

Abstract
Ultrasound (US) produces cavitation‐induced mechanical forces stretching and breaking polymer chains in solution. This type of polymer mechanochemistry is widely used for synthetic polymers, but not biomacromolecules, even though US is biocompatible and commonly used for medical therapy as well as in vivo imaging. The ability to control protein activity by US would thus be a major stepping‐stone for these disciplines. Here, we provide the first examples of selective protein activation and deactivation by means of US. Using GFP as a model system, we engineer US sensitivity into proteins by design. The incorporation of long and highly charged domains enables the efficient transfer of force to the protein structure. We then use this principle to activate the catalytic activity of trypsin by inducing the release of its inhibitor. We expect that this concept to switch “on” and “off” protein activity by US will serve as a blueprint to remotely control other bioactive molecules.
Keywords: enzymes, fluorescence, mechanochemistry, protein engineering, ultrasound
Proteins are genetically engineered and equipped with long supercharged polypeptide handles, rendering them susceptible to ultrasound. The concept is demonstrated by designing ultrasound‐responsive GFP derivatives the fluorescence of which can be switched “off,” while the activation of catalytic activity is switched “on” by ultrasound using trypsin in conjunction with a protein inhibitor.
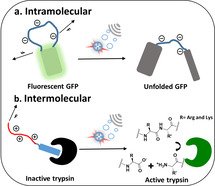
Ultrasound (US) stretches and eventually breaks polymer chains of sufficient contour length [1] by a shear gradient in solution following the collapse of cavitation bubbles.[ 2 , 3 ] By installing predetermined molecular breaking points (mechanophores) into the polymer backbone, these chain‐scission events can be rendered passably selective[ 4 , 5 ] and are popularly exploited to force‐activate functions, such as catalysis,[ 6 , 7 , 8 , 9 , 10 , 11 ] chemical reactions,[ 12 , 13 , 14 ] the release of small molecules,[ 15 , 16 , 17 , 18 , 19 , 20 ] or the optical visualization of material damage.[ 21 , 22 ] However, research in polymer mechanochemistry until now mainly focused on understanding the force‐induced chemical transformations and their impact on material properties. Although force was shown to impact naturally occurring macromolecules both on the single molecule level[ 23 , 24 , 25 ] as well as in cells,[ 26 , 27 ] tissues, [28] and biohybrid materials, [29] the US‐induced activation of biomacromolecules to achieve biological function remained unexplored. We herein display the unprecedented activation of genetically engineered proteins [30] by US firstly on a GFP model system and eventually with the release of an inhibitor of the enzyme trypsin turning “on” its catalytic activity.
To apply the mechanochemical principle to proteins beyond single‐molecule force spectroscopy, we argue that a long and stretchable domain is required, which, we hypothesize, can be genetically engineered into the protein or attached post‐translationally through noncovalent binding. The protein moreover needs to be designed such that either an alteration of the protein structure or the release of the active protein are induced by force.
Extensive US irradiation is destructive to proteins, leading to unfolding, aggregation, or nonspecific bond scission within the peptide backbone.[ 31 , 32 ] Hence, before endowing proteins with the ability to functionally respond to US, we initially identified US‐stable proteins. We selected green fluorescent protein (GFP), as the physical and spectroscopic properties of its many mutants have been characterized extensively. We found, however, that EGFP forms a precipitate when subjected to US at 20 kHz, a power intensity of 7 W cm−2, and a protein concentration of 10 μm (Figure S3). Therefore, we investigated superfolder GFP (sfGFP)—a mutant with increased folding stability.[ 33 , 34 ] For later engineering efforts, we constructed circular permuted variants by inserting a small Glycine–Serine (GS) linker between the native N‐ and C‐termini while introducing the new termini in the loop between the 10th and 11th beta‐strand (GFP–GS).[ 35 , 36 ] This mutant retained both solubility and structure when subjected to US for several minutes, as judged from its absorption, fluorescence, and circular‐dichroism spectra (Figure S4–S6), hence providing a GFP with higher stability against shear forces.
To selectively alter the properties of GFP–GS by US, we replaced the small GS linker with a series of elastin‐like proteins (ELPs), with the pentapeptide repeat unit (VPGXG)n (Scheme 1). [37] Following our hypothesis, these unfolded polypeptide units can be obtained with predictable size and properties and can be stretched akin to a synthetic polymer. [38] The resulting mechanical force would destabilize the 11th beta‐strand that is part of the beta‐barrel, abolishing green fluorescence but retaining the protein secondary structure. We first inserted the neutral (VPGVG)40 domain (GFP–V40), resulting in a protein with a somewhat lower fluorescence quantum yield compared to the parent protein (Table S3). However, US treatment also resulted in precipitation of GFP–V40. Likely, the hydrophobicity of the chain enhanced aggregate formation (Figure S11).
Scheme 1.
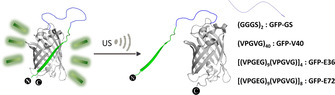
Schematic representation of US‐induced unfolding of GFP–SUPs.
Thus, we replaced the neutral peptide structure with the more hydrophilic anionic supercharged polypeptide (SUP) unit (VPGEG)36 domain (GFP–E36).[ 39 , 40 , 41 , 42 ] Next to higher hydrophilicity and thus solubility, the repulsion between the negative charges increased the hydrodynamic radius of the protein and resulted in higher sensitivity towards US. Indeed, the GFP–E36 did not precipitate during sonication while the absorbance and fluorescence (Figure 1 a) decreased in a defined manner without changing the secondary structure: The absorbance at 280 nm [43] was found to be even more stable than for GFP–GS and the corresponding CD spectra (Figure 1 c) only showed specific changes upon US exposure, but no precipitation. The 470 nm absorbance of the deprotonated fluorophore was reduced up to 50 % after 8 min US while the 395 nm absorbance of the protonated fluorophore did not change. [44] The decreased absorption can be related to the 50 % drop in fluorescence upon 488 nm excitation. When recording the fluorescence excitation spectra at 515 nm emission, we found that the fluorescence decreased for all excitation wavelengths (Figure S12). Together, this implied that the resulting protonated fluorophore in GFP–E36 occupied a different environment within the protein without green fluorescence upon exposure to US.
Figure 1.
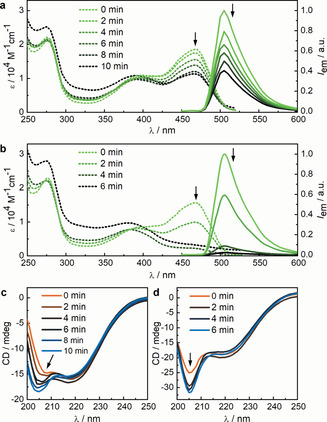
Spectra recorded of GFP–E36 and GFP–E72 at different sonication times. a) Absorption (dashed) and normalized fluorescence emission (solid) spectra of GFP–E36. b) Absorption (dashed) and normalized fluorescence emission (solid) spectra of GFP–E72. c) CD spectra of GFP–E36. d) CD spectra of GFP–E72. The peak at circa 205 nm is attributed to the GFP α‐helices (Figure S21 and S22). All spectra were recorded in PBS buffer (pH 7.4) at room temperature, and protein concentrations were in the range of 10 μm.
To achieve complete quenching of the GFP fluorophore, we increased the contour length of the linker structure between the beta‐sheets by inserting (VPGEG)72, leading to variant GFP–E72. This increase led to a drastic increase in sensitivity: We observed a complete disappearance of the 470 nm absorption and fluorescence (Figure 1 b) upon 4 min of US application. The secondary structure remained, as judged from absorption and CD spectroscopy (Figure 1 d), and the excitation spectra at 515 nm emission disappeared completely (Figure S13). Hence, the GFP–E72 showed comparable but more pronounced susceptibility to US compared to the GFP–E36. [45] Moreover, we found that the absorption band at 395 nm shifted to 380 nm, suggesting a release of the fluorophore from the hydrogen‐bonding network of the beta‐barrel, in accordance with a mechanism where the strand that provided the hydrogen bonding was released from the barrel. [46] Similar to the GFP–E36, US can eventually denature GFP–E72: We observed an increase of the 280 nm band after 6 min sonication, which was indicative of GFP unfolding. To verify that the effect of US on the protein is a consequence of mechanical forces concomitant with the collapse of cavitation bubbles, and not an increase in temperature, we assessed whether the melting temperatures of our proteins were related to the observed sensitivity to US. We found by temperature‐induced unfolding, as monitored with CD spectroscopy, that the melting temperatures of these proteins were similar and did not relate to the effect of US (Figure S7–S10). [47] The reversibility of the process was then investigated by observing the recovery of UV/Vis, fluorescence, and CD spectra of sonicated GFP–E36 over 144 h (Figure S14–S20). A partial but not complete recovery of the optical properties hinted towards reversible and irreversible transitions within the GFP chromophore upon US irradiation. We thus argue to have full control over the protein structure with US, by engineering long charged hydrophilic domains into an otherwise US‐stable protein (Figure 2).
Figure 2.
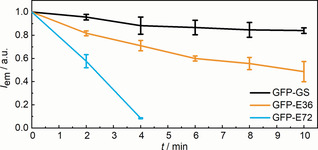
US‐responsive circularly permuted GFP system. Dependence of fluorescence quenching on the size of the SUPs inserted into GFPs and sonication time. Mean values ±SD from the mean, N=3 independent experiments.
Having established proof of concept and identified design criteria, we sought to apply this principle to switch “on” protein activity. Therefore, we fused the long charged domain to a peptide that inhibits an enzyme: Application of US would break the protein–protein interaction between enzyme and inhibitor releasing the active enzyme. We selected the well‐studied combination of trypsin and bovine pancreatic trypsin inhibitor (BPTI). Because BPTI itself has a net positive charge, we fused it to the cationic SUP (VPGKG)36 domain (K36)[ 48 , 49 ] to prevent it from blocking BPTI's function. The disordered domain was placed at the C‐terminus of BPTI. To improve expression and folding, we fused small ubiquitin‐related modifier (SUMO) protein to the N‐terminus of BPTI, which can be removed by SUMO protease. [50] Importantly, both termini are on the opposite side of the trypsin interaction domain of BPTI (Scheme 2).
Scheme 2.
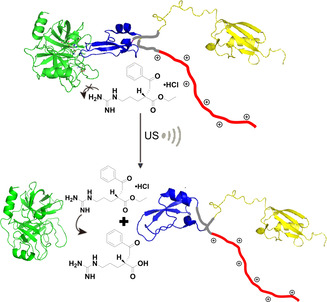
Concept of disassembly of the trypsin (green)‐SUMO (yellow)‐BPTI (blue)‐K36 (red) complex by US, which can be followed with the substrate BAEE, hydrolysis of which yields a product absorbing at 253 nm.
To confirm that the new domains do not influence binding between BPTI and trypsin, we determined the binding affinities by isothermal titration calorimetry (ITC). We observed that both the SUMO and K36 domains reduce the binding affinity, although the affinities with dissociation constants of circa 10−8 m are still higher than trypsin with most of its substrates (Figure S24–S26). [51]
We determined the activity of trypsin from the turnover of its substrate N α‐benzoyl‐l‐arginine ethyl ester (BAEE), which results in absorbance at 253 nm. This leads to complete digestion within 2 min, with a specific activity of 40 BAEE units mL−1 trypsin, while the presence of any of our BPTI derivatives completely inhibits the reaction. We next applied 30 or 60 s of US to the complex of trypsin deactivated with 4 equivalents excess of SUMO‐BPTI‐K36. In these experiments, we added the substrate prior to sonication because trypsin otherwise rebinds the BPTI in competition with the substrate during or after US application. We observed the activation of the trypsin, showing that the mechanical force can remove the inhibitor from the trypsin. The turnover of trypsin after 30 and 60 s US is 4.8 and 6.9 units mL−1 trypsin, respectively, which corresponds to approximately 12 and 17 % of the activity of trypsin in the absence of BPTI (Figure 3). Importantly, trypsin is only negligibly affected by the employed sonication conditions (Figure S28–29). The effect of US is a direct consequence of the K36 domain because the binding of SUMO–BPTI without K36 is not affected by sonication. This confirms our hypothesis that a long hydrophilic polypeptide chain renders proteins active towards shear force in solution. Accordingly, removing the SUMO from the BPTI‐K36 did not change the sensitivity to US, resulting in activities of 10 and 12 % of free trypsin (Figure S32). Concomitantly, increasing BPTI concentration decreased overall turnover (Figure S33). We thus show that by introducing a long hydrophilic domain into a protein inhibitor, we can remotely activate an enzyme by US.
Figure 3.
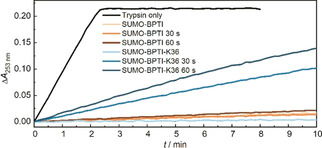
The enzymatic activity of trypsin, trypsin‐SUMO‐BPTI, and trypsin‐SUMO‐BPTI‐K36 complexes for different times of sonication. SUMO–BPTI inhibits the enzymatic reaction without and with sonication (shades of brown). Similarly, SUMO‐BPTI‐K36 abolishes the catalytic activity of trypsin without application of US (light blue). In contrast, the complexes consisting of trypsin and SUMO‐BPTI‐K36 can be disassembled by US within seconds turning on enzyme activity (shades of blue). The x‐axis represents the catalysis time; the individual traces correspond to different preceding sonication times. The plateau of pure trypsin catalytic activity is associated to complete substrate depletion.
Altogether, we established the first molecular concept to activate and deactivate the optical and catalytic activities of genetically engineered proteins by employing US, and thus shear force in solution, as a trigger. Using external stimuli to control protein function has become extremely relevant, as evidenced by the growing field of optogenetics and its impact on neuroscience and other areas.[ 52 , 53 ] In analogy, we established here the first molecular designs towards sonogenetics, the US‐enabled control of protein activity. With genetic engineering as a synthetic tool and the introduction of supercharged, unfolded polypeptide chains, proteins were rendered susceptible to shear force. To demonstrate the broad applicability of this approach, two design concepts to gain control over protein activity were successfully realized employing different enzymes and switching protein activity “on” and “off”. Our ability to tune the sensitivity of proteins towards US holds great promise for future applications that may lie in remote control of protein activity in vivo.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by The European Union (European Research Council Advanced Grant SUPRABIOTICS No. 694610) and China Scholarship Council to Y.Z. R.G. is grateful for support by a Freigeist‐Fellowship of the Volkswagen Foundation (No. 92888). Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine‐Westphalia (No. 300088302). Open access funding enabled and organized by Projekt DEAL.
Y. Zhou, S. Huo, M. Loznik, R. Göstl, A. J. Boersma, A. Herrmann, Angew. Chem. Int. Ed. 2021, 60, 1493.
References
- 1. May P. A., Munaretto N. F., Hamoy M. B., Robb M. J., Moore J. S., ACS Macro Lett. 2016, 5, 177–180. [DOI] [PubMed] [Google Scholar]
- 2. Cravotto G., Gaudino E. C., Cintas P., Chem. Soc. Rev. 2013, 42, 7521–7534. [DOI] [PubMed] [Google Scholar]
- 3. Stratigaki M., Göstl R., ChemPlusChem 2020, 85, 1095–1103. [DOI] [PubMed] [Google Scholar]
- 4. Brown C. L., Craig S. L., Chem. Sci. 2015, 6, 2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akbulatov S., Boulatov R., ChemPhysChem 2017, 18, 1422–1450. [DOI] [PubMed] [Google Scholar]
- 6. Piermattei A., Karthikeyan S., Sijbesma R. P., Nat. Chem. 2009, 1, 133–137. [DOI] [PubMed] [Google Scholar]
- 7. Jakobs R. T. M., Sijbesma R. P., Organometallics 2012, 31, 2476–2481. [Google Scholar]
- 8. Groote R., Jakobs R. T. M., Sijbesma R. P., Polym. Chem. 2013, 4, 4846–4859. [Google Scholar]
- 9. Michael P., Binder W. H., Angew. Chem. Int. Ed. 2015, 54, 13918–13922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14124–14128. [Google Scholar]
- 10. Michael P., Biewend M., Binder W. H., Macromol. Rapid Commun. 2018, 39, 1800376. [DOI] [PubMed] [Google Scholar]
- 11. Funtan S., Funtan A., Paschke R., Binder W. H., Org. Mater. 2020, 02, 116–128. [Google Scholar]
- 12. Ramirez A. L. B., Kean Z. S., Orlicki J. A., Champhekar M., Elsakr S. M., Krause W. E., Craig S. L., Nat. Chem. 2013, 5, 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Gao F., Cao X., Li Y., Xu Y., Weng W., Boulatov R., Angew. Chem. Int. Ed. 2016, 55, 3040–3044; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3092–3096. [Google Scholar]
- 14. Verstraeten F., Göstl R., Sijbesma R. P., Chem. Commun. 2016, 52, 8608–8611. [DOI] [PubMed] [Google Scholar]
- 15. Diesendruck C. E., Steinberg B. D., Sugai N., Silberstein M. N., Sottos N. R., White S. R., Braun P. V., Moore J. S., J. Am. Chem. Soc. 2012, 134, 12446–12449. [DOI] [PubMed] [Google Scholar]
- 16. Larsen M. B., Boydston A. J., J. Am. Chem. Soc. 2013, 135, 8189–8192. [DOI] [PubMed] [Google Scholar]
- 17. Larsen M. B., Boydston A. J., J. Am. Chem. Soc. 2014, 136, 1276–1279. [DOI] [PubMed] [Google Scholar]
- 18. Nagamani C., Liu H., Moore J. S., J. Am. Chem. Soc. 2016, 138, 2540–2543. [DOI] [PubMed] [Google Scholar]
- 19. Hu X., Zeng T., Husic C. C., Robb M. J., J. Am. Chem. Soc. 2019, 141, 15018–15023. [DOI] [PubMed] [Google Scholar]
- 20. Lin Y., Kouznetsova T. B., Craig S. L., J. Am. Chem. Soc. 2020, 142, 99–103. [DOI] [PubMed] [Google Scholar]
- 21. Calvino C., Neumann L., Weder C., Schrettl S., J. Polym. Sci. Part A J. Polym. Sci. A 2017, 55, 640–652. [Google Scholar]
- 22. Göstl R., Clough J. M., Sijbesma R. P., in Mechanochemistry in Materials, Royal Society Of Chemistry, London, 2017, pp. 53–75. [Google Scholar]
- 23. Butera D., Passam F., Ju L., Cook K. M., Woon H., Aponte-Santamaría C., Gardiner E., Davis A. K., Murphy D. A., Bronowska A., Luken B. M., Baldauf C., Jackson S., Andrews R., Gräter F., Hogg P. J., Sci. Adv. 2018, 4, eaaq1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goktas M., Luo C., Sullan R. M. A., Bergues-Pupo A. E., Lipowsky R., Verde A. V., Blank K. G., Chem. Sci. 2018, 9, 4610–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. López-García P., Goktas M., Bergues-Pupo A. E., Koksch B., Silva D. V., Blank K. G., Phys. Chem. Chem. Phys. 2019, 21, 9145–9149. [DOI] [PubMed] [Google Scholar]
- 26. Grashoff C., Hoffman B. D., Brenner M. D., Zhou R., Parsons M., Yang M. T., McLean M. A., Sligar S. G., Chen C. S., Ha T., Schwartz M. A., Nature 2010, 466, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Michels L., Gorelova V., Harnvanichvech Y., Borst J. W., Albada B., Weijers D., Sprakel J., Proc. Natl. Acad. Sci. USA 2020, 117, 18110–18118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zapp C., Obarska-Kosinska A., Rennekamp B., Kurth M., Hudson D. M., Mercadante D., Barayeu U., Dick T. P., Denysenkov V., Prisner T., Bennati M., Daday C., Kappl R., Gräter F., Nat. Commun. 2020, 11, 2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grad E. M., Tunn I., Voerman D., de Léon A. S., Hammink R., Blank K. G., Front. Chem. 2020, 8, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeymer C., Hilvert D., Annu. Rev. Biochem. 2018, 87, 131–157. [DOI] [PubMed] [Google Scholar]
- 31. Stathopulos P. B., Scholz G. A., Hwang Y.-M., Rumfeldt J. A. O., Lepock J. R., Meiering E. M., Protein Sci. 2004, 13, 3017–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Leo V., Catucci L., Di Mauro A. E., Agostiano A., Giotta L., Trotta M., Milano F., Ultrason. Sonochem. 2017, 35, 103–111. [DOI] [PubMed] [Google Scholar]
- 33. Cabantous S., Terwilliger T. C., Waldo G. S., Nat. Biotechnol. 2005, 23, 102–107. [DOI] [PubMed] [Google Scholar]
- 34. Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S., Nat. Biotechnol. 2006, 24, 79–88. [DOI] [PubMed] [Google Scholar]
- 35. Baird G. S., Zacharias D. A., Tsien R. Y., Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heinemann U., Hahn M., Prog. Biophys. Mol. Biol. 1995, 64, 121–143. [DOI] [PubMed] [Google Scholar]
- 37. Roberts S., Dzuricky M., Chilkoti A., FEBS Lett. 2015, 589, 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schaefer M., Icli B., Weder C., Lattuada M., Kilbinger A. F. M., Simon Y. C., Macromolecules 2016, 49, 1630–1636. [Google Scholar]
- 39. Liu K., Pesce D., Ma C., Tuchband M., Shuai M., Chen D., Su J., Liu Q., Gerasimov J. Y., Kolbe A., Zajaczkowski W., Pisula W., Müllen K., Clark N. A., Herrmann A., Adv. Mater. 2015, 27, 2459–2465. [DOI] [PubMed] [Google Scholar]
- 40. Yang H., Ma C., Li K., Liu K., Loznik M., Teeuwen R., van Hest J. C. M., Zhou X., Herrmann A., Wang J., Adv. Mater. 2016, 28, 5008–5012. [DOI] [PubMed] [Google Scholar]
- 41. Han J., Ma C., Wang B., Bender M., Bojanowski M., Hergert M., Seehafer K., Herrmann A., Bunz U. H. F., Chem 2017, 2, 817–824. [Google Scholar]
- 42. Zhang L., Ma C., Sun J., Shao B., Portale G., Chen D., Liu K., Herrmann A., Angew. Chem. Int. Ed. 2018, 57, 6878–6882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6994–6998. [Google Scholar]
- 43. Clérico E. M., Ermácora M. R., Arch. Biochem. Biophys. 2001, 395, 215–224. [DOI] [PubMed] [Google Scholar]
- 44. Chattoraj M., King B. A., Bublitz G. U., Boxer S. G., Proc. Natl. Acad. Sci. USA 1996, 93, 8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miesenböck G., De Angelis D. A., Rothman J. E., Nature 1998, 394, 192–195. [DOI] [PubMed] [Google Scholar]
- 46. Niwa H., Inouye S., Hirano T., Matsuno T., Kojima S., Kubota M., Ohashi M., Tsuji F. I., Proc. Natl. Acad. Sci. USA 1996, 93, 13617–13622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nicholls S. B., Hardy J. A., Protein Sci. 2013, 22, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. te Brinke E., Groen J., Herrmann A., Heus H. A., Rivas G., Spruijt E., Huck W. T. S., Nat. Nanotechnol. 2018, 13, 849–855. [DOI] [PubMed] [Google Scholar]
- 49. Veeregowda D. H., Kolbe A., van der Mei H. C., Busscher H. J., Herrmann A., Sharma P. K., Adv. Mater. 2013, 25, 3426–3431. [DOI] [PubMed] [Google Scholar]
- 50. Sun Z., Lu W., Jiang A., Chen J., Tang F., Liu J.-N., Protein Expression Purif. 2009, 65, 238–243. [DOI] [PubMed] [Google Scholar]
- 51. Gardner Q.-A. A., Younas H., Akhtar M., Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 182–190. [DOI] [PubMed] [Google Scholar]
- 52. Repina N. A., Rosenbloom A., Mukherjee A., Schaffer D. V., Kane R. S., Annu. Rev. Chem. Biomol. Eng. 2017, 8, 13–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim C. K., Adhikari A., Deisseroth K., Nat. Rev. Neurosci. 2017, 18, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary