

Abstract
Objective
Clinical genetic sequencing is frequently utilized to diagnose individuals with neurodevelopmental disorders (NDDs). Here we perform a meta‐analysis and systematic review of the success rate (diagnostic yield) of clinical sequencing through next‐generation sequencing (NGS) across NDDs. We compare the genetic testing yield across NDD subtypes and sequencing technology.
Methods
We performed a systematic review of the PubMed literature until May 2020. We included clinical sequencing studies that utilized NGS in individuals with epilepsy, autism spectrum disorder (ASD), or intellectual disability (ID). Data were extracted, reviewed, and categorized according to the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) guidelines. Two investigators performed clinical evaluation and grouping following the International League Against Epilepsy (ILAE) guidelines. Pooled rates of the diagnostic yield and 95% confidence intervals were estimated with a random‐effects model.
Results
We identified 103 studies (epilepsy, N = 72; ASD, N = 14; ID, N = 21) across 32,331 individuals. Targeted gene panel sequencing was used in 73, and exome sequencing in 36 cohorts. Given highly selected patient cohorts, the diagnostic yield was 17.1% for ASD, 24% for epilepsy, and 28.2% for ID (23.7% overall). The highest diagnostic yield for epilepsy subtypes was observed in individuals with ID (27.9%) and early onset seizures (36.8%). The diagnostic yield for exome sequencing was higher than for panel sequencing, even though not statistically significant (27.2% vs 22.6%, P = .071). We observed that clinical sequencing studies are performed predominantly in countries with a high Inequality‐adjusted Human Development Index (IHDI) (countries with sequencing studies: IHDI median = 0.84, interquartile range [IQR] = 0.09 vs countries without sequencing studies: IHDI median = 0.56, IQR = 0.3). No studies from Africa, India, or Latin America were identified, indicating potential barriers to genetic testing.
Significance
This meta‐analysis and systematic review provides a comprehensive overview of clinical sequencing studies of NDDs and will help guide policymaking and steer decision‐making in patient management.
Keywords: autism, epilepsy, genetics, neurodevelopmental disorders, neurodevelopmental disorders, sequencing
Key Points.
This systematic review evaluated the diagnostic yield of next‐generation sequencing (NGS) in neurodevelopmental disorders and their subtypes
In 103 studies that include 32331 individuals, the overall diagnostic yield was 23.7%‐17.1% for autism spectrum disorder (ASD), 24% for epilepsy, and 28.2% for intellectual disability (ID)
Around one in five neurodevelopmental disorder (NDD) patients will receive a diagnosis using NGS, especially when investigating the whole exome
The highest diagnostic yield for epilepsies was observed in individuals with ID (27.9%) and early onset seizures (36.8%)
1. INTRODUCTION
Neurodevelopmental disorders (NDDs)—including epilepsy, autism spectrum disorder (ASD), and intellectual disability (ID)—represent genetically and clinically heterogeneous groups of disorders that affect about 3% of children worldwide. 1 Advances in sequencing technologies have facilitated the identification of an exponentially growing number of NDD‐associated genes. 2 The identification of disease‐associated genes can improve the understanding of disease pathogenesis and trajectories. It may also guide the identification of more genetically homogeneous subgroups within the spectrum of NDDs. 3 A recent study showed that 33% of children with a molecularly confirmed genetic epilepsy would benefit from precision medicine. 4 However, the proportion of individuals with NDDs who carry a genetic abnormality that can be identified using next‐generation sequencing (NGS) has yet to be well established.
Sequencing studies that report the diagnostic yield (ie, percentage of pathogenic variant carriers identified in a cohort) in NDDs are few but are becoming increasingly common. Estimates of diagnostic yield vary considerably across individual studies (8% 5 –61% 6 ). This likely reflects differences in measurement, reporting, and clinical characteristics such as etiology and disorder type/subtype. Although many literature reviews have been published in the past, only two systematic meta‐analyses of genetic testing in NDDs have been reported to date to the best of our knowledge. A literature review is a descriptive summary of the existing material relating to some topic or area of study. A systematic review is a review of the literature that is conducted methodically based on a pre‐specified protocol. It aims to synthesize the retrieved information often through a meta‐analysis. A systematic review sometimes produces results that, inconveniently, contradict common beliefs. 7
A recent systematic meta‐analysis of 30 NDD genetic testing studies showed that screening all genes with exome sequencing (ES) has a clinical diagnostic yield of 36% for individuals with NDD, 16% for a subset of individuals with autism spectrum disorder (ASD), and 39% for individuals with intellectual disability (ID). 8 Epilepsy was considered in only one systematic meta‐analysis of genetic testing, which focused on assessing different technologies' diagnostic yield. The authors analyzed 23 epilepsy clinical genetic studies and found that ES had the highest diagnostic yield (45%; 6 studies), followed by targeted gene panel sequencing (panel) (23%; 9 studies), and chromosomal microarray testing (8%; 8 studies). 9 Because NGS has only been established as a clinical diagnostic tool within the last decade, previous studies evaluating sequencing strategies have included only 30 studies or less. Furthermore, the diagnostic yield across subtypes of NDDs—including milder and more severe forms of epilepsy—or sequencing technologies has yet to be consistently established.
Here we present the most substantial and up‐to‐date systematic review and meta‐analysis for clinical diagnostic sequencing in NDDs. In our study, we quantify the yield of diagnostic sequencing in different types of NDDs. We also explore heterogeneity sources among studies and perform additional analyses considering the country of origin, type of sequencing test, and adherence to current variant interpretation guidelines. 10
2. METHODS
2.1. Search strategy
The systematic review was conducted following the Preferred Reporting Items for Systematic Review and Meta‐Analysis (PRISMA) protocol, considering all studies contained in PubMed until May 20, 2020. 11 As keywords for the PubMed search, we used disease‐specific terms (“epilepsy”, “epileptic encephalopathy”, “neurodevelopmental disorder”, “seizures”, “autism”, “ASD”, “autism spectrum disorder”, “intellectual disability”, “ID”, and “mental retardation”), each combined with sequencing technology terms (“exome”, “next generation sequencing”, “NGS”, “panel”, “targeted sequencing”, and “whole genome sequencing”) and other content related terms (“cohort”, “diagnostic yield”, “diagnostic test”, and “clinical practice”).
We performed an automated PubMed search using the R package RISmed. 12 Moreover, 40 additional records were identified through other sources (eg, listed as references in studies identified through PubMed screen). We only considered studies written in English. Duplicated studies, including response letters and studies without a title or abstract, were removed. Furthermore, studies that investigated somatic variants from resected brain tissue were also removed. The remaining studies were reviewed in two steps: (a) manual title and abstract screening to remove reviews, other non‐original studies, and studies which did not perform clinical genetic testing using NGS technologies; (b) manual full‐text review to select only sequencing studies, which used NGS technologies, studies focused on germline variants, and studies that screened more than five genes in at least 20 individuals with epilepsy, ASD, or ID. We excluded studies that specifically ascertained individuals for congenital malformations of the brain or any other disorder where epilepsy, ASD, or ID were considered a secondary phenotype. Moreover, we excluded studies that investigated somatic variants from resected brain tissue. The overall screening design is detailed in Figure 1.
Figure 1.
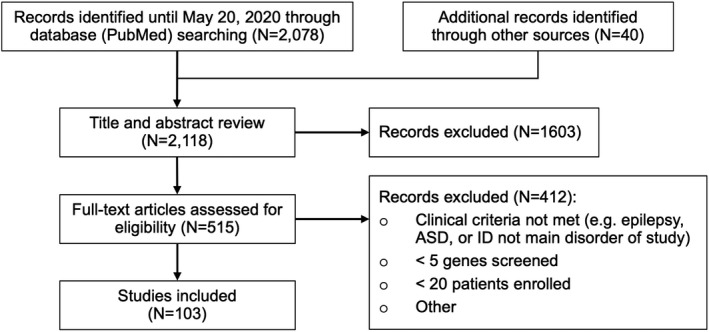
Process of data search, identification, and filtering
2.2. Data synthesis and analysis
The 103 qualifying studies (Table S1 in the Supplement) were divided into cohorts based on three criteria: (a) disorder, (b) disorder subtype, and (c) sequencing method. If a study investigated multiple disorders, disorder subtypes, or sequencing methods, we split these studies into disorder‐specific, disorder subtype‐specific, and method‐specific cohort subsets (Figure 2). Disorder cohorts included: epilepsy, ASD, and ID. Disorder subtype cohorts included: focal epilepsy (FE), generalized epilepsy (GE), combined generalized and focal epilepsy (GE & FE), epilepsy without ID, epilepsy with ID, ASD with ID or developmental delay (DD), west syndrome (WS), and other developmental and epileptic encephalopathies (DEEs). All of the phenotypes above were taken from the corresponding study. Only ASD with ID or DD was manually classified based on the cohort. The minimum requirement for the "ASD with ID or DD" grouping is a moderate severity of ASD or DD. Age at onset subgroups comprised: Neonatal/Infantile, Childhood, Any Age. The group "Any Age" represents studies that report on genetic testing in heterogenous patient groups with variable age at seizure onset (~1–20 years). The age at onset grouping was performed by inferring the age at onset from the reported clinical syndrome of the patient. Subgrouping for the age at onset and epilepsy with and without ID was only performed if the majority of each cohort reported the phenotype. Method‐related cohorts included exome sequencing (ES) and targeted gene panel sequencing (panel). Groups with at least three studies were considered. Epilepsy type and epilepsy syndrome were classified for subjects in every study, according to 2017 ILAE (International League Against Epilepsy) epilepsy classification and www.epilepsydiagnosis.org, a website designed by ILAE as a guide for epilepsy syndrome diagnosis. 13 , 14 The website provides a full list of epilepsy syndromes divided by their typical age at seizure onset. All cohort groups are detailed in Figure 2.
Figure 2.
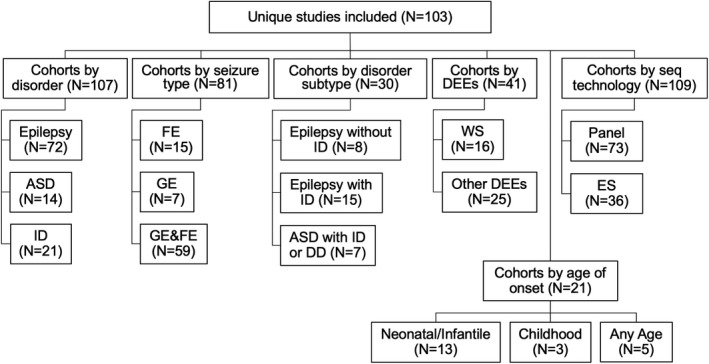
Separation of 103 unique studies included. We collected 103 studies which included heterogeneous types of NDDs. We were able to separate these into 107 distinct disorder cohorts, 81 cohorts by seizure type, 41 DEE cohorts, 21 cohorts by age at onset, and 109 sequencing technology cohorts. Abbreviations: ASD, autism spectrum disorder; DD, developmental delay; DEE, developmental epileptic encephalopathy; ES, exome sequencing; FE, focal epilepsy; GE & FE, combined generalized and focal epilepsy; GE, generalized epilepsy; ID, intellectual disability; panel, targeted gene panel sequencing; WS, West syndrome
Small cohorts are more likely to be biased. Therefore, to enrich for representative studies, we did not consider studies that tested less than five genes or less than 20 individuals. Because only coding variants were reported, studies that employed whole‐genome sequencing were included in the ES group. We only considered diagnostic yield from copy number variants (CNVs) if the variants were called from sequencing reads. Results from classical cytogenic or chromosomal microarray testing were not considered.
In addition, the open database of the United Nations Development Programme was used to obtain the Inequality‐adjusted Human Development Indices (IHDI) for each country. 15 The IHDI is based on the Human Development Index (HDI), a composite index of social and economic achievement that is adjusted by the inequality in the distribution of the HDI within each country. The HDI has four components: A life expectancy index, a mean years of schooling index, an expected years of schooling index, and an income index. 16 The IHDI ranges from 0 to 1, with a higher value indicating a higher socioeconomic level (low: <0.550, medium: 0.550–0.699, high: 0.700–0.799, and very high: >0.800). The IHDI was subsequently utilized to assess the median IHDIs across countries with and without reported diagnostic sequencing studies. We also investigated the number of disease‐associated genes being reported by disorder per year. Finally, we examined whether investigators applied the American College of Human Genetics & Genomics (ACMG) guidelines in NDD sequencing studies.
2.3. Statistical analysis and statistical software
We used R version 3.6 for all the analyses. 17 We performed systematic meta‐analyses across all studies and cohorts (Figures S1 to Figure S25 in the Supplement) using a random‐effects model (REM) with the R package meta. 18 The REM was used considering an expected high degree of heterogeneity between studies. Plots were created using the meta and ggplot2 packages. 18 , 19 The magnitude of between‐study heterogeneity was quantified using the I2 statistic. A priori, we decided to report the pooled, weighted estimate generated by random‐effects models, to account for a potentially high degree of between‐study heterogeneity. We used funnel plots and the Egger method 20 to evaluate potential publication bias. If bias was found, we performed a correction using the Duval and Tweedie trim and fill procedure. 21 The Wilcoxon rank‐sum test was used to determine significant differences between the diagnostic yield of panels and ES.
3. RESULTS
3.1. Study selection
In our staged study selection process, we initially identified 2078 unique studies through an automated PubMed search after inclusion and exclusion criteria were applied (Figure 1). Through other sources (eg, reference lists), we identified 40 more studies. In total, we found 2118 studies that met our criteria. We eliminated 1603 studies after abstract review, and another 412 after full‐text review. A total of 103 studies, representing 32 331 individuals (mean: 314, median: 93, IQR: [50; 169]), were included in the systematic review. We conducted the analysis according to the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (or PRISMA) guidelines. The corresponding flowchart is shown in Figure 1.
3.2. Diagnostic yield overall, by disorder and by disorder subtype
Random‐effects meta‐analysis of all 103 included studies (Figure 3) revealed an overall diagnostic yield for neurodevelopmental disorder sequencing studies of 23.7% (95% confidence interval [CI] 22%–26%), weighted by the number of cases in each study. Heterogeneity existed between estimates (I2 = 93%). In the disorder‐specific analysis, the highest diagnostic yield was observed for ID (28.2%, 95% CI 22%–35%), followed by epilepsy (24%, 95% CI 22%–27%) and ASD (17.1%, 95% CI 11%–25%) (Table 1). Heterogeneity existed between estimates for ID (I2 = 92%), epilepsy (I2 = 93%), and ASD (I2 = 89%).
Figure 3.
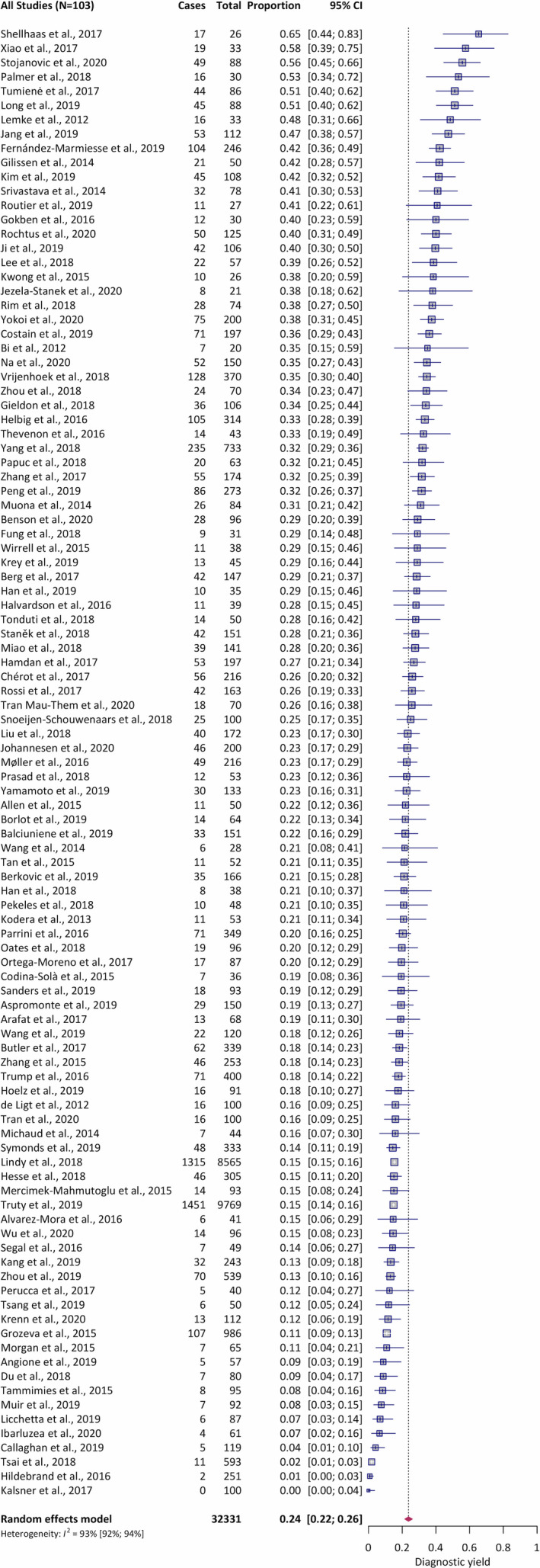
Forest plot of meta‐analysis of the overall diagnostic yield from 103 studies. Abbreviations: CI, confidence interval; I2, estimated proportion of the variance in study estimates that is due to heterogeneity; Proportion, fraction of individuals with a positive genetic test (ie, pathogenic or likely pathogenic variant)
TABLE 1.
Diagnostic yield across different categories
| Grouping | Subgroup |
No. of incl. cohorts |
No. of incl. individuals |
Diagnostic yield (95% CI) |
|---|---|---|---|---|
| Overall | 103 | 32 310 | 23.7% (22%–26%) | |
| By disorder | ASD | 14 | 1530 | 17.1% (11%–25%) |
| Epilepsy | 72 | 27 923 | 24.0% (22%–27%) | |
| ID | 21 | 2863 | 28.2% (22%–35%) | |
| By seizure type | FE | 15 | 1944 | 15.8% (10%–24%) |
| GE | 7 | 1258 | 24.3% (18%–32%) | |
| GE & FE | 59 | 26 888 | 24.8% (22%–28%) | |
| By disorder subtype | Epilepsy without ID | 8 | 1224 | 9.3% (4%–23%) |
| ASD with ID or DD | 7 | 591 | 24.6% (18%–32%) | |
| Epilepsy with ID | 15 | 1290 | 27.9% (24%–33%) | |
| By other DEEs | WS | 16 | 768 | 19.3% (14%–26%) |
| Other DEEs | 8 | 232 | 38.8% (23%–57%) | |
| By age of onset | Any Age | 5 | 1080 | 6.6% (2%–22%) |
| Childhood | 3 | 171 | 14.7% (4–42%) | |
| Neonatal/Infantile | 13 | 986 | 29.3% (23%–36%) | |
| By sequencing technology | Panel | 73 | 28 665 | 22.6% (20%–25%) |
| ES | 36 | 3720 | 27.3% (24%–31%) |
For details of the grouping and subgrouping see Section 2.
Abbreviations: ASD, autism spectrum disorder; CI, confidence interval; DD, developmental delay; DEE, developmental epileptic encephalopathy; ES, exome sequencing; FE, focal epilepsy; GE & FE, combined generalized and focal epilepsy; GE, generalized epilepsy; ID, intellectual disability; panel, targeted gene panel sequencing; WS, West syndrome.
In the seizure type analysis, the diagnostic yield was 15.8% for FE (95% CI 10%–24%), 24.3% for GE (95% CI 18%–32%), and 24.7% for GE & FE (95% CI 22%–28%). Heterogeneity existed between estimates for FE (I2 = 92%), GE (I2 = 87%), and GE & FE (I2 = 94%). In the disorder subtype analysis, the diagnostic yield was 9.3% for epilepsy without ID (95% CI 4%–23%), 24.6% for ASD with ID or DD (95% CI 18%–32%), and 27.9% for epilepsy with ID (95% CI 24%–33%). The highest diagnostic yield was observed in the DEE subgroup analysis. The yield for WS was 19.3% (95% CI 14%–26%) and 36.8% for other DEEs. In the age at onset subgroup analysis, the diagnostic yield for any age was 6.6% (95% CI 2%–22%), 14.7% for childhood (95% CI 4%–42%), and 29.3% for neonatal/infantile (95% CI 23%–36%) (Table 1) (Figures S6 to Figure S17 and Figure S22 to Figure S25 in the Supplement). Heterogeneity existed between estimates for epilepsy without ID (I2 = 94%), ASD with ID or DD (I2 = 73%), epilepsy with ID (I2 = 68%), WS (I2 = 68%), and other DEEs (I2 = 76%). Among the age at onset subgroups, the heterogeneity between estimates for neonatal/infantile was I2 = 76%, I2 = 89% for childhood, and I2 = 95% for any age. Visual inspection of the funnel plots showed that correction for publication bias was not required (see Section 2).
3.3. Diagnostic yield by sequencing technology
Next, we stratified the study cohorts by sequencing technology (ES, N = 36; panels, N = 73). Random‐effects meta‐analysis showed a diagnostic yield of 27.2% for ES (95% CI 24%–31%) and 22.6% for panels (95% CI 20%–25%) (Table 1 and Figure 4). The mean yield in ES was higher, even though the difference was not statistically significant (27.2% vs 22.6%, P = .071). Heterogeneity existed between estimates for ES (I2 = 83%) and panels (I2 = 92%).
Figure 4.
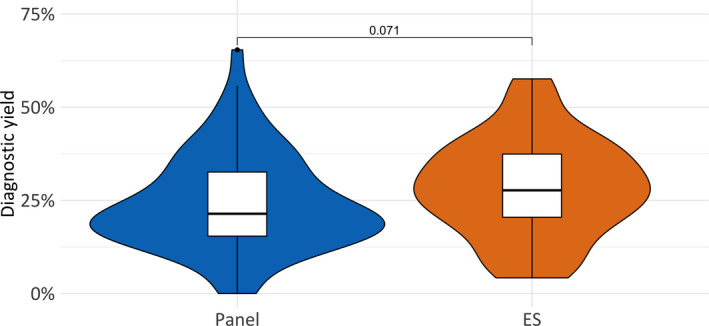
Diagnostic yield by sequencing technology. The mean yield in ES is higher compared to panel testing, even though the difference is not statistically significant‐ 27.2% vs 22.6% (P = .071). Abbreviations: ES, exome sequencing; panel, targeted gene panel sequencing
3.4. Diagnostic sequencing utilization and variant interpretation over time
Diagnostic sequencing in neurodevelopmental disorders has only recently been introduced into clinical practice, and its adaptation across global regions has not been assessed. We grouped all countries with reported diagnostic sequencing studies (N = 25), and all countries without reported diagnostic sequencing studies (N = 125). The first authors' affiliation was considered the studies origin. We then calculated the median IHDI for each group. Countries with sequencing studies had a significantly higher median IHDI, compared to countries without sequencing studies (IHDI median 0.84, IQR = 0.09 vs 0.56, IQR = 0.3, P = 5.8 × 10‐11) (Figure 5). Overall, 83% of all published studies originated from countries with an IHDI of 0.71 and higher, corresponding to countries with a high to very high socioeconomic index. China (IHDI = 0.636), as one of only three countries with a moderate IHDI, has shown rapid growth in sequencing study output and is responsible for over 15% of all published studies.
Figure 5.
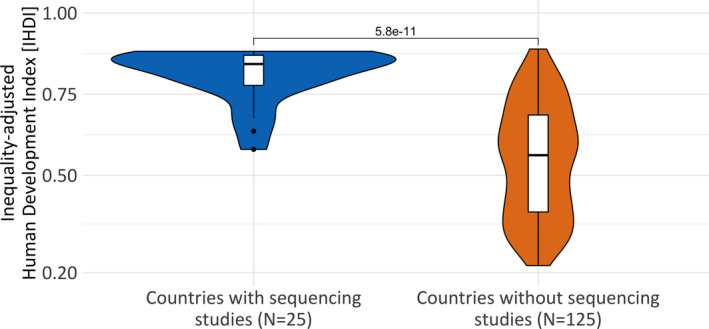
Inequality‐adjusted Human Development Index (IHDI) by country group. Countries with sequencing studies have a significantly higher median Inequality‐adjusted Human Development Index (IHDI), compared to countries without sequencing studies 0.84 vs 0.56 (P = 5.8 × 10‐11)
We determined the number of genes being reported by disorder per year to demonstrate the continual discovery of new genes. We found an upward trend for the number of genes with pathogenic variants being reported (Figure S26 in the Supplement). However, not all genetic variants identified in a diagnostic test are pathogenic. 10 , 22 Interpretation guidelines have been developed by the community, leading to the implementation of the ACMG guidelines in 2015. We examined whether investigators applied the guidelines in NDD sequencing studies. Most studies started to apply the ACMG guidelines in 2016, a year after the original publication (Figure S27A in the Supplement). At the same time, we observed an increase in studies that reported variants of uncertain significance (VUS), starting at around 20% of all reported variants in 2014 and reaching 70% in 2020 (Figure S27B in the Supplement). The number of studies reporting VUS has increased significantly after the introduction of the ACMG guidelines (OR = 38.6, P = 5.2 × 10−14, Fisher's exact test). Only two studies reported on VUS before 2016, whereas 45 studies reported on VUS after 2016 (Figure S27D in the Supplement).
4. DISCUSSION
Here we present the largest systematic review and meta‐analysis of clinical diagnostic sequencing in individuals with neurodevelopmental disorders. We identified 103 studies representing 32,331 individuals with epilepsy, ASD, or ID, and observed a cross‐neurodevelopmental disorder diagnostic yield of 23.7%. Our diagnostic yield of 17.1% for ASD, identified from 14 studies, corresponds to the diagnostic yield of 16% reported in a recent systematic meta‐analysis of five studies. 8 Our 24% combined diagnostic yield from panel and ES for epilepsy (N = 72 studies) is similar to 23% yield from panels (N = 9 studies) reported in a recent systematic meta‐analysis. 9 However, our diagnostic yield for ES was lower than the yield reported in the same study (27.2% vs 45%). 9 Although the observed diagnostic yield of 28.2% for ID was the highest in our disorder analysis, the yield is lower than reported previously (39%). 8 In addition, recent articles report genetic testing diagnostic yields of up to 55%–70% for individuals with ID. 23
The study composition of this systematic review could explain the lower diagnostic yield for ES and epilepsy. Compared to the previous studies, the number of studies included in our systematic review was three to four times larger. Furthermore, we only included studies with at least 20 participants to increase statistical accuracy, which was not done by the previous systematic meta‐analyses. This restriction could explain why our reported diagnostic yield is lower compared to other studies. Finally, we also included panel‐based studies, whereas, the previous systematic meta‐analysis on NDDs focused solely on ES data. 8
In our sequencing method comparison, ES generated only suggestively higher yields than panel testing (27.2% vs 22.6%, P = .071). The difference was less pronounced compared with two previous systematic meta‐analyses. 8 , 9 The observed yield for ES in our meta‐analysis (27.2% in 36 studies) was in line with the estimated yield in a recent smaller systematic meta‐analysis for NDDs without epilepsy (31% in 21 studies). 8 However, the true diagnostic yield of ES may be higher when excluding ES performed in patients tested initially as negative with panel sequencing. In addition, ES enables reanalysis of existent sequencing data to account for new gene discoveries, thus potentially increases in yield over time without resequencing for some patients. Our systematic meta‐analysis showed in between‐study heterogeneity with I2 values ranging between 68% and 95%, in line with previous systematic meta‐analyses. Concerning the high I2 values, our results have to be interpreted with caution because diagnostic yields can vary widely across studies screening patients with apparently similar phenotypes. NDDs represent a clinically heterogeneous group of disorders and differing patient ascertainment criteria could affect the diagnostic yield. Two studies were labeling their cohort as “autism” cohort, could ascertain patients with different subtypes (eg, Asperger syndrome vs Pervasive developmental disorder) without indicating this information in the methods. The heterogeneity in the diagnostic yield of panel testing is likely to reflect the different genes targeted by different panels. 2 , 24 , 25 In addition, it is important to note that the reported yields depend on the patient population and the center in which the data were acquired. The majority of all included studies originated from tertiary care centers where the patient cohorts are frequently highly selected and hence do not represent the general patient population. Such patients are more likely drug‐resistant and can present with an elevated diagnostic yield compared to nonreferred patients. 26 In addition, the high yield for FE is driven by studies that have been performed in pediatric cohorts (top four studies with the highest yields in the FE category), for which higher genetic yields are to be expected.
In the meta‐analysis of all studies by age at onset, only one article represented adolescent‐adult onset epilepsies, Lee et al (2018) 27 , indicating a lack of publications in adolescent‐adult onset epilepsies, which might prevent changes in therapy, prognosis, and better counseling for these patients and their families. 28 , 29 Notably, the Lee et al (2018) 27 study was performed in patients with generalized genetic epilepsy, one of the epilepsy subtypes that is thought to be largely genetic and will accordingly lead to high genetic yields.
Several studies have successfully shown that guidelines are valuable in reanalyzing exome data. 30 , 31 , 32 , 33 , 34 Before variant interpretation guidelines were developed 5 years ago, interpretation was not standardized. We show that, since their implementation, the field is progressively adapting the ACMG guidelines (Figure 3A).
Finally, observe an overall increase in genetic testing, specifically in countries with high and very high socioeconomic indices (IHDI > 0.7). We did not find any study from Latin America, India, or Africa. Apart from the socioeconomic development, a lack in genetic training may add to this disparity in clinical sequencing.
This systematic meta‐analysis should be interpreted in light of several limitations. First, diagnostic yield may be underestimated in some studies that pre‐screened individuals and performed only NGS on patients for which a molecular diagnosis could not be established using standard genetic testing. Second, not all studies used the ACMG variant classification guidelines, which were first implemented in 2015. 10 We also recognize that specific analysis approaches may differ in terms of variant filtering and technical platform (eg, trio‐based ES vs proband‐only ES). Furthermore, the studies included in this comprehensive systematic review and meta‐analysis represent a heterogeneous collection of sampling and data collection methodologies, with sparse descriptive information across all studies. Finally, due to the absence of studies from Africa, India, or Latin America, the generalizability to individuals on a global level remains to be determined. In this study, we focused on the diagnostic yields of particular phenotypes from a disorder perspective. Future studies are needed to compare the phenotypic groups from a gene perspective.
This study represents the largest meta‐analysis investigating diagnostic sequencing yield with three to four times more studies than previous meta‐analyses. In the absence of more extensive studies—excluding non‐systematic reviews—this systematic review and meta‐analysis can guide policymaking and help steer decision‐making in patient management. Alongside policymakers and patients, healthcare providers can also benefit from this comprehensive overview. However, additional randomized controlled studies are still needed. Notably, studies that involve the evidence base for what type of genetic test should be used to provide the best care for the patient. 35
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
AS and DL designed the study. AS, YCL, and EPP analyzed the data. DL supervised the study. AS, CL, and DL wrote the manuscript. All authors interpreted the data and revised the manuscript.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Supplementary Material
Stefanski A, Calle‐López Y, Leu C, Pérez‐Palma E, Pestana‐Knight E, Lal D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta‐analysis. Epilepsia.2021;62:143–151. 10.1111/epi.16755
DATA AVAILABILITY STATEMENT
All relevant data and methods are reported in the article and in the Supporting Information.
REFERENCES
- 1. Shashi V, McConkie‐Rosell A, Rosell B, Schoch K, Vellore K, McDonald M, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next‐generation sequencing for undiagnosed genetic disorders. Genet Med Off J Am Coll Med Genet. 2014;16:176–82. [DOI] [PubMed] [Google Scholar]
- 2. Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50:1048–53. [DOI] [PubMed] [Google Scholar]
- 3. Vissers LELM, van Nimwegen KJM, Schieving JH, Kamsteeg E‐J, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017;19:1055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Truty R, Patil N, Sankar R, Sullivan J, Millichap J, Carvill G, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole‐exome sequencing in children with Autism spectrum disorder. JAMA. 2015;314:895. [DOI] [PubMed] [Google Scholar]
- 6. Xiao B, Qiu W, Ji X, Liu X, Huang Z, Liu H, et al. Marked yield of re‐evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. Am J Med Genet A. 2018;176:107–15. [DOI] [PubMed] [Google Scholar]
- 7. Robinson P, Lowe J. Literature reviews vs systematic reviews. Aust N Z J Public Health. 2015;39(2):103. [DOI] [PubMed] [Google Scholar]
- 8. Srivastava S, Love‐Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta‐analysis and multidisciplinary consensus statement: exome sequencing is a first‐tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21(11):2413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sánchez Fernández I, Loddenkemper T, Gaínza‐Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy. Neurology. 2019;92:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097 Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2707599/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kovalchik S. RISmed: Download Content from NCBI Databases. 2007. Available from https://CRAN.R‐project.org/package=RISmed
- 13.EpilepsyDiagnosis.org [Internet]. [cited 2020]. Available from https://www.epilepsydiagnosis.org/
- 14. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Human Development Data (1990‐2018) . Human Development Reports [Internet]. [cited 2020]. Available from http://hdr.undp.org/en/data
- 16. Human Development Index (HDI) . Human Development Reports [Internet]. [cited 2020]. Available from http://hdr.undp.org/en/content/human‐development‐index‐hdi
- 17. R Core Team . In: R: A Language and Environment for Statistical Computing, version 36 [software] [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2017. Available from https://www.r‐project.org/ [Google Scholar]
- 18. Schwarzer G. meta: An R package for meta‐analysis. R News. 2007;7:40–5. [Google Scholar]
- 19. Wickham H. ggplot2: Elegant Graphics for Data Analysis [Internet]. New York, NY: Springer‐Verlag; 2016. Available from https://ggplot2.tidyverse.org [Google Scholar]
- 20. Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ. 1997;315:629–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duval S, Tweedie R. Trim and Fill: a simple funnel‐plot–based method of testing and adjusting for publication bias in meta‐analysis. Biometrics. 2000;56:455–63. [DOI] [PubMed] [Google Scholar]
- 22. Niestroj L‐M, Du J, Nothnagel M, May P, Palotie A, Daly MJ, et al. Guideline‐based and bioinformatic reassessment of lesion‐associated gene and variant pathogenicity in focal human epilepsies. Epilepsia. 2018;59:2145–52. [DOI] [PubMed] [Google Scholar]
- 23. Vissers LELM, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17:9–18. [DOI] [PubMed] [Google Scholar]
- 24. Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug‐resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017;38:216–25. [DOI] [PubMed] [Google Scholar]
- 25. Yuskaitis CJ, Sheidley BR, Poduri A. Variability among next‐generation sequencing panels for early‐life epilepsies. JAMA Pediatr. 2018;172:779–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, Diadori P, et al. The genetic landscape of infantile spasms. Hum Mol Genet. 2014;23:4846–58. [DOI] [PubMed] [Google Scholar]
- 27. Lee CG, Lee J, Lee M. Multi‐gene panel testing in Korean patients with common genetic generalized epilepsy syndromes. Russo E, editor. PLOS ONE. 2018;13(6):e0199321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lewis‐Smith D, Thomas RH. The prevalence of genetically diagnosable epilepsies in young adulthood: How many should we be looking for? Epilepsia. 2020;61(9):2053–4. [DOI] [PubMed] [Google Scholar]
- 29. Rubboli G, Møller RS, Johannesen KM. Genetic testing in adult epilepsy patients: a call to action for clinicians. Epilepsia. 2020;61(9):2055–6. [DOI] [PubMed] [Google Scholar]
- 30. Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genet Med Off J Am Coll Med Genet. 2018;20:1216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Al‐Nabhani M, Al‐Rashdi S, Al‐Murshedi F, Al‐Kindi A, Al‐Thihli K, Al‐Saegh A, et al. Reanalysis of exome sequencing data of intellectual disability samples: yields and benefits. Clin Genet. 2018;94:495–501. [DOI] [PubMed] [Google Scholar]
- 32. Epilepsy Genetics Initiative . The epilepsy genetics initiative: systematic reanalysis of diagnostic exomes increases yield. Epilepsia. 2019;60:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J, Gao K, Yan H, Xiangwei W, Liu N, Wang T, et al. Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene. 2019;700:168–75. [DOI] [PubMed] [Google Scholar]
- 34. Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med Off J Am Coll Med Genet. 2017;19:209–14. [DOI] [PubMed] [Google Scholar]
- 35. National Academies of Sciences E and Medicine . An Evidence Framework for Genetic Testing [Internet]. Washington, DC: The National Academies Press; 2017. Available from https://www.nap.edu/catalog/24632/an‐evidence‐framework‐for‐genetic‐testing [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
All relevant data and methods are reported in the article and in the Supporting Information.