-treated individuals (Fig. 4d, Extended Data Fig. 5d).
For a formal analysis of proviral IS positioning relative to active transcription units in the host DNA, we performed RNA-Seq-based gene expression profiling in autologous total CD4+ T-cells, as well as autologous central-memory (CM) and effector-memory (EM) CD4+ T-cell subsets which harbor the majority of all HIV-1 IS26. These experiments demonstrated a significantly increased chromosomal distance between IS of IPs and the most proximal host TSS in ECs, relative to long-term ART-treated individuals13 (Fig. 4e). Simultaneously, we calculated the chromosomal distance between IS coordinates of IPs and accessible chromatin, as determined by genome-wide ATAC-Seq data obtained from autologous CD4+ T-cells. Although IS in satellite and microsatellite DNA were excluded from this analysis (and from the subsequent analysis using ChIP-Seq, Hi-C-Seq and methylation-Seq data; see below) due to the reduced ability to map next-generation sequencing reads to repetitive genomic DNA regions27, we noted that IS in cells from ECs were located at significantly increased distances to accessible chromatin, compared to those from ART-treated individuals13 (Fig. 4f). These differences were observed when clonal sequences were counted only once (Fig. 4e–f) but were also notable when all clonal sequences were considered individually (Extended Data Fig. 5e–f).
In a subsequent analysis, we calculated the number of DNA reads associated with defined epigenetic histone marks in the proximity to viral IS using ChIP-Seq data from primary memory CD4+ T-cells available from the ROADMAP Epigenomics Project25. In comparison to ART-treated individuals13, this analysis revealed a marked enrichment for the repressive histone features H3K9me3 (Chr19, Chr7) and/or a de-enrichment for the activating chromatin feature H3K4me1 (Chr19, Chr17) at IS of IPs from ECs (Fig. 4g); a trend for differential expression of additional activating and inhibitory chromatin modifications in proximity to IS of IPs from ECs and ART-treated individuals was also noted (Extended Data Fig. 6a–d). Furthermore, an alignment of IS coordinates to three-dimensional chromosomal contact data generated by Hi-C-Seq28 demonstrated a significantly-increased proportion of IPs from ECs located in compartment B, containing mostly closed chromatin. This effect was particularly obvious for IS in KRAB-ZNF genes on Chromosome 19 in ECs, which were all located in subcompartment B4 (Fig. 4h, Extended Data Fig. 5g). This very small compartment (accounting for approximately 0.3% of the human genome) is known to harbor dense heterochromatin marks28 and represents a highly atypical chromosomal IS location for HIV-1 in non-controller individuals13. A profoundly-increased frequency of IPs from ECs in compartment B was also noted when Hi-C-Seq data from Jurkat cells29 were used for alignment (Extended Data Fig. 6e–f).
Taking advantage of previously-published genome-wide bisulfite sequencing data in CD4+ T-cells30, we observed that the frequency of hypermethylated (>90% methylation) cytosine residues was significantly higher in proximity to IPs from ECs, relative to IS of IPs from long-term ART-treated individuals13 (Fig. 4i). These data suggest that chromosomal regions more susceptible to DNA methyltransferases represent preferential sites for the long-term persistence of IPs in ECs, arguably because the integration into hypermethylated genomic DNA might facilitate deep latency of IPs and protect against immune-cell targeting. Given that closely-neighboring cytosine residues are likely to share the same methylation status31, these results raise the possibility that HIV-1 promoter methylation, previously shown to induce proviral HIV-1 silencing in in-vitro assays32, may contribute to durable transcriptional repression of IPs from ECs. The frequencies of IPs located in lamina-associated domains (LADs), genomic regions that interact with the inner nuclear membrane, contain mostly closed chromatin and represent a rare target for HIV-1 integration33, were not significantly different between IPs from ECs and ART-treated individuals when clonal sequences were counted only once; however, a significant enrichment of IPs from ECs in LADs was noted when clonal IPs were counted as independent proviruses (Extended Data Fig. 7d–e).
Given that non-coding centromeric satellite DNA is a highly disfavored target site for HIV-1 integration18, the disproportionately increased number of IS in satellite DNA described here is a particularly striking feature of ECs. Notably, ECs expressed normal mRNA levels of LEDGF/p75 and CPSF6 (Extended Data Fig. 7f), host factors that interact directly with HIV-1 proteins to bias HIV-1 IS selection to active transcription units34,35. Although protein levels of these molecules were not assessed, these results suggest that there is no increased susceptibility of centromeric satellite DNA to HIV-1 integration in ECs. To further address this, we infected CD4+ T-cells from n=12 ECs from our study cohort and n=9 HIV-1 negative healthy individuals with a GFP-encoding HIV-1 construct, followed by sorting of GFP+ and GFP− CD4+ T-cells and a subsequent IS analysis. These experiments, retrieving >120,000 independent HIV-1 integration coordinates, demonstrated that IS in satellite DNA accounted for extremely low proportions of all integration events (0.04–0.06% in GFP+ and 0.11–0.12% in GFP− CD4+ T-cells), irrespective of the analyzed study cohort (Extended Data Fig. 8a–b, Supplementary Table 2). Moreover, there was no evidence for preferential targeting of non-genic chromosomal regions or genes encoding for KRAB-ZNF proteins in in-vitro infected CD4+ T-cells from ECs (Extended Data Fig. 8b–c).
In conclusion, this work identifies a markedly distinct intact proviral reservoir landscape in PBMC from individuals with durable natural control of HIV-1, characterized by IS features highly suggestive of deep latency. For additional functional validation of this conclusion, we analyzed the frequency of HIV-1 RNA transcripts in ECs and ART-treated individuals; these additional experiments demonstrated that the number of HIV-1 RNA copies, normalized to the corresponding number of IPs, was significantly lower in ECs (Fig. 4j). As such, ECs seem to exemplify attributes of a “block and lock” mechanism36 of viral control, defined by silencing of proviral gene expression through chromosomal integration into repressive chromatin locations37. We propose that the distinct reservoir configuration in ECs is not related to altered IS preferences during acute infection in ECs, but instead represents the result of cell-mediated immune selection forces that preferentially eliminate proviral sequences more permissive to viral transcription, in a process that we suggest referring to as the “autologous shock and kill” mechanism. In contrast, less transcriptionally active proviral sequences with features of deep latency, leading to lower vulnerability to immune recognition, seem to persist long term. In very rare cases, such as in EC1 and EC2, such selection forces may have accomplished near total clearance of all IPs, raising the possibility that a sterilizing cure of HIV-1 infection can, at least in principle, spontaneously occur through natural, immune-mediated mechanisms. Future studies will be necessary to determine whether signs of immune-mediated selection pressure on viral reservoir cells are also visible in IPs from lymphoid tissues, which harbor the majority of viral reservoir cells38.
While our data strongly suggest that deep latency plays a role in maintaining spontaneous, drug-free control of HIV-1 in some ECs, deep viral latency is not completely permanent or irreversible, as reflected by our ability to retrieve replication-competent virus from ECs in in-vitro outgrowth assays. However, in-vitro viral outgrowth assays with maximum stimuli are unlikely to adequately reflect susceptibility to viral reactivation in vivo; indeed, in-vitro viral outgrowth may largely be a stochastic process11,39, and may occur independently of molecular pathways fine-tuning in-vivo viral outgrowth behavior. Nevertheless, it is likely that deep viral latency in ECs is a dynamic process, and that occasional bursts of viral transcription may occur despite genomic and epigenetic IS features restricting viral gene expression. In fact, a proviral landscape with low permissiveness to viral reactivation stimuli may expose the immune system to a tailored viral antigen dose that can maintain a highly-functional antiviral T-cell response, a hallmark of antiviral immunity in ECs3, without supporting high-level viral replication promoting cytotoxic T-cell exhaustion. Therefore, a reciprocal equilibrium between a weakly-inducible viral reservoir and an efficient HIV-1-specific CD8+ T-cell response may represent the cornerstone of natural HIV-1 immune control. Given that evidence for selection of IPs with features of deeper latency was also observed in long-term ART-treated individuals, albeit at weaker degrees13, it is hoped that future longitudinal evaluations will be informative for designing strategies to induce a long-term drug-free remission of HIV-1 infection in larger populations of individuals.
Methods
Study participants
HIV-1-infected study participants were recruited at the Massachusetts General Hospital (MGH), the Brigham and Women’s Hospital (BWH, both in Boston, MA, USA) and at the University of California, San Francisco (UCSF) at the Zuckerberg San Francisco General Hospital (San Francisco, CA, USA). PBMC and tissue samples were obtained according to protocols approved by the respective Institutional Review Boards. Clinical and demographical characteristics of study participants are summarized in Extended Data Table 1.
Droplet digital PCR
PBMC or CD4+ T-cells enriched from total PBMC using CD4 T Cell Isolation Kit (Miltenyi Biotec #130-096-533) were subjected to DNA extraction using commercial kits (Qiagen DNeasy #69504). We amplified total HIV-1 DNA using droplet digital PCR (Bio-Rad), using primers and probes described previously8 (127 bp 5’LTR-gag amplicon; HXB2 coordinates 684–810). PCR was performed using the following program: 95℃ for 10 min, 45 cycles of 94℃ for 30s and 60℃ for 1 min, 72℃ for 1 min. The droplets were subsequently read by the QX200 droplet reader and data were analyzed using QuantaSoft software (Bio-Rad).
Whole genome amplification
Extracted DNA was diluted to single viral genome levels according to ddPCR results, so that 1 provirus was present in approximately 20–30% of wells. Subsequently, DNA in each well was subjected to multiple displacement amplification (MDA) with phi29 polymerase (Qiagen REPLI-g Single Cell Kit #150345), per the manufacturer’s protocol. Following this unbiased whole genome amplification40, DNA from each well was split and separately subjected to viral sequencing and integration site analysis, as described below. If necessary, a second-round MDA reaction was performed to increase the amount of available DNA.
HIV near full-genome sequencing
DNA resulting from full-genome amplification reactions was subjected to HIV-1 near full-genome amplification using a 1-amplicon and/or non-multiplexed 5-amplicon approach, as described before13. PCR products were visualized by agarose gel electrophoresis (Quantify One and ChemiDoc MP Image Lab, BioRad). All near full-length and/or 5-amplicon positive amplicons were subjected to Illumina MiSeq sequencing at the MGH DNA Core facility. Resulting short reads were de novo assembled using Ultracycler v1.0 and aligned to HXB2 to identify large deleterious deletions (<8000bp of the amplicon aligned to HXB2), out-of-frame indels, premature/lethal stop codons, internal inversions, or packaging signal deletions (≥15 bp insertions and/or deletions relative to HXB2), using an automated in-house pipeline written in Python programming language (https://github.com/BWH-Lichterfeld-Lab/Intactness-Pipeline)41, consistent with prior studies8,42,43. Presence/absence of APOBEC-3G/3F-associated hypermutations was determined using the Los Alamos National Laboratory (LANL) HIV Sequence Database Hypermut 2.044 program. Viral sequences that lacked all mutations listed above were classified as “genome-intact” sequences. Sequence alignments were performed using MUSCLE45. Phylogenetic distances between sequences were examined using maximum likelihood trees in MEGA (www.megasoftware.net) and MAFFT (https://mafft.cbrc.jp/alignment/software), and visualized using Highlighter plots (https://www.hiv.lanl.gov/content/sequence/HIGHLIGHT/highlighter_top.html). Viral sequences were considered clonal if they had completely identical consensus sequences; single nucleotide variations in primer binding sites were not considered for clonality analysis. Clades of intact HIV-1 proviral sequences were determined using the LANL HIV Sequence Database Recombinant Identification Program46. Within intact HIV-1 clade B sequences, the proportions of optimal CTL epitopes (restricted by autologous HLA class I alleles) matching the clade B consensus sequence and CTL escape variants restricted by selected HLA class I alleles and supertypes described in the LANL HIV Immunology Database (www.hiv.lanl.gov) were determined.
Integration site analysis
Integration sites associated with each viral sequence were obtained using integration site loop amplification (ISLA), using a protocol previously described by Wagner et al14, or by ligation-mediated PCR15 (Lenti-X™ Integration Site Analysis Kit (Takara Bio #631263)); DNA produced by whole-genome amplification was used as template. For selected clonal sequences, viral-host junction regions were also amplified using primers annealing upstream of the integration site in host DNA and downstream of the integration site in viral DNA. Resulting PCR products were subjected to next-generation sequencing using Illumina MiSeq. MiSeq paired-end FASTQ files were demultiplexed; small reads (142 bp) were then aligned simultaneously to human reference genome GRCh38 and HIV-1 reference genome HXB2 using bwa-mem47. Biocomputational identification of integration sites was performed according to previously-described procedures14,48: Briefly, chimeric reads containing both human and HIV-1 sequences were evaluated for mapping quality based on (i) HIV-1 coordinates mapping to the terminal nucleotides of the viral genome, (ii) absolute counts of chimeric reads, (iii) depth of sequencing coverage in the host genome adjacent to the viral integration site. The final list of integration sites and corresponding chromosomal annotations was obtained using Ensembl (v86, www.ensembl.org), the UCSC Genome Browser (www.genome.ucsc.edu) and GENCODE (v29, www.gencodegenes.org). Repetitive genomic sequences harboring HIV-1 integration sites were identified using RepeatMasker (www.repeatmasker.org).
Cell sorting and flow cytometry
PBMC were stained with monoclonal antibodies to CD4 (1:50, clone RPA-T4, Biolegend #300518), CD3 (1:50, clone OKT3, Biolegend #317332), CD45RO (1:40, clone UCHL1, Biolegend #304236) and CCR7 (1:40, clone G043H7, Biolegend #353216). Afterwards, cells were washed and CD45RO+ CCR7+ (central-memory) and CD45RO+ CCR7− (effector-memory) and CD3+ CD4+ (total) CD4+ T-cells were sorted in a specifically designated biosafety cabinet (Baker Hood), using a FACS Aria cell sorter (BD Biosciences) at 70 pounds per square inch. Cell sorting was performed by the Ragon Institute Imaging Core Facility at MGH and resulted in isolation of lymphocytes with the defined phenotypic characteristics of >95% purity. Data were analyzed using FlowJo software (Treestar).
RNA-Seq
Total RNA was extracted from sorted CD4+ T-cell populations using a PicoPure RNA Isolation Kit (Applied Biosystems #KIT0204). RNA-Seq libraries were generated as previously described49. Briefly, whole transcriptome amplification (WTA) and tagmentation-based library preparation was performed using SMART-seq2, followed by sequencing on a NextSeq 500 Instrument (Illumina). The quantification of transcript abundance was conducted using RSEM software (v1.2.22) supported by STAR aligner software (STAR 2.5.1b) and aligned to the hg38 human genome. Transcripts per million (TPM) values were then normalized among all samples using the upper quantile normalization method.
ATAC-Seq
A previously-described protocol with some modifications50,51 was used. Briefly, 20,000 sorted cells were centrifuged at 1500 rpm for 10 min at 4°C in a pre-cooled fixed-angle centrifuge. All supernatant was removed and a modified transposase mixture (including 25 μl of 2x TD buffer, 1.5 μl of TDE1, 0.5 μl of 1% digitonin, 16.5 μl of PBS, 6.5 μl of nuclease-free water) was added to the cells and incubated in a heat block at 37°C for 30 min. Transposed DNA was purified using a ChIP DNA Clean & Concentrator Kit (Zymo Research #D5205) and eluted DNA fragments were used to amplify libraries. The libraries were quantified using an Agilent Bioanalyzer 2100 and the Q-Qubit™ dsDNA High Sensitivity Assay Kit. All Fast-ATAC libraries were sequenced using paired-end, single-index sequencing on a NextSeq 500/550 instrument with v2.5 Kits (75 Cycles). The quality of reads was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk). Low quality DNA end fragments and sequencing adapters were trimmed using Trimmomatic (http://www.usadellab.org). Sequencing reads were then aligned to the human reference genome hg38 using a short-read aligner (Bowtie2, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) with the non-default parameters “X2000”, “non-mixed” and “non-discordant”. Reads from mitochondrial DNA were removed using Samtools (http://www.htslib.org). Peak calls were made using MACS2 with the callpeak command (https://pypi.python.org/pypi/MACS2), with a threshold for peak calling set to FDR-adjusted p<0.05.
Viral outgrowth assays
CD4+ T-cells were isolated from PBMC using EasySep™ Human CD4 Positive Selection Kit II (STEMCELL Technologies #17852). Cells were plated in limiting dilutions based on the intact provirus reservoir size determined through FLIP-Seq. Irradiated feeder PBMC were added at 1×105 cells/well. Cells were activated with 1 μg/mL PHA for four days, which was subsequently washed away and 10,000 MOLT-4 CCR5+ cells (NIH AIDS Reagent Program #4984) were added to propagate infection. On the thirteenth and twentieth days, culture supernatants from each well were individually incubated with 10,000 TZM-bl cells (NIH AIDS Reagent Program #8129) to drive Tat-dependent luciferase production. On the fifteenth and twenty-second days, TZM-bl cells were lysed, and luciferase activity was measured using Britelite Plus (PerkinElmer #6066761). Luciferase positive wells were defined as having signal levels >3 fold higher than negative controls. Cells from positive wells were then harvested and plated into lower compartments of Transwell tissue culture inserts (Costar® 6.5 mm Transwell®, 0.4 μm Pore Polyester Membrane Inserts, STEMCELL #38024), while 1×106 MOLT-4 cells were placed in upper compartments. After five additional days of culture, MOLT-4 cells from the upper wells were harvested, subjected to FLIP-Seq. Large scale quantitative viral outgrowth measurements on EC2 were performed by a similar standard method52 with a p24 ELISA assay used to detect outgrowth.
IPDA
The intact proviral DNA assay (IPDA) uses digital droplet PCR to quantitate proviruses lacking overt fatal defects, especially large deletions and hypermutation, and was performed as previously described53.
In-vitro infection assays
CD4+ T-cells were stimulated in RPMI medium supplemented with 10% fetal calf serum, recombinant IL-2 (50 U/ml), and an anti-CD3/CD8 bispecific antibody (0.5 μg/ul, NIH AIDS Reagent Program #12277). Cells were infected on day 5 with a GFP-encoding NL4–3 construct with a BAL-derived R5-tropic envelope54 at a multiplicity of infection (MOI) of 0.1 for 4 h at 37°C. After 2 washes, cells were resuspended in medium and plated at 5 × 105 cells/well in a 24-well plate. On day 5, GFP+ and GFP− CD4+ T-cells were sorted. Cells were processed to DNA extraction and integration site analysis using ligation-mediated PCR according to a previously-described protocol48.
Analysis of cell-associated HIV-1 RNA
Total cell-associated RNA and DNA was extracted in parallel from the same PBMC sample, using the GenElute RNA/DNA/Protein Purification Plus Kit (Sigma #RDP300) according to the manufacturer`s protocol. RNA was reverse transcribed into cDNA using a polyadenylation-RT reaction55 to efficiently detect HIV-1 RNA transcripts, followed by ddPCR-based amplification with primers and probes spanning the HIV-1 TAR region, as described before55. Simultaneously, cell-associated DNA was subjected to ddPCR-based amplification of the RPP30 gene to determine cell counts in PBMC samples, using probes and primers described previously56. HIV-1 RNA copies per million PBMC were normalized to the corresponding number of intact proviruses per million PBMC (determined by FLIP-Seq).
Statistics
Data are presented as pie charts, bar charts, scatter plots with individual values or heatmaps. Differences were tested for statistical significance using Mann-Whitney U test (two-tailed), Fisher’s exact test (two-tailed), or Chi-squared test (two-tailed), as appropriate. p-values of <0.05 were considered significant, false discovery rate (FDR) correction was performed using the Benjamini-Hochberg method57. Analyses were performed using Prism (GraphPad Software, Inc.), SPICE58 and R (R Foundation for Statistical Computing59).
Study approval
Study participants gave written informed consent to participate in accordance with the Declaration of Helsinki. The study was approved by the institutional review boards of MGH, BWH and UCSF.
Extended Data
Extended Data Figure 1: Viral sequence analysis of intact HIV-1 proviruses from ECs.
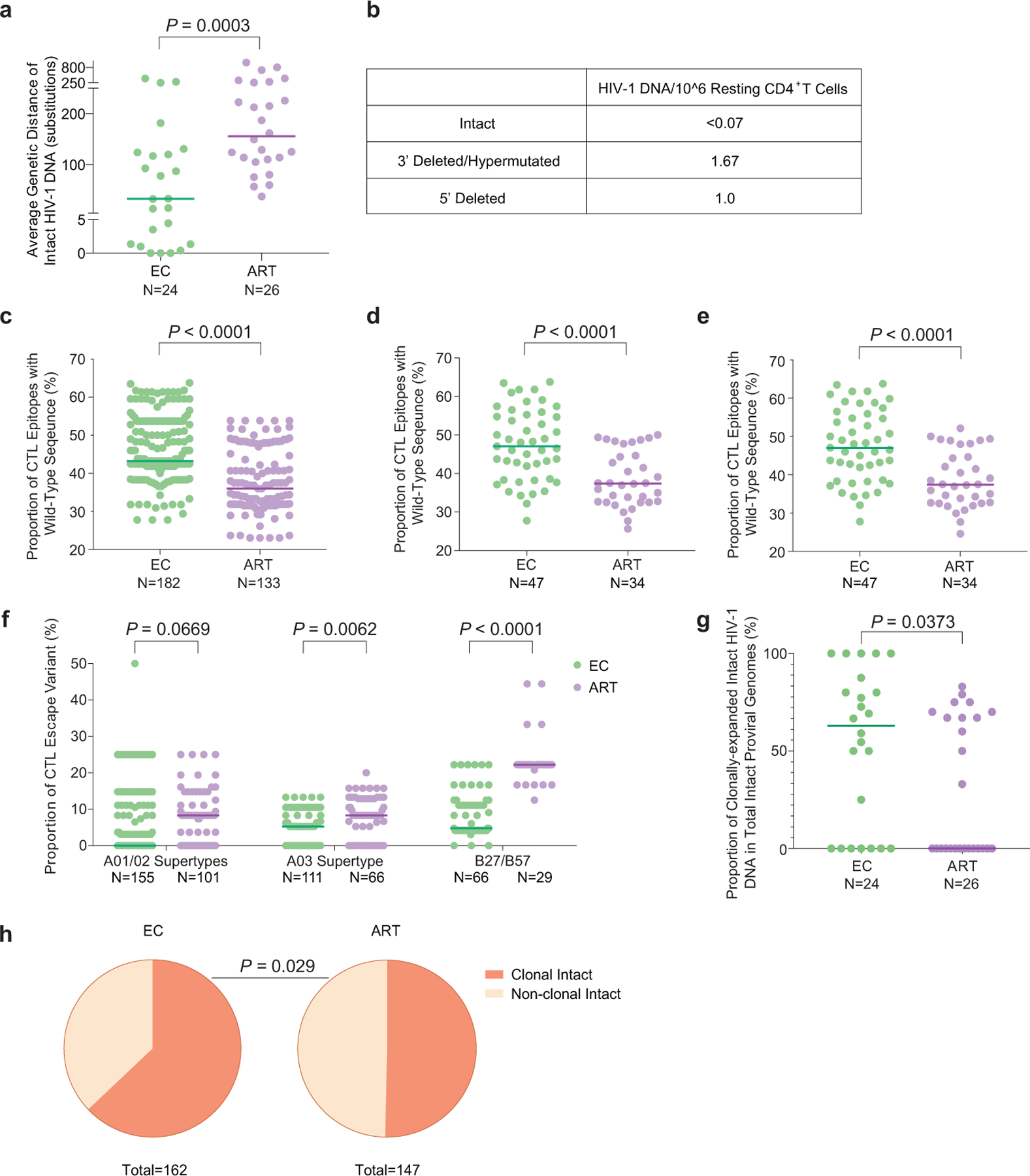
(a): Genetic distance (expressed as average number of base pair substitutions) among all intact near full-length proviral sequences obtained from each study participant. Clonal sequences were considered as individual sequences; participants with at least two intact proviruses are included (n=175 intact proviral sequences from 24 ECs and n=147 intact proviral sequences from 26 ART-treated individuals). (b): Frequencies of proviral species (copies per million resting CD4+ T-cells) detected by IPDA from EC2. (c): Proportion of optimal CTL epitopes (restricted by autologous HLA class I isotypes) with wild-type sequences. Each dot represents one intact proviral sequence. N=182 and N=133 HIV-1 clade B intact sequences from 47 ECs and 34 ART-treated individuals are included, respectively. Optimal CTL epitopes matching the clade B consensus sequences were considered as wild-type sequences. Clonal sequences were considered as individual sequences. (d-e): Average frequencies of autologous HLA-restricted optimal CTL epitopes with wild-type sequences calculated from intact proviruses in each study participant. Clonally-expanded sequences were counted either once (d) or individually (e). Each dot represents one study participant. (f): Proportion of CTL escape variants (restricted by HLA-A01/A02 supertypes, HLA-A03 supertype, or HLA-B*27/B*57). Each dot represents one intact proviral sequence. Clonal sequences were counted individually. (g-h): Proportion of clonal intact proviruses among all intact proviruses within each study participant (g) or within all intact proviruses from ECs and ART-treated individuals(h). Study participants in whom at least two intact proviruses were detected are included in (g) and (h). (Two-tailed Mann Whitney U tests were used for panels a, c-g; two-sided Fisher’s exact test was used for panel h).
Extended Data Figure 2: Longitudinal evolution of CD4+ T-cell counts and HIV-1 viral loads in EC1-EC13.
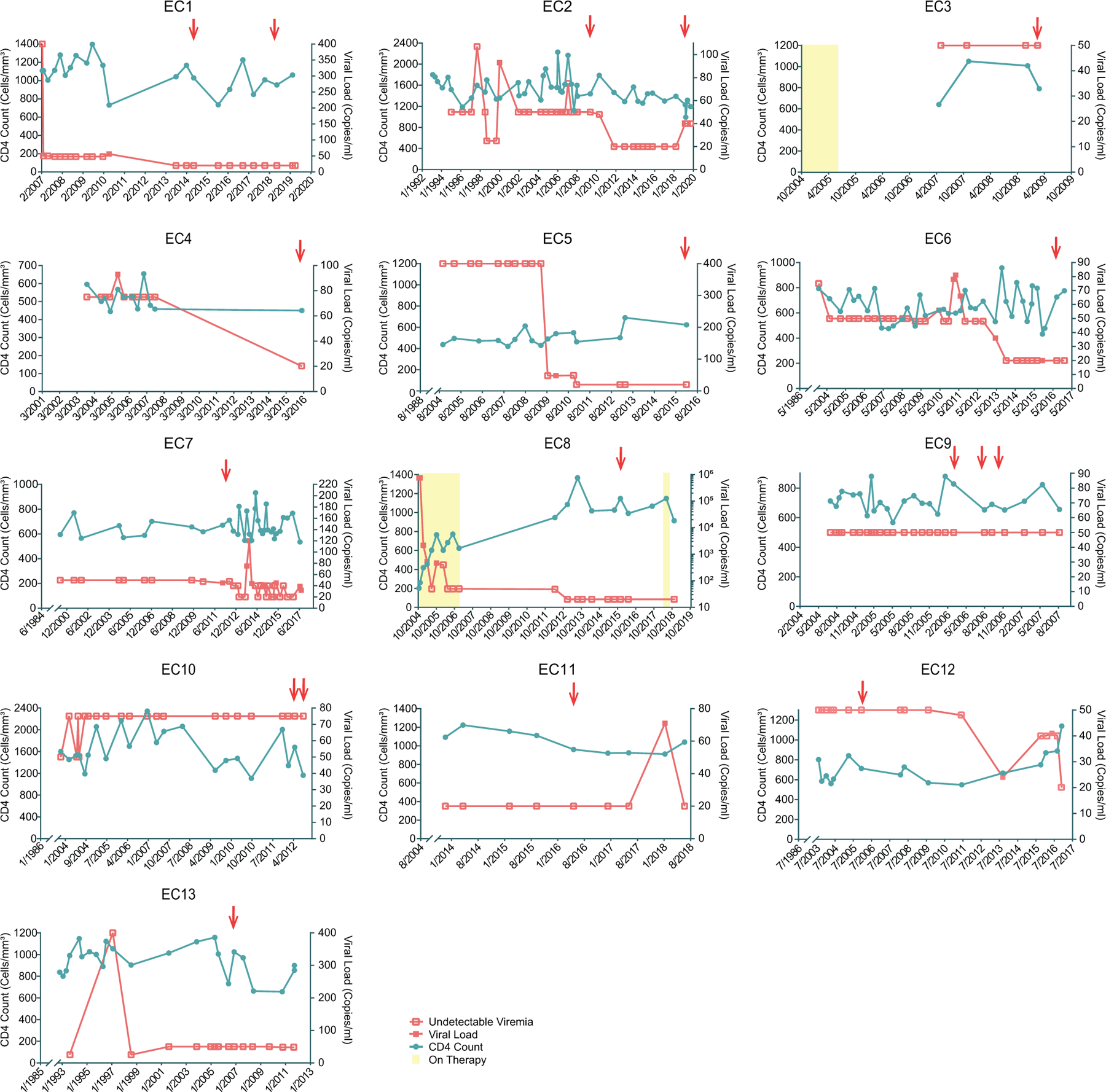
The recorded diagnosis date of HIV-1 infection for each study participant is shown as the first date on x-axis. PBMC sampling time points are indicated by red arrows.
Extended Data Figure 3: Diagrams reflecting the structural composition of proviral reservoirs in ECs.
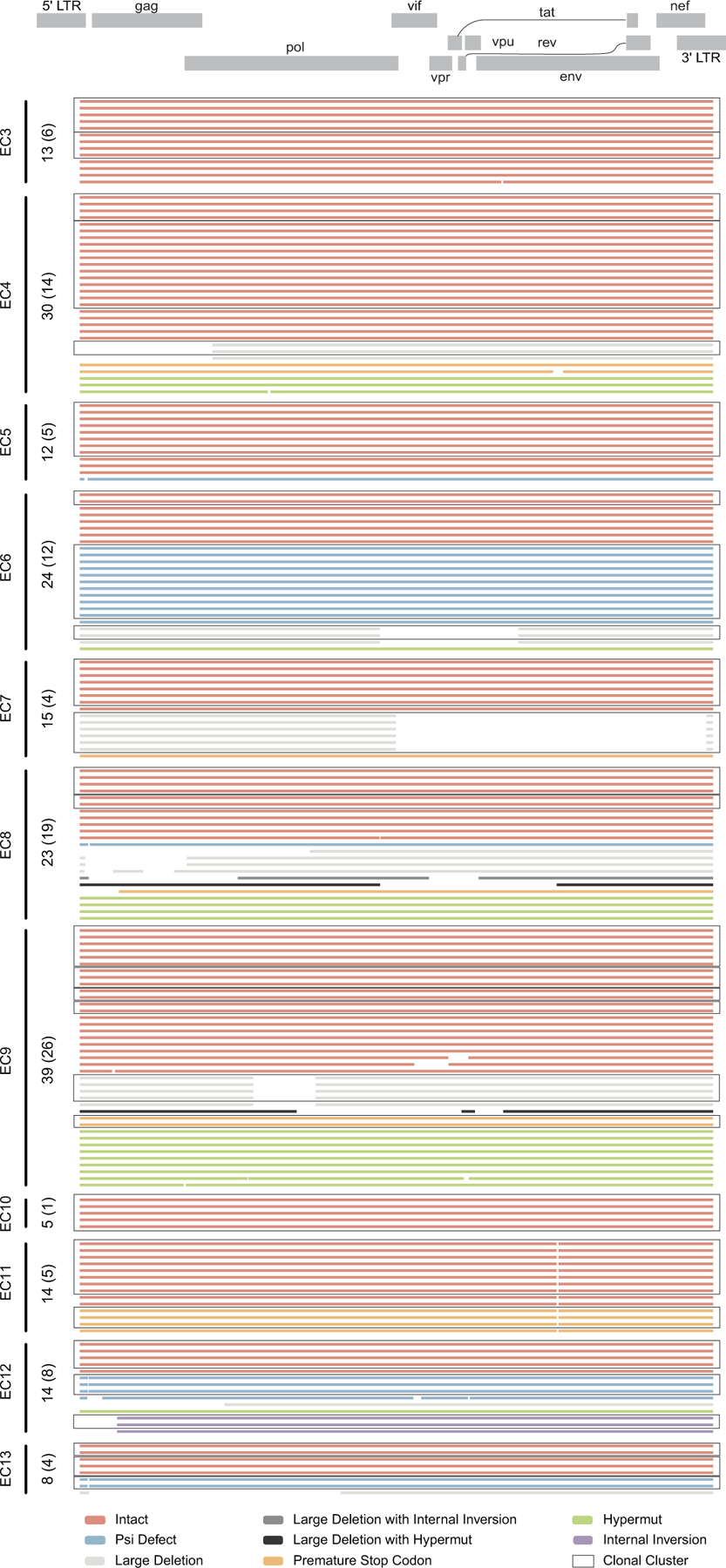
Virograms reflect the genetic coverage of individual sequences of proviral genomes analyzed in EC3-EC13. Numbers of total near full-length proviral sequences obtained from each individual are shown on the vertical axis; numbers of independent sequences are indicated in brackets. Open boxes indicate clonal clusters.
Extended Data Figure 4: Highlighter plot reflecting variations in HIV-1 DNA sequences in 5’ LTR regions from intact proviruses isolated from indicated ECs, relative to HXB2.
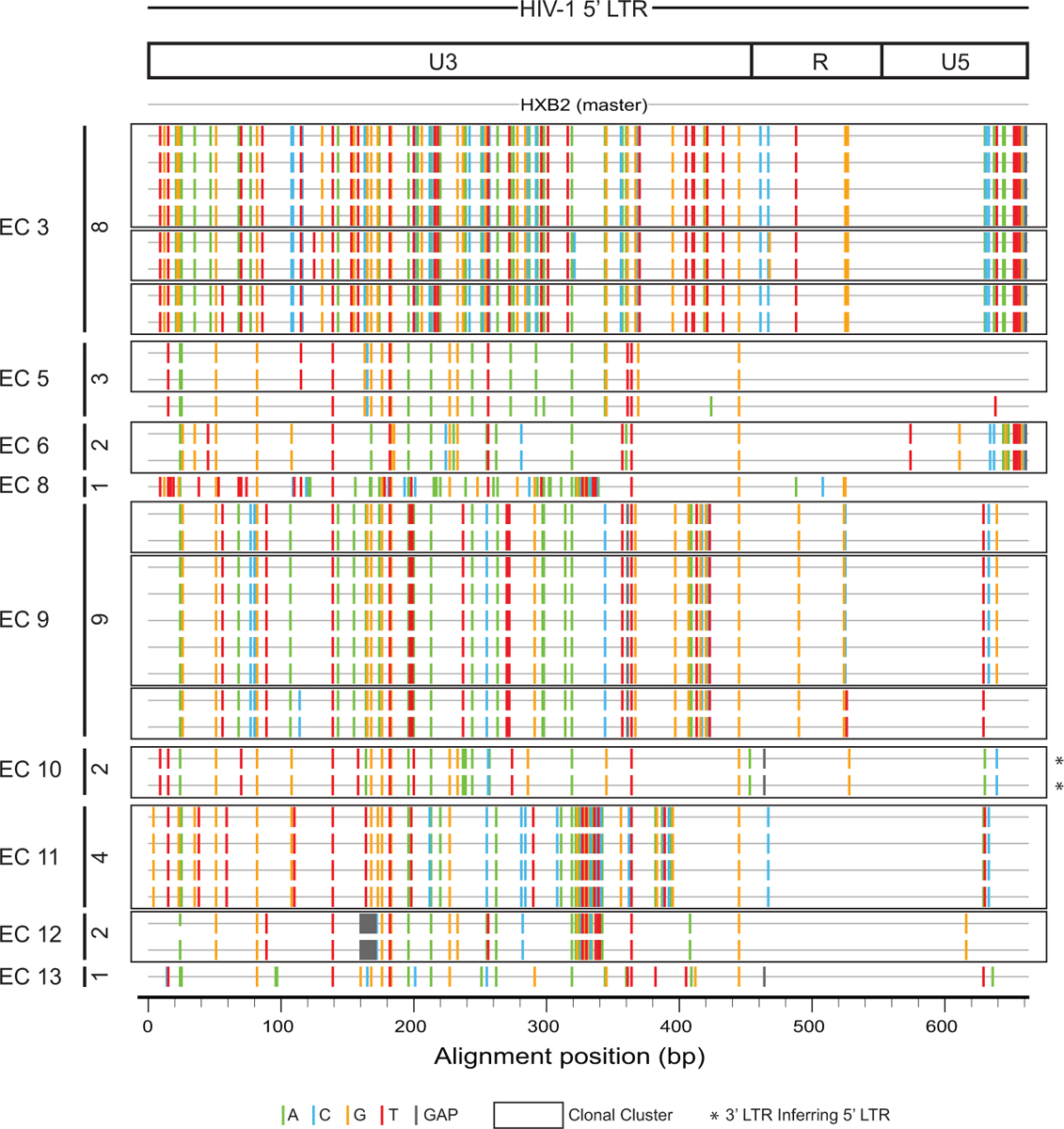
Numbers of 5’ LTR sequences of intact proviruses obtained from each individual are shown on the vertical axis. Open boxes indicate clonal clusters.
Extended Data Figure 5: Chromosomal integration site features of intact proviruses from ECs after counting clonal sequences individually.
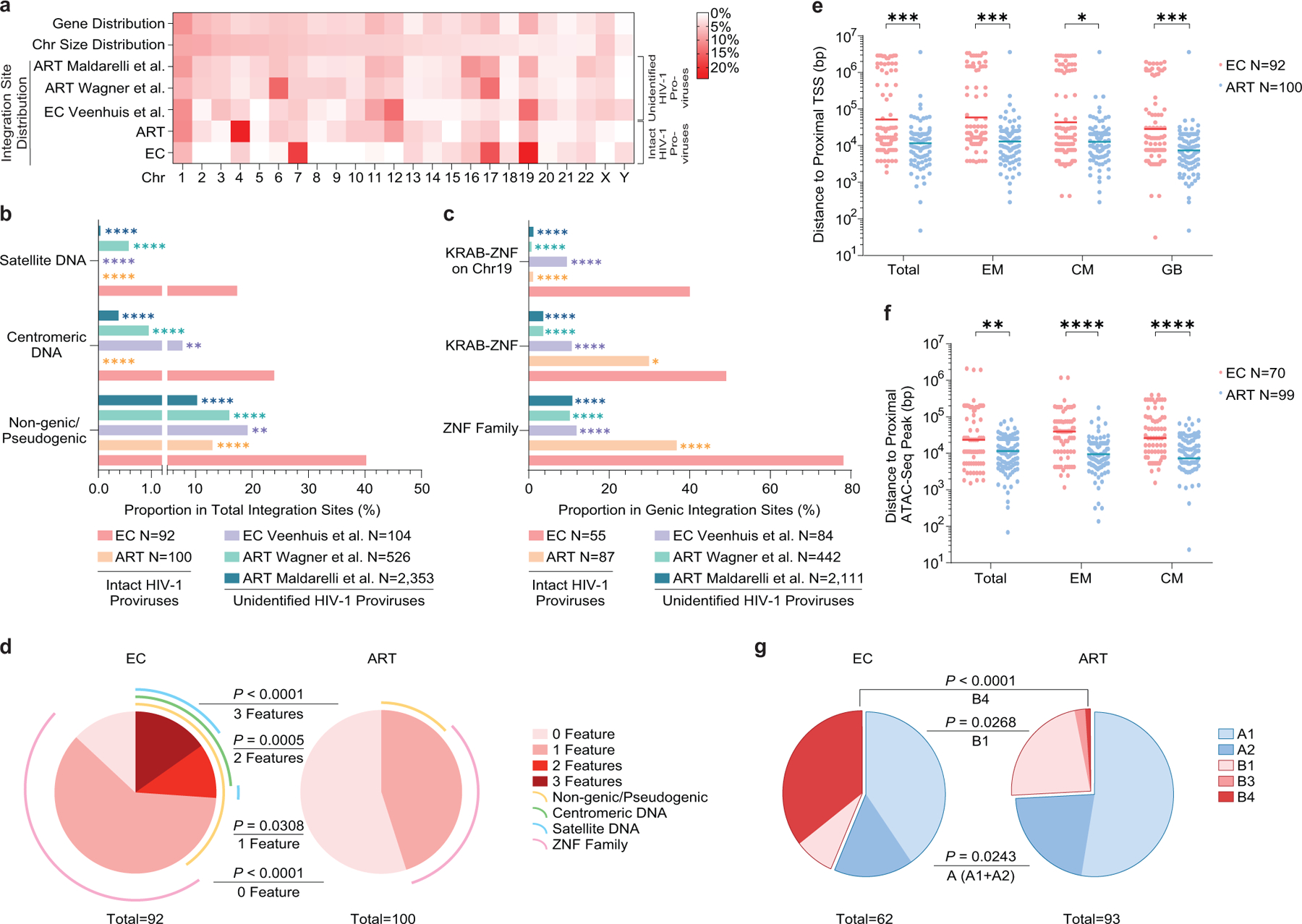
(a): Heatmap indicating the relative proportion of proviral integration sites of intact proviruses in each chromosome in ECs, relative to corresponding data from long-term ART treated individuals13. Proviral integration site data from prior publications are shown for comparative purposes (Veenhuis et al.7, Maldarelli et al16, Wagner et al.14); integration sites from intact and defective proviruses were not distinguished in these studies. Contributions of each chromosome to total number of genes (first row) and to total size of human genome (second row) are included as references. (b-c): Proportion of near full-length intact proviruses located in indicated genomic regions. Data from near full-length intact proviral sequences in long-term ART-treated individuals (ART) are shown for reference purpose13; chromosomal integration sites from unselected (intact and defective) proviral sequences in ECs (Veenhuis et al.7) and in ART-treated individuals (Maldarelli et al16, Wagner et al.14) are also shown for comparison. (d): SPICE diagrams58 demonstrating proportion of intact proviruses with indicated chromosomal integration site features in ECs and ART-treated individuals. (e-f): Chromosomal distance between integration sites of intact proviruses and the most proximal transcriptional start sites (TSS, determined by RNA-Seq) (e) or to the most proximal ATAC-Seq peak (f) in autologous total, central-memory and effector-memory CD4+ T-cells and in GB. Horizontal lines reflect the geometric mean. (g): Proportions of proviral sequences located in structural compartments A and B, as determined based on Hi-C-Seq data published by Rao et al28. Chromosomal integration regions not covered in the study by Rao et al. were excluded from analysis. (f-g): Sequences in genomic regions included in the blacklist for functional genomics analysis identified by the ENCODE and modENCODE consortia27 were excluded due to absence of reliable ATAC-Seq and Hi-C-Seq reads in such repetitive regions. (a-g): All members of clonal clusters were included as individual sequences. (****p<0.0001, ***p<0.001, **p<0.01, *p<0.05, FDR-adjusted two-sided Fisher’s exact tests were used for panels b and c; two-sided Fisher’s exact tests were used for panel d and g, FDR-adjusted two-tailed Mann Whitney U tests were used for panels e and f; all comparisons were made between ECs and reference groups).
Extended Data Figure 6: Epigenetic features of chromosomal integration sites of intact proviruses from ECs.
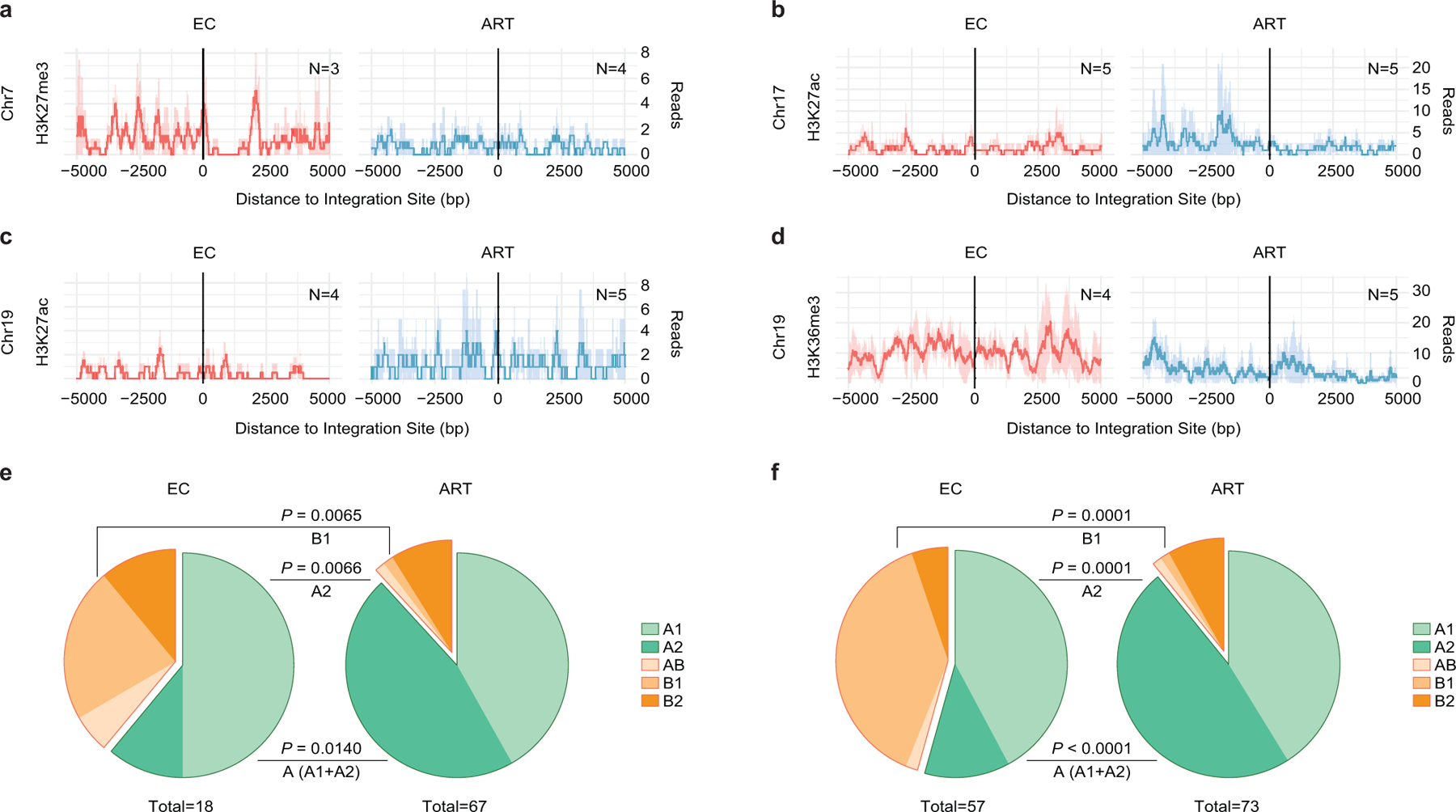
(a-d): Numbers of DNA sequencing reads associated with activating (H3K27ac) or repressive (H3K27me3) histone protein modifications in proximity to integration sites from ECs and long-term ART-treated individuals; median and confidence intervals (defined by one standard deviation) of ChIP-Seq data from primary memory CD4+ T-cells included in the ROADMAP repository25 are shown. Negative distances indicate genomic regions upstream of the HIV 5’ LTR host-viral junction, while positive distances indicate regions downstream of the 3’ LTR viral-host junction. DNA sequencing reads associated with H3K36me3, an activating chromatin mark that is atypically enriched in KRAB-ZNF genes on Chromosome 19, are also shown28. (e-f): Proportions of intact proviral sequences located in structural compartments A and B (and associated sub-compartments) by counting clonal sequences once (e) or by counting clonal sequences individually (f), as determined based on alignment of chromosomal integration sites of intact proviruses to Hi-C-Seq data from Jurkat cells29. Chromosomal integration regions not covered in the Jurkat cell study were excluded from the analysis. Compartment B4 was not assessed in the source data29 for this analysis. Two-sided Fisher’s exact tests were used for statistical comparisons, nominal p-values are reported. (a-f): Sequences in genomic regions included in the blacklist for functional genomics analysis identified by the ENCODE and modENCODE consortia27 were excluded due to absence of reliable ChIP-Seq and Hi-C-Seq reads in such repetitive regions.
Extended Data Figure 7: Accessory chromosomal integration site features of intact proviral sequences from ECs.
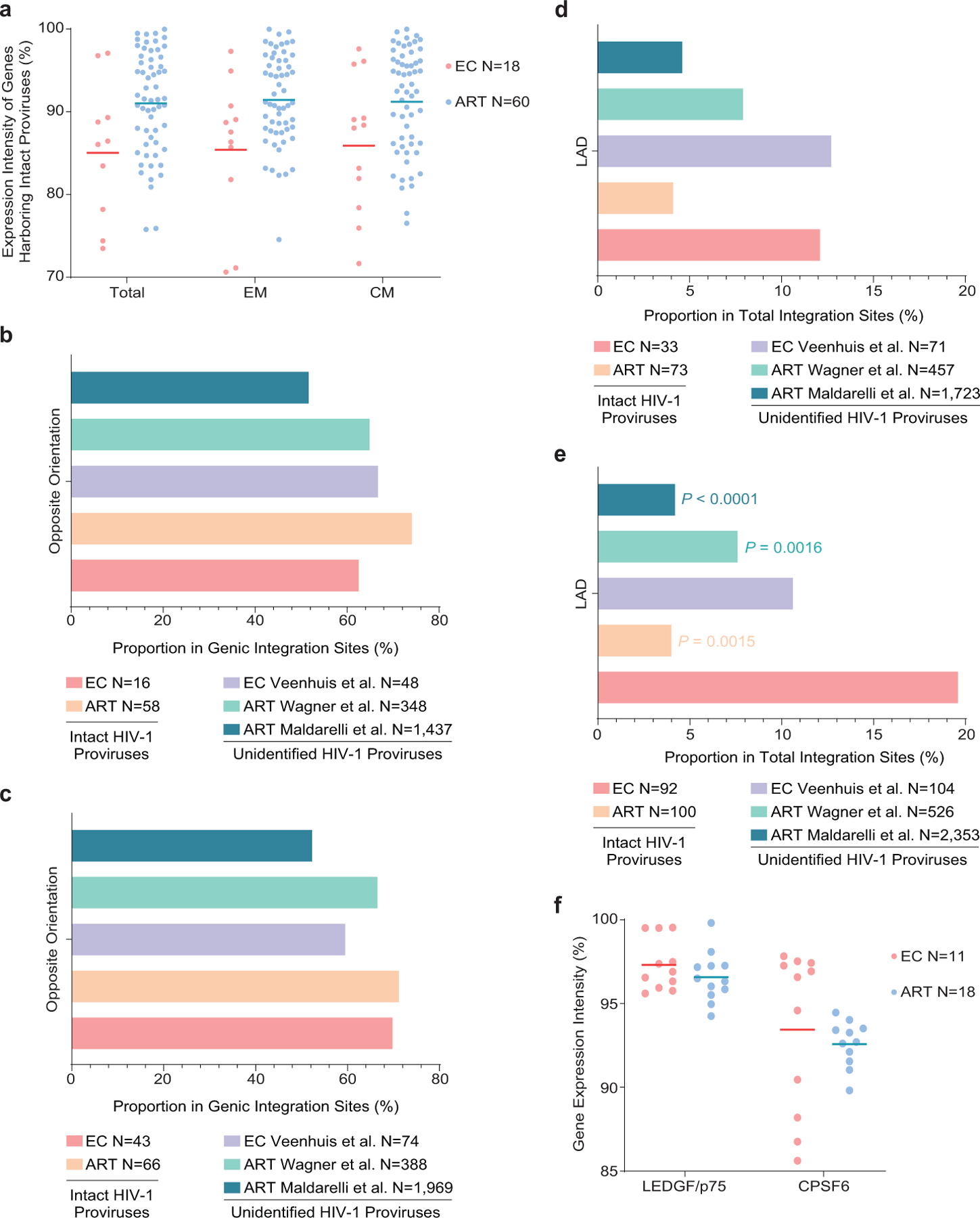
(a): Expression of host genes harboring intact proviral sequences in ECs and long-term ART-treated individuals, as determined by autologous RNA-Seq data in total, central-memory (CM) and effector-memory (EM) CD4+ T-cells. Gene expression percentiles are indicated. (b-c): Orientation of intact proviruses relative to host genes in ECs and long-term ART-treated individuals. All data for genic integration sites with exclusive orientation towards host genes are included. Integration site data from prior studies involving ECs (Veenhuis et al.7) and ART-treated individuals (Maldarelli et al.16, Wagner et al.14) are shown for comparative purposes. (d-e): Proportion of intact proviruses from ECs and long-term ART-treated individuals in lamina-associated domains (LAD), determined using Lamin B1-DNA adenine methyltransferase Identification (DamID) by Robson et al.60 for resting Jurkat cells. Integration site data from prior studies involving ECs (Veenhuis et al.7) and ART-treated individuals (Maldarelli et al.16, Wagner et al.14) are shown for comparative purposes. (b, d): Clonal proviruses were counted once; (c, e): clonal proviruses were counted as individual sequences (FDR-adjusted two-sided Fisher’s exact tests). (f): Expression of LEDGF/p75 and CPSF6 mRNA in autologous total CD4+ T-cells from ECs and long-term ART-treated individuals, as determined by RNA-Seq. Gene expression percentiles are indicated. (a, f): Horizontal lines reflect the geometric mean. All comparisons were made between ECs and reference groups.
Extended Data Figure 8: Chromosomal integration site features of in-vitro infected CD4+ T-cells from ECs and HIV-1 negative study participants.
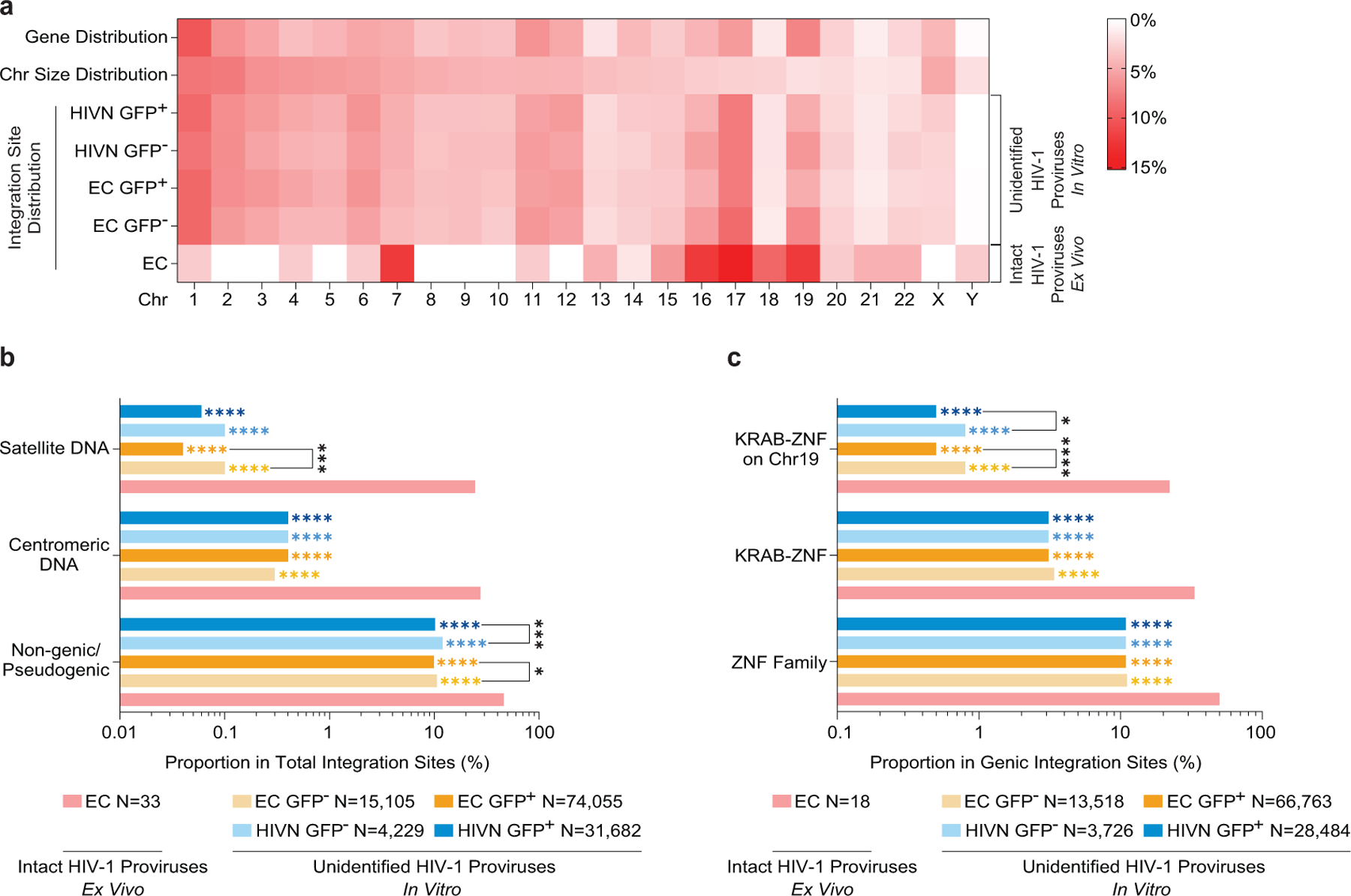
(a): Heatmap indicating the relative proportion of proviral integration sites in sorted GFP+/GFP− in-vitro infected CD4+ T-cells (determined by LM-PCR48) from ECs and HIV-1 negative study participants (HIVNs), relative to proviral integration sites of intact proviruses in each chromosome in ECs; integration sites from intact and defective proviruses were not distinguished in in-vitro infection studies. Data from GFP+ (n=74,055) and GFP− (n=15,105) CD4+ T-cell populations from ECs and from GFP+ (n=31,682) and GFP− (n=4,229) CD4+ T-cell populations from HIVNs were included. Contributions of each chromosome to total number of genes (first row) and to total size of human genome (second row) are included as references. (b-c): Proportion of proviral integration sites located in indicated genomic regions (b) or defined genes (c). Data from near full-length intact proviral sequences in ECs are indicated for reference. (****p<0.0001, ***p<0.001, *p<0.05, FDR-adjusted two-sided Fisher’s exact tests or two-tailed Chi-square tests were used as appropriate; p-values indicating comparisons made between ECs and each in-vitro infection group are shown in corresponding colors).
Extended Data Table 1:
Demographical and clinical characteristics of all study participants.
| Elite Controllers (EC) | ART-treated Participants (ART) | |
|---|---|---|
| Number of participants | 64 | 41 |
| Age in years* | 57 (31 – 75) | 55 (34 – 73) |
| Female (%) | 18.75% | 21.95% |
| CD4 counts* | 908† (450 – 2,282) | 726 (316 – 1,649) |
| Viral loads | Under limit of detection | Under limit of detection |
| Number of viral load tests* | 18 (3 – 91) | 32.5 (4 – 73) |
| HLA-B*27/B*57 (%) | 27.34%‡ | 8.75% |
| Time since diagnosis (year)* | 17 (1 – 34) | 17 (5 – 35) |
| Recorded duration of undetectable viremia (year)* | 9 (1 – 24) | 9 (2 – 19) |
median with range;
P = 0.0006, tested using two-tailed Mann-Whitney U test;
P = 0.0012, tested using two-sided Fisher’s exact test.
Supplementary Material
Acknowledgement
XGY is supported by NIH grants HL134539, AI116228, AI078799, DA047034 and the Bill and Melinda Gates Foundation (INV-002703). ML is supported by NIH grants AI098487, AI135940 AI114235, AI117841, AI120008, DK120387. ML and XGY are Associated Members of the BEAT-HIV Martin Delaney Collaboratory (UM1 AI126620). ANE is supported by NIH grant AI052014. Support was also provided by the Harvard University (HU) and University of California at San Francisco (UCSF)/Gladstone Institute for HIV Cure Research Centers for AIDS Research (P30 AI060354 and P30 AI027763, respectively) which are supported by the following institutes and centers co-funded by and participating with the U.S. National Institutes of Health: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR, and by HU CFAR Developmental Awards (SH). The authors gratefully acknowledge the MGH DNA core facility. RFS and JMS are supported by the NIH Martin Delaney I4C (UM1 AI126603), BEAT-HIV (UM1 AI126620), and DARE (UM1 AI126611) Collaboratories and by the Howard Hughes Medical Institute and the Bill and Melinda Gates Foundation (OPP1115715). Additional support for the SCOPE cohort was provided by the Delaney AIDS Research Enterprise (DARE; AI096109, AI127966) and the amfAR Institute for HIV Cure Research (amfAR 109301). The International HIV Controller Cohort is supported by the Bill and Melinda Gates Foundation (OPP1066973), the Ragon Institute of MGH, MIT and Harvard, the NIH (R37 AI067073 to BDW) and the Mark and Lisa Schwartz Family Foundation. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Footnotes
Data availability statement
RNA-Seq and ATAC-Seq data have been deposited in a public repository (NCBI GEO, accession number GSE144334). Due to study participant confidentiality concerns, full-length viral sequencing data cannot be publicly released, but will be made available to investigators upon reasonable request and after signing a coded tissue agreement. The Los Alamos HIV Sequence Database Hypermut 2.0 and the Los Alamos HIV Immunology Database 2.0 are available at www.hiv.lanl.gov. The iMethyl database is available at http://imethyl.iwate-megabank.org. ROADMAP epigenomic data are available at http://www.roadmapepigenomics.org.
ANE has received fees from ViiV Healthcare Co. within the past year for work unrelated to this project. All other authors declare that conflicts of interest do not exist.
References
- 1.Saez-Cirion A & Pancino G HIV controllers: a genetically determined or inducible phenotype? Immunol Rev 254, 281–294 (2013). [DOI] [PubMed] [Google Scholar]
- 2.McLaren PJ & Carrington M The impact of host genetic variation on infection with HIV-1. Nat Immunol 16, 577–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Migueles SA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29, 1009–1021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaiha GD, et al. Structural topology defines protective CD8(+) T cell epitopes in the HIV proteome. Science 364, 480–484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migueles SA & Connors M Success and failure of the cellular immune response against HIV-1. Nat Immunol 16, 563–570 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Blankson JN, et al. Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J Virol 81, 2508–2518 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veenhuis RT, et al. Long-term remission despite clonal expansion of replication-competent HIV-1 isolates. JCI Insight 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee GQ, et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 127, 2689–2696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yukl SA, et al. Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog 9, e1003347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Popper K Die Logik der Forschung Zur Erkenntnistheorie der modernen Naturwissenschaft. , (Springer-Verlag, Wien, 1935). [Google Scholar]
- 11.Ho YC, et al. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 155, 540–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melamed A, et al. Genome-wide determinants of proviral targeting, clonal abundance and expression in natural HTLV-1 infection. PLoS Pathog 9, e1003271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Einkauf KB, et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J Clin Invest 129, 988–998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner TA, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345, 570–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohn LB, et al. HIV-1 integration landscape during latent and active infection. Cell 160, 420–432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maldarelli F, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McNulty SM & Sullivan BA Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res 26, 115–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carteau S, Hoffmann C & Bushman F Chromosome structure and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J Virol 72, 4005–4014 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan A, Bisgrove D & Verdin E HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22, 1868–1877 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewinski MK, et al. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol 79, 6610–6619 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder AR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Ecco G, Imbeault M & Trono D KRAB zinc finger proteins. Development 144, 2719–2729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukic S, Nicolas JC & Levine AJ The diversity of zinc-finger genes on human chromosome 19 provides an evolutionary mechanism for defense against inherited endogenous retroviruses. Cell Death Differ 21, 381–387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel MJ, et al. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res 16, 1493–1504 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roadmap Epigenomics C, et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15, 893–900 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amemiya HM, Kundaje A & Boyle AP The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucic B, et al. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat Commun 10, 4059 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komaki S, et al. iMETHYL: an integrative database of human DNA methylation, gene expression, and genomic variation. Hum Genome Var 5, 18008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Affinito O, et al. Nucleotide distance influences co-methylation between nearby CpG sites. Genomics 112, 144–150 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Kauder SE, Bosque A, Lindqvist A, Planelles V & Verdin E Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog 5, e1000495 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marini B, et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 521, 227–231 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Ciuffi A, et al. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11, 1287–1289 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Achuthan V, et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 24, 392–404 e398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Debyser Z, Vansant G, Bruggemans A, Janssens J & Christ F Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 11(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battivelli E, et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. Elife 7(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Estes JD, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med 23, 1271–1276 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP & Schaffer DV Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 122, 169–182 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Burtt NP Whole-genome amplification using Phi29 DNA polymerase. Cold Spring Harb Protoc 2011, pdb prot5552 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Lee GQ, et al. HIV-1 DNA sequence diversity and evolution during acute subtype C infection. Nat Commun 10, 2737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiener B, et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4(+) T Cells from Effectively Treated Participants. Cell Rep 21, 813–822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinzone MR, et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat Commun 10, 728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rose PP & Korber BT Detecting hypermutations in viral sequences with an emphasis on G --> A hypermutation. Bioinformatics 16, 400–401 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siepel AC, Halpern AL, Macken C & Korber BT A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retroviruses 11, 1413–1416 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serrao E, Cherepanov P & Engelman AN Amplification, Next-generation Sequencing, and Genomic DNA Mapping of Retroviral Integration Sites. J Vis Exp (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trombetta JJ, et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing Current protocols in molecular biology / edited by Ausubel Frederick M. … [et al.] 107, 4 22 21–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corces MR, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corces MR, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laird GM, Rosenbloom DI, Lai J, Siliciano RF & Siliciano JD Measuring the Frequency of Latent HIV-1 in Resting CD4(+) T Cells Using a Limiting Dilution Coculture Assay. Methods Mol Biol 1354, 239–253 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Bruner KM, et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 566, 120–125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J Clin Invest 121, 1549–1560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yukl SA, et al. HIV latency in isolated patient CD4(+) T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci Transl Med 10(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuo HH, et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4(+) T Cells. Immunity (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini YH, Yosef. Controlling the false discovery rate: A practical and powerful approach to multiple testing. . Journal of the Royal Statistical Society, Series B: Methodological 57, 289–300 (1995). [Google Scholar]
- 58.Roederer M, Nozzi JL & Nason MC SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A 79, 167–174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.team, R.c. R: A language and environment for statistical computing. (ed. R Foundation for Statistical Computing, V., Austria: ) (http://www.R-project.org, 2019). [Google Scholar]
- 60.Robson MI, et al. Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Res 27, 1126–1138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.