

Abstract
During the last decades intermediate filaments (IFs) have emerged as important regulators of cellular signaling events, ascribing IFs with functions beyond the structural support they provide. The organ and developmental stage‐specific expression of IFs regulate cell differentiation within developing or remodeling tissues. Lack of IFs causes perturbed stem cell differentiation in vasculature, intestine, nervous system, and mammary gland, in transgenic mouse models. The aberrant cell fate decisions are caused by deregulation of different stem cell signaling pathways, such as Notch, Wnt, YAP/TAZ, and TGFβ. Mutations in genes coding for IFs cause an array of different diseases, many related to stem cell dysfunction, but the molecular mechanisms remain unresolved. Here, we provide a comprehensive overview of how IFs interact with and regulate the activity, localization and function of different signaling proteins in stem cells, and how the assembly state and PTM profile of IFs may affect these processes. Identifying when, where and how IFs and cell signaling congregate, will expand our understanding of IF‐linked stem cell dysfunction during development and disease.
Keywords: differentiation, stem cells, cytoskeleton, cell signaling, regeneration
Abbreviations
- BEC
biliary epithelial cells
- CNS
central nervous system
- Dll
Delta‐like ligand
- EC
endothelial cell
- ECM
extracellular matrix
- EMDM
Emery‐Dreifuss muscular dystrophy
- EMT
epithelial‐mesenchymal transition
- GFAP
glial fibrillary acidic protein
- HES
hairy and enhancer of split
- HEY
hairy/E(spl)‐related with YRPW motif
- HGPS
Hutchinson‐Gilford Progeria Syndrome
- IBD
inflammatory bowel disease
- IF
intermediate filaments
- KO
knockout
- MMEC
mouse mammary epithelial cell
- MSC
mesenchymal stem cell
- NF
neurofilament
- NICD
Notch intracellular domain
- NPC
neural progenitor cell
- NSC
neural stem cell
- PC
pachyonychia congenital
- TA
transit amplifying cell
- VSMC
vascular smooth muscle cell
1. BACKGROUND
Cells, just like humans, contain a skeleton providing them with shape, support, and mechanical rigidity. The cellular skeleton, called the cytoskeleton, is made up of different types of fibers such as the actin filaments, microtubules, and intermediate filaments. This review focuses on the intermediate filaments and how they interact with developmental signaling pathways to regulate cell fate decisions. Intermediate filaments (IFs) have tissue‐specific expression profiles that change according to developmental stage and many IFs are upregulated in regenerating tissues. It is known that mechanical forces and intercellular interactions regulate cell fate decisions, and there IFs play a major role. 1 How this cell fate regulation is mediated remains mainly unresolved, despite recent enlightening findings. Lim et al 2 recently demonstrated that cytoskeletal organization directs cell fate already prior to formation of the morula. Keratins are asymmetrically distributed between blastomeres to the outermost cells, promoting apical polarization, and controlling developmental cell signaling, and thereby directing trophectoderm cell fate commitment. 2 These findings provide intriguing early evidence of IF‐mediated cell fate specification during embryonic development. Further, the prevalence of disease‐causing mutations in genes encoding for different IFs have revealed them as cellular structures impacting functions connected to cell signaling, organelle architecture, and transcriptional regulation. 3 , 4 Yet, the underlying molecular mechanisms linking IFs to the diseases are still mostly unknown. In this review, we want to highlight IFs and their role in steering developmental processes and cell fate decisions, by modulating cell signaling dynamics (Figure 1). We will emphasize the role of IFs in regulation of mesenchymal, neuronal, epithelial, and myogenic cell determination and discuss the role of nuclear IFs in relation to disease.
FIGURE 1.
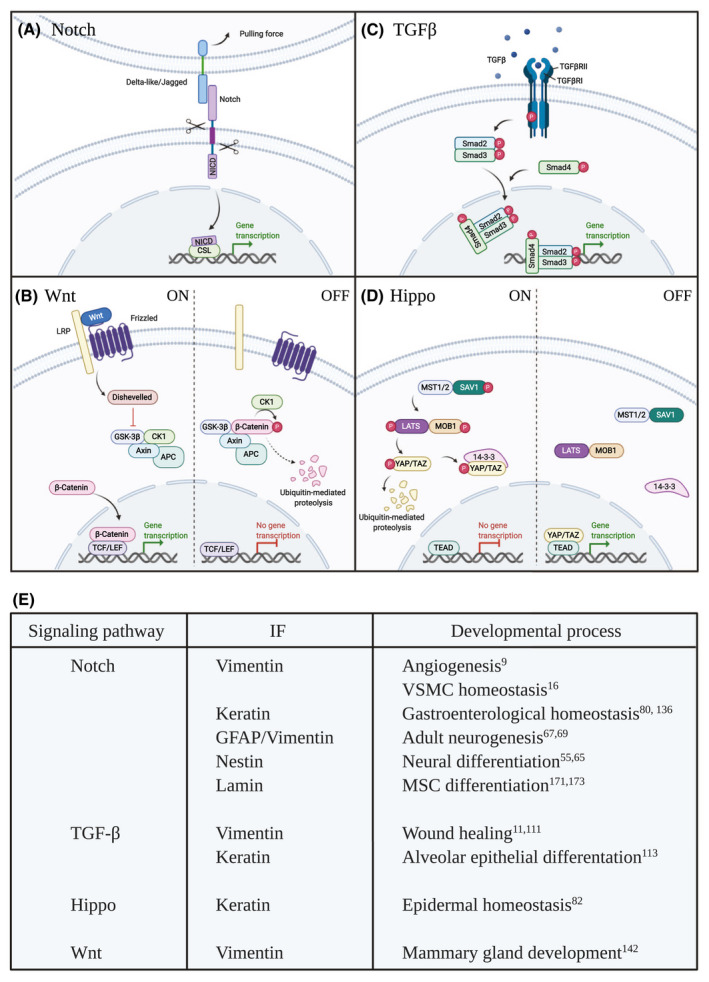
Intermediate filaments cross talk with different signaling pathways. A, The Notch signaling pathway is activated when a Notch ligand on a contacting cell binds to the Notch receptor. This initiates trans‐endocytosis of the ligand‐receptor complex and generates a pulling force, leading to proteolytic processing of the Notch receptor and release of the Notch intracellular domain (NICD). NICD translocates to the nucleus and interacts with the transcriptional regulator CSL (CBF‐1, Suppressor of Hairless, Lag1) activating transcription of target genes. B, Wnt signaling is activated as soluble Wnt molecules bind to the Frizzled and LRP receptors. Frizzled recruits Dishevelled and inhibits the formation of degradation complex consisting of GSK‐3β, CK1, APC, and Axin, allowing β‐Catenin to translocate to the nucleus and bind TCF/LEF initiating transcription of target genes. In the absence of a Wnt signal, GSK‐3β, CK1, APC, and Axin forms a degradation complex that phosphorylates β‐Catenin. This phosphorylation targets β‐Catenin for ubiquitination and proteasomal degradation. C, TGFβ initiates assembly of a tetrameric receptor complex (two copies of both TGFβRI and TGFβRII) at the cell membrane, followed by receptor phosphorylation and activation of the receptor kinase domain. Thereby the TGFβ receptor complex further phosphorylates Smad proteins, which subsequently form dimers and recruit Smad4. The formed Smad complex is transferred into the nucleus to control the transcription of target genes. D, The Hippo signaling pathway is made up of a kinase cascade where MST1/2 kinases (denoted Hippo in Drosophila melanogaster) and SAV1 form a complex to phosphorylate and activate LATS. LATS forms a complex with MOB1 to further phosphorylate and inhibit the function of the transcriptional activator YAP/TAZ (eg, through degradation or interaction with other proteins such as 14‐3‐3). Non‐phosphorylated YAP/TAZ enters the nucleus and binds TEAD transcription factors initiating expression of target genes. E, Overview of the interaction between signaling pathways and intermediate filaments (IFs) during different developmental processes
2. MESENCHYMAL CELL FATE
2.1. Vimentin is the major IF of mesenchymal cells
Vimentin is a ubiquitously expressed protein and the major IF of mesenchymal cells. One of the main roles of vimentin is to provide cells with mechanical stability. In addition to its mechanical properties vimentin is an important regulator of epithelial to mesenchymal transition (EMT) both during normal development and disease, and is therefore, often used as a marker for EMT. Vimentin is also known to be involved in processes that involve migratory behavior of cells during wound healing and tissue repair.
Originally vimentin knockout (KO) mice were considered to display a very mild, if any, phenotype. 5 Still, recent findings have linked vimentin deficiency with several types of developmental deficiencies, some of which may not become evident until the animal ages or is exposed to stress. 6 , 7 , 8 , 9 , 10 , 11 Already in 1989, Capetanaki et al 12 found that overexpression of vimentin in mice interferes with the development of the eye lens. During early development of the mouse embryo, vimentin expression is observed already during E8.5 in cells of the primitive mesoderm and its expression in neuronal cells also precedes the expression of tissue‐specific IFs. 13 Recently, Cogné et al 14 discovered a vimentin mutation (L387P) that has been connected to a human disease phenotype. The mutation gives rise to a less stable vimentin variant, which is unable to form functional filaments. The patient carrying this mutation shows severe developmental defects manifesting as lipodystrophy, peripheral neuropathy, and frontonasal dysostosis among others. 14
2.2. Vimentin regulates cardiovascular tissue development and homeostasis
Vimentin has been associated with angiogenesis and vascular remodeling. 8 , 9 , 15 , 16 Halfway through embryonic development all blood vessels express vimentin. 13 An intact vimentin network is important for maintenance of endothelial barrier integrity 17 and ablation of vimentin in embryonic stem cells impairs their ability to differentiate toward an endothelial phenotype. 10 In ECs, vimentin has been shown to bind the Notch ligand Jagged1 and provide the force required for proper activation of the Notch signaling pathway. 9 , 16 Interestingly, the transmembrane domain of the Notch receptor has been linked to endothelial barrier stability through interaction with, for example, VE‐cadherin, Lar, and the Rac1 GEF Trio 18 and it is intriguing to speculate whether this complex also interacts with vimentin at the endothelial cell junctions. 19 During formation of new blood vessels vimentin interacts with calpain and MT‐MMP‐1 facilitating degradation of the extracellular matrix (ECM), pawing way for new endothelial sprouts. 20 Endothelial sprouting is characterized by tip vs stalk cell selection, during which binding of vimentin to the Notch ligand Jagged1 balances and maintains the ratio between these cell identities. In absence of vimentin, Jagged1‐mediated signaling through Notch is impaired, leading to reduced endothelial branching 9 in an antiangiogenic fashion characteristic for prevalent Delta‐like (Dll) signaling (Figure 2). Schiffers et al 15 found that carotid artery ligation resulted in media hypertrophy in vimentin KO mice. Additionally, lack of vimentin leads to thickening of the endothelial basement membrane resulting in increased carotid stiffness. 8 Both endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) express vimentin, and absence of vimentin leads to a dedifferentiated VSMC phenotype. 16 Differentiation of the VSMCs layer starts at the endothelial cell layer and propagates outward through a mechanism called lateral induction, where Jagged1 activates Notch signaling through a positive feedback loop. In light of the effects of vimentin deficiency on endothelial functions 9 one must consider that lack of vimentin may disrupt Jagged activation of Notch and thereby lateral induction, resulting in reduced VSMC differentiation and enhanced proliferation of synthetic VSMCs. 16 The tuning of Jagged‐mediated Notch signaling by vimentin in both endothelia and VSMCs provides direct evidence of intermediate filaments as potent cell fate mediators.
FIGURE 2.

Lack of vimentin delays vascular development. A, At E12.5 embryos lacking vimentin display a less refined vascular network due to disrupted angiogenesis. In absence of vimentin, the function of the proangiogenic Notch ligand Jagged1 is perturbed, leading to reduced vascular sprouting. Modified from Ref.9 B, Summary of the molecular processes connected to the Vim‐/‐ phenotype
2.3. Vimentin and EMT
Vimentin has been well established as a regulator of EMT and is frequently used as a marker to identify cells that have undergone transition to a mesenchymal phenotype. 21 , 22 , 23 EMT is characterized by an altered cell shape with front‐rear polarity, an increased expression of vimentin at the expense of keratins, and a loosening of cell‐cell junctions with a concomitant increase in motility and invasion. 24 , 25 EMT is critical for tissue morphogenesis and development and is also implicated in cancer metastasis. 26 , 27 EMT is first observed during gastrulation when mesodermal cells are generated from the epiblast. 26 Vimentin is considered a central player in the EMT process and vimentin expression alone has been reported to induce the phenotypic changes associated with a mesenchymal cell fate. 22 Vimentin allows for cell flexibility through its filament network, which also forms interactions with actin filaments and microtubules to further aid in the motility and shape of the cell. These interactions are both direct and indirect through adaptor proteins such as plectins. 28 , 29 , 30 , 31 , 32 The cooperation of the cytoskeleton in this context has been shown to increase migration through elongation of invadopodia, and by influencing focal adhesions and actomyosin contractility. 33 , 34 , 35 , 36 , 37
Vimentin further interacts with various EMT‐linked signaling proteins, transcription factors, and adaptor proteins, which collectively affect multiple signaling pathways. These include regulation of the EMT‐related transcription factor SLUG and oncogenic H‐Ras through the tyrosine kinase Axl. 38 Vimentin also binds to phosphorylated ERK (pERK) protecting it from dephosphorylation. 39 Further, vimentin has been shown to act as a scaffold recruiting SLUG to ERK in order to induce SLUG phosphorylation. 40 The direct interaction between Akt1 and vimentin induces phosphorylation of serine 39 in the head domain of vimentin, protecting vimentin from proteolysis and leading to enhanced migration and invasion in vivo. 41 Other direct interactions with vimentin that can influence the EMT phenotype include the tumor suppressor APC, integrin receptors, NLRP3 inflammasomes, and the scaffolding protein Scribble. 42 , 43 , 44 , 45 Vimentin can also bind 14‐3‐3 and this interaction has been demonstrated to prevent the MAPK pathway component Raf from binding to 14‐3‐3. 46 Similarly, vimentin interacts with the tumor suppressor Beclin through 14‐3‐3. 47 As 14‐3‐3 is a multifunctional adaptor, vimentin may influence multiple other 14‐3‐3 interacting proteins by regulating 14‐3‐3 availability.
Overexpression of vimentin is frequently reported in various epithelial cancers and is often correlated with invasive and migratory metastatic tumors and poor prognosis. 48 , 49 , 50 , 51 , 52 This topic is under intense investigation as the targeting of vimentin in cancer implies a promise of antimetastatic effects, but this complex area is outside the scope of this review and we refer the reader to multiple excellent reviews on vimentin in cancer and EMT. 23 , 24 , 53
3. NEURONAL CELL FATE
3.1. Different IFs conjoin in the CNS
In a process called neurogenesis neural stem cells (NSCs) give rise to neurons and glial cells, the main constituents of the central nervous system (CNS). Development of the nervous system is characterized by alternating expression of different IFs. Undifferentiated cells express vimentin, neuroepithelial cells and neuroblasts express nestin, peripherin, and α‐internexin. 54 , 55 As neuronal differentiation is initiated, the cells start expressing tissue‐specific intermediate filaments called neurofilaments (NF). NF‐light (NF‐L) is the first NF to be expressed followed by NF‐medium (NF‐M) during neurite sprouting. NF‐high (NF‐H) is mainly expressed postnatally at the later stages of neuronal differentiation (reviewed in Ref. 54). Neurogenesis is initiated early during development when neural stem cells from the neural tube give rise to progenitor cells called radial glia. The radial glia will generate neurons during the later stages of neurogenesis as well as astrocytes and oligodendrocytes further during gliogenesis and oligodendrocytogenesis, respectively. 56 In the adult mammalian brain NSCs are found in the ventricular‐subventricular zone and the subgranular zone. In addition to NFs, neurons may also express other intermediate filaments such as nestin and vimentin. The main intermediate filament in astrocytes is glial fibrillary acidic protein (GFAP), 57 but immature astrocytes express both nestin and vimentin. 58 Interestingly, mice devoid of nestin, vimentin, or GFAP develop normally and do not show signs of CNS aberrations. 5 , 57 , 59 The changes in IF expression during neuronal development coincides with cell differentiation and specification toward distinct cellular lineages. Degradation of nestin in NSCs marks the initial steps of neuronal differentiation and this degradation has been linked to simultaneous loss of Notch signaling, resulting in proteosomal degradation of nestin. Thus, loss of Notch leads to subsequent degradation of nestin, as ectopic expression of the Notch intracellular domain (NICD) maintains nestin levels and sustains the undifferentiated cellular state. 55 Vimentin expressed by astrocytes prior to differentiation, is to some extent replaced by GFAP as the astrocytes mature. 60
3.2. Astrocytic IFs regulate neuronal differentiation and regeneration
GFAP is described as the last IF to be expressed during development. 13 GFAP is predominantly expressed by the astrocytes of the CNS, which switch from expressing vimentin to expressing GFAP during the differentiation process. Mice lacking GFAP develop normally and are capable of producing offspring. 57 , 61 , 62 GFAP deficiency has been linked to aberrant long‐term potentiation and long‐term depression of hippocampal and cerebellar neurons, respectively, 61 , 63 together with a reduced eye blinking response. 63 Notch‐induced nuclear factor 1A (NF1A) drives GFAP expression through demethylation of the Gfap promoter. 64 Mice lacking nestin are also viable. 59 Nestin does not appear to be required for the proliferation of NSCs. Still, nestin KO mice display increased neurogenesis in the hippocampal dentate gyrus. The nestin‐mediated negative effects on neuronal differentiation are not NSC intrinsic, but rather mediated through astrocyte‐initiated Notch signaling. 65 Nestin is involved in regulating vesicle dynamics in astrocytes and thereby also steers Notch signal activation from astrocytes, 65 , 66 as Notch ligands are endocytosed by the ligand‐presenting cell upon interaction with Notch receptors. In absence of nestin, the number of Jagged containing vesicles in astrocytes is reduced and the distribution of Jagged into different vesicular compartments is perturbed, leading to reduced Notch signaling and enhanced neural differentiation. 65
Reactive gliosis, a response by astrocytes to injury, is characterized by enhanced expression of intermediate filaments in astrocytes. Mice lacking vimentin and GFAP show an altered response to trauma, increased neuronal differentiation and better survival of neural grafts. 67 Astrocytes are the supporting cells of the CNS and they regulate the neurogenic niche through cell‐contact and paracrine signaling. 68 Wilhelmsson et al 69 found that the increase in neuronal differentiation in mice lacking vimentin and GFAP was due to reduced Notch signaling between astrocytes and neuronal precursors (Figure 3). Neuronal differentiation is regulated by Notch signaling in an inhibitory manner. 70 Thus, lack of vimentin and GFAP enhances neuronal differentiation as a direct consequence of reduced Notch signaling. The effects are, however, not as severe as in CSL (CBF‐1, Suppressor of Hairless, Lag‐1) KO mice. CSL is a part of the Notch transcriptional complex and mediates activation of Notch target gene expression (Figure 1A). In these mice the Notch signaling response is completely inhibited leading to exaggerated neuronal differentiation and stem cell depletion. 71 Such a difference in phenotype severity could be explained by compensatory Dll‐mediated Notch signaling in the mice lacking vimentin and GFAP. This notion is supported by the fact that vimentin has been shown to specifically interact with Jagged1, but not Dll ligands. 9 Interestingly, the phosphorylation status of vimentin has been linked to neuronal differentiation. Neurospheres extracted from mice with mutated vimentin phosphorylation sites (mitotic phosphorylation sites mutated from serine to alanine: S6A, S24A, S38A, S46A, S55A, S64A, S65A, S71A, S72A, S82A, and S86A) show enhanced neuronal differentiation. The increase in differentiation is not caused by disturbed cell‐cell communication between astrocytes and progenitors cells, suggesting perturbation of a cell intrinsic signaling mechanism within the neurosphere cell population. 72 Still, it is tempting to speculate that hampering with vimentin dynamics affects the interaction with Jagged1. Interaction of vimentin with other proteins, such as 14‐3‐3, has been shown to be phosphorylation dependent 46 and vimentin phosphorylation plays a role in proper localization of some cell surface proteins. 73 Further, Hagemann et al 74 showed disrupted adult neurogenesis in a mouse model for Alexander disease, a syndrome caused by mutations on the gene coding for GFAP. 75 In these mice, protein aggregation exhausts the proteasomal degradation machinery of the cell, disrupting degradation of NICD in GFAP expressing neural progenitors, and perturbing the balance between neurogenesis and gliogenesis. 74 , 76 , 77
FIGURE 3.

Astrocytic intermediate filaments regulate Notch‐mediated neurogenesis. A, Expression of GFAP and vimentin by astrocytes is required for Notch‐mediated inhibition of neuronal differentiation. In absence of GFAP and vimentin the expression of the Notch ligand Jagged1 is reduced, leading to decreased Notch activation and subsequent neural differentiation. B, Summary of the molecular mechanisms deregulated in absence of GFAP and vimentin. Modified from Ref. 69
4. EPITHELIAL CELL FATE
4.1. Keratins govern epithelial tissues
Keratins are the major IFs of epithelial tissues. With 54 different genes encoding for keratins, these IFs constitute the largest family of IF proteins. Keratins are divided into acidic type I and neutral‐basic type II proteins and they form heteropolymers consisting of type I and type II filaments. Keratins are found in the squamous epithelial cells of the epidermis (K1, K2, K5, K9, K10, & K14), simple type epithelial cells (K7, K8, K18, K19, and K20) and hair follicle epithelial cells (K31, K35, K81, K85, and K86). 78 The endothelium, a specialized type of epithelial tissue lining the walls of blood and lymphatic vessels, expresses minor amounts of keratins 79 with vimentin being the major IF expressed. Endothelial cells are, therefore, described previously in this review. Upon stress, epithelial cells induce expression of specific stress resilient keratins (K6, K16, & K17), which possess‐specific characteristics beneficial, for example, wound healing and inflammation. Keratins are thought to provide cells with mechanical stability and tissues with appropriate architecture, but recently keratins are emerging as signaling platforms or modulators regulating several different cellular processes. 80 , 81 , 82 Mutations affecting keratins, cause an array of diseases called keratinopathies that mainly manifest trough disturbances in skin and nails, for example, epidermolysis bullosa simplex, ichthyosis hystrix, epidermolytic hyperkeratosis, and pachyonychia congenita 83 (full list of keratin‐related diseases found on interfil.org). Meesmann epithelial corneal dystrophy 84 and some gastroenterological disorders have also been linked to keratins. 85 , 86 , 87
4.2. Keratins maintain the protective barrier of the skin
In the stratified epithelia of the skin different keratin pairs are expressed depending on the differentiation status. K5/K14 is expressed by the basal proliferating keratinocytes, and becomes replaced by K1/K10 in the differentiated cells of skin. 88 Loss of K14 is associated with severe skin blistering in mice around postnatal day 2. 89 Ablation of K14 in keratinocytes in vitro causes increased expression of keratinocyte differentiation markers K1 and involucrin, 88 indicating that K14 maintains keratinocytes in an undifferentiated state. Notch signaling is usually known to maintain stem cell fate and counteract cell differentiation. In skin, however, the current view is that Notch activation promotes differentiation of keratinocytes, 90 but some contradicting opinions have also been raised. 91 Blanpain et al 92 showed that Notch is involved in specifying spinous vs basal cell fate. Depletion of K14 decreased proliferation and increased NICD levels in keratinocytes. 88 Correspondingly, inhibition of Notch activity in keratinocytes led to suppressed expression of K1 and involucrin 93 in line with reduced differentiation of keratinocytes. In these cells Notch induces p21Cip expression and withdrawal from the cell cycle, 93 whereas ablation of K14 reduces Akt phosphorylation accompanied by reduced cell cycle progression. 88 p21Cip is an Akt target, thus, activation of Akt through phosphorylation may affect p21Cip and lead to enhanced proliferation, through inhibition of the antiproliferative effects of p21Cip. 94 The signal inducing enhanced Notch expression and promoting keratinocyte differentiation is not yet known, 93 but Notch and K14 intertwine in a complex regulatory feedback system together with p63. p63 regulates K14 expression, 95 and thereby keratinocyte differentiation. Notch‐induced differentiation is regulated by p63, but, on the contrary, Notch also suppresses p63 expression. 96
Mice lacking K16 or K9 display footpad lesions mimicking palmoplantar keratoderma, a common feature of pachyonychia congenital (PC). 97 , 98 In humans PC is caused by mutations in K6a, K6b, K16, & K17, whereas K9 mutations are linked to epidermolytic palmoplantar keratoderma (EPPK). K9 is found in terminally differentiating keratinocytes of the palms and soles, and interestingly ablation of K16 in mice disrupts K9 expression and keratinocyte differentiation. The phenotype can be reversed by reactivation of K9 expression trough topical application of sulforaphane, a known activator of Nrf2 signaling, 99 but Nrf2 does not seem to directly regulate K9 expression. 100 Analysis of paw skin from K16 null mice revealed enhanced expression of several signaling pathway components, for example, Notch and Wnt, involved in epithelial differentiation, 100 but the molecular mechanism causing the aberrant differentiation remains elusive.
14‐3‐3σ, a keratinocyte‐specific isoform of the 14‐3‐3 protein family, docks to different proteins and may also function as a signaling hub, bringing together various signaling molecules. 14‐3‐3σ is expressed by the differentiated cells of the skin and loss of 14‐3‐3σ allows the keratinocytes to escape terminal differentiation by maintaining telomerase activity and reducing p16INK4a expression. 101 14‐3‐3 specifically binds keratins 82 , 102 , 103 , 104 and it is tempting to hypothesize that the interaction with keratin would be essential for bringing 14‐3‐3 into proximity of its other interaction partners. The downstream effector of the Hippo kinase cascade, YAP, interacts with 14‐3‐3 maintaining its cytoplasmic localization (Figure 1D). Recently, Guo et al 82 found that impaired K14 disulfide bond formation distorts the localization of 14‐3‐3σ in keratinocytes. The displacement of 14‐3‐3σ results in enhanced nuclear entry of YAP together with increased proliferation and defected terminal differentiation. 82 YAP signaling has also been linked to Notch suppression in the epidermis, which would further counteract keratinocyte terminal differentiation. 105
When the epidermis is subjected to damage it requires efficient repair and regeneration to minimize blood loss and recreate the tissue barrier to the outside. This restoration process is called wound healing and it activates different cellular processes such as proliferation, migration, and differentiation of cells in the vicinity. This topic has recently been covered in an extensive review, 106 thus, we will only beriefly touch upon some of the key findings. IFs are instrumental for efficient wound closure and involved already in clot formation. 107 K6, K16, and K17 respond to stresses and their expression is enhanced at the injury site while the expression of differentiation‐specific keratins, K1 and K10, is reduced (reviewed in Ref. 108). During embryonic wound closure, which is mediated by fibroblast driven forces, absence of vimentin is detrimental. When it comes to closure of adult wounds, lack of vimentin leads to delayed transdifferentiation of myofibroblasts and their inaccurate localization within the wounded tissue due to migratory defects. 109 In lens epithelial tissue, vimentin is secreted into the extracellular space upon injury. There the extracellular vimentin regulates the differentiation of mesenchymal repair cells into myofibroblasts. 110 Lack of vimentin also perturbs the proliferation of fibroblasts together with altered transformation of keratinocytes toward a migratory phenotype. These complications are linked to impaired TGFβ1 signaling (Figure 1C) and consequently delayed reepithelization. 11 In the alveolar epithelia of the lung, the vimentin promoter contains a SMAD‐binding element, thus, TGFβ directly induces expression of vimentin. 111 In myoblasts, on the contrary, direct SMAD‐binding elements are lacking and the TGFβ regulated vimentin expression is mediated by tandem AP‐1 elements. 112 Similarly, TGFβ has been linked to K8 and K18 expression in differentiating alveolar epithelial cells and their reciprocal expression appears to regulate terminal differentiation. 113 However, the mechanisms behind this cross talk still remain unknown.
4.3. IFs maintain gastroenterological homeostasis
The intestinal simple type epithelial cell layer is constantly renewed, and thus, controlled cell proliferation and differentiation is a prerequisite for maintaining intestinal homeostasis. Both the small and large intestine contain an array of different cell types, and in these cells, keratins constitute the major IFs. Absorptive enterocytes, secretory goblet cells, enteroendocrine cells, and intestinal stem cells are found both in the small and large intestine. Paneth cells and tuft cells are specialized secretory cells of the small intestine with immunoprotective functions. 114 , 115 K8 and K19 is found uniformly throughout the intestine. K20 localizes to the differentiated cells of the villi and K18 is expressed uniformly by the epithelial cells of the colon, but restricted to enterocytes and goblet cells in the small intestine. 116
Notch signaling regulates the proliferation and differentiation of intestinal epithelial cells and is essential for stem cell maintenance. 117 The general view is that Notch activity antagonizes goblet and enteroendocrine cell fates promoting differentiation of enterocytes. 118 It has also been proposed that the dose of Notch activity is essential for further specification between goblet or paneth cell fate vs enteroendocrine cell fate. Notch signaling may regulate cell fate specification in two distinct steps in the intestinal epithelium. First, in uncommitted progenitor cells Notch directs the decision between absorptive progenitors vs secretory progenitors, favoring differentiation along the absorptive lineage. Later, Notch is involved in further specification of secretory progenitors inducing goblet cell fate at the expense of paneth cells. 117 , 119 Wnt signaling is another central player regulating intestinal cell fate and this signaling pathway cross talks with Notch in maintaining gut homeostasis. 120 Wnts are secreted molecules that act in their local environment (Figure 1B). In the intestinal crypts, the paneth cells and stromal cells produce Wnt signals in order to induce proliferation and maintain the pool of the intestinal stem cells. 121 , 122 On the contrary, Wnt also controls the terminal differentiation of the paneth cells. 123
The colon is responsible for water retention and vitamin absorption. Mice lacking K8 show hyper proliferation of colonic epithelial cells, increased crypt length and a phenotype resembling colitis. 124 , 125 , 126 Lack of K8 is linked to reduced Notch levels in colonic epithelial cells. This reduction is followed by decreased mRNA levels of Hey1 and Hey2 (Hairy/E(spl)‐related with YRPW motif), accompanied by a shift in differentiation toward the secretory lineage 80 (Figure 4). This is in line with other studies showing a transition toward secretory cells upon Notch inhibition. 127 , 128 In the K8‐/‐ colon, differentiation toward goblet and enteroendocrine cell fates is promoted at the expense of enterocytes. The number of transit amplifying (TA) cells is also increased in the absence of K8, 80 a finding that contradicts the complete block of proliferation observed upon Notch inhibition. 127 There are indications of a regulatory interaction between Notch and K8, but the possible interacting domains remain under investigation. 80 In addition, K8‐/‐ mice show elevated levels of IL‐22 in the colon. 129 IL‐22 has recently been shown to promote TA cell proliferation and to inhibit expression of Notch and Wnt pathway components, 130 providing further diversity to the disturbed colonic homeostasis in the absence of K8.
FIGURE 4.

Keratins maintains cellular homeostasis in the colon. A, Lack of keratins 8 increases the proliferation of colonic epithelial cells and leads to elongated crypts. B, In absence of keratin 8 the differentiation of intestinal stem cells is shifted toward secretory cell fate due to reduced expression of Notch in colonic epithelial cells. C, Summary of the molecular processes related to the K8‐/‐ phenotype. Modified from Ref. 80
In Drosophila, the Notch target gene Hey controls cellular identity through regulation of different signaling pathways and specific regulation of nuclear lamins. Brodsly et al 131 recently demonstrated that Hey maintains the cell fate of differentiated enterocytes and loss of Hey results in epithelial hyperplasia together with disturbed gut homeostasis. Loss of Hey in enterocytes perturbs the balanced expression of stem cell‐related B‐type lamins (lamDm) and lamins characteristic of differentiated cells (lamin C). Further, lamin C negatively regulates expression of the Notch ligand Delta and enhances surface localization of the Wnt pathway component Armadillo (β‐catenin in mammals), in polyploid cells of the Drosophila midgut. 131 Petrovsky and Großhans 132 interestingly showed that B‐type lamins regulate cytokine‐induced proliferation of intestinal stem cells in the Drosophila midgut. In this study B‐type lamins were found to anatagonise Jak/Stat signaling, 132 providing further evidence of intermediate filaments as regulators of gut homeostasis. Additionally, lamin expression in T‐cells has been shown to play a role in the outcome of inflammatory bowel disease (IBD). Lack of lamin A/C shifts the differentiation of T‐cells toward regulatory T‐cells at the expense of T helper (type 1) cells in a TGFβ and retinoic acid dependent manner, mitigating the IBD phenotype. 133
K19 is not only found in the intestine, but also expressed by biliary epithelial cells (BEC) and liver progenitor cells. 134 Still, ablation of K19 does not cause an evident epithelial phenotype in the liver. 135 However, Chen et al 136 found that induction of cholestasis by introducing a 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC) supplemented diet perturbed the proliferation of liver progenitor cells in K19 KO mice. The number of liver progenitor cells as well as BECs was reduced in absence of K19. 136 Notch2 is known to drive BEC fate 137 and interestingly immunofluorescent analysis of liver samples revealed loss of Notch2 in BECs of K19 KO mice. Bile duct ligation resulted in a similar loss of Notch2 in BECs. 136 It is intriguing to speculate how K19 may regulate Notch2 expression in BECs, as one of the main symptoms of the pediatric disease Alagille syndrome is intrahepatic bile duct paucity. 138 The disease is caused by mutations on either NOTCH2 or JAGGED1, 139 , 140 which implicates that the phenotype displayed by K19 KO mice could be caused by direct deregulation of Notch2 function or through the perturbed activation of Notch2 by the ligand Jagged1.
4.4. Vimentin regulates mammary gland development
The mammary glands develop sequentially during female growth and development starting with quiescence after birth, continuing during puberty and reaching maturity upon pregnancy and lactation. Further, the mammary glands regress upon weaning through apoptosis and ECM remodeling. 141 Vimentin is expressed by basal mammary epithelial cells and stromal cells, whereas luminal cells are devoid of vimentin. 142 Mammary stem cells, on the contrary, express both vimentin and K15, an epithelial stem cell marker, thereby displaying a mixed epithelial‐mesenchymal phenotype. 142 , 143
Mice lacking vimentin are fertile and capable of nurturing their offspring. Nevertheless, vimentin appears to be important for mammary gland development. Peuhu et al 142 found that mammary ductal outgrowth is delayed in mice lacking vimentin (Figure 5). Vimentin regulates the ratio between basal and luminal mouse mammary epithelial cells (MMECs) and loss of vimentin led to loss of basal cell identity followed by increased mammary ductal lumen size. In addition, the epithelial regeneration capacity was impaired in absence of vimentin, as indicated by reduced mammosphere formation by MMECs isolated from Vim‐/‐ mice. Gene expression analysis revealed perturbed expression of several developmentally important players in the basal MMECs, for example, proteins belonging to the Wnt and Hedgehog protein families. 142 However, the functions of the upregulated Wnt isoforms, Wnt11 and Wnt9a, in mammary gland development are still elusive. 144 Notch signaling is involved in the regulation of mammary gland development, especially during luminal lineage commitment. 145 Interestingly, Wnt and Notch signaling pathways come together during mammary stem cell differentiation. Pygo2, a Wnt coactivator, localizes with β‐catenin at the Notch3 locus, inhibiting Notch3 promoted luminal cell fate. 146 Jagged is expressed in the developing mammary gland, mainly in the luminal cells. 145 , 147 Thus, it is tempting to speculate whether vimentin may regulate Jagged function similarly in mammary gland differentiation as in angiogenesis.
FIGURE 5.

Vimentin is required for balanced development of the mammary gland. Lack of vimentin delays mammary ductal outgrowth in mice at 10 weeks of age. In absence of vimentin the ratio of basal vs luminal cells is perturbed resulting in loss of basal cells and enlargement of the ductal lumen. In absence of vimentin expression of Wnt signaling components is deregulated, possibly relating to the loss in basal cell fate. The molecular mechanisms leading to aberrant Wnt signaling are still unknown. Modified from Ref. 142
5. MYOGENIC CELL FATE IS CONTROLLED BY NESTIN
Myogenesis involves proliferation and differentiation of single nucleated myoblasts to form multinucleated contractile myotubes, characteristic of muscle tissue. 148 Nestin is a type VI intermediate filament containing a long tail domain that makes up the majority of the protein, combined with an unusually short head domain. 149 As previously stated, nestin is expressed during development with high expression in neural progenitor cells (NPCs), and it is commonly used as a marker for NPCs. 150 Several nestin KO models have been published. Park et al 151 first described an embryonically lethal phenotype with brain damage as a result of low numbers of NPCs. Another nestin KO model, as well as a nestin RNAi mouse model were both viable without a significant neuronal phenotype, but with defects in myogenesis and neuromuscular junctions. 59 , 152 Regulation of myoblast differentiation is governed by Cdk5 activity levels, and nestin has been detailed to regulate Cdk5‐mediated myogenesis. The tail domain of nestin has been shown to function as a scaffold for active Cdk5 by binding to the Cdk5 activator p35. 152 , 153 , 154 , 155 , 156 The scaffolding of Cdk5/p35 protects the calpain‐mediated cleavage of p35 to its hyperactive form p25, and knockdown of nestin results in hyperactivation of Cdk5/p25 and enhanced differentiation of myoblasts. 155 Indeed, downregulation of either Cdk5 or p35 has been shown to hamper normal muscle development. 157 During muscular injury, nestin is reexpressed during regeneration. 158 Depletion of nestin leads to a similar phenotype in NSCs as in myoblasts, with abnormally increased differentiation. However, in NSCs the regulation is considered to be through Numb/Notch regulation, not Cdk5. 65 K8 and K19 are also expressed in striated muscle 159 and mice devoid of K19 display myopathy with a loss of contractile force in the muscle. 160 This muscle phenotype is related to the redistribution of mitochondria and is not a consequence of deregulated stem cell signaling.
6. NUCLEAR INTERMEDIATE FILAMENTS AND CELL FATE
6.1. Nuclear lamins and disease
Lamins are intermediate filament proteins forming the nuclear lamina. More than 100 different mutations in LMNA have been identified and linked to human disorders, such as Emery‐Dreifuss muscular dystrophy, congenital muscular dystrophy and familial partial lipodystrophy, Hutchinson‐Gilford Progeria Syndrome (HGPS), Mandibuloacral dysplasia type A (MADA), Cardiomyopathy, dilated with hypergonadotropic hypogonadism (CMDHH), and Charcot‐Marie‐Tooth disease 2B1 (CMT2B1) (reviewed in Ref. 161).
Lamins are divided into two families. A‐type lamins, consist of the major isoforms lamin A & C and two minor isoforms C2 & AΔ10. All of these are alternative splicing isoforms of the gene product encoded by LMNA. 162 , 163 B‐type lamins contain the three isoforms B1, B2, & B3. B1 is encoded by LMNB1, whereas Lamin B2 and B3 are alternative splicing isoforms encoded by LMNB2. 164 , 165 , 166 Like other intermediate filaments, lamins consist of an N‐terminal head and a coiled‐coil central domain, which form dimers due to the interchain ionic interactions. 167 One unique feature of lamin A is a C‐terminal tail containing a CaaX (C = Cystein, a = Aliphatic, X = any amino acid) motif important for association with the inner nuclear membrane. 168 This CaaX domain is missing in lamin C. The CaaX domain of the immature form of lamin A, called prelamin A, is farnesylated and this farnesylation eventually gives rise to the mature form of lamin A through proteolytic processing by the zinc metalloprotease ZMPSTE24. 169 B‐type lamins are expressed in most cells throughout development. Lamin A/C, on the contrary, is found mainly in differentiated cells and adult stem cells of mesenchymal origin. 170 Zhang et al 171 summarized the effects of lamin A/C overexpression and silencing in different types of mesenchymal stem cells (MSCs) induced toward adipogenic or osteogenic fates. Knockdown of lamin A/C impaired osteogenic differentiation mainly through perturbation of Runx2‐related mechanisms. The enhanced osteogenic differentiation caused by lamin A/C overexpression is linked to enhanced Wnt and Notch signaling. 171 , 172 , 173 Lamin A/C promotes translocation of β‐catenin into the nucleus. This results in a positive feedback loop as β‐catenin induces the expression of Wnt7b and Wnt10b which further stimulates osteogenic differentiation. 172 The same Wnts are involved in suppressing adipogenic differentiation as a result of lamin A/C overexpression. Wnt10b reduces PPARγ expression, thereby inhibiting adipogenesis. 174 Recently, lamins have been found to protect the nucleus from DNA damage and cell cycle arrest caused by mechanical overload. 175 Swift et al 176 connected lamin A expression and changes in Ig domain conformation to mechanical cues in MSCs. Furthermore, LMNA mutant myoblasts show disturbed responses to mechanical stress. 177 In different studies, lamin mechanosensitivity has been linked to cell fate determination trough cross talk with different signaling pathways, for example, YAP/TAZ, 176 , 177 , 178 serum response factor, 176 , 179 & retinoic acid. 176 , 180 , 181 Vimentin has also been connected to disturbed accumulation of body fat and lipid droplet stability, 14 , 182 , 183 but these findings have not implicated a role for vimentin in cell fate determination during adipogenesis, and therefore, falls outside the scope of this review.
Production of progerin, a pathological truncated variant of Lamin A, is the main cause of Hutchinson‐Gilford Progeria Syndrome (HGPS) a disease in which the patients experience premature‐aging symptoms. Individuals with HGPS have a point mutation in the LMNA gene which gives rise to a 50 amino acid deletion in the lamin A protein. The deleted region contains the site for processing mediated by ZMPSTE24. Lack of this processing site results in accumulation of farnesylated mutant prelamin A. 184 , 185 Progerin perturbs the structural integrity of the nucleus, resulting in deformation of the nuclear envelope. 186 , 187 The deformation has also been related to disrupted localization of nuclear pores, which may affect molecular trafficking to and from the nucleus and thereby perturb the communication between the nucleus and cytoplasm. 188 In addition, progerin also causes loss of peripheral heterochromatin and may therefore alter the gene expression profile. 187 Increased DNA damage, represented by γH2AX containing foci, is yet another consequence of cellular progerin accumulation. 189 , 190
6.2. Laminopathies and stem cell signaling
Stem cell depletion has been suggested to dislodge tissue homeostasis and cause the aging phenotype in HGPS patients. The compromised nuclear structure caused by progerin may prevent signaling molecules from entering the nucleus and thereby obstruct stem cell signaling. The Wnt signaling pathway is implicated in the maintenance of different stem cell populations. Nuclear localization of β‐catenin is reduced in both patient derived fibroblasts and in cells of the inter follicular epidermis in a mouse model of HGPS (tetop‐LAG608G+; K5‐tTA+mice) 188 and in odontoblasts expressing progerin. 190 Similarly, Choi et al 191 demonstrated defective osteogenesis of patient derived progerin expressing induced pluripotent stem cells (iPSCs) and human bone marrow derived mesenchymal stem cells (hBM‐MSCs), and that the faulty bone formation is due to suppressed β‐catenin signaling.
Notch signaling relies on nuclear translocation of the proteolytically processed intracellular signaling fragment, NICD, and aberrant Notch function has been linked to HGPS. 189 In cells derived from HGPS patients the expression of the Notch signaling effectors HES1, HES5 (hairy and enhancer of split), and HEY1 is enhanced, which is unrelated to changes in the NICD levels. On the contrary, expression of progerin alters the cellular identity of immortalized MSCs through Notch‐related mechanisms. Induction of differentiation along mesenchymal lineages in the presence of either progerin or NICD is similar, leading to enhanced osteogenic but reduced adipogenic differentiation. 173
Emerin is a constituent of the nuclear envelope, which connects the cytoplasm to nucleus. Emerin interacts with both the actin and β‐tubulin networks and binds the nuclear lamins. 192 , 193 In the nucleus emerin and prelamin A regulate each other's localization. 194 Mutations in the gene coding for emerin results in a disease called Emery‐Dreifuss muscular dystrophy type‐1 (X‐linked EMDM), a type of striated muscle laminopathy. 195 Mutation of the LMNA gene causes the related autosomal dominant type of EMDM (AD‐EMDM). Due to the close regulatory relationship between emerin and prelamin A, perturbed emerin function might be involved in several laminopathies. Emerin is known to interact with different signaling pathways, for example, Notch and Wnt signaling. Downregulation of emerin enhances signaling downstream of Notch and β‐catenin. Nuclear entry of β‐catenin is restricted by emerin, whereas NICD localizes together with emerin at the nuclear rim. 196 , 197 Thus, expression of mutant Lamin A/C variants may hamper with the functionality of these signaling pathways, because of disrupted emerin localization.
7. MECHANISMS OF IF‐MEDIATED CELL FATE DECISIONS
IFs utilize an array of different mechanisms to steer cell fate decisions. This can be achieved through direct interaction with signaling proteins or by regulating the localization of specific signaling molecules. Anchorage of keratins to the apical domain, followed by asymmetric division of keratins in daughter cells together with YAP dependent expression of CDX2, dictates cell fate already in a 16‐cell embryo. 2 Vimentin, on the contrary, is, for example, known to regulate integrin trafficking and membrane localization. 73 In addition, vimentin may control cell fate specification by connecting extracellular mechanical cues with the intracellular transcriptional control machinery. 16 Similarly, nuclear lamins are essential in governing nuclear entry of certain signaling moieties 172 , 188 and presumably regulates nucleocytoplasmic shuttling of other transcription factors as well. Interestingly, keratins have also been found to shuttle in and out of the nucleus. Keratins K7, K8, K17, and K18 were found to enter and exit the nucleus in HeLa cells 198 and K17 was subsequently identified as a regulator of gene expression in tumor epithelial cells from skin and cervical tumors. 199 , 200 The IF‐mediated regulation of signaling proteins is often dependent on the phosphorylation and assembly state of the IFs. The vimentin‐mediated regulation of the Notch ligand Jagged1 is phosphorylation dependent and enhanced vimentin phosphorylation promotes Notch activation. 9 On the contrary, inhibition of vimentin phosphorylation sequesters integrins in intracellular vesicles and thereby attenuates integrin signaling. 73 The importance of phosphorylation‐mediated control of IFs is attested during mitosis. Mutation of serine residues on vimentin provokes failure of cytokinesis and upregulation of p21 and other senescence‐related genes. 201 Inter‐keratin disulfide bonding is essential for the assembly and organization of the keratin network 202 , 203 and disruption of these bonds not only affects the structure of the keratin network, but also displaces 14‐3‐3 and YAP in keratinocytes leading to perturbed epidermal homeostasis. 82 The dynamic regulation of IFs in this manner allows context dependent cell fate choices. For example, shear stress experienced by endothelial and epithelial cells induces phosphorylation of both keratins and vimentin. Shear stress reduces the levels of soluble keratin in alveolar cells subjected to shear stress. 204 Shear activates protein kinase C ζ, leading to enhanced K18 phosphorylation and intensified interaction between K18 and 14‐3‐3. 205 Similarly, shear stress enhanced the phosphorylation of vimentin and enhanced the Jagged1‐mediated activation of Notch signaling. 16 IFs also sense and respond to other types of mechanical stimuli 206 , 207 and could thereby facilitate mechano‐regulation of cell fate. 208 Cell stress in the form of hypoxia also alters IF expression. Hypoxia alters vimentin phosphorylation and assembly through activation of p21 activated kinase (PAK), 209 whereas keratin stability is compromised upon hypoxia. 210 Furthermore, IF phosphorylation, and thereby IF‐associated signaling, is regulated by hormones. Vitamin D and thyroid hormones induces phosphorylation of vimentin in rat testes, 211 , 212 whereas the same hormones regulate phosphorylation of vimentin, GFAP, and NFs in rat cerebral cortex. 213 , 214 IFs may also be involved in generating oscillatory signaling cascades, characteristic for tissue patterning during development through interactions with different signaling molecules. 39 , 215 , 216 Cell behavior may be coordinated by IFs through regulation of cell‐cell signaling in multicellular tissues. It is also worth noting that IFs and developmental signaling pathways may come together in a reciprocal regulatory network, where IFs regulate developmental transcriptional programs which in turn may control IF expression, a hypothesis supported by the highly developmental stage‐specific expression profiles of different IFs.
8. CONCLUDING REMARKS
Coordination of cell specification and organization is essential for tissue and organ development. The cellular microenvironment critically regulates cell fate, tissue development, and homeostasis. IFs provide a unique scaffolding framework that orchestrates cell‐cell interactions and cell signaling dynamics, and thereby modulates the subsequent cell fate decisions. As the IFs respond to external stimuli and stressors, 217 IFs may integrate external and internal cues to support functional cell fate decisions in a fluctuating environment. The dynamic regulation of IFs allows context dependent cell fate decisions. 218 , 219 Interaction of IFs with specific signaling proteins allows IFs to specify signaling output, for example, through modulation of Jagged1, but not Dll4, mediated Notch signaling. 9 IF proteins are expressed in developmental stage and cell type‐specific manner 13 and the differences in regulation, turnover rates, and mechanical properties between IF proteins are likely optimized for different cell and tissue‐specific functions. IFs can also be considered to play a role in generating subcellular signaling compartments through the localization of filamentous and soluble forms of the IFs to different parts of the cell and thereby affect signaling at these local sites. 220 Proper localization of IFs is instrumental for maintaining cell polarity and organizing polarized cell signaling. 42 , 153 , 221 , 222 Complete understanding of how IF dynamics are regulated by changes in the cellular microenvironment and how this integrates with the cellular decision machinery is also lacking. Further, new innovative methods will have to be developed to tackle the details of how IFs are regulated in terms of turnover, dynamic exchange, and other remodeling events in vivo (current open questions summarized in Box 1). Recently, novel therapeutic tools modifying IFs have emerged and vimentin serves as a promising potential target in cancer therapy. Disruption of vimentin can reduce migration and invasion of highly motile metastatic cancer cells and several compounds targeting vimentin have shown anticancer properties (reviewed in Ref. 53). These include compounds such as fluvastatin leading to proteolysis of vimentin, 223 ajoene that disrupts the vimentin network, 224 simvastatin leading to reorganization and bundling of the vimentin network, 225 the specific vimentin targeting small molecule FiVe1 causing disorganization, phosphorylation, and mitotic catastrophe, 226 and an antibody targeting cell surface vimentin. 227 Although the specific mechanisms by which IFs regulate cell fate decisions and developmental signaling are emerging, there is still a fundamental knowledge gap in this area and more detailed knowledge on how IFs regulate signaling proteins is required. We need to identify the interaction sites of IFs and signaling proteins, study how the interactions are regulated by IF dynamics and posttranslational modifications. Enhanced understanding of the cross talk between IFs and developmental signaling pathways could clarify our perception of perturbances in these signaling pathways and how they relate to disease etiology. Several developmental diseases have puzzling phenotypes, where similar mutations have various disease outcomes. In such cases tissue‐specific expression of IFs and their regulation of cell signaling dynamics may be a contributing factor for disease severity.
Box 1. Current questions on the mechanisms of IF mediated cell fate decisions.
Is the expression of IFs regulated by developmental signaling pathways?
What is the role of IFs in coordinating cell behaviour in multicellular tissues?
How is the interaction between IFs and IF‐interacting proteins regulated?
Are IFs regulated locally at the subcellular level and how does that affect cell signaling?
How do post‐translational modifications (PTMs) regulate IFs in different cellular contexts?
Can we target IFs pharmacologically to steer cell fate?
Can stem cell signaling be controlled through mechanical regulation of IFs?
How is stem cell signaling affected when IFs respond to changes in the cellular microenvironment (e.g. metabolism, inflammation, fibrosis)?
Are there still unidentified IF mutations that cause developmental diseases?
-
How can the context dependent function of IFs be studied? Potential approaches include:
Tools for controlling IF assembly and function (IF targeting drugs, peptide microinjections, optogenetics).
Transgenic models for cell isolation and engineering (PTM mutants, spatio‐temporal control of gene expression, lineage tracing).
Techniques for visualization of IF functions (Single molecule resolution imaging, AFM for filament nano‐dissection).
Biomimetic approaches (3D cultures, organoids, organ‐on‐chip technology).
Mathematical modeling and generation of computational tools.
AUTHOR CONTRIBUTIONS
M. Sjöqvist, D. Antfolk, F. Suarez Rodriquez, and C. M. Sahlgren wrote the review.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Kai Lan Lin for help with the figures. Images were created using Biorender.com. This work was supported by the ForceMorph project, funded by the ERC, project number 771168 (CS, FSR), by the ÅA CoE BACE (MS), the Swedish Cultural Foundation in Finland (MS, DA) and by K. Albin Johanssons stiftelse (DA).
Sjöqvist M, Antfolk D, Suarez‐Rodriguez F, Sahlgren C. From structural resilience to cell specification—Intermediate filaments as regulators of cell fate. The FASEB Journal. 2021;35:e21182 10.1096/fj.202001627R
REFERENCES
- 1. Discher D, Dong C, Fredberg JJ, et al. Biomechanics: cell research and applications for the next decade. Ann Biomed Eng. 2009;37(5):847‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lim HYG, Alvarez YD, Gasnier M, et al. Keratins are asymmetrically inherited fate determinants in the mammalian embryo. Nature. 2020;585(7825):404‐409. [DOI] [PubMed] [Google Scholar]
- 3. Steen K, Chen D, Wang F, et al. A role for keratins in supporting mitochondrial organization and function in skin keratinocytes. Mol Biol Cell. 2020;31(11):1103‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bera M, Sengupta K. Nuclear filaments: role in chromosomal positioning and gene expression. Nucleus. 2020;11(1):99‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colucci‐Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79(4):679‐694. [DOI] [PubMed] [Google Scholar]
- 6. Terzi F, Henrion D, Colucci‐Guyon E, et al. Reduction of renal mass is lethal in mice lacking vimentin. Role of endothelin‐nitric oxide imbalance. J Clin Invest. 1997;100(6):1520‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mendez MG, Restle D, Janmey PA. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys J. 2014;107(2):314‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Langlois B, Belozertseva E, Parlakian A, et al. Vimentin knockout results in increased expression of sub‐endothelial basement membrane components and carotid stiffness in mice. Sci Rep. 2017;7(1):11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Antfolk D, Sjoqvist M, Cheng F, et al. Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc Natl Acad Sci U S A. 2017;114(23):E4574‐E4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boraas LC, Ahsan T. Lack of vimentin impairs endothelial differentiation of embryonic stem cells. Sci Rep. 2016;6:30814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng F, Shen Y, Mohanasundaram P, et al. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF‐β–Slug signaling. Proc Natl Acad Sci U S A. 2016;113(30):E4320‐E4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Capetanaki Y, Smith S, Heath JP. Overexpression of the vimentin gene in transgenic mice inhibits normal lens cell differentiation. J Cell Biol. 1989;109(4 Pt 1):1653‐1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duprey P, Paulin D. What can be learned from intermediate filament gene regulation in the mouse embryo. Int J Dev Biol. 1995;39(3):443‐457. [PubMed] [Google Scholar]
- 14. Cogné B, Bouameur JE, Hayot G, et al. A dominant vimentin variant causes a rare syndrome with premature aging. Eur J Hum Genet. 2020;28(9):1218‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schiffers PM, Henrion D, Boulanger CM, et al. Altered flow‐induced arterial remodeling in vimentin‐deficient mice. Arterioscler Thromb Vasc Biol. 2000;20(3):611‐616. [DOI] [PubMed] [Google Scholar]
- 16. van Engeland NCA, Suarez Rodriguez F, Rivero‐Müller A, et al. Vimentin regulates Notch signaling strength and arterial remodeling in response to hemodynamic stress. Sci Rep. 2019;9(1):12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu T, Ghamloush MM, Aldawood A, et al. Modulating endothelial barrier function by targeting vimentin phosphorylation. J Cell Physiol. 2014;229(10):1484‐1493. [DOI] [PubMed] [Google Scholar]
- 18. Polacheck WJ, Kutys ML, Yang J, et al. A non‐canonical Notch complex regulates adherens junctions and vascular barrier function. Nature. 2017;552(7684):258‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778(3):794‐809. [DOI] [PubMed] [Google Scholar]
- 20. Kwak HI, Kang H, Dave JM, et al. Calpain‐mediated vimentin cleavage occurs upstream of MT1‐MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis. 2012;15(2):287‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chung BM, Rotty JD, Coulombe PA. Networking galore: intermediate filaments and cell migration. Curr Opin Cell Biol. 2013;25:600‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24(6):1838‐1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033‐3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kokkinos MI, Wafai R, Wong MK, Newgreen DF, Thompson EW, Waltham M. Vimentin and epithelial‐mesenchymal transition in human breast cancer ‐ observations in vitro and in vivo. Cells Tissues Organs. 2007;185:191‐203. [DOI] [PubMed] [Google Scholar]
- 25. Messica Y, Laser‐Azogui A, Volberg T, et al. The role of vimentin in regulating cell invasive migration in dense cultures of breast carcinoma cells. Nano Lett. 2017;17(11):6941‐6948. [DOI] [PubMed] [Google Scholar]
- 26. Nakaya Y, Sheng G. EMT in developmental morphogenesis. Cancer Lett. 2013;341:9‐15. [DOI] [PubMed] [Google Scholar]
- 27. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009;139:871‐890. [DOI] [PubMed] [Google Scholar]
- 28. Esue O, Carson AA, Tseng Y, Wirtz D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J Biol Chem. 2006;281(41):30393‐30399. [DOI] [PubMed] [Google Scholar]
- 29. Duarte S, Viedma‐Poyatos Á, Navarro‐Carrasco E, Martínez AE, Pajares MA, Pérez‐Sala D. Vimentin filaments interact with the actin cortex in mitosis allowing normal cell division. Nat Commun. 2019;10(1):4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiche G, Osmanagic‐Myers S, Castañón MJ. Networking and anchoring through plectin: a key to IF functionality and mechanotransduction. Curr Opin Cell Biol. 2015;32:21‐29. [DOI] [PubMed] [Google Scholar]
- 31. Burgstaller G, Gregor M, Winter L, Wiche G. Keeping the vimentin network under control: cell‐matrix adhesion‐associated plectin 1f affects cell shape and polarity of fibroblasts. Mol Biol Cell. 2010;21(19):3362‐3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135(4):991‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schoumacher M, Goldman RD, Louvard D, Vignjevic DM. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J Cell Biol. 2010;189(3):541‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sutoh Yoneyama M, Hatakeyama S, Habuchi T, et al. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur J Cell Biol. 2014;93(4):157‐169. [DOI] [PubMed] [Google Scholar]
- 35. Lynch CD, Lazar AM, Iskratsch T, Zhang X, Sheetz MP. Endoplasmic spreading requires coalescence of vimentin intermediate filaments at force‐bearing adhesions. Mol Biol Cell. 2013;24(1):21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gregor M, Osmanagic‐Myers S, Burgstaller G, et al. Mechanosensing through focal adhesion‐anchored intermediate filaments. FASEB J. 2014;28(2):715‐729. [DOI] [PubMed] [Google Scholar]
- 37. Battaglia RA, Delic S, Herrmann H, Snider NT. Vimentin on the move: new developments in cell migration. F1000Res. 2018;7:F1000 Faculty Rev‐1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vuoriluoto K, Haugen H, Kiviluoto S, et al. Vimentin regulates EMT induction by Slug and oncogenic H‐Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30(12):1436‐1448. [DOI] [PubMed] [Google Scholar]
- 39. Perlson E, Michaelevski I, Kowalsman N, et al. Vimentin Binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364(5):938‐944. [DOI] [PubMed] [Google Scholar]
- 40. Virtakoivu R, Mai A, Mattila E, et al. Vimentin‐ERK signaling uncouples Slug gene regulatory function. Cancer Res. 2015;75(11):2349‐2362. [DOI] [PubMed] [Google Scholar]
- 41. Zhu QS, Rosenblatt K, Huang KL, et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011;30(4):457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Phua DC, Humbert PO, Hunziker W. Vimentin regulates scribble activity by protecting it from proteasomal degradation. Mol Biol Cell. 2009;20(12):2841‐2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J, Yang C, Kim EJ, et al. Vimentin filaments regulate integrin‐ligand interactions by binding to the cytoplasmic tail of integrin β3. J Cell Sci. 2016;129(10):2030‐2042. [DOI] [PubMed] [Google Scholar]
- 44. Sakamoto Y, Boëda B, Etienne‐Manneville S. APC binds intermediate filaments and is required for their reorganization during cell migration. J Cell Biol. 2013;200(3):249‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dos Santos G, Rogel MR, Baker MA, et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6:6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tzivion G, Luo ZJ, Avruch J. Calyculin A‐induced vimentin phosphorylation sequesters 14‐3‐3 and displaces other 14‐3‐3 partners in vivo. J Biol Chem. 2000;275(38):29772‐29778. [DOI] [PubMed] [Google Scholar]
- 47. Wang RC, Wei Y, An Z, et al. Akt‐mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei J, Xu G, Wu M, et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via Src regulation. Anticancer Res. 2008;28(1A):327‐334. [PubMed] [Google Scholar]
- 49. Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial‐mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2011;3(1):90‐99. [PMC free article] [PubMed] [Google Scholar]
- 50. Hu L, Lau SH, Tzang CH, et al. Association of Vimentin overexpression and hepatocellular carcinoma metastasis. Oncogene. 2004;23(1):298‐302. [DOI] [PubMed] [Google Scholar]
- 51. Korsching E, Packeisen J, Liedtke C, et al. The origin of vimentin expression in invasive breast cancer: epithelial‐ mesenchymal transition, myoepithelial histogenesis or histogenesis from progenitor cells with bilinear differentiation potential? J Pathol. 2005;206(4):451‐457. [DOI] [PubMed] [Google Scholar]
- 52. Kidd ME, Shumaker DK, Ridge KM. The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol. 2014;50(1):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Strouhalova K, Přechová M, Gandalovičová A, Brábek J, Gregor M, Rosel D. Vimentin intermediate filaments as potential target for cancer treatment. Cancers. 2020;12(1):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58(1):131‐148. [DOI] [PubMed] [Google Scholar]
- 55. Mellodew K, Suhr R, Uwanogho DA, et al. Nestin expression is lost in a neural stem cell line through a mechanism involving the proteasome and Notch signalling. Dev Brain Res. 2004;151(1–2):13‐23. [DOI] [PubMed] [Google Scholar]
- 56. Zahr SK, Kaplan DR, Miller FD. Translating neural stem cells to neurons in the mammalian brain. Cell Death Differ. 2019;26(12):2495‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pekny M, Levéen P, Pekna M, et al. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J. 1995;14(8):1590‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zerlin M, Levison SW, Goldman JE. Early patterns of migration, morphogenesis, and intermediate filament expression of subventricular zone cells in the postnatal rat forebrain. J Neurosci. 1995;15(11):7238‐7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mohseni P, Sung HK, Murphy AJ, et al. Nestin is not essential for development of the CNS but required for dispersion of acetylcholine receptor clusters at the area of neuromuscular junctions. J Neurosci. 2011;31(32):11547‐11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dahl D. The vimentin‐GFA protein transition in rat neuroglia cytoskeleton occurs at the time of myelination. J Neurosci Res. 1981;6(6):741‐748. [DOI] [PubMed] [Google Scholar]
- 61. McCall MA, Gregg RG, Behringer RR, et al. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci U S A. 1996;93(13):6361‐6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gomi H, Yokoyama T, Fujimoto K, et al. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron. 1995;14(1):29‐41. [DOI] [PubMed] [Google Scholar]
- 63. Shibuki K, Gomi H, Chen L, et al. Deficient cerebellar long‐term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16(3):587‐599. [DOI] [PubMed] [Google Scholar]
- 64. Namihira M, Kohyama J, Semi K, et al. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16(2):245‐255. [DOI] [PubMed] [Google Scholar]
- 65. Wilhelmsson U, Lebkuechner I, Leke R, et al. Nestin regulates neurogenesis in mice through notch signaling from astrocytes to neural stem cells. Cereb Cortex. 2019;29(10):4050‐4066. [DOI] [PubMed] [Google Scholar]
- 66. Lasič E, Trkov Bobnar S, Wilhelmsson U, et al. Nestin affects fusion pore dynamics in mouse astrocytes. Acta Physiol. 2020;228(3):e13399. [DOI] [PubMed] [Google Scholar]
- 67. Widestrand Å, Faijerson J, Wilhelmsson U, et al. Increased neurogenesis and astrogenesis from neural progenitor cells grafted in the hippocampus of GFAP−/− Vim−/− mice. Stem Cells. 2007;25(10):2619‐2627. [DOI] [PubMed] [Google Scholar]
- 68. Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417(6884):39‐44. [DOI] [PubMed] [Google Scholar]
- 69. Wilhelmsson U, Faiz M, de Pablo Y, et al. Astrocytes negatively regulate neurogenesis through the Jagged1‐mediated Notch pathway. Stem Cells. 2012;30(10):2320‐2329. [DOI] [PubMed] [Google Scholar]
- 70. Louvi A, Artavanis‐Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev. 2006;7(2):93‐102. [DOI] [PubMed] [Google Scholar]
- 71. Imayoshi I, Sakamoto M, Yamaguchi M, Mori K, Kageyama R. Essential roles of Notch signaling in maintenance of neural stem cells in developing and adult brains. J Neurosci. 2010;30(9):3489‐3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen M, Puschmann TB, Marasek P, et al. Increased neuronal differentiation of neural progenitor cells derived from phosphovimentin‐deficient mice. Mol Neurobiol. 2018;55(7):5478‐5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. PKCepsilon‐mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005;24(22):3834‐3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hagemann TL, Paylor R, Messing A. Deficits in adult neurogenesis, contextual fear conditioning, and spatial learning in a Gfap mutant mouse model of Alexander disease. J Neurosci. 2013;33(47):18698‐18706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hsiao VC, Tian R, Long H, et al. Alexander‐disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J Cell Sci. 2005;118(9):2057‐2065. [DOI] [PubMed] [Google Scholar]
- 76. Liu Y, Namba T, Liu J, Suzuki R, Shioda S, Seki T. Glial fibrillary acidic protein‐expressing neural progenitors give rise to immature neurons via early intermediate progenitors expressing both glial fibrillary acidic protein and neuronal markers in the adult hippocampus. Neuroscience. 2010;166(1):241‐251. [DOI] [PubMed] [Google Scholar]
- 77. Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes, and the small heat shock protein alphaB‐crystallin reverses the inhibition. J Biol Chem. 2010;285(14):10527‐10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Strnad P, Usachov V, Debes C, Gräter F, Parry DAD, Omary MB. Unique amino acid signatures that are evolutionarily conserved distinguish simple‐type, epidermal and hair keratins. J Cell Sci. 2011;124(Pt 24):4221‐4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Miettinen M, Fetsch JF. Distribution of keratins in normal endothelial cells and a spectrum of vascular tumors: implications in tumor diagnosis. Hum Pathol. 2000;31(9):1062‐1067. [DOI] [PubMed] [Google Scholar]
- 80. Lahdeniemi IAK, Misiorek JO, Antila CJM, et al. Keratins regulate colonic epithelial cell differentiation through the Notch1 signalling pathway. Cell Death Differ. 2017;24(6):984‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vijayaraj P, Kroeger C, Reuter U, Hartmann D, Magin TM. Keratins regulate yolk sac hematopoiesis and vasculogenesis through reduced BMP‐4 signaling. Eur J Cell Biol. 2010;89(4):299‐306. [DOI] [PubMed] [Google Scholar]
- 82. Guo Y, Redmond CJ, Leacock KA, et al. Keratin 14‐dependent disulfides regulate epidermal homeostasis and barrier function via 14‐3‐3σ and YAP1. Elife. 2020;9:e53165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vijayaraj P, Söhl G, Magin TM. Keratin transgenic and knockout mice: functional analysis and validation of disease‐causing mutations. Methods Mol Biol. 2007;360:203‐251. [DOI] [PubMed] [Google Scholar]
- 84. Allen EHA, Courtney DG, Atkinson SD, et al. Keratin 12 missense mutation induces the unfolded protein response and apoptosis in Meesmann epithelial corneal dystrophy. Hum Mol Genet. 2016;25(6):1176‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Owens DW, Wilson NJ, Hill AJM, et al. Human keratin 8 mutations that disturb filament assembly observed in inflammatory bowel disease patients. J Cell Sci. 2004;117(10):1989‐1999. [DOI] [PubMed] [Google Scholar]
- 86. Tao GZ, Strnad P, Zhou Q, et al. Analysis of keratin polypeptides 8 and 19 variants in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5(7):857‐864. [DOI] [PubMed] [Google Scholar]
- 87. Ku N‐O, Lim JK, Krams SM, et al. Keratins as susceptibility genes for end‐stage liver disease. Gastroenterology. 2005;129(3):885‐893. [DOI] [PubMed] [Google Scholar]
- 88. Alam H, Sehgal L, Kundu ST, Dalal SN, Vaidya MM. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol Biol Cell. 2011;22(21):4068‐4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lloyd C, Yu QC, Cheng J, et al. The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J Cell Biol. 1995;129(5):1329‐1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nickoloff BJ, Qin JZ, Chaturvedi V, Denning MF, Bonish B, Miele L. Jagged‐1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF‐kappaB and PPARgamma. Cell Death Differ. 2002;9(8):842‐855. [DOI] [PubMed] [Google Scholar]
- 91. Palazzo E, Morandi P, Lotti R, et al. Notch cooperates with survivin to maintain stemness and to stimulate proliferation in human keratinocytes during ageing. Int J Mol Sci. 2015;16(11):26291‐26302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Blanpain C, Lowry WE, Pasolli HA, Fuchs E. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006;20(21):3022‐3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rangarajan A, Talora C, Okuyama R, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20(13):3427‐3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17(3):590‐603. [DOI] [PubMed] [Google Scholar]
- 95. Romano R‐A, Ortt K, Birkaya B, Smalley K, Sinha S. An active role of the ΔN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS One. 2009;4(5):e5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nguyen B‐C, Lefort K, Mandinova A, et al. Cross‐regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006;20(8):1028‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lessard JC, Coulombe PA. Keratin 16–null mice develop palmoplantar keratoderma, a hallmark feature of pachyonychia congenita and related disorders. J Invest Dermatol. 2012;132(5):1384‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fu DJ, Thomson C, Lunny DP, et al. Keratin 9 is required for the structural integrity and terminal differentiation of the palmoplantar epidermis. J Invest Dermatol. 2014;134(3):754‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dinkova‐Kostova AT, Holtzclaw WD, Cole RN, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99(18):11908‐11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zieman AG, Poll BG, Ma J, Coulombe PA. Altered keratinocyte differentiation is an early driver of keratin mutation‐based palmoplantar keratoderma. Hum Mol Genet. 2019;28(13):2255‐2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dellambra E, Golisano O, Bondanza S, et al. Downregulation of 14‐3‐3sigma prevents clonal evolution and leads to immortalization of primary human keratinocytes. J Cell Biol. 2000;149(5):1117‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Benzinger A, Muster N, Koch HB, Yates JR, Hermeking H. Targeted proteomic analysis of 14‐3‐3 sigma, a p53 effector commonly silenced in cancer. Mol Cell Proteomics. 2005;4(6):785‐795. [DOI] [PubMed] [Google Scholar]
- 103. Margolis SS, Perry JA, Forester CM, et al. Role for the PP2A/B56delta phosphatase in regulating 14‐3‐3 release from Cdc25 to control mitosis. Cell. 2006;127(4):759‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441(7091):362‐365. [DOI] [PubMed] [Google Scholar]
- 105. Totaro A, Castellan M, Battilana G, et al. YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat Commun. 2017;8:15206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cheng F, Eriksson JE. Intermediate filaments and the regulation of cell motility during regeneration and wound healing. Cold Spring Harb Perspect Biol. 2017;9(9):a022046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cerecedo D, Martínez‐Vieyra I, Mondragõn R, Mondragõn M, González S, Galván IJ. Haemostatic role of intermediate filaments in adhered platelets: importance of the membranous system stability. J Cell Biochem. 2013;114(9):2050‐2060. [DOI] [PubMed] [Google Scholar]
- 108. Depianto D, Coulombe PA. Intermediate filaments and tissue repair. Exp Cell Res. 2004;301(1):68‐76. [DOI] [PubMed] [Google Scholar]
- 109. Eckes B, Colucci‐Guyon E, Smola H, et al. Impaired wound healing in embryonic and adult mice lacking vimentin. J Cell Sci. 2000;113(13):2455‐2462. [DOI] [PubMed] [Google Scholar]
- 110. Walker JL, Bleaken BM, Romisher AR, Alnwibit AA, Menko AS. In wound repair vimentin mediates the transition of mesenchymal leader cells to a myofibroblast phenotype. Mol Biol Cell. 2018;29(13):1555‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rogel MR, Soni PN, Troken JR, Sitikov A, Trejo HE, Ridge KM. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011;25(11):3873‐3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wu Y, Zhang X, Salmon M, Lin X, Zehner ZE. TGFβ1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP‐1 and Sp1 family members. Biochim Biophys Acta ‐ Mol Cell Res. 2007;1773(3):427‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jiang P, Gil de Rubio R, Hrycaj SM, et al. Ineffectual AEC2‐to‐AEC1 differentiation in IPF: persistence of KRT8 hi transitional state. Am J Respir Crit Care Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75(1):289‐311. [DOI] [PubMed] [Google Scholar]
- 115. Ting H‐A, von Moltke J. The immune function of tuft cells at gut mucosal surfaces and beyond. J Immunol. 2019;202(5):1321‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhou Q, Toivola DM, Feng N, Greenberg HB, Franke WW, Bishr OM. Keratin 20 helps maintain intermediate filament organization in intestinal epithelia. Mol Biol Cell. 2003;14(7):2959‐2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Noah TK, Shroyer NF. Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol. 2013;75:263‐288. [DOI] [PubMed] [Google Scholar]
- 118. Jensen J, Pedersen EE, Galante P, et al. Control of endodermal endocrine development by Hes‐1. Nat Genet. 2000;24(1):36‐44. [DOI] [PubMed] [Google Scholar]
- 119. Li HJ, Kapoor A, Giel‐Moloney M, Rindi G, Leiter AB. Notch signaling differentially regulates the cell fate of early endocrine precursor cells and their maturing descendants in the mouse pancreas and intestine. Dev Biol. 2012;371(2):156‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42(9):705‐710. [DOI] [PubMed] [Google Scholar]
- 121. Sato T, Van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kabiri Z, Greicius G, Madan B, et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Dev. 2014;141(11):2206‐2215. [DOI] [PubMed] [Google Scholar]
- 123. van Es JH, Jay P, Gregorieff A, et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7(4):381‐386. [DOI] [PubMed] [Google Scholar]
- 124. Baribault H, Penner J, Iozzo RV, Wilson‐Heiner M. Colorectal hyperplasia and inflammation in keratin 8‐deficient FVB/N mice. Genes Dev. 1994;8(24):2964‐2973. [DOI] [PubMed] [Google Scholar]
- 125. Toivola DM, Krishnan S, Binder HJ, Singh SK, Omary MB. Keratins modulate colonocyte electrolyte transport via protein mistargeting. J Cell Biol. 2004;164(6):911‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Asghar MN, Silvander JSG, Helenius TO, et al. The amount of keratins matters for stress protection of the colonic epithelium. PLoS One. 2015;10(5):e0127436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. van Es JH, de Geest N, van de Born M, Clevers H, Hassan BA. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat Commun. 2010;1(2):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Milano J, McKay J, Dagenais C, et al. Modulation of notch processing by gamma‐secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82(1):341‐358. [DOI] [PubMed] [Google Scholar]
- 129. Misiorek JO, Lähdeniemi IAK, Nyström JH, et al. Keratin 8‐deletion induced colitis predisposes to murine colorectal cancer enforced by the inflammasome and IL‐22 pathway. Carcinogenesis. 2016;37(8):777‐786. [DOI] [PubMed] [Google Scholar]
- 130. Zha JM, Li HS, Lin Q, et al. Interleukin 22 expands transit‐amplifying cells while depleting Lgr5 + stem cells via inhibition of Wnt and Notch signaling. CMGH. 2019;7(2):255‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Brodsly NF, Bitman‐Lotan E, Boico O, et al. The transcription factor Hey and nuclear lamins specify and maintain cell identity. Elife. 2019;8:e44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Petrovsky R, Großhans J. Expression of lamina proteins lamin and kugelkern suppresses stem cell proliferation. Nucleus. 2018;9(1):104‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Toribio‐Fernández R, Herrero‐Fernandez B, Zorita V, et al. Lamin A/C deficiency in CD4 + T‐cells enhances regulatory T‐cells and prevents inflammatory bowel disease. J Pathol. 2019;249(4):509‐522. [DOI] [PubMed] [Google Scholar]
- 134. Strnad P, Stumptner C, Zatloukal K, Denk H. Intermediate filament cytoskeleton of the liver in health and disease. Histochem Cell Biol. 2008;129(6):735–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Tao GZ, Toivola DM, Zhong B, et al. Keratin‐8 null mice have different gallbladder and liver susceptibility to lithogenic diet‐induced injury. J Cell Sci. 2003;116(Pt 22):4629‐4638. [DOI] [PubMed] [Google Scholar]
- 136. Chen Y, Guldiken N, Spurny M, et al. Loss of keratin 19 favours the development of cholestatic liver disease through decreased ductular reaction. J Pathol. 2015;237(3):343‐354. [DOI] [PubMed] [Google Scholar]
- 137. Tchorz JS, Kinter J, Müuller M, Tornillo L, Heim MH, Bettler B. Notch2 signaling promotes biliary epithelial cell fate specification and tubulogenesis during bile duct development in mice. Hepatology. 2009;50(3):871‐879. [DOI] [PubMed] [Google Scholar]
- 138. Vajro P, Ferrante L, Paolella G. Alagille syndrome: an overview. Clin Res Hepatol Gastroenterol. 2012;36(3):275‐277. [DOI] [PubMed] [Google Scholar]
- 139. Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16(3):235‐242. [DOI] [PubMed] [Google Scholar]
- 140. Kamath BM, Bauer RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49(2):138‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Inman JL, Robertson C, Mott JD, Bissell MJ. Mammary gland development: cell fate specification, stem cells and the microenvironment. Development. 2015;142(6):1028‐1042. [DOI] [PubMed] [Google Scholar]
- 142. Peuhu E, Virtakoivu R, Mai A, Warri A, Ivaska J. Epithelial vimentin plays a functional role in mammary gland development. Development. 2017;144(22):4103‐4113. [DOI] [PubMed] [Google Scholar]
- 143. Soady KJ, Kendrick H, Gao Q, et al. Mouse mammary stem cells express prognostic markers for triple‐negative breast cancer. Breast Cancer Res. 2015;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Alexander CM, Goel S, Fakhraldeen SA, Kim S. Wnt signaling in mammary glands: plastic cell fates and combinatorial signaling. Cold Spring Harb Perspect Biol. 2012;4(10):a008037 10.1101/cshperspect.a008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Raouf A, Zhao Y, To K, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008;3(1):109‐118. [DOI] [PubMed] [Google Scholar]
- 146. Gu B, Watanabe K, Sun P, Fallahi M, Dai X. Chromatin effector Pygo2 mediates Wnt‐notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell Stem Cell. 2013;13(1):48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bouras T, Pal B, Vaillant F, et al. Notch signaling regulates mammary stem cell function and luminal cell‐fate commitment. Cell Stem Cell. 2008;3(4):429‐441. [DOI] [PubMed] [Google Scholar]
- 148. Bentzinger CF, Wang YX, Rudnicki MA. Building muscle: molecular regulation of myogenesis. EMBO Rep. 2013;14(12):1062‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Steinert PM, Chou YH, Prahlad V, et al. A high molecular weight intermediate filament‐associated protein in BHK‐ 21 cells is nestin, a type VI intermediate filament protein: limited co‐ assembly in vitro to form heteropolymers with type III vimentin and type IV α‐internexin. J Biol Chem. 1999;274(14):9881‐9890. [DOI] [PubMed] [Google Scholar]
- 150. Dahlstrand J, Lardelli M, Lendahl U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Dev Brain Res. 1995;84(1):109‐129. [DOI] [PubMed] [Google Scholar]
- 151. Park D, Xiang AP, Mao FF, et al. Nestin is required for the proper self‐renewal of neural stem cells. Stem Cells. 2010;28(12):2162‐2171. [DOI] [PubMed] [Google Scholar]
- 152. Yang J, Dominguez B, De Winter F, Gould TW, Eriksson JE, Lee KF. Nestin negatively regulates postsynaptic differentiation of the neuromuscular synapse. Nat Neurosci. 2011;14(3):324‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sahlgren CM, Mikhailov A, Vaittinen S, et al. Cdk5 Regulates the Organization of Nestin and Its Association with p35. Mol Cell Biol. 2003;23(14):5090‐5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sahlgren CM, Pallari HM, He T, Chou YH, Goldman RD, Eriksson JE. A nestin scaffold links Cdk5/p35 signaling to oxidant‐induced cell death. EMBO J. 2006;25(20):4808‐4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Pallari HM, Lindqvist J, Torvaldson E, et al. Nestin as a regulator of Cdk5 in differentiating myoblasts. Mol Biol Cell. 2011;22(9):1539‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lindqvist J, Torvaldson E, Gullmets J, et al. Nestin contributes to skeletal muscle homeostasis and regeneration. J Cell Sci. 2017;130(17):2833‐2842. [DOI] [PubMed] [Google Scholar]
- 157. Philpott A, Porro EB, Kirschner MW, Tsai LH. The role of cyclin‐dependent kinase 5 and a novel regulatory subunit in regulating muscle differentiation and patterning. Genes Dev. 1997;11(11):1409‐1421. [DOI] [PubMed] [Google Scholar]
- 158. Íková D, Soukup T, Mokrý J. Nestin expression reflects formation, revascularization and reinnervation of new myofibers in regenerating rat hind limb skeletal muscles. Cells Tissues Organs. 2009;189(5):338‐347. [DOI] [PubMed] [Google Scholar]
- 159. Ursitti JA, Lee PC, Resneck WG, et al. Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle: interaction with the dystrophin glycoprotein complex. J Biol Chem. 2004;279(40):41830‐41838. [DOI] [PubMed] [Google Scholar]
- 160. Stone MR, O'Neill A, Lovering RM, et al. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J Cell Sci. 2007;120(22):3999‐4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Butin‐Israeli V, Adam SA, Goldman AE, Goldman RD. Nuclear lamin functions and disease. Trends Genet. 2012;28(9):464‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Dittmer TA, Misteli T. The lamin protein family. Genome Biol. 2011;12(5):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268(22):16321‐16326. [PubMed] [Google Scholar]
- 164. Lin F, Worman HJ. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics. 1995;27(2):230‐236. [DOI] [PubMed] [Google Scholar]
- 165. Biamonti G, Giacca M, Perini G, et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S‐phase. Mol Cell Biol. 1992;12(8):3499‐3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Furukawa K, Hotta Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 1993;12(1):97‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Parry DA, Conway JF, Steinert PM. Structural studies on lamin. Similarities and differences between lamin and intermediate‐filament proteins. Biochem J. 1986;238(1):305‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kitten GT, Nigg EA. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J Cell Biol. 1991;113(1):13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Davies BSJ, Fong LG, Yang SH, Coffinier C, Young SG. The posttranslational processing of prelamin A and disease. Annu Rev Genomics Hum Genet. 2009;10(1):153‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Meshorer E, Gruenbaum Y. Gone with the Wnt/Notch: stem cells in laminopathies, progeria, and aging. J Cell Biol. 2008;181(1):9‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Zhang B, Yang Y, Keyimu R, Hao J, Zhao Z, Ye R. The role of lamin A/C in mesenchymal stem cell differentiation. J Physiol Biochem. 2019;75(1):11‐18. [DOI] [PubMed] [Google Scholar]
- 172. Bermeo S, Vidal C, Zhou H, Duque G. Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/β‐catenin pathway. J Cell Biochem. 2015;116(10):2344‐2353. [DOI] [PubMed] [Google Scholar]
- 173. Scaffidi P, Misteli T. Lamin A‐dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10(4):452‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102(9):3324‐3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Cho S, Vashisth M, Abbas A, et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell‐cycle arrest. Dev Cell. 2019;49(6):920‐935.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin‐A scales with tissue stiffness and enhances matrix‐directed differentiation. Science. 2013;341(6149):1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Bertrand AT, Ziaei S, Ehret C, et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA‐mutated muscle precursors. J Cell Sci. 2014;127(13):2873‐2884. [DOI] [PubMed] [Google Scholar]
- 178. Zhang J, Yang H, Abali BE, Li M, Xia Y, Haag R. Dynamic mechanics‐modulated hydrogels to regulate the differentiation of stem‐cell spheroids in soft microniches and modeling of the nonlinear behavior. Small. 2019;15(30):e1901920. [DOI] [PubMed] [Google Scholar]
- 179. Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1‐SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Pellegrini C, Columbaro M, Capanni C, et al. All‐trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget. 2015;6(30):29914‐29928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ivanovska IL, Swift J, Spinler K, Dingal D, Cho S, Discher DE. Cross‐linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol Biol Cell. 2017;28(14):2010‐2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wilhelmsson U, Stillemark‐Billton P, Borén J, Pekny M. Vimentin is required for normal accumulation of body fat. Biol Chem. 2019;400(9):1157‐1162. [DOI] [PubMed] [Google Scholar]
- 183. Lieber JG, Evans RM. Disruption of the vimentin intermediate filament system during adipose conversion of 3T3‐L1 cells inhibits lipid droplet accumulation. J Cell Sci. 1996;109(13):3047‐3058. [DOI] [PubMed] [Google Scholar]
- 184. Pendás AM, Zhou Z, Cadiñanos J, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase–deficient mice. Nat Genet. 2002;31(1):94‐99. [DOI] [PubMed] [Google Scholar]
- 185. Bergo MO, Gavino B, Ross J, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99(20):13049‐13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson‐Gilford progeria syndrome. Nature. 2003;423(6937):293‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson‐Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101(24):8963‐8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Sola‐Carvajal A, Revêchon G, Helgadottir HT, et al. Accumulation of progerin affects the symmetry of cell division and is associated with impaired Wnt signaling and the mislocalization of nuclear envelope proteins. J Invest Dermatol. 2019;139(11):2272‐2280.e12. [DOI] [PubMed] [Google Scholar]
- 189. Scaffidi P, Misteli T. Lamin A‐dependent nuclear defects in human aging. Science. 2006;312(5776):1059‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Choi H, Kim T‐H, Jeong J‐K, Strandgren C, Eriksson M, Cho E‐S. Expression of the hutchinson‐gilford progeria mutation leads to aberrant dentin formation. Sci Rep. 2018;8(1):15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Choi JY, Lai JK, Xiong Z‐M, et al. Diminished canonical β‐catenin signaling during osteoblast differentiation contributes to osteopenia in progeria. J Bone Miner Res. 2018;33(11):2059‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Holaska JM, Kowalski AK, Wilson KL. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2004;2(9):e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Salpingidou G, Smertenko A, Hausmanowa‐Petrucewicz I, Hussey PJ, Hutchison CJ. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J Cell Biol. 2007;178(6):897‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Capanni C, Coco R, Mattioli E, et al. Emerin‐prelamin A interplay in human fibroblasts. Biol Cell. 2009;101(9):541‐554. [DOI] [PubMed] [Google Scholar]
- 195. Bione S, Maestrini E, Rivella S, et al. Identification of a novel X‐linked gene responsible for Emery‐Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323‐327. [DOI] [PubMed] [Google Scholar]
- 196. Lee B, Lee TH, Shim J. Emerin suppresses Notch signaling by restricting the Notch intracellular domain to the nuclear membrane. Biochim Biophys Acta ‐ Mol Cell Res. 2017;1864(2):303‐313. [DOI] [PubMed] [Google Scholar]
- 197. Markiewicz E, Tilgner K, Barker N, et al. The inner nuclear membrane protein Emerin regulates β‐catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25(14):3275‐3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kumeta M, Hirai Y, Yoshimura SH, Horigome T, Takeyasu K. Antibody‐based analysis reveals “filamentous vs. non‐filamentous” and “cytoplasmic vs. nuclear” crosstalk of cytoskeletal proteins. Exp Cell Res. 2013;319(20):3226‐3237. [DOI] [PubMed] [Google Scholar]
- 199. Hobbs RP, Depianto DJ, Jacob JT, et al. Keratin‐dependent regulation of Aire and gene expression in skin tumor keratinocytes. Nat Genet. 2015;47(8):933‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Escobar‐Hoyos LF, Shah R, Roa‐Peña L, et al. Keratin‐17 promotes p27KIP1 nuclear export and degradation and offers potential prognostic utility. Cancer Res. 2015;75(17):3650‐3662. [DOI] [PubMed] [Google Scholar]
- 201. Matsuyama M, Tanaka H, Inoko A, et al. Defect of mitotic vimentin phosphorylation causes microophthalmia and cataract via aneuploidy and senescence in lens epithelial cells. J Biol Chem. 2013;288(50):35626‐35635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Feng X, Coulombe PA. A role for disulfide bonding in keratin intermediate filament organization and dynamics in skin keratinocytes. J Cell Biol. 2015;209(1):59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Feng X, Coulombe PA. Complementary roles of specific cysteines in keratin 14 toward the assembly, organization, and dynamics of intermediate filaments in skin keratinocytes. J Biol Chem. 2015;290(37):22507‐22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Flitney EW, Kuczmarski ER, Adam SA, Goldman RD. Insights into the mechanical properties of epithelial cells: the effects of shear stress on the assembly and remodeling of keratin intermediate filaments. FASEB J. 2009;23(7):2110‐2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Sivaramakrishnan S, Schneider JL, Sitikov A, Goldman RD, Ridge KM. Shear stress induced reorganization of the keratin intermediate filament network requires phosphorylation by protein kinase C ζ. Mol Biol Cell. 2009;20(11):2755‐2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Patteson AE, Vahabikashi A, Pogoda K, et al. Vimentin protects cells against nuclear rupture and DNA damage during migration. J Cell Biol. 2019;218(12):4079‐4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. De Pascalis C, Pérez‐González C, Seetharaman S, et al. Intermediate filaments control collective migration by restricting traction forces and sustaining cell‐cell contacts. J Cell Biol. 2018;217(9):3031‐3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wickström SA, Niessen CM. Cell adhesion and mechanics as drivers of tissue organization and differentiation: local cues for large scale organization. Curr Opin Cell Biol. 2018;54:89‐97. [DOI] [PubMed] [Google Scholar]
- 209. Liu T, Guevara OE, Warburton RR, Hill NS, Gaestel M, Kayyali US. Regulation of vimentin intermediate filaments in endothelial cells by hypoxia. Am J Physiol Physiol. 2010;299(2):C363‐C373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Na N, Chandel NS, Litvan J, Ridge KM. Mitochondrial reactive oxygen species are required for hypoxia‐induced degradation of keratin intermediate filaments. FASEB J. 2010;24(3):799‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Zamoner A, Pierozan P, Vidal LF, et al. Vimentin phosphorylation as a target of cell signaling mechanisms induced by 1α,25‐dihydroxyvitamin D3 in immature rat testes. Steroids. 2008;73(14):1400‐1408. [DOI] [PubMed] [Google Scholar]
- 212. Zamoner A, Corbelini PF, Funchal C, Menegaz D, Barreto Silva FRM, Pessoa‐Pureur R. Involvement of calcium‐dependent mechanisms in T3‐induced phosphorylation of vimentin of immature rat testis. Life Sci. 2005;77(26):3321‐3335. [DOI] [PubMed] [Google Scholar]
- 213. Zamoner A, Funchal C, Heimfarth L, Silva FRMB, Pessoa‐Pureur R. Short‐term effects of thyroid hormones on cytoskeletal proteins are mediated by GABAergic mechanisms in slices of cerebral cortex from young rats. Cell Mol Neurobiol. 2006;26(2):209‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Zanatta L, Goulart PB, Gonçalves R, et al. 1α,25‐Dihydroxyvitamin D3 mechanism of action: modulation of L‐type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim Biophys Acta ‐ Mol Cell Res. 2012;1823(10):1708‐1719. [DOI] [PubMed] [Google Scholar]
- 215. Hiratsuka T, Bordeu I, Pruessner G, Watt FM. Regulation of ERK basal and pulsatile activity control proliferation and exit from the stem cell compartment in mammalian epidermis. Proc Natl Acad Sci U S A. 2020;117(30):17796‐17807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Seymour PA, Collin CA, Egeskov‐Madsen ALR, et al. Jag1 modulates an oscillatory Dll1‐Notch‐Hes1 signaling module to coordinate growth and fate of pancreatic progenitors. Dev Cell. 2020;52(6):731‐747.e8. [DOI] [PubMed] [Google Scholar]
- 217. Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20(2):79‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Hyder CL, Pallari HM, Kochin V, Eriksson JE. Providing cellular signposts–post‐translational modifications of intermediate filaments. FEBS Lett. 2008;582(14):2140‐2148. [DOI] [PubMed] [Google Scholar]
- 219. Pallari HM, Eriksson JE. Intermediate filaments as signaling platforms. Sci STKE. 2006;2006(366):pe53. [DOI] [PubMed] [Google Scholar]
- 220. Perlson E, Hanz S, Ben‐Yaakov K, Segal‐Ruder Y, Seger R, Fainzilber M. Vimentin‐dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45(5):715‐726. [DOI] [PubMed] [Google Scholar]
- 221. Liu CY, Lin HH, Tang MJ, Wang YK. Vimentin contributes to epithelial‐mesenchymal transition ancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget. 2015;6(18):15966‐15983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Runembert I, Queffeulou G, Federici P, et al. Vimentin affects localization and activity of sodium‐glucose cotransporter SGLT1 in membrane rafts. J Cell Sci. 2002;115(4):713‐724. [DOI] [PubMed] [Google Scholar]
- 223. Kanugula AK, Dhople VM, Völker U, Ummanni R, Kotamraju S. Fluvastatin mediated breast cancer cell death: a proteomic approach to identify differentially regulated proteins in MDA‐MB‐231 cells. PLoS One. 2014;9(9):e108890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Kaschula CH, Tuveri R, Ngarande E, et al. The garlic compound ajoene covalently binds vimentin, disrupts the vimentin network and exerts anti‐metastatic activity in cancer cells. BMC Cancer. 2019;19(1):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Trogden KP, Battaglia RA, Kabiraj P, Madden VJ, Herrmann H, Snider NT. An image‐based small‐molecule screen identifies vimentin as a pharmacologically relevant target of simvastatin in cancer cells. FASEB J. 2018;32(5):2841‐2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Bollong MJ, Pietilä M, Pearson AD, et al. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc Natl Acad Sci U S A. 2017;114(46):E9903‐E9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Noh H, Yan J, Hong S, et al. Discovery of cell surface vimentin targeting mAb for direct disruption of GBM tumor initiating cells. Oncotarget. 2016;7(44):72021‐72032. [DOI] [PMC free article] [PubMed] [Google Scholar]