

Abstract
The reaction of [Cp′′′Ni(η3‐P3)] (1) with in situ generated phosphenium ions [RR′P]+ yields the unprecedented polyphosphorus cations of the type [Cp′′′Ni(η3‐P4R2)][X] (R=Ph (2 a), Mes (2 b), Cy (2 c), 2,2′‐biphen (2 d), Me (2 e); [X]−=[OTf]−, [SbF6]−, [GaCl4]−, [BArF]−, [TEF]−) and [Cp′′′Ni(η3‐P4RCl)][TEF] (R=Ph (2 f), tBu (2 g)). In the reaction of 1 with [Br2P]+, an analogous compound is observed only as an intermediate and the final product is an unexpected dinuclear complex [{Cp′′′Ni}2(μ,η3:η1:η1‐P4Br3)][TEF] (3 a). A similar product [{Cp′′′Ni}2(μ,η3:η1:η1‐P4(2,2′‐biphen)Cl)][GaCl4] (3 b) is obtained, when 2 d[GaCl4] is kept in solution for prolonged times. Although the central structural motif of 2 a–g consists of a “butterfly‐like” folded P4 ring attached to a {Cp′′′Ni} fragment, the structures of 3 a and 3 b exhibit a unique asymmetrically substituted and distorted P4 chain stabilised by two {Cp′′′Ni} fragments. Additional DFT calculations shed light on the reaction pathway for the formation of 2 a–2 g and the bonding situation in 3 a.
Keywords: nickel, phosphenium cations, phosphorus, polyphosphorus cations, ring expansion
Rings and chains with P: The reaction of [Cp′′′Ni(η3‐P3)] with in situ generated phosphenium cations [R2P]+ yields unprecedented cationic bent P4R2 rings and P4R3 chains coordinated to {Cp′′′Ni} fragments. These results suggest that the formation of polyphosphorus cations from phosphenium cations is not restricted to polyphosphines and P4 as substrates, but opens the way to polyphosphorus ligands fixed in the coordination sphere of transition metals.
Introduction
The structural diversity observed in organic chemistry is immense, and it is based on the high tendency of carbon to form homoatomic bonds, owing to the high C−C single and multiple bond energies. [1] Owing to its diagonal relationship to carbon and the isolobality of CH fragments to P, [2] a similar chemical behaviour is predicted for phosphorus. Thus, a broad variety of anionic and neutral polyphosphorus compounds was reported. [3] In contrast, the area of polyphosphorus cations is still underdeveloped. Important efforts in view of the synthesis of organosubstituted polyphosphorus cations were made by the groups of Burford and Weigand, respectively, who were able to isolate catenated [4] as well as ring‐ [5] and cage‐type (I, III) [6] polyphosphorus cations (Scheme 1), the latter representing mostly unsubstituted polyphosphorus moieties. The Krossing group was able to extend these results by isolating halogen‐substituted cages (II), [7] and they even succeeded in demonstrating the formation of the first homoleptic polyatomic phosphorus cation (IV) in solution. [8] The syntheses of the cations I, II and III could be achieved by the reaction of P4 with phosphenium cations [RR′P]+, which have proven to be very useful electrophiles for this purpose. Although donor‐substituted derivatives of these heavy carbene analogues can be isolated, [9] phosphenium cations bearing alkyl, aryl or halogen substituents are too reactive [10] so that they can only be generated in situ by the reaction of halogenophosphines with a suitable halide‐abstracting agent. A general reactivity pattern of these phosphenium cations is their insertion into P−P bonds, affording expanded polyphosphorus structures.
Scheme 1.
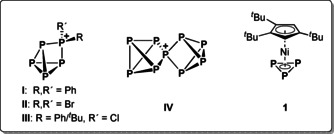
Left: Selected polyphosphorus cations (I, II and III) derived from the reaction of P4 with suitable phosphenium ions. Middle: The first homoatomic polyphosphorus cation IV generated upon the reaction of P4 with [NO][TEF]. Right: The starting compound 1, which is isolobal to P4.
On the other hand, polypnictogen units can also serve as ligands (En ligands; E=P, As, Sb, Bi) stabilised in the coordination sphere of transition metals. Reductive [11] and nucleophilic [11e] functionalisations of these units have proven to be promising routes to obtain large polypnictogen frameworks. Furthermore, initial studies on their oxidation chemistry revealed the potential in the formation of larger cationic polypnictogen structures.[ 11a , 12 ] Although the reaction of such complexes with electrophilic pnictinidenes [Cp*E{W(CO)5}2] (E=P, As) [13] and the condensation of an anionic bimetallic complex with halogenophosphines [14] lead to neutral En ring‐expanded products, their functionalisation with cationic electrophiles has been scarcely explored. Examples of the latter are alkylation reactions of [(L)Co(η3‐P3)] [15] or [(L)Rh(η1:η2‐P4R)] [16] (L=MeC(CH2PPh2)3). Furthermore, it was shown only recently that derivatives of I can be reacted with highly reduced metal units to yield cationic polyphosphorus compounds. [17] An alternative approach, however, the reactivity of polyphosphorus complexes towards phosphenium cations, has not been investigated so far. Therefore, the question arises as to whether this would be a general way to the synthesis of a large variety of novel cationic polyphosphorus frameworks. This way would open a huge variety of functionalisation possibilities by the used phosphenium cations as well as afterwards by having introduced suitable functional groups. Moreover, also the question as to a possible mechanism between addition and insertion on the one hand or the immediate insertion on the other came up. Inspired by the results obtained on the electrophilic functionalisation of P4 by the Krossing group (see above), we envisioned the P4 isolobal polyphosphorus complex [Cp′′′Ni(η3‐P3)] (1, Cp′′′=1,2,4‐tBu3C5H2)[ 11e , 11g ] (Scheme 1) as a suitable starting material for such investigations. Although the recently reported coordinative transformation of derivatives of I is so far limited to P5R2 moieties, [17] our approach would allow for the preparation of yet unknown cyclo‐P4R2 units. Moreover, if we could demonstrate this reactivity on 1 with success, this approach would potentially open a general way to cationic polyphosphorus ligands of different sizes, as a large variety of polyphosphorus ligand complexes already exists. Additionally, this approach allows for the simple exchange of substituents in the final products, if, for example, halogen substituents are present and, therefore, the obtained compounds could serve as valuable starting materials in the preparation of novel functionalised polyphosphorus compounds.
Results and Discussion
When 1 is reacted with [Ph2P][OTf] (Ph=phenyl, [OTf]−=[SO3CF3]−), in situ generated by the reaction of Tl[OTf] as the halide‐abstracting agent and Ph2PCl in o‐DFB (1,2‐difluorobenzene), a colour change from bright orange to dark red and the formation of a white precipitate (TlCl) can be observed over the course of 20 h. Upon workup and recrystallisation, [Cp′′′Ni(η3‐P4Ph2)][OTf] (2 a[OTf]) can be isolated in 63 % crystalline yield (Scheme 2).
Scheme 2.
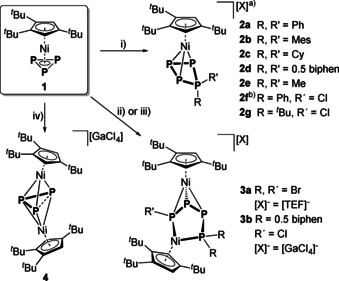
Reaction of 1 with phosphenium and halogenophosphenium ions: i) “[RR′P][X]” in situ generated from RR′PCl and Tl[TEF] (for other abstracting agents, see the Supporting Information); ii) “[Br2P][TEF]” from PBr3 and Tl[TEF]; iii) “[biphenP][GaCl4]” from biphenPCl and GaCl3 with prolonged reaction time (14 d); iv) GaCl3. a) All compounds 2 a–g were prepared as their [TEF]− salts and 2 a–d additionally as their [GaCl4]− salts; b) Isolated as an isomeric mixture of 2 f endo/2 f exo=7:1 with endo and exo referring to the position of the Ph group at the folded P4 ring.
As we had anticipated the halide‐abstracting agent to have a crucial impact on this reaction, we evaluated other compounds applicable for this purpose. Performing the same reaction in the presence of Ag[SbF6], M[TEF] (M+=Ag+ or Tl+, [TEF]−=[Al(OC(CF3)3)4]−) [18] or [(Et3Si)2(μ‐H)][BArF] [19] ([BArF]−=[B(C6F5)4]−) yielded 2 a[X] ([X]−=[SbF6]−, [TEF]−, [BArF]−) in comparably lower yields (for details see the Supporting Information). The exchange of these ionic halide‐abstracting agents for the neutral GaCl3, however, gave 2 a[GaCl4] in 71 % yield after only 4 h of reaction. With these results in hand, we turned our interest towards exchanging the Ph substituents at the phosphenium ion for other organic groups, by performing the reactions either with GaCl3 or Tl[TEF] (enhanced stabilisation). Notably, replacing Ph2PCl with the sterically very demanding tBu2PCl (tBu=tert‐butyl) or the electronically deactivated (Et2N)2PCl does not lead to any reaction with 1. Instead, using Mes2PCl, Cy2PCl or (biphen)PCl (Mes=mesityl, Cy=cyclohexyl, biphen=2,2′‐biphenyl) gives the products [Cp′′′Ni(η3‐P4R2)][X] (R=Mes (2 b: 53 % [X]−=[GaCl4]− or 46 % [X]−=[TEF]−), Cy (2 c: 59/57 %); R2=biphen (2 d: 70/55 %; [X]−=[GaCl4]−/[TEF]−)) in moderate to good yields. When employing the highly reactive Me2PCl, only the [TEF]− salt is stable enough to isolate [Cp′′′Ni(η3‐P4Me2)][TEF] (2 e: 52 %). The insertion of multiple [Ph2P]+ entities into 1 is not observed, not even when using two or more equivalents of Ph2PCl and Tl[TEF]. Expanding the scope of our approach, we replaced halogenophosphines with dihalogenophosphines RPCl2. When 1 is reacted with the respective dihalogenophosphine (PhPCl2 or tBuPCl2) under similar conditions, a colour change from bright orange to brownish red and again the formation of a white precipitate (TlCl) are observed. After workup, the products [Cp′′′Ni(η3‐P4RCl)][TEF] (R=Ph (2 f: 60 %), R=tBu (2 g: 50 %)) can be isolated as brownish red powders. Although for 2 g the formation of a single product is observed, NMR data (see below) suggest the formation of two isomers of 2 f (Ph‐substituent in endo/exo positions; see Figures S2 and S3 in the Supporting Information). Furthermore, when we implemented PBr3 as the phosphenium ion precursor, the expected [Cp′′′Ni(η3‐P4Br2)]+ (2 h) was only observed as an intermediate (see Figures S8–S10 in the Supporting Information for the 31P{1H} NMR spectra), which cannot be isolated. However, stirring the reaction mixture for three days at room temperature leads to the formation of the dinuclear complex [{Cp′′′Ni}2(μ,η3:η1:η1‐P4Br3)][TEF] (3 a, Scheme 2). The additional detection of P4 hints at a fragmentation pathway for the formation of 3 a, which, after workup and crystallisation, is isolated as dark‐brown blocks in high yields (90 %, based on 1). Intriguingly, we discovered that keeping 2 d[GaCl4] in o‐DFB solution at room temperature for two weeks yields a similar dinuclear complex [{Cp′′′Ni}2(μ,η3:η1:η1‐P4(biphen)Cl)][GaCl4] (3 b) in 74 % yield (based on 1). When we tried to further increase the yield of reactions involving GaCl3 by varying the order of addition, we found that 1 also reacts with GaCl3 alone in the absence of R2PCl. Investigating this new reaction pattern revealed that the formation of the novel triple‐decker complex [{Cp′′′Ni}2(μ,η3:η3‐P3)][GaCl4] (4) can be achieved that way. To the best of our knowledge, 4 represents the first cationic representative of a Ni/Ni triple‐decker complex with an η3:η3‐P3 middle deck. Thus, it completes the series of previously reported anionic and neutral complexes [{Cp′′′Ni}2(μ,η3:η3‐P3)]−/0. [11e] Except for 2 b and 2 g, all obtained products could be crystallised as their [GaCl4]− (2 a, 2 c, 2 d) or [TEF]− (2 e, 2 f) salt, allowing their X‐ray crystallographic characterisation (Figure 1). For 2 f, only the endo‐Ph (regarding the P4 ring) isomer could be characterised crystallographically. Additional solid‐state structures of 2 a[X] ([X]=[OTf]−, [SbF6]− or [TEF]−) hint at a negligible influence of the anion on the structure of 2 a (see Figures S12–S15 in the Supporting Information). The central structural motif of 2 consists of a bent cyclo‐P4 unit, which is attached to a {Cp′′′Ni} fragment, carrying two substituents at one of the P atoms. This structural motif arises from the formal insertion of the corresponding phosphenium cation into one of the P−P bonds of 1 and represents a rare example of a ring expansion reaction from a strained three‐membered ring to a four‐membered one.[ 5b , 20 ] The P−P bond lengths in the P4 ring (2 a: 2.173(1)–2.194(2) Å, 2 c: 2.174(1)–2.195(1) Å, 2 d: 2.162(1)–2.208(1) Å, 2 e: 2.149(2)–2.195(2) Å, 2 f: 2.146(1)–2.204(2) Å) are all well within the range of P−P single bonds, [21] and comparably shorter than found in I or III.[ 6a , 6b ] Along the lines of the latter, the shortest P−P bonds are those involving the atom P1. These bond lengths are the shortest in 2 f (2.146(1)/2.147(1) Å), increase according to the steric bulk of the substituents (2 f<2 e<2 d<2 a<2 c) and are the longest in 2 c (2.174(1)/2.183(1) Å). A similar trend is not observed for the dihedral angle P1‐P2‐P4‐P3, which is the smallest in 2 e (40.75(9)°) but the largest in 2 f (49.48(7)°). The former P−P bond of 1 is clearly broken in these compounds, which is indicated by the elongated P2−P4 distances (2 a: 3.028(5) Å, 2 c: 3.020(1) Å, 2 d: 3.044(2) Å, 2 e: 3.031(3) Å, 2 f: 3.078(1) Å).[ 21 , 22 ] The solid‐state structures of the cations 3 a and 3 b could be established as their [TEF]− and [GaCl4]− salts, respectively (Figure 2). The central structural motif within both cations is the unprecedented, unsymmetrically substituted P4 chain, which is coordinated to two {Cp′′′Ni} fragments. The structure of these products can be formally described by the insertion of a second {Cp′′′Ni} fragment into a P−P bond of the cations 2 and the addition of a halide substituent to the P4 unit. The P−P bond lengths in 3 a (2.162(1)–2.223(1) Å) and 3 b (2.144(2)–2.239(2) Å) match the expected values for P−P single bonds, [21] where the longest ones are those between P1 and P2. Although the Ni1−P bond lengths (3 a: 2.221(1)–2.269(1) Å, 3 b: 2.217(1)–2.262(1) Å) are close to the values found for the starting material 1 (2.236(1)–2.246(1) Å), [11e] the Ni2−P1/P4 distances are comparably shorter (3 a: 2.104(1)–2.157(1) Å, 3 b: 2.142(1)–2.147(1) Å). The formulation as a P4 chain is clearly manifested in the large P1−P4 distances of 2.878(1) Å and 2.919(2) Å in 3 a and 3 b, respectively. [21] With P3‐P4‐P2‐P1 dihedral angles of 135.30(6)° (3 a) and 130.58(9)° (3 b) the P1 atom is located below the plane spanned by the other P atoms, and the halide substituents are situated above that plane (P2‐P4‐P3‐X=163.09(5) (3 a), 162.14(8)° (3 b)). The solid‐state structure of 4 reveals an allylically distorted P3 ligand in‐between two {Cp′′′Ni} fragments (Figure 2). The P1−P2 (2.202(1) Å) and P2−P3 (2.198(1) Å) bonds are within common P−P single bonds, [21] whereas the P1−P3 (2.539(1) Å) distance is clearly elongated. The elongation of the P1−P3 distance is more pronounced than in the neutral (2.398(1) Å) and anionic (2.241(1) Å) derivatives of 4, [11e] with the anion showing a nearly triangular P3 ligand. Furthermore, the P1‐P2‐P3 angles in the anionic (61.16(2)°) and neutral (66.60(2)°) derivatives [11e] are comparably smaller than the one in 4 (70.48(5)°). According to DFT calculations, the increasing allylic distortion going from the anion [{Cp′′′Ni}2(μ,η3:η3‐P3)]− over the neutral species [{Cp′′′Ni}2(μ,η3:η3‐P3)] to the cation 4 is caused by the stepwise depopulation of the HOMO of the anion, which has a large P−P bonding contribution. [11e]
Figure 1.
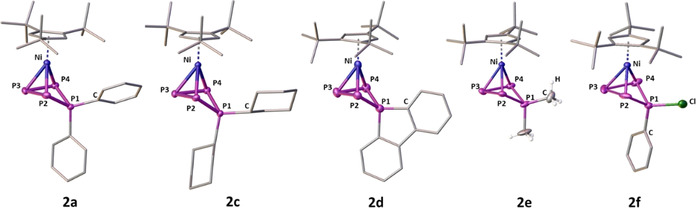
Solid‐state structures of 2 a and 2 c–f. Hydrogen atoms (except Me in 2 e), solvent molecules (0.5 o‐DFB (2 c), 0.4 n‐hexane (2 d) and counter anions ([GaCl4]− (2 a, 2 c, 2 d), [TEF]− (2 e, 2 f)) are omitted for clarity and ADPs are drawn at the 50 % probability level. The cation denoted with Ni1 is displayed for the structures of 2 d and 2 e, as the respective crystal structures contain five and four molar equivalents in the asymmetric unit, respectively.
Figure 2.
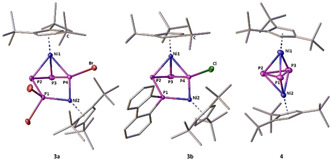
Solid‐state structures of the cations in 3 a, 3 b and 4. H atoms and the counter anions are omitted for clarity; ADPs are drawn at the 50 % probability level.
The 31P and 31P{1H} NMR spectra of the isolated product 2 in CD2Cl2 show AA′MX (2 b, 2 d, 2 f), AMM′X (2 a, 2 e, 2 g) or AMXX′ (2 c) spin systems (Figure 3), which are in agreement with the cyclic P4R2 ligand found in these compounds. For 2 f, an additional set of signals is found, as both isomers (endo‐Ph and exo‐Ph) are present in a 7:1 ratio (see Figures S2 and S3 in the Supporting Information). The chemical shifts of the signals are found between δ=−10 and 120 ppm (exact values are provided in the Supporting Information) and their assignment is simplified by additional P−H coupling of the signal corresponding to the former phosphenium cation in the 31P NMR spectrum. The 1 J P–P coupling constants in 2 a–g (see the Supporting Information) are slightly larger compared with the isolobal I/III,[ 6a , 6b ] and the varying sequence of signals (in addition to electronic effects) may be attributed to similar “cross‐ring through space” interactions as reported for derivatives of III. [6b] The 1H NMR spectrum of 3 a in CD2Cl2 shows five signals for the tBu groups and four signals for the residual protons of the Cp′′′ ligands, respectively. The 1H NMR spectrum of 3 b in CD2Cl2 is similar but shows six signals for the tBu groups and additional resonances for the 2,2′‐biphen substituent. Thus, a hindered rotation of the Cp′′′ ligands can be assumed in both cases at ambient temperatures. The 31P NMR spectrum of 3 a in CD2Cl2 reveals an AMNX spin system with signals at δ=−1.1 (PX), 128.0 (PN), 134.2 (PM) and 182.0 (PA) ppm, which is consistent with an asymmetric P4 chain. An unexpectedly large 2 J P–P coupling constant (281.7 Hz) of the P atoms at each end of the chain may, however, hint at possible orbital interactions between those atoms in 3 a (see below). The 31P NMR spectra of 3 b in CD2Cl2 recorded at room temperature only reveal two moderately resolved and two very broad resonances at δ=−31.9 (br), 80.6 (br), 84.1 and 146.6 ppm. Upon cooling to −80 °C, these signals split into two separate, well‐resolved sets indicating the presence of two isomers, which we assume to arise from a hindered rotation of the Cp′′′ ligands at this temperature. Again, unusually high 2 J P–P coupling constants are found between the P atoms at each end of the chain for both isomers (3 b1: 2 J P–P=108.2 Hz, 3 b2: 2 J P–P=113.2 Hz). The 1H NMR spectrum of 4 in CD2Cl2 shows three signals consistent with two symmetrical Cp′′′ ligands. Accordingly, its 31P NMR spectrum reveals only one singlet at δ=139.3 ppm, which does not change even upon cooling to −80 °C. Thus, we assume a rapid dynamic behaviour for the allylic P3 ligand.
Figure 3.
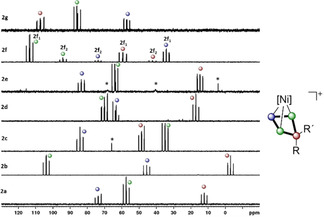
31P{1H} NMR spectra of isolated 2 a–g in CD2Cl2 recorded at 298 K with signal assignment according to the colour code. The two sets of signals observed for 2 f are labelled with 2 f1 and 2 f2, with the former being most probably the endo‐Ph isomer owing to steric reasons. *=unidentified minor impurities.
ESI(+) mass spectrometry studies on salts of the cations 2 a–g reveal their high stability towards fragmentation, as the only observed peaks are those attributed to the molecular cations (m/z=569.1 (2 a), 653.2 (2 b), 581.2 (2 c), 567.1 (2 d), 445.1 (2 e), 527.1 (2 f), 507.1 (2 g)). Only for 2 d[GaCl4], a second peak at m/z=895.2 (3 b) is observed, which is in agreement with our experimental finding of a (slow) fragmentation/rearrangement process of 2 d. When isolated samples of 3 a or 3 b in o‐DFB are subjected to ESI(+) MS experiments, only the molecular ion peaks are found (m/z=947.0 (3 a), 895.2 (3 b)). The ESI(+) mass spectrum of 4 also shows the molecular ion peak (m/z=675.2).
To obtain insight into the formation of the cations 2, we carried out DFT calculations (BP86/def2‐TZVP, PCM solvent correction for CH2Cl2, see the Supporting Information for details) on the model system consisting of [CpNi(η3‐P3)] (1′) and [Me2P]+ (Figure 4). Although the crucial impact of the [GaCl4]− anion on the formation of the isolobal compounds III has been pointed out for reactions involving GaCl3 as the halide‐abstracting agent, [6b] this seems unfeasible for reactions involving the weakly coordinating [TEF]− anion. Furthermore, investigations of the solution chemistry of the PBr3/Ag[TEF] couple showed that the formation of free phosphenium cations is unlikely, [23] but, to our knowledge, similar reactivity has not been studied for Tl+. Thus, we assume the formation of a free phosphenium cation ([Me2P]+) in the first step of the reaction. Compound 1′ reacts with [Me2P]+ in an exergonic reaction (ΔG 298K=−24.08 kcal mol−1) and without a transition state to yield the intermediate I2 in which [Me2P]+ is coordinated to the cyclo‐P3 ligand of 1′ in an η1 fashion. This reaction proceeds via TS2 (ΔG 298K=−18.31 kcal mol−1, ΔΔG ≠ 298K=5.77 kcal mol−1) in which the [Me2P]+ fragment is coordinated to one of the P−P bonds of 1′ and finally gives the product P2, which shows the complete insertion of [Me2P]+ into the respective P−P bond. The overall reaction is exergonic (ΔG 298K=−42.26 kcal mol−1), which is caused by the assumption of free [Me2P]+ being initially formed.
Figure 4.
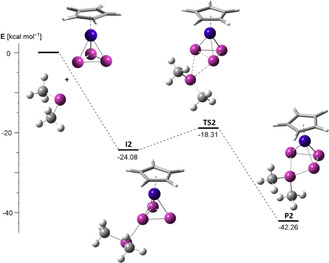
Calculated (BP86/def2TZVP, PCM solvent correction for CH2Cl2) energy profile of the reaction between the model compound 1′ and [Me2P]+. Stationary points were verified by analytical frequency calculations, energies are given in kcal mol−1 and the couple 1′+[Me2P]+ was arbitrarily set to an energy of 0 kcal mol−1.
Finally, we also investigated the electronic and bonding properties of 3 a/b computationally (B3LYP/def2SVP, PCM solvent correction for CH2Cl2). The observed geometric parameters for 3 a and 3 b obtained from these calculations match well with the experimental values, which corroborates the formulation of the P4X3 ligands as chains. Furthermore, the Wiberg Bond Indices (WBIs) for both compounds, obtained from natural bond orbital (NBO) analyses, are in line with this formulation (3 a: 0.87 (P1−P2), 1.12 (P2−P3), 1.04 (P3−P4); 3 b: 0.84 (P1−P2), 1.14 (P2−P3), 1.05 (P3−P4)). When we examined the canonical frontier orbitals of 3 a, we found an allylic‐type interaction between P1, Ni2 and P4 within the HOMO‐4 (see Figure S24 in the Supporting Information for details). The comparably small but significant orbital contributions of both P1 (7 %) and P4 (6 %) may explain the large 2 J P–P coupling constant found in the 31P NMR spectra of 3 a and 3 b.
Conclusion
The reaction of 1 with a variety of phosphenium cations yields numerous cationic polyphosphorus scaffolds, which are stabilised by coordination to {Cp′′′Ni} moieties. Whereas the cations 2 contain novel P4R2 ligands, 3 a and 3 b display a P4X3 chain motif that is unprecedented to date. DFT calculations revealed the reaction pathway leading to 2 as an addition/insertion mechanism and gave insight into the electronic structure of 3 a/b. Thus, we could show that the combination of phosphenium cations with polyphosphorus ligand complexes (such as 1) is an additional possibility to design polyphosphorus cations of the desired size in a rational way. We demonstrate this for the cyclo‐P3 complex 1, and we expect our approach to be transferrable to other polyphosphorus ligand complexes as well. The presented simple synthetic route allows for large diversification regarding the substitution pattern of the product by simply exchanging the phosphenium ion precursor. Although the discussed reactivity is exemplified by the P3 ligand complex 1, our approach should also be transferrable to other neutral polyphosphorus and other polypnictogen ligand complexes, and investigations in this direction are in progress. Furthermore, the obtained products hold great potential for functionalisations by reduction, nucleophilic functionalisation, or displacement of the {Cp′′′Ni} fragments, paving the way for even more elaborate substituted polyphosphorus compounds.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG Sche 384/36‐1). Open access funding enabled and organized by Projekt DEAL.
C. Riesinger, L. Dütsch, G. Balázs, M. Bodensteiner, M. Scheer, Chem. Eur. J. 2020, 26, 17165.
Dedicated to Professor Peter Klüfers on the occasion of his 70th birthday
References
- 1.
- 1a. Luo Y.-R., Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, 2007; [Google Scholar]
- 1b. Benson S. W., J. Chem. Educ. 1965, 42, 502. [Google Scholar]
- 2. Hoffmann R., Angew. Chem. Int. Ed. Engl. 1982, 21, 711–724; [Google Scholar]; Angew. Chem. 1982, 94, 725–739. [Google Scholar]
- 3.
- 3a. Baudler M., Glinka K., Chem. Rev. 1993, 93, 1623–1667; [Google Scholar]
- 3b. Baudler M., Glinka K., Chem. Rev. 1994, 94, 1273–1297. [Google Scholar]
- 4.
- 4a. Burford N., Cameron T. S., Ragogna P. J., Ocando-Mavarez E., Gee M., McDonald R., Wasylishen R. E., J. Am. Chem. Soc. 2001, 123, 7947–7948; [DOI] [PubMed] [Google Scholar]
- 4b. Burford N., Ragogna P. J., McDonald R., Ferguson M. J., J. Am. Chem. Soc. 2003, 125, 14404–14410; [DOI] [PubMed] [Google Scholar]
- 4c. Burford N., Dyker C. A., Decken A., Angew. Chem. Int. Ed. 2005, 44, 2364–2367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2416–2419; [Google Scholar]
- 4d. Possart J., Martens A., Schleep M., Ripp A., Scherer H., Kratzert D., Krossing I., Chem. Eur. J. 2017, 23, 12305–12313. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Burford N., Dyker C. A., Lumsden M., Decken A., Angew. Chem. Int. Ed. 2005, 44, 6196–6199; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6352–6355; [Google Scholar]
- 5b. Weigand J. J., Burford N., Lumsden M. D., Decken A., Angew. Chem. Int. Ed. 2006, 45, 6733–6737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6885–6889; [Google Scholar]
- 5c. Dyker C. A., Burford N., Chem. Asian J. 2008, 3, 28–36. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Weigand J. J., Holthausen M., Fröhlich R., Angew. Chem. Int. Ed. 2009, 48, 295–298; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 301–304; [Google Scholar]
- 6b. Holthausen M. H., Feldmann K. O., Schulz S., Hepp A., Weigand J. J., Inorg. Chem. 2012, 51, 3374–3387; [DOI] [PubMed] [Google Scholar]
- 6c. Holthausen M. H., Weigand J. J., Chem. Soc. Rev. 2014, 43, 6639–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Krossing I., Raabe I., Angew. Chem. Int. Ed. 2001, 40, 4406–4409; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 4544–4547; [Google Scholar]
- 7b. Gonsior M., Krossing I., Müller L., Raabe I., Jansen M., von Wüllen L., Chem. Eur. J. 2002, 8, 4475–4492. [DOI] [PubMed] [Google Scholar]
- 8. Köchner T., Engesser T. A., Scherer H., Plattner D. A., Steffani A., Krossing I., Angew. Chem. Int. Ed. 2012, 51, 6529–6531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6635–6637. [Google Scholar]
- 9.
- 9a. Cowley A. H., Cushner M. C., Lattman M., McKee M. L., Szobota J. S., Wilburn J. C., Pure Appl. Chem. 1980, 52, 789–797; [Google Scholar]
- 9b. Cowley A. H., Kemp R. A., Chem. Rev. 1985, 85, 367–382; [Google Scholar]
- 9c. Spinney H. A., Korobkov I., DiLabio G. A., Yap G. P. A., Richeson D. S., Organometallics 2007, 26, 4972–4982. [Google Scholar]
- 10. Slattery J. M., Hussein S., Dalton Trans. 2012, 41, 1808–1815. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Butovskiy M. V., Balázs G., Bodensteiner M., Peresypkina E. V., Virovets A. V., Sutter J., Scheer M., Angew. Chem. Int. Ed. 2013, 52, 2972–2976; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3045–3049; [Google Scholar]
- 11b. Li T., Gamer M. T., Scheer M., Konchenko S. N., Roesky P. W., Chem. Comm. 2013, 49, 2183–2185; [DOI] [PubMed] [Google Scholar]
- 11c. Arleth N., Gamer M. T., Köppe R., Pushkarevsky N. A., Konchenko S. N., Fleischmann M., Bodensteiner M., Scheer M., Roesky P. W., Chem. Sci. 2015, 6, 7179–7184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Arleth N., Gamer M. T., Köppe R., Konchenko S. N., Fleischmann M., Scheer M., Roesky P. W., Angew. Chem. Int. Ed. 2016, 55, 1557–1560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1583–1586; [Google Scholar]
- 11e. Mädl E., Balázs G., Peresypkina E. V., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 7702–7707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7833–7838; [Google Scholar]
- 11f. Schoo C., Bestgen S., Schmidt M., Konchenko S. N., Scheer M., Roesky P. W., Chem. Comm. 2016, 52, 13217–13220; [DOI] [PubMed] [Google Scholar]
- 11g. Schmidt M., Konieczny D., Peresypkina E. V., Virovets A. V., Balázs G., Bodensteiner M., Riedlberger F., Krauss H., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 7307–7311; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7413–7417. [Google Scholar]
- 12.
- 12a. Fleischmann M., Dielmann F., Gregoriades L. J., Peresypkina E. V., Virovets A. V., Huber S., Timoshkin A. Y., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 13110–13115; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13303–13308; [Google Scholar]
- 12b. Dütsch L., Fleischmann M., Welsch S., Balázs G., Kremer W., Scheer M., Angew. Chem. Int. Ed. 2018, 57, 3256–3261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3311–3317. [Google Scholar]
- 13. Piesch M., Seidl M., Stubenhofer M., Scheer M., Chem. Eur. J. 2019, 25, 6311–6316. [DOI] [PubMed] [Google Scholar]
- 14. Ziegler C. G. P., Maier T. M., Pelties S., Taube C., Hennersdorf F., Ehlers A. W., Weigand J. J., Wolf R., Chem. Sci. 2019, 10, 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Capozzi G., Chiti L., Di Vaira M., Peruzzini M., Stoppioni P., Chem. Comm. 1986, 24, 1799–1800; [Google Scholar]
- 15b. Barth A., Huttner G., Fritz M., Zsolnai L., Angew. Chem. Int. Ed. Engl. 1990, 29, 929–931; [Google Scholar]; Angew. Chem. 1990, 102, 956–958; [Google Scholar]
- 15c. Peruzzini M., Abdreimova R. R., Budnikova Y., Romerosa A., Scherer O. J., Sitzmann H., J. Organomet. Chem. 2004, 689, 4319–4331. [Google Scholar]
- 16. Barbaro P., Ienco A., Mealli C., Peruzzini M., Scherer O. J., Schmitt G., Vizza F., Wolmershäuser G., Chem. Eur. J. 2003, 9, 5195–5210. [DOI] [PubMed] [Google Scholar]
- 17. Adhikari A. K., Ziegler C. G. P., Schwedtmann K., Taube C., Weigand J. J., Wolf R., Angew. Chem. Int. Ed. 2019, 58, 18584–18590; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18757–18763. [Google Scholar]
- 18.
- 18a. Krossing I., Chem. Eur. J. 2001, 7, 490–502; [DOI] [PubMed] [Google Scholar]
- 18b. Gonsior M., Krossing I., Mitzel N., Z. Anorg. Allg. Chem. 2002, 628, 1821–1830. [Google Scholar]
- 19. Connelly S. J., Kaminsky W., Heinekey D. M., Organometallics 2013, 32, 7478–7481. [Google Scholar]
- 20. Chitnis S. S., Musgrave R. A., Sparkes H. A., Pridmore N. E., Annibale V. T., Manners I., Inorg. Chem. 2017, 56, 4521–4537. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186–197; [DOI] [PubMed] [Google Scholar]
- 21b. Pyykkö P., J. Phys. Chem. A 2015, 119, 2326–2337. [DOI] [PubMed] [Google Scholar]
- 22. Mantina M., Chamberlin A. C., Valero R., Cramer C. J., Truhlar D. G., J. Phys. Chem. A 2009, 113, 5806–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Gonsior M., Krossing I., Dalton Trans. 2005, 11, 2022–2030; [DOI] [PubMed] [Google Scholar]
- 23b. Gonsior M., Krossing I., Dalton Trans. 2005, 7, 1203–1213; [DOI] [PubMed] [Google Scholar]
- 23c. Gonsior M., Krossing I., Matern E., Chem. Eur. J. 2006, 12, 1703–1714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary