

Abstract
Atropisomerism is a fundamental feature of substituted biaryls resulting from rotation around the biaryl axis. Different stereoisomers are formed due to restricted rotation about the single bond, a situation often found in substituted porphyrins. Previously NMR determination of porphyrin atropisomers proved difficult, if not almost impossible to accomplish, due to low resolution or unresolvable resonance signals that predominantly overlapped. Here, we shed some light on this fundamental issue found in porphyrinoid stereochemistry. Using benzenesulfonic acid (BSA) for host‐guest interactions and performing 1D, 2D NMR spectroscopic analyses, we were able to characterize all four rotamers of the nonplanar 5,10,15,20‐tetrakis(2‐aminophenyl)‐2,3,7,8,12,13,17,18‐octaethylporphyirin as restricted H‐bonding complexes. Additionally, X‐ray structural analysis was used to investigate aspects of the weak host–guest interactions. A detailed assignment of the chemical signals suggests charge‐assisted complexation as a key to unravel the atropisomeric enigma. From a method development perspective, symmetry operations unique to porphyrin atropisomers offer an essential handle to accurately identify the rotamers using NMR techniques only.
Keywords: atropisomers, crystallography, NMR, porphyrinoids, supramolecular chemistry
Porphyrin atropisomers: Upon full assignment of all NMR signals symmetry elements were found to play a crucial part in the atropisomeric characteristics. A detailed study on nonplanar porphyrin atropisomers highlights new means to detect and identify individual rotamers in solution while X‐ray crystallographic analyses identified new supramolecular binding modes and specified aspects of weak interactions.
Introduction
Atropisomers as a subclass of conformers arise from restricted rotation about a C−C single bond predominantly due to steric hindrance. The atropisomerism observed in biphenyl derivatives [1] is very similar to the rotation of phenyl rings in 5,10,15,20‐tetraphenylporphyrins (Figure 1 a). For a phenyl group to rotate, it must pass through the plane of the porphyrin. This is similar to the situation for two aryl rings in a biphenyl moiety, that must become coplanar in the transition state. However, the porphyrin ring becomes considerably deformed in the transition state, which contributes to the overall potential‐energy barrier. [2] Typically, increasing the steric bulk on ortho‐substituted phenyls [3] or meta‐substituted phenyls [4] increases the barrier of rotation and reduces interconvertibility (see Figure 2 a for porphyrin positions). Therefore, the rotational barrier to convert one rotamer into another, is usually sufficiently large enough to isolate separate stereoisomers. [5] The seminal study of atropisomerism in planar porphyrins was reported by Gottwald and Ullman, showing that 5,10,15,20‐tetrakis(o‐hydroxyphenyl)porphyrin could be separated into four stereoisomers. [6]
Figure 1.
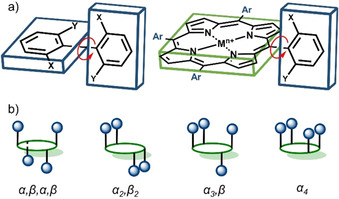
Representation of rotamers: a) comparison of biphenyl and porphyrin derivatives, b) possible porphyrin atropisomers based on meso‐substitution.
Figure 2.
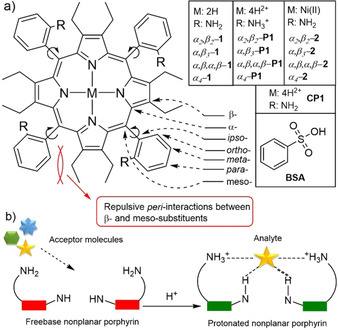
a) Atropisomers discussed in this study; b) graphical representation of interlocked porphyrin system by analyte coordination. [11]
The four porphyrin atropisomers are distinguished by their position with respect to the location of the meso‐substituent above (α) or below (β) the 24‐atom macrocycle plane. The rotamer with all four groups on one side is denoted as α4 (↑↑↑↑), while the others are α 2, β2 (↑↑↓↓), α 3, β (↑↑↑↓) and α,β,α,β (↑↓↑↓), respectively (Figure 1 b). Since the aryl groups in planar meso‐arylporphyrins are roughly perpendicular to the porphyrin plane, with limited rotation around the porphyrin‐aryl C−C bond, each atropisomer can be described by symmetry point group notations: α4 (C 4v), α,β,α,β (D 2d), α 2, β2 (C 2h), α 3, β (Cs). [7]
The phenomenon of atropisomerism has played an important role in the development of porphyrin chemistry. In the mid‐1970s, Collman et al. utilized the α4‐rotamer bearing steric groups in the ortho‐positions of 5,10,15,20‐tetraarylporphyrin as a structural scaffold to create “picket fence” porphyrins to act as biological mimics for dioxygen‐binding hemoproteins. [8] Porphyrin atropisomers have been utilized in applications as diverse as induced chirality, [9] supramolecular strapped systems, [10] molecular recognition [11] and coordination chemistry. [12] Moreover, atropisomers in general have attracted significant attention in drug development. [13]
Atropisomerism and conformational aspects in planar porphyrins have been previously reviewed. [14] However, only a handful of studies have been undertaken to identify the factors contributing to the ease of arene rotation in nonplanar porphyrins. [15] Nonplanar porphyrins, also called highly substituted porphyrins, [16] are a quickly growing research field in sensing,[ 11 , 17 ] organocatalysis [18] and regioselective reactions. [19] The well‐defined macrocycle deformations in nonplanar porphyrins can be used as a model system to elucidate the precise mechanism of atropisomer interconversion. In a prior study of dodecaarylporphyrin rotamers, Medforth et al. showed an unusual increase in rotational barrier upon protonation. [15] The ongoing transition to enantioselective systems, with its requirement to position peripheral substituents in specific facial orientations, [9] can only be achieved through access to specific and fully characterized rotamers. Therefore, a thorough structural analysis of nonplanar porphyrin atropisomers is of timely importance.
The identification of individual porphyrin atropisomers is a tedious and exhaustive process. This challenging analytical task has been a topic of interest for more than five decades. [6] There are few methods that suggest a particular orientation of porphyrin rotamers. Firstly, statistically speaking, α,β,α,β; α 2, β2, α 3, β, and α4 atropisomers would form in a 1:2:4:1 ratio; therefore, identification of individual rotamers is possible upon separation and quantification.[ 6 , 8 , 11 ] However, this is the least reliable method due to possible enrichments via atropisomer interconversions during the separation process. A change in the statistical mixture can be induced by various factors: steric bulk, [4] self‐organization [20] or guest‐assisted coordination, [21] thermally‐driven orientation in different solvents, [22] or central metal [23] and ligand [24] effects. A more reliable method to identify a particular rotamer is by its retention time in liquid chromatography. Usually, the smallest retention time is observed for α,β,α,β; followed by α 2, β2 and α 3, β with α4 being the most polar entity.[ 6 , 8 , 11 , 25 ] However, this is applicable only to completely separable atropisomeric mixtures. In terms of IR and UV‐vis spectroscopies, only marginal differences are usually observed between individual stereoisomers.[ 5 , 11 , 26 ]
By far the most popular porphyrin atropisomer characterization technique is NMR spectroscopy. The first observation of atropisomerism in porphyrins by NMR was reported by Walker et al. almost 50 years ago. [27] The proton NMR spectrum was found to show multiple resonances for the methyl signal in 5,10,15,20‐tetrakis(o‐tolyl)porphyrin, which were attributed to porphyrin atropisomerism and a full 1H and 13C characterization of individual porphyrin rotamers carried out by Abraham et al. [28] Atropisomeric effects have been described not only for the meso‐substituents but also for the porphyrin macrocycle, with distinct NMR signals observed for β‐position protons. However, in the case of α,β,α,β and α4, the insignificant spectral differences, makes using only 1D or even most 2D [29] NMR techniques inadequate to identify all isomers due to the high symmetry.
To date, X‐ray crystallographic study has been the most reliable approach to pinpoint the conformation of a particular porphyrin atropisomer and has been extensively used since the mid‐1970s. [8] However, X‐ray crystallography has its limitations. The microscopic sized crystal mounted in the diffractometer only represents a fraction of the isolated product; moreover, crystal growth is a complicated process that depends on a plenitude of physical and chemical properties. To the best of our knowledge thus far, only in a recent report by us have all four porphyrin rotamers been identified by X‐ray crystallography. [11]
In this report, we explore the 1D and 2D NMR spectroscopy of nonplanar porphyrin P1 rotamers complexed with benzenesulfonic acid (BSA) (Figure 2 a). The studies highlight a new avenue of atropisomer identification by NMR, while X‐ray crystallographic studies provide a better understanding on coordination of the charged entities by P1.
Results and Discussion
A detailed NMR analysis of porphyrin atropisomers has, in the past, been hampered by the high symmetry of the planar porphyrin macrocycle. To reduce porphyrin symmetry, we chose to induce nonplanarity by a highly substituted molecular strategy (porphyrins carrying a high number of β‐ and meso‐substituents). As shown in our previous research, the interplay between all peripheral substituents (i.e. peri‐interactions and peripheral control) of P1 results in a well‐defined active site directing to the inner core system for analyte binding and recognition (Figure 2 b). [11] A counter anion locks in the anion‐selective inner core system and reduces the proton exchange rate, generating static proton signals for higher resolution NMR analysis. Sterically overcrowded porphyrins are generally known to often adopt the saddle 3D shape conformation demonstrated by P1. [11] Molecular engineering of a porphyrin with peripheral binding sites allows us to report an ideal case study in which P1 atropisomers can stand in rotation‐restricted states for which high‐resolution NMR may be available.
NMR analysis of the inner core system
Typically, the 1H NMR spectrum of the nonplanar porphyrin inner core system is distinguished by a relatively broad singlet. However, coordination of BSA to the center of free base P1 porphyrin produced sharp, fingerprint‐like 1H NMR proton signals of the inner N‐H region. One singlet (4 H) in ↑↓↑↓, three (2H:1H:1 H) in ↑↑↓↓, four (1H:1H:1H:1 H) in ↑↑↑↓ and two (2H:2 H) in the ↑↑↑↑ atropisomer (Figure 3). The same 1H NMR integration ratios of the inner core system is observed in P1 rotamers complexed with methanesulfonic acid (MSA), [11] thus highlighting the anion‐independence of the fingerprint‐like systems. Distinctive inner core N−H signals can be utilized for swift and quantitative analysis of atropisomeric porphyrin mixtures, as showcased for P1‐MSA complexes in Figure S39. Sharp, well defined inner core signals are important to identify and completely assign the orientation of each rotamer using various 2D NMR techniques, as discussed in the 2D NMR section.
Figure 3.
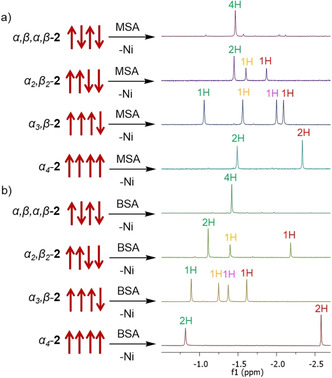
Fingerprint‐like 1H NMR N−H signals observed with two different complexing acids: a) methanesulfonic acid (MSA); [11] b) benzenesulfonic acid (BSA) in CD3CN.
As the corresponding atropisomers in their complexed form P1‐BSA showed prominent 1H NMR signals, further investigations were carried out using the 15N–1H HSQC technique. The 15N resonances were observed in a range of 125.1–127.3 ppm, correlating to the 1H of the inner core system (Figure 4).
Figure 4.
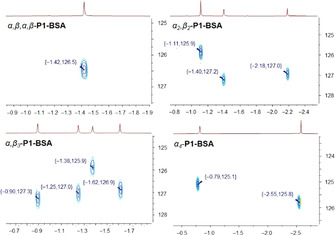
15N–1H HSQC spectra (ppm) of P1‐BSA atropisomers in CD3CN.
Mainly there are two ways to obtain a 15N NMR spectrum. First, isotopic enrichment of the sample to achieve the requisite signal sensitivity. [30] Multiple reports have investigated the porphyrin inner core nitrogen atoms by 15N NMR techniques, predominantly using 15N enriched porphyrins due to its low natural abundance (0.37 %). [31] Second, detection of the inner nitrogen atoms can be accomplished indirectly using heteronuclear correlation 15N–1H HSQC. [32] However, to obtain a 15N–1H HSQC spectrum, the N−H proton signals must be sharp and undergo a very slow exchange. [33] The host being “locked” with the guest analyte is a valuable asset for 15N NMR investigations of porphyrins without isotope enrichment due to the minimized exchange rates.
Overall, fingerprint‐like 1H and 15N inner core systems are exciting toolboxes providing intrinsic porphyrin atropisomer characteristics and could be used as a fast identification method for a particular orientation.
Full characterization of atropisomers using 1D and 2D NMR
The full 1H, 13C, and 15N assignments have been delineated for all atropisomers and are listed in Table S1. Beginning with the 1H spectrum, all eight CH3 group resonances of P1‐BSA rotamers were readily identified as triplets in the region between −0.19 and 0.74 ppm. The observed CH2 resonance signals (1.78–2.63 ppm) were split in two distinct doublet of quartets due to the diastereotopic methylene protons (outer) that point away from each other, toward the phenyl rings and (inner) protons that point toward each other (see Figure 6). As described above, the inner core proton signals were found between −2.58 and −0.82 ppm as sharp singlets (Figure 3 b). Moving to the aryl groups, clearly visible ortho‐proton signals were obtained as shifted downfield (8.63–8.92 ppm) doublets due to the proximity to the porphyrin ring current. In comparison, other aromatic region signals (meta‐ and para‐) were moderately overlapping (7.85–8.13 ppm). Peripheral NH3 + groups were not observed due to the rapid proton exchange.
Figure 6.
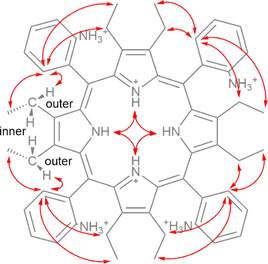
Key 1H–1H ROESY correlations of P1‐BSA in CD3CN and representation of inner and outer CH2 protons.
Basic assignment of the structure was carried out using a combination of 13C, 13C‐DEPT135, 1H–1H TOCSY, 1H–13C HSQC, and 1H–13C HMBC taken from the Bruker pulse library. This provided adequate information to identify positional meso‐Ar fragments. In HMBC, with a combination from ethyl groups, inner N−H showed three‐ and two‐bond correlations identifying the corresponding pyrrole units (Figure 5). Unfortunately, the long‐range heteronuclear correlation between N−H and Cmeso was not observed, which would have assisted to identify the order that pyrrole/Ar fragments follow in the porphyrin macrocycle.
Figure 5.
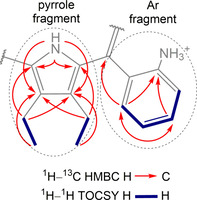
Key 1H–13C HMBC and 1H–1H TOCSY correlations of P1‐BSA in [D3]acetonitrile and representation of identified pyrrole and Ar fragments.
The rotating frame Overhauser effect spectra (1H–1H ROESY) were recorded to determine which signals arise from protons that are close to each other in space. This was vital to obtain the correct order of the identified Ar and pyrrole fragments (Figure 6). The two key observations are: firstly, the inner N−H showed a clear correlation between the adjacent N−H protons; secondly, ortho‐position protons showed a strong correlation to neighboring ethyl groups. Moreover, the split CH2 inner and outer protons were successfully assigned. Through‐space correlations to CH2 units were only observed for the outer protons (that point toward the phenyl rings) (Figure 6). The observations from 1H–1H ROESY NMR provided intrinsic information on the linking of pyrrole/Ar fragments to the porphyrin platform.
The last piece to the puzzle was to identify the above or below macrocycle plane orientation of the pyrrole/Ar fragments. As evidence by the crystal structures discussed below, the terminal CH3 groups of the ethyl fragment freely rotate. Therefore, 1H–1H ROESY correlation of the ortho‐positions to the two neighboring CH3 groups was observed. As an example, the ↑↑↑↑ atropisomer showed two different 1H–1H ROESY Cortho−H to CH3 signal intensities. The stronger signal corresponds to the CH3 facing the same side as the proton on the ortho‐position of the phenyl ring due to the closer proximity and favored interactions, while the weaker correlation can be attributed to the CH3 units predominantly facing away from the ortho‐position protons (Figure 7 a). An additional feature derived from the clear chemical shifts of the inner core protons was the observation of long‐distance through‐space correlation between peripheral Cortho−H and inner N−H units suggesting the conformational positions in these atropisomers. For instance, in the ↑↑↓↓ isomer one inner core proton that is facing below the plane correlates to the two down facing Cortho−H (Figure 7 b, green), while the two above the plane inner N−H protons correlate to the facing up Cortho−H protons (Figure 7 b, yellow).
Figure 7.
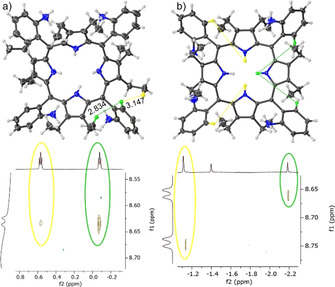
The key examples of 1H–1H ROESY correlations of P1‐BSA: a) in ↑↑↑↑ the ortho‐ to CH3 correlation strength dependent on the distance (Å) shown by the α4‐P1‐BSA crystal structure; b) illustration of ↑↑↓↓ atropisomer highlighting the long‐distance ortho‐ to N−H correlation.
All signals (1H, 13C, and 15N) with the highlighted orientation of P1‐BSA atropisomers are listed in Table S1. The symmetric units and corresponding point‐group notations of rotamers observed from NMR analysis are illustrated in Figure 8. The rotational symmetry of order in P1‐BSA was observed two‐fold smaller as compared to the planar porphyrin atropisomers presented by Gust and co‐workers. [7] This is due to the nonplanarity of the dodecasubstituted porphyrin and ≈45° angle phenyl rotation. [34] The highest order [35] 4 symmetry marked in α4‐P1‐BSA as C 2v (with twofold symmetry axis and two mirror planes) and α,β,α,β‐P1‐BSA as S4 with improper rotation (rotation‐reflection) axis (Figure S47). Order 2 symmetry with only one mirror plane assigned in α 2, β2‐P1‐BSA as CS and the unsymmetrical, chiral α 3, β‐P1‐BSA system as C1 point‐group. Overall, to accurately identify conformers by using only NMR techniques, it is crucial to follow symmetry operations as a basis in porphyrin atropisomers.
Figure 8.
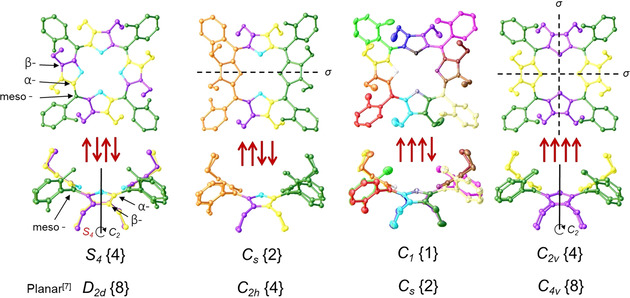
Representation of symmetry in P1 atropisomers with corresponding point‐group notations {order number} compared to their planar counterparts. [7] In black: dashed lines are mirror planes (σ), solid lines represent two‐fold rotation around the principal axis (C 2) in atropisomers. In red: representation of improper rotation (S 4), first a 90° rotation (C 4) around C 2 axis followed by reflection in a perpendicular plane (see Figure S47). X‐ray crystal structures of 2 were used to visualize the symmetrical groups observed in NMR analysis. The matching colors in the structures represent identical signals originating from the symmetry elements as indicated by NMR analysis, e.g., in α,β,α,β‐P1 four meso‐position carbon signals are identical (highlighted in green) whereas, in α 3, β‐P1 they resonated as four different ones.
Single‐crystal X‐ray crystallographic analysis
To further investigate the structural aspects and interactions of porphyrins described in Figure 2, crystallization of the individual atropisomers allowed X‐ray structural determinations. The structure of the core protonated (CP) porphyrin CP1 hydrogen bonded complex with BF4 − as well as crystal structures of each of the α4 and α 3, β rotamers in their free base 1, NiII 2 and protonated forms P1 (with two different complexing acids H2SO4 and BSA) are reported (Figure 9, Figures S40 and S41).
Figure 9.
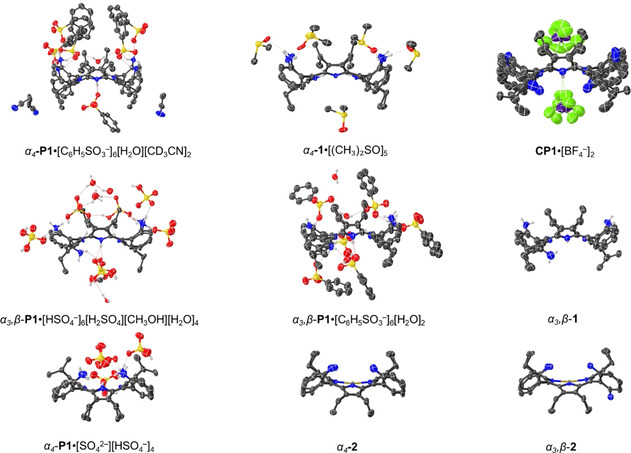
Side views of the single‐crystal X‐ray structures determined as single porphyrin units (see Figure S40 and S41 for the top view, or access the structures from CCDC 2012195–2012200, see Experimental Section). Non‐essential hydrogen atoms were omitted for clarity and thermal ellipsoids give 50 % probability.
The majority of crystal structure determinations of porphyrin dication salts have two counter anions hydrogen bonded to two opposing outward tilted pyrrole N‐H protons on each of the porphyrin faces, forming a linear “I‐shaped” complex (Table S4). [36] There are only two prior examples of counter anions being separated from the core system by intermediate water molecules in 5,10,15,20‐tetraphenylporphyrin (TPP) and 2,3,7,8,12,13,17,18‐octafluoro‐5,10,15,20‐tetraphenylporphyrin (OFTPP) forming “H‐shaped” complexes (Figure S44). In H4TPP4+⋅[BF4 −]2[H2O]2[CHCl3] and H4OFTPP4+⋅[ClO4 −]2[H2O]2[CH2Cl2]2 two water molecules bridge the dications and corresponding counter anions above and below the porphyrin mean plane. [37]
In the α4‐P1⋅[C6H5SO3 −]6[H2O][CD3CN]2 and α3,β‐P1⋅[C6H5SO3 −]6[H2O]2 structures, a water molecule interacting with inner core system is observed only on the side with a predominant number of ammonium groups forming a new “T‐shaped” hydrogen‐bonding complex feature (Figure 10 a). Similarly, in α4‐P1⋅[SO4 2−][HSO4 −]4 and α3,β‐P1⋅[HSO4 −]6[H2SO4][CH3OH][H2O]4 structures, a face selectivity in the porphyrin‐anion interactions is observed without the core bridging water molecule. This interaction forms a Y‐shaped hydrogen bonded complex in which anions form a semi‐symmetrical H‐bond (Figure 10 b). The distance between two of the closest sulfur atoms decreases by approximately 2 Å from “T‐shaped” to “Y‐shaped” complexes due to removal of the inter spaced water molecule. The opposite side of the macrocyclic system in all cases has direct N−H interactions with only one of the following counter anions interacting with the core system, comparable to the H4TPP4+⋅[HSO4 −]2[CH3OH]2 structure with a linear coordination to both sides of the plane. [36b]
Figure 10.
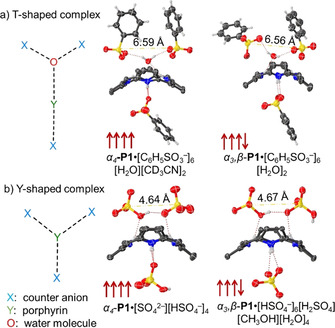
Core interactions observed in the crystal structures of ↑↑↑↑ and ↑↑↑↓ P1 with H2SO4 and BSA: a) T‐shaped and b) Y‐shaped complexes. The dashed yellow line represents the distance between two closest sulfur atoms. Non‐essential hydrogen atoms, meso‐ and β‐substituents were omitted for clarity.
To test if face selectivity can be obtained with counter anions giving ‘H‐shaped“ complexes such as BF4 −, [37b] α4‐1 was recrystallized with an excess of HBF4 using methanol and dichloromethane slow liquid–liquid diffusion. To our surprise, structure CP1⋅[BF4 −]2 [38] was obtained displaying the regular ”I‐shaped“ complexation pattern. Only two of the BF4 − units coordinated perpendicular to the N−H of the inner core system. Two key observations arise: protonation happens first in the core rather than at the peripheral amines as no peripheral amine protonation was observed with HBF4. [39] The selective core protonation over the periphery strengthens evidence for the binding mechanism proposed for α4‐P1‐PPi (α4‐1→α4‐CP1→α4‐P1‐PPi). [11] Secondly, the combination of peripheral charge‐assisted donors and acceptors appears to be necessary for ”unusual“ anion‐binding motifs to occur. From this, we can speculate that symmetrical ”H‐shaped“ type hydrogen bonding complexes could be formed using a two‐sided system with BSA or a new ”X‐shaped“ complex, by using H2SO4 (Figure S44). Solvent and counter anion operations in porphyrin scaffold to arrange in different complexed forms calls for a separate, sophisticated crystal engineering project.
Our understanding of the conformational flexibility of the tetrapyrroles usually arises from chemically altering the macrocycle and core conformation of porphyrins. [16] Shelnutt and co‐workers introduced NSD (normal‐coordinate structural decomposition) as an easy method to delineate, quantify and illustrate the various distortions modes present in the tetrapyrrole macrocycles. [40] Upon protonation, planar porphyrins undergo significant saddle distortion while already nonplanar dodecasubstituted porphyrins show only a marginal increase in distortion. [41] However, the counter anion itself is typically a neglected aspect of increasing dodecasubstituted porphyrin macrocycle deformations. The most noticeable feature in the NSD profile of the α4‐P1⋅[C6H5SO3 −]6[H2O][CD3CN]2 structure is considerably ruffled, this is not present in the other compounds under study (Figure 11). The macrocycle evidently adapts to the overcrowded face of the porphyrin. Apparently, bulky counter anions concentrated on one side of the porphyrin forces the flexible plane to deform in a ruffling manner. Moreover, the intermediary water molecule at the core of the system increases space the counter anion assembly occupies. If bulky BSA counter anions are substituted by HSO4 − without intermediate H2O, no significant ruffling deformation is observed. Similarly, the orientation of the peripheral amines influences the steric repulsion. The degree of ruffling of α3,β‐P1⋅[HSO4 −]6[H2SO4][CH3OH][H2O]4 is less than a third of that observed for ↑↑↑↑.
Figure 11.
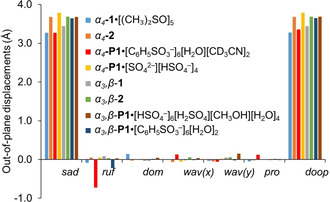
Illustration of the out‐of‐plane normal‐coordinate structural decomposition results for the ↑↑↑↑ and ↑↑↑↓ atropisomers of free base 1, NiII 2 and protonated P1 with H2SO4 and BSA forms.
Macrocycle deformations presented in linear skeletal plots illustrate similar sterically induced conformations (Figure S45). The linear skeletal plot of the unique α4‐P1⋅[C6H5SO3 −]6[H2O][CD3CN]2 structure is comparable to that of the previously determined highly ruffled and saddled structure of 5,10,15,20‐tetrakis(tert‐butyl)porphyrindi‐ium bis(trifluoroacetate) [H4TtBP][CF3COO−]2⋅2 CF3COOH (Figure 12). [36c] In solution studies, α4‐P1‐BSA was determined to have C 2v symmetry. Therefore, the ruffling profile is either not significant enough for NMR observations, or the α4‐P1‐BSA average structure in solution has no deformations additional to the dominant saddle one. Either way, one can determine the dominant porphyrin deformations solely by thorough NMR analysis.
Figure 12.
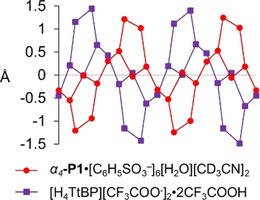
Linear display of the skeletal deviations of α4‐P1⋅[C6H5SO3 −]6[H2O][CD3CN]2 (red) and [H4TtBP][CF3COO−]2⋅2 CF3COOH (purple). [36c]
The crystal packing diagrams (Figure S42, S43) for the protonated porphyrin α4‐P1⋅[C6H5SO3 −]6[H2O][CD3CN]2 indicate the formation of what appear to be large arrays of parallel channels. These molecular interstices are primarily filled with counter‐ions while the porphyrin component α4‐P1 occupies only 31.35 % of the unit cell. This prearrangement of porphyrins with bulky anions increases the intermolecular distances significantly in the solid state, and has implications in the design of molecular sponges incorporating porphyrin components, [42] as well as photophysical properties where control of intermolecular distances is important. [43]
Conclusions
In this study, we have detailed our insight into porphyrin atropisomers and proposed new means for their detection and analysis. Moreover, X‐ray crystallographic studies provided detail of new supramolecular binding modes and highlighted the flexibility of the porphyrin macrocycle. The highly saddled porphyrin P1 rotamers were used for extensive NMR analysis. The weak interactions between P1 and BSA is a valuable asset allowing for sharp and well‐defined inner N−H proton signals. The fingerprint‐like 1H signals were found to be specific for a given atropisomer. The rationale of the explicit signals for all rotamers is the interlocked systems by the counter anions that reduces proton exchange rates. Further investigation using the 15N–1H HSQC technique in natural abundance showed 15N signals correlating well to the inner core protons of the corresponding atropisomers. The symmetry elements and point‐group notions were applied after assigning all the 1H and 13C signals using a variety of 2D NMR techniques such as 1H–1H TOCSY, ROESY, and 1H–13C HSQC, HMBC. The highest symmetry marked in ↑↑↑↑ C 2v and ↑↓↑↓ S4 rotamers, followed by less symmetrical ↑↑↓↓ CS and then the completely unsymmetrical ↑↑↑↓ C 1 system. The symmetry order is two‐fold lower in comparison to the suggested planar porphyrin point‐group notations. The basis for this decrease in symmetry lies in the saddle‐shaped deformations of the porphyrins as determined by NSD analysis, making distortion a molecular engineering tool to achieve symmetry reduction in macrocycles.
The symmetry control aspects are potentially useful for applications in catalysis and detection of specific molecules, e.g., using symmetry manipulation to achieve chirality for asymmetric catalysis or detection of chiral pollutants. The method of interlocking the macrocyclic system with a well‐fitting analyte is extremely appealing not only for detail characterization of dodecasubstituted porphyrins but also for detailed solution‐based structural analysis of specialized receptor systems. Porphyrin atropisomers, upon differentiation, are a promising addition to the molecular engineering toolbox for supramolecular assembly for example, variety of interlinked stereoisomers. Moreover, the accurate determination of atropisomers is relevant in drug design and quality monitoring in pharmaceuticals, as different peripheral orientation can have a significant impact in drug activity.
Experimental Section
Porphyrin 2 rotamers were prepared and characterized by previously described methods. [11] All NMR studies were carried out in [D3]acetonitrile with the addition of BSA as a demetalating and complexing agent. 1H NMR (600 MHz) spectra, 13C NMR (151 MHz) spectra, and 15N NMR (61 MHz) spectra were collected at room temperature.
General information, instrumentation, NMR spectra, crystallographic studies, and complete structural details of all isolated compounds are given in the Supporting Information.
Deposition numbers 2012195, 2012196, 2012197, 2012198, 2012199, and 2012200 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was prepared with the support of the Technical University of Munich—Institute for Advanced Study through a Hans Fischer Senior Fellowship and has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska‐Curie Grant Agreement No. 764837, the Irish Research Council (GOIPG 2017/1172) and from Science Foundation Ireland (IvP 13/IA/1894).
K. Norvaiša, J. E. O'Brien, D. J. Gibbons, M. O. Senge, Chem. Eur. J. 2021, 27, 331.
References
- 1. Bott G., Field L. D., Sternhell S., J. Am. Chem. Soc. 1980, 102, 5618–5626. [Google Scholar]
- 2.
- 2a. Okuno Y., Kamikado T., Yokoyama S., Mashiko S., Comput. Theor. Chem. 2002, 594, 55–60; [Google Scholar]
- 2b. Zhou Z., Zhang X., Liu Q., Yan Z., Lv C., Long G., Inorg. Chem. 2013, 52, 10258–10263. [DOI] [PubMed] [Google Scholar]
- 3. Crossley M. J., Field L. D., Forster A. J., Harding M. M., Sternhell S., J. Am. Chem. Soc. 1987, 109, 341–348. [Google Scholar]
- 4. Arai T., Tsukuni A., Kawazu K., Aoi H., Hamada T., Nishino N., J. Chem. Soc., Perkin Trans. 2 2000, 1381–1390. [Google Scholar]
- 5. Moss G. P., Pure Appl. Chem. 1996, 68, 2193–2222. [Google Scholar]
- 6. Gottwald L. K., Ullman E. F., Tetrahedron Lett. 1969, 10, 3071–3074. [Google Scholar]
- 7. Dirks J. W., Underwood G., Matheson J. C., Gust D., J. Org. Chem. 1979, 44, 2551–2555. [Google Scholar]
- 8. Collman J. P., Gagne R. R., Reed C., Halbert T. R., Lang G., Robinson W. T., J. Am. Chem. Soc. 1975, 97, 1427–1439. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Groves J. T., Myers R. S., J. Am. Chem. Soc. 1983, 105, 5791–5796; [Google Scholar]
- 9b. Hatano K., Chem. Pharm. Bull. 1985, 33, 4116–4119; [Google Scholar]
- 9c. Kuroda Y., Kato Y., Higashioji T., Hasegawa J.-Y., Kawanami S., Takahashi M., Shiraishi N., Tanabe K., Ogoshi H., J. Am. Chem. Soc. 1995, 117, 10950–10958; [Google Scholar]
- 9d. Campbell L. A., Kodadek T., J. Mol. Catal. A: Chem. 1996, 113, 293–310; [Google Scholar]
- 9e. Reginato G., Bari L. D., Salvadori P., Guilard R., Eur. J. Org. Chem. 2000, 1165–1171; [Google Scholar]
- 9f. Boitrel B., Baveux-Chambenoît V., Richard P., Eur. J. Org. Chem. 2001, 4213–4221. [Google Scholar]
- 10.
- 10a. Wagner R. W., Johnson T. E., Lindsey J. S., Tetrahedron 1997, 53, 6755–6790; [Google Scholar]
- 10b. Gerothanassis I. P., Kalodimos C. G., Hawkes G. E., Haycock P., J. Magn. Reson. 1998, 131, 163–165; [DOI] [PubMed] [Google Scholar]
- 10c. Kalodimos C. G., Gerothanassis I. P., Pierattelli R., Troganis A., J. Inorg. Biochem. 2000, 79, 371–380. [DOI] [PubMed] [Google Scholar]
- 11. Norvaiša K., Flanagan K. J., Gibbons D., Senge M. O., Angew. Chem. Int. Ed. 2019, 58, 16553–16557; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16705–16709. [Google Scholar]
- 12.
- 12a. Walker F. A., Buchler J., West J. T., Hinds J. L., J. Am. Chem. Soc. 1983, 105, 6923–6929; [Google Scholar]
- 12b. Imai H., Kyuno E., Inorg. Chim. Acta 1988, 153, 175–182; [Google Scholar]
- 12c. Heier P., Förster C., Schollmeyer D., Boscher N., Choquet P., Heinze K., Dalton Trans. 2013, 42, 906–917; [DOI] [PubMed] [Google Scholar]
- 12d. Sun H., Guo K., Gan H., Li X., Hunter C. A., Org. Biomol. Chem. 2015, 13, 8053–8066. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Clayden J., Moran W. J., Edwards P. J., LaPlante S. R., Angew. Chem. Int. Ed. 2009, 48, 6398–6401; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6516–6520; [Google Scholar]
- 13b. LaPlante S. R., Edwards P. J., Fader L. D., Jakalian A., Hucke O., ChemMedChem 2011, 6, 505–513; [DOI] [PubMed] [Google Scholar]
- 13c. LaPlante S. R., Fader L. D., Fandrick K. R., Fandrick D. R., Hucke O., Kemper R., Miller S. P. F., Edwards P. J., J. Med. Chem. 2011, 54, 7005–7022; [DOI] [PubMed] [Google Scholar]
- 13d. Glunz P. W., Bioorg. Med. Chem. Lett. 2018, 28, 53–60; [DOI] [PubMed] [Google Scholar]
- 13e. Toenjes S. T., Gustafson J. L., Future Med. Chem. 2018, 10, 409–422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Donohoe C., Scharbele F., Gonçalves N. P. F., Pereira M. M., Gomes-da-Silva L. C., Arnaut L. G., Proc. SPIE 2019, 11070, 110703Z. [Google Scholar]
- 14.
- 14a. Kottas G. S., Clarke L. I., Horinek D., Michl J., Chem. Rev. 2005, 105, 1281–1376; [DOI] [PubMed] [Google Scholar]
- 14b. Tomé A. C., Silva A. M. S., Alkorta I., Elguero J., J. Porphyrins Phthalocyanines 2011, 15, 1–28. [Google Scholar]
- 15. Medforth C. J., Haddad R. E., Muzzi C. M., Dooley N. R., Jaquinod L., Shyr D. C., Nurco D. J., Olmstead M. M., Smith K. M., Ma J.-G., Shelnutt J. A., Inorg. Chem. 2003, 42, 2227–2241. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Senge M. O., in The Porphyrin Handbook, Vol. 1 (Eds.: Kadish K. M., Smith K. M., Guilard R.), Academic Press, 2000, pp. 239–347; [Google Scholar]
- 16b. Senge M. O., Chem. Commun. 2006, 243–256. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Chaudhri N., Sankar M., RSC Adv. 2015, 5, 3269–3275; [Google Scholar]
- 17b. Kumar R., Chaudhri N., Sankar M., Dalton Trans. 2015, 44, 9149–9157; [DOI] [PubMed] [Google Scholar]
- 17c. Kielmann M., Senge M. O., Angew. Chem. Int. Ed. 2019, 58, 418–441; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 424–448; [Google Scholar]
- 17d. Norvaiša K., Kielmann M., Senge M. O., ChemBioChem 2020, 21, 1793–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Roucan M., Kielmann M., Connon S. J., Bernhard S. S. R., Senge M. O., Chem. Commun. 2018, 54, 26–29; [DOI] [PubMed] [Google Scholar]
- 18b. Aoki E., Suzuki W., Kotani H., Ishizuka T., Sakai H., Hasobe T., Kojima T., Chem. Commun. 2019, 55, 4925–4928; [DOI] [PubMed] [Google Scholar]
- 18c. Dar T. A., Uprety B., Sankar M., Maurya M. R., Green Chem. 2019, 21, 1757–1768; [Google Scholar]
- 18d. Kielmann M., Grover N., Kalisch W. W., Senge M. O., Eur. J. Org. Chem. 2019, 2448–2452; [Google Scholar]
- 18e. Suzuki W., Kotani H., Ishizuka T., Kojima T., J. Am. Chem. Soc. 2019, 141, 5987–5994. [DOI] [PubMed] [Google Scholar]
- 19. Kielmann M., Flanagan K. J., Senge M. O., J. Org. Chem. 2020, 85, 7603–7610. [DOI] [PubMed] [Google Scholar]
- 20. Kuroda Y., Kawashima A., Hayashi Y., Ogoshi H., J. Am. Chem. Soc. 1997, 119, 4929–4933. [Google Scholar]
- 21.
- 21a. Lindsey J., J. Org. Chem. 1980, 45, 5215; [Google Scholar]
- 21b. Hayashi T., Asai T., Hokazono H., Ogoshi H., J. Am. Chem. Soc. 1993, 115, 12210–12211. [Google Scholar]
- 22.
- 22a. Rose E., Quelquejeu M., Pochet C., Julien N., Kossanyi A., Hamon L., J. Org. Chem. 1993, 58, 5030–5031; [Google Scholar]
- 22b. Rose E., Cardon-Pilotaz A., Quelquejeu M., Bernard N., Kossanyi A., Desmazieres B., J. Org. Chem. 1995, 60, 3919–3920. [Google Scholar]
- 23. Norikazu N., Keisuke K., Hisakazu M., Tsutomu F., Chem. Lett. 1992, 21, 1991–1994, DOI: 10.1246/cl.1992.1991. [DOI] [Google Scholar]
- 24. Wang H., Li J., Scheidt W. R., J. Porphyrins Phthalocyanines 2018, 22, 981–988. [Google Scholar]
- 25.
- 25a. Keiichiro H., Kazunori A., Tadashi K., Shoko T., Bull. Chem. Soc. Jpn. 1981, 54, 3518–3521, DOI: 10.1246/bcsj.54.3518; [DOI] [Google Scholar]
- 25b. Keiichiro H., Kazunori A., Akemi N., Keiko F., Bull. Chem. Soc. Jpn. 1985, 58, 3653–3654, DOI: 10.1246/bcsj.58.3653; [DOI] [Google Scholar]
- 25c. Amaravathi M., Rajitha B., Rao M. K., Sitadevi P., J. Heterocycl. Chem. 2007, 44, 821–825. [Google Scholar]
- 26.
- 26a. Anzai K., Hatano K., Chem. Pharm. Bull. 1984, 32, 1273–1278; [Google Scholar]
- 26b. Zhu L., Himmel L., Merkes J. M., Kiessling F., Rueping M., Banala S., Chem. Eur. J. 2020, 26, 4232–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker F. A., Avery G. L., Tetrahedron Lett. 1971, 12, 4949–4952. [Google Scholar]
- 28. Abraham R. J., Plant J., Bedford G. R., Org. Magn. Reson. 1982, 19, 204–210. [Google Scholar]
- 29. Monteiro C. J. P., Pereira M. M., Gonçalves N. P. F., Carvalho C. G., Neves Â. C. B., Abreu A. R., Arnaut L. G., Silva A. M. S., J. Porphyrins Phthalocyanines 2012, 16, 316–323. [Google Scholar]
- 30. Radek M., Antonin L., Erkki K., Elina S., Jaromir T., Curr. Org. Chem. 2007, 11, 1154–1205, DOI: 10.2174/138527207781662519. [Google Scholar]
- 31.
- 31a. Gust D., Roberts J. D., J. Am. Chem. Soc. 1977, 99, 3637–3640; [DOI] [PubMed] [Google Scholar]
- 31b. Schlabach M., Rumpel H., Limbach H.-H., Angew. Chem. Int. Ed. Engl. 1989, 28, 76–79; [Google Scholar]; Angew. Chem. 1989, 101, 84–87. [Google Scholar]
- 32. García-Ortega H., Crusats J., Feliz M., Ribó J. M., J. Org. Chem. 2002, 67, 4170–4176. [DOI] [PubMed] [Google Scholar]
- 33. Martin G. E., Hadden C. E., J. Nat. Prod. 2000, 63, 543–585. [DOI] [PubMed] [Google Scholar]
- 34. Medforth C. J., Senge M. O., Smith K. M., Sparks L. D., Shelnutt J. A., J. Am. Chem. Soc. 1992, 114, 9859–9869. [Google Scholar]
- 35.The order of rotational symmetry of a geometric figure is the number of times you can rotate the geometric figure so that it looks exactly the same as the original Figure.
- 36.
- 36a. Stone A., Fleischer E. B., J. Am. Chem. Soc. 1968, 90, 2735–2748; [Google Scholar]
- 36b. Senge M. O., Kalisch W. W., Z. Naturforsch. B 1999, 54, 943–959; [Google Scholar]
- 36c. Senge M. O., Z. Naturforsch. B 2000, 55, 336–344. [Google Scholar]
- 37.
- 37a. Porhiel E., Toupet L. C., Leroy J., Bondon A., Tetrahedron Lett. 2002, 43, 8293–8296; [Google Scholar]
- 37b. Rayati S., Zakavi S., Ghaemi A., Carroll P. J., Tetrahedron Lett. 2008, 49, 664–667. [Google Scholar]
- 38.The orientation of the ammonium groups was not conclusive due to 50 % occupancy in both sides.
- 39.Determined by C−N distance and the absence of electron densities corresponding to the peripheral counter anions in the structure.
- 40.
- 40a. Jentzen W., Song X.-Z., Shelnutt J. A., J. Phys. Chem. B 1997, 101, 1684–1699; [Google Scholar]
- 40b. Senge M. O., MacGowan S. A., O'Brien J. M., Chem. Commun. 2015, 51, 17031–17063; [DOI] [PubMed] [Google Scholar]
- 40c. Schindler J., Kupfer S., Ryan A. A., Flanagan K. J., Senge M. O., Dietzek B., Coord. Chem. Rev. 2018, 360, 1–16. [Google Scholar]
- 41. Senge M. O., Forsyth T. P., Nguyen L. T., Smith K. M., Angew. Chem. Int. Ed. Engl. 1995, 33, 2485–2487; [Google Scholar]; Angew. Chem. 1994, 106, 2554–2557. [Google Scholar]
- 42.
- 42a. Byrn M. P., Curtis C. J., Goldberg I., Hsiou Y., Khan S. I., Sawin P. A., Tendick S. K., Strouse C. E., J. Am. Chem. Soc. 1991, 113, 6549–6557; [Google Scholar]
- 42b. Goldberg I., Chem. Commun. 2005, 1243–1254; [DOI] [PubMed] [Google Scholar]
- 42c. Lipstman S., Goldberg I., Cryst. Growth Des. 2013, 13, 942–952; [Google Scholar]
- 42d. Flanagan K. J., Twamley B., Senge M. O., Inorg. Chem. 2019, 58, 15769–15787. [DOI] [PubMed] [Google Scholar]
- 43. Sakuma T., Sakai H., Araki Y., Wada T., Hasobe T., Phys. Chem. Chem. Phys. 2016, 18, 5453–5463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary